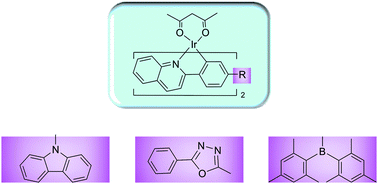
The molecular geometries, electronic structures, photophysical properties, charge-injection and -transporting abilities of a series of Ir(III) complexes with different carrier-transporting substituents, such as carbazole, oxadiazole and dimesitylboryl groups, are investigated theoretically using density functional theory (DFT) and time-dependent DFT (TDDFT) calculations to understand the influence of these substituents on the optical and electronic properties of Ir(III) complexes and to explore how to improve the optoelectronic properties of the complexes. It is found that the introduction of substituents can stabilize both HOMOs and LUMOs and induce variations in the energy gap between HOMO and LUMO. The introduction of hole-transporting carbazole substituent induces the blue-shift of absorption spectrum and improves the hole-injection and -transporting performances of complex. The introduction of electron-transporting oxadiazole substituent and electron-accepting dimesitylboryl substituent induces the red-shift in absorption spectra of complexes, improves their charge transfer abilities and leads to the better balance between the hole- and electron-transporting abilities. Through Lewis acid/base interactions between B atom and F−, the electronic properties of 4 show dramatic changes in the presence of F− and thus 4 can also be used as selective phosphorescent F− probe.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


