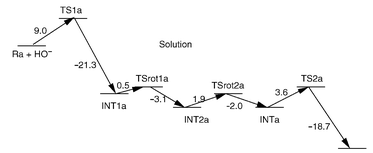
Ab initio molecular orbital calculations were carried out to study reaction pathways and evaluate energy barriers for alkaline hydrolysis of O,O-diethyl p-nitrophenyl phosphate (paraoxon), diisopropyl phosphorofluoridate (DFP), O-isopropyl methylphosphonofluoridate (sarin), O-pinacolyl methylphosphonofluoridate (soman) and O,O-dimethyl phosphorofluoridate. This is the first computational study on the reaction pathways for hydrolysis of phosphotriesters and their structural variants that have small pKa values for the leaving groups. The reaction coordinate calculations reveal that the hydrolysis of DFP, sarin, soman and O,O-dimethyl phosphorofluoridate proceeds through the attack of hydroxide ion at the phosphorus center to form a pentacoordinate intermediate and the decomposition of the intermediate through F leaving. The hydrolysis of paraoxon was found to occur by
a one-step process, which strongly supports the conclusion, based on the previously observed primary and secondary 18O isotope effects, of an SN2-like transition state with the absence of a stable pentacoordinate intermediate during the paraoxon hydrolysis. The energy barriers calculated in the gas phase for the pentacoordinate intermediate decomposition step are slightly lower than for the formation step for DFP and sarin, and are higher for soman and O,O-dimethyl phosphorofluoridate. Solvent effects significantly increase the energy barriers for the pentacoordinate intermediate formation step and significantly decrease the energy barriers for the decomposition step. Thus, the pentacoordinate intermediate formation step of hydrolysis in aqueous solution is always rate-determining. For the hydrolysis of all the compounds examined in this study, the energy barrier, 0.8 kcal mol−1, calculated for the paraoxon hydrolysis is the lowest
in the gas phase, whereas the corresponding barrier, 10.1 kcal mol−1, is the highest in aqueous solution. For the hydrolysis of sarin in aqueous solution, for which the experimental activation energy is available, the calculated energy barrier of 8.6 kcal mol−1 for the pentacoordinate intermediate formation (i.e. the rate-determining step) is in good agreement with the experimental activation energy of 9.1 kcal mol−1. Finally, the relative magnitudes of the energy barriers calculated for the alkaline hydrolysis were qualitatively compared with the relative magnitudes of the available experimental rate constants reported for the corresponding phosphotriesterase-catalyzed hydrolysis, and were employed to discuss substituent effects.

 Please wait while we load your content...
Please wait while we load your content...