
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile construction of an anthracene-decorated highly luminescent coordination polymer for the selective detection of explosive nitroaromatics and the mutagenic pollutant TNP†
Ersad Hossaina,
Abhijit Hazra bc,
Sourav Dattabd,
Samim Khand,
Samit Pramanika,
Priyabrata Banerjee*bc,
Mohammad Hedayetullah Mir*d and
Subrata Mukhopadhyaya
bc,
Sourav Dattabd,
Samim Khand,
Samit Pramanika,
Priyabrata Banerjee*bc,
Mohammad Hedayetullah Mir*d and
Subrata Mukhopadhyaya
aDepartment of Chemistry, Jadavpur University, Kolkata 700 032, India
bElectric Mobility & Tribology Research Group, CSIR-Central Mechanical Engineering Research Institute, Mahatma Gandhi Avenue, Durgapur 713 209, India. E-mail: pr_banerjee@cmeri.res.in
cAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201 002, India
dDepartment of Chemistry, Aliah University, New Town, Kolkata 700 160, India. E-mail: chmmir@gmail.com
First published on 2nd January 2024
Abstract
Explosive nitroaromatic compounds (epNACs) are a group of chemicals that have caused significant human casualties through terrorist attacks and they also pose health risks. For the benefit of homeland security and environmental health, there is room for advancing research on the precise detection of epNACs. Coordination polymers (CPs) successfully serve this purpose because of their binding abilities and quenching capabilities. In this regard, a one-dimensional (1D) CP [Zn(bdc)(avp)2(H2O)]n (1; H2bdc = 1,4-benzenedicarboxylic acid and avp = 4-[2-(9-anthryl)vinyl]pyridine) was synthesized, which remarkably demonstrated extremely efficient ratiometric and selective sensing capacity toward epNACs and the mutagenic pollutant 2,4,6-trinitrophenol (TNP) with a quick response. Density functional theory (DFT) calculations provided a thorough analysis of the mechanistic routes behind the quenching reaction. Herein, geometrically accessible interaction sites were strategically decorated using anthracene moieties, allowing the quick and precise detection of explosive nitro derivatives and the carcinogenic pollutant TNP with increased sensitivity.
Introduction
Over the past few decades, application-based research has received great demand for the fabrication of next-generation materials. Following Alfred Werner's groundbreaking investigation, coordination compounds have become a dynamic player in the world of materials.1 Additionally, coordination polymers (CPs)/metal–organic frameworks (MOFs), which are polymeric forms of coordination motifs, have attracted significant attention due to their exciting structural features and potential multifunctional applications.2–7 In this regard, there is a relation between the structural layout, properties, and applications of the CPs, as the structural architectures can be modified to impose the desired qualities by carefully combining organic ligands with inorganic metal ions. In recent years, a large number of CPs have been developed as chemosensors for their suitability in the detection of molecules, anions, or cations that are present in a given medium, such as a solution, air, drinking water, blood, tissue, and waste effluents.8–16 Several methodologies are available in the field of recognition involving chemosensors, such as atomic absorption spectroscopy (AAS), spectrophotometry, chromatography, titrimetry, inductively coupled plasma atomic emission spectrometry (ICP-AES), inductively coupled plasma mass spectrometry (ICP-MS), fluorescence spectrometry, fluorimetry, potentiometry, and X-ray fluorescence spectrometry. Of these, the fluorescence approach rises to the top to serve sensing activities due to its quick response, high sensitivity, low cost, user-friendly operation, and capacity to evaluate genuine samples.17–19 In this aspect, luminous CPs/MOFs play a pivotal role in serving as chemosensors because of their binding capabilities with compounds that can be quenched via the energy or electron transfer processes.20–23Explosive nitroaromatic compounds (epNACs) are significant chemical reagents and have several uses in the chemical industry; however, they have the potential to produce explosions and cause environmental harm. Therefore, a quick and accurate detection of epNACs is essential to protect us from terrorist attacks.24–26 In addition, the NACs produced by industry and chemical research laboratories pose a serious risk to living beings and environmental systems. The long-term accumulation of NACs may cause a number of health problems, including lung ailments, asphyxiation, and cancer.27–31 Therefore, a new search is also underway to detect these NACs with appropriate size, shape, morphology, and surface area of the chemosensors based on CPs.32–34 In this regard, anthracene-based CPs are particularly important and have advantages over other luminescent CPs for sensing traces of NACs because of their capacity to exhibit receptor–analyte interactions via π⋯π stacking leading to enhanced photophysical properties.35–41 However, the design of anthracene-based CPs acting as chemosensors remains a challenge owing to their steric hindrances with surrounding species. Very few anthracene-based CPs are available that exhibit sensitivity toward NACs.42
Accordingly, an effective strategy for producing luminescent CPs involves the utilization of highly conjugated ligands to attain a stable polymeric network. An illustration of such a desirable ligand system, 4-[2-(9-anthryl)vinyl]pyridine (avp) can be a potential contender where the pyridyl ring of avp may be coordinated to the metal node with a free anthracene moiety. Further, the interdigitation of these anthracene moieties could assist in achieving higher dimensional CP through various supramolecular interactions. Therefore, adhering to these facts, avp containing an explicit one-dimensional (1D) luminescent CP [Zn(bdc)(avp)2(H2O)]n (1; H2bdc = 1,4-benzenedicarboxylic acid) was developed. The structural analysis suggests that the contrived CP 1 generates a 1D chain propagating along the c axis, while the avp ligands dangle from the Zn-centre at both sides of the chain, providing suitable spatial orientation for π⋯π interactions among the adjacent anthracene ring. This intricate structural feature leads to distinct photophysical properties, thereby prompting an investigation into the recognition ability of epNACs. Besides, the instances of luminescent CPs proficient at distinctive, selective detection of the trace-level TNP within a pool of epNACs for point-of-care testing applications are exceedingly infrequent in the literature. The synthesized chemosensor material 1 exhibits remarkably sharp and expeditious responses, indicating its potential for real-time detection of TNP among various epNACs.
Experimental section
Materials and methods
All reagent-grade chemicals were purchased for the synthesis and were used as received. A PerkinElmer 240C elemental analyzer was used for carbon, hydrogen, and nitrogen elemental analysis. A PerkinElmer FT-IR spectrum RX1 spectrometer was used to record the infrared spectrum (IR) of the material using the KBr method (4000–500 cm−1). Fluorescence spectra were recorded using a PerkinElmer LS-45 fluorometer. With 1 mg of CP 1 dispersed in 5 mL of acetonitrile (ACN) solvent, the luminous properties were examined in a dispersive state for further investigation of the photophysical characteristics and sensitivity investigations. Different NACs were used in the sensing study, including 4-nitrophenol (NP), nitrobenzene (NB), 3,5-dinitrobenzoic acid (3,5-DNBA), 2,4,6-trinitrophenol (TNP), 4-nitrobenzoic acid (NBA), nitromethane (NM), 2,4-dinitrophenol (2,4-DNP) and 2,4-dinitrotoluene (2,4-DNT) in the solution of 10−4 M can.Synthesis of compound 1
First, the avp ligand was synthesized following the literature method.43 Then, in a test tube, a solution of avp (0.113 g, 0.4 mmol) in DMF (2 mL) was slowly and carefully layered to MeOH (2 mL) using 2 mL 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) solution of MeOH and H2O in Zn(NO3)·6H2O (0.059 g, 0.2 mmol). Afterwards, a solution of H2bdc (0.023 g, 0.2 mmol) was neutralized with Et3N (0.021 g, 0.2 mmol) and a 2 mL EtOH was layered on it. After a few days, light yellow coloured block-shaped crystals of {[Zn(bdc)(avp)2(H2O)]}n (1) were obtained (0.094 g, yield 60%). Elemental analysis (%) calcd for C50H36N2O5Zn: C, 74.12; H, 4.48; N, 3.46; found: C, 74.3; H, 4.2; N, 3.7. IR (KBr pellet, cm−1): 1557 νas (COO−), 1386 νsys (COO−).
:1 (v/v) solution of MeOH and H2O in Zn(NO3)·6H2O (0.059 g, 0.2 mmol). Afterwards, a solution of H2bdc (0.023 g, 0.2 mmol) was neutralized with Et3N (0.021 g, 0.2 mmol) and a 2 mL EtOH was layered on it. After a few days, light yellow coloured block-shaped crystals of {[Zn(bdc)(avp)2(H2O)]}n (1) were obtained (0.094 g, yield 60%). Elemental analysis (%) calcd for C50H36N2O5Zn: C, 74.12; H, 4.48; N, 3.46; found: C, 74.3; H, 4.2; N, 3.7. IR (KBr pellet, cm−1): 1557 νas (COO−), 1386 νsys (COO−).
Single crystal X-ray crystallography
A Bruker SMART APEX II diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å) was used for obtaining single crystal data on compound 1 with acceptable dimensions. The molecular structure was solved using the SHELXT 2014/4 structure solution program.44 Least squares refinements of all reflections within the hkl range −7 ≤ h ≤ 7, −19 ≤ k ≤ 19, −27 ≤ l ≤ 27 were used to figure out the unit cell parameters and crystal-orientation matrices. The collected data (I > 2σ(I)) was integrated using the SAINT45 program, and the absorption correction was performed using SADABS.46 Anisotropic thermal parameters were used to refine non-hydrogen atoms. Each hydrogen atom was positioned in its geometrically ideal location and forced to ride on its parent atom. Table S1 (ESI†) provides a summary of the crystallographic data for compound 1, and Table S2 (ESI†) lists the specific bond lengths and bond angles.Results and discussion
Structural descriptions
X-ray single crystal diffraction study revealed that the compound 1 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 2. The Zn(II) centre adopts a distorted octahedral coordination environment with the ZnO4N2 core. Here, two O atoms from a chelating bdc (Zn1–O1, 2.175(4); Zn1–O2, 2.342(4) Å), one O from a monodentate bdc (Zn1–O3, 1.965(3) Å) and one O atom from an aqua ligand (Zn1–O3, 1.020(3) Å) form the equatorial plane, while the two N atoms from the two avp ligands occupy the axial positions (Zn1–N1, 2.205(3); Zn1–N2, 2.208(3) Å) to complete the octahedral geometry (Fig. 1a). The Zn(II) centres are connected with the 1,4-bdc ligands via the carboxylate group resulting in a 1D chain polymer propagated along the c axis (Fig. 1b). The 1D chains are assembled via the intermolecular hydrogen bonding between the aqua ligand and the O atom of a monodentate bdc ligand (Fig. 1c) with the O⋯O separation in the range of 2.612(3)–2.694(3) Å, thus fabricating a 2D layer in ac plane. The avp ligands attached to Zn(II) centre in the axial positions are aligned in parallel among the adjacent chains and undergo π⋯π stacking interactions with centroid⋯centroid distance of 3.629(3)–3.664(3) Å among the aromatic rings. The avp ligands of the neighbouring layers further undergo C–H⋯π interactions along the b-axis to generate a 3D supramolecular network structure (Fig. S1, ESI†). Here, the distance between the centres of the neighbouring C
with Z = 2. The Zn(II) centre adopts a distorted octahedral coordination environment with the ZnO4N2 core. Here, two O atoms from a chelating bdc (Zn1–O1, 2.175(4); Zn1–O2, 2.342(4) Å), one O from a monodentate bdc (Zn1–O3, 1.965(3) Å) and one O atom from an aqua ligand (Zn1–O3, 1.020(3) Å) form the equatorial plane, while the two N atoms from the two avp ligands occupy the axial positions (Zn1–N1, 2.205(3); Zn1–N2, 2.208(3) Å) to complete the octahedral geometry (Fig. 1a). The Zn(II) centres are connected with the 1,4-bdc ligands via the carboxylate group resulting in a 1D chain polymer propagated along the c axis (Fig. 1b). The 1D chains are assembled via the intermolecular hydrogen bonding between the aqua ligand and the O atom of a monodentate bdc ligand (Fig. 1c) with the O⋯O separation in the range of 2.612(3)–2.694(3) Å, thus fabricating a 2D layer in ac plane. The avp ligands attached to Zn(II) centre in the axial positions are aligned in parallel among the adjacent chains and undergo π⋯π stacking interactions with centroid⋯centroid distance of 3.629(3)–3.664(3) Å among the aromatic rings. The avp ligands of the neighbouring layers further undergo C–H⋯π interactions along the b-axis to generate a 3D supramolecular network structure (Fig. S1, ESI†). Here, the distance between the centres of the neighbouring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds is 6.011 Å, indicating no possibility of a photochemical [2 + 2] cycloaddition reaction to occur according to Schmidt's criteria (<4.20 Å).47
C bonds is 6.011 Å, indicating no possibility of a photochemical [2 + 2] cycloaddition reaction to occur according to Schmidt's criteria (<4.20 Å).47
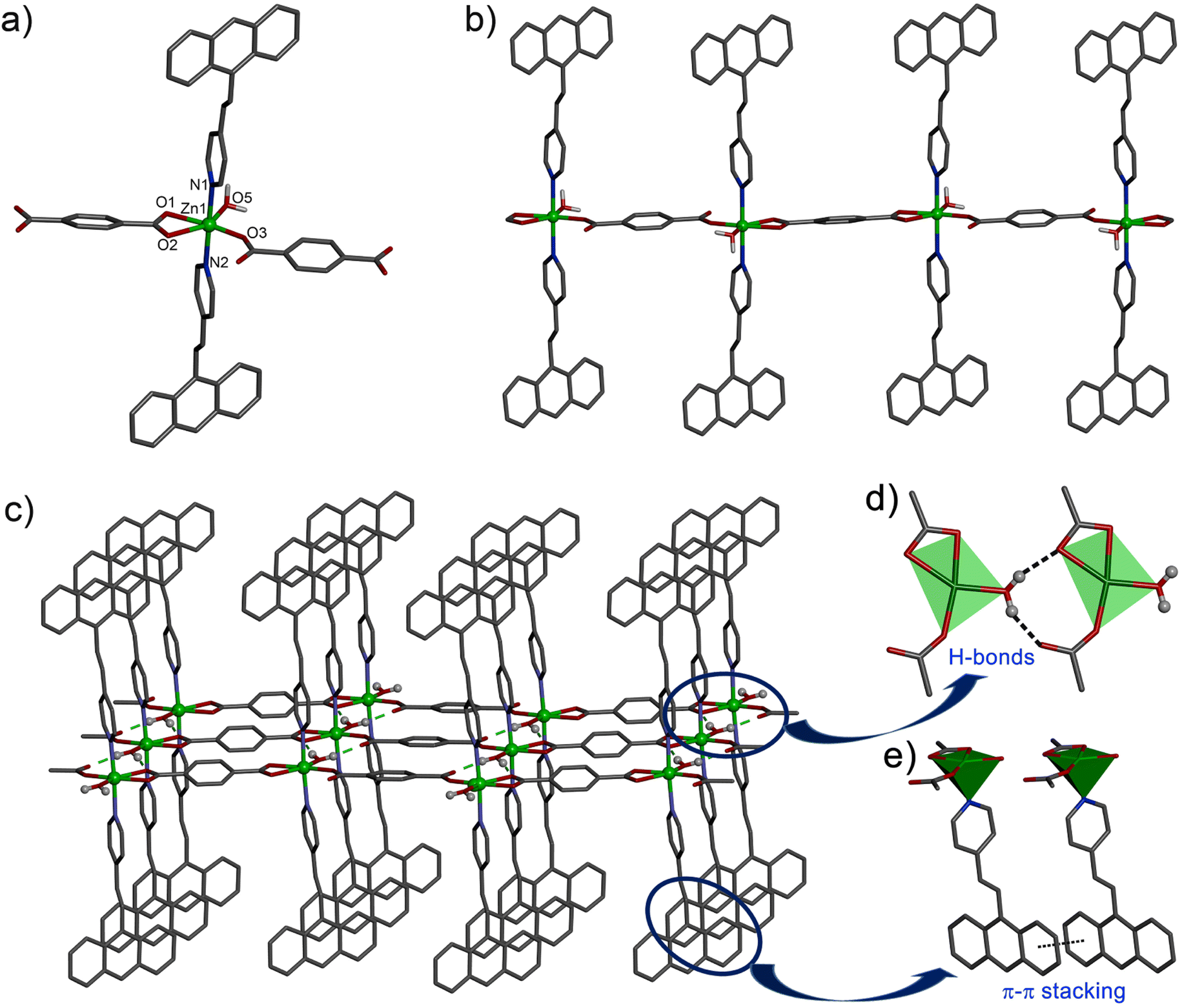 | ||
| Fig. 1 (a) A perspective view of compound 1 showing the coordination environment around the Zn centre. (b) A portion of the 1D polymeric chain of 1. (c) 2D layer of 1 formed via intermolecular hydrogen bonding and π⋯π interactions in 1. Only selected atoms are shown for clarity. (d) and (e) Schematic diagrams of hydrogen bonding and π⋯π interactions, respectively. | ||
Photo-physical properties
The structural architecture reveals that the synthesized CP 1 is featured with anthracene-based avp ligands appended to Zn(II) metal centres with a monodentate binding fashion. In this context, the bdc ligand adopts the role of a co-ligand augmenting the propagation of the coordination chain (Fig. 1b). This spatial distribution of the conjugated anthracene moieties indulges in extended π⋯π interactions with adjacent anthracene moieties, which in turn assist in constituting a higher-order stable framework. This type of supramolecular association may be responsible for closed-packed solid-state structure and high luminescent characteristics. In this regard, the photo-physical characteristic of CP 1 was investigated wherein a broad absorption band was observed at approximately 360 nm of well-dispersed ACN solution and different from the absorption band constituting anthracene moiety (Fig. S3a, ESI†). Similarly, the luminescent spectra also exhibited a broad emission profile around 490 nm in the ACN solvent, which is quite different from the emission spectra of the associated anthracene moiety of the avp ligand upon excitation at 360 nm (Fig. S3b, ESI†). This fluorescence characteristic is preferably subjected to intra-ligand charge transfer among the coordinated conjugated ligands through π⋯π interactions. The charge transfer mechanism for enhanced fluorescence is also evident from the broad emission spectra of the uncoordinated avp ligand at 487 nm (ex ∼ 360 nm). However, there is a very slight shift in emission maxima, which may be attributed to the inclusion of the bdc ligand in the polymeric chain. The bdc ligand may influence the charge transfer phenomenon with variation in the distribution of conjugation along the polymer in comparison to the free avp ligands. In order to understand the influence of bdc ligand, cyclic voltammetric (CV) studies of the free H2bdc and avp ligands were performed at ambient conditions. The CV showed two irreversible reduction peaks, Ic at E = 0.679 V and Ia at E = 0.285 V, corresponding to a quasi-reversible redox process for the oxidation–reduction mechanism of bdc acid. The peak potential indicates Ia = 0.506 V for oxidation and Ic = 0.435 V for reduction of the anthracene-type ligand. Therefore, the centre point of the transition region of the electron provides a reasonable estimation of the standard reduction potential (RP) of 0.941 V for the anthracene-type ligand (Fig. S4, ESI†). This RP value accounts for the unified mechanism in between the phases of anthracene-type ligands via the dissociative electron transfer mechanisms. Therefore, the increased electron density at the hetero atom and the anthracene system is reflected by the shift of the appropriate redox potentials towards lower E° values. In addition to the increased electron density, the conjugation between the anthryl units provides higher stability of the anthracene due to its planar environment. This unequivocally demonstrates the plausible augmentation of conjugation within the luminescent probes, correlating to the promotion of the energetically favorable electronic transitions, which justifies the enhanced emission characteristics of CP 1.Further, utilizing density functional theory (DFT) calculations within the B3LYP/LANLDZ theoretical framework, the band gap of the coordination polymer CP 1 was computationally determined. This assessment aligns with the heightened fluorescence intensity observed in CP 1, a phenomenon ascribed to its comparably narrower band gap. Consequently, the distinctive spatial arrangement, characterized by the viable anthracene moiety conjugation emerges as a pivotal determinant contributing to the augmented photo-physical characteristics exhibited by CP 1. The intensified conjugation inherent to the anthracene core in conjunction with a vinyl backbone engenders an elevated electron density envelopment encircling the anthracene rings. This generates a favorable environment for interactions, effectively serving as an interface for engagements with incoming analytes. The resultant configuration markedly amplifies the potential for enriched sensing capabilities of this fluorescent probe.
Sensing activity of compound 1 toward epNACs
The elevated luminescence intensity of CP 1 and its inherent propensity for interactions have prompted an exploration of its potential utility in the realm of the trace-level detection of epNACs. Therefore, an exhaustive investigation into the luminescence quenching phenomenon of CP 1 induced by various epNACs was performed. The primary objective was to discern the efficacy of CP 1 selectively and adeptly to detect trace concentrations of TNP. To scrutinize the nuanced effects of the disparate analytes, a well-defined dispersion of 1 mg of CP 1 in 5 mL of the ACN solvent was prepared. Subsequently, a series of distinct epNACs solutions were individually introduced into the CP 1 dispersion, following identical stoichiometric ratios. Intriguingly, despite the introduction of a range of epNACs, the emission intensity of CP 1 exhibited minimal alteration except TNP and 2,4-DNP. However, a distinctive quenching response manifested solely in the presence of TNP, wherein the emission intensity underwent complete attenuation, which was evident in the fluorescence quenching phenomena. Notably, the extent of fluorescence quenching for 2,4-DNP was comparatively lower than that for TNP (Fig. 2). | ||
| Fig. 2 Subsequent luminescent responses of CP 1 with different epNACs (10−4 M in ACN). | ||
In order to investigate the extent of quenching, fluorescence titration was performed with the dispersive solution of CP 1 upon excitation at 360 nm. The change in the fluorescence intensity upon progressive addition of epNAC solution was measured at 490 nm. Interestingly, upon incremental addition of 0–845 μL of TNP solution, a sharp and maximum quenching efficiency (∼78% quenching) of the inherent fluorescence intensity of CP 1 (Fig. 3) was observed for the series of epNACs. Noticeably, the initial fluorescence intensity of CP 1 remained mostly unaffected in response to other analytes, with the exception of 2,4-DNP; however, the quenching efficiency (∼48%) was not substantial compared to that of TNP (Fig. S5, ESI†). Therefore, apart from naked eye TNP detection, CP 1 exhibited a sharp and selective quenching response towards TNP. Further investigation on the limit of detection (LOD) and quenching constant rationalizes its efficiency in the context of efficacious luminescent probes in the realm of epNACs detection.
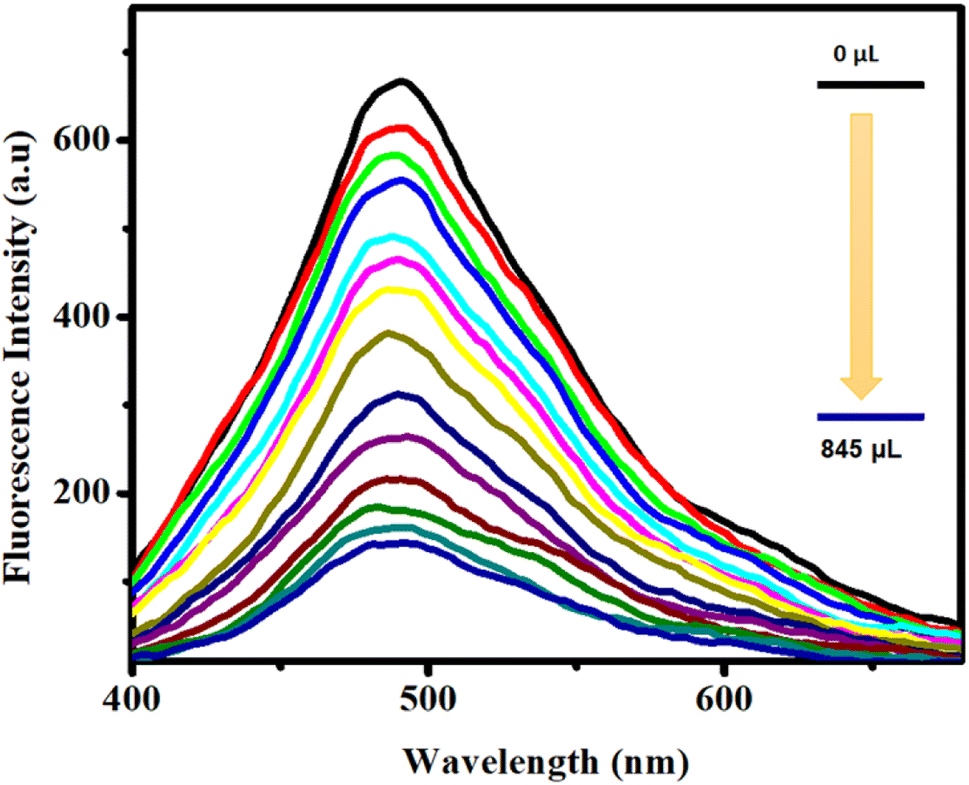 | ||
| Fig. 3 Emission profile of CP 1 upon incremental addition of TNP (10−4 M ACN). | ||
The measured LOD of CP 1 was ∼0.58 μM, calculated using 3σ/k (see the section for the calculation of the detection limit; Fig. S6–S7 and Table S5, ESI†), which exhibited elevated sensitivity of CP 1 among the recently reported luminescent probes for the detection of TNP (Table S6, ESI†). The time-dependent fluorescence study showed that the addition of 846 μL of 10−4 M TNP solution to the CP 1 quenched the initial fluorescence intensity of CP 1 with a sharp quenching response within 1 min (Fig. S8, ESI†). Moreover, the response time determination plot remained stable for a continuous observation for 30 min, which depicts the rapidity and stability of the sensory response of this fluorescent probe toward TNP. The Stern–Volmer (S–V) plot (I0/I vs. [C]; where I0 and I represent luminescence intensities without and with the presence of the analytes, respectively, and [C] denotes the molar concentration of the analyte) assists in assessing the quenching constant (Ksv) utilizing S–V equation: (I0/I) = 1 + Ksv[C]. However, the S–V plot for CP 1 suggested non-linear fitting, therefore, in order to calculate the quenching constant (Ksv), a modified S–V equation i.e., [(I0/I) = 1.16 + (−0.46× [C] + 39.53× [C]2)] was introduced. The S–V plot illustrates the formation of a nonlinear plot (Fig. 4). The calculated Ksv (18.18 × 104 M−1) signifies the high quenching efficiency of CP 1 toward TNP over other epNACs. The remarkable sensitivity and the extensively low LOD of CP 1 distinctly showcase its competency as a prominent and robust chemosensor within the context of contemporary literature (Table S6, ESI†).
 | ||
| Fig. 4 Stern–Volmer (S–V) plot of TNP for CP 1. | ||
Insight into the sensing mechanism
The crystallographic configuration delineates the dangling position of the anthracene moiety within the crystal lattice. This structural arrangement gives rise to a discernible host–guest interaction, where the highly π electron-rich nature of the anthracene segment interacts with the significantly electron-deficient characteristics of TNP, leading to the observed quenching response. Among the plethora of potential sensing mechanisms, two predominant phenomena, such as (i) photo-induced electron transfer (PET) and (ii) resonance energy transfer (RET) are assumed to be responsible for the fluorescence quenching of CP 1. Within the confinement of the fundamentals of the PET quenching mechanism, an electron in an excited state of the fluorophore is transferred to the lower-energy unoccupied π*-orbitals of the quencher. This intricate process precipitates the quenching of inherent fluorescence.48,49 Consequently, the efficacy of fluorescence quenching is intricately tied to the magnitude of electron transfer that occurs between the electron-rich luminescent CP and electron-deficient epNACs.41,50 Therefore, in the foundation of this mechanism, the driving force for the feasible electron transfer considerably relies on the energy level of the lowest unoccupied molecular orbital (LUMO) of the analyte. Additionally, the extent of electron transfer from the electron-rich CP 1 to the electron-deficient NACs results in the quenching of the fluorescence of CP 1 and also assists in rationalizing the relative magnitude/order of quenching efficiency of different epNACs.51,52 In this regard, the highest electron-deficient analyte possesses the lowest LUMO energy providing the most possible electron transfer, subsequently more prominent quenching (electron deficient order; TNP > DNP > DNT > DNBA > NBA > NB > NP > NM). Therefore, TNP being the most electron-deficient epNAC shows the maximum quenching of fluorescence (Fig. S9, ESI†).In order to evaluate the energy gradient for the electron transfer process, DFT calculations were performed with B3LYP and LANLDZ theory levels. These computations offer insights into the energy profile of the highest occupied molecular orbitals (HOMOs) and LUMOs for both the sensory probe CP 1 and epNACs (Fig. 5).24 The analysis of the energy profile for the analyte suggests that the most diminished LUMO energy level is distinctively possessed by TNP among the epNACs. This finding implies that upon excitation, the electron residing in the LUMO of the CP 1 encounters the most propitious pathway for migration to the LUMO of TNP, yielding the highest quenching efficiency. However, an intriguing facet surface with respect to the assorted quenching efficiencies and the corresponding LUMO energies was observed among the epNACs. Nevertheless, a captivating aspect comes into the context that the varied quenching efficiencies and the corresponding LUMO energies observed for the electron-deficient epNACs were imperatively not converging. This serves to underscore a pertinent aspect that the PET quenching mechanism is not exclusively responsible for the quenching phenomenon.
 | ||
| Fig. 5 Energy profile diagram of CP 1 and epNACs with calculated HOMO (black) and LUMO (red) energies by DFT. | ||
Consequently, it becomes imperative to deliberate on the feasibility of the energy transfer alongside electron transfer, which originates from the excited state of the fluorophore towards suitable energy levels of the analytes. However, there are some distinct compelling factors to be considered for the plausibility of RET. Firstly, molecular interactions within the close spatial proximity between CPs and analytes, and secondly, the potential overlap between the emission band of CPs and the absorption band of the analytes, involving non-covalent interactions. In this context, the degree of this spectral overlap emerges as a conceivable interpretation for the observed variation in quenching efficiencies.
In order to facilitate propitious proximity, the spatial configuration of anthracene moieties in the inherent CP 1 confers a strategically favourable spatial orientation for fostering conducive molecular–level interactions with the encountered analytes. Notably, these interactions are predominantly mediated through C–H⋯π and π⋯π stacking interactions. Herein, TNP exhibits a sharp inclination for π⋯π interactions with the available anthracene moieties of the compound due to its highest electron deficiency nature.49 These closer proximal interactions facilitate the potential for energy transfer, however, the overlap between the emission band of CP 1 with the absorption band of epNAC represents the extent of energy transfer in the RET process from the CP fluorophore to the encountered acceptor. Significantly, it is worth noting that a maximum degree of spectral overlap is evident between the fluorescence spectrum of CP and the absorption spectrum of TNP, which is not quite frequently observed for other electron-deficient epNAC (Fig. 6). It is interesting to observe that a significant overlap was also observed for 2,4-DNP, which elucidates the comparatively higher quenching efficiency in compared to other epNACs. In this context, the S–V plot associated with CP 1 exhibits a conspicuous non-linear characteristic. This characteristic provides additional support for the viability of RET mechanism emerging between the anthracene subunit of CP 1 and TNP. Additionally, the non-linear nature of the S–V plot endures crucial implications, suggesting that the observed quenching phenomena are the result of an intricate combination of static and dynamic quenching.53,54
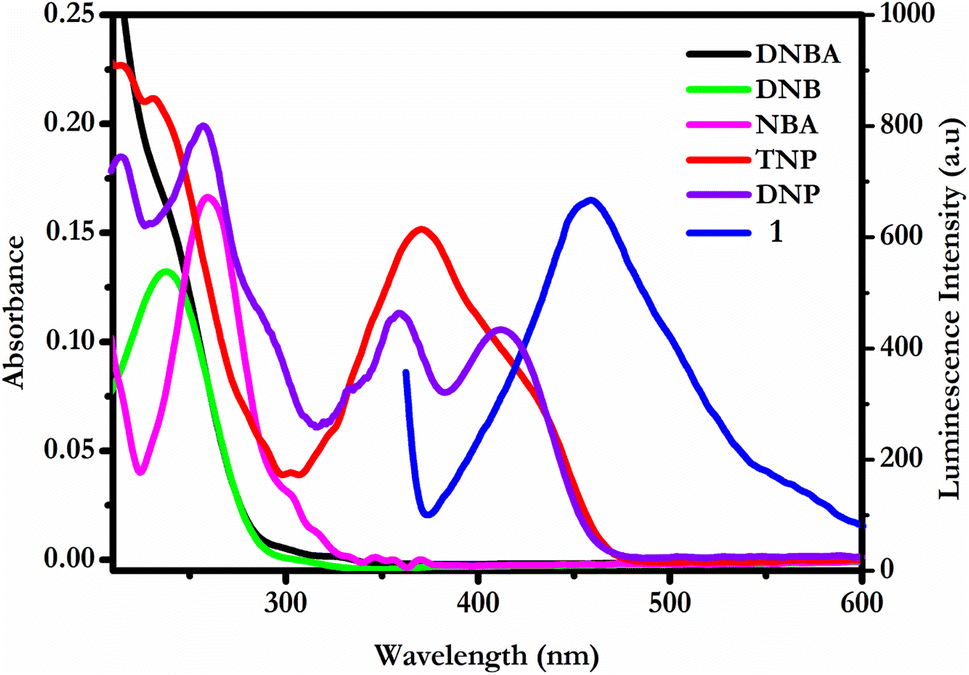 | ||
| Fig. 6 Overlapping of absorption spectra of epNACs with emission spectra CP 1. | ||
In order to elucidate the static and dynamic nature of quenching, the time-resolved fluorescence quenching experiments are beneficial to monitor the fluorescence decays of CP 1 without and with TNP. The time-resolved fluorescence quenching investigations provide further corroboration regarding the plausibility of dynamic quenching. This assertion is substantiated by the noticeable alteration in the average lifetime values observed in the decay profiles of CP 1 when comparing its behavior in the absence and presence of TNP (Fig. S10, ESI†). This impedes the plausibility of static quenching, lending credence to the collisional quenching theory. Such a disposition underscores the potential participation of a fluorescence quenching phenomenon within the realm of dynamic quenching. Consequently, the ascribed quenching mechanism adheres to the intricate pathways of PET and RET, implying a possible host–guest interaction associated with the luminophore CP 1 and TNP. This confers the distinct nature of being the universal luminescent CP 1 chemosensor for the discerning detection of TNP in aqueous media. Because of its robust nature, the CP 1 was subjected to testing reversibility, wherein it showed excellent reversibility (Fig. S11, ESI†) of its fluorescence intensity, and the revival was performed by centrifugation followed by washing several times with the organic solvent.
Conclusions
In conclusion, we have synthesized a new 1D CP based on Zn(II) ion that contains an anthracene chromophore. The 1D CP creates 3D supramolecular architectures by engaging significant intermolecular hydrogen bonding and π⋯π interactions. It is interesting to note that the existence of a free electron-rich anthracene moiety with a good spatial orientation makes it easier for the sensor and analyte to be closer to one another for a reasonable molecular-level interaction through C–H⋯π and π⋯π stacking interactions. As a result, the synthesized CP was used to detect epNACs with a high selectivity for TNP and may be recognized as having a greater future use for the detection of epNACs. Therefore, the present CP with spatially ordered avp ligand provides a new pathway for generating functional materials that may be used for selective TNP sensing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by SERB India (Grant No. CRG/2022/001842, dated 19/12/2022) and E. H. thanks Ministry of Minority Affairs, Govt. of West Bengal for providing a Swami Vivekananda merit-cum-means fellowship. P. B. acknowledges DST SERB-CRG sponsored project, GAP-240712, vide reference no. CRG/2022/001679. A. H. is thankful to CSIR, India for awarding a Senior Research Fellowship (Sanction No. 31/019(0023)/2020-EMR-I). S. D. is thankful to the Department of Science and Technology, Government of India for the fellowship (IF200416). We thank the referees for helpful suggestions.References
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. Khan, Akhtaruzzaman, R. Medishetty, A. Ekka and M. H. Mir, Chem.–Asian J., 2021, 16, 2806–2816 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- B. Dutta, S. Maity, S. Ghosh, C. Sinha and M. H. Mir, New J. Chem., 2019, 43, 5167–5172 RSC.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Cryst. Growth Des., 2017, 17, 2893–2910 CrossRef CAS.
- W. M. Chen, X. L. Meng, G. L. Zhuang, Z. Wang, M. Kurmoo, Q. Q. Zhao, X. P. Wang, B. Shan, C. H. Tung and D. Sun, J. Mater. Chem. A, 2017, 5, 13079–13085 RSC.
- Y. Hasegawa and Y. Kitagawa, J. Photochem. Photobiol., C, 2022, 51, 100485 CrossRef CAS.
- A. M. Downs, J. Gerson, K. L. Ploense, K. W. Plaxco and P. Dauphin-Ducharme, Anal. Chem., 2020, 92, 14063–14068 CrossRef CAS PubMed.
- X. Z. Guo, S. S. Chen, W. D. Li, S. S. Han, F. Deng, R. Qiao and Y. Zhao, ACS Omega, 2019, 4, 11540–11553 CrossRef CAS PubMed.
- B. Ding, M. B. Solomon, C. F. Leong and D. M. D'Alessandro, Coord. Chem. Rev., 2021, 439, 213891 CrossRef CAS.
- Y. Salinas, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- Q. Wang, S.-L. Dong, D.-D. Tao, Z. Li and Y.-B. Jiang, Coord. Chem. Rev., 2021, 432, 213717 CrossRef CAS.
- M. Karmakar, S. Roy and S. Chattopadhyay, New J. Chem., 2019, 43, 10093–10102 RSC.
- B. Dutta, K. Debsharma, S. Dey and C. Sinha, Appl. Organomet. Chem., 2023, 37, e6919 CrossRef CAS.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS.
- M. Park, BioChip J., 2019, 13, 82–94 CrossRef CAS.
- J. Q. Liu, Z. D. Luo, Y. Pan, A. K. Singh, M. Trivedi and A. Kumar, Coord. Chem. Rev., 2020, 406, 213145 CrossRef CAS.
- B. Dutta, R. Jana, A. K. Bhanja, P. P. Ray, C. Sinha and M. H. Mir, Inorg. Chem., 2019, 58, 2686–2694 CrossRef CAS PubMed.
- A. K. Ghosh, A. Hazra, A. Mondal and P. Banerjee, Inorg. Chim. Acta, 2019, 488, 86–119 CrossRef CAS.
- N. Varnaseri, R. Ali, M. Ali and R. Farzaneh, J. Solid State Chem., 2020, 291, 121622 CrossRef CAS.
- S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881–2885 CrossRef CAS PubMed.
- A. Hazra, S. Bej, A. Mondal, N. C. Murmu and P. Banerjee, ACS Omega, 2020, 5, 15949–15961 CrossRef CAS PubMed.
- P. Ghorai, A. Dey, A. Hazra, B. Dutta, P. Brandão, P. P. Ray, P. Banerjee and A. Saha, Cryst. Growth Des., 2019, 19, 6431–6447 CrossRef CAS.
- B. Dutta, A. Hazra, A. Dey, C. Sinha, P. P. Ray, P. Banerjee and M. H. Mir, Cryst. Growth Des., 2020, 20, 765–776 CrossRef CAS.
- B. Parmar, K. K. Bisht, Y. Rachuri and E. Suresh, Inorg. Chem. Front., 2020, 7, 1082–1107 RSC.
- X. Z. Guo, S. S. Chen, W. D. Li, S. S. Han, F. Deng, R. Qiao and Y. Zhao, ACS Omega, 2019, 4, 11540–11553 CrossRef CAS PubMed.
- Z. Tang, H. Chen, Y. Zhang, B. Zheng, S. Zhang and P. Cheng, Cryst. Growth Des., 2019, 19, 1172–1182 CrossRef CAS.
- W. H. Huang, J. Ren, Y. H. Yang, X. M. Li, Q. Wang, N. Jiang, J. Q. Yu, F. Wang and J. Zhang, Inorg. Chem., 2019, 58, 1481–1491 CrossRef CAS PubMed.
- K. Maiti, A. K. Mahapatra, A. Gangopadhyay, R. Maji, S. Mondal, S. S. Ali, S. Das, R. Sarkar, P. Datta and D. Mandal, ACS Omega, 2017, 2, 1583–1593 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- S. Tarasi, A. Tehrani and A. Morsali, Sens. Actuators, B, 2020, 305, 127341 CrossRef CAS.
- F. Bigdeli, F. Rouhani, A. Morsali and A. Ramazani, Ultrason. Sonochem., 2020, 62, 104862 CrossRef CAS PubMed.
- S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC.
- G. X. Wen, M. L. Han, X. X. Wu, Y. P. Wu, W. W. Dong, J. Zhao, D. S. Li and L. F. Ma, Dalton Trans., 2016, 45, 15492–15499 RSC.
- X. X. Wu, H. R. Fu, M. L. Han, Z. Zhou and L. F. Ma, Cryst. Growth Des., 2017, 17, 6041–6048 CrossRef CAS.
- J. H. Qin, H. R. Wang, M. L. Han, X. H. Chang and L. F. Ma, Dalton Trans., 2017, 46, 15434–15442 RSC.
- A. Ma, J. Wu, Y. Han, F. Chen, B. Li, S. Cai, H. Huang, A. Singh, A. Kumar and J. Liu, Dalton Trans., 2018, 47, 9627–9633 RSC.
- A. Lan, K. Li, H. Wu, D. H. Olson, T. j. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed.
- S. Mukherjee, W. P. Lustig, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- S. Khan, A. Hazra, B. Dutta, Akhtaruzzaman, M. J. Raihan, P. Banerjee and M. H. Mir, Cryst. Growth Des., 2021, 21, 3344–3354 CrossRef CAS.
- C.-W. Chan, T.-F. Lai, C.-M. Che and S.-M. Peng, J. Am. Chem. Soc., 1993, 115, 11245–11253 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- SMART and SAINT, Bruker AXS Inc., Madison, WI, 1998 Search PubMed.
- SADAB, Bruker AXS Inc., Madison, Wisconsin, USA, 2014 Search PubMed.
- G. M. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- Y. Rachuri, B. Parmar and E. Suresh, Cryst. Growth Des., 2018, 18, 3062–3072 CrossRef CAS.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- S. Pramanik, C. Zheng, X. Zhang, T. j. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
- X. G. Yang, Z. M. Zhai, X. M. Lu, Y. Zhao, X. H. Chang and L. F. Ma, Dalton Trans., 2019, 48, 10785–10789 RSC.
- V. Kumar, B. Maiti, M. K. Chini, P. De and S. Satapathi, Sci. Rep., 2019, 9, 7269 CrossRef PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- L. N. Wang, Y. H. Zhang, S. Jiang and Z. Z. Liu, CrystEngComm, 2019, 21, 4557–4567 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S12, Tables S1–S6, and X-ray crystallographic data in CIF format for compound 1. CCDC 2271850. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra06926e |
| This journal is © The Royal Society of Chemistry 2024 |