
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monolith-based 68Ga processing: a new strategy for purification to facilitate direct radiolabelling methods†
Ping
He
ab,
Benjamin P.
Burke
ab,
Gonçalo S.
Clemente
b,
Nathan
Brown
bc,
Nicole
Pamme
a and
Stephen J.
Archibald
*ab
aDepartment of Chemistry, University of Hull, Cottingham Road, Hull HU6 7RX, UK. E-mail: S.J.Archibald@Hull.ac.uk
bPositron Emission Tomography Research Centre, University of Hull, Cottingham Road, Hull HU6 7RX, UK
cSchool of Engineering, University of Hull, Cottingham Road, Hull HU6 7RX, UK
First published on 29th June 2016
Abstract
The post-processing of 68Ga generator eluate by means of a novel high capacity cation-exchange silica monolith column has been validated in this work. Quantitative release of a purified 68Ga solution in high concentration can be achieved using weak acidic solutions which can be directly used for chelator or conjugate labelling in an injectable form with improved radiochemical characteristics. The system has the potential to be incorporated in a flow based microfluidic system for dose-on-demand radiotracer synthesis.
68Ga radiopharmaceuticals are of growing importance in clinical positron emission tomography (PET) with the success of octreotide derivatives for imaging neuroendocrine tumours,1,2 and prostate specific membrane antigen (PSMA) derivatives receiving significant recent interest.3,468Ga generators have simultaneously been developed towards routine clinical use5,6 with the Eckert and Ziegler generator receiving recommendation from the EMA for pharmaceutical approval and FDA documents filed in 2014. This generator is based on a TiO2 solid phase in which the produced ionic 68Ga3+ is eluted as GaCl3 from the generator using a solution of 0.1 N HCl.7 However, current 68Ge/68Ga radionuclide generators do not have optimal properties for the direct synthesis of 68Ga-labeled radiopharmaceuticals. The generator eluate has a large volume (5–10 mL) with a high concentration of H+ (pH < 1) and can contain metal ion impurities (commonly aluminium(III), iron(III) and zinc(II)).6,8 All of which can potentially hinder complexation formation and reduce specific activity (SA). Thus, the eluate from a 68Ge/68Ga generator cannot currently be directly used for preparing radiopharmaceuticals that are in routine clinical practice.
This can be overcome by either; (a) advanced chelator design to allow for direct labelling of eluate, a trait which is so far only proven successful with the highly promising tris(hydroxypyridinone) ligands developed by Blower, Ma and co-workers9,10 or; (b) post-processing of the generator eluate to allow for complexation, the approach nearly exclusively followed by groups worldwide and the only method possible for current clinically used tracers.
Several methods for the processing of the generator eluate have been attempted in order to overcome the issues mentioned above including ion-exchange chromatography and fractionation.7,8,11–13 All of which individually mitigate some of the limitations of 68Ga complexation. However, to date, no method delivers on all requirements to release the 68Ga purified from other metal ions into small volumes of low H+ concentration using a directly injectable solvent. Rösch and co-workers use a particle-packed cation-exchange resin column and ethanol (instead of the commonly used acetone) in the elution mixture for post-processing of 68Ga eluate.14 Ethanol-based post-processing provides a more acceptable reaction mixture for the preparation and formulation of directly injectable 68Ga-radiopharmaceuticals. However, the requirement for a higher concentration HCl solution (0.9 N) can suppress the reactivity of chelators and also needs higher concentrations of buffer to balance the pH.
Recently, miniaturised microfluidic-based systems have attracted much attention in PET tracer synthesis.15,16 Such microfluidic systems are easily shielded and therefore reduce external impact and have significant potential to facilitate PET tracer synthesis, reduce consumption of expensive reagents and improve reactivity. Most importantly, microfluidic-based systems are compatible with automated dose-on-demand requirements. However, it is difficult to incorporate the current post-processing into microfluidic systems because particle-packed resin columns are challenging to integrate with microfluidic systems due to the relative high backpressures generated. This is an obstacle in the design of an integrated microfluidic system for the dose-on-demand tracer synthesis.
Owing to their small-size skeletons and mesoporous structures, monolith columns provide elevated permeability and favourable mass transfer compared to particle-packed columns.17–20 Due to these characteristics monolith columns have shown their applications in a variety of chromatographic modes.21–24 Importantly, the integration of monolith columns with microfluidic systems has already been demonstrated in an enzymatic reaction module.25 If a cation-exchange monolith can purify 68Ga generator eluate, it would therefore be possible to build an integrated system comprising a monolith for ion-exchange and a microfluidic system for performing flow-based 68Ga preparation and radiolabelling in an automated dose-on-demand system. To our knowledge there has been no reported use of a cation-exchange monolith for the separation of 68Ga directly from the 68Ge/68Ga generator eluate.
The aim of this work is to fabricate a cation-exchange silica monolith and evaluate its capability for efficient separation of 68Ga directly from the generator eluate. The preparation of injectable 68Ga-labeling pharmaceuticals can then be investigated to determine whether the characteristics are improved when compared to the best current method.
The silica monolith columns used in this work are porous rods consisting of silica skeletons and interconnecting mesopores that give much higher permeability and favourable mass transfer compared to particle-packed columns.26,27 They were produced by the sol–gel method described in our previous study26 followed by surface modification with thiol groups and oxidation to transform –SH to –SO3H (see ESI†). Therefore forming a sulfonic acid functionalised monolith capable of selectively trapping cations. BET nitrogen adsorption/desorption isotherm (see ESI† S5 and Fig. S3) shows typical hysteresis between adsorption and desorption, which is consistent with the disordered mesoporous structure displayed in the SEM (see Fig. 1) giving a specific surface area of 238 m2 g−1 and pore volume of 0.75 cm3 g−1, with an average pore diameter of ∼13 nm. The cation exchange capacity (CEC) quantifies the ability of a material to adsorb cations. Analysis of our –SO3H monoliths gives a value of 180 μeq. g−1, which is 30 times higher than a commercial solid phase extraction (SPE) cation-exchange resin (for further CEC discussion see ESI† S4).
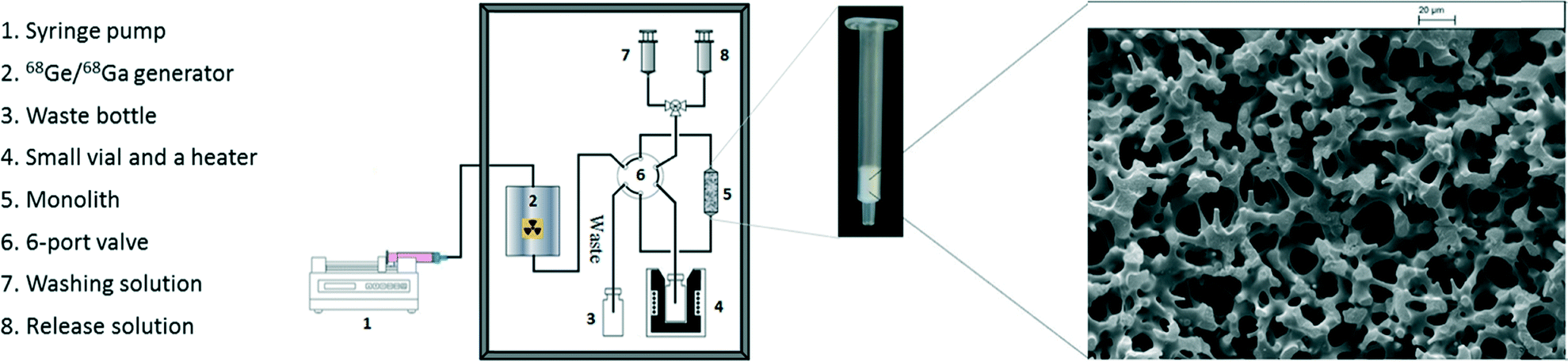 | ||
| Fig. 1 Schematic representation of the monolith based system for 68Ga processing and radiolabelling (left), photograph of cation-exchange silica-monolith column (centre) and SEM image of SiO2 monolith–SO3H (right, scale bar: 20 μm). | ||
Due to shrinkage during the sol–gel processing it is challenging to make a monolith column within a conventional stainless steel column without a void volume. Therefore, commercially available monolith columns are usually confined in heat-shrinkable polymer tubing.17,28 To avoid damage to the functionalised monolith from the heating required to mount the column tightly inside heating-shrinkable tubing, the SiO2 monolith–SO3H rod was fixed in an empty Sep-Pak Vac tube and a void volume between the monolith rod and tube wall was packed with powdered functionalised monolith (ca. 10% wt of total monolith mass) and plastic frits fitted at both ends to form the SiO2 monolith–SO3H column (see Fig. 1).
As mentioned previously, the direct synthesis of 68Ga tracers from the generator eluate is problematic. The most common approach to overcome this is the transfer of the initial 68Ga eluate to a cation-exchange resin column. As it has a high cation-exchange distribution coefficient, 68Ga can be quantitatively adsorbed on the resin from the acidic generator eluate and then eluted with HCl/acetone solutions.7,29,30 Although the use of HCl/acetone solutions can efficiently purify and concentrate the 68Ga eluate, SPE is required for the removal of acetone and the recovery of the 68Ga-labeled compounds. The use of ethanol in place of acetone provides the potential for preparation of injectable 68Ga-radiopharmaceuticals. However, the reactivity of the 68Ga solution released via current methodologies is inadequate for high SA tracer formation due to metal ion impurities and currently used technologies are not compatible with microfluidic reactors, preventing the design of automated compact dose-on-demand systems.
The SiO2 monolith–SO3H columns used in this work not only quantitatively (98 ± 1%) adsorbed 68Ga3+ but also exhibited highly efficient release of the 68Ga3+ in comparison to commercial polymeric cation-exchange resins. For the elution methods (see Table 1), a multi-fraction elution process showed a small advantage compared to single step method.
| Entry | Activity%a | Washing solutionb | Activity%c | Elution solution | Volume | Activity%d |
|---|---|---|---|---|---|---|
| a Activity trapped in monolith. b Volume of washing solution was 1 mL. c Activity in washing solution. d Activity in elution solution. e Release activity increases to ≥98% with 1 mL. f Commercial cation-exchange resin (SCX). Solvent compositions are given in v/v. | ||||||
| 1 | 98 | 85% EtOH/0.2 N HCl | 8.0 | 93% EtOH/0.9 N HCl | 1 mL | 90 |
| 2 | 99 | 80% EtOH/0.1 N HCl | 0.3 | 96% EtOH/0.5 N HCl | 1 mL | 98 |
| 3 | 99 | 80% EtOH/0.1 N HCl | 0.3 | 90% EtOH/0.9 N HCl | 0.4 mL | 94e |
| 4 | 99 | 80% EtOH/0.1 N HCl | 0.3 | 90% EtOH/0.5 N HCl | 0.4 mL | 93e |
| 5 | 99 | 80% EtOH/0.1 N HCl | 0.3 | 90% EtOH/0.5 N HCl | 4 × 0.2 mL | 98 |
| 6 | 99 | 80% EtOH/0.1 N HCl | 0.6 | 90% EtOH/0.5 N HCl | 0.5 mL | 98 |
| 7 | 99 | 80% EtOH/0.1 N HCl | 0.6 | 90% EtOH/0.2 N HCl | 1 mL | 94 |
| 8f | 98 | 80% EtOH/0.2 N HCl | 0.1 | 90% EtOH/0.9 N HCl | 1 mL | 46 |
| 9f | 98 | 80% EtOH/0.1 N HCl | 0.1 | 90% EtOH/0.2 N HCl | 1 mL | 16 |
Larger amounts of ethanol and hydrochloric acid in the washing solution (i.e. 85% EtOH/0.2 N HCl) result in a modest amount (8%) of 68Ga being lost in the washing step. When using 80% EtOH/0.1 N HCl and 90% EtOH/0.5 N HCl as washing and elution solutions respectively, the SiO2 monolith–SO3H does not only efficiently purify 68Ga from other metal ions (see Table 2) but also almost quantitatively (93%) recovers 68Ga from the eluate. This compares to only 25% recovery for the AG50W column using the same conditions.14
| Al3+ (μg L−1) | Fe3+ (μg L−1) | Zn2+ (μg L−1) | Ti4+ (μg L−1) | Ge4+ (μg L−1) | |
|---|---|---|---|---|---|
| a 80% EtOH/0.1 N HCl and 90% EtOH/0.5 N HCl were used as washing and elution solutions respectively. | |||||
| Before | 1140 | 74 | 320 | 420 | 25 |
| After | 35 | 52 | 6 | 1 | Undetected |
It is postulated that the lower release efficiencies for the resin indicate the strong affinity for 68Ga3+ within the cation-exchange resin pores.31 These results suggest that the micro-environment of the monolith columns used in this work favours ion-exchange processing for adsorption and desorption of 68Ga3+.
A main benefit of our method is that the 68Ga can be concentrated up to ten-fold and the 68Ga elution requires a lower concentration of hydrochloric acid (0.5 N) when compared with the best published example.14 Decreasing the concentration of H+ allows lower concentration buffers to be used in radiolabelling reactions. Metallic radiolabelling characteristics are generally directly proportional to the concentration of the two reagents in solution.9 Increase of 68Ga concentration in this manner could have significant impact on future tracer production by decreasing the amount of precursor required for synthesis, therefore dramatically increasing the SA. In order to ensure the purified 68Ga could still be used in standard labelling protocols and behaved analogously to reported chelator reactions, two common gallium(III) chelators (DOTA and NOTA) were selected for comparison of radiolabelling characteristics (see Table 3 and Scheme 1). The monolith-based system for 68Ga processing and labelling is shown in Fig. 1. A yield of 97% was achieved for DOTA (95 °C, 10 min, 5 μM) which offers a two-fold decrease in the concentration required for near quantitative conversion. The results are more pronounced for NOTA, with 95% incorporation at 1 μM (room temperature, 5 min). This is a ten-fold improvement over the reported concentrations of 10 μM giving 80% RCY.9 Finally, a demonstration of clinical applicability was provided by 68Ga radiolabelling of the licensed somatostatin targeted agent DOTATOC,1 which produced near quantitative yields within 10 minutes using standard kit precursor amounts.14
| Entry | Trap (%) | Release (%) | Labelling conditions | Yield (%) | |||
|---|---|---|---|---|---|---|---|
| Chelator | Concentration (μM) | Temperature (°C) | Time (min) | ||||
| a Washing solution was 1 mL of 80% ethanol/0.1 N HCl, elution solution was 0.5 mL of 90% ethanol/0.5 N HCl, buffer was 0.5 mL of NH4OAc (1 M, pH 5). b The 68Ga elution was preheated for 10 min at 95 °C. c The monolith was reused (SiO2 monolith–SO3H). d Room temperature. | |||||||
| 1 | 99 | 98 | DOTA | 1 | 95 | 10 | 0 |
| 2 | 99 | 98 | DOTA | 5 | 95 | 10 | 97 |
| 3 | 99 | 98 | DOTA | 10 | 95 | 10 | 98 |
| 4 | 99 | 98 | DOTA | 20 | 95 | 5 | 98b |
| 5c | 99 | 97 | DOTA | 10 | 95 | 10 | 99 |
| 6 | 99 | 98 | NOTA | 5 | RTd | 5 | 99 |
| 7 | 98 | 97 | NOTA | 1 | RTd | 5 | 95 |
| 8 | 99 | 97 | NOTA | 0.5 | RTd | 5 | 56 |
| 9 | 99 | 95 | DOTATOC | 14 | 95 | 10 | 97 |
| 10 | 99 | 95 | DOTATOC | 14 | 95 | 15 | >99 |
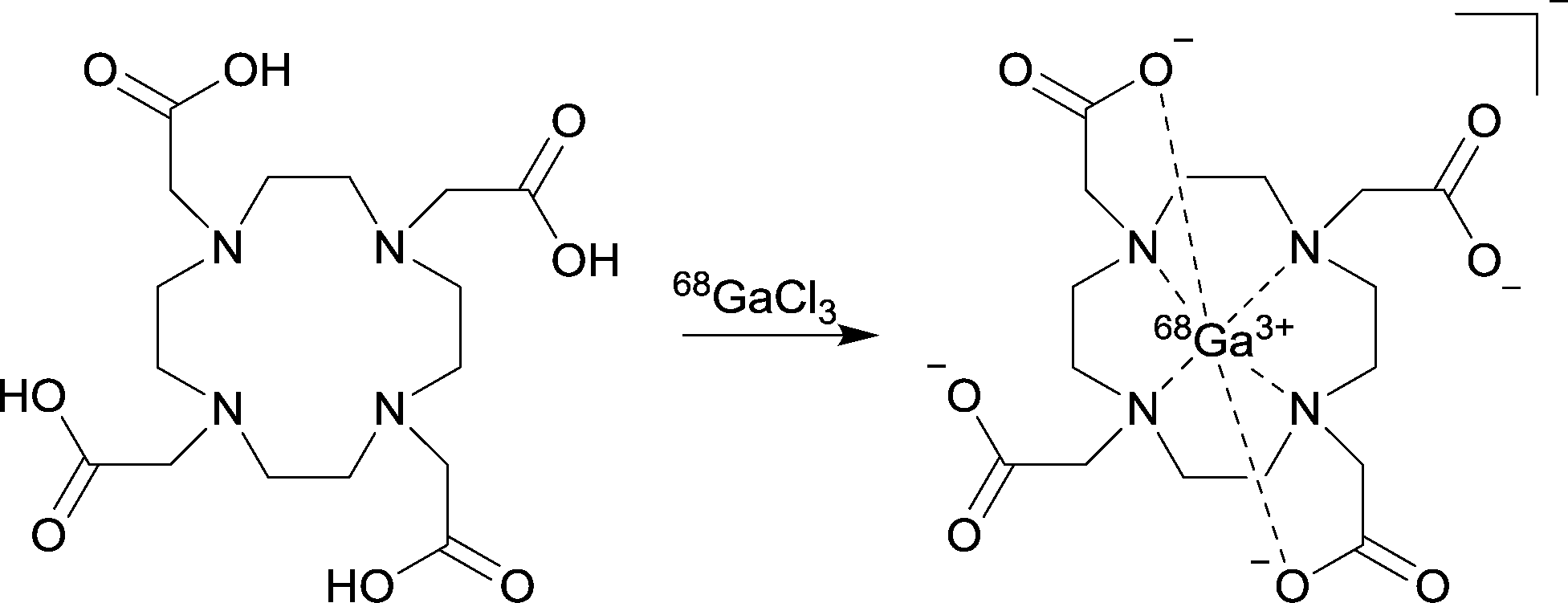 | ||
| Scheme 1 Radiochemical formation of 68GaDOTA. | ||
Complete synthesis of 68Ga-DOTATOC can be carried out, from elution of the generator to delivery of the tracer, in 25 minutes. If all the generator eluate is used (740 MBq), the tracer can be produced with a final SA of 41 GBq μmol−1, significantly higher than commonly reported values of 10–22 GBq μmol−1.32,33
These improved radiolabelling characteristics and shorter reaction times will permit clinical application following a simple pre-injection dilution of the radiopharmaceutical when using ethanol as a solvent.12 The application of the novel monolith column in this work showed that the 68Ga radiolabelling characteristics of the processed solution could be significantly improved, reducing the amount of chelator and the reaction time required. It was also found that the monolith could be reused with no significant reduction in labelling yield observed. This has not been reported for commercial polymeric cation-exchange resin in ethanol-based processing of 68Ga generator eluate.
In conclusion, a rapid, simple and efficient processing of TiO2-based 68Ge/68Ga generator eluate was developed utilising a novel cation-exchange silica-monolith column. It was demonstrated that with an ethanol/HCl eluent the cation-exchange silica-monolith could not only quantitatively recover the 68Ga but also efficiently purify the 68Ga from other metal ion impurities present in the initial generator eluate. The advantage of monolith-based processing compared to the optimum method reported in literature14 is a ca. 50% reduction in both the eluent volume and the HCl concentration required. The higher 68Ga concentration and purity facilitates efficient radiolabelling with the potential for significant increases in specific activity which could dramatically improve image quality for some tracers. The processed 68Ga was directly used for radiolabelling of commonly used chelators DOTA and NOTA, showing significant improvement in reactivity compared to other 68Ga3+ purification methods. Comparative radiolabelling of DOTATOC was performed to demonstrate the utility of this method with a clinically relevant example. The low back pressure of monolith based systems compared to polymeric cation-exchange resins allows, for the first time, the possibility to efficiently integrate an ion-exchange component with a microfluidic reactor and the production of automated modules for continuous 68Ga elution, preparation and labelling. These applications and integrated synthesis unit designs are under investigation.
We gratefully acknowledge the Daisy Appeal Charity for funding (Grant: DAhul0211) and the University of Hull for PET infrastructure support. We thank Dr Assem Allam and his family for their generous donation to help found the PET Research Centre at the University of Hull.
Notes and references
- M. Hofmann, H. Maecke, A. R. Borner, E. Weckesser, P. Schoffski, M. L. Oei, J. Schumacher, M. Henze, A. Heppeler, G. J. Meyer and W. H. Knapp, Eur. J. Nucl. Med., 2001, 28, 1751–1757 CrossRef CAS PubMed.
- I. Velikyan, Theranostics, 2014, 4, 47–80 CrossRef CAS PubMed.
- A. Afshar-Oromieh, A. Malcher, M. Eder, M. Eisenhut, H. G. Linhart, B. A. Hadaschik, T. Holland-Letz, F. L. Giesel, C. Kratochwil, S. Haufe, U. Haberkorn and C. M. Zechmann, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, 486–495 CrossRef CAS PubMed.
- M. Eiber, T. Maurer, M. Souvatzoglou, A. J. Beer, A. Ruffani, B. Haller, F.-P. Graner, H. Kuebler, U. Haberhorn, M. Eisenhut, H.-J. Wester, J. E. Gschwend and M. Schwaiger, J. Nucl. Med., 2015, 56, 668–674 CrossRef PubMed.
- M. Fani, J. P. Andre and H. R. Maecke, Contrast Media Mol. Imaging, 2008, 3, 67–77 Search PubMed.
- B. P. Burke, G. S. Clemente and S. J. Archibald, J. Labelled Compd. Radiopharm., 2014, 57, 239–243 CrossRef CAS PubMed.
- K. P. Zhernosekov, D. V. Filosofov, R. P. Baum, P. Aschoff, H. Bihl, A. A. Razbash, M. Jahn, M. Jennewein and F. Rösch, J. Nucl. Med., 2007, 48, 1741–1748 CrossRef CAS PubMed.
- A. A. Larenkov, A. B. Bruskin and G. E. Kodina, Radiochemistry, 2014, 56, 57–65 CrossRef CAS.
- D. J. Berry, Y. Ma, J. R. Ballinger, R. Tavare, A. Koers, K. Sunassee, T. Zhou, S. Nawaz, G. E. D. Mullen, R. C. Hider and P. J. Blower, Chem. Commun., 2011, 47, 7068–7070 RSC.
- M. T. Ma, C. Cullinane, C. Imberti, J. B. Torres, S. Y. A. Terry, P. Roselt, R. J. Hicks and P. J. Blower, Bioconjugate Chem., 2016, 27, 309–318 CrossRef CAS PubMed.
- W. A. P. Breeman, M. de Jong, E. de Blois, B. F. Bernard, M. Konijnenberg and E. P. Krenning, Eur. J. Nucl. Med. Mol. Imaging, 2005, 32, 478–485 CrossRef CAS PubMed.
- J. Seemann, E. Eppard, B. P. Waldron, T. L. Ross and F. Rösch, Appl. Radiat. Isot., 2015, 98, 54–59 CrossRef CAS PubMed.
- G. J. Meyer, H. Macke, J. Schuhmacher, W. H. Knapp and M. Hofmann, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 1097–1104 CrossRef CAS PubMed.
- E. Eppard, M. Wuttke, P. L. Nicodemus and F. Rösch, J. Nucl. Med., 2014, 55, 1023–1028 CrossRef CAS PubMed.
- P. Y. Keng, S. Chen, H. Ding, S. Sadeghi, G. J. Shah, A. Dooraghi, M. E. Phelps, N. Satyamurthy, A. F. Chatziioannou, C.-J. C. J. Kim and R. M. van Dama, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 690–695 CrossRef CAS PubMed.
- V. Arima, G. Pascali, O. Lade, H. R. Kretschmer, I. Bernsdorf, V. Hammond, P. Watts, F. De Leonardis, M. D. Tarn, N. Pamme, B. Z. Cvetkovic, P. S. Dittrich, N. Vasovic, R. Duane, A. Jaksic, A. Zacheo, A. Zizzari, L. Marra, E. Perrone, P. A. Salvadori and R. Rinaldi, Lab Chip, 2013, 13, 2328–2336 RSC.
- K. Cabrera, J. Sep. Sci., 2004, 27, 843–852 CrossRef CAS PubMed.
- N. Tanaka, H. Kobayashi, N. Ishizuka, H. Minakuchi, K. Nakanishi, K. Hosoya and T. Ikegami, J. Chromatogr. A, 2002, 965, 35–49 CrossRef CAS.
- M. D. Tarn, D. Maneuski, R. Alexander, N. J. Brown, V. O'Shea, S. L. Pimlott, N. Pamme and S. J. Archibald, Chem. Commun., 2016, 52, 7221–7224 RSC.
- J. H. Smith and H. M. McNair, J. Chromatogr. Sci., 2003, 41, 209–214 CAS.
- E. Klodzinska, D. Moravcova, P. Jandera and B. Buszewski, J. Chromatogr. A, 2006, 1109, 51–59 CrossRef CAS PubMed.
- C. Legido-Quigley, N. D. Marlin, V. Melin, A. Manz and N. W. Smith, Electrophoresis, 2003, 24, 917–944 CrossRef CAS PubMed.
- M. P. Taggart, M. D. Tarn, M. M. N. Esfahani, D. M. Schofield, N. J. Brown, S. J. Archibald, T. Deakin, N. Pamme and L. F. Thompson, Lab Chip, 2016, 16, 1605–1616 RSC.
- F. Svec, J. Sep. Sci., 2005, 28, 729–745 CrossRef CAS PubMed.
- P. He, J. Davies, G. Greenway and S. J. Haswell, Anal. Chim. Acta, 2010, 659, 9–14 CrossRef CAS PubMed.
- P. D. I. Fletcher, S. J. Haswell, P. He, S. M. Kelly and A. Mansfield, J. Porous Mater., 2011, 18, 501–508 CrossRef CAS.
- K. Nakanishi and N. Soga, J. Non-Cryst. Solids, 1992, 139, 1–13 CrossRef CAS.
- P. He, S. J. Haswell, P. D. I. Fletcher, S. M. Kelly and A. Mansfield, J. Flow Chem., 2012, 2, 47–51 CrossRef CAS.
- J. S. Fritz and T. A. Rettig, Anal. Chem., 1962, 34, 1562–1566 CrossRef CAS.
- F. W. E. Strelow, A. H. Victor, C. R. Vanzyl and C. Eloff, Anal. Chem., 1971, 43, 870–876 CrossRef CAS.
- S. D. Chambers, K. M. Glenn and C. A. Lucy, J. Sep. Sci., 2007, 30, 1628–1645 CrossRef CAS PubMed.
- M. Luisa Soto-Montenegro, S. Pena-Zalbidea, J. Maria Mateos-Perez, M. Oteo, E. Romero, M. Angel Morcillo and M. Desco, PLoS One, 2014, 9 Search PubMed.
- Y. Menda, L. L. B. Ponto, M. K. Schultz, G. K. D. Zamba, G. L. Watkins, D. L. Bushnell, M. T. Madsen, J. J. Sunderland, M. M. Graham, T. M. O'Dorisio and M. S. O'Dorisio, Pancreas, 2013, 42, 937–943 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of monolith preparation, analysis and radiochemistry. See DOI: 10.1039/c6re00081a |
| This journal is © The Royal Society of Chemistry 2016 |