
Acylation or phosphorylation of hydroxyurea unexpectedly takes place on N rather than on O, leading to the formation of amides instead of the expected esters†
Natalie Pariente-Cohen,
Michal Weitman,
Nassdyuk Tania,
Dan T. Major,
Hugo E. Gottlieb,
Shmaryahu Hoz and
Abraham Nudelman*
Chemistry Department, Bar-Ilan University, Ramat Gan 52900, Israel. E-mail: nudelman@biu.ac.il
First published on 24th February 2015
Abstract
Attempted acylation of the anticancer agent hydroxyurea (HU) with acyl chlorides or anhydrides led to acylation on the NH group rather than on the OH. The structures of the products were confirmed by 15N-HMBC NMR. An analogous reaction conducted with hydroxamic acids (RCONHOH) or N-hydroxycarbamates (ROCONHOH) led to acylation on the OH. Surprisingly, despite the established affinity of phosphorous to O, phosphorylation of HU also took place at the NH group instead of the OH. These results are rationalized based on the different dominant resonance structures of HU, the hydroxamic acids or the N-hydroxycarbamates.
Results and discussion
Previous studies from our laboratory have dealt with the synthesis and biological evaluation of various mutual prodrugs, where the main goal of those studies was to examine possible synergistic effects between the components of the prodrugs. The investigations included the synthesis and biological evaluations of the following derivatives: (a) all-trans-retinoic acid (ATRA), an anti-acne agent, linked to butyric acid, a histone deacetylase (HDAC) inhibitor;1 (b) tegafur, a prodrug of the anticancer agent 5-fluorouracyl (5-FU), linked to butyric acid;2 (c) a derivative of two HDAC inhibitors, 4-phenylbutyric acid and butyric acid, as an anticancer agent;3 (d) perphenazine, a dopamine antagonist bound to γ-aminobutyric acid (GABA), as an anti-schizophrenic agent;4 (e) 5-aminolevulinic acid (5-ALA) linked to butyric acid as an anticancer agent for photodynamic therapy;5 (f) the GABA amide of nortriptyline, an antidepressant, for the treatment of neuropathic pain.6 Herein we describe studies related to a mutual prodrug of hydroxyurea (HU), an agent used in the treatment of myelocytic leukemia, and the HDAC inhibitors, valproic and butyric acids.Although a large number of hydroxamic acid derivatives have been reported in the literature,7 and many patents claim various uses for these compounds, only a few compounds that possess hydroxamic acid groups (R–CONHOH) are currently in clinical use, including: vorinostat, as a HDAC inhibitor, and HU as an anticancer agent. Thus, we intended to prepare and study the esters of HU connected to acidic HDAC inhibitors. A literature search of acylated HU derivatives revealed that very few publications claim the synthesis of HU esters, described as being synthesized by O-acylation of HU8 or via the reaction of isocyanates with hydroxylamine.9 Whereas Exner reported10 that the benzoylation of HU with benzoic anhydride led to the benzoate ester 1 (Scheme 1), in our studies we found that the acylation took place easily on the secondary nitrogen rather than on the hydroxyl group. Herein we report on various products obtained from the reaction of activated acids with HU. To prove our claim that the acylation takes place preferentially on the NH group, we repeated Exner's procedure10 and found that the obtained product was the amide 2 (ref. 11) rather than the claimed ester 1, or the acylated enolic form of HU 3 (Scheme 1).
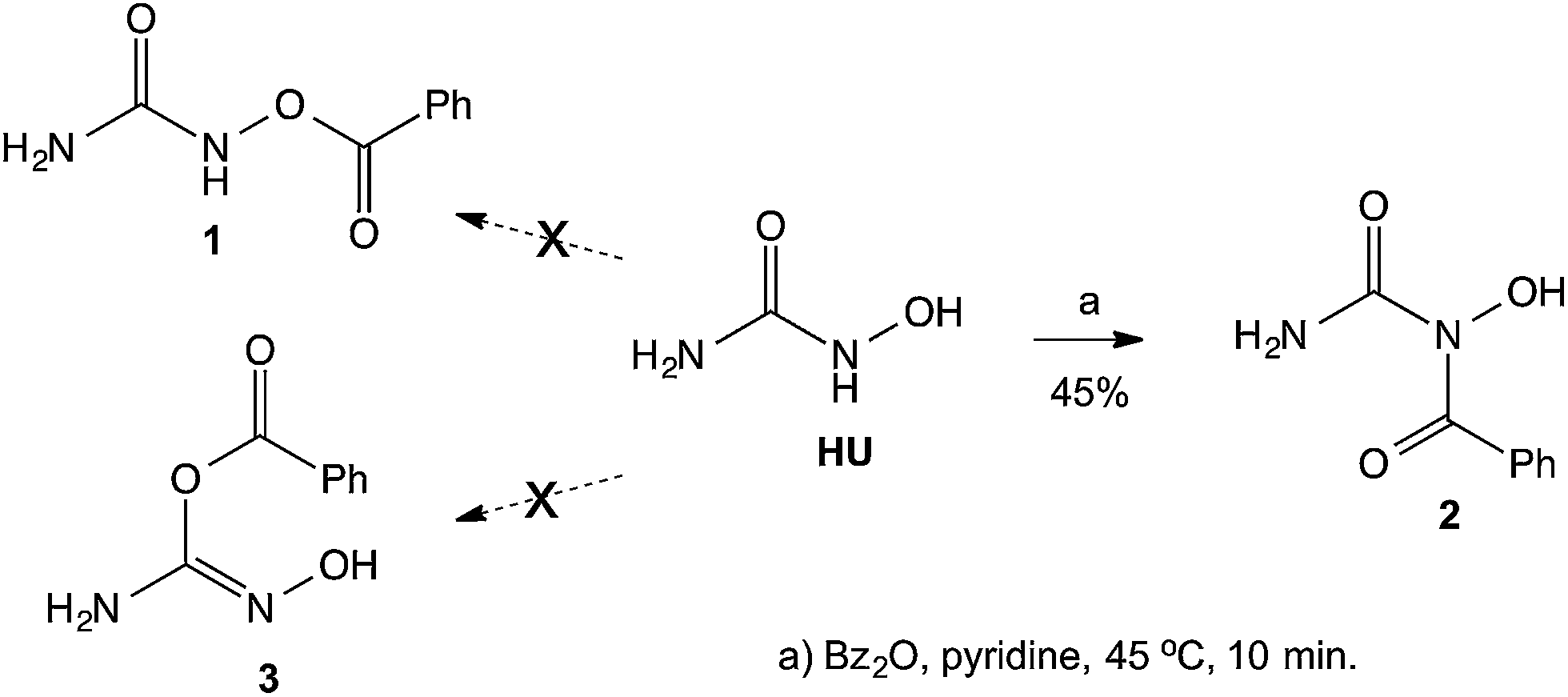 | ||
| Scheme 1 Benzoylation of HU. | ||
Since the 1H NMR spectrum of the product is inconclusive as to the site of acylation, whether it takes place on the N or on either of the O's, and in the 13C NMR the chemical shift of the carbonyl of the acyl group does not readily distinguish between a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O that belongs to an ester or to an amide, we resorted to determine the 15N-HMBC NMR spectrum of the product. This experiment, based on the 15N-1H coupling identifies unambiguously, NH or NH2 groups, as the 15N signal of these is split in the 1H dimension into a doublet (J = ca. 90 Hz) due to the one-bond coupling in the presence of –NH– or –NH2. When the H is not directly attached to the N, but is found 2 or three atoms away, the longer-range coupling constants (2J and 3J) are much smaller, and even if their correlation peaks appear, they are not visibly split. Furthermore, the chemical shift of the N in NH2 groups in ureas is usually found at ∼–300 ppm, that of amides NCOR at ∼–200 ppm, and that of oximes N–OH at ∼–50 ppm.12 By examination of the NMR spectrum, we can identify which of the protons on a heteroatom are connected to a N atom and by exclusion, those that are connected to O. In the spectrum of the benzoylated product of HU the following observations are made: (a) the 15N-HMBC showed an N signal at −305.8 ppm, which was attached through one bond to the two H's at 6.36 ppm, indicating the presence of an amide –CONH2; (b) a second N that appears at ∼−200 ppm indicative of an NCOR; (c) no N is found to have chemical shift of ∼−50 ppm; and (e) the other 1H on a heteroatom (δ 9.24) is not connected to a N and is therefore assigned to the OH group. Based on these observations, the assignment of the structure of the product is that of compound 2 (Fig. 1).
O that belongs to an ester or to an amide, we resorted to determine the 15N-HMBC NMR spectrum of the product. This experiment, based on the 15N-1H coupling identifies unambiguously, NH or NH2 groups, as the 15N signal of these is split in the 1H dimension into a doublet (J = ca. 90 Hz) due to the one-bond coupling in the presence of –NH– or –NH2. When the H is not directly attached to the N, but is found 2 or three atoms away, the longer-range coupling constants (2J and 3J) are much smaller, and even if their correlation peaks appear, they are not visibly split. Furthermore, the chemical shift of the N in NH2 groups in ureas is usually found at ∼–300 ppm, that of amides NCOR at ∼–200 ppm, and that of oximes N–OH at ∼–50 ppm.12 By examination of the NMR spectrum, we can identify which of the protons on a heteroatom are connected to a N atom and by exclusion, those that are connected to O. In the spectrum of the benzoylated product of HU the following observations are made: (a) the 15N-HMBC showed an N signal at −305.8 ppm, which was attached through one bond to the two H's at 6.36 ppm, indicating the presence of an amide –CONH2; (b) a second N that appears at ∼−200 ppm indicative of an NCOR; (c) no N is found to have chemical shift of ∼−50 ppm; and (e) the other 1H on a heteroatom (δ 9.24) is not connected to a N and is therefore assigned to the OH group. Based on these observations, the assignment of the structure of the product is that of compound 2 (Fig. 1).
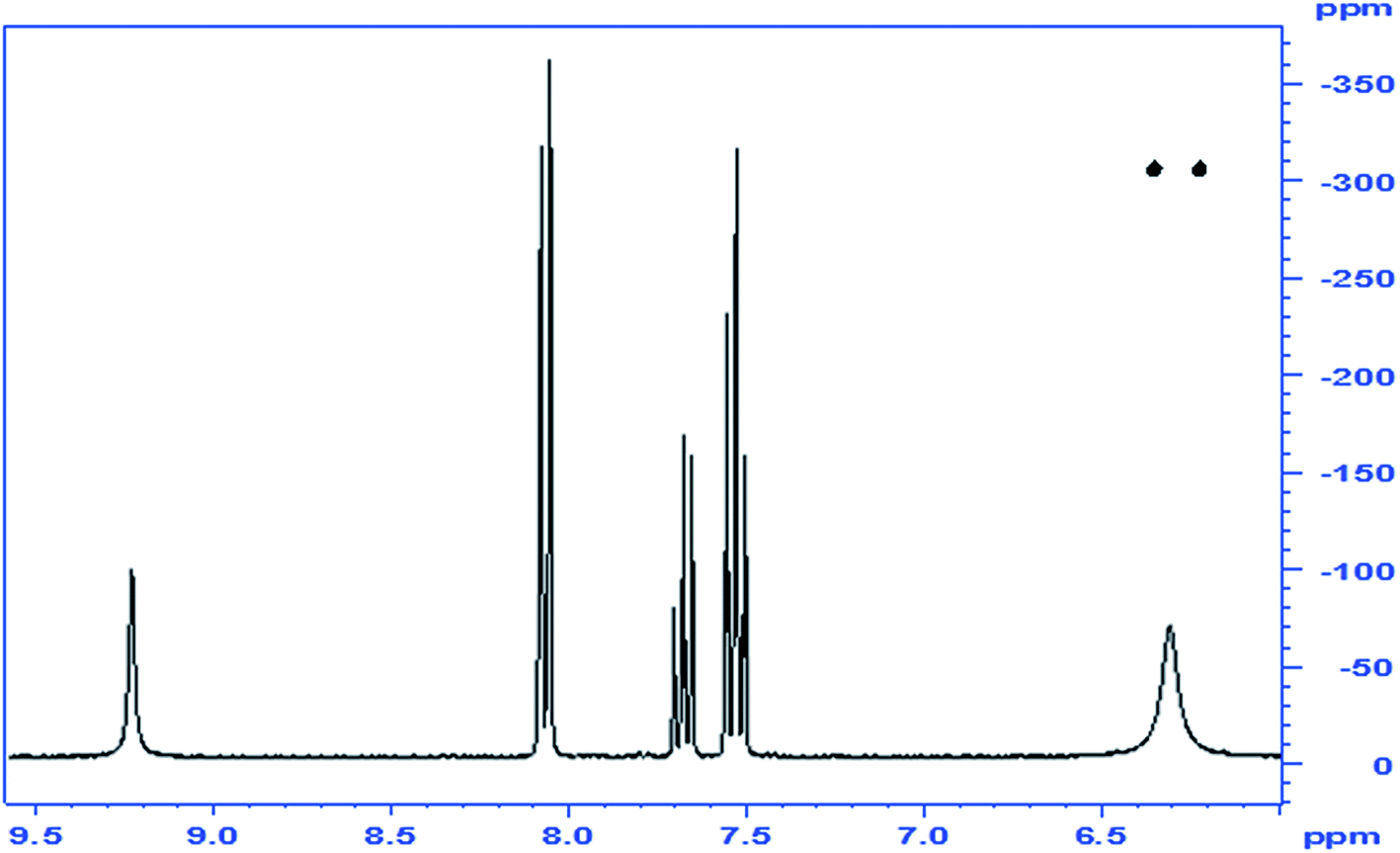 | ||
| Fig. 1 1Hx15N NMR correlation spectrum of compound 2, the N-benzoylated product of HU; inset: 1H-1D spectrum. | ||
When analogous acylations of HU were carried out with valproyl chloride the main product 4 was again found to be that of N-acylation. In addition, the acylation led to the formation of a small amount of the N,O-bis-valproylated hydroxylamine 5, the formation of which may be accounted for by decomposition of an N,O-bis-valproylated HU intermediate with concomitant release of HNCO, or by initial breakdown of 4 to give the N-acylated hydroxylamine, which underwent further O-acylation with another equivalent of the valproyl chloride to give compound 5 (Scheme 2). A similar reaction was reported by Exner10 involving the exclusive formation of the analog of 5 (R = Ph)13 upon treatment of HU with benzoyl chloride (Fig. 2).
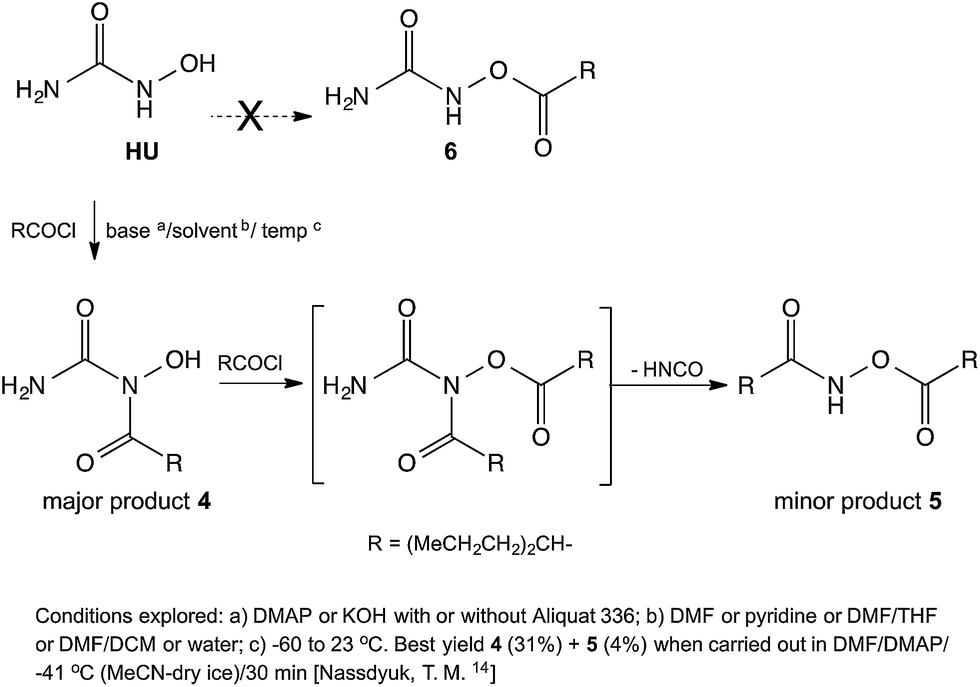 | ||
| Scheme 2 Reaction of HU with valproyl or butyryl chloride. | ||
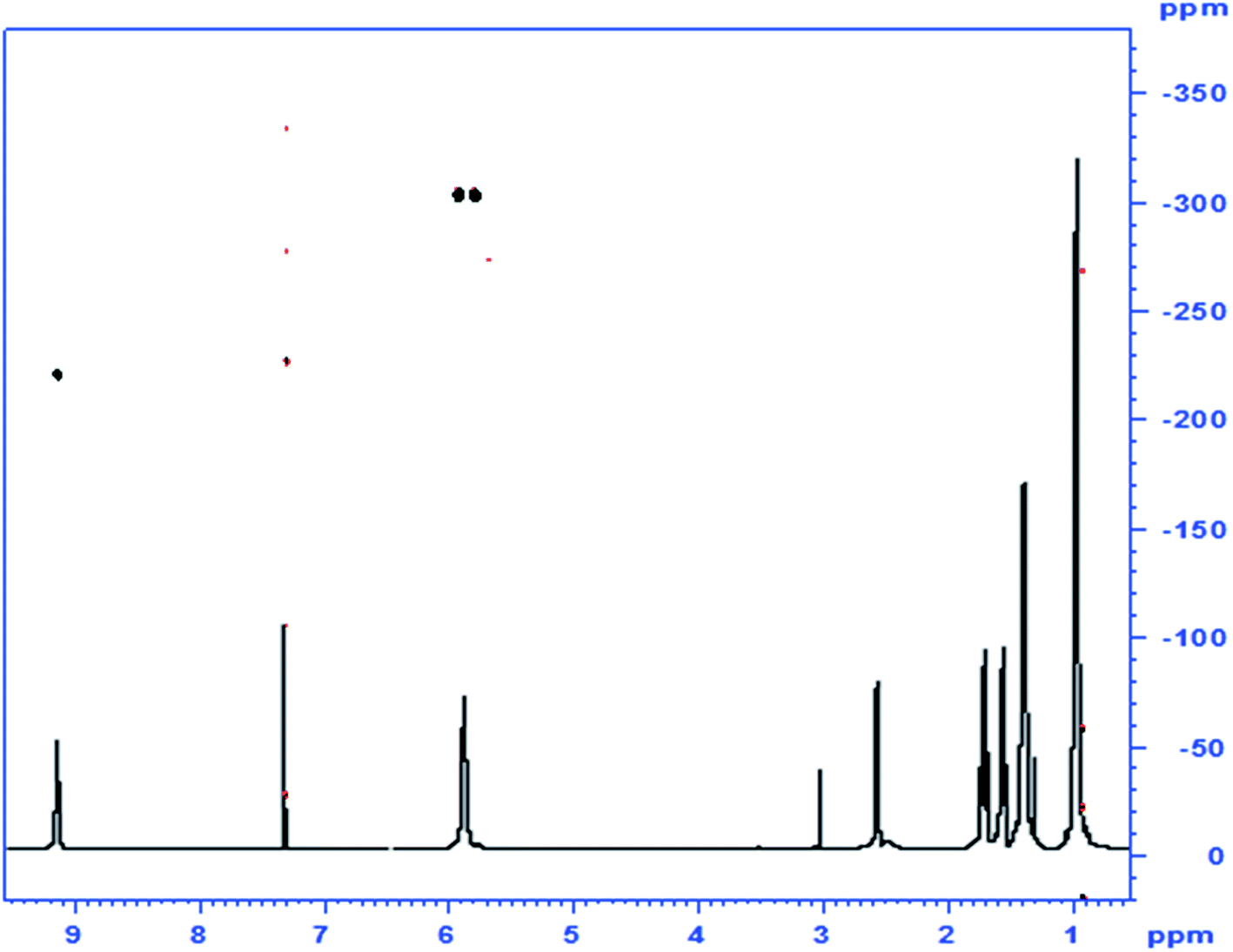 | ||
| Fig. 2 1Hx15N NMR correlation spectrum of compound 4, the N-valproylated product of HU; inset: 1H-1D spectrum. | ||
Since HU possess a –CONHOH group, analogous to that found in hydroxamic acids, we examined the reactions of N-hydroxypivalamide 7 (ref. 15) and the N-hydroxycarbamate 10 (ref. 16) with valproyl chloride. In these cases the products of O-acylation 8 and 11 formed readily, and no N-acylated products 9 or 12 were detected. This outcome shows that HU is an unusual hydroxamic acid and the presence of the NH2 group instead of alkyl or alkoxy groups caused the HU to react differently (Scheme 3).
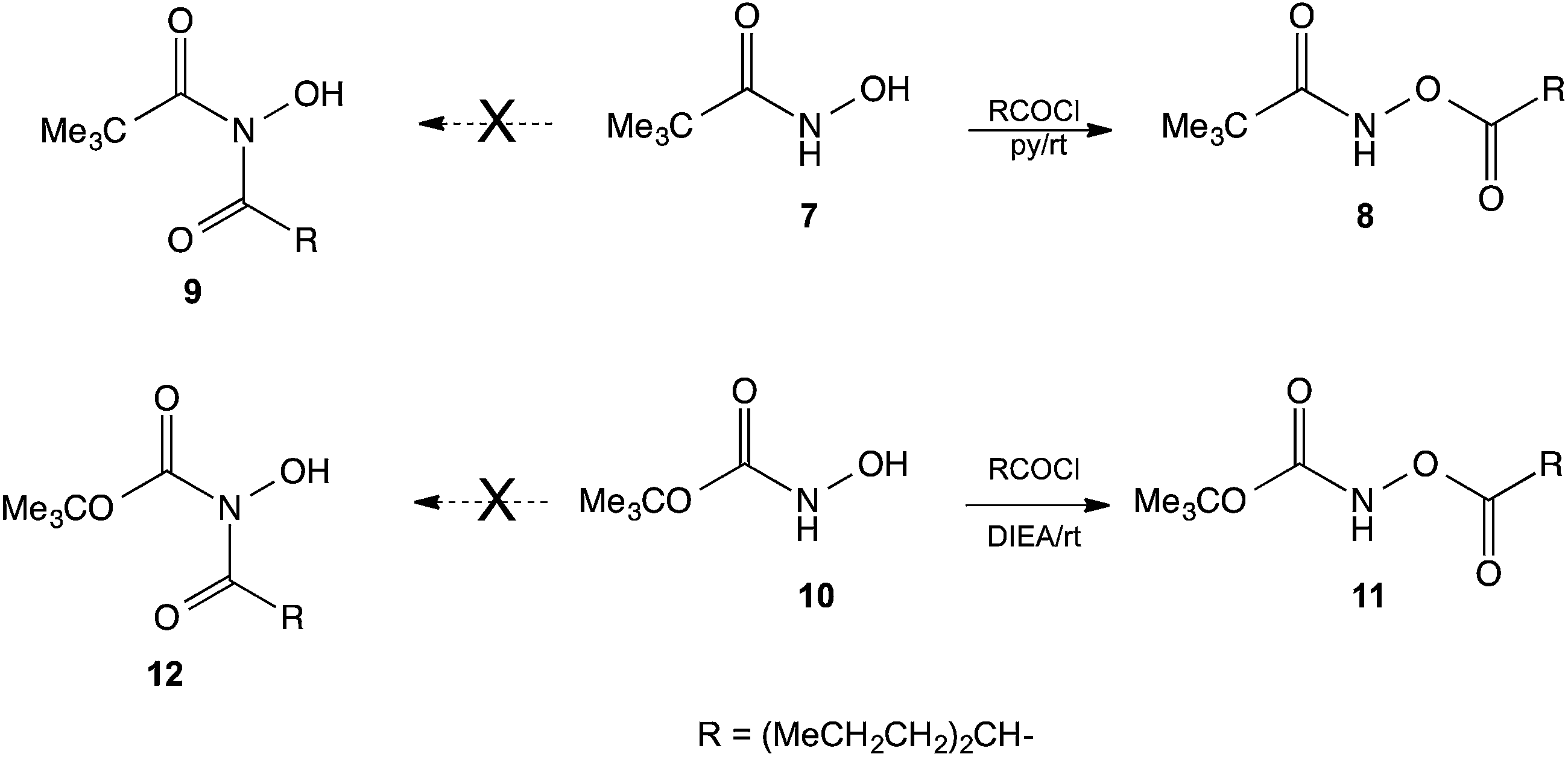 | ||
| Scheme 3 O-Acylation of a hydroxamic acid and an N-hydroxycarbamate. | ||
In an attempt to understand the different behavior of HU toward acylation in comparison to other hydroxamic acid analogs we compared the resonance structures expected for these compounds. In the case of HU, we suggest that the primary resonance takes place between the unshared electrons on the NH2 nitrogen and the oxygen of the carbonyl, whereas in the hydroxamic acid and the N-hydroxycarbamate the main resonance takes place between the unshared electrons on the NH nitrogen and the oxygen of the carbonyl (Scheme 4). Thus, in the case of the HU the unshared electrons on the NH group are readily available for nucleophilic attack leading to the found N-hydroxyimide. Further support for this suggestion is based on the fact that X-ray crystallography of HU reveals that the length of the C–NH2 bond is 1.328 Å whereas that of the C–NHOH is 1.347 Å.17 This length difference may indicate that the C–NH2 contributes the dominant resonance structure and might have more sp2 hybridization, making the N of the –NHOH group more basic-nucleophilic leading to the observed N-acylation. This observation, however does not clarify why the acylation takes place on the N and not on the O of the NHOH, and moreover it does not rationalize why the phosphorylation also takes place on the N rather than on the O, despite the well-established affinity of P to O. Whereas in the hydroxamic acid and the N-hydroxycarbamate the electron concentration of the NH is diminished, leading to the acylation of the OH groups.
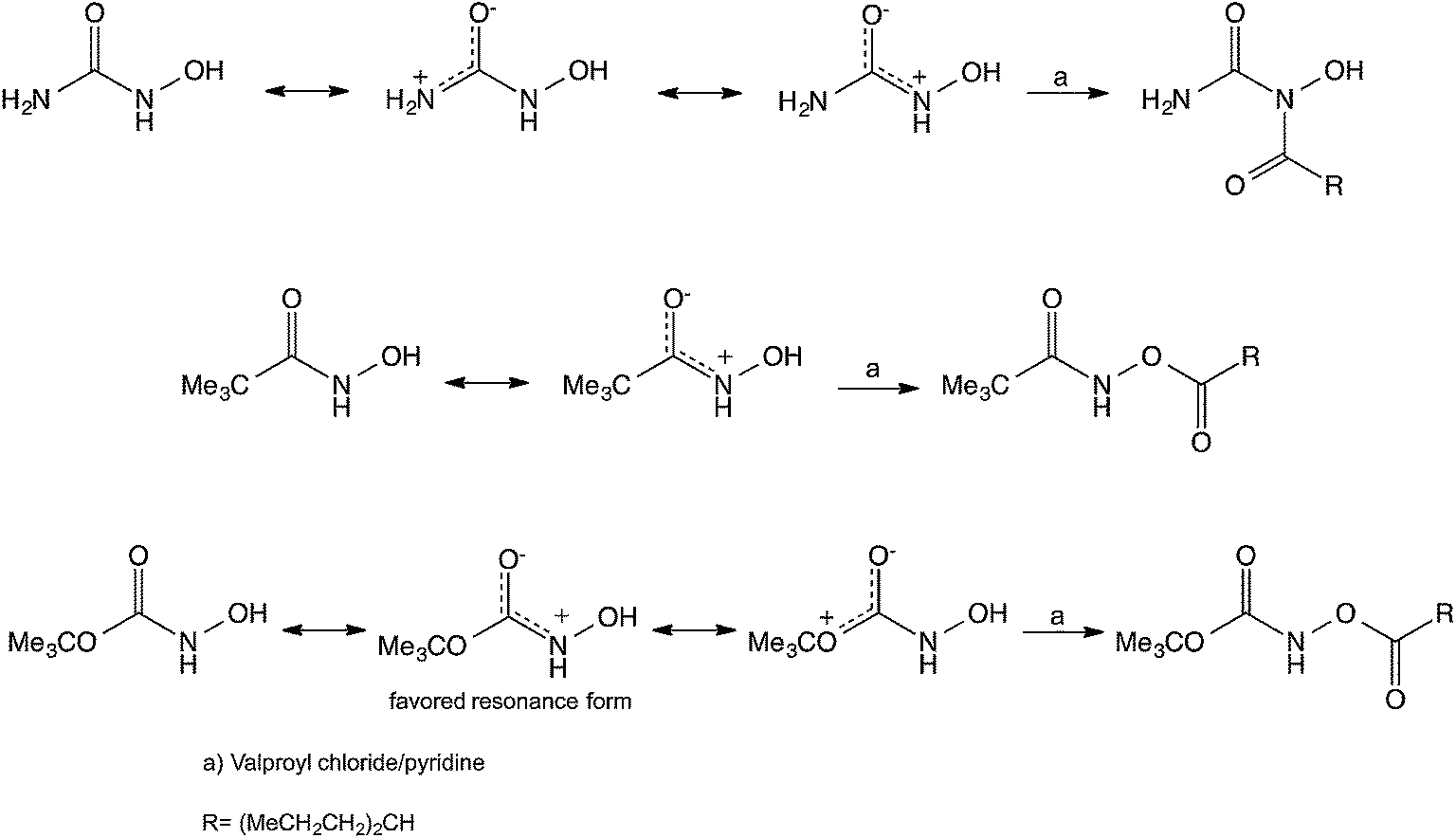 | ||
| Scheme 4 Some suggested resonance structures and the obtained acylation products of HU, a hydroxamic acid and an N-hydroxycarbamate. | ||
An additional cause for the preference of NH over the NH2 as the nucleophilic site comes from the α-effect.18 This effect is observed in cases where a lone pair, carrying atom resides α to the nucleophilic atom. The anion of hydrogen peroxide is a classic example of this group of nucleophiles, which exhibit an enhanced nucleophilicity mainly towards compounds with a low lying LUMO.18e Hence, because of the neighboring oxygen atom, the NH will be more nucleophilic than the NH2 group.
In view of the unexpected course of HU acylation described above, we proceeded with an attempted phosphorylation of HU. In this case, based on the well established, high-affinity of phosphorous to oxygen, it was expected that the phosphorylation would take place on the oxygen of HU. Surprisingly, when HU was reacted with diethyl chlorophosphate the phosphorylation again took place on the NH and not on the OH to give the corresponding diethyl phosphoramidate 13 instead of the phosphates 14 or 15 (Scheme 5). The structure of the isolated product 13 was also established by its 15N-HMBC NMR and by the chemical shifts in the 15N NMR spectrum, where the N's have chemical shifts of ∼−300 ppm and ∼−200 ppm, and not ∼−50 ppm as would have been expected had the compound contain an N–OH group as that shown in structure 15 (see experimental). It appears that based on the above suggested resonance argument, the N in the NH group is of sufficiently nucleophilic character that it overcomes the well-established affinity of O to P, leading to the N-phosphorylated product.
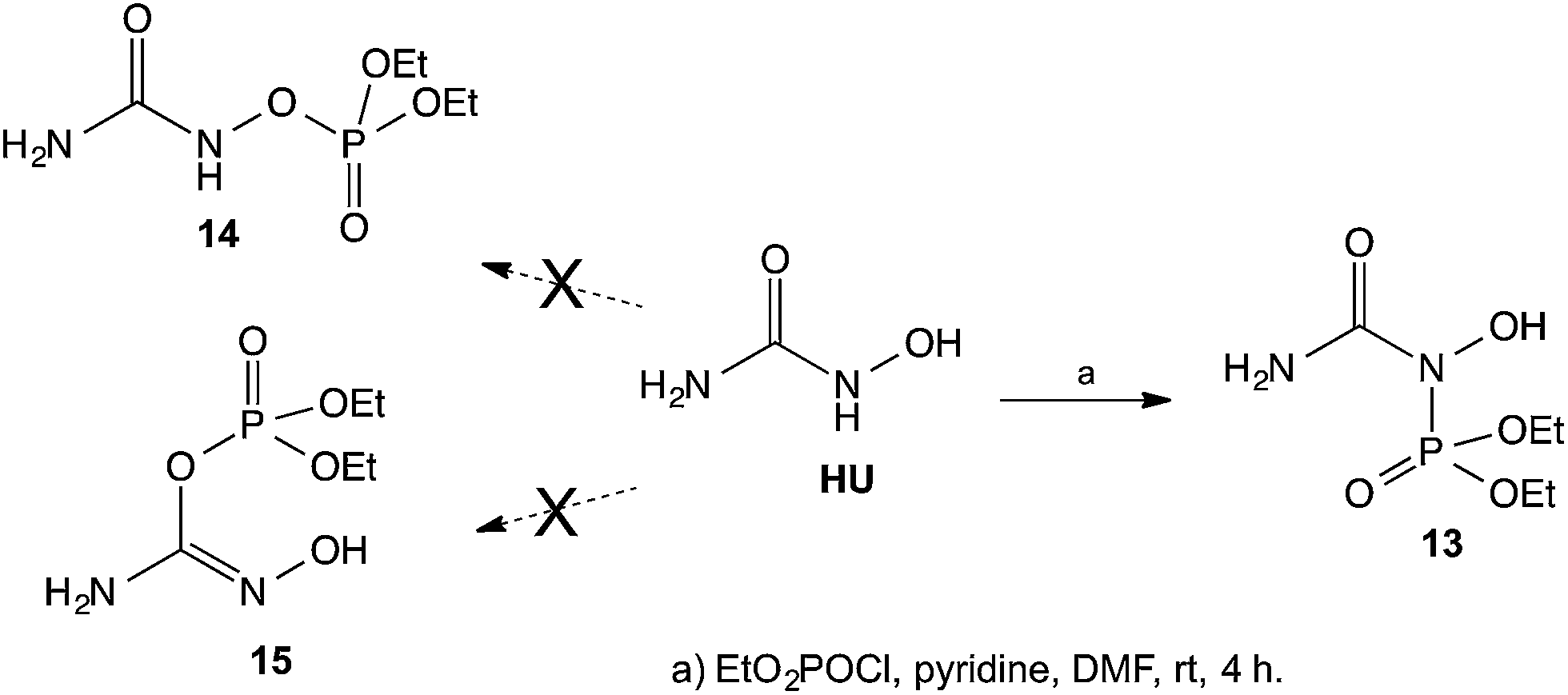 | ||
| Scheme 5 Phosphorylation of HU. | ||
Conclusion
In conclusion, several attempts were made to synthesize the valproate ester of HU, 6, as a potential mutual prodrug of the HDAC inhibitor valproic acid and the established anticancer agent HU. A literature search of acylated HU derivatives revealed that very few publications claim the synthesis of HU esters. Whereas an early publication claimed that the benzoylation of HU led to the benzoate ester, 1, in our studies we found that the acylation took place easily on the secondary N to give compound 2 rather than on the hydroxyl group. Moreover, and surprisingly, in the course of further investigations it was found that even phosphorylation of HU took place on the N rather than on the O, leading to the phosphoramide 13. This result was highly unexpected based on the well established, high-affinity of phosphorous to oxygen. The analogous acylation reaction of the hydroxamic acid 7 or the N-hydroxycarbamate 10, gave the O-acylated products 8 and 11. The structural assignments of the products were based on their 15N-HMBC NMR spectra. These results support our experimental findings and show that HU is an unusual hydroxamic acid and the presence of the NH2 group rather than alkyl or alkoxy groups, found in hydroxamic acids, caused it to react differently. We rationalized these results based on the different resonance structures of HU, the hydroxamic acid and an N-hydroxycarbamate.General information
1H, 13C and 15N NMR spectra were obtained on Bruker Avance-II-200, DPX-300, DMX-600 and Avance-III-700 spectrometers. Chemical shifts for 1H and 13C are expressed in ppm downfield from Me4Si (TMS) used as internal standard and for 15N are related to neat nitromethane as an external standard; the chemical shifts for 15N were determined indirectly from the 15N-HMBC spectra. The values are given in δ scale. LRMS were also obtained on a QToF micro (Waters UK) spectrometer in ESI (= Electronspray Ionization), HRMS were obtained on an AutoSpec Premier (Waters UK) spectrometer in EI+ mode, and SYNAPT spectrometer (Waters UK). Progress of the reactions was monitored by TLC on silica gel (Merck, Art. 5554). Flash chromatography was carried out on silica gel (Merck, Art. 9385). Melting points were determined on a Fisher-Johns apparatus. The commercially available valproic acid and HU were used without further purification. The nomenclature of the compounds was assigned according to ChemDraw Ultra v. 11.0.1 (CambridgeSoft). Valproyl chloride19 and N-hydroxypivalamide15 and N-Boc-hydroxylamine16 were prepared according to known procedures.N-Carbamoyl-N-hydroxybenzamide, (2)11
The following procedure is analogous to that described by Exner10 where the chromatographic purification has been added. To a solution of hydroxyurea (0.5 g, 6.57 mmol) in dry pyridine at 45 °C, benzoic anhydride (1.48 g, 6.57 mmol) was added slowly. The solution was then heated for 10 min at 45 °C. The solvent was then evaporated and the residue was purified by flash chromatography silica gel, eluent EtOAc–n-hexane 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to give 2 (45% yield, mp 125–127 °C).
:1 to give 2 (45% yield, mp 125–127 °C).
1H NMR (300 MHz, acetone-D6): δ: 6.36 (bs, 2H), 7.56 (t, J = 15.93 Hz, 2H), 7.71 (t, J = 15.93 Hz, 2H), 8.10 (d, J = 6.37 Hz, 2H), 9.24 (bs, 1H).
13C NMR (75 MHz, acetone-D6): δ: 128.68, 129.60, 130.48, 134.74, 160.48, 166.19.
15N NMR (indirectly from HMBC spectrum): δ: −305.8 (NH2).
MS (ES+) m/z 181 (MH+, 100); 203 ([M + Na]+, 96).
Anal. calcd for C8H8N2O3: C, 53.33; H, 4.48; N, 15.55; O, 26.64. found: C, 53.48; H, 4.38; N, 15.35; O, 26.14%.
N-Carbamoyl-N-hydroxy-2-propylpentanamide (4) and 2-propyl-N-((2-propylpentanoyl)oxy)pentanamide (5)13
To a solution of hydroxyurea (0.5 g, 6.57 mmol) in dry DMF under N2 was added DMAP (0.8 g, 6.57 mmol) followed by valproyl chloride (1.07 g, 6.57 mmol). The mixture was stirred at −41 °C (acetonitrile/dry ice bath) for 30 min and was then evaporated. The residue, which contained a mixture of 4 and 5 was purified by chromatography (EtOAc–n-hexane 1:20 -> 1:1), to give 5 as a white solid, mp 45–47 °C, (4% yield) and 4 as a yellowish oil (31% yield).
1H NMR of compound 4 (700 MHz, CDCl3): δ: 0.91 (t, J = 7 Hz, 6H), 1.33 (m, 4H), 1.51 (m, 2H), 1.66 (m, 2H), 2.52 (m, 1H), 5.85 (bs, 2H), 9.14 (bs, 1H).
13C NMR (176 MHz, CDCl3): δ: 13.90, 20.54, 34.40, 43.40, 160.09, 174.66.
15N NMR (obtained indirectly from the HMBC spectrum): δ: −221 (N–OH), −303 (NH2).
MS (ES+) m/z 203 (MH+, 100); 225 ([M + Na]+, 96). HRMS calcd for C9H18N2O3Na (M+, Na+) 225.1222, found 225.1215.
1H NMR of compound 5 (700 MHz, CDCl3): δ: 0.91 (m, 12H), 1.28–1.45 (m, 10H), 1.51 (m, 2H), 1.67 (m, 4H), 2.15 (m, 1H), 2.57 (m, 1H), 8.95 (s, 1H).
13C NMR (176 MHz, CDCl3): δ: 13.94, 14.05, 20.51, 20.65, 34.43, 34.86, 43.19, 44.24, 174.40, 174.98.
15N NMR (obtained indirectly from the HMBC spectrum): δ: −208.8.
MS (ES+) m/z 286 (MH+, 100); 308 ([M + Na]+, 9).
HRMS (APPI+) calcd for C16H31NO3Na (M+, Na+) 308.2202, found 308.2209.
N-(2-Propylpentanoyloxy)pivalamide (8)
To a solution of N-hydroxypivalamide 7 (ref. 15) (0.3 g, 2.56 mmol) in dry pyridine under N2 was added valproyl chloride (0.4 g, 2.56 mmol). The mixture was stirred at ∼23 °C for 4 h, was then concentrated and the residue was purified by column chromatography (EtOAc–n-hexane 1:20), to give 8 as a white solid, mp 60–62 °C (90% yield).
1H NMR (700 MHz, CDCl3): δ: 0.91 (t, J = 7 Hz, 6H), 1.28 (bs, 9H), 1.38 (m, 4H), 1.51 (m, 2H), 1.68 (m, 2H), 2.57 (m, 1H), 9.04 (s, 1H).
13C NMR (176 MHz, CDCl3): δ: 13.94, 20.49, 27.21, 34.42, 38.68, 43.22, 175.17, 176.85.
15N NMR (obtained indirectly from the HMBC spectrum, 700 MHz, CDCl3): δ: −212.2.
MS (ES+) m/z 244 (MH+, 100), 266 ([M + Na]+, 28.86).
Anal. calcd for C13H25NO3: C, 64.16; H, 10.36; N, 5.76; found: C, 63.46; H, 10.75; N, 5.81%.
tert-Butyl((2-propylpentanoyl)oxy)carbamate (11)
To a solution of N-Boc-hydroxylamine 10 (ref. 16) (0.1 g, 0.75 mmol) in dry DCM under N2 were added DIEA (0.1 g, 0.75 mmol) followed by valproyl chloride (0.12 g, 0.75 mmol). The mixture was stirred at room temperature for 30 min, concentrated, diluted with EtOAc and acidified 1 M KHSO4. The layers separated and the organic phase was washed with NaHCO3, dried over Na2SO4, filtered and evaporated. The residue was purified by chromatography (EtOAc–n-hexane 1:10) to give 11 as a clear oil (81% yield).
1H NMR (700 MHz, CDCl3): δ: 0.84 (m, 6H), 1.28 (m, 4H), 1.40 (m, 9H), 1.60 (s, 2H), 2.46 (m, 1H), 7.98 (s, 1H).
13C NMR (176 MHz, CDCl3): δ: 13.74, 20.30, 27.87, 34.19, 42.97, 82.74, 155.54, 175.64.
15N NMR (obtained indirectly from the HMBC spectrum, 700 MHz, acetone-D6): δ: −240.0.
MS (ES+) m/z 282 ([M + Na]+, 40).
HRMS (APPI−) calcd for C13H25NO4 (M−) 258.1705, found 258.1718.
Diethyl carbamoyl(hydroxy)phosphoramidate (13)
To a solution of hydroxyurea, (0.2 g, 2.63 mmol) in dry pyridine at ice bath temperature, was dropwise added diethyl chlorophosphate (0.45 g, 2.63 mmol). The mixture was stirred at ∼23 °C for 4 h, a precipitate that formed was filtered, the filtrate was evaporated and the residue was purified by chromatography (DCM–MeOH 30:1) to give 13 as a white solid, 115–117 °C (25% yield).
1H NMR (700 MHz, CDCl3): δ: 1.37 (t, J = 8 Hz, 6H), 4.26 (m, 4H), 6.22 (s, 2H), 8.98 (s, 1H).
13C NMR (176 MHz, CDCl3): δ: 16.03 (d, J = 6 Hz), 65.72 (d, J = 6 Hz), 161.47 (d, J = 4 Hz).
15N NMR (indirectly from HMBC spectrum): δ: −220.1 (N–OH), −300.8 (NH2CO).
TOF MS (ES+) m/z 235 ([M + Na]+, 100).
31P NMR (400 MHz, CDCl3): δ: 1.16.
Anal. calcd for C5H13N2O5P: C, 28.31; H, 6.18; N, 13.21; found: C, 28.84; H, 6.15; N, 13.06%.
Acknowledgements
This work was partially supported by the “Marcus Center for Pharmaceutical and Medicinal Chemistry” in Bar-Ilan University.References
- (a) A. Nudelman and A. Rephaeli, J. Med. Chem., 2000, 43, 2962–2966 CrossRef CAS PubMed; (b) K. K. Mann, A. Rephaeli, A. L. Colosimo, Z. Diaz, A. Nudelman, I. Levovich, Y. Jing, S. Waxman and W. H. Miller Jr, Mol. Cancer Res., 2003, 12, 903–912 Search PubMed.
- D. Engel, A. Nudelman, N. Tarasenko, I. Levovich, I. Makarovsky, S. Sochotnikov, I. Tarasenko and A. Rephaeli, J. Med. Chem., 2008, 51, 314–323 CrossRef CAS PubMed.
- M. Entin-Meer, A. Rephaeli, X. Yang, A. Nudelman and D. A. Hass-Kogan, Cancer Lett., 2007, 253, 205–214 CrossRef CAS PubMed.
- (a) A. Nudelman, I. Gil-Ad, N. Shpaisman, I. Tarasenko, H. Ron, K. Savitsky, Y. Geffen, A. Weizman and A. Rephaeli, J. Med. Chem., 2008, 51, 2858–2862 CrossRef CAS PubMed; (b) Y. Geffen, A. Nudelman, I. Gil-Ad, A. Rephaeli, M. Huang, K. Savitsky, L. Klapper, I. Winkler, H. Y. Meltzer and A. Weizman, Eur. Neuropsychopharmacol., 2009, 19, 1–13 CrossRef CAS PubMed.
- G. Berkovitch-Luria, M. Weitman, A. Nudelman, A. Rephaeli and Z. Malik, Invest. New Drugs, 2012, 30, 1028–1038 CrossRef CAS PubMed.
- A. Rephaeli, I. Gil-Ad, A. Aharoni, I. Tarasenko, N. Tarasenko, Y. Geffen, E. Halbfinger, Y. Nisemblat, A. Weizman and A. Nudelman, J. Med. Chem., 2009, 52, 3010–3017 CrossRef CAS PubMed.
- H. Weinmann and E. Ottow, Annu. Rep. Med. Chem., 2004, 39, 185–196 CAS.
- (a) C. D. Hurd and L. U. Spence, J. Am. Chem. Soc., 1927, 49, 266–274 CrossRef CAS; (b) L. Wang, T. Peng, H. Jiang, E. Wang, X. Wen, L. Li, J. Liu, S. Zhang and X. Pan, Faming Zhuanli Shenqing, CN 1785972 A 20060614, 2006; (c) O. Exner, Collect. Czech. Chem. Commun., 1957, 22, 335–336 CrossRef CAS; (d) G. Zinner and R. Stoffel, Arch. Pharm., 1969, 302, 838–847 CrossRef CAS.
- (a) R. L. Dannley and M. Lukin, J. Org. Chem., 1956, 21, 1036–1037 CrossRef CAS; (b) K. C. Murddock and R. B. Angier, J. Org. Chem., 1962, 27, 3317–3319 CrossRef.
- (a) O. Exner, Chem. Listy, 1956, 50, 2025–2026 CAS; (b) O. Exner, Chem. Listy, 1957, 51, 497–504 Search PubMed.
- R. J. Yu and E. J. Van Scott, J. Invest. Dermatol., 1974, 63, 279–283 CAS.
- S. Berger, S. Braun and H. O. Kalinowski, NMR Spectroscopy of the Non-Metallic Elements, Wiley, Chichester, 1997 Search PubMed.
- M. Levi, B. Yagen and M. Bialer, Pharm. Res., 1997, 14, 213–217 CrossRef CAS.
- T. Nassdyuk, M. Sc. thesis, Bar Ilan University, 2007.
- C. Tsukano, M. Okuno and Y. Takemoto, Angew. Chem., Int. Ed., 2012, 51, 2763–2766 CrossRef CAS PubMed.
- A. C. Willis and M. D. McLeod, J. Org. Chem., 2012, 77, 8480–8491 CrossRef PubMed.
- The authors thank one of the referees for pointing out to us this relevant reference, W. E. Thiessen, H. A. Levy and B. D. Flaig, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2495–2502 CrossRef.
- (a) E. Buncel and I. H. Um, Tetrahedron, 2004, 60, 7801–7825 CrossRef CAS PubMed; (b) S. Hoz and E. Buncel, Isr. J. Chem., 1985, 26, 313–319 CrossRef CAS; (c) A. P. Grekov and V. Y. Beselov, Russ. Chem. Rev., 1978, 47, 631–648 CrossRef PubMed; (d) N. J. Fina and J. O. Edwards, Int. J. Chem. Kinet., 1973, 5, 1–26 CrossRef CAS; (e) S. Hoz, J. Org. Chem., 1982, 47, 3545–3547 CrossRef CAS.
- M. S. Wolfe, Synth. Commun., 1997, 27, 2975–2984 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01016k |
| This journal is © The Royal Society of Chemistry 2015 |