
One-pot protocol to synthesize N-(β-nitro)amides by tandem Henry/Ritter reaction†
Wensi Aib,
Ronghua Shib,
Liyan Zhua,
Dehong Jiangb,
Xiaobo Maa,
Jilan Yuana and
Zhouyu Wang*ab
aDepartment of Chemistry, Xihua University, Chengdu, 610039, China. E-mail: zhouyuwang@mail.xhu.edu.cn; zhouyuwang77@gmail.com; Fax: +86-28-87723006
bDepartment of Pharmaceutics Engineering, Xihua University, Chengdu, 610039, China
First published on 25th February 2015
Abstract
A novel, efficient and atom economical one pot protocol for the synthesis of N-(β-nitro)amides has been described by combining the Henry reaction with the Ritter reaction. The designed products could be obtained from easily available aldehydes, nitroalkanes and nitriles with 60–83% overall yields under mild reaction conditions. In addition, the product can easily be transformed into diamine or protective amine derivatives.
Introduction
N-(β-nitro)amides are important organic intermediates that have been widely used in the synthesis of natural products, drugs and agrochemicals since the nitro group can be easily converted to other useful functional groups such as amine, carboxylic acid, or oxime.1,2 Thus, studies for the development of efficient methods to synthesize N-(β-nitro)amides have attracted considerable attention both from organic chemists and medicinal chemists. The first method to synthesize N-(β-nitro)amides was reported by Jelena and co-workers in 1952,3,4 which use nitro methane or ethyl nitro acetate to substitution of the amide group of bisamides to form the designed products. From then on, a variety of synthetic methods have been reported to synthesize N-(β-nitro)amides such as aza-Michael additions of amides to nitro alkenes,5–8 aza-Henry reaction of nitro protected imines9–11 and nitroacetamidation12–14 of styrene (Fig. 1). However, most of the present methods for synthesize N-(β-nitroalkyl)amides are not atom economical and suffer from limited substrates. Therefore, the development of an efficient and atom economical method for the synthesis of N-(β-nitro)amides with sufficient diversity is still highly desirable and of significant value. | ||
| Fig. 1 Selected methods to synthesize N-(β-nitro)amides. | ||
Ritter reaction is a classical acid-catalyzed reaction between a carbocation precursor, such as a substituted olefin or alcohol, and nitriles to generate amides.15–38 This is an atom economical reaction. It has versatile applications for the synthesis of active pharmaceutical intermediates and heterocycles, particularly for the preparation of bulky amides, which can be precursors of hindered amines. In recent years, it has been developed greatly both in the catalyst system and the substrate scope. Variously amides, such as unsymmetrical di- and trisubstituted ureas, 3-substituted-3-aminooxindoles, 4-acylam-inotetrahydroindazoles, N-(4-iodo-1,3-diarylbutyl)acetamides, have been synthesized with this reaction. In principle, Ritter reaction could be an efficient method to synthesize N-(β-nitro)amides by reacting the corresponding β-nitro alcohols or nitro alkenes with nitriles. However, to the best of knowledge, the synthesis of N-(β-nitro)amides by Ritter reaction has been rarely explored. Only one example was reported by Bach,21 which use 2-nitro-1-phenylpropan-1-ol 1 to react with benzonitrile under the catalysis of trifluoromethanesulfonic acid to get the N-(β-nitro)amides 3 with good yield and moderate diastereoselectivity (Scheme 1). Up to now, a general preparation of N-(β-nitro)amides by Ritter reaction is still underdeveloped.
 | ||
| Scheme 1 Bach reported N-(β-nitro)amides by Ritter reaction. | ||
It is obvious that the alcohol 1 could be synthesized by the Henry reaction between benzylaldehyde and nitro ethane.39–44 Thus, we are wondering whether the N-(β-nitro)amide product 3 could be synthesized by reacting acetonitrile directly with the in situ formed alcohol 1 that prepared by the nucleophilic addition of nitro ethane to benzylaldehyde under the catalysis of base. Herein, we wish to report a novel and efficient protocol to synthesize N-(β-nitro)amides by tandem Henry/Ritter reaction, which has a versatile substrates scope and an environment-friendly reaction conditions.
Results and discussion
Initially, benzylaldehyde and nitro methane were chosen as the standard substrates to optimize the reaction conditions of Henry reaction. It was found that 1,4-diazabicyclo[2.2.2]octane (DABCO) was a proper base for the Henry reaction, and 67% yield could be obtained when one equivalent of DABCO was added into the dichloromethane solution of the substrates at 20 °C (entries 1–5, Table 1). To the best of our knowledge, there is no report about DABCO was used as a promoter in Henry reaction. Highest yield could be obtained when set up the reaction in acetonitrile under otherwise identical conditions, which clearly shown that the polar solvent system was helpful to get better yield for the reaction (entries 5–8, Table 1). The yield would drop to 71% from 83% when the reaction temperature was increased to 40 °C, and 90% yield could be obtained when the reaction temperature was decreased to 0 °C. Decreasing the reaction temperature to −20 °C could not clearly improve the yield (entries 8–11, Table 1). Thus, 0 °C was used in the following experiments. Next, we found 95% yield could be achieved when 0.5 equivalent of DABCO was added into the reaction. Further decreasing the loading of DABCO or reaction time resulted poor yield (entries 13–16, Table 1). The same yield was obtained when the reaction time was prolonged to 48 hours (entry 17, Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Base (eq.) | Temp. (°C) | Sol. | Time (h) | Yieldb (%) |
| a 1.0 mmol of benzaldehyde and 4.0 mmol of nitromethane were used for each reaction.b Isolated yield. DABCO = 1,4-diazabicyclo[2.2.2]octane. | |||||
| 1 | TEA (1) | 20 | DCM | 24 | 25 |
| 2 | DEA (1) | 20 | DCM | 24 | 20 |
| 3 | t-BuOK (1) | 20 | DCM | 24 | 44 |
| 4 | EtONa (1) | 20 | DCM | 24 | 20 |
| 5 | DABCO (1) | 20 | DCM | 24 | 67 |
| 6 | DABCO (1) | 20 | CHCl3 | 24 | 65 |
| 7 | DABCO (1) | 20 | Toluene | 24 | 10 |
| 8 | DABCO (1) | 20 | MeCN | 24 | 83 |
| 9 | DABCO (1) | 40 | MeCN | 24 | 71 |
| 10 | DABCO (1) | 0 | MeCN | 24 | 90 |
| 11 | DABCO (1) | −20 | MeCN | 24 | 91 |
| 12 | DABCO (0.5) | 0 | MeCN | 24 | 95 |
| 13 | DABCO (0.2) | 0 | MeCN | 24 | 45 |
| 14 | DABCO (0.1) | 0 | MeCN | 24 | 30 |
| 15 | DABCO (0.5) | 0 | MeCN | 6 | 50 |
| 16 | DABCO (0.5) | 0 | MeCN | 12 | 70 |
| 17 | DABCO (0.5) | 0 | MeCN | 48 | 95 |
| 18 | DABCO (0.5) | 0 | PhCN | 24 | 87 |
| 19 | DABCO (0.5) | 0 | BnCN | 24 | 90 |

With the optimized reaction conditions for Henry reaction in hand, we started to test whether Ritter reaction would happen by directly adding Lewis acid or Brønst acid to the reaction mixture. At the beginning, three equivalents of pure trifluoromethanesulfonicacid (TfOH) were added to the reaction mixture of Henry reaction, and no designed Ritter reaction product was detected no matter the reaction temperature was kept at 0 °C or increased to 80 °C for 24 hours (entries 1 and 2, Table 2). Fortunately, we found moderate yield could be obtained when 1 mL of acetonitrile was replaced with equal volume of other solvents such as toluene, chloroform and dichloromethane (entries 3–5, Table 2). Lewis acid such as Bi(OTf)3, BF3·Et2O and FeCl3 were not efficient catalyst for the current reaction system, and no more than 10% yield could be obtained when these acids were used in the reaction (entries 15–18, Table 2). Dichloromethane (DCM) was found to be the best solvent for the reaction since highest yield could be obtained albeit at low reaction temperature. Next, we found 82% yield could be achieved when 0.5 mL acetonitrile and 1.5 mL DCM were used as the solvent for the reaction. It seems that the ratio of the two solvents was important to get high yield since adding more DCM or acetonitrile into the reaction would cause a lower yield (entries 6–9, Table 2). 40 °C was the best temperature for the reaction, lowering the temperature would cause a clearly decline of the yield. Three equivalents of TfOH were enough for the reaction. We didn't found obviously impact on the yield when more TfOH was added.
|
|||||
|---|---|---|---|---|---|
| Entry | MeCN (mL) | Sol. (mL) | Temp. (°C) | Acid (eq.) | Yieldb (%) |
| a 1.0 mmol of benzaldehyde and 4.0 mmol of nitromethane were used for each reaction.b Isolated yield. DNBSA = 2,4-dinitrobenzene sulfonic acid. | |||||
| 1 | 2.0 | — | 0 | TfOH (3.0) | ND |
| 2 | 2.0 | — | 80 | TfOH (3.0) | ND |
| 3 | 1.0 | Toluene (1.0) | 100 | TfOH (3.0) | 34 |
| 4 | 1.0 | CHCl3 (1.0) | 60 | TfOH (3.0) | 25 |
| 5 | 1.0 | DCM (1.0) | 40 | TfOH (3.0) | 35 |
| 6 | 0.5 | DCM (1.5) | 40 | TfOH (3.0) | 82 |
| 7 | 0.5 | DCM (2.0) | 40 | TfOH (3.0) | 78 |
| 8 | 0.5 | DCM (4.0) | 40 | TfOH (3.0) | 50 |
| 9 | 0.5 | DCM (6.0) | 40 | TfOH (3.0) | 20 |
| 10 | 0.5 | DCM (1.0) | 40 | TfOH (3.0) | 55 |
| 11 | 0.5 | DCM (1.5) | 20 | TfOH (3.0) | 60 |
| 12 | 0.5 | DCM (1.5) | 0 | TfOH (3.0) | 35 |
| 13 | 0.5 | DCM (1.5) | 40 | TfOH (2.0) | 47 |
| 14 | 0.5 | DCM (1.5) | 40 | TfOH (4.0) | 80 |
| 15 | 0.5 | DCM (1.5) | 40 | Bi(OTf)3 (3.0) | <5 |
| 16 | 0.5 | DCM (1.5) | 40 | DNBSA (3.0) | <5 |
| 17 | 0.5 | DCM (1.5) | 40 | BF3·Et2O (3.0) | <5 |
| 18 | 0.5 | DCM (1.5) | 40 | FeCl3 (3.0) | <5 |
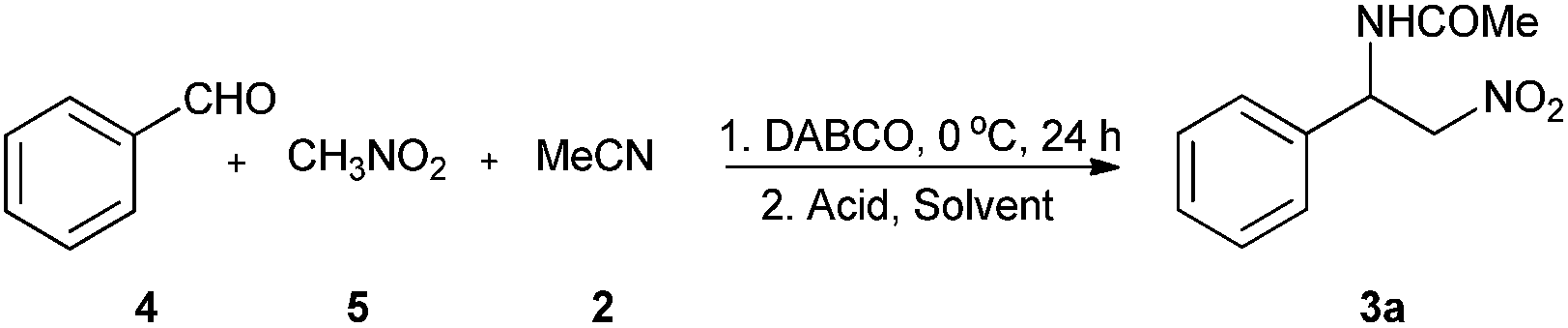
With the optimized reaction conditions in hand, the substrate scope of the reaction was explored. Various aldehydes and nitriles were used and a series of N-(β-nitro)amides were synthesized through this protocol. We found the structure of nitrile was important for the reaction. As shown in Table 3, moderate to good yields could be obtained when acetonitrile, benzonitrile and phenylacetonitrile were used in the reaction, but no product could be isolated when acrylonitrile was used under otherwise identical conditions. All the tested aromatic aldehydes were good substrates for the reaction. Nevertheless, aliphatic aldehydes were not good substrate for the reaction, and no designed product could be isolated when they were used. The yield of the reaction was barely impacted by the electron donating or withdrawn groups on the phenyl ring of the aromatic aldehydes. The stereohindrance of the aldehyde would not decline the activity significantly. As shown in 3r–w, 62–71% yield could be obtained also when 2-bromobenzaldehyde and 2-naphthaldehyde were used in the reaction. Nitroethane was still a proper substrate for the reaction, and more than 70% yield and 69% dr value could be obtained. Heteroaromatic aldehyde such as thiophene-2-carbaldehyde could be used as good substrate for the current reaction system, and 80% of 3aa was obtained.
|
|---|
| a 1.0 mmol of benzaldehyde and 4.0 mmol of nitromethane were used for each reaction, isolated yield. |
 |

To illustrate the synthetic utility of the present method, the conversion of N-(2-nitro-1-phenylethyl) benzamide 3b into 1,2-diamine 7 and double protective derivatives 8 were conducted by known methods, respectively (Scheme 2). The corresponding products 7 and 8 were obtained in good yields.
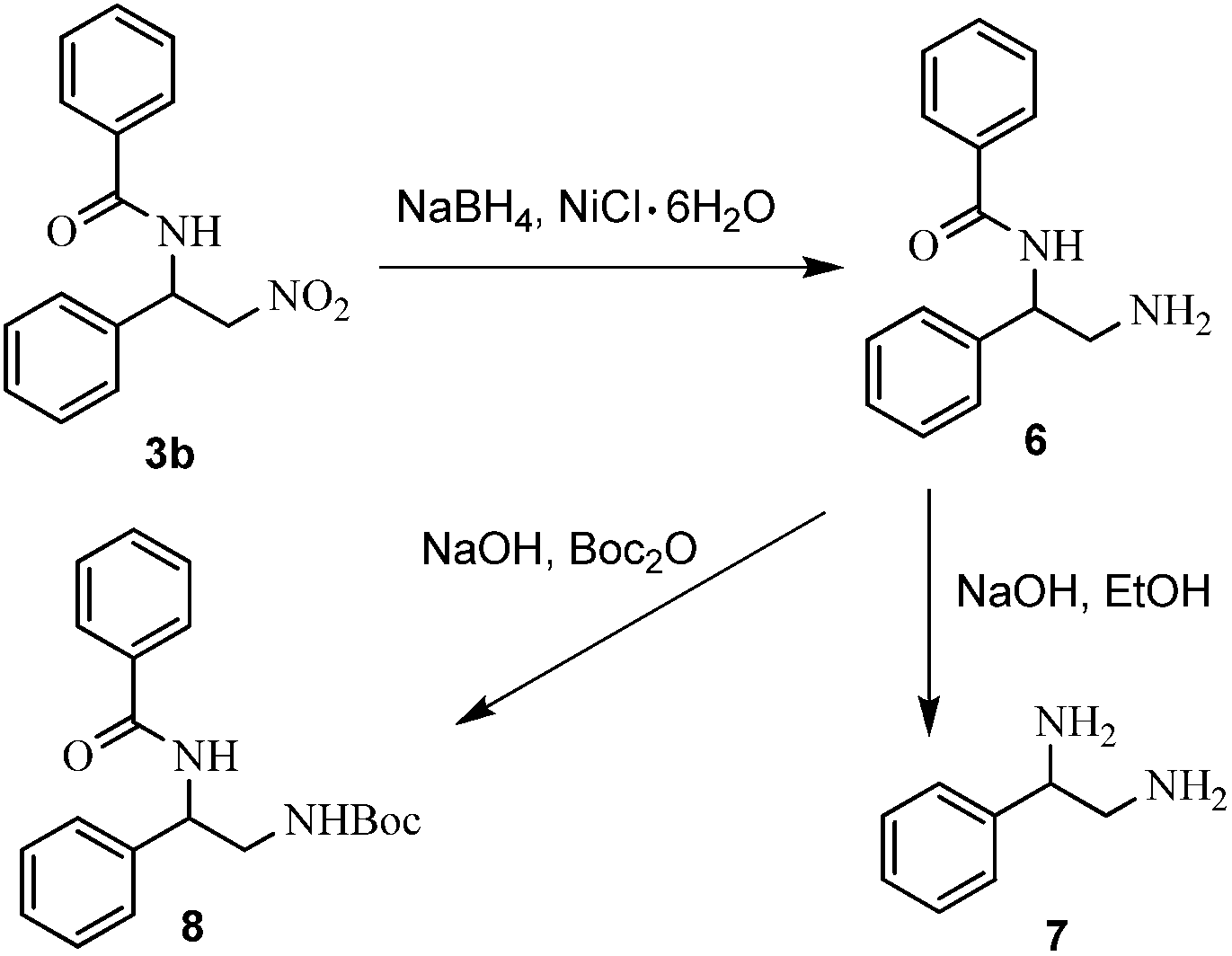 | ||
| Scheme 2 Conversion of N-(2-nitro-1-phenylethyl) benzamide 3b into 1,2-diamine 7 and double protective derivatives 8. | ||
Conclusions
In conclusion, a novel, efficient and atom economical one pot protocol for the synthesis of N-(β-nitro)amides has been described by combining the Henry reaction with Ritter reaction. The DABCO was firstly found to be an efficient promoter in the Henry reaction. The designed N-(β-nitro)amides could be easily obtained with 60–83% over all yields by directly adding TfOH to the solution of Henry reaction under mild reaction conditions. The electron rich and deficient aromatic and heteroaromatic aldehydes were tolerable in the current system, and high yield could be obtained for all the tested aromatic aldehydes. The protocol provided a simple and environment-friendly alternative to prepare N-(β-nitro)amides from easily available aldehydes, nitroalkane and nitriles. Furthermore, the product can be transformed into diamine or protective amine derivatives.Experimental section
General procedures for the synthesis of 2-nitro-1-phenyl-ethanol
The mixture of benzaldehyde (1.0 mmol), nitromethane (4.0 mmol) and base were stirred in the solvent at designed temperature for designed time. Then the solvent was removed under reduced pressure and the residue was purified by silicagel column chromatography (petroleum ether/ethyl acetate = 10/1) to give the corresponding 2-nitro-1-phenylethanol.General procedures for the synthesis of N-(β-nitroalkyl)amides
The 2-nitro-1-arylethanol (1.0 mmol), nitrile (4 mmol), and 1,4-diazabicyclo[2.2.2]octane (DABCO, 0.5 mmol) were dissolved in nitrile (0.5 mL). The reaction mixture was stirred at 0 °C for 24 hours. Then the trifluoromethanesulfonic acid (3 mmol) was added and the mixture was stirred at 40 °C for 24 hours. The reaction was then quenched by the addition of sodium bicarbonate at room temperature. The solvent was removed under reduced pressure and the residue was purified by silicagel column chromatography (petroleum ether/ethyl acetate = 10/1) to give the corresponding N-(β-nitroalkyl)amides.The procedures for the synthesis of 7
To a well-stirred mixture of N-(2-nitro-1-phenylethyl) benzamide (130.1 mg, 0.5 mmol) and nickel chloride hexahydrate (119 mg, 0.5 mmol) in absolute ethyl alcohol (5 mL) at 0 °C, sodium borohydride (90 mg, 5 mmol) was added. The mixture was stirred at 0 °C for 2 hours and then quenched with saturated aqueous NH4Cl. The mixture was diluted with dichloromethane. The organic layer was separated and aqueous layer was extracted with CH2Cl2 (10 mL × 3). The combined extracts were washed with water (10 mL × 2), dried over magnesium sulfate and concentrated under vacuum to afford crude amine intermediate 6. The crude amine 6 was dissolved in absolute ethyl alcohol (5 mL). Sodium hydroxide was introduced slowly which made the pH exceed 12. The mixture was stirred at 90 °C for 5 hours and then concentrated under reduced pressure. After purification by flash chromatography with neutral alumina (dichloromethane/methyl alcohol = 10/1), the product 7 was obtained (82% yield).The procedures for the synthesis of 8
To a well-stirred mixture of N-(2-nitro-1-phenylethyl) benzamide (130.1 mg, 0.5 mmol) and nickel chloride hexahydrate (119 mg, 0.5 mmol) in absolute ethyl alcohol (5 mL) at 0 °C, sodium borohydride (90 mg, 5 mmol) was added. The mixture was stirred at 0 °C for 2 hours and then quenched with saturated aqueous NH4Cl. The mixture was diluted with dichloromethane. The organic layer was separated and aqueous layer was extracted with CH2Cl2 (10 mL × 3). The combined extracts were washed with water, dried over magnesium sulfate and concentrated under vacuum to afford crude amine intermediate 4. The crude amine 4 and were dissolved in H2O/1,4-dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mL). The (Boc)2O (130.8 mg, 0.6 mmol) was added. Then sodium hydroxide was introduced slowly which made the pH exceed 12. The mixture was stirred at room temperature for 2 hours and then extracted with EtOAc (10 mL × 3). The combined organic extracts were dried over MgSO4. The solvent was removed under reduced pressure. After purification by flash chromatography (petroleum ether/ethyl acetate = 5/1), the product 8 were obtained (72% yield).
:2 mL). The (Boc)2O (130.8 mg, 0.6 mmol) was added. Then sodium hydroxide was introduced slowly which made the pH exceed 12. The mixture was stirred at room temperature for 2 hours and then extracted with EtOAc (10 mL × 3). The combined organic extracts were dried over MgSO4. The solvent was removed under reduced pressure. After purification by flash chromatography (petroleum ether/ethyl acetate = 5/1), the product 8 were obtained (72% yield).
Acknowledgements
We are grateful for the financial supports from the National Natural Science Foundation of China (21102115), the ChunhuiProject (Z2012020), the Great Cultivate Project from the Education Department of SichuanProvince (14CZ0014), the Undergraduate Scientific and Technological Innovation Project, and the Innovation Foundation of Graduate Student of Xihua University.Notes and references
- P. M. O'Brien, D. R. Sliskovic, C. J. Blankley, B. D. Roth, M. W. Wilson, K. L. Hamelehle, B. R. Krause and R. L. Stanfield, J. Med. Chem., 1994, 37, 1810–1822 CrossRef.
- R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS PubMed.
- G. Stefanovic and J. Bojanovic, J. Org. Chem., 1952, 17, 816–822 CrossRef CAS.
- G. Stefanović, J. Bojanović and K. Sirotanović, J. Org. Chem., 1952, 17, 1110–1113 CrossRef.
- Z. G. Chen, Y. Wang, J. F. Wei, P. F. Zhao and X. Y. Shi, J. Org. Chem., 2010, 75, 2085–2088 CrossRef CAS PubMed.
- S. Zhi, H. Mei, G. Zhang, H. Sun, J. Han, G. Li and Y. Pan, Sci. China: Chem., 2010, 53, 1946–1952 CrossRef CAS PubMed.
- Y. Qian, X. Ji, W. Zhou, J. Han, G. Li and Y. Pan, Tetrahedron, 2012, 68, 6198–6203 CrossRef CAS PubMed.
- S. Ma, L. Wu, M. Liu, Y. Huang and Y. Wang, Tetrahedron, 2013, 69, 2613–2618 CrossRef CAS PubMed.
- K. Komura, Y. Taninaka, Y. Ohtaki and Y. Sugi, Appl. Catal., A, 2010, 388, 211–215 CrossRef CAS PubMed.
- N. R. Amarasinghe, P. Turner and M. H. Todd, Adv. Synth. Catal., 2012, 354, 2954–2958 CrossRef CAS.
- A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS PubMed.
- A. J. Bloom, M. Fleischmann and J. M. Mellor, Tetrahedron Lett., 1984, 25, 4971–4974 CrossRef CAS.
- A. J. Bloom, M. Fleischmann and J. M. Mellor, J. Chem. Soc., Perkin Trans. 1, 1984, 2357–2362 RSC.
- C. J. Easton, P. D. Roselt and E. R. T. Tiekink, Tetrahedron, 1995, 51, 7809–7822 CrossRef CAS.
- For recent reviews, see: (a) A. Guérinot, S. Reymond and J. Cossy, Eur. J. Org. Chem., 2012, 19–28 CrossRef; (b) D. H. Jiang, T. He, L. Ma and Z. Y. Wang, RSC Adv., 2014, 4, 64936–64946 RSC.
- L. R. Jefferies and S. P. Cook, Tetrahedron, 2014, 70, 4204–4207 CrossRef CAS PubMed.
- M. M. Sa, M. Ferreira, G. F. Caramori, L. Zaramello, A. J. Bortoluzzi, D. Faggion Jr and J. B. Domingos, Eur. J. Org. Chem., 2013, 2013, 5180–5187 CrossRef CAS.
- V. Panduranga, H. Basavaprabhu and V. V. Sureshbabu, Tetrahedron Lett., 2013, 54, 975–979 CrossRef CAS PubMed.
- F. Zhou, M. Ding and J. Zhou, Org. Biomol. Chem., 2012, 10, 3178–3181 CAS.
- S. Q. Jiang, Z. Y. Wang, Z. J. Jiang and J. H. Li, Lett. Org. Chem., 2012, 9, 24–28 CrossRef CAS.
- P. Rubenbauer and T. Bach, Chem. Commun., 2009, 2130–2132 RSC.
- E. Callens, A. J. Burton and A. G. M. Barrett, Tetrahedron Lett., 2006, 47, 8699–8701 CrossRef CAS PubMed.
- R. Sanz, A. Martinez, V. Guilarte, J. M. Alvarez-Gutierrez and F. Rodriguez, Eur. J. Org. Chem., 2007, 4642–4645 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and A. Khoshnood, Catal. Commun., 2008, 9, 529–531 CrossRef CAS PubMed.
- B. Anxionnat, A. Guerinot, S. Reymond and J. Cossy, Tetrahedron Lett., 2009, 50, 3470–3473 CrossRef CAS PubMed.
- B. V. S. Reddy, N. S. Reddy, C. Madan and J. S. Yadav, Tetrahedron Lett., 2010, 51, 4827–4829 CrossRef PubMed.
- P. Theerthagiri, A. Lalitha and P. N. Arunachalam, Tetrahedron Lett., 2010, 51, 2813–2819 CrossRef CAS PubMed.
- S. Khaksar, E. Fattahi and E. Fattahi, Tetrahedron Lett., 2011, 52, 5943–5946 CrossRef CAS PubMed.
- N. Ibrahim, A. S. K. Hashmi and F. Rominger, Adv. Synth. Catal., 2011, 353, 461–468 CrossRef CAS.
- M. H. Al-huniti and S. D. Lepore, Adv. Synth. Catal., 2013, 355, 3071–3076 CrossRef CAS PubMed.
- D. Mondal, L. Bellucci and S. D. Lepore, Eur. J. Org. Chem., 2011, 7057–7061 CrossRef CAS PubMed.
- H. Basavaprabhu and V. V. Sureshbabu, Org. Biomol. Chem., 2012, 10, 2528–2533 CAS.
- M. Turks, I. Strakova, K. Gorovojs, S. Belyakov, Y. A. Piven, T. S. Khlebnicova and F. A. Lakhvich, Tetrahedron, 2012, 68, 6131–6140 CrossRef CAS PubMed.
- Y. V. Shklyaev, T. S. Vshivkova, O. A. Maiorova and A. A. Gorbunov, Russ. J. Org. Chem., 2012, 48, 257–267 CrossRef CAS.
- J.-M. Huang, Z.-J. Ye, D.-S. Chen and H. Zhu, Org. Biomol. Chem., 2012, 10, 3610–3612 CAS.
- S. Stavber, T. S. Pecan, M. Papez and M. Zupan, Chem. Commun., 1996, 2247–2248 RSC.
- M. B. Gawande, A. K. Rathi, I. D. Nogueira, R. S. Varma and P. S. Branco, Green Chem., 2013, 15, 1895–1899 RSC.
- N. Hazarika and G. Baishya, Eur. J. Org. Chem., 2014, 5686–5690 CrossRef CAS.
- For review, see: C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561–2574 CrossRef CAS.
- D. H. Jhuo, B. C. Hong, C. W. Chang and G. H. Lee, Org. Lett., 2014, 16, 2724–2727 CrossRef CAS PubMed.
- H. Huang, H. Zong, G. Bian, H. Yue and L. Song, J. Org. Chem., 2014, 79, 9455–9464 CrossRef CAS PubMed.
- Y. Zhou, Y. Zhu, S. Yan and Y. Gong, Angew. Chem., Int. Ed., 2013, 52, 10265–10269 CrossRef CAS PubMed.
- M. Tsakos, M. R. J. Elsegood and C. G. Kokotos, Chem. Commun., 2013, 49, 2219–2221 RSC.
- C. C. J. Loh, I. Atodiresei and D. Enders, Chem.–Eur. J., 2013, 19, 10822–10826 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01536g |
| This journal is © The Royal Society of Chemistry 2015 |