
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Processes for non-destructive transfer of graphene: widening the bottleneck for industrial scale production
Aliaksandr V.
Zaretski
and
Darren J.
Lipomi
*
Department of NanoEngineering, University of California, San Diego, 9500 Gilman Drive Mail Code 0448, La Jolla, CA 92093-0448, USA. E-mail: dlipomi@ucsd.edu
First published on 30th April 2015
Abstract
The exceptional charge-transport, mechanical, and barrier properties of graphene are well known. High-quality films of single-layer graphene produced over large areas, however, are extremely expensive. The high cost of graphene precludes its use in industries—such as transparent electrodes and flexible packaging—that might take full advantage of its properties. This minireview presents several strategies for the transfer of graphene from the substrates used for growth to substrates used for the final application. Each strategy shares the characteristic of being non-destructive: that is, the growth substrate remains reusable for further synthesis of new graphene. These processes have the potential to lower significantly the costs of manufacturing graphene, to increase production yields, and to minimize environmental impact. This article is divided into sections on (i) the synthesis of high-quality single-layer graphene and (ii) its non-destructive transfer to a host substrate. Section (ii) is further divided according to the substrate from which graphene is transferred: single-crystalline wafers or flexible copper foils. We also comment, wherever possible, on defects produced as a result of the transfer, and potential strategies to mitigate these defects. We conclude that several methods for the green synthesis and transfer of graphene have several of the right characteristics to be useful in industrial scale production.
1. Introduction.
While many of the most exciting potential applications of graphene exist at the nanoscale—e.g., all-graphene integrated circuits—this versatile material has equally exciting applications on the very large scale. For example, graphene can substitute for indium-tin oxide in transparent conducting electrodes,1,2 can serve as a gas-separation membrane,3 and exhibits barrier properties for the encapsulation of organic electronic devices.4 All these applications require large areas of this material. For graphene to constitute any considerable segment of the global markets for these applications, the outstanding performance and properties of graphene have to be accompanied by large-volume manufacturability at low cost and with minimal environmental impact. Manufacturing for large-area applications can be subdivided into two steps: synthesis and transfer.Methods for synthesizing graphene have been under investigation since 2004.5 The intensity of this work has resulted in the ability to synthesize large areas of graphene (100 m sheets) in a roll-to-roll fashion6 at high speeds (0.6 m min−1).7 Nevertheless, there appears to be a bottleneck in the industrial-scale production of graphene, that is, inexpensive, non-destructive transfer from the substrate used for synthesis to the final substrate used for the application. The current state-of-the-art techniques for transferring high-quality single layers are slow (i.e., they cost a minimum of 30 min per batch because of etching the growth substrate) and deleterious to the environment.8 To enable proliferation of large-area graphene to every industry for which the material has potential—and potentially great—value, the ability to transfer graphene rapidly and in a non-destructive manner is required. While there exist methodologies of inexpensive large-volume production of suspensions of exfoliated graphene that can be spray-coated onto a substrate to generate conducting surfaces9 or mixed in with polymers to make conductive composites10 and barrier films,11 this minireview focuses only on processes for transfer that preserve the desirable characteristics of high-quality single-layer graphene, including high transparency, high electron mobility, and low sheet resistance.12
2. Synthesis of large-area graphene
There are two principal methodologies for the synthesis of large-area single-layer graphene: silicon-carbide (SiC) epitaxy13 and chemical vapor deposition (CVD).14 While neither of the techniques guarantees the formation of a single-layer of graphene, both can be modified to do so, and both have their advantages and limitations.2.1 SiC epitaxy
In SiC epitaxy, a single crystalline SiC wafer is heated in a vacuum or argon atmosphere to the temperatures above 2000 °C. At this temperature, silicon atoms sublimate from the (0001) face of the crystal. The remaining carbon (which has a lower vapor pressure than silicon) rearranges to form graphene (Fig. 1a).13 By tight control of the parameters of this process, synthesis of single-, few-, and multilayer graphene has been demonstrated.15 One particular advantage of this method is that it produces graphene covering the entire surface of an insulating wafer, and thus allows the fabrication of circuitry directly on the growth substrate. Such graphene possesses exceptionally high electron mobilities, which is desirable for high-performance components of integrated circuits. We return to this process in section 3.1.1 when we discuss the process of transferring single layer graphene from the SiC wafer onto flexible host substrates.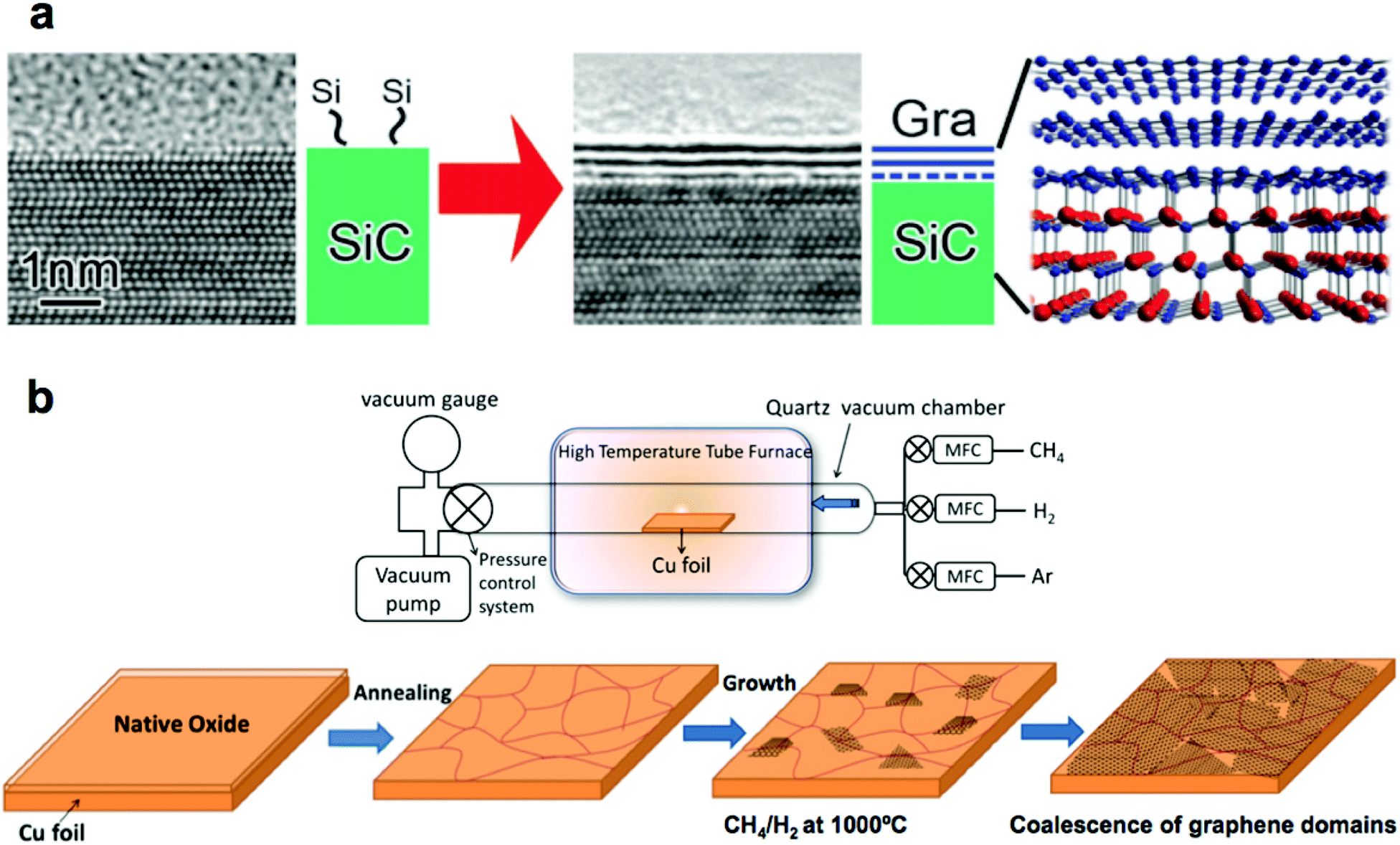 | ||
| Fig. 1 Methods for synthesizing high-quality large-area graphene. (a) Epitaxial synthesis of graphene on a SiC wafer. Reproduced with permission from ref. 13. Copyright 2014, Royal Society of Chemistry. (b) Chemical vapor deposition (CVD) of graphene on copper foil. Reproduced with permission from ref. 16, Copyright 2011, American Chemical Society, and from ref. 20, Copyright 2013, InTech. | ||
2.2 CVD of graphene
The process of chemical vapor deposition (CVD) of graphene offers great versatility as it allows synthesis of single- or multi-layer graphene on a large number of substrates (usually refractory metals,16Fig. 1b). The use of copper foil is by far the best studied, because copper is relatively inexpensive. Moreover, the low solubility of carbon in copper permits synthesis of predominantly single layer graphene of high quality.16 CVD of graphene can be performed in vacuum17 or atmospheric pressure18 and at temperatures as low as 300 °C.19 Additionally, as compared to SiC epitaxy, CVD of graphene is not limited to the small dimensions of a wafer.3. Graphene transfer methods
In order to be usable, the graphene synthesized on copper foil by CVD has to be transferred to the substrate of interest, which usually requires supporting of graphene by a polymeric film and etching the copper foil in a corrosive medium8,20 (Fig. 2). Such transfer processes are time consuming: it takes at least 30 min to dissolve 25 μm-thick copper foil. This process is also wasteful: it requires 300 kg of copper to produce 1 g of graphene,21 and thus produces an outsized amount of toxic waste. These disadvantages currently limit the fabrication of graphene to small amounts mostly for research and development purposes and need to be overcome in order to make graphene over large areas at low costs.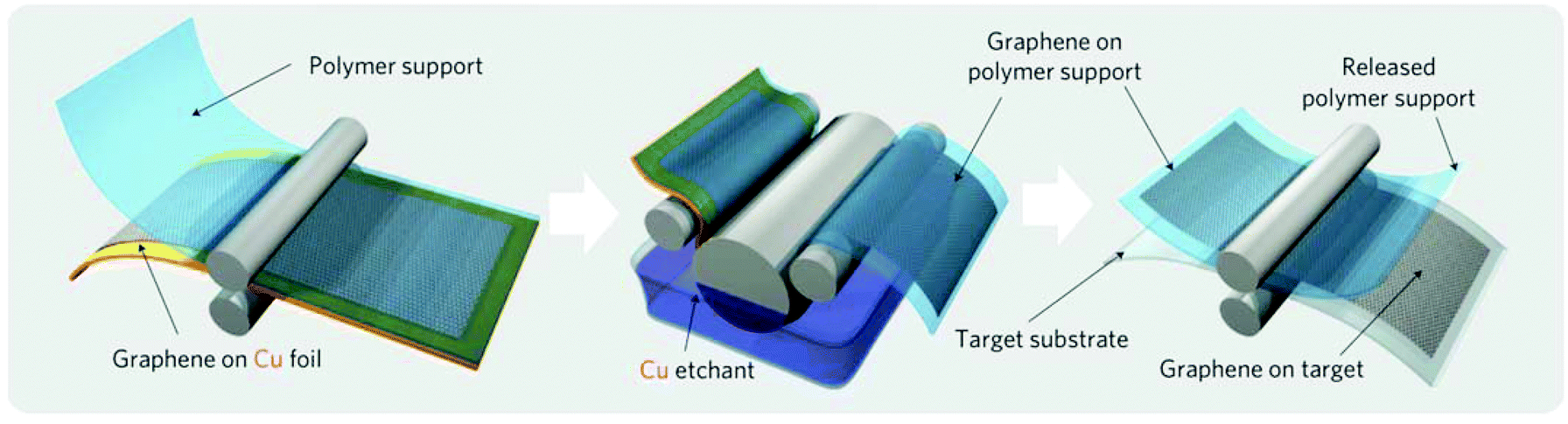 | ||
| Fig. 2 Schematic of the wet roll-to-roll graphene transfer from copper foils to polymeric substrates. Reproduced with permission from ref. 8. Copyright 2010, Nature Publishing Group. | ||
3.1 Non-destructive graphene transfer
In order to remove graphene from the synthesis substrate non-destructively so that the substrate can be recycled, the adhesion between graphene and the growth substrate needs to be overcome. Despite its mechanical strength, a single-layer of graphene is too delicate to be exfoliated unsupported, or manipulated as a free-standing film, and thus requires a relatively rigid backing. Typically, the rigid support is a thicker polymeric sheet or foil that, in some cases, is the final receiving substrate, or is an intermediate substrate used only for mechanical support during transfer. In any case, the strength of adhesion of graphene to the supporting substrate has to be greater than to the substrate used for growth. The supporting substrate can provide strong adhesion in either its native state or it can be modified with adhesion layers whose effect is produced by covalent or van der Waals bonding. Knowing both values of adhesion energy, or at least their relative magnitudes, allows the design of the graphene exfoliation process from a given growth substrate to the final receiving substrate either directly or using a multi-step manipulation. The strengths of adhesion of graphene to various media are listed in Table 1.The design of a process to exfoliate graphene is not limited to the relative strengths of adhesion between graphene and the substrate used for synthesis and the substrate used for exfoliation. For example, the roughness of the substrate used for growth and the parameters of processing—e.g., the temperature and pressure at which the supporting substrate is deposited, which affects the interfacial area between the graphene and the supporting substrate—also plays a key role. A hot press is often used to apply the supporting substrate to increase its adhesion to the graphene. Another consideration is the chemical modification of graphene by the supporting/adhesion layer (covalent bonding, doping), which can potentially change the electronic properties of graphene.29 Not least important are the mechanical aspects of the transfer process such as the distribution of stress during the exfoliation, which may be especially problematic with roll-to-roll transfer. For example, a film that is 200 microns thick when bent to the radius of 5 millimeters experiences a tensile strain on the top surface of 2% and an equal in magnitude compressive strain on the bottom surface.30 Such strains can cause catastrophic failures in a number of thin-film materials.31
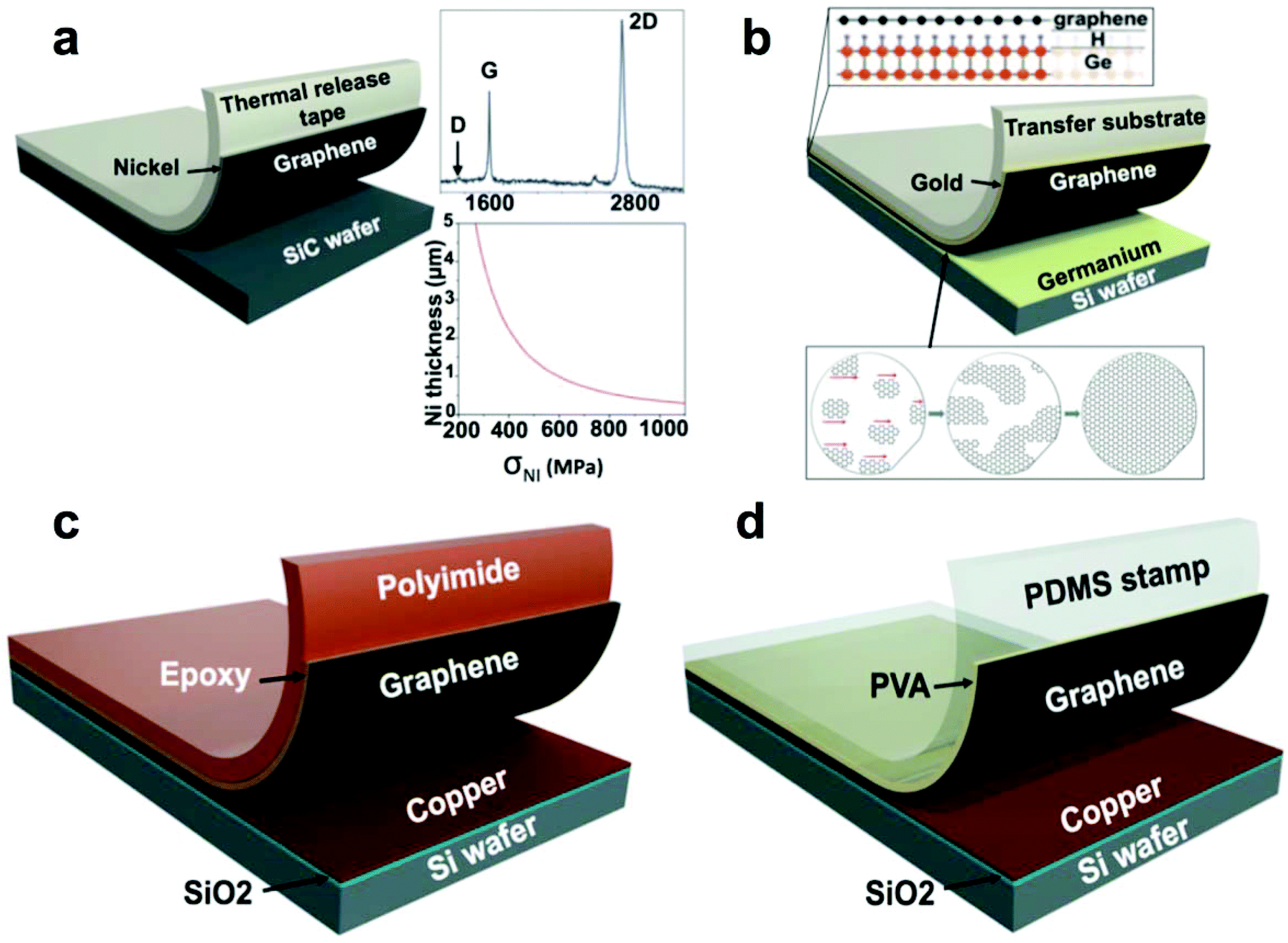 | ||
| Fig. 3 Summary of methods used to transfer graphene from single-crystalline wafers. (a) Schematic diagram of graphene transfer from SiC wafer after SiC epitaxy. Reproduced with permission from ref. 33. Copyright 2013, American Association for the Advancement of Science. (b) Graphene transfer from a silicon wafer bearing a film of germanium. Reproduced with permission from ref. 35. Copyright 2014, American Association for the Advancement of Science. (c) Graphene transfer from evaporated copper substrate using epoxy. Adapted from ref. 25. (d) Graphene transfer from evaporated copper substrate using a PDMS/PVA stamp. Adapted from ref. 36. | ||
Repetitive synthesis by CVD and subsequent exfoliation of a wafer-scale single-crystalline single-layer graphene from a hydrogen-terminated germanium-coated silicon wafer has been demonstrated by Lee et al.35 This process was facilitated by three characteristics (1) the low solubility of carbon in germanium, (2) its catalytic activity toward methane cracking, and (3) its perfect crystalline order as deposited on the surface of the silicon wafer. The hydrogen-terminated germanium, which exhibited a weak adhesion to graphene, allowed the authors to exfoliate the graphene from the wafer using a thin film of evaporated gold (Fig. 3b). This group also demonstrated a repetitive sequential graphene synthesis and exfoliation from the same wafer without any apparent deterioration of the graphene.35
Copper evaporated onto a silicon wafer bearing a layer of thermally grown oxide can also support CVD of graphene. Yoon et al.25 demonstrated the effective direct exfoliation of graphene from the copper-coated wafer to a flexible polyimide film (PI) by binding them with epoxy as the permanent adhesion layer (Fig. 3c). The authors stated that this approach helped to avoid doping of graphene with metals and the detrimental acidic treatments. They demonstrated the utility of the graphene prepared in this manner by fabricating a flexible field-effect transistor (FET) with good gate modulation of the graphene channel conductivity on the PI film. Regrowth of graphene on the same substrate was performed and generated graphene with a somewhat increased defect density as judged by the Raman D-peak of graphene on copper.25 This defectiveness could be explained in principle by the evaporative deterioration of the thin copper layer during graphene synthesis.
Similarly, Yang et al.36 employed an adhesion layer, albeit water soluble—polyvinyl alcohol—to exfoliate graphene from copper-coated Si/SiO2 wafer and to transfer it to arbitrary substrates (Fig. 3d). During the transfer, graphene sustained minimal physical damage, as indicated by the D/G peak ratio of 0.06. A polydimethylsiloxane (PDMS) stamp served as a support substrate for the transfer and provided a kinetically controlled adhesion necessary for the graphene transfer: rapidly peeling the stamp away from the copper-coated wafer promoted exfoliation of the PVA-coated graphene. In the next step, the PVA-coated graphene was pressed against the final receiving substrate and by peeling the stamp away slowly was released. After the transfer, the PVA adhesion layer could be dissolved in deionized water. As with the exfoliation process that did not involve metal etching, developed by Yoon et al.25 above, the graphene produced by this method was free of doping and preserves its charge neutrality with the value of Vdirac close to 0 V in the FETs fabricated with the exfoliated graphene.
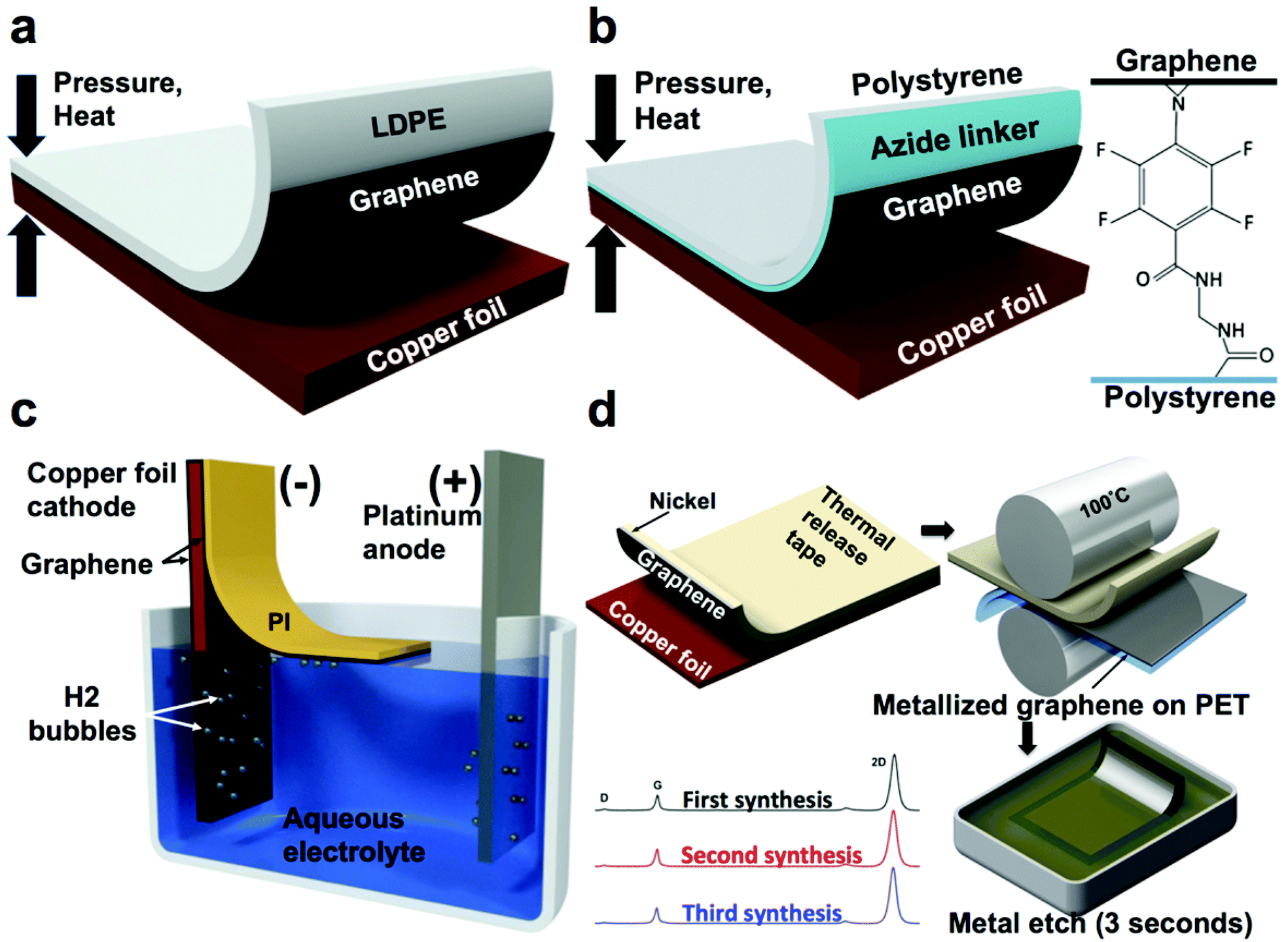 | ||
| Fig. 4 Summary of methods used to transfer graphene from flexible copper foils. (a) Direct dry transfer of graphene from copper foil to polymeric substrates using a hot press. Adapted from ref. 38. (b) Adhesion molecule assisted dry transfer of graphene from copper foil to polymeric substrates using a hot press. Adapted from ref. 39. (c) Electrochemical delamination of PI-supported graphene from copper foil. Adapted from ref. 43. (d) Metal-assisted exfoliation of graphene from copper foil. Adapted from ref. 21. | ||
A similar approach enhanced with a molecule that covalently bonds graphene to polystyrene—N-ethylamino-4-azidotetrafluorobenzoate (TFPA-NH2)—was employed by Lock et al.39–41 and resulted in graphene with a sheet resistance of about 1 kΩ sq−1 (Fig. 4b). TFPA is known to form strong covalent bonds with graphene42 and thus the polystyrene substrate was dip-coated in TFPA-NH2 solution after plasma treatment to promote the adhesion to graphene. It is noteworthy that without the TFPA treatment, polystyrene demonstrated poor adhesion, which resulted in only partial (19%) transfer.38
A noteworthy process to transfer graphene non-destructively by “electrochemical delamination” has been demonstrated by Wang et al.43 In this process, the final receiving substrate (PI) is spin–coated on the CVD graphene on copper foil, which serves as a cathode in an electrochemical cell with 0.5 M sodium hydroxide as an electrolyte and platinum wire mesh as an anode. By applying a 15 V bias between the electrodes, hydrolysis of water produced hydrogen bubbles between the copper foil and graphene and promoted separation of this interface (Fig. 4c). Graphene supported by 9 μm-thick PI was observed to float away from the copper foil. It was then collected, and its conductivity was measured under various bending radii. The process reportedly took 5 min for a 50 × 15 mm size sheet and generated graphene with a sheet resistance of 459 Ω sq−1.
Our laboratory reported a process for exfoliating single-layer graphene from copper foils without the use of a hot press, nicknamed metal-assisted exfoliation (MAE, Fig. 4d).21 This method employed the evaporation of a thin film (>20 nm) of nickel or cobalt onto the CVD-grown graphene on copper foil. The preferential adhesion of graphene to nickel and cobalt ensured delamination of graphene from the copper during exfoliation, while the use of a thermal-release tape as the supporting substrate enabled the transfer of the metalized exfoliated graphene onto flexible transparent polymeric films bearing a thermoplastic adhesive by quick processing through a commercial document laminator. Further, the nanoscopic metal film (nickel or cobalt) could be etched rapidly in an acidic solution to yield single-layer graphene on a flexible polymeric film.
The mechanical stresses induced in thin films during exfoliation process need to be considered as even small forces may cause large deformations. Evidence of the effects of anisotropic stresses induced in roll-to-roll processing is the anisotropic sheet resistances of the graphene resulting from MAE when excessively large tensile strains are imposed onto the metallized graphene during the process of exfoliation. The bending radii of the metallized graphene supported by thermal release tape during the manual exfoliation were small enough to cause metal film cracking that propagates through graphene and produced sheet resistances on the order of 0.8 kΩ sq−1 parallel to the cracks and 8 kΩ sq−1 in the perpendicular orientation. This problem can in principle be solved by avoiding excessively small bending radii during the exfoliation process or by using very thin supporting substrates.
4. Conclusion
With more than 8000 patents filed to date, graphene has been proposed for a multitude of possible applications in a number of various industries. It is likely that different approaches of graphene synthesis and transfer will be used for different applications, considering the strong dependence of the quality, yield, and cost of graphene based on the process selected. Some of the processes discussed in this minireview may be better suited for research and development and small chip-scale production, while others are more amenable to industrial-scale roll-to-roll fabrication schemes. Further developments are needed in order to overcome the still remaining shortcomings: mechanical damage during exfoliation, the use of expensive sacrificial layers (evaporated metal films) and transfer substrates (thermal-release tapes and thermoplastic adhesives). In particular, routes of mechanical degradation of the graphene should be characterized more fully than they currently are, so that these pathways can be mitigated. We conclude that non-destructive, economically feasible, and environmentally friendly and sustainable processes for manufacturing graphene at scales demanded by industry will need to be further developed, though several potentially significant strategies have been proposed. Progress in these areas is required if this material is to make a truly significant impact on society.Acknowledgements
This work was supported by the laboratory startup funds from the University of California, San Diego. A. Z. acknowledges a fellowship from SoCal Clean Energy Technology Acceleration Program from the von Liebig Center at UCSD sponsored by the US Department of Energy, and a fellowship from the National Science Foundation Graduate Research Fellowship Program, DGE-1144086.References
- I. Khrapach, F. Withers, T. H. Bointon, D. K. Polyushkin, W. L. Barnes, S. Russo and M. F. Craciun, Adv. Mater., 2012, 24, 2844–2849 CrossRef CAS PubMed
.
- J. Ryu, Y. Kim, D. Won, N. Kim, J. S. Park, E.-K. Lee, D. Cho, S.-P. Cho, S. J. Kim, G. H. Ryu, H.-A.-S. Shin, Z. Lee, B. H. Hong and S. Cho, ACS Nano, 2014, 8, 950–956 CrossRef CAS PubMed
- D. E. Jiang, V. R. Cooper and S. Dai, Nano Lett., 2009, 9, 4019–4024 CrossRef CAS PubMed
- Z. Liu, J. Li and F. Yan, Adv. Mater., 2013, 25, 4296–4301 CrossRef CAS PubMed
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- T. Kobayashi, M. Bando, N. Kimura, K. Shimizu, K. Kadono, N. Umezu, K. Miyahara, S. Hayazaki, S. Nagai, Y. Mizuguchi, Y. Murakami and D. Hobara, Appl. Phys. Lett., 2013, 102, 023112 CrossRef PubMed
- T. Yamada, J. Kim, M. Ishihara and M. Hasegawa, J. Phys. D: Appl. Phys., 2013, 46, 063001 CrossRef
- S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim, Y. Il Song, Y.-J. Kim, K. S. Kim, B. Ozyilmaz, J.-H. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed
- D.-Y. Kim, S. Sinha-Ray, J.-J. Park, J.-G. Lee, Y.-H. Cha, S.-H. Bae, J.-H. Ahn, Y. C. Jung, S. M. Kim, A. L. Yarin and S. S. Yoon, Adv. Funct. Mater., 2014, 24, 4986–4995 CrossRef CAS PubMed
- Y. Li, Y. A. Samad, K. Polychronopoulou, S. M. Alhassan and K. Liao, Sci. Rep., 2014, 4, 4652 Search PubMed
- Y. Su, V. G. Kravets, S. L. Wong, J. Waters, A. K. Geim and R. R. Nair, Nat. Commun., 2014, 5, 4843 CrossRef CAS PubMed
- J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. Van Der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano Lett., 2008, 8, 2458–2462 CrossRef CAS PubMed
- W. Norimatsu and M. Kusunoki, Phys. Chem. Chem. Phys., 2014, 16, 3501–3511 RSC
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2008, 9, 30–35 CrossRef PubMed
- W. Norimatsu and M. Kusunoki, Chem. Phys. Lett., 2009, 468, 52–56 CrossRef CAS PubMed
- C. Mattevi, H. Kim and M. Chhowalla, J. Mater. Chem., 2011, 21, 3324 RSC
- N. S. Mueller, A. J. Morfa, D. Abou-Ras, V. Oddone, T. Ciuk and M. Giersig, Carbon N. Y., 2014, 78, 347–355 CrossRef CAS PubMed
- I. Vlassiouk, P. Fulvio, H. Meyer, N. Lavrik, S. Dai, P. Datskos and S. Smirnov, Carbon N. Y., 2013, 54, 58–67 CrossRef CAS PubMed
- P. Procházka, J. Mach, D. Bischoff, Z. Lišková, P. Dvořák, M. Vaňatka, P. Simonet, A. Varlet, D. Hemzal, M. Petrenec, L. Kalina, M. Bartošík, K. Ensslin, P. Varga, J. Čechal and T. Šikola, Nanotechnology, 2014, 25, 185601 CrossRef PubMed
-
A. Kumar and C. H. Lee, in Advances in Graphene Science, ed. M. Aliofkhazraei, 2013, pp. 55–75. DOI:10.5772/55728
- A. V. Zaretski, H. Moetazedi, C. Kong, E. J. Sawyer, S. Savagatrup, E. Valle, T. F. O'Connor, A. D. Printz and D. J. Lipomi, Nanotechnology, 2015, 26, 45301 CrossRef CAS PubMed
- D. J. Henry, G. Yiapanis, E. Evans and I. Yarovsky, J. Phys. Chem. B, 2005, 109, 17224–17231 CrossRef CAS PubMed
- J. Lahiri, T. S. Miller, A. J. Ross, L. Adamska, I. I. Oleynik and M. Batzill, New J. Phys., 2011, 13, 025001 CrossRef
- Z. Xu and M. J. Buehler, J. Phys.: Condens. Matter, 2010, 22, 485301 CrossRef PubMed
- T. Yoon, W. C. Shin, T. Y. Kim, J. H. Mun, T.-S. Kim and B. J. Cho, Nano Lett., 2012, 12, 1448–1452 CrossRef CAS PubMed
- I. Hamada and M. Otani, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 153412 CrossRef
- A. Mattausch and O. Pankratov, Phys. Rev. Lett., 2007, 99, 086802 CrossRef
- G. H. Wells, T. Hopf, K. V. Vassilevski, E. Escobedo-Cousin, N. G. Wright, A. B. Horsfall, J. P. Goss, A. G. O'Neill and M. R. C. Hunt, Appl. Phys. Lett., 2014, 105, 193109 CrossRef PubMed
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335 RSC
- N. Lu, Z. Suo and J. J. Vlassak, Acta Mater., 2010, 58, 1679–1687 CrossRef CAS PubMed
- S. Savagatrup, A. D. Printz, T. F. O'Connor, A. V. Zaretski, D. Rodriquez, E. J. Sawyer, K. M. Rajan, R. I. Acosta, S. E. Root and D. J. Lipomi, Energy Environ. Sci., 2015, 8, 55–80 CAS
- T. Moldt, A. Eckmann, P. Klar, S. V. Morozov, A. A. Zhukov, K. S. Novoselov and C. Casiraghi, ACS Nano, 2011, 5, 7700–7706 CrossRef CAS PubMed
- J. Kim, H. Park, J. B. Hannon, S. W. Bedell, K. Fogel, D. K. Sadana and C. Dimitrakopoulos, Science, 2013, 342, 833–836 CrossRef CAS PubMed
- Y. He, W. F. Chen, W. B. Yu, G. Ouyang and G. W. Yang, Sci. Rep., 2013, 3, 2660 CAS
- J.-H. Lee, E. K. Lee, W.-J. Joo, Y. Jang, B.-S. Kim, J. Y. Lim, S.-H. Choi, S. J. Ahn, J. R. Ahn, M.-H. Park, C.-W. Yang, B. L. Choi, S.-W. Hwang and D. Whang, Science, 2014, 344, 286–289 CrossRef CAS PubMed
- S. Y. Yang, J. G. Oh, D. Y. Jung, H. Choi, C. H. Yu, J. Shin, C.-G. Choi, B. J. Cho and S.-Y. Choi, Small, 2015, 11, 175–181 CrossRef CAS PubMed
- D. Lee, G. D. Kwon, J. H. Kim, E. Moyen, Y. H. Lee, S. Baik and D. Pribat, Nanoscale, 2014, 6, 12943–12951 RSC
- G. J. M. Fechine, I. Martin-Fernandez, G. Yiapanis, R. Bentini, E. S. Kulkarni, R. V. Bof de Oliveira, X. Hu, I. Yarovsky, A. H. Castro Neto and B. Özyilmaz, Carbon N. Y., 2015, 83, 224–231 CrossRef CAS PubMed
- E. H. Lock, M. Baraket, M. Laskoski, S. P. Mulvaney, W. K. Lee, P. E. Sheehan, D. R. Hines, J. T. Robinson, J. Tosado, M. S. Fuhrer, S. C. Hernández and S. G. Walton, Nano Lett., 2012, 12, 102–107 CrossRef CAS PubMed
- E. H. Lock, D. M. Delongchamp, S. W. Schmucker, B. Simpkins, M. Laskoski, S. P. Mulvaney, D. R. Hines, M. Baraket, S. C. Hernandez, J. T. Robinson, P. E. Sheehan, C. Jaye, D. A. Fisher and S. G. Walton, Carbon N. Y., 2015, 86, 288–300 CrossRef CAS PubMed
- E. H. Lock, S. C. Hernández, T. J. Anderson, S. W. Schmucker, M. Laskoski, S. P. Mulvaney, F. J. Bezares, J. D. Caldwell, P. E. Sheehan, J. T. Robinson, B. N. Feygelson and S. G. Walton, Surf. Coat. Technol., 2014, 241, 118–122 CrossRef CAS PubMed
- L. H. Liu and M. Yan, Nano Lett., 2009, 9, 3375–3378 CrossRef CAS PubMed
- X. Wang, L. Tao, Y. Hao, Z. Liu, H. Chou, I. Kholmanov, S. Chen, C. Tan, N. Jayant, Q. Yu, D. Akinwande and R. S. Ruoff, Small, 2014, 10, 694–698 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |