Homochiral supramolecular polymerization of bowl-shaped chiral macrocycles in solution†
Kohei
Sato
a,
Yoshimitsu
Itoh
*a and
Takuzo
Aida
*ab
aDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan. E-mail: itoh@macro.t.u-tokyo.ac.jp; aida@macro.t.u-tokyo.ac.jp; Fax: +81 3-5841-7310; Tel: +81 3-5841-7251
bRIKEN Center for Emergent Matter Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 18th September 2013
Abstract
Chiral monomers 1 and 2, carrying C4- and C3-symmetric bowl-shape peptidic macrocycle cores, respectively, undergo supramolecular polymerization in solution via van der Waals and hydrogen bonding interactions. Size-exclusion chromatographic studies, using UV and CD detectors, on the supramolecular copolymerization of their enantiomers demonstrated that these monomers are the first chiral macrocycles that polymerize enantioselectively with a strong preference for chiral self-sorting.
Introduction
Since the discovery of spontaneous optical resolution of chiral sodium ammonium tartrate by crystallization in the mid-19th century,1 crystallization has been the most convenient and important method for chiral separation. However, despite long-term efforts, only 5–10% of chiral compounds are known to be optically resolved by this conventional method.2,3 This is most likely because the enthalpic gain hardly compensates its large entropic loss upon homochiral crystallization. Needless to say, chiral separation, which can be referred to as “chiral self-sorting”, is far more difficult in the solution phase.4–11 Meanwhile, in synthetic polymer chemistry, homochiral polymerization of racemic monomers can be regarded as a particular example of chiral self-sorting, where there are many successful examples, owing to the development of excellent enantioselective catalysts.12 In contrast, chiral self-sorting in supramolecular polymerization has not been well explored to date. This is surprising considering the fundamental importance of this subject but has certainly been caused by a preconceived notion that a high level of chiral discrimination can hardly be attained in such dynamically assembling systems. The other reason, which is more practical, is the lack of a direct method to evaluate the enantioselectivity of supramolecular polymerization. For these reasons, only two successful examples have been reported to date.5,11 In 2002, we reported that a rigid S-shaped chiral diketopiperazine dimer undergoes homochiral supramolecular polymerization via multiple hydrogen-bonding (H-bonding) interactions.5 For substantiating the homochiral nature of the polymerization, we developed a conceptually versatile analytical strategy (vide infra) using size-exclusion chromatography (SEC) in conjunction with a circular dichroism (CD) detector.5 More recently, Schenning, Meijer and coworkers reported that a chiral star-shaped discotic molecule with 24 stereogenic centers shows a thermodynamic bias for chiral self-sorting in its supramolecular polymerization.11Here we report the first chiral macrocyclic monomers 1 and 2 that undergo homochiral supramolecular polymerization in solution. Monomers 1 and 2 (Fig. 1) carry C4- and C3-symmetric bowl-shaped chiral cores, respectively, which are appended with paraffinic wedges via an alkyl amide spacer. Because of their particular ring shapes, columnar polymerization of 1 and 2 in a head-to-tail manner should provide nanotubes with pore diameters of 5.7 and 4.0 Å, respectively. Since organic nanotubes are attractive for many potential applications in chemistry, biotechnology, and material sciences,13–20 one-dimensional columnar assembly (polymerization) of shape-persistent macrocycles has extensively been studied.21–24 However, before the present work, no chiral macrocycle has been reported to polymerize enantioselectively in a non-covalent fashion.
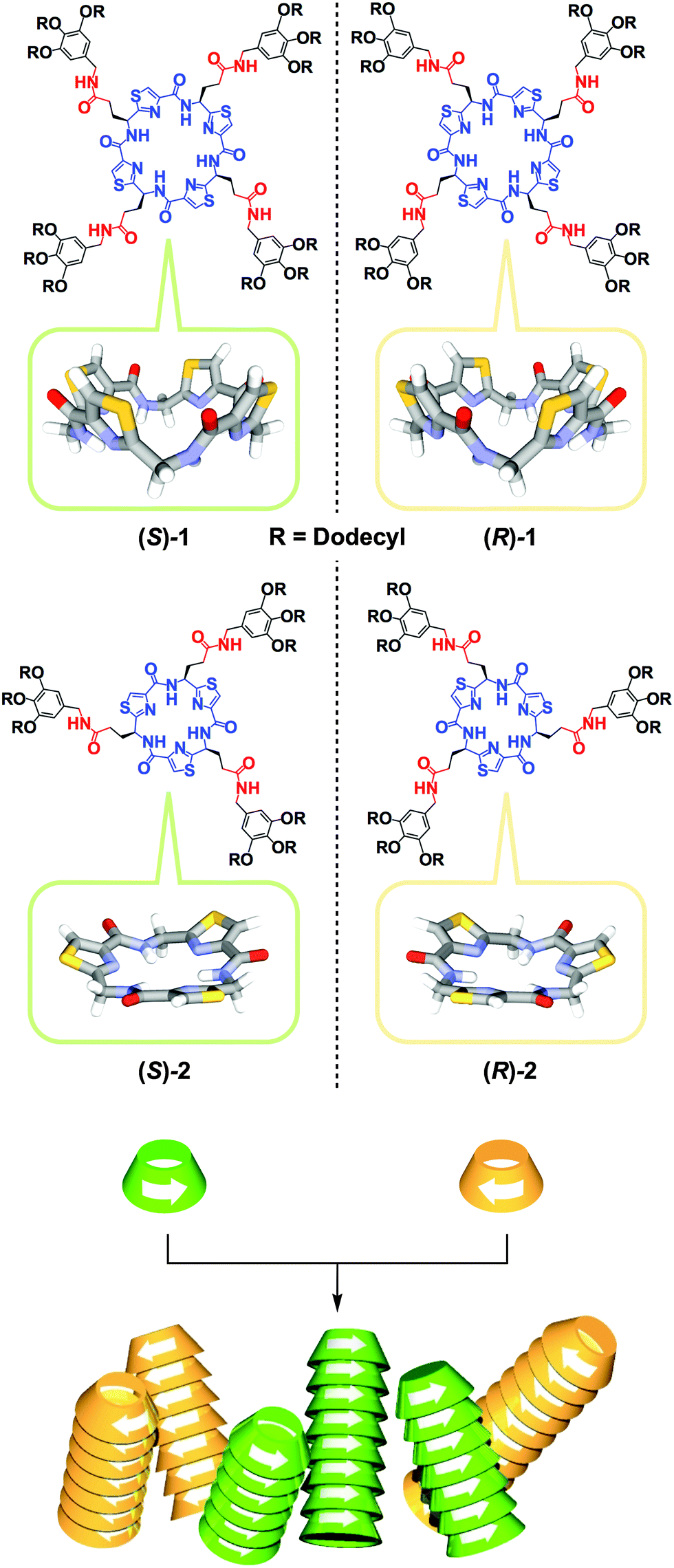 | ||
| Fig. 1 Schematic illustrations for the structures of bowl-shaped chiral peptidic macrocycles 1 and 2 and their homochiral columnar assembly. | ||
The core structures of 1 and 2 were designed with inspiration from naturally occurring bioactive peptidic macrocycles isolated from marine organisms.25–28 Their paraffinic wedge-appended derivatives were intended to self-assemble into a functional nanoporous soft material by their columnar assembly using van der Waals and H-bonding interactions at the bowl-shaped core and side-chain amide groups, respectively. In fact, we confirmed that these compounds self-assemble into nanoporous columnar liquid crystals over a wide temperature range including room temperature.29 In the course of this study, we wondered whether the assembling events of 1 and 2 are stereochemically affected by the bowl chirality at their peptidic macrocycle cores. In order to address this issue, we decided to investigate the stereochemical supramolecular copolymerization of their enantiomers in solution (Fig. 1). Compared with two previous examples,5,11 the chiral cores of 1 and 2 are macrocyclic and unlikely to exert a strong appearance of structural asymmetry. Therefore, we initially thought that stereoselective copolymerization of their enantiomers is unlikely. However, on the contrary to our expectation, the copolymerization exhibited a strong preference for chiral self-sorting.
Results and discussion
According to a method analogous to that reported previously,29 (R)-1 and (R)-2 (Fig. 1), opposite enantiomers to our previously reported ones, were newly synthesized and unambiguously characterized by 1H and 13C NMR spectroscopy and MALDI–TOF mass spectrometry (Fig. S1–S6†).30 For chiral self-sorting to occur in the stereochemical copolymerization of the enantiomers of 1 and 2 (Fig. 1), an appreciable energetic difference should exist between their homo- and heterochiral stacking modes. At first, we carried out a computational study on dimerized core models of 1. In a previous paper, we reported that the exocyclic amide units form intermolecular H-bonds for stabilizing its columnar assembly, whereas endocyclic ones are involved only in an intracyclic H-bonding network.29 Thus, the initial geometries of the homo- and heterochiral dimer models of 1 for the computational calculation were constructed based on these structural features (Fig. S7†).30 As can be seen from Fig. 2, the homochiral stacking (Fig. 2a) was calculated to be favored by 2.3 kcal mol−1 over its heterochiral stacking (Fig. 2b). The observed energy difference is rather surprising, considering the absence of core-to-core H-bonding interactions among the stacked peptidic macrocycles of 1.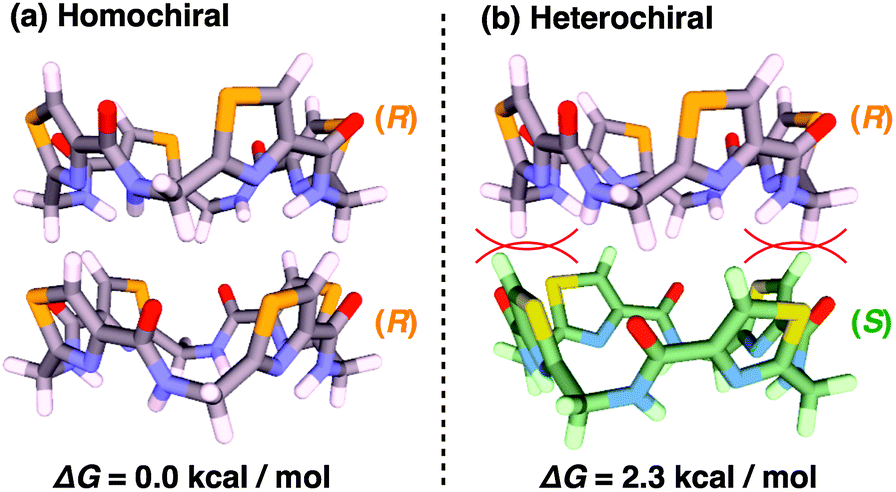 | ||
| Fig. 2 Energy minimized (a) homo- and (b) heterochiral dimeric core models of 1 calculated based on M06-2X/6-31G**//B3LYP/6-31G*. Red arches represent steric repulsion. Side chains are omitted for clarity. | ||
As shown in Fig. 3a (left), the SEC trace of (R)-1, monitored by a UV (λ = 254 nm) detector (SEC-UV: [(R)-1] loaded; 1.00 mM, eluent; CHCl3–C6H12 (1/1 in v/v)),31 displayed an elution peak in a relatively high molecular weight region. As expected, when monitored by a circular dichroism (CD) detector (SEC-CD), a fraction corresponding to this elution peak was optically active (Fig. 3a, right). When [(R)-1] loaded was decreased stepwise from 1.00 mM to 0.06 mM, the elution volume of the produced polymeric assembly was increased from 15.9 mL to 23.8 mL (Fig. 3a–f, left). Such a concentration-dependent SEC profile is typical of dynamically assembled supramolecular polymers.32–34 Accordingly, upon 2-fold dilution of a C6D12 solution of (R)-1 (1.00 mM), the change in 1H NMR chemical shift, associated with the H-bonding interaction at the side-chain amide units, became less explicit (Fig. S8†).30 Likewise, trimeric (R)-2 also undergoes supramolecular polymerization in solution. However, the assembly event seems more dynamic than that of tetrameric (R)-1 with a larger number of the H-bonding amide units, since an elution peak due to the polymeric assembly of (R)-2 was not observed unless the volume content of C6H12 in the SEC eluent was increased to, e.g., 70% (Fig. S9 and S10†).30
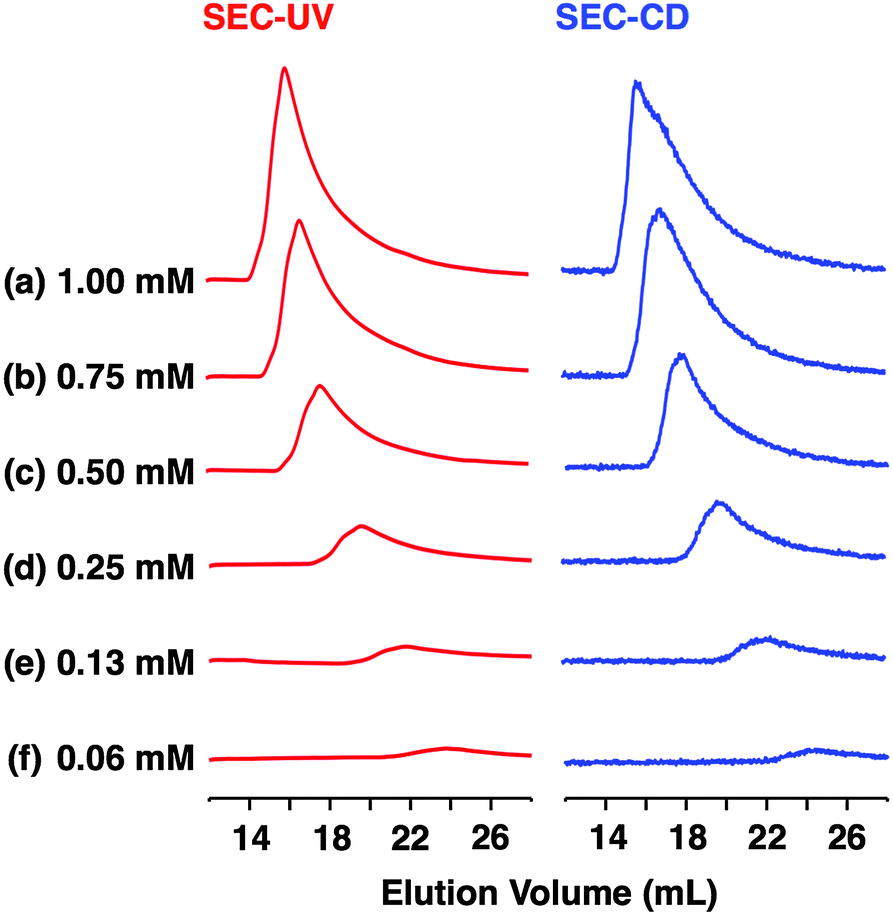 | ||
| Fig. 3 SEC-UV (red curves) and SEC-CD (blue curves) traces (25 °C, 254 nm) of supramolecular polymerization of (R)-1 at [(R)-1] = (a) 1.00, (b) 0.75, (c) 0.50, (d) 0.25, (e) 0.13 and (f) 0.06 mM. Loading solvent; C6H12, eluent; CHCl3–C6H12 (1/1 in v/v), column; TOSOH TSKgel G3000HHR. | ||
Fig. 4a shows CD spectra at 25 °C of mixtures of (S)-1 and (R)-1 in C6H12 at different molar ratios [(S)-1]/[(R)-1],31 where the CD band at 236 nm became less intense monotonically as the enantiomeric excess (ee) of 1 was lower (Fig. 4b). The same held true for (R)-2 (Fig. S11†).30 Before investigating the possibility of chiral self-sorting, we hereafter discuss the possible correlations between polymer molecular weight and monomer ee when [1] is kept constant. Unless any stereoselection occurs in the polymerization of 1, the polymer molecular weight should remain intact to an applied change in monomer ee. However, if chiral self-sorting (homochiral polymerization) occurs, this is not the case (Fig. 5a). Namely, decreasing the monomer ee should result in lowering the highest attainable polymer molecular weight. In particular, when the ee value of 1 is 0%, both [(S)-1] and [(R)-1] are at a minimum. Therefore, the molecular weights of poly((S)-1) and poly((R)-1) should be minimum and identical to one another. The schematic illustration in Fig. 5a also suggests an interesting possibility that the first eluted SEC fraction at [(S)-1] ≠ [(R)-1] may give enantiopure 1. On the other hand, if the polymerization of 1 takes place heterochirally, the polymer molecular weight should certainly change with the monomer ee but in a different manner from the case of homochiral chain growth, i.e., the enantiomer in excess at [(S)-1] ≠ [(R)-1], in principle, should remain unpolymerized and elute last (Fig. 5b).
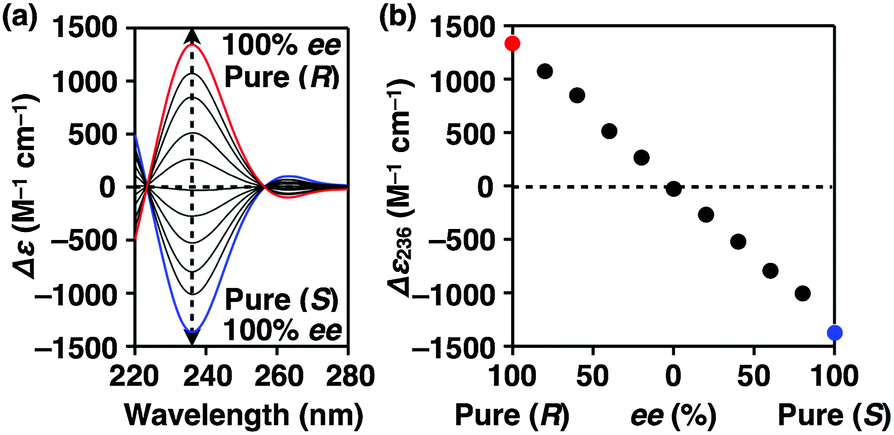 | ||
| Fig. 4 Stereochemical supramolecular copolymerization of (S)-1 and (R)-1 ([1] = 1.00 mM) in cyclohexane (C6H12) at different enantiomeric excess (ee) values. (a) Circular dichroism (CD) spectra in C6H12 at 25 °C and (b) plots of Δε at 236 nm versus ee. | ||
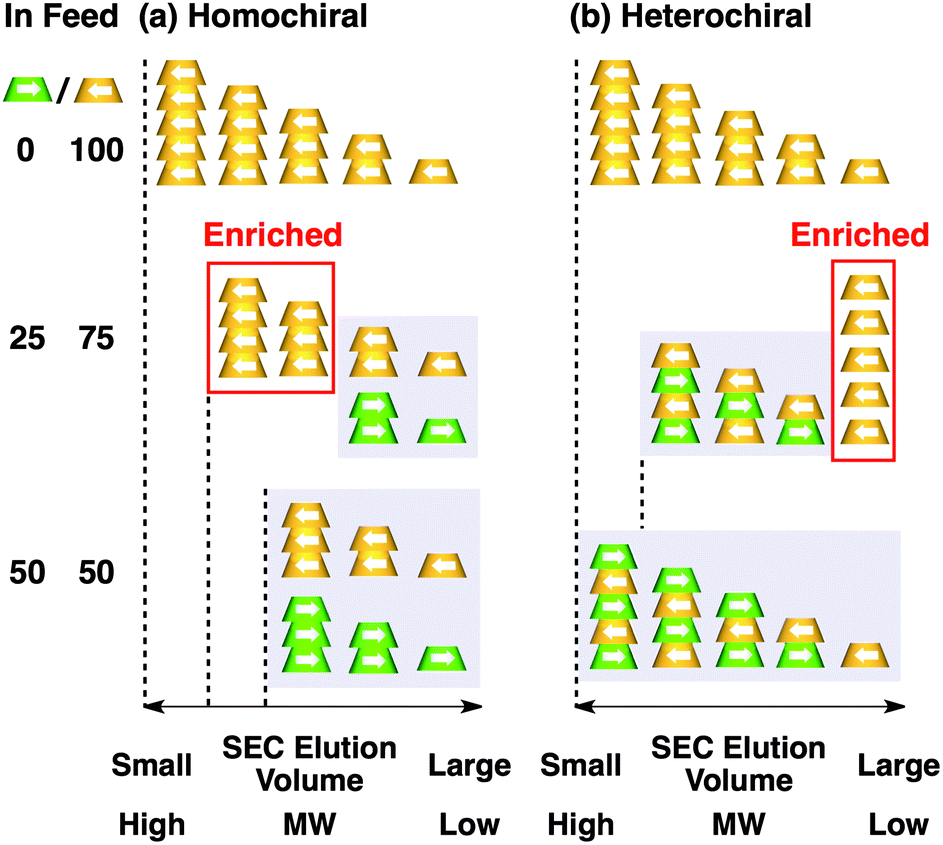 | ||
| Fig. 5 Stereochemical basis for supramolecular copolymerization of enantiomers MS and MR. Schematic illustrations for size-exclusion chromatography (SEC) profiles expected for ideal (a) homo- and (b) heterochiral chain growth events. | ||
Fig. 6 shows SEC-UV traces at 25 °C of supramolecular copolymerization of the enantiomers of 1 at different molar ratios [(S)-1]/[(R)-1] ([1] = 1.00 mM). Obviously, the elution profile of the polymeric assembly depended on the ee value of the monomer. In particular, at [(S)-1]/[(R)-1] = 50/50 (0% ee), a polymeric assembly with the smallest molecular weight formed. As discussed in the above paragraph (Fig. 5a), this trend is characteristic of homochiral chain growth. In order to confirm whether or not enantiomerically enriched 1 is isolable from a higher molecular weight fraction, the SEC-CD trace of the polymeric assembly formed at [(S)-1]/[(R)-1] = 25/75 (blue curve) was compared with that of the enantiopure one (red curve), normalized by the peak areas of their SEC-UV traces (Fig. 7a, left). Obviously, two SEC-CD traces, after the normalization, were different at their tail parts, where the ee value of 1 in the first-eluted polymer fraction, as estimated by dividing the former SEC trace by the latter, was close to 100% ((R)-1) (Fig. 7b, left). However, this value dropped abruptly and reached 0% in the last-eluted polymer fraction. The same held true when the loading molar ratio [(S)-1]/[(R)-1] was 75/25 (Fig. 7a and b, right). All of these observations are consistent with the theoretical correlation between polymer molecular weight and monomer ee as expected for homochiral chain growth (Fig. 5a). Therefore, the enantiomers of 1 were hardly copolymerized but self-sorted. Nevertheless, we must point out that the observed change in SEC elution volume with the monomer ee (Fig. 6) is less explicit than the ideal one as expected from Fig. 3. Most symbolically, the molecular weight of a polymeric assembly of racemic 1 ([(S)-1] = [(R)-1] = 0.50 mM) is slightly larger than that of enantiopure 1 at [(R)-1] = 0.50 mM.35 Therefore, a non-selective assembly of its enantiomers, where the polymer molecular weight is not affected by the monomer ee, cannot be excluded perfectly.
 | ||
| Fig. 6 SEC-UV (red curves) and SEC-CD (blue curves) traces (25 °C, 254 nm) of stereochemical supramolecular copolymerization of (S)-1 and (R)-1 at molar ratios [(S)-1]/[(R)-1] of (a) 0/100, (b) 25/75, (c) 50/50, (d) 75/25 and (e) 100/0. Loading solvent; C6H12, loading concentration; [1] = 1.00 mM, eluent; CHCl3–C6H12 (1/1 in v/v), column; TOSOH TSKgel G3000HHR. | ||
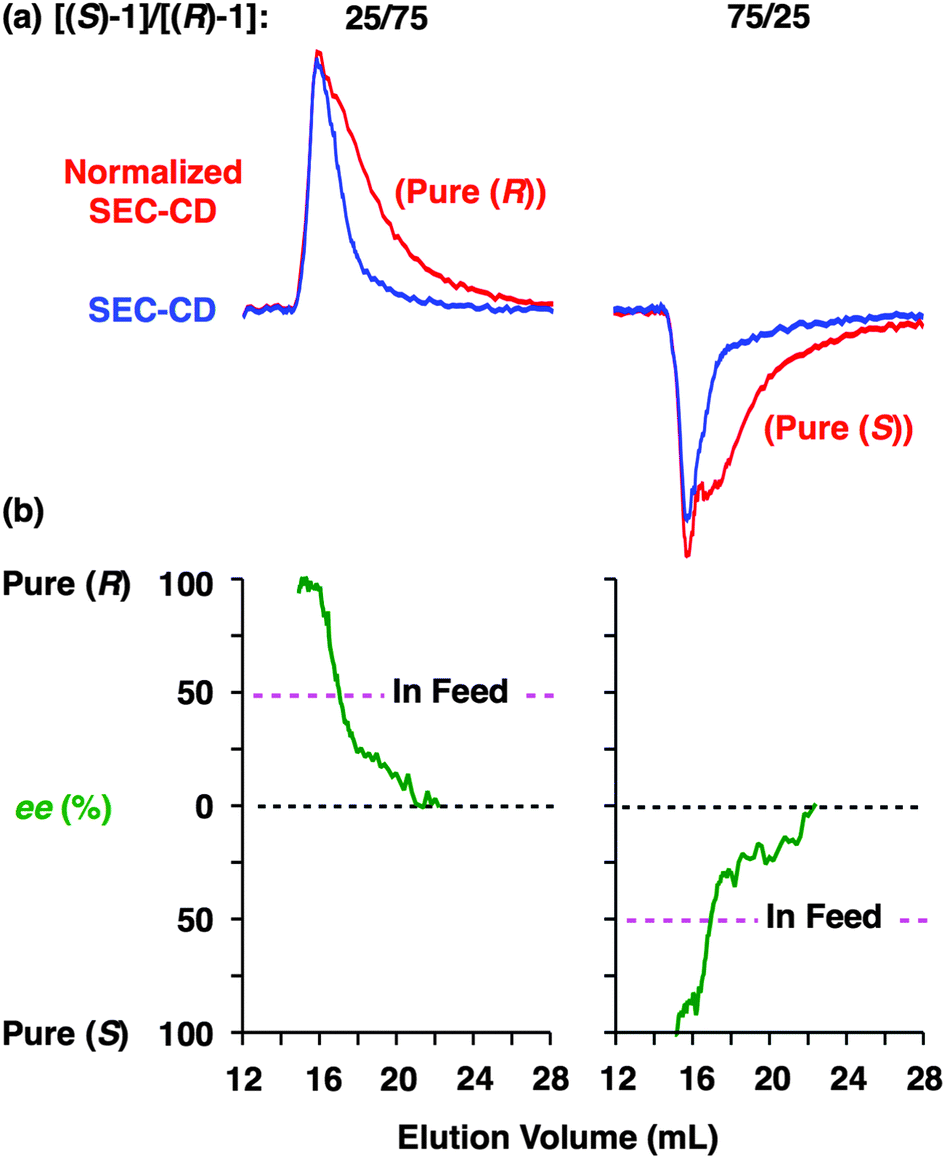 | ||
| Fig. 7 Stereochemical supramolecular copolymerization of (S)-1 and (R)-1. (a) SEC-CD traces (25 °C, 254 nm) at [(S)-1]/[(R)-1] of 0/100 (red curve) and 25/75 (blue curve) [left] and 100/0 (red curve) and 75/25 (blue curve) [right], normalized by their total SEC-UV peak areas. Loading solvent; C6H12, loading concentration; [1] = 1.00 mM, eluent; CHCl3–C6H12 (1/1 in v/v), column; TOSOH TSKgel G3000HHR. (b) Changes in enantiomeric excess (ee) with SEC elution volume obtained from (a). | ||
With these results in mind, we also investigated the stereochemical supramolecular copolymerization of the enantiomers of 2. Although the assembly event is more dynamic than that of 1 (vide ante), its SEC-CD traces in CHCl3–C6H12 (3/7 in v/v) at 25 °C were similar to those of 1 (Fig. S12 and S13†).30 Namely, the enantiomers are self-sorted in the supramolecular polymerization.
Conclusions
In conclusion, through stereochemical SEC studies on supramolecular copolymerization of enantiomers of Cn-symmetric 1 and 2, we demonstrated that these monomers are the first chiral macrocycles that undergo homochiral supramolecular polymerization in solution (Fig. 1). Despite our initial reservation that such ring-shaped chiral compounds are hard to be recognized stereochemically, almost enantiopure 1 and 2 were successfully isolated from the first-eluted SEC fractions of their polymeric assemblies at [(S)]/[(R)] = 25/75 or 75/25 (Fig. 7 and S13†).30 Covalent transformation of the tube-like assemblies of these macrocycles and utilization of their confined chiral inner space for asymmetric recognition and synthesis is one of the subjects worthy of further investigations.Acknowledgements
Generous allotment of computational time from the Institute for Molecular Science, Okazaki, Japan is greatly acknowledged. The present work was supported by the Japan Society for the Promotion of Science (JSPS) through its “Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program)”. K. S. thanks JSPS for a Young Scientist Fellowship. Y. I. thanks the Ministry of Education, Science, Sports and Culture, Japan, Grant-in-Aid for Challenging Exploratory Research (Grant no. 24655095).Notes and references
- L. Pasteur, C. R. Hebd. Seances Acad. Sci., 1848, 26, 535–539 Search PubMed.
- J. Jacquws, A. Collet and S. H. Wilen, in Enantiomers, Racemates, and Resolutions, Krieger Publishing Company, Malabar, FL, 1994 Search PubMed.
- A. Collet, in Comprehensive Supramolecular Chemistry, ed. D. N. Reinhoudt, Pergamon, Oxford, 1996, vol. 10, pp. 113–149 Search PubMed.
- X. Shi, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2001, 123, 6738–6739 CrossRef CAS.
- Y. Ishida and T. Aida, J. Am. Chem. Soc., 2002, 124, 14017–14019 CrossRef CAS PubMed.
- L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342–356 RSC.
- H. Katagiri, T. Miyagawa, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 1741–1744 CrossRef CAS PubMed.
- L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2007, 36, 941–967 RSC.
- M. M. S-Sempere, P. Osswald, M. Stolte, M. Grüne, M. Renz, M. Kaupp, K. Radacki, H. Braunschweig and F. Würthner, J. Am. Chem. Soc., 2011, 133, 9580–9591 CrossRef PubMed.
- M. M. S-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef PubMed.
- M. Wolffs, J. L. J. van Dongen, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2012, 18, 15057–15064 CrossRef CAS PubMed.
- Y. Okamoto and T. Nakano, Chem. Rev., 1994, 94, 349–372 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1993, 364, 516–518 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS.
- V. Percec, A. E. Dulcey, V. S. K. Balagurusamy, Y. Miura, J. Smidrkal, M. Peterca, S. Nummelin, U. Edlund, S. D. Hudson, P. A. Heiney, H. Duan, S. N. Magonov and S. A. Vinogradov, Nature, 2004, 430, 764–768 CrossRef CAS PubMed.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Itoh, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- S. Biswas, K. Kinbara, N. Oya, N. Ishii, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2009, 131, 7556–7557 CrossRef CAS PubMed.
- Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521–1526 CrossRef CAS PubMed.
- A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019–1027 CAS.
- S. Höger, Chem.–Eur. J., 2004, 10, 1320–1329 CrossRef PubMed.
- Y. Yang, W. Feng, J. Hu, S. Zou, R. Gao, K. Yamato, M. Kline, Z. Cai, Y. Gao, Y. Wang, Y. Li, Y. Yang, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2011, 133, 18590–18593 CrossRef CAS PubMed.
- M. Fritzsche, A. Bohle, D. Dudenko, U. Baumeister, D. Sebastiani, G. Richardt, H. W. Spiess, M. R. Hansen and S. Höger, Angew. Chem., Int. Ed., 2011, 50, 3030–3033 CrossRef CAS PubMed.
- P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS.
- J. P. Michael and G. Pattenden, Angew. Chem., Int. Ed. Engl., 1993, 32, 1–23 CrossRef.
- A. Bertram and G. Pattenden, Nat. Prod. Rep., 2007, 24, 18–30 RSC.
- Á. Pintér and G. Haberhauer, Synlett, 2009, 3082–3098 Search PubMed.
- K. Sato, Y. Itoh and T. Aida, J. Am. Chem. Soc., 2011, 133, 13767–13769 CrossRef CAS PubMed.
- See ESI†.
- Samples for CD and SEC measurements were prepared by mixing cyclohexane solutions of (S) and (R) monomers, and the resulting mixtures were annealed at 75 °C for 5 min and then allowed to cooled to 25 °C.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CAS.
- T. F. A. D-Dreef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- Fig. 3c shows that the elution volume of a homochiral polymer formed with (R)-1 at [(R)-1] = 0.50 mM is 17.7 mL. If chiral self-sorting indeed occurs ideally, the elution volume should remain unchanged even when the same amount of its enantiomer ([(S)-1] = 0.50 mM) is added. However, the observed value (16.4 mL), read out from Fig. 6c at [(S)-1] = [(R)-1] = 0.50 mM for the stereochemical copolymerization, is 1.3 mL smaller than 17.7 mL.
Footnote |
| † Electronic supplementary information (ESI) available: Details of characterization of chiral macrocycles, 1H and 13C NMR spectra and MALDI–TOF mass spectra, computational simulations, and SEC profiles. See DOI: 10.1039/c3sc52449c |
| This journal is © The Royal Society of Chemistry 2014 |