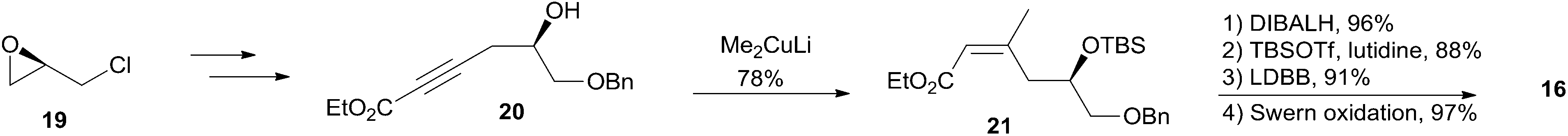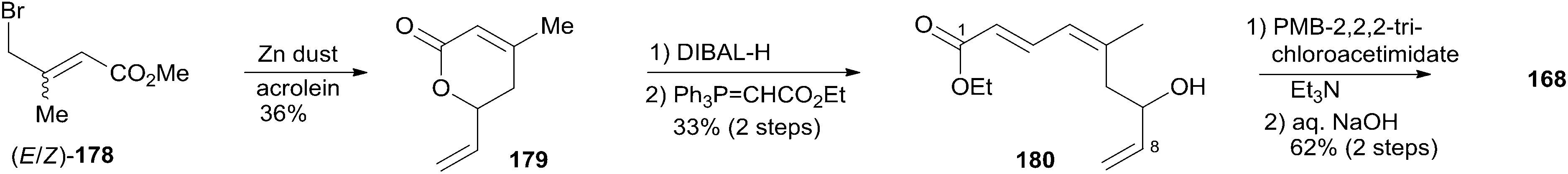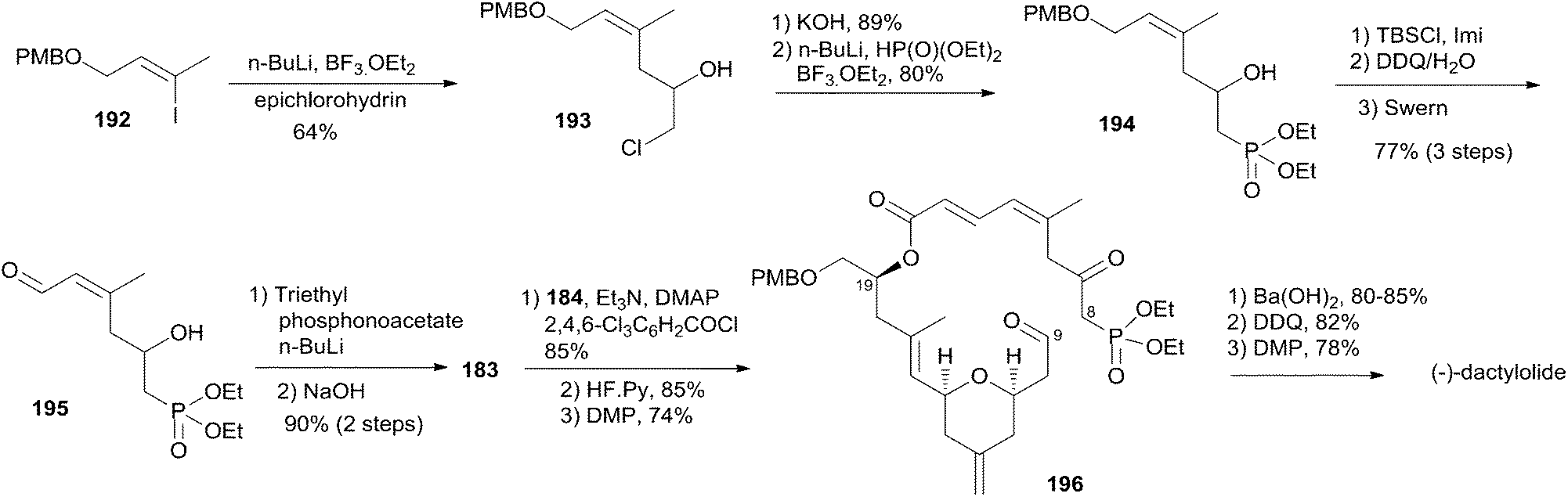Zampanolide and dactylolide: cytotoxic tubulin-assembly agents and promising anticancer leads
Qiao-Hong
Chen
*a and
David G. I.
Kingston
*b aDepartment of Chemistry, California State University, Fresno, 2555 E. San Ramon Avenue, M/S SB70, Fresno, CA 93740, USA. E-mail: qchen@csufresno.edu; Fax: +1 559 2784402; Tel: +1 559 2782394 bDepartment of Chemistry and Virginia Tech Center for Drug Discovery, M/C 0212, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: dkingston@vt.edu; Fax: +1 540 2313255; Tel: +1 540 2316570
Received
2nd March 2014
First published on 19th June 2014
Abstract
Covering: through January 2014
Zampanolide is a marine natural macrolide and a recent addition to the family of microtubule-stabilizing cytotoxic agents. Zampanolide exhibits unique effects on tubulin assembly and is more potent than paclitaxel against several multi-drug resistant cancer cell lines. A high-resolution crystal structure of αβ-tubulin in complex with zampanolide explains how taxane-site microtubule-stabilizing agents promote microtubule assemble and stability. This review provides an overview of current developments of zampanolide and its related but less potent analogue dactylolide, covering their natural sources and isolation, structure and conformation, cytotoxic potential, structure–activity studies, mechanism of action, and syntheses.
Qiao-Hong Chen
Qiao-Hong Chen received her Ph.D. degree from Sichuan University, China. Appointed as a Lecturer in 2001, she was promoted to the position of Full Professor in 2003 at Sichuan University. She was a Postdoctoral Fellow for three years at the University of Alberta in Canada and a Senior Research Fellow for six years at Virginia Tech in the USA. She started her current position as an Assistant Professor of Chemistry at California State University, Fresno, in 2012. With her research interests focused on natural products-based anticancer agents, she is working to engineer drug-like analogs of zampanolide, curcumin, and genistein.
David G. I. Kingston
David Kingston received his Ph.D. degree from Cambridge University in 1964. He is currently a University Distinguished Professor of Chemistry and Director of the Virginia Tech Center for Drug Discovery. He has worked extensively on the chemistry and microtubule binding of paclitaxel and the epothilones, and he has carried out drug discovery work in Madagascar. He is a recipient of the ACS Ernest Guenther Award in the Chemistry of Natural Products, and is a member of the NIH National Advisory Council for Complementary and Alternative Medicine. He also serves as one of several lay pastors in his local church, the Blacksburg Christian Fellowship.
1. Introduction
The year 1979 marked a significant turning point in understanding the mechanism of action of anticancer agents, when Susan Horwitz published her pivotal paper indicating that paclitaxel (Taxol®) promoted the polymerization of tubulin to microtubules.1,2 The discovery of this mechanism, an unprecedented mechanism of action for an anticancer drug at the time, was a crucial factor in paclitaxel's development as one of the most important drugs available for the treatment of breast and ovarian cancers.3,4 The clinical success of paclitaxel in first-line treatment of cancer spurred the interest of the natural products community in the discovery of other microtubule stabilizing agents (MSAs) with a diversity of chemical structures and sources, and MSAs now represent one of the most important classes of cytotoxic agents. At this time the epothilone derivative ixabepilone5 is the only MSA besides the taxanes paclitaxel, docetaxel, and cabazitaxel4 that has entered clinical use. It is however very likely that other MSAs will enter clinical use in the future, especially as novel MSAs with improved pharmacologic properties and potent activities against multi-drug resistant cancers continue to be discovered from nature,6 and our understanding of the binding sites of these compounds continues to advance.
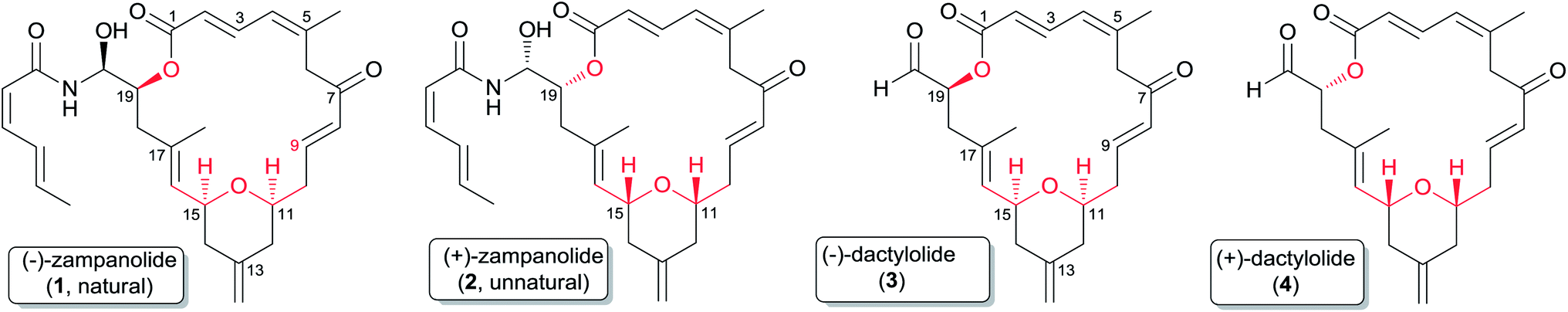
(−)-Zampanolide (1) (Fig. 1)7 is a 20-membered polyketide first isolated from a marine sponge in 1996, and is a unique and recent addition to the class of MSAs.8 It exhibits low nanomolar cytotoxicity against both drug sensitive and multi-drug resistant cancer cell lines, and induces microtubule bundle formation.8 Its potent activity combined with its low number of chiral centres makes it an attractive compound for large-scale synthetic preparation for further study or clinical applications. As described in more detail below, the unique effects of zampanolide on tubulin assembly and its superior activity compared with that of paclitaxel against several multi-drug resistant cancer cell lines suggests that this natural macrolide could exhibit a unique spectrum of cytotoxic activities in a clinical setting and could be a valuable addition to existing anticancer drugs.
Fig. 1 Structures of zampanolide and dactylolide.
The related compound dactylolide was isolated from the marine sponge Dactylospongia sp. in 2001 by Riccio and co-workers,9 and its core structure is the same (excluding stereochemistry) as the macrocyclic core of zampanolide. Its absolute configuration has not been established, but as discussed in Section 2.3 it seems likely on biogenetic grounds that it is the same as that of (−)-zampanolide, which would make natural dactylolide the (−) isomer 3 in spite of its low (+) rotation (Fig. 1).
This review summarizes current knowledge about zampanolide and its structural relative dactylolide, covering their sources and isolation, structures, anticancer potential, mechanism of action, and syntheses.
2. Natural sources, isolation, and structure determination
2.1 Natural sources and isolation
(+)-Zampanolide (1) was first isolated in 1996 in 0.001% yield by Tanaka and Higa from 480 g of the marine sponge Fasciospongia rimasa collected off Cape Zampa on Okinawa.7 It was isolated by repeated chromatography of the acetone extract of the sponge over silica gel and by ODS HPLC. It was re-isolated thirteen years later by Northcote and Miller and co-workers in an even lower 0.0005% yield from 341 g of the marine sponge Cacospongia mycofijiensis collected from an underwater cave off the Tongan coast.8 Zampanolide was obtained from the 80% acetone–H2O fraction of the methanolic extract of the frozen sponge by sequential chromatography over a silica gel column and by HPLC.
Dactylolide was isolated in 2001 by Riccio from 353 g of a marine sponge of the genus Dactylospongia collected off the coast of Vanuatu.9 It was obtained from the CCl4 fraction of the methanolic extract through purification by medium pressure liquid chromatography over silica gel and by C-18 reversed-phase HPLC. Smith and co-workers found that thermolysis of synthetic (+)-zampanolide (2) in benzene provided (+)-dactylolide (4),10 confirming the stereochemical relationship between (+)-zampanolide and (+)-dactylolide, and raising the possibility that dactylolide might be an artefact produced from decomposition of 2 during isolation. To test this hypothesis, Uenishi and co-workers exposed (−)-zampanolide and the sponge to the extraction conditions reported by Riccio and co-workers, and detected no trace of (−)-dactylolide (3) in either case.11 They thus concluded that dactylolide is a naturally occurring entity instead of a degradation artefact of 1 or 2.11 However, this does not exclude the possibility that zampanolide could be a biosynthetic precursor of dactylolide.
2.2 Structure and conformation
The structure and partial stereochemistry of zampanolide were elucidated by Tanaka and Higa based on extensive interpretation of its 2D NMR spectra.7 The relative configurations of the three chiral centres at C11, C15, and C19 were confirmed by observation of critical nOe correlations, but the relative configuration of C20 and the absolute configuration remained undetermined.7 The complete stereochemistry was ultimately assigned by Smith and co-workers on the basis of the total synthesis of the unnatural antipode of zampanolide (2) in 2001.10,12 The relative and absolute configurations of (+)-zampanolide were assigned as 11R, 15R, 19R, and 20R (Fig. 1). Zampanolide features a unique 20-membered macrolactone containing a cis-2,6-tetrahydropyran (THP), four olefinic bonds, and three chiral centres, and an uncommon N-acyl hemiaminal side chain with one chiral centre. Zampanolide is relatively hydrophobic in comparison with other macrolide MSAs and paclitaxel, as evidenced by its higher mobility on normal-phase TLC.
The planar structure of natural dactylolide was reported by Riccio and co-workers in 2001, also based on extensive spectroscopic analysis.9
The planar structures of zampanolide and dactylolide are very similar to each other, with the only difference being that the side chain in zampanolide is replaced by an aldehyde group in dactylolide. The chirality at C19 and the absolute configuration of natural dactylolide were not determined, but were assigned by Smith and co-workers on the basis of a total synthesis of 4 and the earlier conversion of 2 to 4.10,13 This work allowed assignment of the relative and absolute configurations of (+)-dactylolide as 11R, 15R, and 19R.10 So far, no (+)-zampanolide has been isolated from nature.
Taylor and co-workers investigated the solution conformation behaviour of the macrolide core of (−)-zampanolide and (−)-dactylolide through a combination of high-field ROESY NMR experiments and computational modeling.14 Three interconverting conformational families, one of which bears a strong resemblance to zampanolide's tubulin-bound conformation, were found in solution for both molecules.14 A rigid C15–C19 region and an s-trans northern diene are the common features shared by the three conformational families. The primary difference between the two major families [“hooked” (33%) and “flat” (35%)] is the orientation of the mostly inflexible northern fragment relative to the plane of the macrolide ring. The third conformational family (20%) features a twisted shape due to a 180° rotation around the C6–C7 bond. Although the western and diene segments are conformationally rigid, the macrolide has a flexible C6–C11 region, in part due to the ability of the C5–C6–C7–C8 torsion to adopt both 60° and 300° dihedral angles.14
2.3 The optical rotations of zampanolide and dactylolide
As shown in Table 1, there are significant deviations in optical rotations reported for the same compound from different sources. Keck15 opined that the deviation may be caused by the enolization of a proton at C6, C10, or even the C5 methyl group. The enol content could vary with the use of different batches of solvent and/or different sources of the sample. For such highly conjugated enols, the measured optical rotation could be significantly different due to even small variations in enol content, and the large negative rotation of zampanolide is consistent with the large but variable negative rotations of the synthetic products with structure 1. The configuration of naturally occurring zampanolide has thus been established as 1.
Table 1Optical rotations for zampanolide and dactylolide
The case of dactylolide is more complex, since the optical rotation value of the natural product is relatively small, and the rotations for natural and synthetic dactylolides are very different. Natural dactylolide has a rotation of +30, which is between the values for synthetic (+)-dactylolide, which range from +134 to +235, and synthetic (−)-dactylolide, which range from −128 to −258. Because the natural product has such a different rotation from both (+) and (−) synthetic enantiomers, the absolute configuration of the natural product cannot be considered to be established, and indeed the original paper on the isolation of dactylolide did not assign a configuration to the aldehyde at C19 or any absolute configuration to the other chiral centers.9 Consequently, given the variation in optical rotations caused by varying amounts of enol form, and also given the fact that Hoye and co-workers noted the propensity of the aldehyde of dactylolide to form a stable hemiacetal with methanol,18 it is possible that naturally occurring dactylolide possesses the same absolute configuration as naturally occurring (−)-zampanolide and has a (−) rotation. Its small observed (+) rotation could be an artefact of enolization and/or hemiacetal formation. The strong probability that (−)-dactylolide is an intermediate in the biosynthesis of (−)-zampanolide (or vice versa) strengthens the assignment of natural dactylolide as 3.
3. Biological activities and SAR
3.1 Cytotoxicity and MDR susceptibility
(−)-Zampanolide was initially found to exhibit potent cytotoxicity (IC50 = 2–10 nM) against the P388, A549, HT29, and Mel28 cell lines.7,22 Follow-up investigations demonstrated its low-nanomolar cytotoxicity against HL-60,8 1A9,8 A2780,8,23 OVCAR 8,24 SKM-1,11 and U937 (ref. 11) cell lines. More importantly, it also exhibits low nanomolar cytotoxicity against multi-drug resistant cancer cells that overexpress the P-gp multidrug resistance (MDR) pump, as shown in Table 2.8,24 As noted by Diaz and his co-workers,23 zampanolide is an interesting lead compound because of this diminished susceptibility. Such compounds may also have the potential for oral administration.25 Covalent binding of a drug to its target can effectively inhibit the ability of P-gp to pump the drug out of the cell, and this has been demonstrated to be a method of avoiding P-gp mediated drug resistance in preclinical settings.6 Since zampanolide binds covalently to tubulin, it has the potential to treat multi-drug resistant cancer.8 Interestingly, (−)-dactylolide (3) also exhibits similar cytotoxicity towards both drug sensitive (A2780) and drug resistant cancer cells (A2780AD), even though it possesses much lower potency against these two cell lines than (−)-zampanolide (Table 2).
Table 2IC50 values (nM) for zampanolide and dactylolide against drug-sensitive and drug-resistant cancer cells
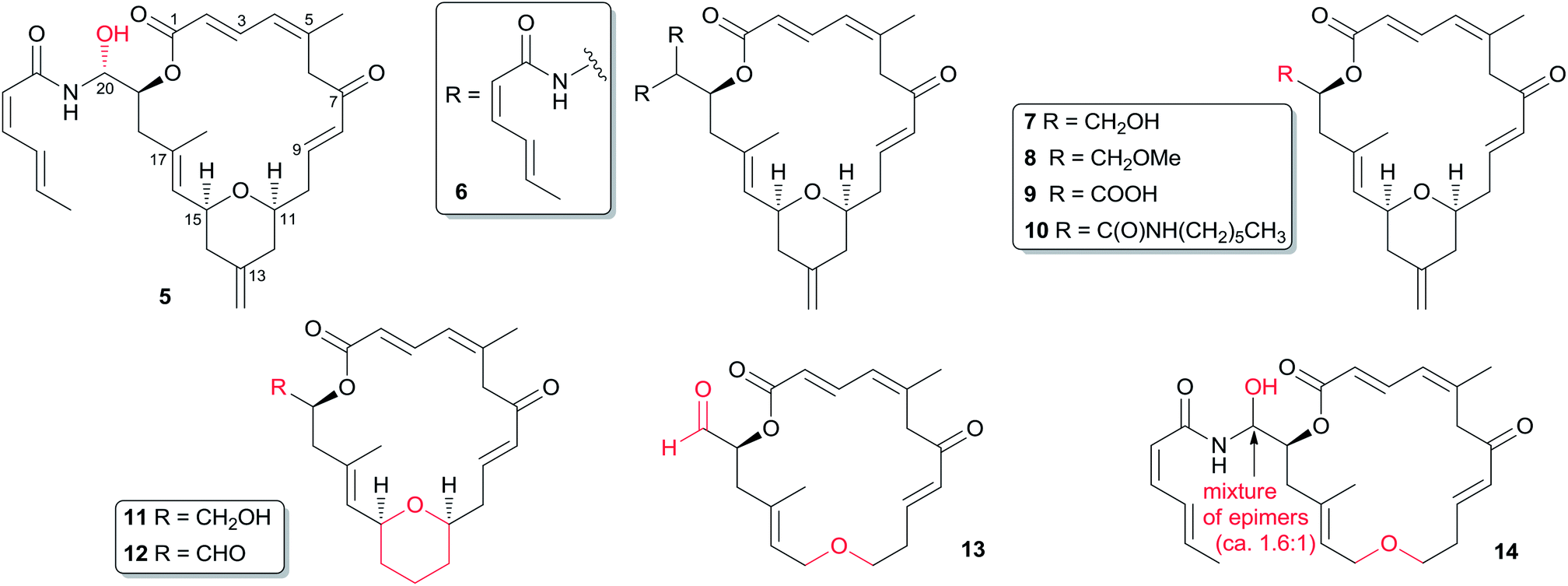
Only a small amount of structure–activity work has been reported on zampanolide (1). So far, all the available analogues (Fig. 2) for SAR investigation have come from synthetic by-products or from work from the Altmann laboratory. As shown in Table 3, its N-acyl hemiaminal side chain plays a dramatic role in the nanomolar IC50 values against the four cell lines tested.19 Jennings' synthetic (−)-dactylolide (3) was 10- to 1000-fold less potent (GI50) than 1 against A549, HT29, and SK-Mel-28 cell lines. Interestingly, synthetic 3 is slightly more active than natural dactylolide against the SK-OV-3 cancer cell line.
Fig. 2 Zampanolide analogs.
Table 3Antiproliferative activities of 1, 3, and 4a
Cell line
1
3
4
aGI50 values: data from ref. 19 unless otherwise stated.bIC50 or GI50 value.cGI40 value.
The C20-epimer of 1 (5) and the bis-amide by-product (6)11,17,24 are about one order of magnitude less potent than 1 in all the cell lines examined. Altmann's analogs 7, 11, and 12 showed comparable activity to that of 3, suggesting that neither the aldehyde functionality nor the 13-methylene group are essential for the relatively weak antiproliferative activity of 3.17 Conversion of the C20-hydroxyl group in 7 to the C20-methoxyl group in 8 leads to a 14-fold loss in antiproliferative potency. Carboxylic acid 9 is significantly less potent than the corresponding primary alcohol 7. Interestingly, amide 10 is about 10-fold more potent than acid 9, although the latter compound is several hundred-fold less potent than zampanolide. The encouraging activity of 10 suggests that fine-tuning of the substituent moiety on the amide nitrogen might be a way to develop improved analogs. The most intriguing analog from the Altmann laboratory is compound 14, with submicromolar activity and a simple ether instead of the THP ring of the parent compound. Although the compound is 25 to 80-fold less potent than zampanolide, its activity is still very good in light of the removal of half the chiral centres and a rigidifying structural element. The corresponding (−)-dactylolide derivative (13) is about an order of magnitude less active than 14. It is important to note that 14 is a ca. 1.6:1 mixture of diastereomers, suggesting that the IC50 values for 18S-14 could in fact be lower than observed (Table 4). These structure–cytotoxicity relationships are consistent with the results expected from the X-ray binding structure of zampanolide and β-tubulin, as discussed in Section 4.3.
Table 4IC50 values (nM) for zampanolide and its analogs against four cell linesa
As noted above, zampanolide is a novel and potent MSA with properties similar to those of paclitaxel and docetaxel, stoichiometrically inducing tubulin assembly. It arrests cells in the G2/M phase of the cell cycle, induces microtubule bundles in interphase cells and spindle asters in dividing cells, and causes a dose-dependent shift of soluble tubulin to polymerized tubulin in cells and with purified tubulin.8,26 It competes with paclitaxel for binding to microtubules,23 but its mechanism of action is very different from paclitaxel's, as it involves covalent binding to β-tubulin, whereas paclitaxel binds non-covalently. The following sections describe its binding site and its mechanism of action in more detail.
4.2 Binding site on tubulin
There are three distinct binding sites for MSAs on microtubules: the taxane luminal site, the taxane pore site, and the peloruside site. The taxane luminal site is the classical taxane binding site that has been mapped to β-tubulin on the interior lumen of the microtubule.27 Such a luminal location makes access to the binding site difficult in assembled microtubules. Kinetic data suggested that MSAs are able to access this site only after initially binding to a low-affinity site on the exterior of the microtubule.28 The exterior site was later identified as the Thr220 taxane pore binding site by Diaz and co-workers when they investigated cyclostreptin, the first MSA known to bind covalently to microtubules.29,30 The third MSA binding site is entirely distinct from the taxane site and is targeted by the natural products laulimalide and peloruside A. Mass shift perturbation analysis in combination with data-directed computational docking were used to demonstrate that peloruside A and laulimalide bind to β-tubulin on the exterior surface of the microtubule.31,32
The first direct evidence for the binding site of zampanolide on tubulin was provided by Diaz and co-workers, who used mass spectrometry to determine the binding sites of both zampanolide and dactylolide on β-tubulin,23 and provided a step-by-step protocol for this procedure.33 Both zampanolide (1) and synthetic (−)-dactylolide (3) bind with a 1:1 stoichiometry, and compete with paclitaxel for binding to microtubules.23 High resolution mass spectrometry showed that zampanolide binds to β-tubulin by irreversible alkylation of residues Asn228 and His229 at the taxane luminal site on the microtubule, with binding at His229 yielding a significantly more abundant fragment ion than binding at Asn228. Zampanolide was able to bind to these amino acids in both dimeric tubulin and intact microtubules, indicating that the taxane luminal site is accessible in intact microtubules and also exists in tubulin dimers. In addition, since the binding to the luminal site occurs with a 1:1 stoichiometry, zampanolide does not bind at the pore site. Zampanolide strongly inhibited paclitaxel binding without interfering with peloruside A binding, and its specific binding to the taxane luminal site can impede or modify the taxane pore site. Thus hexaflutax (a fluorescent taxane probe that binds to the taxane pore site34) did not bind to tubulin in the presence of zampanolide even at stoichiometric concentrations with tubulin.23 The apparent binding constants of both 1 and 3 increased with temperature, indicating possible covalent binding to microtubules,35,36 similar to that of cyclostreptin.37
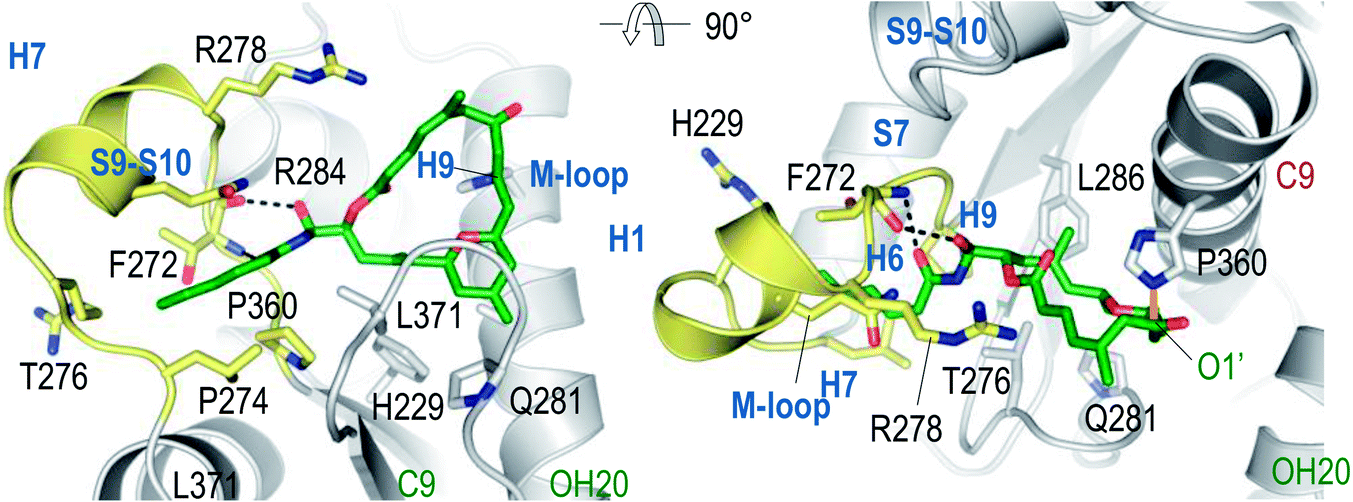
In an important publication Steinmetz et al. reported the structure of α,β-tubulin complexed with the stathmin-like protein RB3, tubulin–tyrosine ligase, and zampanolide at 1.8 Å resolution (Fig. 3).38 This structure clearly showed that zampanolide was deeply buried in the taxane luminal binding pocket formed by hydrophobic residues of helix H7, β strand S7, and the M-loop, and that its C9 atom was covalently bound to the NE2 atom of His229 of β-tubulin, consistent with the major fragment ion in the mass spectrometric results described above.23
Fig. 3 Two views of the tubulin–zampanolide complex at 1.8 Å resolution. Adapted from Science, 2013, 339, 587–590 with permission.
Kinetic binding studies of 1 and 3 using HPLC and fluorescence methods indicated that (−)-zampanolide reacted covalently with the taxane luminal site on both assembled microtubules and unassembled tubulin at a faster rate and more extensively than (−)-dactylolide.23 (−)-Zampanolide has similar binding modes with both microtubules and unassembled tubulin, but (−)-dactylolide has three binding modes with microtubules and two binding modes with unassembled tubulin.23
4.3 Bioactive conformation on tubulin
The bioactive conformation of dactylolide bound to the tubulin α,β-heterodimer and MTs was deduced by analysis of TR-NOESY crosspeaks. This was then used to build a putative zampanolide bioactive conformation.23 This binding conformation was then confirmed by the crystal structure of the zampanolide tubulin complex noted above.38 In this conformation, the 20OH group and the 1′O atom of zampanolide are hydrogen bonded to the main-chain carbonyl oxygen and the NH group of Thr276, respectively. The helical conformation of the M-loop is induced by the hydrophobic and polar contacts between the side chain of zampanolide and residues of the M-loop. This bound conformation closely resembles the major solution conformation found by Taylor et al., albeit with a change to an s-cis orientation of the C7–C9 enone.14
This bioactive conformation is consistent with the cytotoxicities of zampanolide analogs summarized in Section 3.2. The cytotoxicity for the C20-epimer of 1 is decreased due to the lack of hydrogen bonding between the 20OH group of zampanolide and the main-chain carbonyl oxygen of Thr276. The side chain of zampanolide is a major contributor to its potent cytotoxicity, due to its interaction with the M-loop, and the potent cytotoxicity of Altmann's simplified compound 14 can be explained by the fact that no critical interaction was observed between the THP ring of zampanolide and β-tubulin.
4.4 Mechanism of microtubule stabilization
As a covalent binding MSA, zampanolide induces microtubule polymerization through an allosteric effect in which the modified tubulin has a higher affinity towards polymerized microtubules than the unmodified fraction of tubulin.23 The two types of contacts between tubulin subunits in the polymerization process are longitudinal contacts within protofilaments and lateral contacts between protofilaments.39 It has been demonstrated that the M-loops of both α- and β-tubulin play a central role in the lateral tubulin contacts in the absence of ligands.39,40 The structure of the tubulin–zampanolide complex confirmed the existence of a short helix, consisting of residues Arg278-Tyr283, in the M-loop of β-tubulin.38 Steinmetz et al. found that the MSA-stabilized helical M-loop conformation of β-tubulin agrees with the corresponding density of electron microscopy reconstructions of microtubules, and that Tyr283 of the M-loop is inserted across protofilaments into a pocket shaped by the S2′–S2′′ β-hairpin and the H2–H3 loop of a neighbouring β-tubulin subunit. They also showed that the secondary structure elements of the taxane binding pocket were not significantly affected by the curved-to-straight tubulin conformational transition, and that the favourable positions of the M-loop residues Ser280, Gln282, Arg284, and Ala285 provide additional contacts with the neighbouring β-tubulin. Based on these results, it was proposed that the binding of a MSA results in tubulin preorganization and subsequent reduction of the entropy loss associated with microtubule polymerization.38
Modeling of the helical conformation of the M-loop in the microtubule lattice, using the observed tubulin structure and cryogenic electron microscopy reconstructions of microtubules at ∼8 Å resolution, led Steinmetz et al. to propose that zampanolide and other taxane-site MSAs promote tubulin polymerization by binding to the taxane luminal site, converting the disordered M-loop to a helix, and thus favouring polymerization. Microtubules that are formed spontaneously can also be stabilized by binding of the MSA to tubulin in the microtubule.
A recent computational docking study supported the paclitaxel site as the tubulin-binding site of zampanolide, and also indicated that His227 is in a position to attack zampanolide and form a covalent bond.41
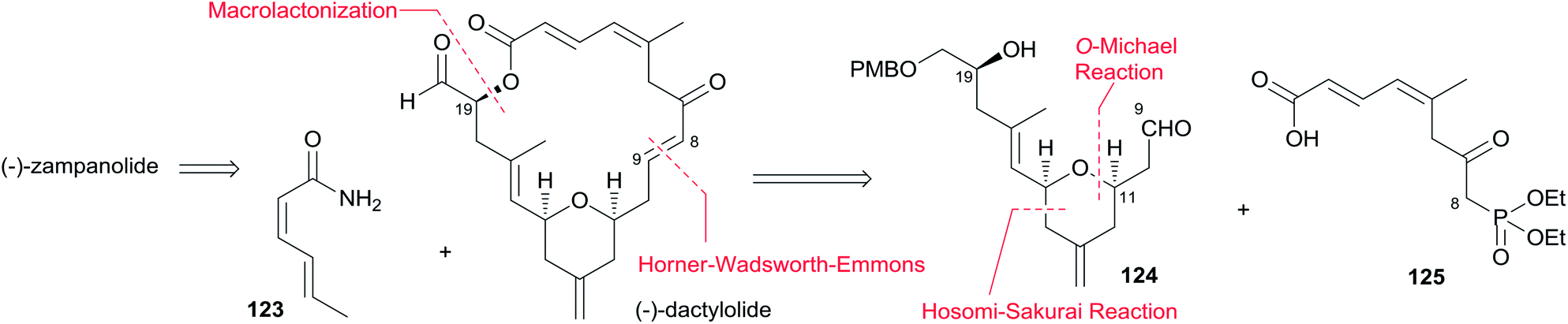
5. Total synthesis of zampanolide and dactylolide
5.1 Introduction
Twelve independent total syntheses of zampanolide, dactylolide and related macrolactone core have been achieved to date. Five synthetic approaches to zampanolide and dactylolide published prior to 2005 have been included in an earlier review,42 and Sharif and O'Doherty43 provided a condensation of and commentary on Smith's and Jennings' syntheses.
5.2 Smith's syntheses of (+)-zampanolide and (+)-dactylolide
The first total synthesis of unnatural (+)-zampanolide (2) was achieved by the Smith group. Since neither the relative configuration at C20 nor the absolute configuration of 1 had been defined at that time, they arbitrarily set 2 as their initial target.10 Subsequently, the Smith group employed a similar synthetic strategy to synthesize (+)-dactylolide (4).10,13
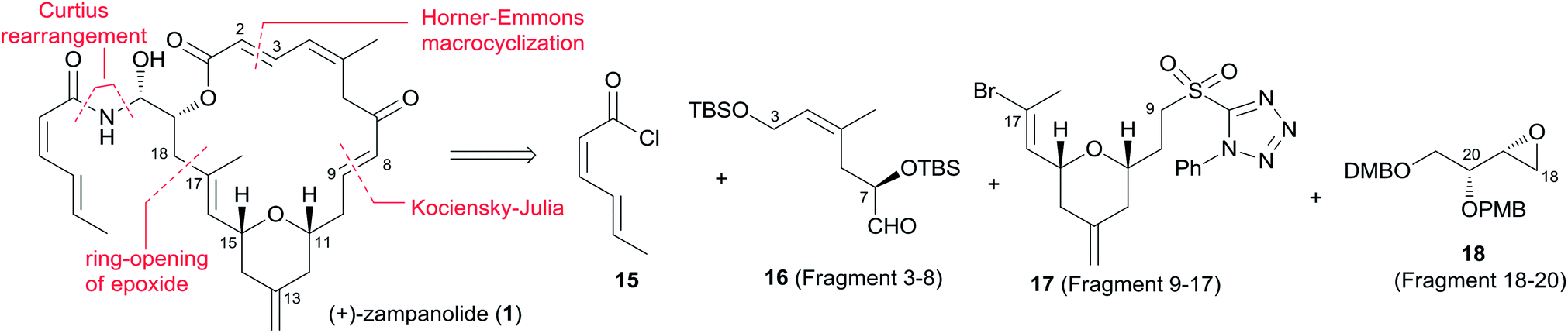
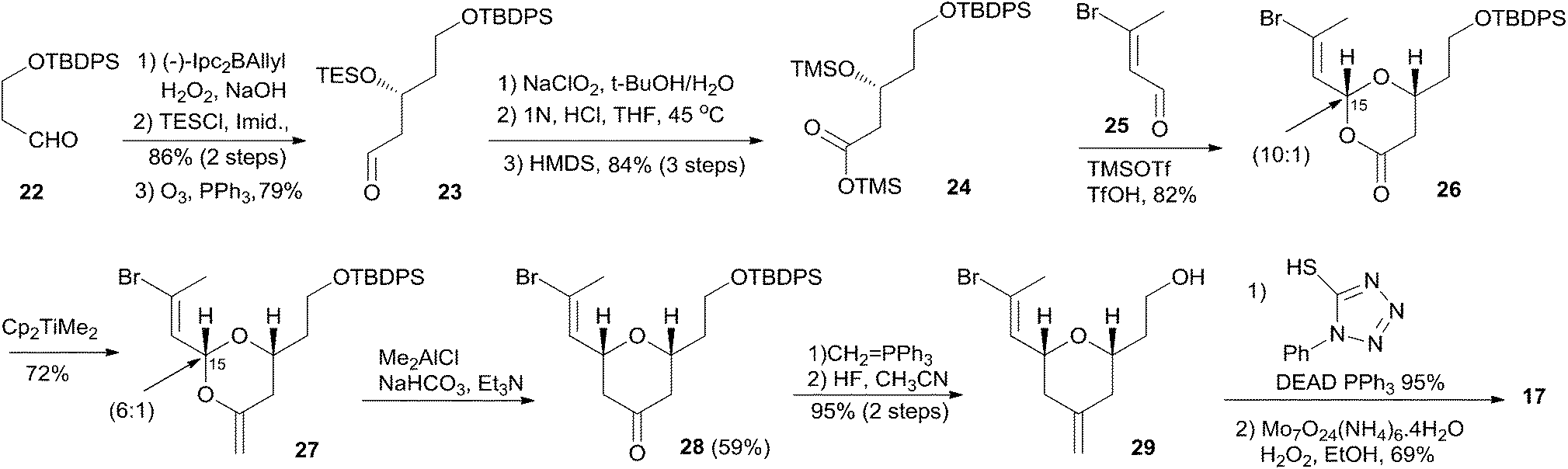
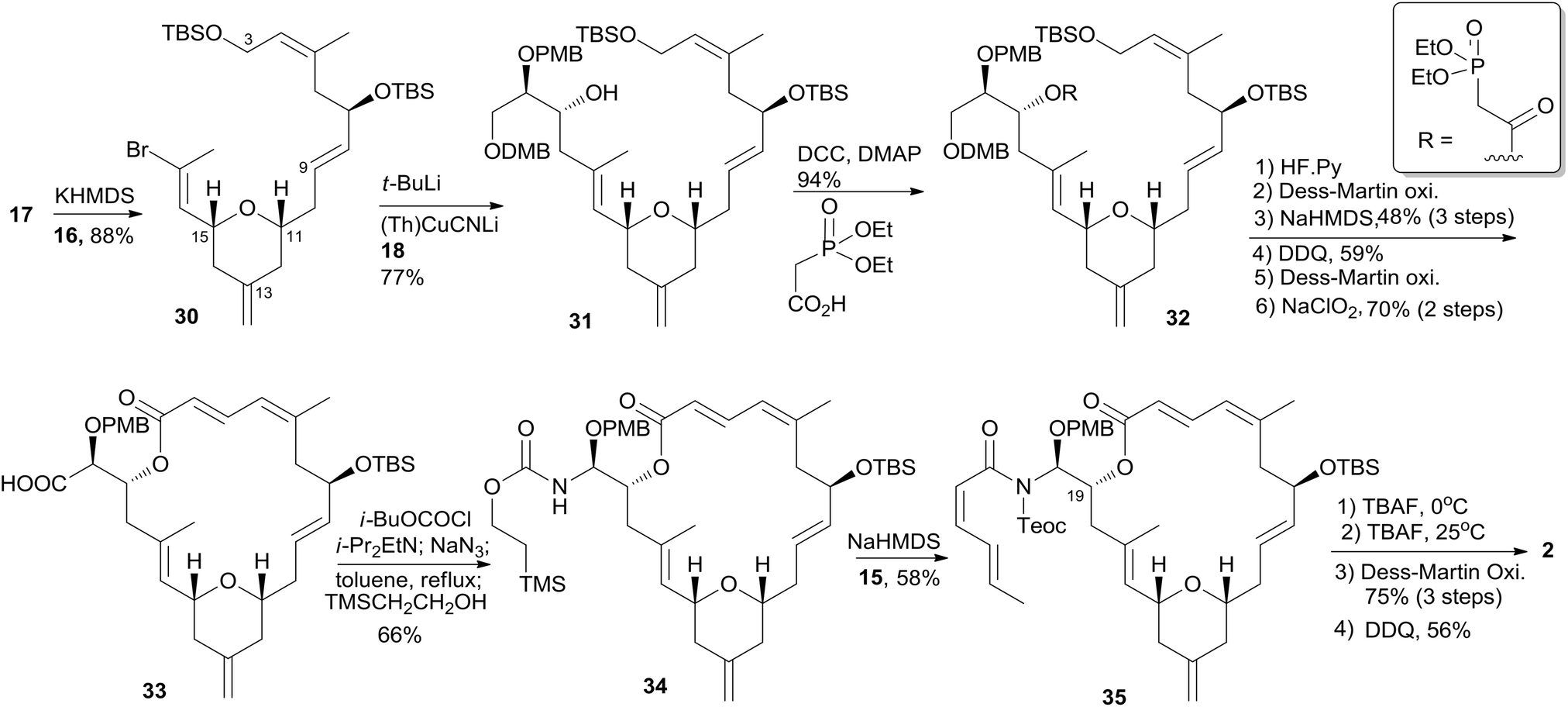
Smith's retrosynthetic analysis is outlined in Scheme 1.10,12,13 A stereoselective Curtius rearrangement44 was used to install the N-acyl hemiaminal side chain and Horner–Emmons macrocyclization45 was employed to close the macrolactone ring at C2–C3. The macrocyclization precursor was constructed using nucleophilic epoxide ring-opening and Kocienski–Julia olefination as key reactions. The Petasis–Ferrier rearrangement was developed by Smith as a powerful and stereoselective approach to cis-2,6-disubstituted THP groups, and it was elegantly applied to the construction of the same unit in Smith's synthesis of zampanolide.46 As shown in Scheme 2, fragment C3–C8 (16) was prepared from known alkynolate 20 (ref. 47) via a Michael-type carbometallation followed by four sequential reactions: reduction, protection, deprotection, and oxidation. The Smith synthesis of fragment C9–C20 is shown in Scheme 3. The aldehyde 23 was prepared via Brown asymmetric allylation of aldehyde 22,48 followed by silylation and oxidative cleavage of the terminal alkene. The aldehyde was readily converted to β-hydroxy ester 24 in two steps, and this was condensed with aldehyde 25 mediated by TMSOTf to give a mixture of dioxanones 26 (10:1 at C-5). This cyclization was presumed to proceed via a transition state wherein the aldehyde side chain adopts a pseudoequatorial orientation. A mixture of the cyclic acetals 27 (6:1 at C-15) was achieved via the Petasis–Tebbe methylenation of 26, which was subjected to the Petasis-Ferrier rearrangement with Me2AlCl to yield 2,6-cis-pyranone 28. Wittig methylenation of ketone 28 followed by desilylation gave alkene 29. Sulfone 17 was achieved by incorporation of the thiotetrazole via the Mitsunobu protocol and oxidation. Fragment C18–C21 (18) was prepared using (+)-diethyl tartrate as starting material through a seven-step sequence. As shown in Scheme 4, the trans-C8–C9 double bond in 30 was built by treating aldehyde 16 with sulfone 17 through the Kocienske–Julia olefination reaction. Fragment C3–C21 (31) was achieved by reaction of the mixed cyano-Gilman cuprate, derived from vinyl bromide 30 and lithium 2-thienylcyanocuprate, with epoxide 18. Phosphonoacetate 32, prepared by condensing diethylphosphonoacetic acid with 31, was subjected to Horner–Emmons macrocyclization reaction and a following three-step sequence to provide carboxylic acid 33. Carbamate 34 was prepared via Curtius rearrangement followed by treatment with trimethylsilylethanol. Transformation of carbamate 34 to (+)-zampanolide (2) was achieved by acylation with acid chloride 15 followed by a four-step, deprotection–oxidation sequence.
Scheme 1 Smith's retrosynthetic analysis of (+)-zampanolide.
Scheme 2 Smith's synthesis of fragment C3–C8 of (+)-zampanolide.
Scheme 3 Smith's synthesis of fragment C9–C17 of (+)-zampanolide.
Scheme 4 Smith's total synthesis of (+)-zampanolide.
5.3 Hoye's total synthesis of (−)-dactylolide and (−)-zampanolide
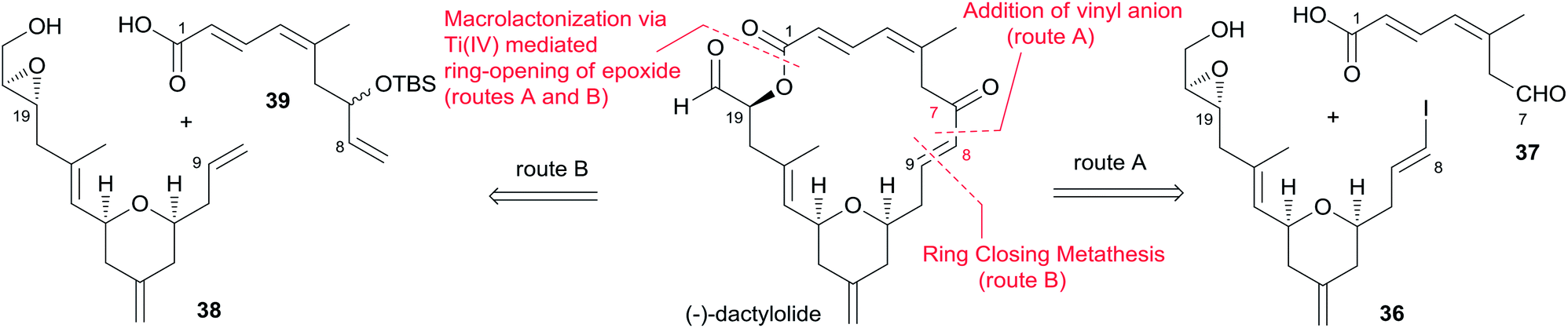
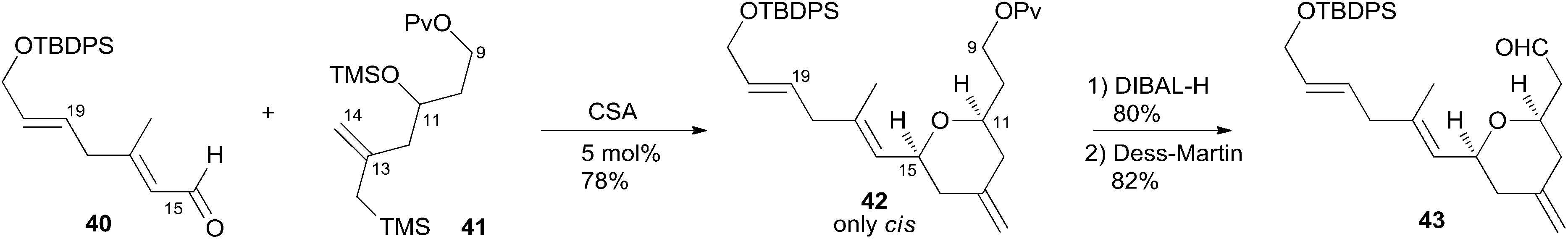
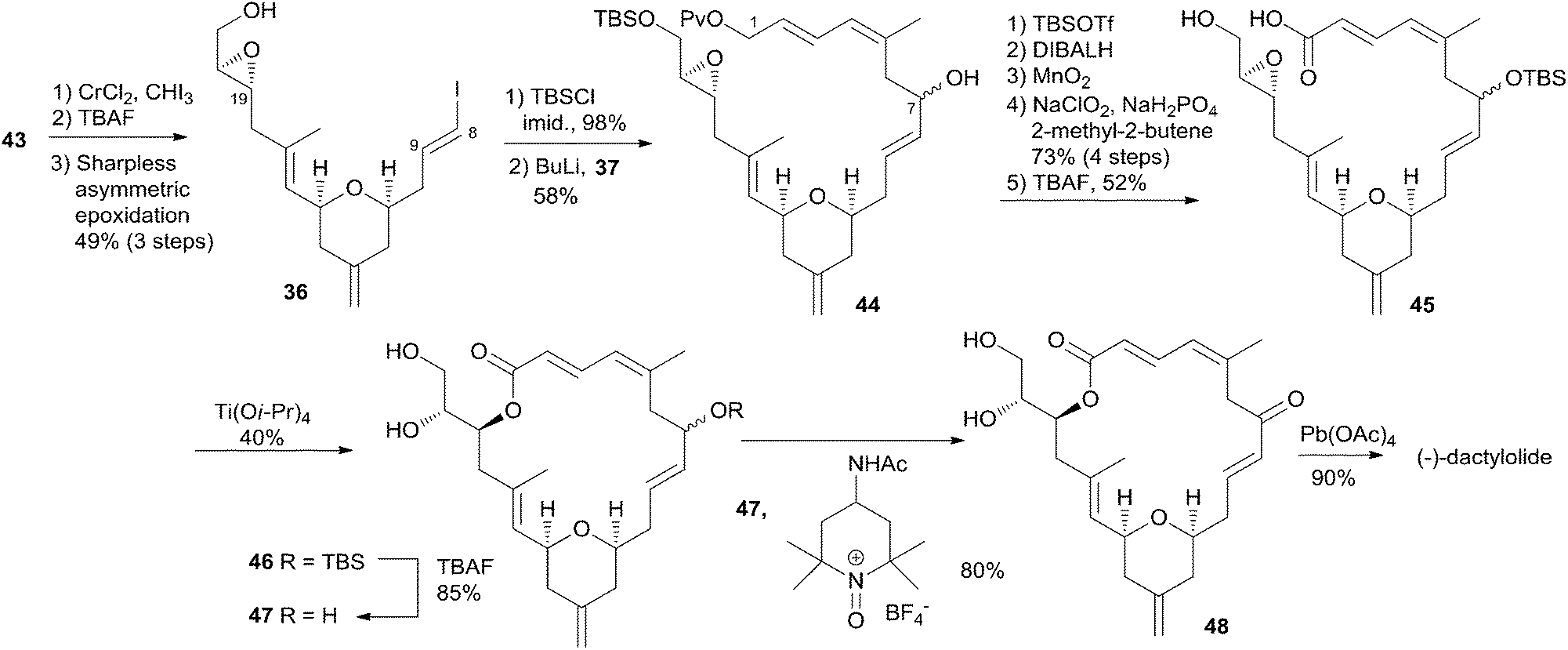
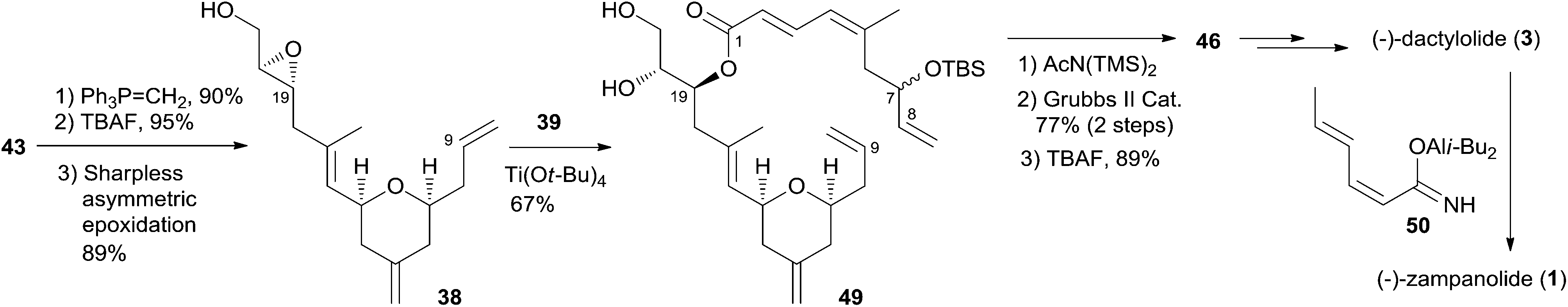
Hoye and co-workers completed their syntheses of (−)-dactylolide (3)18 using two strategies: (i) a novel Ti(IV)-mediated macrolactonization of an epoxy-acid (route A); and (ii) a complementary ring-closing metathesis (RCM) macrocyclization (route B) (Scheme 5). The fragment C9–C20 was constructed using Hosomi–Sakurai–Prins (HSP) cyclization as a key reaction (Scheme 6).49 The 2,6-cis-THP 42, which was readily converted into aldehyde 43, was exclusively generated by the HSP cyclization reaction49 of enal 40 with allyl silane 41 (ref. 50) catalyzed by a protic acid. As shown in Scheme 7, aldehyde 43 was converted to epoxide 36 through a three-step sequence including Takai iodoalkenylation, desilylation, and Sharpless asymmetric epoxidation. Reaction of the alkenyllithium, prepared from epoxide 36 by protection followed by halogen–lithium exchange reaction, with aldehyde 37 generated fragment C1–C20 (44), which was converted into epoxy-carboxylic acid 45 in a five-step sequence. Macrolactone 46 was achieved by treating 45 with Ti(Oi-Pr)4 under high dilution conditions. Sequential desilylation, oxidation of allylic alcohol, and oxidative cleavage of the C20–C21 diol generated 3. Hoye's alternative and more convergent synthesis of 3 as well as its subsequent conversion to 1 is shown in Scheme 8. Olefin 38, prepared from aldehyde 43, was coupled with trienoic acid 39 catalyzed by Ti(Ot-Bu)4 to give diene precursor 49. After protection of the vicinal diol in situ with excess bis-trimethylsilylacetamide, Grubbs RCM generated macrolactone 46 with an E-geometry of the C8–C9 alkene.
Scheme 5 Hoye's retrosynthetic analysis of (−)-dactylolide.
Scheme 6 Hoye's synthesis of fragment C9–C20 of (−)-dactylolide.
Scheme 7 Hoye's total synthesis of (−)-dactylolide.
Scheme 8 Hoye's alternative total synthesis of (−)-dactylolide and (−)-zampanolide.
Compound 3 was converted to zampanolide via the aluminum aza-aldol addition of 50 in an unreported yield.
5.4 Floreancig's total synthesis of (+)-dactylolide
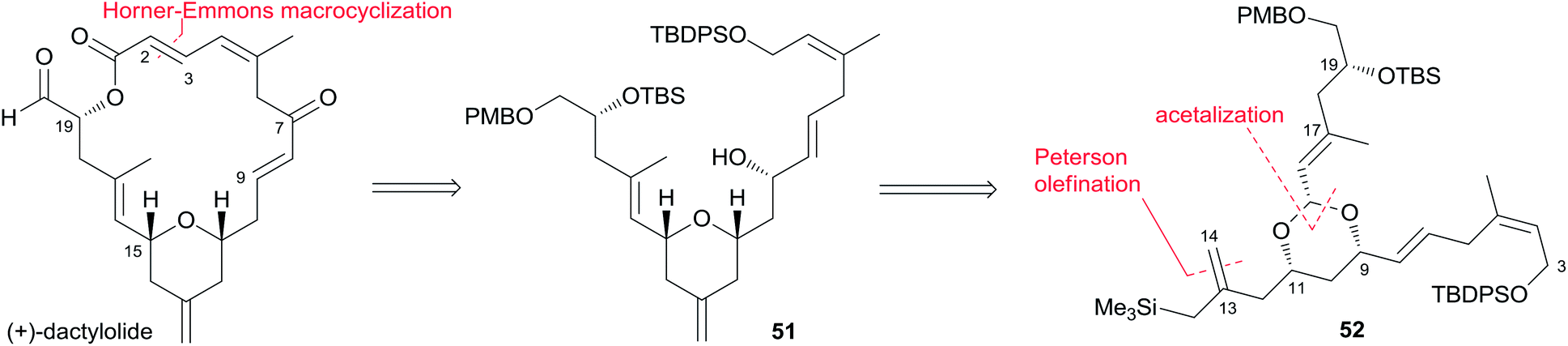
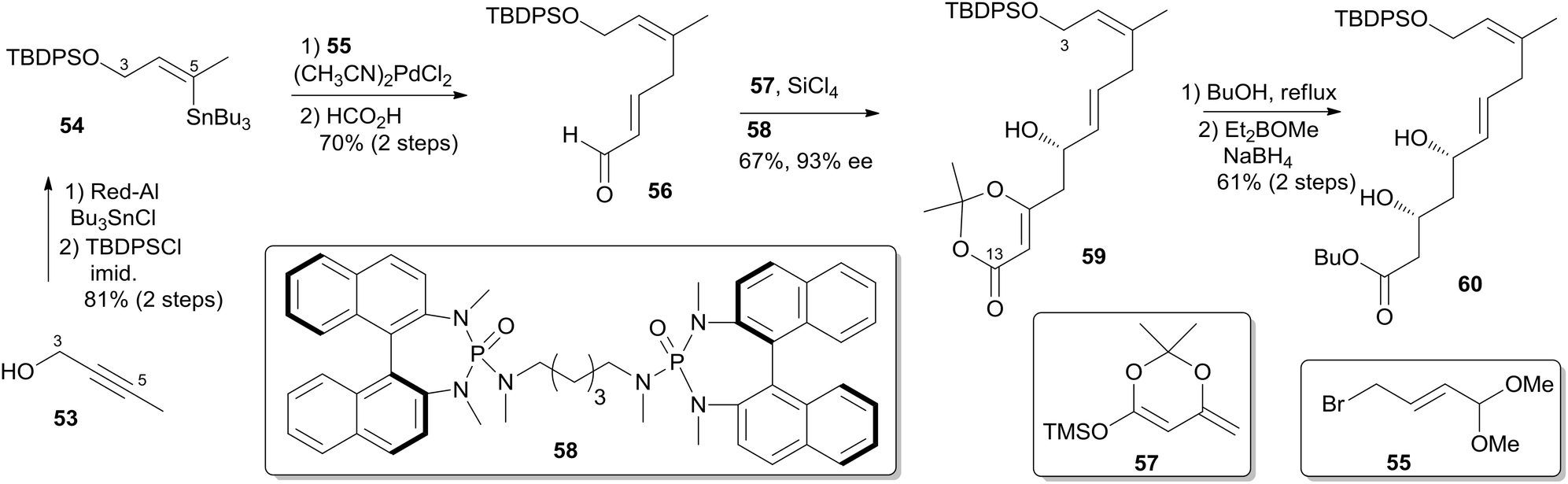
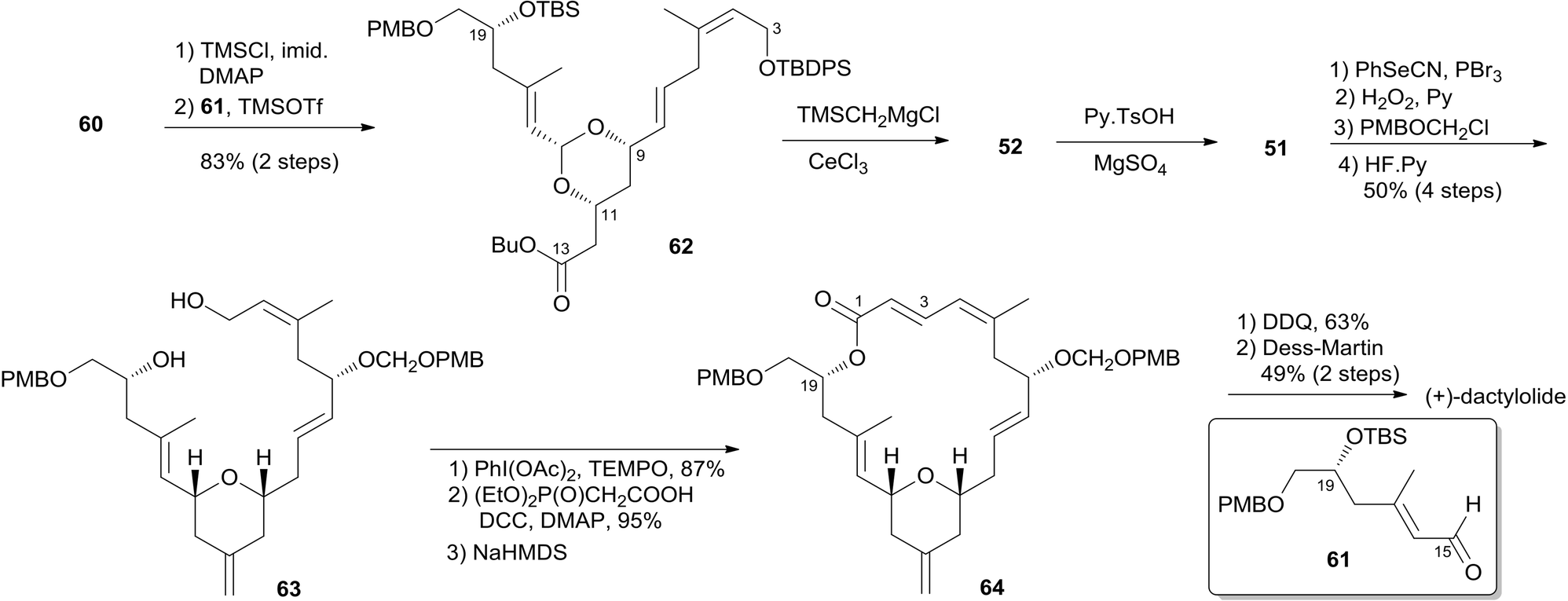
Two key objectives to Floreancig's total synthesis of dactylolide were to maximize convergency and to minimize the number of carbon–carbon bond-forming reactions.21 They postulated that the former objective could be achieved through the union of two advanced fragments by an acetal linkage. They envisioned asymmetric vinylogous Mukaiyama aldol reactions to be effective vehicles for achieving the latter objective. As shown in Floreancig's retrosynthetic analysis (Scheme 9), macrocyclization could be achieved by a Horner–Wadsworth–Emmons (HWE) reaction, the same strategy used in Smith's synthesis.10,12,13 2,6-cis-THP in fragment C3–C20 (51) could be constructed via the sequential cyclization starting from acetal 52. Fragment C15–C20 (61) was prepared using the same procedure as described by Evans.51 Fragment C3–C13 (60) was synthesized using the procedure as illustrated in Scheme 10. The (Z)-vinyl stannane 54 was prepared from hydroalumination of alkyne 53 followed by stannylation. Coupling of stannane 54 with bromide 55 followed by hydrolysis generated enal 56. Ketal 59 was achieved in 93% ee through the asymmetric Mukaiyama aldol reaction of enal 56 with acetal 57 catalysed by Denmark's catalyst 58 and SiCl4. Fragment C3–C13 (60) was readily obtained by esterification of 59 followed by stereoselective reduction. Treatment of bis-TMS ether of 60 with aldehyde 61 mediated by TMSOTf provided the key acetal 62. 2,6-cis-THP 51 was prepared by Peterson olefination of 62 with excess TMSMgCl and CeCl3 followed by HSP cyclization catalysed by pyridinium triflate and MgSO4. The observed selectivity of kinetically facile 6-endo pathway over 8-endo pathway suggested that cyclization instead of ionization might be the product-determining step. Rearrangement of the C9 hydroxy group to C7 was achieved by a selenium variant of the Mislow–Evans rearrangement. Transformation from 51 to 63 was achieved in four steps. The corresponding phosphonoacetate, prepared by selective oxidation of allylic alcohol in 63 followed by esterification of C19 hydroxy group with phosphono acetic acid, underwent intramolecular HWE reaction mediated by NaHMDS to generate macrolactone 64. Finally, (+)-dactylolide was obtained through a deprotection–oxidation procedure (Scheme 11).
Scheme 9 Floreancig's retrosynthetic analysis of (+)-dactylolide.
Scheme 10 Floreancig's synthesis of fragment C3–C13 of (+)-dactylolide.
Scheme 11 Floreancig's total synthesis of (+)-dactylolide.
5.5 Jennings' total synthesis of (−)-dactylolide
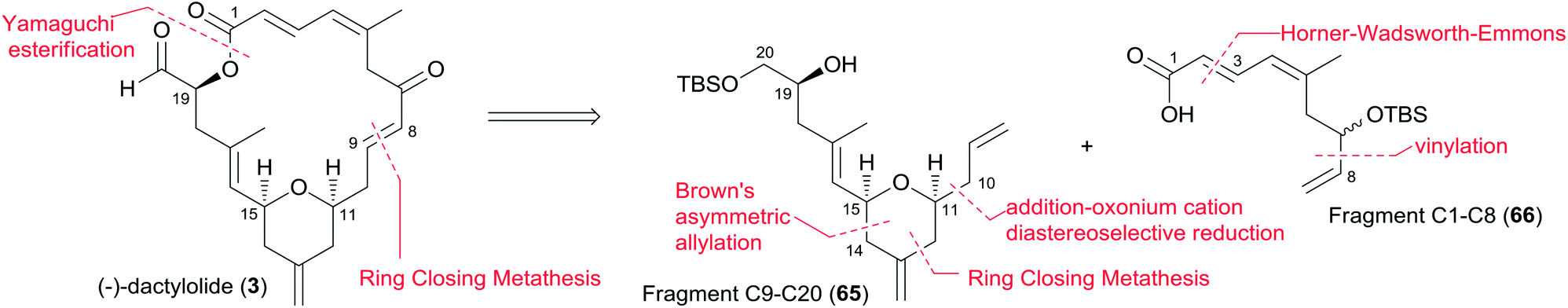
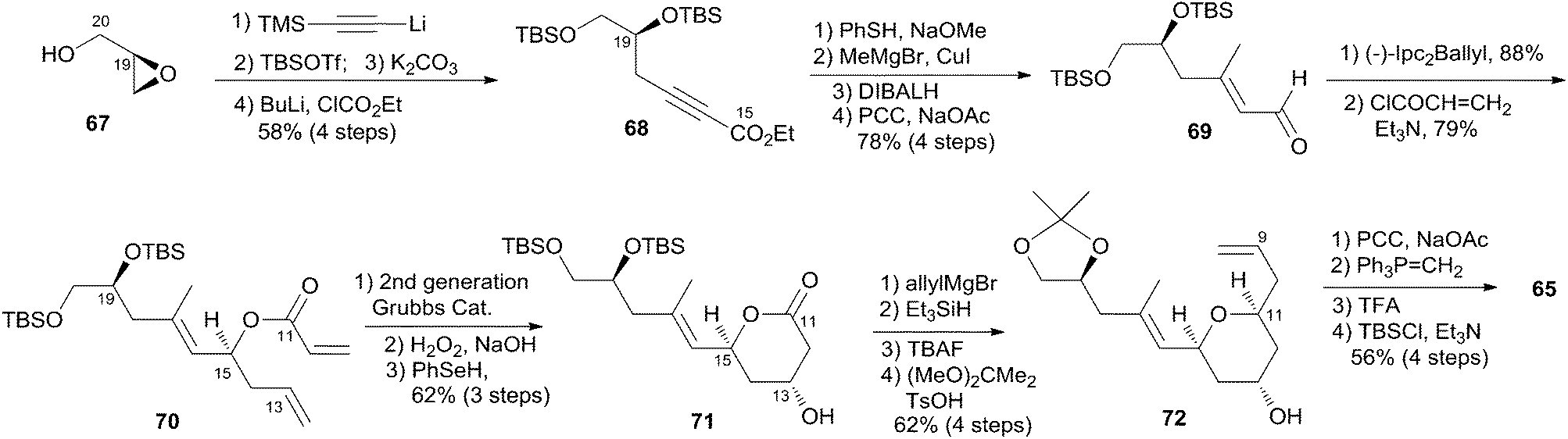
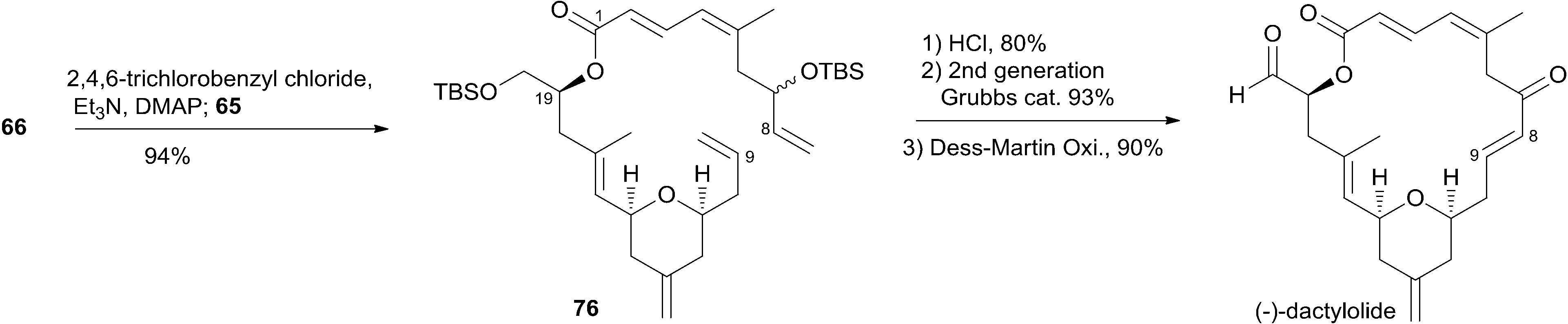
Jennings and co-workers constructed the macrolactone core structure by reaction of fragment C9–C20 (65) and fragment C1–C8 (66) by Yamaguchi esterification and RCM (Scheme 12).19,52 Fragment C9–C20 (65) was constructed via diastereoselective axial reduction of an oxonium cation, RCM of a divinyl ester, and introduction of an asymmetric centre by Brown's asymmetric allylation. As delineated in Scheme 13, the α,β-alkynyl ester 68, prepared from glycidol 67 in four steps, was converted to enal 69 through a 4-step sequence: Michael addition of thiolate, copper-mediated 1,4-addition–elimination, reduction, and oxidation. Brown's asymmetric allylation of 69 followed by esterification furnished acrylate ester 70. RCM of 70 followed by oxidation and reduction with PhSeH provided lactone 71. 2,6-cis-THP 72 was prepared by the nucleophilic addition of ester 71 followed by diastereoselective reduction of oxonium cation. Fragment C9–C20 (65) was obtained in a four-step sequence: oxidation, methylenation, deprotection, and protection. Jennings' approach to fragment C1–C8 features a diastereoselective HWE, as shown in Scheme 14. The diene precursor 76 was made by Yamaguchi esterification of alcohol 65 with carboxylic acid 66 (Scheme 15). After removal of the protecting groups at C7 and C20, the corresponding diene was subjected to Grubbs' RCM to generate the desired macrolactone. Finally, (−)-dactylolide was obtained by oxidation with Dess–Martin reagent.
Scheme 12 Jenning's retrosynthetic analysis of (−)-dactylolide.
Scheme 13 Jenning's synthesis of fragment C9–C20 of (−)-dactylolide.
Scheme 14 Jenning's synthesis of fragment C1–C8 of (−)-dactylolide.
Scheme 15 Jenning's total synthesis of (−)-dactylolide.
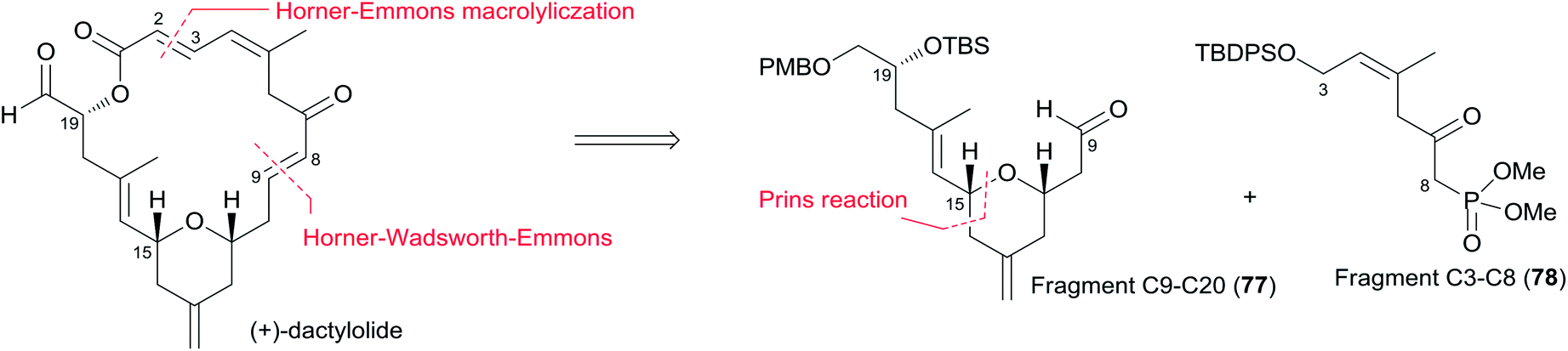
5.6 Keck's synthesis of (+)-dactylolide
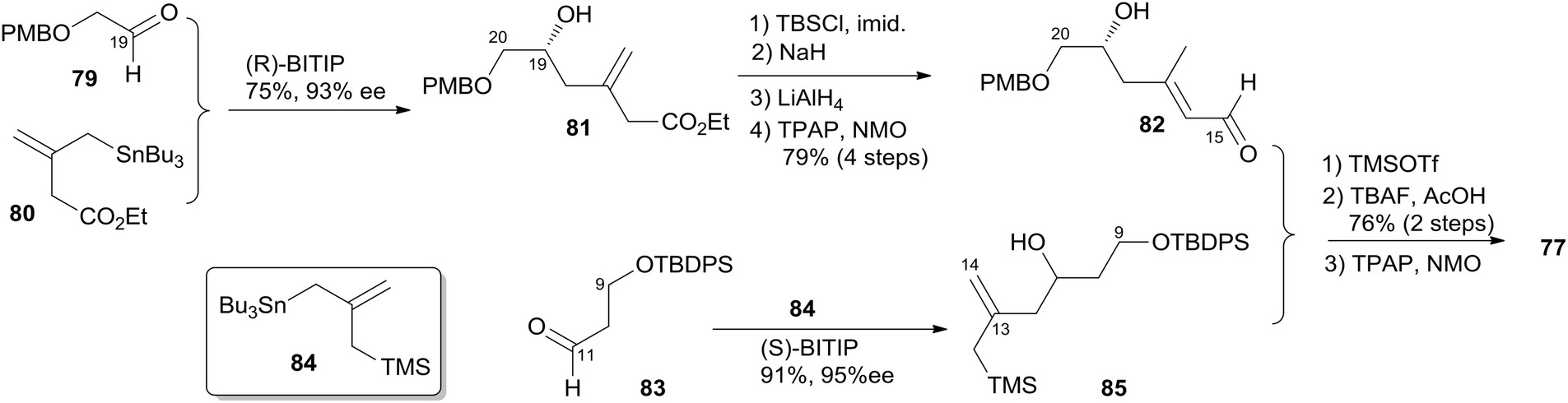
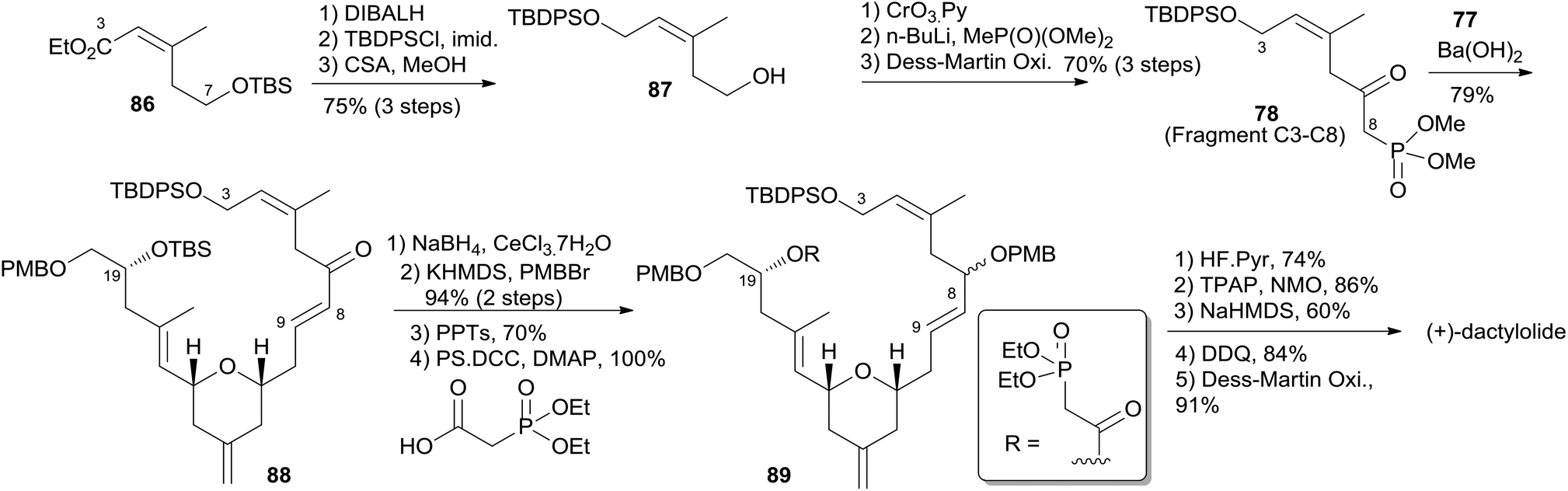
Keck constructed the macrolactone core using HWE macrocyclization and the HSP reaction, featuring construction of two asymmetric centres via his catalytic asymmetric allylation reaction and a diastereoselective pyran annulation (Scheme 16).53 The preparation of fragment C9–C20 (77) is shown in Scheme 17. Asymmetric allylation of aldehyde 79 with allylstannane 80 catalyzed by BINOL titanium tetraisopropoxide (BITIP) gave (R)-homoallylic alcohol 81 in 93% ee. Isomerization to the (E)-unsaturated aldehyde 82 (32:1) was achieved by protection of the hydroxyl group, treatment with NaH, and a two-step reduction (Scheme 17). Keck and co-workers believed that the observed high stereoselectivity is a kinetic phenomenon associated with a preferred pathway for formation of the U shaped internally chelated enolate anion with sodium as counterion. Fragment C9–C14 (85) was prepared by asymmetric allylation of aldehyde 83 and allyl stannane 84 catalyzed by (S)-BITIP in 95% ee (Scheme 17). Fragment C9–C20 (77) was prepared by the HSP reaction between 82 and 85 followed by a deprotection–oxidation procedure. The synthesis of fragment C3–C8 (78) began with intermediate 86 (ref. 54) (Scheme 18). Transformation of ester 86 to silyl ether 87 was carried out via a three-step sequence. The desired β-ketophosphonate 78 was achieved via another three-step sequence including oxidation, addition of the lithiate of methyl dimethyl phosphonate into this aldehyde, and oxidation of the resulting alcohol. HWE olefination of 77 with 78 under Peterson's conditions provided (E)-enone 88. To avoid the possibility that the acidity of C6–H might cause C7 enolization, the ketone at C7 was converted to its PMB ether, which was selected by Keck because both PMB groups (at C7 and C20) could be deprotected and both alcohols oxidized in the same operation at the later stage. Next, phosphonoacetate 89 was prepared by selective deprotection and modified Keck–Boden macrolactonization.55 A polymer-bound DCC (PS-DCC) rather than DCC itself was used to simplify the workup and purification due to the polarity of the phosphonate. Intramolecular HWE macrolactonization followed by PMB deprotection and double oxidation afforded 4 (Scheme 18).
Scheme 16 Keck's retrosynthetic analysis of (+)-dactylolide.
Scheme 17 Keck's synthesis of fragment C9–C20 of (+)-dactylolide.
Scheme 18 Keck's total synthesis of (+)-dactylolide.
5.7 McLeod's total synthesis of (−)-dactylolide
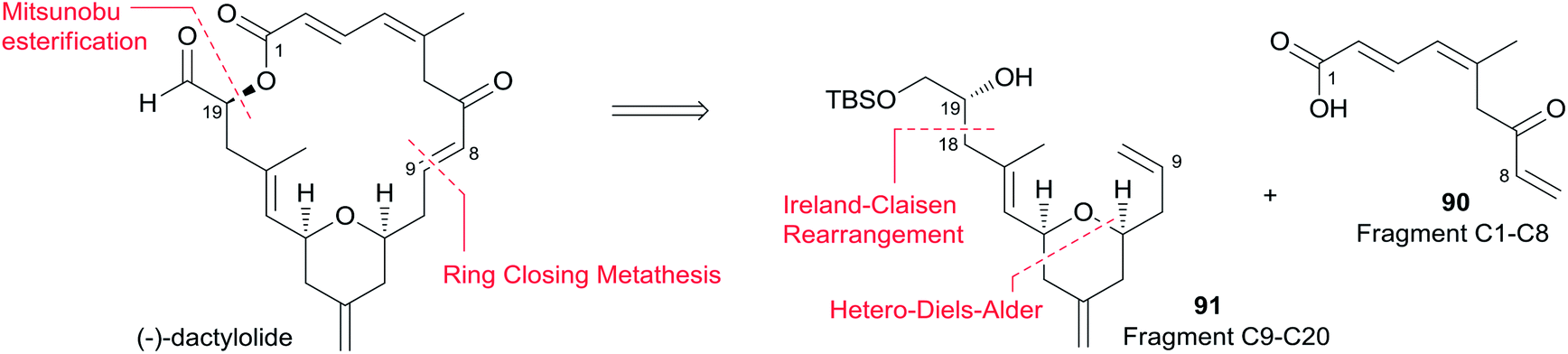
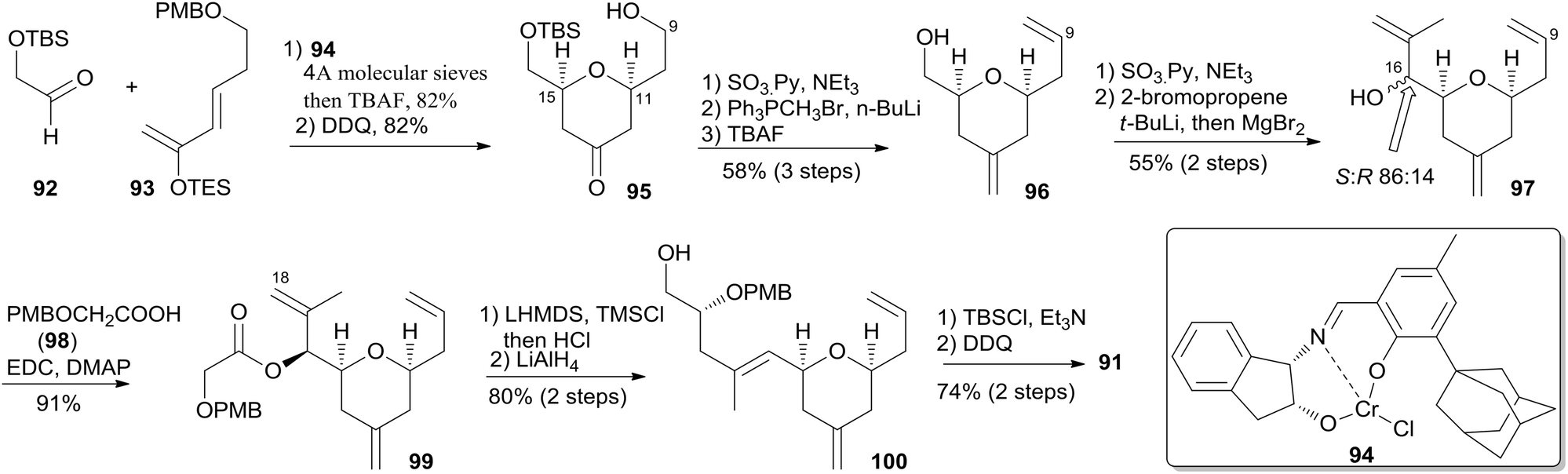
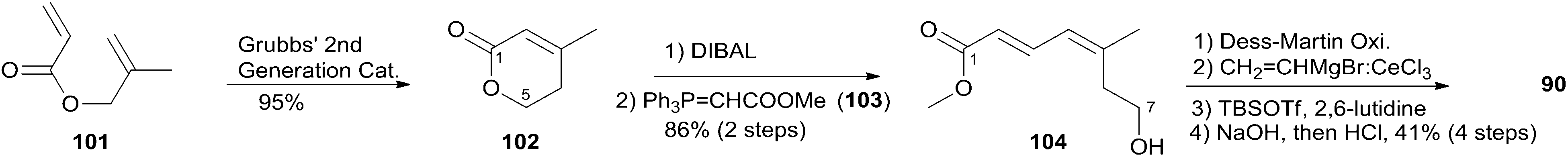
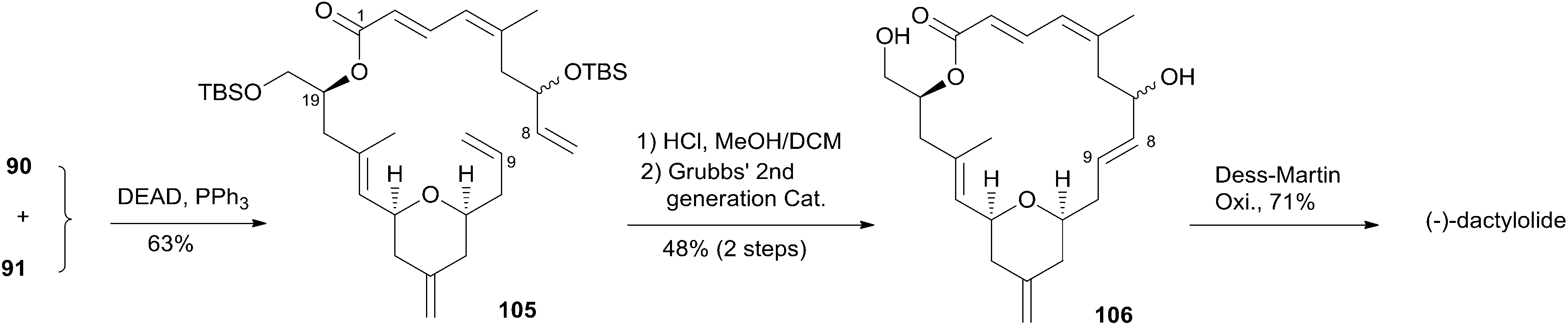
McLeod reported an enantioselective synthesis of (−)-dactylolide, featuring the construction of the 2,6-cis-THP by catalytic asymmetric Jacobsen hetero-Diels–Alder reaction,56 and the remote C19 chiral centre by a sequence of chelation-controlled Grignard addition and Ireland–Claisen rearrangement.20 As shown in the retrosynthetic analysis (Scheme 19), the macrolactone core was constructed by convergent coupling of fragment C1–C8 (90) and fragment C9–C20 (91) using a sequence of Mitsunobu esterification followed by Grubbs' RCM. The synthesis of 1 (Scheme 20) began with the linkage of aldehyde 92 and silyl enol ether 93 mediated by Jacobsen's chiral tridentate chromium(III) catalyst 94. THP 95 can be synthesized on a multigram scale by careful workup followed by PMB removal. Diene 96 was synthesized through a three-step procedure including oxidation, Wittig methylenation, and silyl ether deprotection. Nucleophilic addition of isopropenyl Grignard to the aldehyde obtained by oxidation of 96 proceeded with chelation control to favour formation of 97 with the 16S-configuration. Alcohol 97 was then converted to ester 99via esterification with PMB protected glycolic acid 98. Direct reduction of a crude product of the polar carboxylic acid derived from the Ireland–Claisen [3,3] sigmatropic rearrangement of ester 99 gave alcohol 100 as a single diastereomer. Alcohol 100 was converted to alcohol 91 through a simple protection–deprotection procedure. Synthesis of trienoic acid 90 was completed (Scheme 21) in seven steps from acrylate ester 101 (ref. 57) with trisubstituted Z-alkene established via a six-membered lactone intermediate 102. Direct Wittig reaction of the lactol derived from partial reduction of lactone 102 with ylide 103 afforded (2E, 4Z)-diene ester 104. The aldehyde, derived from 104, reacted with vinylcerium reagent, followed by a protection–hydrolysis procedure, to afford fragment C1–C8 (90). The final stage synthesis is shown in Scheme 22. Mitsunobu esterification provided ester 105. The diene precursor, derived from 105, was subjected to a Grubbs RCM reaction to afford macrocyclic diols 106, which were subjected to global oxidation to give (−)-dactylolide 3.
Scheme 19 McLeod's retrosynthetic analysis of (−)-dactylolide.
Scheme 20 McLeod's synthesis of fragment C9–C20 of (−)-dactylolide.
Scheme 21 McLeod's synthesis of fragment C1–C8 of (−)-dactylolide.
Scheme 22 McLeod's total synthesis of (−)-dactylolide.
5.8 Porco's synthesis of the macrolactone core of (−)-zampanolide
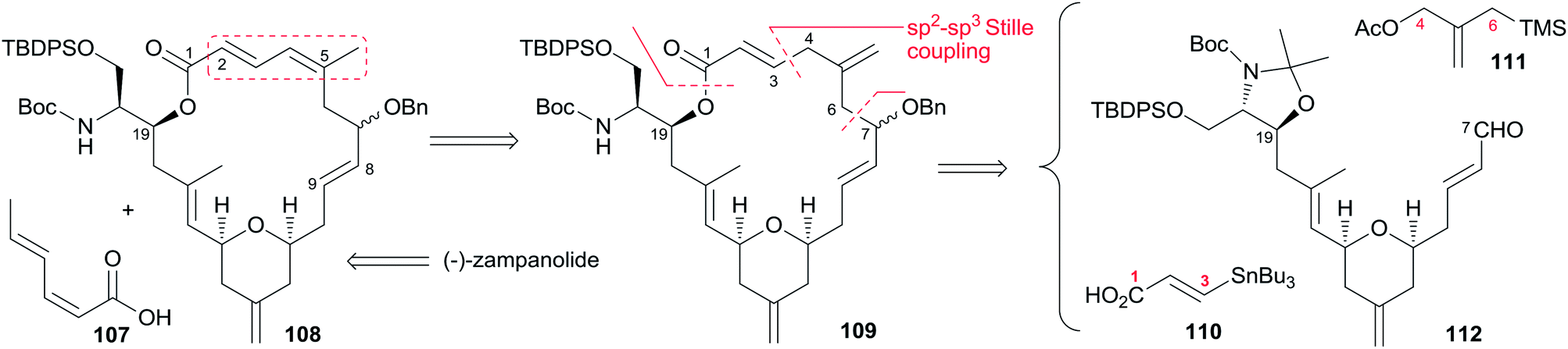
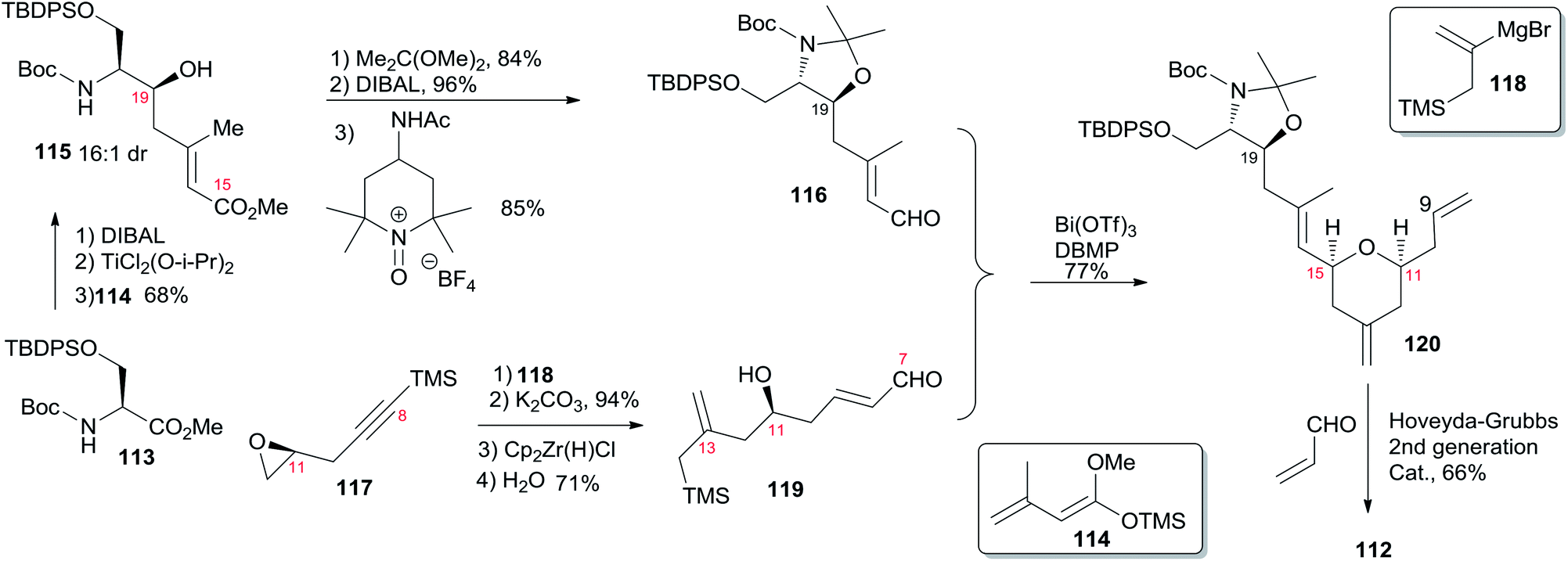
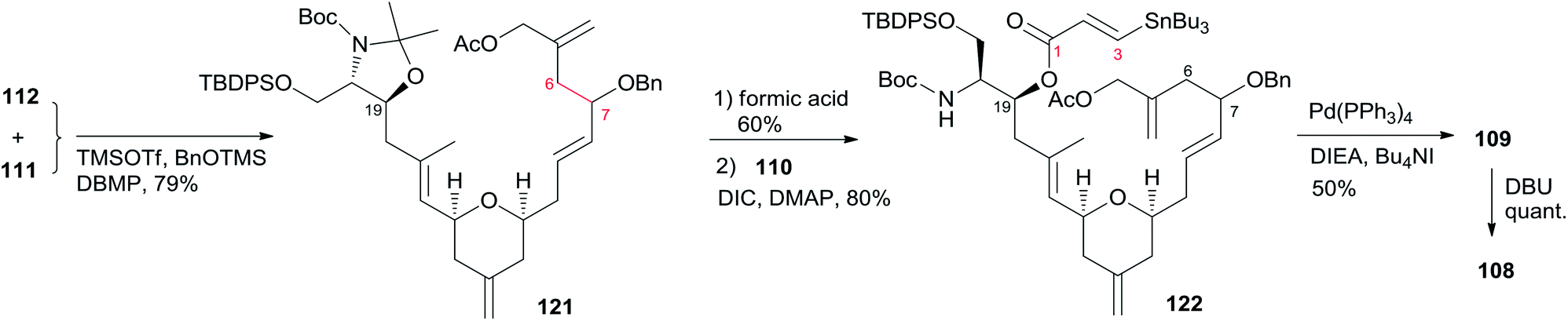
Porco's synthesis of the macrocyclic core of 1 was initiated by interest in the N-acyl hemiaminal side chain and its apparent stabilization through an intramolecular hydrogen bond network, and entailed preparation of an N-acyl hemiaminal model system.58,59 The synthetic strategy is characterised by construction of fragment C15–C20 by a one-pot reduction/vinylogous aldol reaction, construction of the 2,6-cis-THP through an intramolecular silyl-modified Sakurai (ISMS) reaction,60 and closing the macrolactone ring via an sp2–sp3 Stille reaction. As outlined in Scheme 23, Porco's synthetic strategy for zampanolide is to install the N-acyl hemiaminal side chain from the protected amino alcohol-bearing macrolide 108 at a late stage using their previously reported oxidative decarboxylation–hydrolysis approach.59 In order to install the potentially sensitive 1,3-dienoate portion of the macrolide at a late stage, these workers investigated a less conventional disconnection by targeting 1,4-dienoate 109 instead of 1,3-dienoate 108. They expected that subsequent isomerization would occur under macrocyclic control and installation of the dienoate in a “masked” format would allow for unveiling after construction of the macrolide. Further disconnection of macrolide 109 afforded three precursors: β-stannylacrylic acid 110, allylic acetate 111, and the pyran containing the C7–C20 fragment 112. The C3–C4 bond would be constructed via an intramolecular Stille reaction, representing a pioneer example of using an sp2–sp3 Stille macrocyclization in a complex synthesis. Synthesis of the C7–C20 fragment is shown in Scheme 24. The two-step process developed by Kiyooka61 using DIBAL-H and TiCl2(O-iPr)2 provided the desired vinylogous aldol product 115 (dr = 16:1) from methyl ester 113. Vinylogous aldol substrate 115 was next advanced to the α,β-unsaturated aldehyde 116 through a three-step sequence including protection of amino alcohol as the N,O-acetonide, reduction of the methyl ester, and subsequent oxidation with Bobbit's reagent.62 The requisite allylsilane 119 for an intramolecular Sakurai cyclisation (IMSC)60 reaction was obtained from Cu(I)-mediated addition of vinyl Grignard reagent 118 (ref. 63) to chiral epoxide 117 (ref. 64) (Scheme 24). The IMSC reaction of 116 and 119 mediated by Bi(OTf)3 in conjunction with 2,6-di-tert-butylpyridine as triflic acid scavenger afforded pyran 120 as a single diastereomer. Elaboration of the terminal olefin by Grubbs–Hoveyda cross-metathesis provided α,β-unsaturated aldehyde 112 (Scheme 24). Allylation of 112 with the Trost trimethylenemethane (TMM) reagent 111 (ref. 65) in the presence of BnOTMS afforded allylic acetate 121 (dr = 1:1) (Scheme 25). Removal of the acetonide protecting group followed by esterification with β-stannyl acrylate 110 (ref. 66) provided the macrocyclization precursor 122. Macrocyclization of vinylstannane 122 was mediated by Pd(PPh3)4-Bu4NI-DIEA to afford macrolactone 109. Porco indicated that only 1,4-diene product was observed, likely due to the constraints of the macrolide on olefin isomerization. Subsequent isomerization by DBU resulted in formation of 1,3-diene 108 in quantitative yield as a 1:1 mixture of E,Z-isomers.
Scheme 23 Porco's retrosynthetic analysis of (−)-zampanolide.
Scheme 24 Porco's synthesis of fragment C7–C20 of (−)-zampanolide.
Scheme 25 Porco's synthesis of the macrolactone core of (−)-zampanolide.
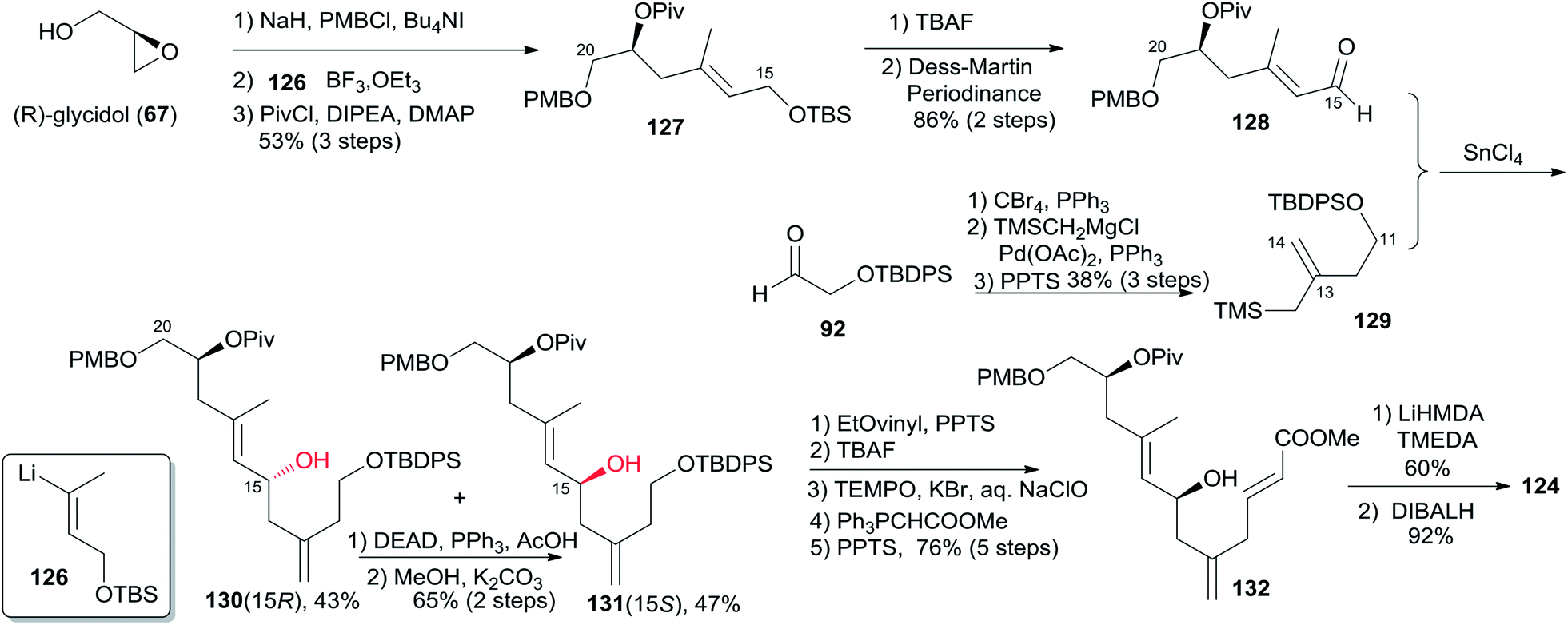
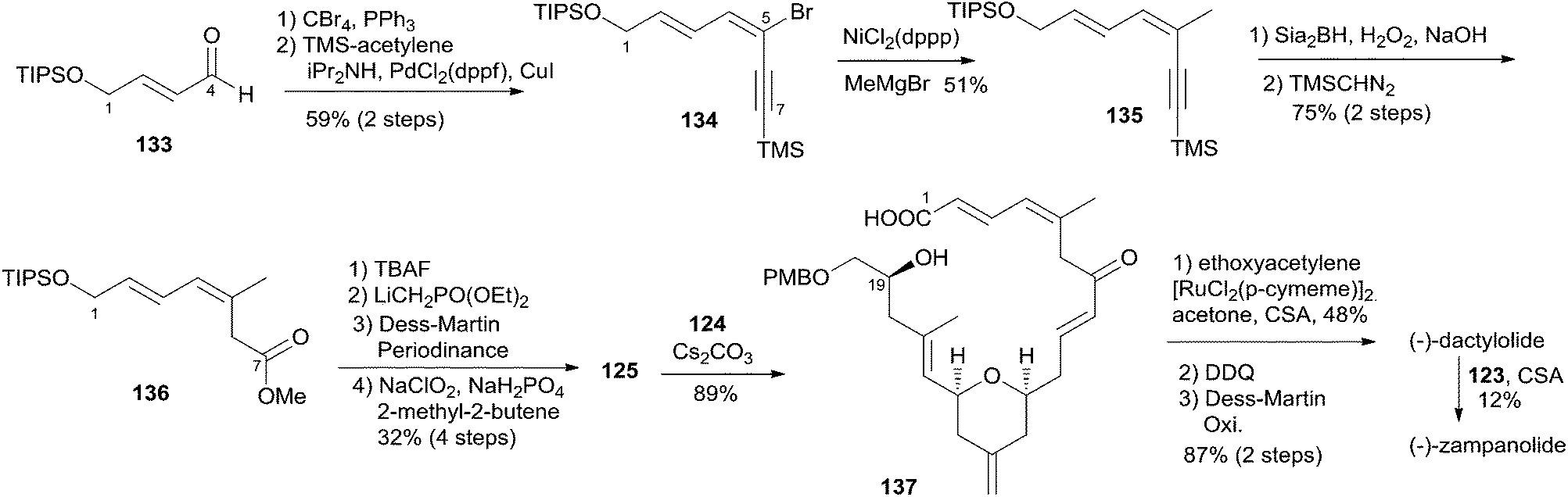
5.9 Uenishi's synthesis of (−)-zampanolide and (−)-dactylolide
Uenishi's retrosynthetic strategy is illustrated in Scheme 26.11 The side chain of 1 could be introduced by acid-catalyzed N-hemiacetalization of 3 with amide 123. The macrolactone ring could be constructed using HWE reaction and macrolactonization as critical reactions. As shown in Scheme 27, the synthesis of the C9–C20 fragment commenced from ring opening of PMB protected (R)-glycidol with vinyllithium 126 (prepared from the corresponding stannane67 and BuLi). The subsequent product was protected as its pivaloate ether and converted to enal 128 by a two-step procedure. Allylsilane 129 was prepared from aldehyde 92 through the three-step sequence of dibromomethylenation, Kumada–Tamao–Corriu coupling with TMSCH2MgCl, and protodesilylation. The Hosomi–Sakurai reaction of 128 with 129 promoted by SnCl4 afforded the desired 131 (15S) in 47% yield as well as its isomer 130 (15R) in 42% yield; the latter could be converted to 131 in 65% yield by Mitsunobu reaction followed by methanolysis. Transformation of 131 to 132 proceeded through an efficient five-step sequence in 76% overall yield. The THP ring in 124 was built by intramolecular O-Michael reaction; the subsequent ester was reduced with DIBALH, with concomitant removal of the pivaloate group, to provide the C9–C20 fragment (124). The preparation of the C1–C8 fragment (125) started with dibromomethylenation of aldehyde 133 followed by stereoselective Sonogashira coupling with TMS-acetylene (Scheme 28). Introduction of a methyl group to alkyne 134via the Kumada–Tamao–Corriu coupling proceeded with the anticipated inversion of olefin geometry.68 Transformation of the terminal TMS-acetylene to ester 136 proceeded through a two-step sequence. Another four-step sequence including TIPS ether removal, diethyl methylphosphonate introduction, and oxidations completed the synthesis of 125. The syntheses of 3 and 1 were completed as shown in Scheme 28. The HWE reaction of aldehyde 124 and β-ketophosponate 125 provided seco acid 137, which was subjected to cyclization using the Trost–Kita method to generate a macrolactone. Dactylolide (3) was readily prepared from the macrolactone via a two-step sequence. Treatment of 3 with hexadienoylamide 123 catalyzed by CSA afforded 1 in 12% yield.
Scheme 26 Uenishi's retrosynthetic analysis of (−)-zampanolide.
Scheme 27 Uenishi's synthesis of fragment C9–C20 of (−)-zampanolide.
Scheme 28 Uenishi's total synthesis of (−)-zampanolide.
5.10 Lee's synthesis of (−)-dactylolide
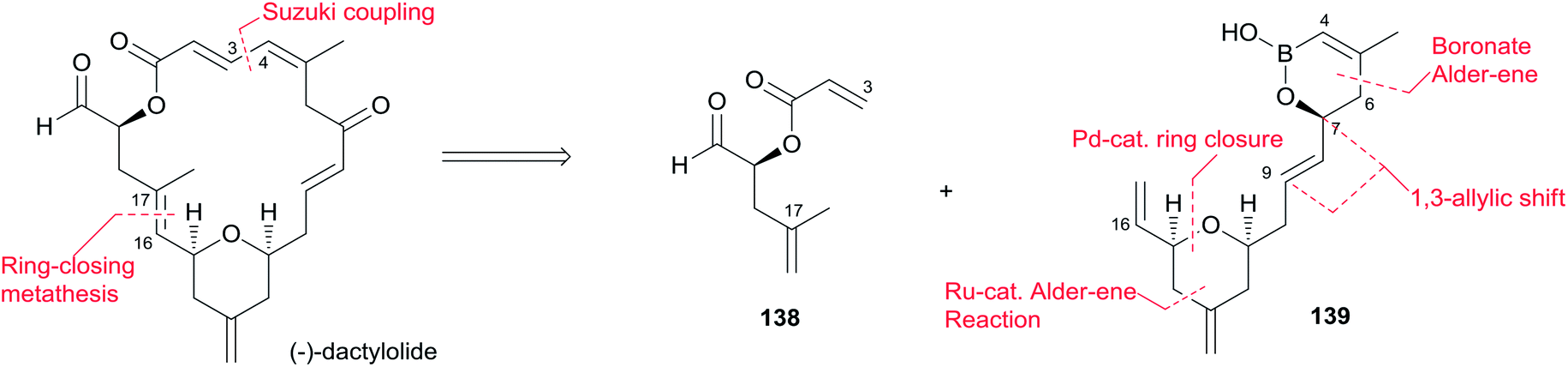
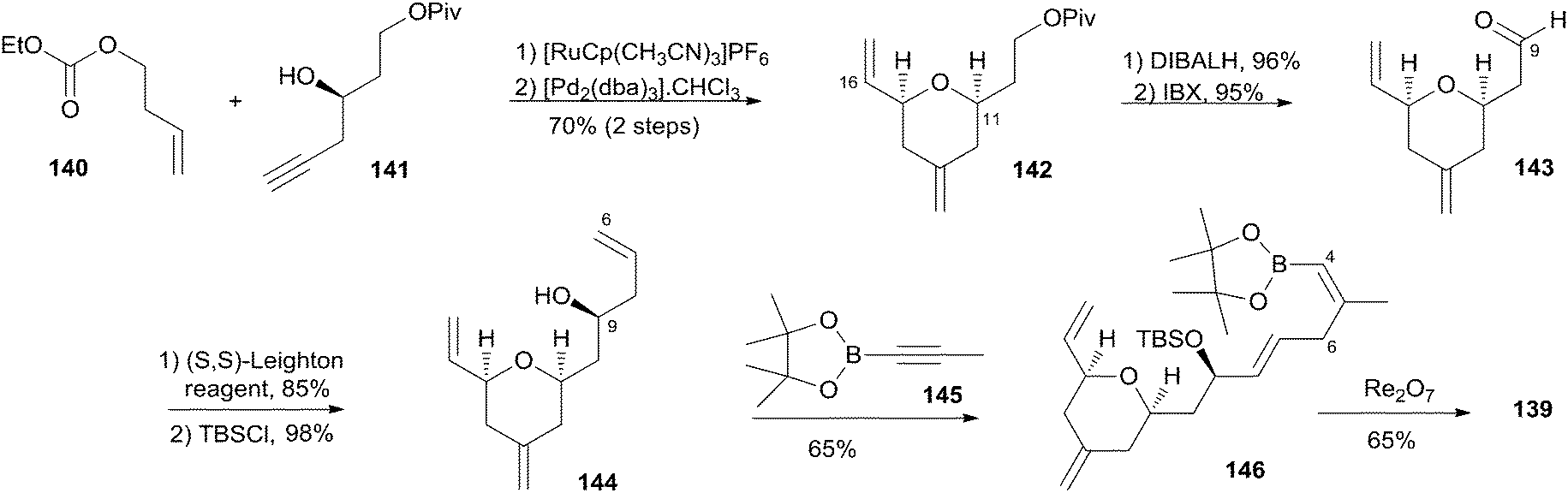
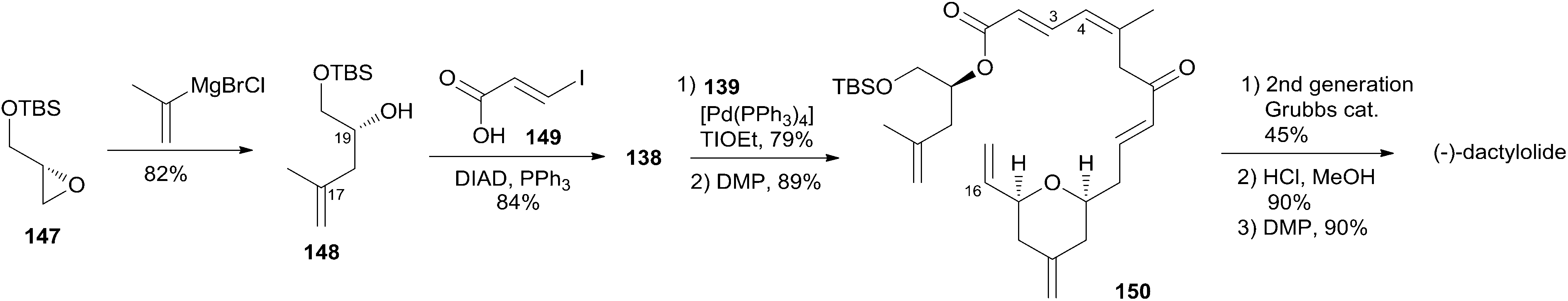
Lee's retrosynthetic analysis, as shown in Scheme 29, features the construction of C–C and C–O bonds by transition-metal-catalyzed reactions in the hope of minimizing the need for functional group transformations and protection–deprotection steps.69,70 They envisioned closing the macrolactone ring through a late-stage RCM reaction at C16–C17. The C3–C4 bond was planned to be synthesized through a Suzuki–Miyaura coupling of iodoacylate 138 and a cyclic boronic acid half ester 139. This could be accessed through the ruthenium-catalyzed Alder-ene reaction (RCAER)71 and allylic [1,3]-transposition.72 The pyran subunit in 139 would be installed using a tandem RCAER and palladium-catalyzed ring closure. The synthesis of the C4–C16 fragment 139 commenced with the RCAER between ethyl carbonate 140 and homopropargylic alcohol 141 (Scheme 30).73 The product was treated with palladium catalyst in the presence of Trost's chiral (+)-DPPBA ligand to afford 2,6-cis-disubstituted THP 142, which was readily converted to aldehyde 143 through a deprotection–oxidation sequence. Leighton allylation followed by TBS protection afforded homoallyl silyl ether 144. The RCAER of alkynyl boronate 145 occurred selectively with the least hindered double bond at C7 of 144, and afforded vinyl boronate 146. Cyclic boronic acid half ester 139 was obtained by the 1,3-transposition of the allyl alcohol of 146 with rhenium oxide.74 2-Iodoacrylate 138, the Suzuki coupling partner, was prepared from TBS-protected (S)-glycidol 147 through the sequence shown in Scheme 31. Ester 138 was synthesized by ring opening with 2-propenylmagnesium chloride followed by Mitsunobu reaction of 148 with 2-iodoacrylic acid 149. Suzuki coupling between 138 and 139 followed by a four-step sequence delivered (−)-dactylolide.
Scheme 29 Lee's retrosynthetic analysis of (−)-dactylolide.
Scheme 30 Lee's synthesis of fragment C4–C16 of (−)-dactylolide.
Scheme 31 Lee's total synthesis of (−)-dactylolide.
5.11 Hong's total synthesis of (+)-dactylolide
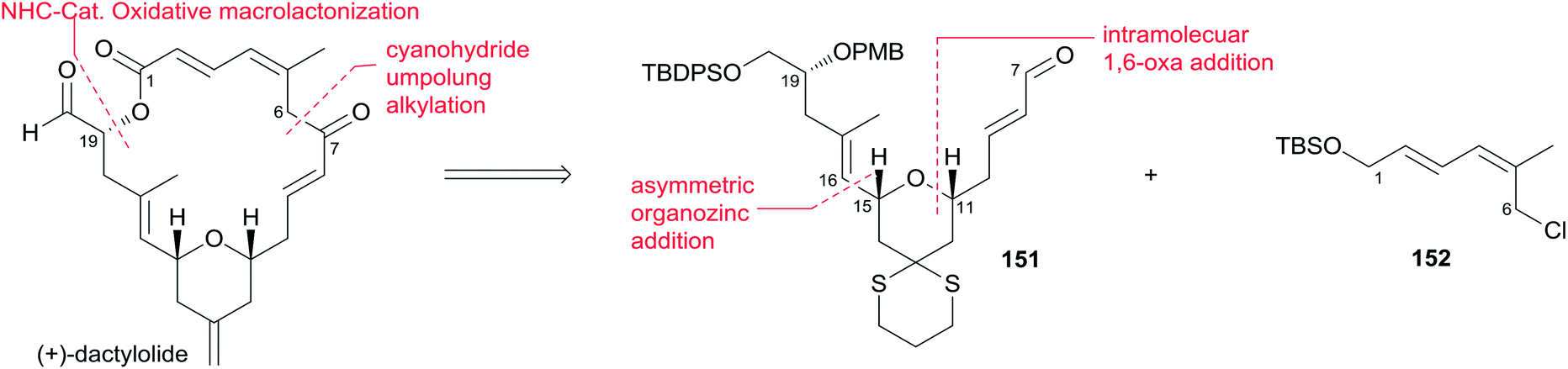
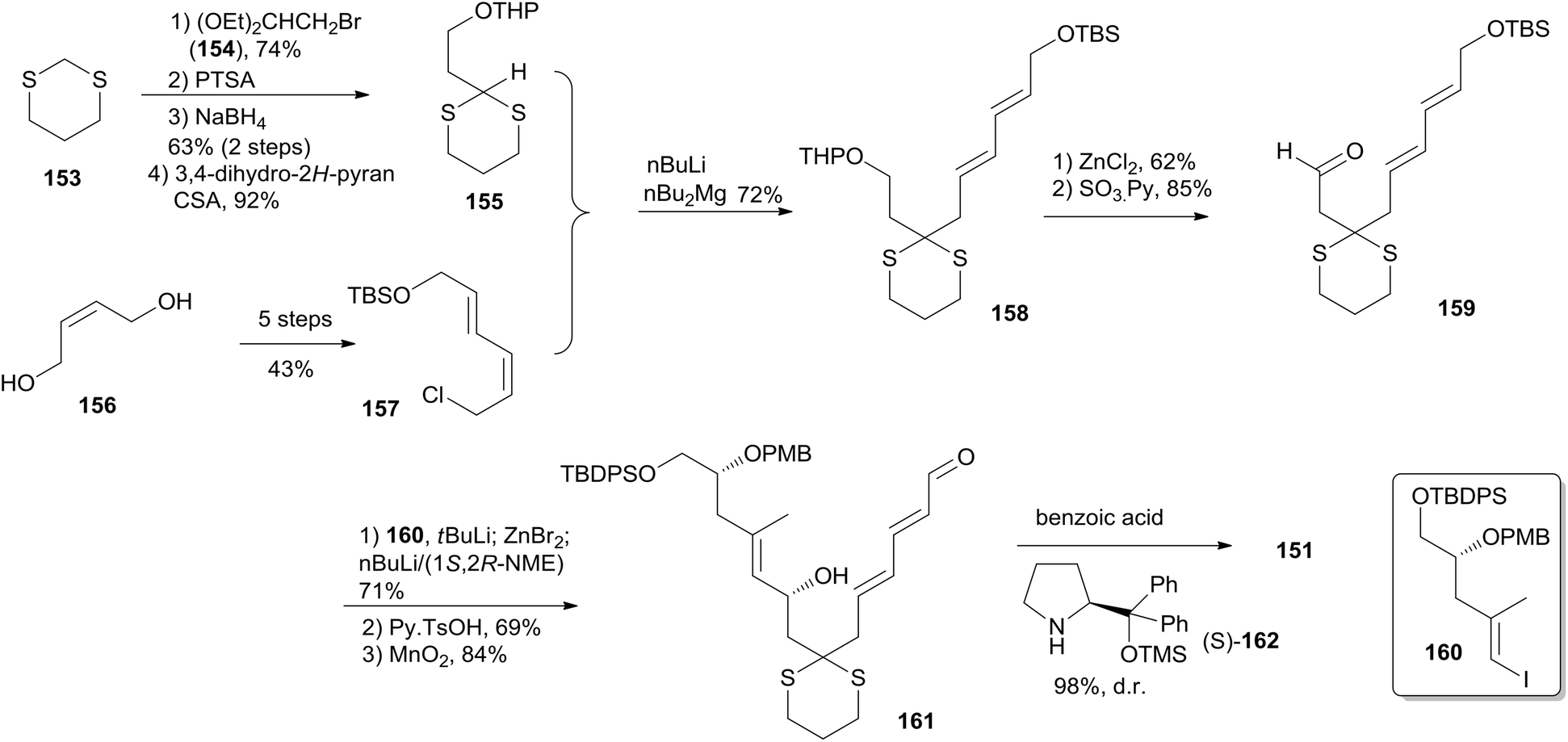
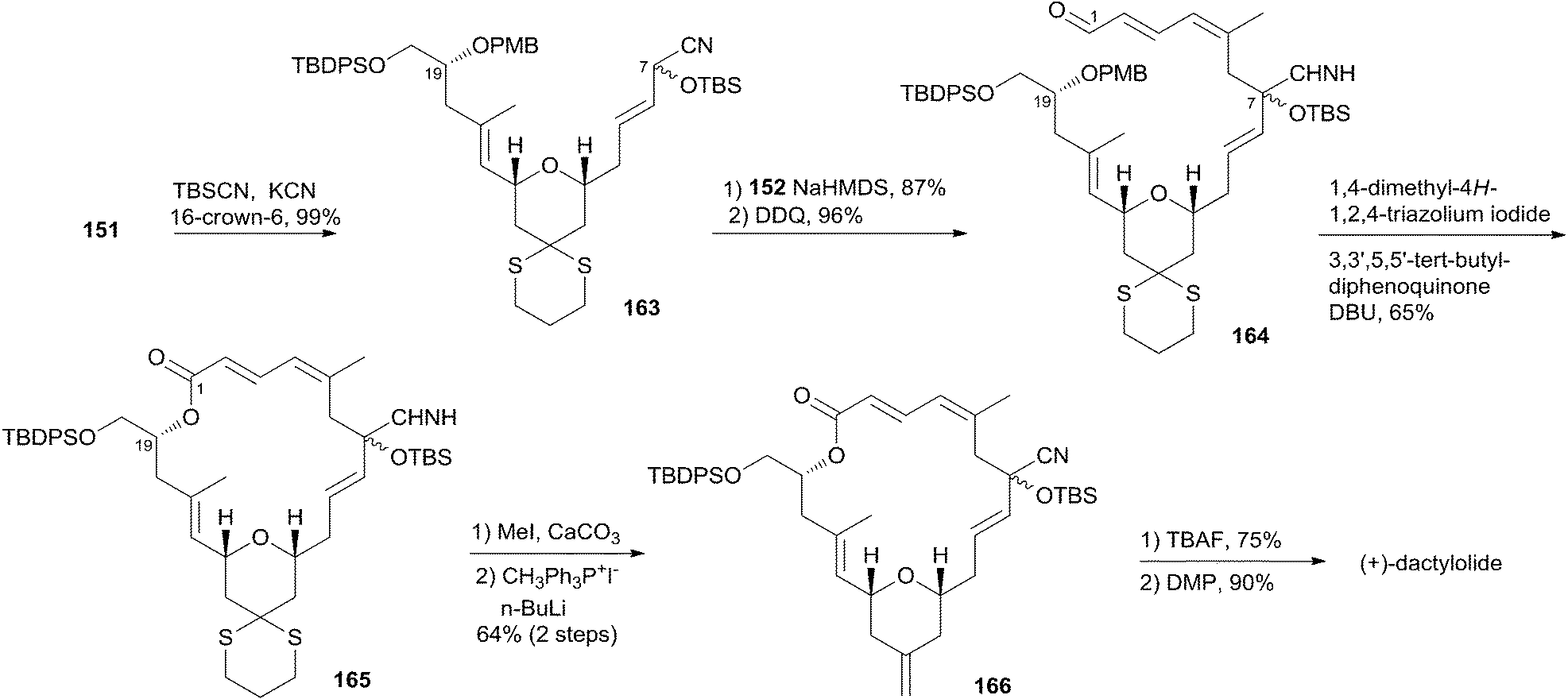
(+)-Dactylolide (4) was synthesized by the Hong group from 1,3-dithiane, featuring the 1,6-oxa conjugate addition reaction of a 2,4-dienal for the facile synthesis of the 2,6-cis-THP subunit, the umpolung alkylation reaction of a cyanohydrin, and the N-heterocyclic carbene (NHC)-catalyzed oxidative macrolactonization reaction.75 Hong's retrosynthetic plan for 4 is outlined in Scheme 32. The macrolactone core could be constructed from the C1–C6 fragment (152) and the C7–C20 fragment (151) by intramolecular oxidative macrolactonization of ω-hydroxy aldehyde catalyzed by an NHC and umpolung alkylation of cyanohydrin. The synthesis of the C7–C20 fragment is shown in Scheme 33. The coupling of the dithiane 155, prepared from dithiane 153,76 with dienyl chloride 157 (ref. 77) mediated by nBuLi–nBu2Mg provided 158, which was converted to aldehyde 159 through a deprotection–oxidation sequence. Stereoselective installation of the C15 secondary carbinol by an asymmetric organozinc addition, avoiding the possible chelation of the oxygen atoms to zinc, required slow addition of 159 to a mixture of the corresponding bromozinc reagent of 160 and lithiated (1S, 2R)-NME. This procedure provided 161 in good stereoselectivity (dr = 7.7:1), and this was subjected to organocatalytic 1,6-oxa conjugate addition reaction catalyzed by (S)-162 to provide the desired 2,6-cis-THP 151 with excellent stereoselectivity and yield, representing the first successful example of THP construction through an intramolecular 1,6-oxa conjugate addition reaction. Hong and co-workers proceeded to install the C1–C6 fragment using an acyl anion equivalent (Scheme 34). The TBS-protected cyanohydrin 163, prepared from 151, was coupled with dienyl chloride 152 followed by concomitant PMB removal and oxidation at C1 to generate ω-hydroxy aldehyde 164. NHC-catalyzed oxidative macrolactonization of 164 provided macrolactone 165. This represents the first example of NHC-catalyzed oxidative macrolactonization of a ω-hydroxy aldehyde. The synthesis of 4 was completed by elaborating the C13 exo-methylene group, unveiling the C7 carbonyl group, and oxidizing the C20 hydroxyl group.
Scheme 32 Hong's retrosynthetic analysis of (+)-dactylolide.
Scheme 33 Hong's synthesis of fragment C7–C20 of (+)-dactylolide.
Scheme 34 Hong's total synthesis of (+)-dactylolide.
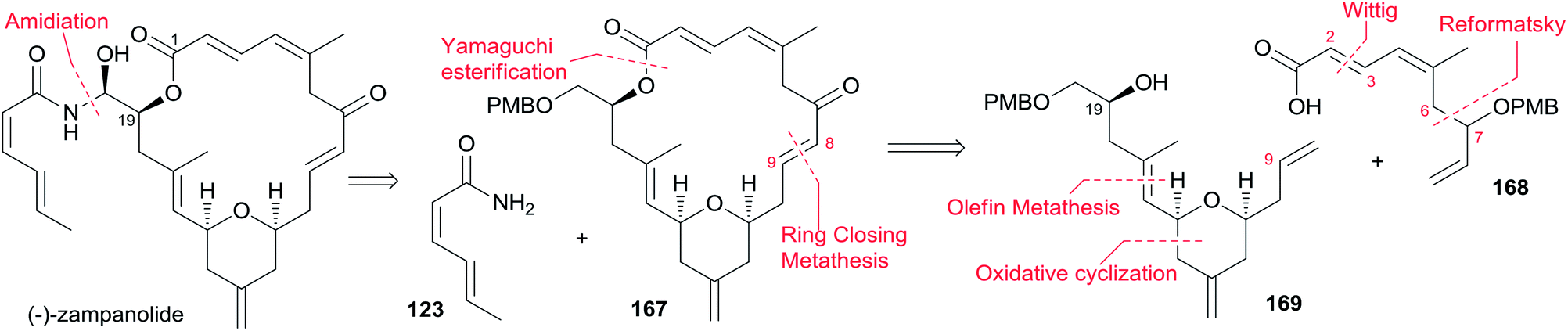
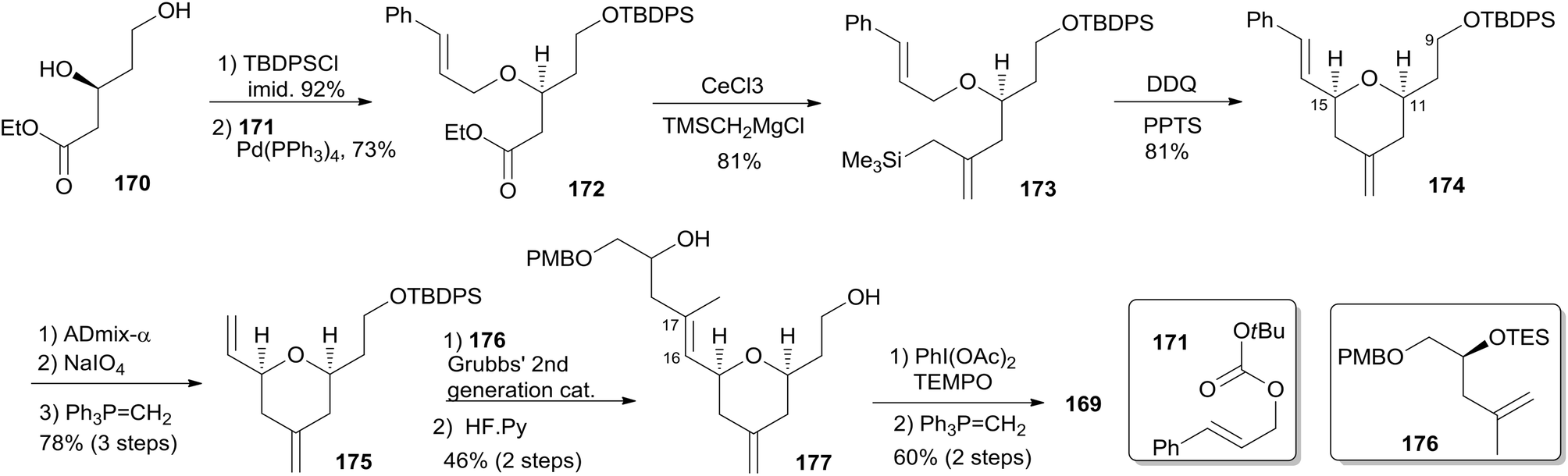
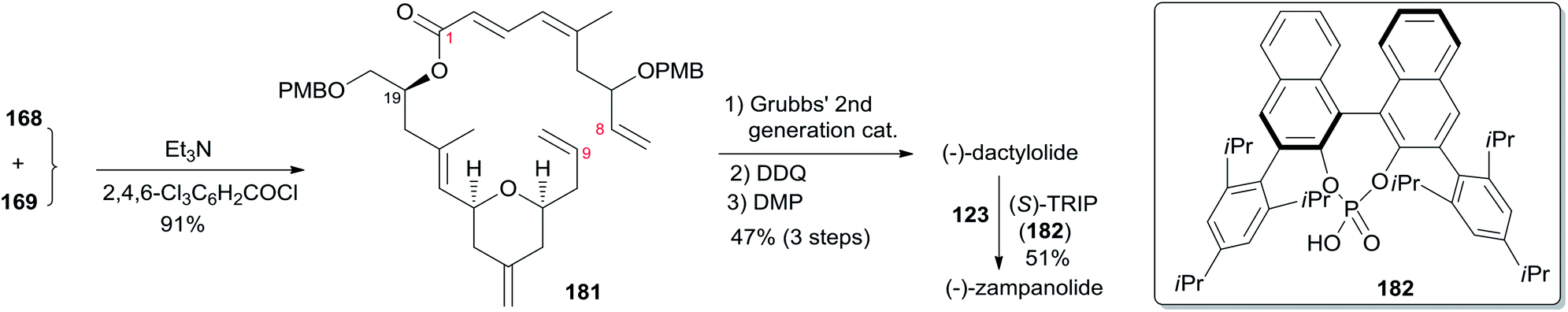
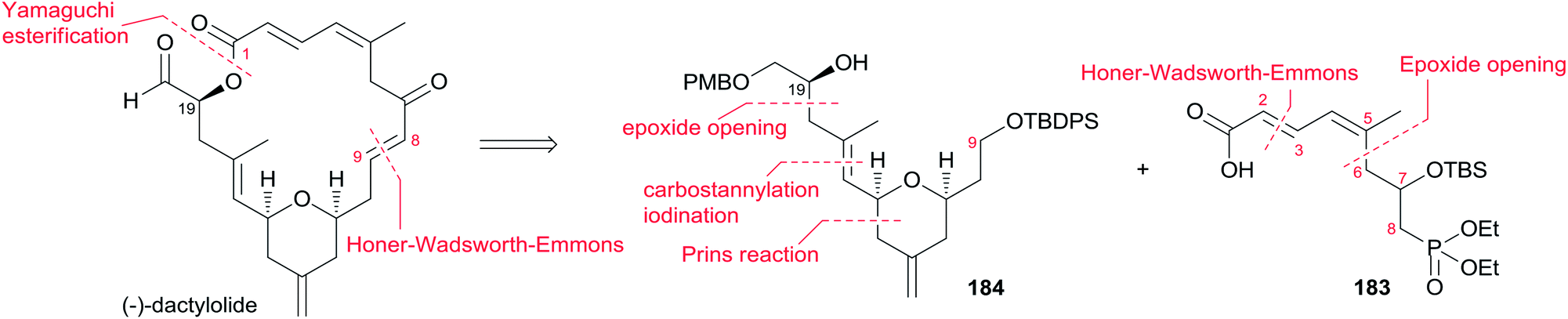
The Ghosh group accomplished an enantioselective total synthesis of 1 using a novel DDQ/Bronsted acid promoted cyclization as the key reaction.78 Their retrosynthetic analysis is shown in Scheme 35.16,24 The strategic bond disconnection of the side chain at C20 provides macrolactone 167, which can be constructed from the C9–C20 fragment 169 and the C1–C8 fragment 168 by Yamaguchi esterification followed by RCM. A similar RCM strategy was first employed by Hoye.18 The double bond at C16–C17 in the C9–C20 fragment 169 could be installed by a cross metathesis reaction, and the THP ring could be constructed by an oxidative cyclization reaction. Fragment C1–C8 (168) could be built by Reformatsky reaction followed by Wittig olefination. As shown in Scheme 36, the synthesis commenced with known ester 170.79 Selective protection followed by esterification with tert-butyl cinnamyl carbonate (171) catalyzed by Pd(PPh3)4 afforded cinnamyl ether 172, which was converted to allylsilane 173 employing the procedure modified by Narayanan and Bunnelle.80 Oxidative cyclization of 173 with DDQ catalyzed by PPTS constructed the 4-methylene-THP ring in 174 stereoselectively and efficiently, presumably due to the Zimmerman–Traxler transition state where all substituents are equatorially oriented. Disubstituted olefin 174 was converted to monosubstituted olefin 175via a three-step sequence. Olefin 176 was obtained by opening PMB-protected glycidol with isopropenylmagnesium bromide followed by alcohol protection as TES ether. Grubbs cross-metathesis of 175 with 176 provided an E/Z olefin mixture (1.7:1). Removal of all silyl groups followed by chromatographic purification provided trisubstituted olefin 177. Selective oxidation followed by Wittig olefination generated 169. The synthesis of the C1–C8 fragment (Scheme 37) began with the preparation of allyl bromide 178,81 which was subjected to Reformatsky reaction with acrolein to give unsaturated δ-lactone 179. DIBAL-H reduction followed by Wittig reaction afforded allylic alcohol 180. Fragment C1–C8 (168) was prepared from 180 through a protection-saponification sequence. The completion of the synthesis (Scheme 38) started with Yamaguchi esterification of acid 168 with alcohol 169 to furnish 181, which was subjected to Grubbs RCM reaction. The subsequent macrolactone was converted to 3 by a deprotection–oxidation sequence. The conversion of 3 to 1 was completed by treatment of aldehyde 3 with amide 123 in the presence of (S)-TRIP (182) in 51% yield.
Scheme 35 Ghosh's retrosynthetic analysis of (−)-zampanolide.
Scheme 36 Ghosh's synthesis of fragment C9–C20 of (−)-zampanolide.
Scheme 37 Ghosh's synthesis of fragment C1–C8 of (−)-zampanolide.
Scheme 38 Ghosh's total synthesis of (−)-zampanolide.
5.13 Altmann's total synthesis of (−)-zampanolide17,82
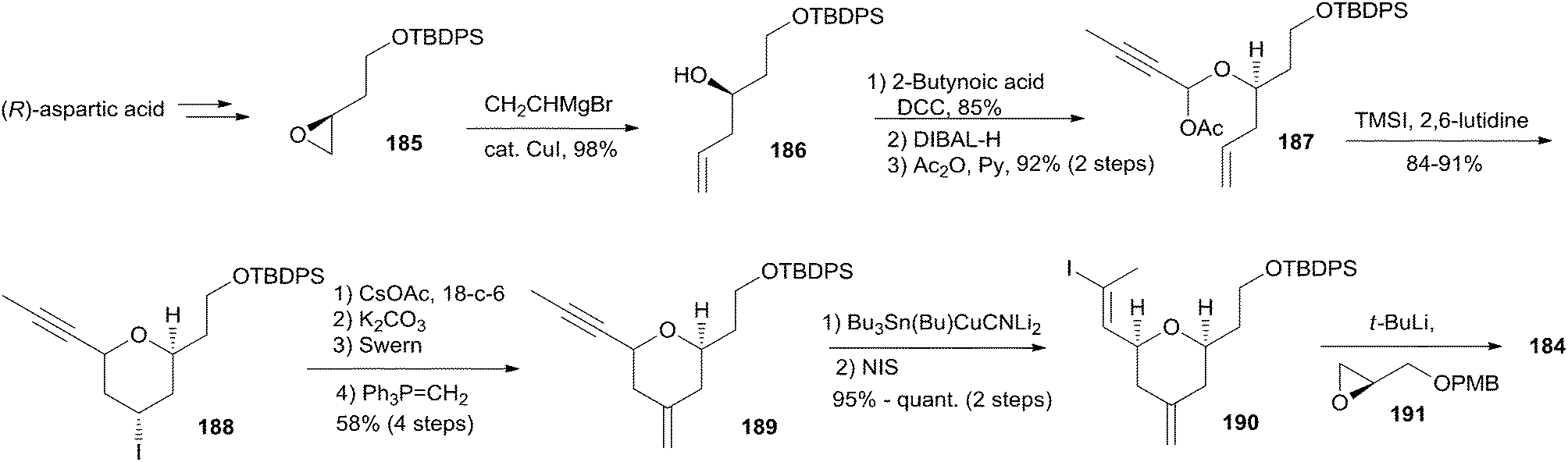
Altmann's retrosynthetic analysis for 3 (Scheme 39) is centred on HWE macrocyclization involving the formation of the C8–C9 double bond.17,82 The requisite precursor would be obtained via ester formation between the C1–C8 fragment (183) and the C9–C20 fragment (184). The synthesis of the C9–C20 fragment 184 started with the Cu-mediated regioselective epoxide opening of 185, prepared from (R)-aspartic acid in three steps, with vinyl-MgBr (Scheme 40). Alcohol 186 was then elaborated into THP 188 in a highly stereoselective Prins-type reaction employing a segment coupling approach as developed by Rychnovsky.83 In a first step, this involved esterification of 186 with 2-butynoic acid, which was followed by DIBAL-H reduction of the ester and trapping of the aluminated intermediate at −78 °C with Ac2O to furnish 187. Treatment of 187 with TMSI gave substituted THP 188 with the desired 2,6-syn relationship and the iodine located anti to the substituents in the 2- and 6-positions. This is in line with previous observations by Rychnovsky for related transformations. Conversion of 188 to 189 was achieved by a four-step sequence: iodide displacement with CsOAc/18-crown-6, hydrolysis, Swern oxidation, and Wittig reaction. Conversion of 189 to the desired E-vinyl iodide 190 was achieved with Bu3Sn(Bu)CuCNLi2 generated in situ followed by Sn–I exchange with NIS. Reaction of vinyl iodide 190 with PMB-protected (R)-glycidol 191 generated fragment C9–C20 (184). Unsaturated acid 183 was prepared by coupling of lithiated Z-vinyl iodide 192 (obtained in two steps from 2-butynol) and epichlorohydrin mediated by BF3·OEt2 (Scheme 41). Treatment of 193 with base followed by BF3-mediated epoxide opening with lithiated diethylphosphite gave β-hydroxyphosphonate 194, which was converted to aldehyde 195 through a protection–deprotection–oxidation sequence. HWE reaction followed by basic hydrolysis then afforded the desired acid 183. The final assembly of (−)-dactylolide commenced with esterification of alcohol 184 with acid 183 (Scheme 41) under Yamaguchi conditions to give an ester which was converted to HWE precursor 196 through a global desilylation–oxidation sequence. Ring-closure step was achieved with Ba(OH)2 as an optimized base and the synthesis of 3 was completed by PMB removal and DMP oxidation.
Scheme 39 Altmann's retrosynthetic analysis of (−)-dactylolide.
Scheme 40 Altmann's synthesis of fragment C9–C20 of (−)-dactylolide.
Scheme 41 Altmann's total synthesis of (−)-dactylolide.
Conversion of 3 to 1 was brought about by Hoye's aza-aldol reaction with (Z,E)-sorbamide,18 which gave a mixture of 1 and epi-1 in 18% and 12% yield respectively.
5.14 Summary and evaluation
The common feature for all syntheses is to begin with construction of the macrolactone core and to end with installation of the N-acyl hemiaminal side chain. The longest linear steps together with the overall yields for these total syntheses are listed in Table 5. The Hoye and Uenishi routes are the most convergent and efficient of the dactylolide syntheses, and the Porco partial synthesis is also efficient, although it has yet to be elaborated to dactylolide. The approaches by Altmann and Jennings are strategically attractive due to their efficiency, even though they have longer linear sequences.
Table 5Summary of total syntheses
Compound
Approach
Linear steps
Overall yield
Ref.
aThe approach used a known fragment; the linear steps and overall yield were counted from the known fragment as a starting material.bThe yield for the last step was not reported.
The Achilles' heel of the approaches to zampanolide is the stereospecific construction of the N-acylhemiaminal group, and this was a challenging step in all the synthetic routes to this compound. Smith and co-workers constructed the N-acyl hemiaminal in their synthesis of 2 using a stereospecific Curtius rearrangement as a key step.12 Hoye and co-workers used an aluminium aza-aldol addition reaction to construct the N-acyl hemiaminal, but did not report their yield. C20 epimerization is a key issue for these two methods. The conversion of 3 to 1 was completed in Uneshi's synthesis in only 12% yield. To improve this reaction, Ghosh and co-workers investigated N-acyl aminal formation between aldehyde 3 and amide 123 in the presence of matched chiral phosphoric acid (S)-TRIP (182, 20 mol%). These reaction conditions furnished 1 in 51% yield, so at this point this is the most attractive and efficient approach.16,24
The general strategies to close macrolactone rings are the Yamaguchi esterification, the Horner–Emmons macrocyclization, and ring-closing metathesis. Three additional methods were developed for this reaction: NHC-catalyzed oxidative macrolactonization (Hong),75 macrocyclization of a vinylstannane (Porco),58 and macrolactonization via Ti(IV) mediated ring-opening of an epoxide (Hoye).18 The macrolactone contains three chiral centres at C11, C15, and C19. The C19 chiral centre was generally obtained from the chiral pool (e.g. (S)-glycidol). The reported syntheses focus on diastereoselective construction of the 2,6-cis-disubstituted THP subunit containing the C11 and C15 chiral centres and on the efficient construction of the macrolactone core.
6. Other synthetic studies
6.1 Construction of cis 2,6-disubstituted THP rings
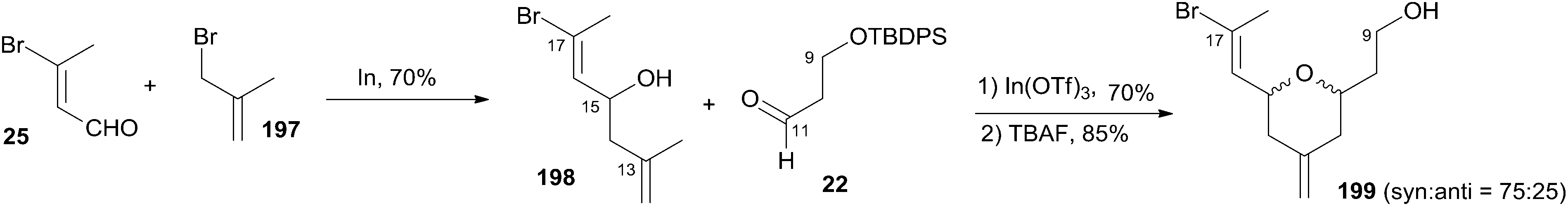
As shown in Scheme 42, Loh and co-workers reported a diastereoselective construction of fragment C9–C17 of zampanolide via In(OTf)3-catalyzed intramolecular 2,5-oxonium-ene cyclization from enal 25.84 In(OTf)3-catalyzed intramolecular 2,5-oxonium-ene cyclization of 198 and aldehyde 22, followed by desilylation with TBAF, provided the desired cyclization product 199 with good syn diastereoselectivity (75:25) as two inseparable isomers. The observed predominant 2,6-syn selectivity can be rationalized by the cyclic six-membered chair-like transition state favouring an all-equatorial substitution pattern.
Scheme 42 Loh's synthesis of fragment C9–C17 of zampanolide.
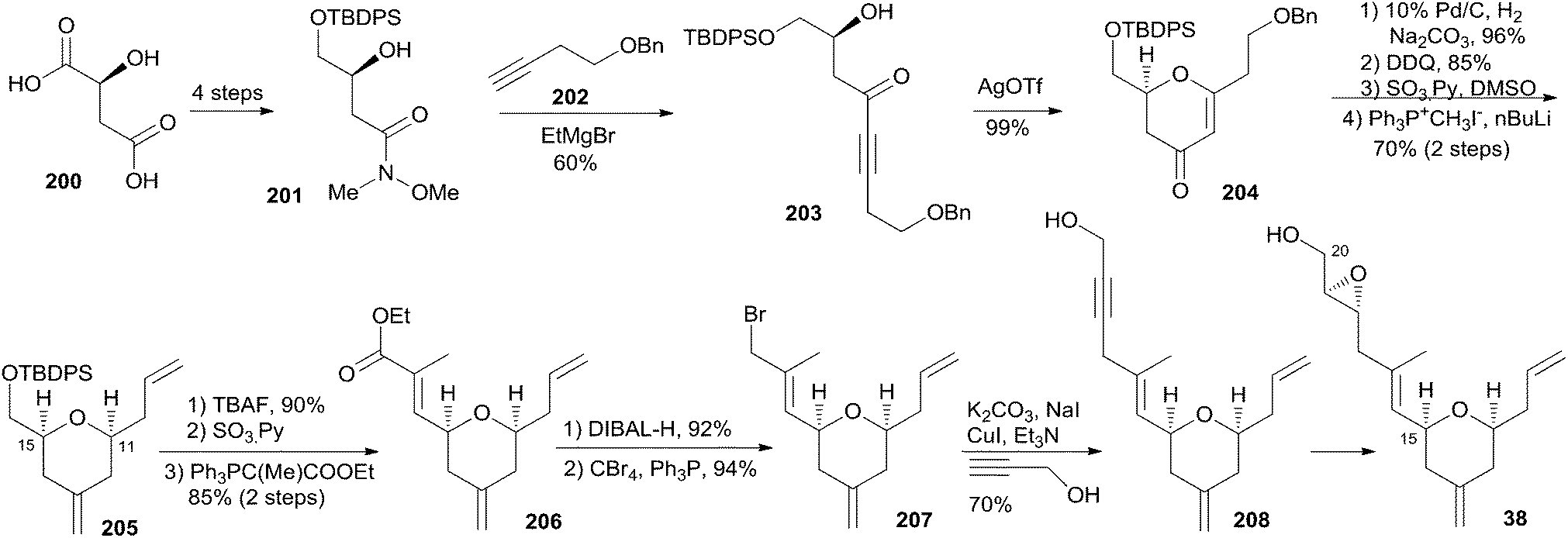
Reddy and co-workers reported a new synthetic route for the known pyran-containing subunit 38 (ref. 85) that had been synthesized by Hoye and co-workers.18 The synthesis began with the known Weinreb amide 201 (Scheme 43), which was obtained in four steps from L-malic acid following a reported protocol.86 The initial four-carbon extension was achieved by the addition of homopropargyl benzyl ether (202) to Weinreb amide 201, which gave the desired alkynone 203. Key cyclization of the alkynone gave pyranone 204. THP 205 was obtained from 204via sequential reduction, DDQ benzyl removal, oxidation, and Wittig olefination. Conversion of 205 to ester 206 was achieved by a deprotection–oxidation–Wittig reaction sequence. Bromide 207 was achieved by reduction of ester 206 and subsequent treatment with CBr4 and PPh3. A three-carbon unit was introduced to compound 207 through a CuI/K2CO3/NaI-mediated coupling reaction to give 208. Reduction of the alkyne to the desired trans-olefin followed by Sharpless asymmetric epoxidation gave target compound 38.
Scheme 43 Reddy's synthesis of fragment C8–C20 of (−)-zampanolide.
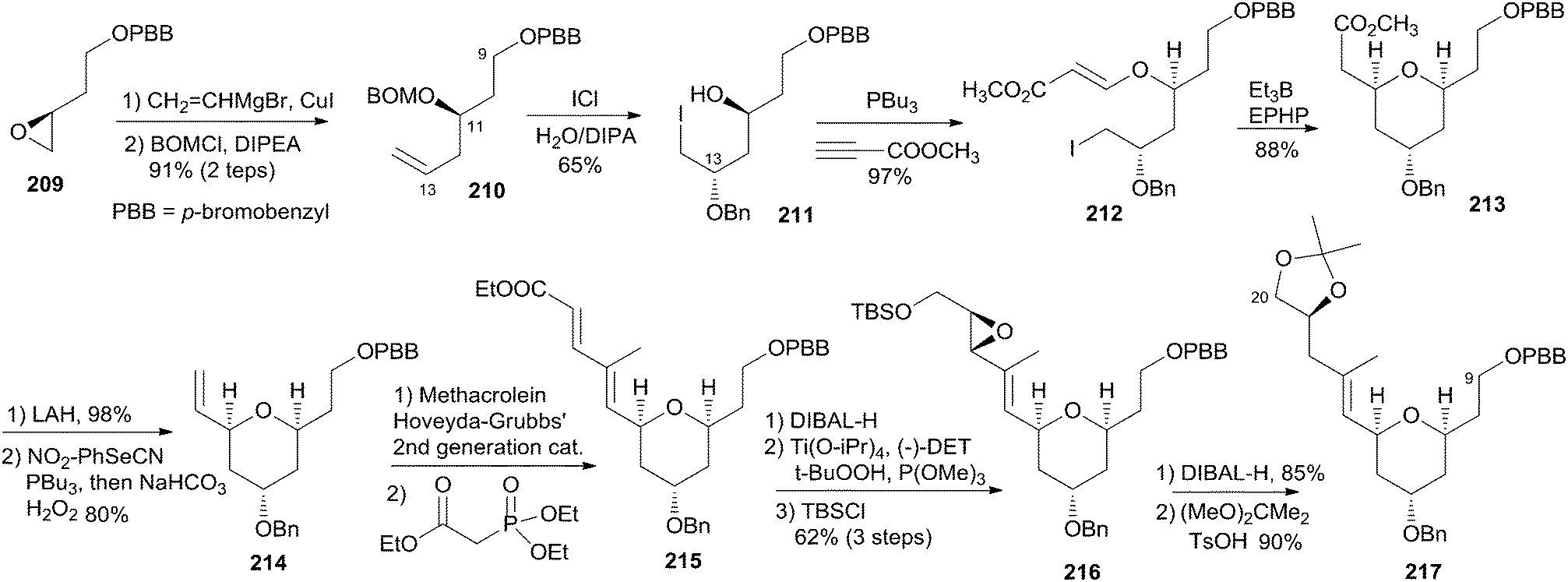
Taylor and co-workers applied their recently developed methodology87 to assemble the 4-alkoxy-2,6-cis-THP 217 from an advanced sulfonyl pyran fragment. The synthesis commenced with preparation of 210 as illustrated in Scheme 44. The epoxide 209 was prepared from (R)-aspartic acid via modification of the literature's procedure.88 Regioselective epoxide opening with vinyl magnesium bromide in the presence of catalytic CuI followed by p-bromobenzyloxy methyl ether alkylation gave the PBB ether 210. Electrophilic activation of the resulting ether with ICl at low temperature provided exclusively 1,3-syn diol monoether. PBu3-catalyzed conjugate addition of alcohol 211 to methyl propiolate yielded β-alkoxyacrylate 212. Intramolecular radical cyclization promoted by Et3B in the presence of 1-ethylpiperidinium hypophosphate gave THP 213 in good diastereoselectivity (>20:1). Reduction followed by Grieco–Sharpless olefination provided 214. Exposure of terminal olefin 214 to excess methacrolein mediated by Hoveyda–Grubbs' second-generation catalyst followed by HWE olefination gave dienonate 215. Reduction of the dienoate with DIBAL-H followed by Sharpless epoxidation and in situ silyl protecting gave alcohol 216. Completion of the C9–C20 fragment was accomplished via regioselective opening of the vinyl epoxide with DIBAL-H at low temperature. Desilylation followed by acetonide formation provided 217.
Scheme 44 Taylor's synthesis of fragment C9–C20 of (−)-zampanolide.
6.2 Preparation of N-acyl hemiaminal model systems
Porco and Troast reported59 the synthesis of N-acyl hemiaminal model systems related to the side chain of zampanolide. Key steps involve oxidative decarboxylation of N-acyl-α-amino acid intermediates, followed by ytterbium triflate mediated solvolysis. This method has not yet been applied to the installation of the hemiaminal side chain of zampanolide.
7. Conclusions
Zampanolide has been demonstrated as a very promising MSA with potent cytotoxicity against both drug sensitive and multi-drug resistant cancer cell lines. It can promote tubulin assembly in a paclitaxel-like manner. Its mechanism of action has been shown to involve covalent binding to β-tubulin at residues Asn228 and His229 at the taxane luminal site, and the use of its side chain to induce structuring of the M-loop into a short helix. Despite isolation from two marine organisms and several total syntheses, samples of zampanolide remain scarce. Moreover, only a few analogs have been reported to date. Future work on this promising compound needs to include both more efficient and practical approaches to the synthesis of (−)-zampanolide to provide larger quantities for in-depth investigation of its cytotoxic activity, and the design and construction of novel (−)-zampanolide analogues to further investigate the SAR of this very promising class of natural products.
Acknowledgements
California State University (CSU), Fresno and the NIH RIMI program at CSU Fresno (P20MD002732) are acknowledged for partial support of this work.
P. B. Schiff, J. Fant and S. B. Horwitz, Nature, 1979, 277, 665–667 CrossRefCAS.
D. G. I. Kingston, J. Nat. Prod., 2009, 72, 507–515 CrossRefCASPubMed.
D. G. I. Kingston, in Anticancer Agents from Natural Products, eds. G. M. Cragg, D. G. I. Kingston and D. J. Newman, CRC press, Boca Raton, FL, 2nd edn, 2012, pp. 123–176 Search PubMed.
R. M. Borzilleri and G. D. Vite, in Ann. Rep. Med. Chem., 2009, vol. 44, ch. 15, pp. 301–322 Search PubMed.
C. C. Rohena and S. L. Mooberry, Nat. Prod. Rep., 2014, 31, 335–355 RSC.
J. Tanaka and T. Higa, Tetrahedron Lett., 1996, 37, 5535–5538 CrossRefCAS.
J. J. Field, A. J. Singh, A. Kanakkanthara, T. i. Halafihi, P. T. Northcote and J. H. Miller, J. Med. Chem., 2009, 52, 7328–7332 CrossRefCASPubMed.
A. Cutignano, I. Bruno, G. Bifulco, A. Casapullo, C. Debitus, L. Gomez-Paloma and R. Riccio, Eur. J. Org. Chem., 2001, 775–778 CrossRefCAS.
A. B. Smith, I. G. Safonov and R. M. Corbett, J. Am. Chem. Soc., 2002, 124, 11102–11113 CrossRefCASPubMed.
J. Uenishi, T. Iwamoto and J. Tanaka, Org. Lett., 2009, 11, 3262–3265 CrossRefCASPubMed.
A. B. Smith III, I. G. Safonov and R. M. Corbett, J. Am. Chem. Soc., 2001, 123, 12426–12427 CrossRef.
A. B. Smith III and I. G. Safonov, Org. Lett., 2002, 4, 635–637 CrossRefPubMed.
E. M. Larsen, M. R. Wilson, J. Zajicek and R. E. Taylor, Org. Lett., 2013, 15, 5246–5249 CrossRefCASPubMed.
C. C. Sanchez and G. E. Keck, Org. Lett., 2005, 7, 3053–3056 CrossRefCASPubMed.
A. K. Ghosh and X. Cheng, Org. Lett., 2011, 13, 4108–4111 CrossRefCASPubMed.
D. Zurwerra, F. Glaus, L. Betschart, J. Schuster, J. Gertsch, W. Ganci and K.-H. Altmann, Chem.–Eur. J., 2012, 18, 16868–16883 CrossRefCASPubMed.
T. R. Hoye and M. Hu, J. Am. Chem. Soc., 2003, 125, 9576–9577 CrossRefCASPubMed.
F. Ding and M. P. Jennings, J. Org. Chem., 2008, 73, 5965–5976 CrossRefCASPubMed.
I. Louis, N. L. Hungerford, E. J. Humphries and M. D. McLeod, Org. Lett., 2006, 8, 1117–1120 CrossRefCASPubMed.
D. L. Aubele, S. Wan and P. E. Floreancig, Angew. Chem., Int. Ed., 2005, 44, 3485–3488 CrossRefCASPubMed.
T. Higa, J. Tanaka and D. Garcia Gravalos, International Pat., WO9710242 (A1) 1997.
J. J. Field, B. Pera, E. Calvo, A. Canales, D. Zurwerra, C. Trigili, J. Rodriguez-Salarichs, R. Matesanz, A. Kanakkanthara, S. J. Wakefield, A. J. Singh, J. Jimenez-Barbero, P. Northcote, J. H. Miller, J. A. Lopez, E. Hamel, I. Barasoain, K.-H. Altmann and J. F. Diaz, Chem. Biol., 2012, 19, 686–698 CrossRefCASPubMed.
A. K. Ghosh, X. Cheng, R. Bai and E. Hamel, Eur. J. Org. Chem., 2012, 2012, 4130–4139 CrossRefCASPubMed.
C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CrossRefCASPubMed.
J. H. Miller, A. J. Singh and P. T. Northcote, Mar. Drugs, 2010, 8, 1059–1079 CrossRefCASPubMed.
E. Nogales, S. G. Wolf, I. A. Khan, R. F. Luduena and K. H. Downing, Nature, 1995, 375, 424–427 CrossRefCASPubMed.
F. Diaz Jose, I. Barasoain and M. Andreu Jose, J. Biol. Chem., 2003, 278, 8407–8419 CrossRefPubMed.
R. M. Buey, E. Calvo, I. Barasoain, O. Pineda, M. C. Edler, R. Matesanz, G. Cerezo, C. D. Vanderwal, B. W. Day, E. J. Sorensen, J. A. Lopez, J. M. Andreu, E. Hamel and J. F. Diaz, Nat. Chem. Biol., 2007, 3, 117–125 CrossRefCASPubMed.
E. Calvo, I. Barasoain, R. Matesanz, B. Pera, E. Camafeita, O. Pineda, E. Hamel, C. D. Vanderwal, J. M. Andreu, J. A. Lopez and J. F. Diaz, Biochemistry, 2012, 51, 329–341 CrossRefCASPubMed.
J. T. Huzil, J. K. Chik, G. W. Slysz, H. Freedman, J. Tuszynski, R. E. Taylor, D. L. Sackett and D. C. Schriemer, J. Mol. Biol., 2008, 378, 1016–1030 CrossRefCASPubMed.
M. J. Bennett, K. Barakat, J. T. Huzil, J. Tuszynski and D. C. Schriemer, Chem. Biol., 2010, 17, 725–734 CrossRefCASPubMed.
J. J. Field, E. Calvo, P. T. Northcote, J. H. Miller, K.-H. Altmann and J. F. Diaz, Methods Cell Biol., 2013, 115, 303–325 CAS.
I. Barasoain, A. M. Garcia-Carril, R. Matesanz, G. Maccari, C. Trigili, M. Mori, J.-Z. Shi, W.-S. Fang, J. M. Andreu, M. Botta and J. F. Diaz, Chem. Biol., 2010, 17, 243–253 CrossRefCASPubMed.
R. M. Buey, I. Barasoain, E. Jackson, A. Meyer, P. Giannakakou, I. Paterson, S. Mooberry, J. M. Andreu and J. F. Diaz, Chem. Biol., 2005, 12, 1269–1279 CrossRefCASPubMed.
R. Matesanz, I. Barasoain, C.-G. Yang, L. Wang, X. Li, C. de Ines, C. Coderch, F. Gago, J. J. Barbero, J. M. Andreu, W.-S. Fang and J. F. Diaz, Chem. Biol., 2008, 15, 573–585 CrossRefCASPubMed.
M. C. Edler, R. M. Buey, R. Gussio, A. I. Marcus, C. D. Vanderwal, E. J. Sorensen, J. F. Diaz, P. Giannakakou and E. Hamel, Biochemistry, 2005, 44, 11525–11538 CrossRefPubMed.
A. E. Prota, K. Bargsten, D. Zurwerra, J. J. Field, J. F. Diaz, K.-H. Altmann and M. O. Steinmetz, Science, 2013, 339, 587–590 CrossRefCASPubMed.
E. Nogales, M. Whittaker, R. A. Milligan and K. H. Downing, Cell, 1999, 96, 79–88 CrossRefCAS.
F. J. Fourniol, C. V. Sindelar, B. Amigues, D. K. Clare, G. Thomas, M. Perderiset, F. Francis, A. Houdusse and C. A. Moores, J. Cell Biol., 2010, 191, 463–470 CrossRefCASPubMed.
S.-Y. Liao, G.-Q. Mo, J.-C. Chen and K.-C. Zheng, J. Mol. Model., 2014, 20, 2070 CrossRefPubMed.
M. Shindo, Top. Heterocycl. Chem., 2006, 5, 179–254 CAS.
E. Sharif and G. A. O'Doherty, Chemtracts, 2009, 22, 67–79 CAS.
C. M. Rojas, in Name Reactions for Homologations, Part 2, ed. L. J. Li, Wiley, Hoboken, NJ, 2009, pp. 136–163 Search PubMed.
M. L. Morin-Fox and M. A. Lipton, Tetrahedron Lett., 1993, 34, 7899–7902 CrossRefCAS.
A. B. Smith III, R. J. Fox and T. M. Razler, Acc. Chem. Res., 2008, 41, 675–687 CrossRefPubMed.
M. Oizumi, M. Takahashi and K. Ogasawara, Synlett, 1997, 1111–1113 CrossRefCASPubMed.
P. Somfai and R. Olsson, Tetrahedron, 1993, 49, 6645–6650 CrossRefCAS.
L. E. Overman and L. D. Pennington, J. Org. Chem., 2003, 68, 7143–7157 CrossRefCASPubMed.
D. R. Williams, M. P. Clark and M. A. Berliner, Tetrahedron Lett., 1999, 40, 2287–2290 CrossRefCAS.
D. A. Evans, E. Hu, J. D. Burch and G. Jaeschke, J. Am. Chem. Soc., 2002, 124, 5654–5655 CrossRefCASPubMed.
F. Ding and M. P. Jennings, Org. Lett., 2005, 7, 2321–2324 CrossRefCASPubMed.
G. E. Keck, J. A. Covel, T. Schiff and T. Yu, Org. Lett., 2002, 4, 1189–1192 CrossRefCASPubMed.
W. C. Still, C. Gennari, J. A. Noguez and D. A. Pearson, J. Am. Chem. Soc., 1984, 106, 260–262 CrossRefCAS.
G. E. Keck, C. Sanchez and C. A. Wager, Tetrahedron Lett., 2000, 41, 8673–8676 CrossRefCAS.
A. G. Dossetter, T. F. Jamison and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 2398–2400 CrossRefCAS.
A. Fuerstner, O. R. Thiel, L. Ackermann, H.-J. Schanz and S. P. Nolan, J. Org. Chem., 2000, 65, 2204–2207 CrossRefCAS.
D. M. Troast, J. Yuan and J. A. Porco Jr, Adv. Synth. Catal., 2008, 350, 1701–1711 CrossRefCASPubMed.
D. M. Troast and J. A. Porco Jr, Org. Lett., 2002, 4, 991–994 CrossRefCASPubMed.
B. Leroy and I. E. Marko, Tetrahedron Lett., 2001, 42, 8685–8688 CrossRefCAS.
S. Kiyooka, K. Suzuki, M. Shirouchi, Y. Kaneko and S. Tanimori, Tetrahedron Lett., 1993, 34, 5729–5732 CrossRefCAS.
J. M. Bobbitt, J. Org. Chem., 1998, 63, 9367–9374 CrossRefCAS.
S. Takano, T. Kamikubo, T. Sugihara, M. Suzuki and K. Ogasawara, Tetrahedron: Asymmetry, 1993, 4, 201–204 CrossRefCAS.
T. Hanazawa, K. Sasaki, Y. Takayama and F. Sato, J. Org. Chem., 2003, 68, 4980–4983 CrossRefCASPubMed.
B. M. Trost, S. A. King and T. Schmidt, J. Am. Chem. Soc., 1989, 111, 5902–5915 CrossRefCAS.
H. Oda, T. Kobayashi, M. Kosugi and T. Migita, Tetrahedron, 1995, 51, 695–702 CrossRefCAS.
J. Uenishi, R. Kawahama, O. Yonemitsu, A. Wada and M. Ito, Angew. Chem., Int. Ed., 1998, 37, 320–323 CrossRefCAS.
J. Uenishi, K. Matsui and M. Ohmi, Tetrahedron Lett., 2005, 46, 225–228 CrossRefCASPubMed.
Y. Yun Sang, C. Hansen Eric, I. Volchkov, J. Cho Eun, Y. Lo Wai and D. Lee, Angew. Chem., Int. Ed., 2010, 49, 4261–4263 CrossRefPubMed.
D. Lee and I. Volchkov, Strategies Tactics Org. Synth., 2012, 8, 171–197 CAS.
B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630–6666 CrossRefCASPubMed.
S. Bellemin-Laponnaz, H. Gisie, J. P. Le Ny and J. A. Osborn, Angew. Chem., Int. Ed. Engl., 1997, 36, 976–978 CrossRefCAS.
G. Pattenden, M. A. Gonzalez, P. B. Little, D. S. Millan, A. T. Plowright, J. A. Tornos and T. Ye, Org. Biomol. Chem., 2003, 1, 4173–4208 CAS.
E. C. Hansen and D. Lee, J. Am. Chem. Soc., 2006, 128, 8142–8143 CrossRefCASPubMed.
K. Lee, H. Kim and J. Hong, Angew. Chem., Int. Ed., 2012, 51, 5735–5738 CrossRefCASPubMed.
M. J. Gaunt, A. S. Jessiman, P. Orsini, H. R. Tanner, D. F. Hook and S. V. Ley, Org. Lett., 2003, 5, 4819–4822 CrossRefCASPubMed.
F. Caussanel, P. Deslongchamps and Y. L. Dory, Org. Lett., 2003, 5, 4799–4802 CrossRefCASPubMed.
Y. Hayashi and T. Mukaiyama, Chem. Lett., 1987, 1811–1814 CrossRefCAS.
M. M. Claffey, C. J. Hayes and C. H. Heathcock, J. Org. Chem., 1999, 64, 8267–8274 CrossRefCASPubMed.
B. A. Narayanan and W. H. Bunnelle, Tetrahedron Lett., 1987, 28, 6261–6264 CrossRefCAS.
B. Lei and A. G. Fallis, Can. J. Chem., 1991, 69, 1450–1456 CrossRefCAS.
D. Zurwerra, J. Gertsch and K.-H. Altmann, Org. Lett., 2010, 12, 2302–2305 CrossRefCASPubMed.
S. D. Rychnovsky, Y. Hu and B. Ellsworth, Tetrahedron Lett., 1998, 39, 7271–7274 CrossRefCAS.
T.-P. Loh, J.-Y. Yang, L.-C. Feng and Y. Zhou, Tetrahedron Lett., 2002, 43, 7193–7196 CrossRefCAS.
C. R. Reddy and B. Srikanth, Synlett, 2010, 1536–1538 CrossRefCASPubMed.
T. Shioiri, N. McFarlane and Y. Hamada, Heterocycles, 1998, 47, 73–76 CrossRefCASPubMed.
M. R. Wilson and R. E. Taylor, Org. Lett., 2012, 14, 3408–3411 CrossRefCASPubMed.
J. A. Frick, J. B. Klassen, A. Bathe, J. M. Abramson and H. Rapoport, Synthesis, 1992, 621–623 CrossRefCAS.
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: