 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural products containing ‘decalin’ motif in microorganisms
Gang
Li†
,
Souvik
Kusari†
* and
Michael
Spiteller
*
Institute of Environmental Research (INFU), Department of Chemistry and Chemical Biology, Chair of Environmental Chemistry and Analytical Chemistry, TU Dortmund, Otto-Hahn-Str.6, 44221 Dortmund, Germany. E-mail: souvik.kusari@infu.tu-dortmund.de; m.spiteller@infu.tu-dortmund.de; Fax: +49-231-755-4084; Fax: +49-231-755-4085; Tel: +49-231-755-4086 Tel: +49-231-755-4080
First published on 2nd July 2014
Abstract
Covering: up to February 2014
Microorganisms are well-known producers of a wide variety of bioactive compounds that are utilized not only for their primary metabolism but also for other purposes such as defense, detoxification, or communication with other micro- and macro-organisms. Natural products containing a ‘decalin ring’ occur often in microorganisms. They exhibit diverse and remarkable biological activities, including antifungal, antibacterial, anticancer and immunosuppressive activities, to name a few. This review surveys the natural decalin-type compounds that have been isolated from microorganisms, with emphasis on both chemical and biological implications. Total syntheses of some important decalin moiety-containing natural products are also highlighted.
 Gang Li | Gang Li received his B.Sc. degree (2009) at Shandong University of Traditional Chinese Medicine and M.Sc. degree (2012) under the supervision of Prof. Hong-Xiang Lou from Shandong University, China. He is presently working at INFU, TU Dortmund, Germany for his doctorate with a fellowship from the China Scholarship Council (CSC). His research focuses on bioactive secondary metabolites from endophytic fungi. |
 Souvik Kusari | Souvik Kusari received bachelor and master degrees in Biotechnology from Bangalore University, India. He then earned a doctorate in Natural Sciences from INFU, TU Dortmund, Germany in 2010. After working as a scientist in the same institute for over two years, he became a visiting researcher at the Department of Plant Sciences, University of Oxford, UK for a year in 2013. He is also a visiting lecturer at the University of Yaoundé 1, Cameroon, Africa since 2012 within the scope of the DAAD “Welcome to Africa” initiative. His research interests include characterization of fungal and bacterial endophytes to evaluate their diversity, chemical ecology and biosynthetic pathways. |
 Michael Spiteller | Michael Spiteller received his Diploma in Chemistry (1976) from the Georg-August-Universität in Göttingen, Germany where he earned a doctorate in Chemistry in 1979. After Habilitation in Soil Science, he joined the Institute for Metabolism and Environmental Fate, Bayer Corp., Germany for 8 years as a Group Leader. He then joined the University of Kassel as a full Professor and is now Professor and Head of INFU, TU Dortmund, Germany. He is involved in various projects dealing with natural product drug discovery from plants and endophytes, structural identification of unknown xenobiotics and their metabolites, metabolism of pesticides in the environment, and veterinary drugs. |
1 Introduction
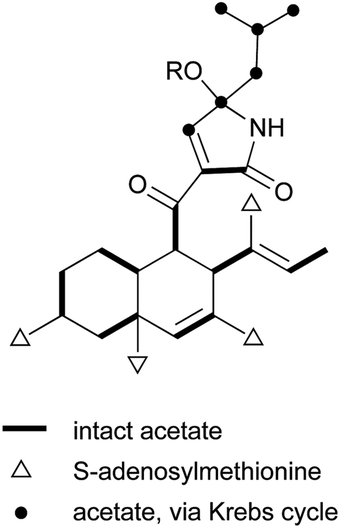
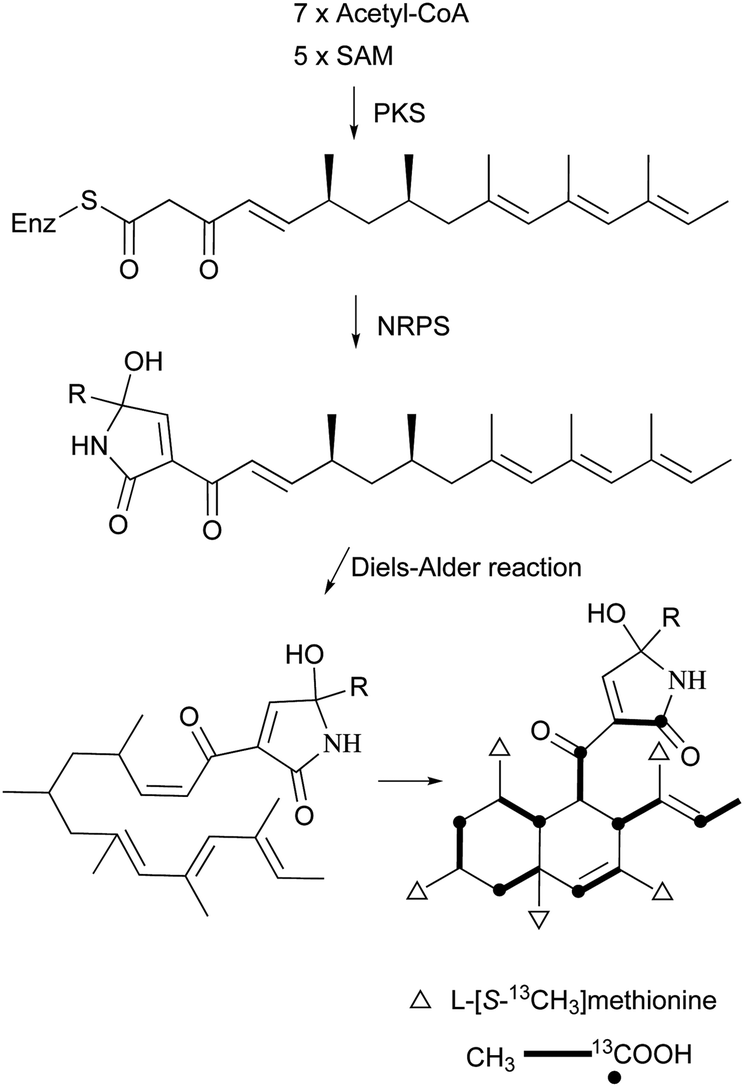
The ‘decalin’ motif is found in an array of secondary metabolites produced by microorganisms, mainly fungi and actinomycetes. It is usually correlated with highly multi-functionalized or architecturally complex groups, thereby demonstrating surprising structural and functional diversity. Their intricate structures and diverse biological activities have attracted researchers around the world to investigate their biosynthesis, chemical synthesis, and the various facets of the functionalized decalin skeleton.1–3The decalin scaffold is primarily biosynthesized by microorganisms based on two biosynthetic derivations, isoprenoids (mevalonate) and polyketides (acetate). Isoprenoid-derived decalins typically belong to natural sesquiterpenoids and diterpenoids, and have been thoroughly covered in a series of reports by Fraga4 and Hanson.5,6 However, there is a dearth of comprehensive reviews on polyketide decalin-derived secondary metabolites, either about their isolation and biological activities, or related syntheses. Furthermore, no critical observation about the differences or similarities of these decalin moiety-containing compounds from a microbial origin has been reported.
The polyketide decalin skeleton in some fungal or bacterial secondary metabolites is proposed to be biosynthesized by an enzymatic intramolecular Diels–Alder (IMDA) cycloaddition.1,7 However, only five potential or possible Diels–Alderase enzymes have been purified until now,7,8 of which lovastatin nonaketide synthase (LovB) and solanapyrone synthase (SPS) are proposed to be related to the decalin formation of lovastatin and solanapyrone A.9,10 It should be noted that the non-enzymatic catalytic synthesis of decalin in biological systems is also possible, as suggested in a biomimetic total synthesis of the decalin compound (±)-UCS1025A.11 Based on the IMDA reaction of a substrate,9 all possible cyclization types to decalin are shown in Scheme 1. There are four comprehensive reviews on the syntheses of trans- or cis-decalins mainly in isoprenoids (mevalonate)-derived natural products.3,12–14 However, to the best of our knowledge, reviews compiling different facets of the polyketide decalin have not been reported.
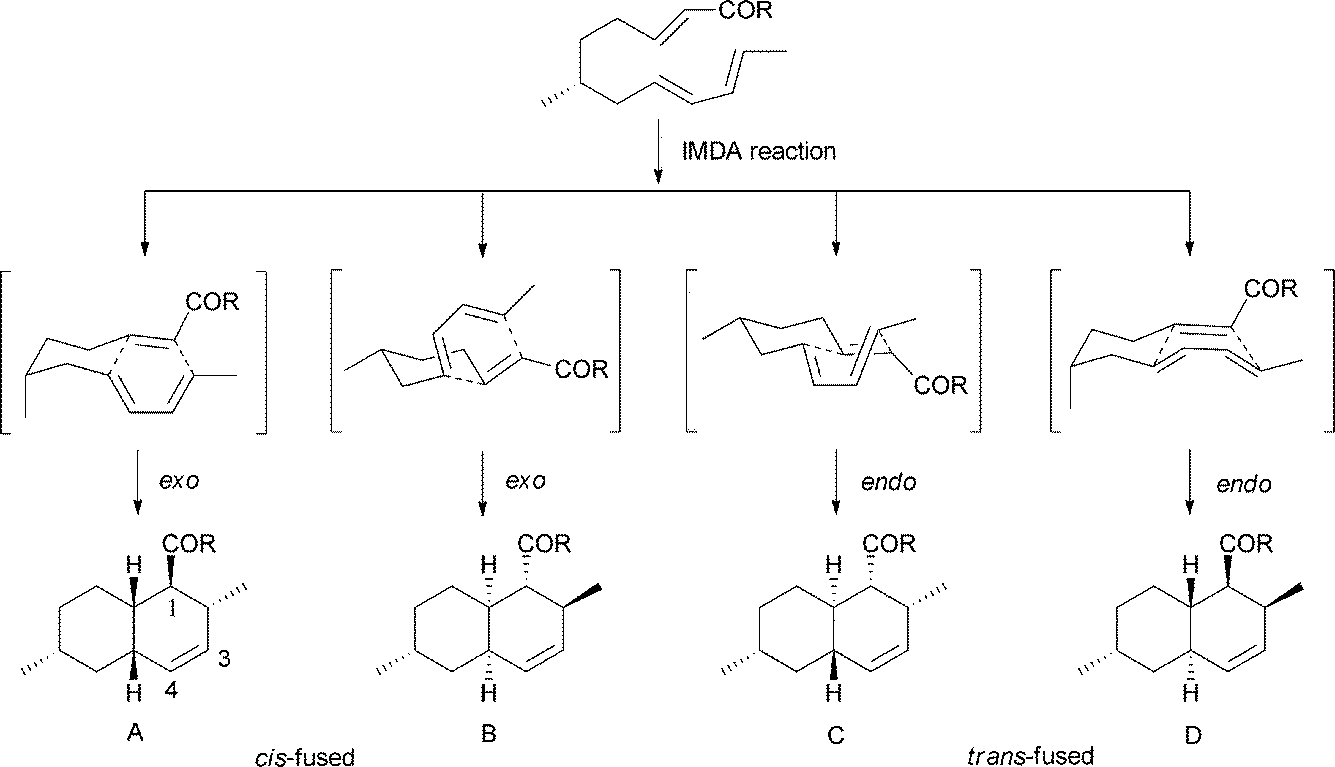 | ||
| Scheme 1 The IMDA reaction to decalin. | ||
Therefore, given the present gaps in a comprehensive elaboration of the ‘decalin’ system and considering their intriguing structural and biological features, the target of this review is to provide an informative overview of the topic that can serve as a point of reference for an understanding of the functions and applications of decalin.
2 Structural classification
Decalin, as a ring system or scaffold, can be greatly modified by many functional groups, such as a side chain, or diverse moieties. The side chains are usually substituted by many functional groups such as hydroxyl, carboxyl, or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO double bonds. Many other moieties such as small lactone ring, pyrone, tetramic acid, unusual sugars, pyridone, tetronic acid, or pyrrolizidine act as diverse moieties, and are usually found in this type of compound. With the occurrence and interaction of these functional units in structures, a complex macrocycle or polycycle that is often fused with the decalin ring, is formed in microbial secondary metabolites. In this manuscript, those secondary metabolites are mainly classified according to the proposed biosynthesis of the decalin moiety-containing compounds: polyketide or isoprenoid biosynthetic derivations. The compounds with polyketide or isoprenoid decalin motifs are further classified according to the features of the remaining side chain or diverse moieties.
C and CO double bonds. Many other moieties such as small lactone ring, pyrone, tetramic acid, unusual sugars, pyridone, tetronic acid, or pyrrolizidine act as diverse moieties, and are usually found in this type of compound. With the occurrence and interaction of these functional units in structures, a complex macrocycle or polycycle that is often fused with the decalin ring, is formed in microbial secondary metabolites. In this manuscript, those secondary metabolites are mainly classified according to the proposed biosynthesis of the decalin moiety-containing compounds: polyketide or isoprenoid biosynthetic derivations. The compounds with polyketide or isoprenoid decalin motifs are further classified according to the features of the remaining side chain or diverse moieties.
3 Polyketide decalin
Fungi and bacteria are the major microbial resources that produce the decalin moiety-containing secondary metabolites with a broad spectrum of biological activities. Most of these compounds have a polyketidic decalin scaffold. Frequently, they are highly functionalized through the substitution of methyl, hydroxyl, or CC and CO double bonds on the decalin skeleton, or through a three-, five-, or seven-membered side chain with carboxyls (or its ester), several double bonds, or via ring formation. Furthermore, a pyrone moiety connected to a poly-substituted decalin nucleus by a carbon–carbon bond contributes to an important class of polyketide natural products. In addition, the pyrrolidine-2-one moieties such as tetramic acids and the 4-hydroxy-2-pyridone group are important functionalized units found in many polyketide decalin natural products. The promising biosynthetic potential of fungi is also elaborated here with several examples of macrocyclic or polycyclic compounds.
The biosynthesis of secondary metabolites with a polyketide decalin system is attributed to single or mixed biosynthetic pathways. Specifically, these compounds are proposed to be assembled by a linear polyketide unit, which are then cyclized by an enzymatic or non-enzymatic IMDA cycloaddition to form the decalin scaffold.1,7 Prior to or after cyclization, many functionalized substituted groups are joined to the above polyketide skeletons which undergo some inter- or intra-molecular reactions to form the complete structure. Amino acids or other unusual units are also occasionally integrated into such a polyketide system to form diverse structures with intriguing structural features.
3.1 Monacolins
Monacolins are produced by microorganisms, and show a remarkable inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase that catalyzes the key step in cholesterol biosynthesis.15 Lovastatin (1), a cyclic nonaketide acylated by a diketide, and its semisynthetic derivative simvastatin act as potent inhibitors of HMG-CoA reductase and are widely prescribed in the treatment of hypercholesterolemia.16 Potent monacolin-type inhibitors of HMG-CoA reductase have been isolated from several fungal strains, mainly Monascus ruber, Aspergillus terreus, and Penicillium citrinum. They share a HMG-like moiety which is linked to the rigid and hydrophobic decalin system, and occupy a portion of the binding site of HMG-CoA, thus blocking access of this substrate (HMG-CoA) to the active site when these potent decalin-type inhibitors are bound.15 Until now, about 27 monacolins have been isolated and identified from fungal resources. The decalin skeleton is most likely formed via a biological Diels–Alder reaction by polyketide synthase (PKS) enzymes, as suggested earlier for lovastatin nonaketide synthase (LovB).9 This kind of compound is often isolated as an inactive lactone or active hydroxy-acid form. In this review, we will use the word “acid” to describe some lactone-opened monacolins for easy readability.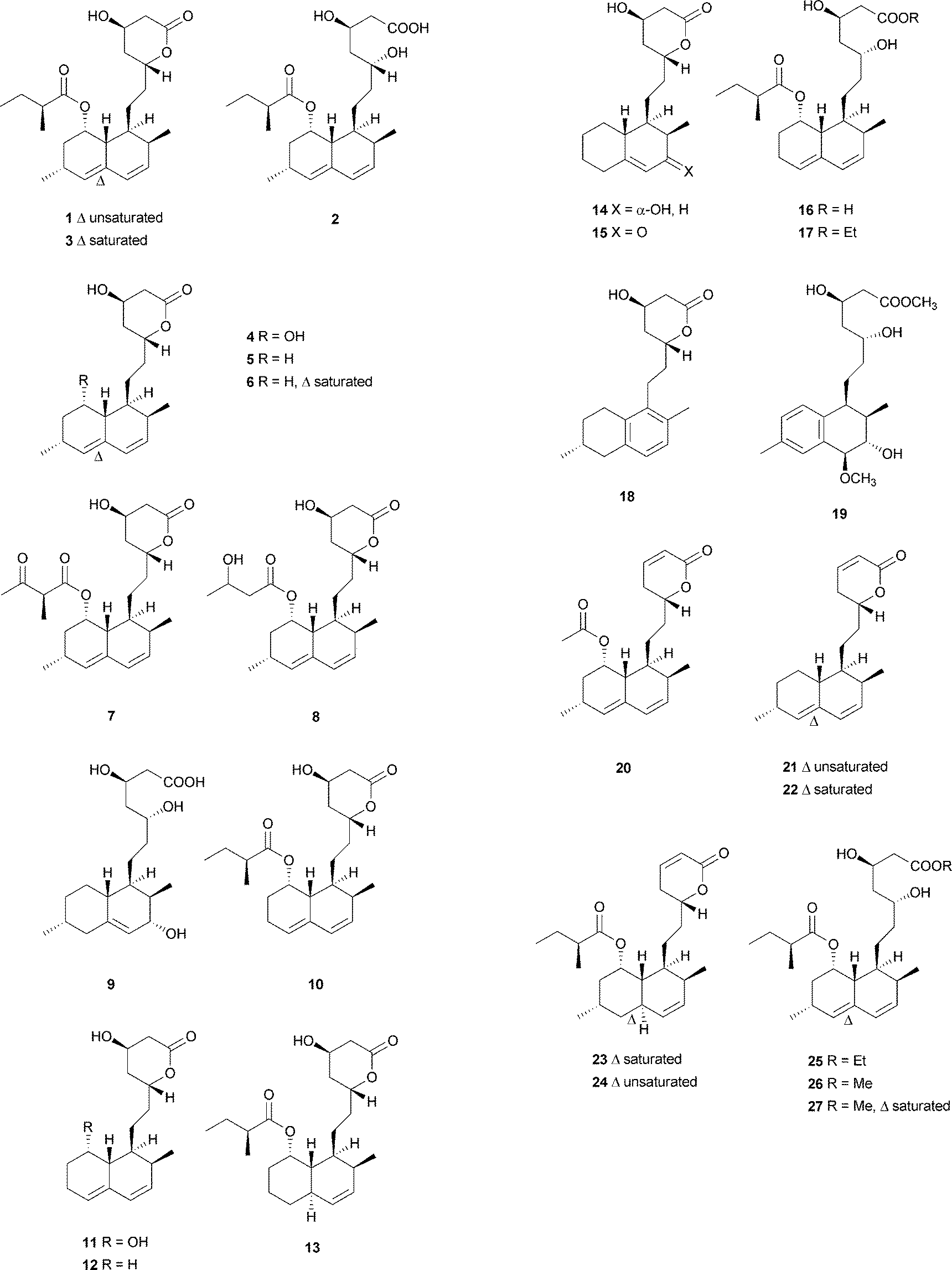
Lovastatin (1) (also called monacolin K or mevinolin) was first isolated in 1979 as an active inhibitor of HMG-CoA reductase (Endo reported 1 as monacolin K isolated from M. ruber in 1979;17,18 Alberts et al., isolated and reported the same compound as mevinolin isolated from A. terreus19). The lovastatin biosynthetic gene cluster was first identified in 1999 by the groups of Vederas and Hutchinson. After this work, more attention was paid towards identification of the complete biosynthetic pathway leading to 1. Recently, the group of Tang16 briefly summarized important findings and recent advances on the investigation of the lovastatin (1) biosynthesis,9,20–23 and proposed that LovG from the lovastatin (1) gene cluster is responsible for the LovB protein (lovastatin nonaketide synthase, LNKS) turnover and release of dihydromonacolin L (6) (Scheme 2). From the cultures of A. terreus, in addition to 1, its lactone-opened form, lovastatin acid (2) (designated as mevinolinic acid) was obtained.19 Another lovastatin analog, 4a,5-dihydromevinolin (3), a potent hypocholesterolemic agent, was isolated from this fungus.24 Two metabolites related to 1 were isolated from M. ruber and designated as monacolins J (4) and L (5) by Endo et al.25 From a mutant strain of M. ruber, dihydromonacolin L (6) and monacolin X (7) were further discovered.26 Endo and co-workers also isolated a β-hydroxybutyryl ester of 4, named monacolin M (8).27 A hydrolysis derivative of 6, identified as 3α-hydroxy-3,5-dihydromonacolin L (9), was found to be produced by A. terreus.28 Compactin (10) was first isolated from Penicillium brevicompactum in 1976 by Brown et al. as an antifungal metabolite.29 In the same year, 10 (designated ML-236B) was also isolated as a hypercholesterolemic agent from cultures of the fungus P. citrinum.30 ML-236A (11) and ML-236C (12) were further isolated from this fungus. Of the three compounds, 10 was most active in inhibiting cholesterol synthesis to 50% of control at a concentration of 26 nM, compared to 280 nM for 12 and 590 nM for 11.30 However, the sodium acid of 1 was found to be more than twice as active as the sodium acid of 10 as an inhibitor of HMG-CoA reductase, with respective Ki values of 0.64 nM and 1.4 nM.19 As also shown in an acute assay for rats, the orally administered sodium salt of 1 inhibited cholesterol biosynthesis at a concentration of 46 μg kg−1 in 50% inhibitory dose compared to a 50% inhibitory dose of 290 μg kg−1 for the sodium salt of 10.19 In 1981, Lam et al. discovered 4a,5-dihydrocompactin (13) from the fungus P. citrinum.31 3α-Hydroxy-3,5-dihydro-ML-236C (14) was isolated as a white amorphous solid, in its sodium salt form, from Paecilomyces viridis.32 Compound 14 along with 3,5-didydro-3-oxo-ML-236C (15), compactin acid (16), and the ethyl ester of compactin (17) were isolated and identified from Eupenicillium javanicum.33 Monacophenyl (18) and aromonacolin A (19), two unusual aromatic monacolin analogs, were isolated from Monascus purpureus-fermented rice (red yeast rice).34,35 In the course of further investigation using red yeast rice, five cytotoxic dehydromonacolins, namely dehydromonacolin N (20), dehydromonacolin L (21), α,β-dehydrodihydromonacolin L (22), α,β-dehydrodihydromonacolin K (23), and dehydromonacolin K (24), together with the ethyl ester of 1 (25), the methyl ester of 1 acid (26) and the methyl ester of 3 (27) were isolated and characterized.36
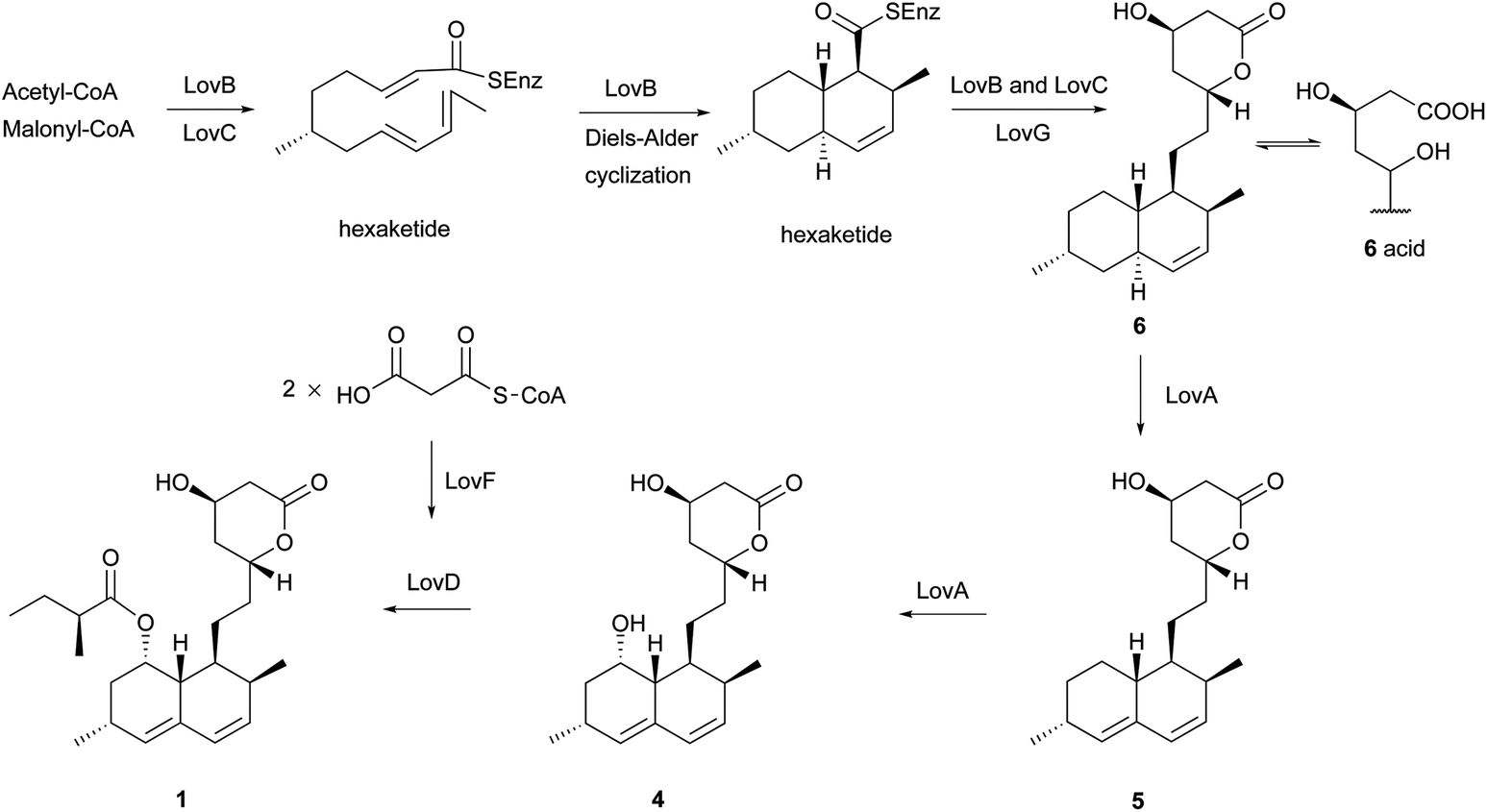 | ||
| Scheme 2 The proposed biosynthetic pathway for 1. | ||
3.2 Monacolin derivatives
Simple decalin derivatives are usually found in monacolin-producing fungi. From the biosynthetic point of view, they are presumed to be formed by β-oxidation or/and dehydrogenation of monacolins.37Antifungal activity-guided fractionation led to the isolation of the two decalin derivatives, eujavanoic acids A (28) and B (29) from E. javanicum.33 A heptaketide (30) and a decalin derivative, monascusic acid A (31) were isolated from red yeast rice fermented by M. purpureus.38 Further chemical investigation using red yeast rice provided five new decalin derivatives, monascusic acids B–E (32–35) and monascusic lactone A (36).3730–34 showed the immunosuppressive effect on human T cell proliferation in a dose-dependent manner from 10 to 100 μM.37Compound 36, the first reported naturally-occurring decalin derivative possessing a spiro lactone at the C-1 position, is biosynthetically related to monacolin L (Scheme 3).
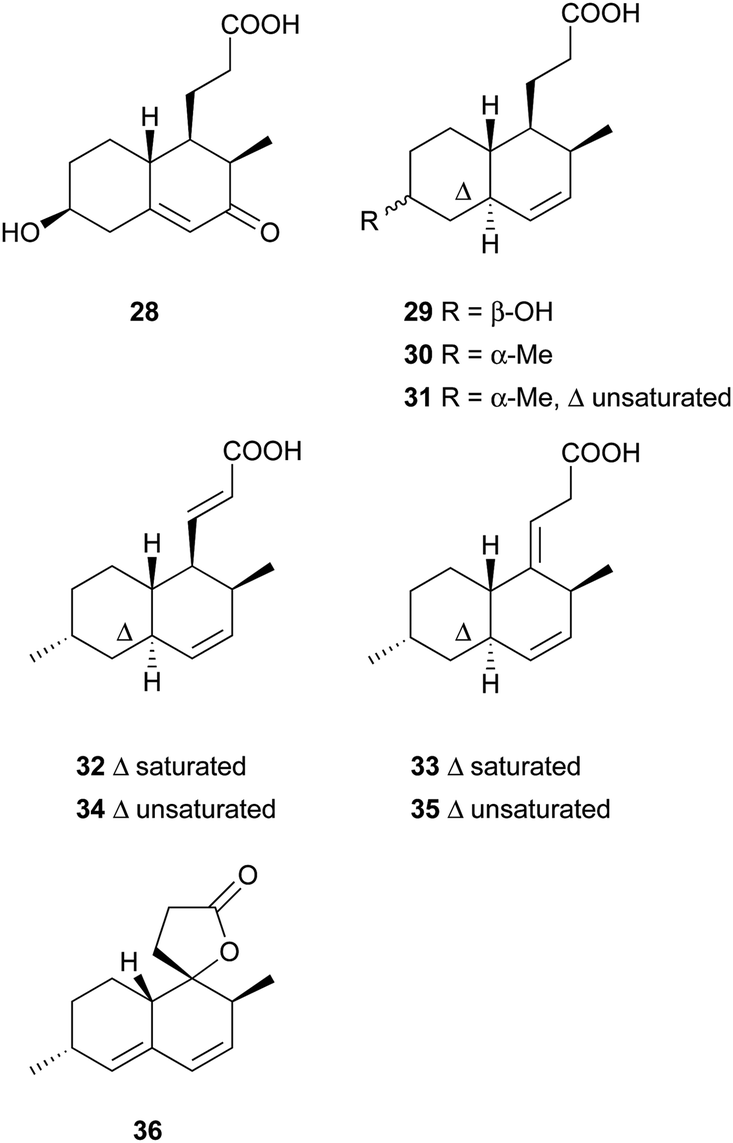
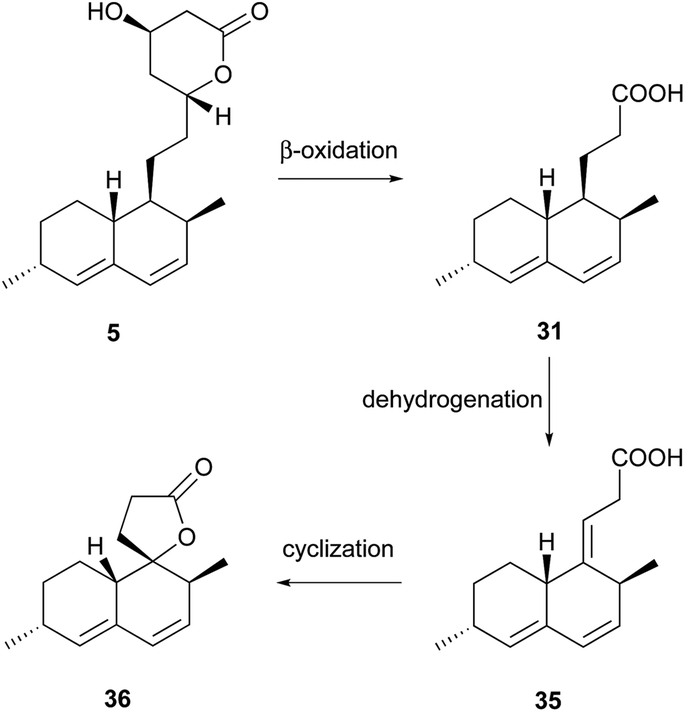 | ||
| Scheme 3 The proposed biosynthesis of 36. | ||
3.3 Side chains with a 3-oxopropanol or its derivative
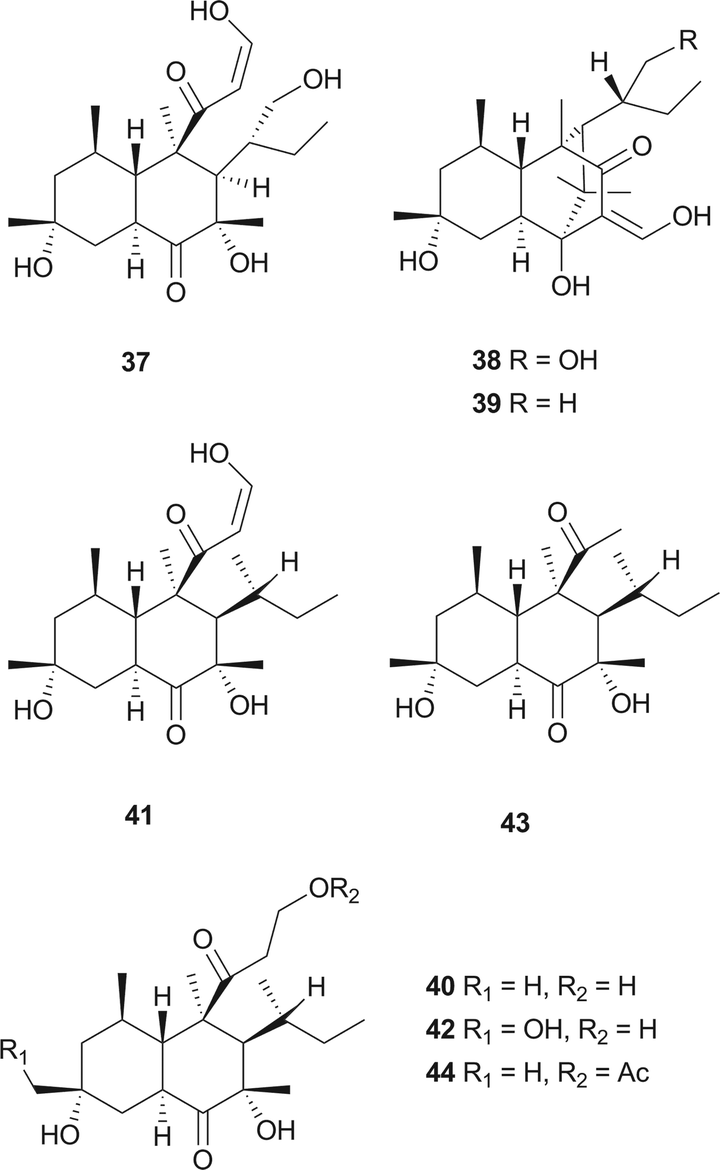
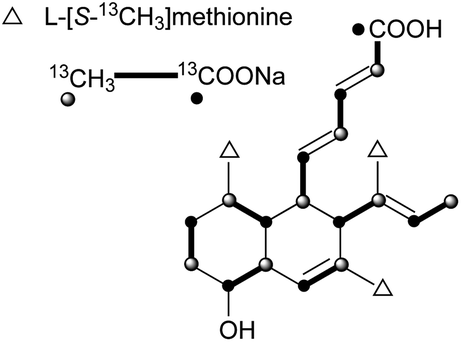
The skeleton of these compounds could have the same construction pattern from some acetate units via a polyketide pathway, and like lovastatin (1) biosynthesis, an intramolecular Diels–Alder reaction seems to be responsible for their decalin scaffold formation. They share a highly substituted polyfunctionalized trans-decalin, which is modified by methyls, hydroxyl, double bond and side chain groups or moieties. More importantly, the side chain with a β-ketoaldehyde seems to be crucial for some biological activities.Stemphyloxin I (37) and II (38), two nonspecific phytotoxic ferric ion chelates, were isolated from the plant pathogenic fungus Stemphylium botryosum.39–41 They are highly functionalized trans-decalin derivatives and exhibited high toxicity towards tomato and eggplant. The presence of β-ketoaldehyde functional group appears to be crucial for both toxicity and chelation of iron.42 Six phytotoxins, betaenones A–F (39–44), have been isolated from the cultures of Phoma betae, a fungus causing leaf spot disease of sugar beet.43,44 The biosynthesis of 40 involving an intramolecular Diels–Alder reaction was studied using feeding experiments (Fig. 1).45,46
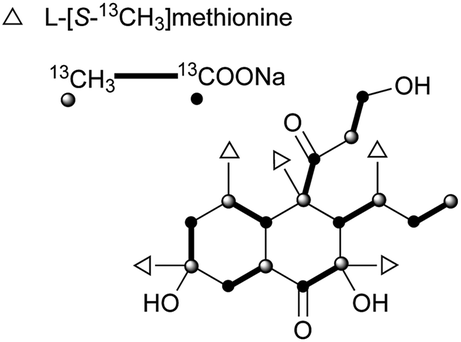 | ||
| Fig. 1 Biosynthetic origins of carbons in 40. | ||
From the marine sponge-derived fungus Trichoderma harzianum, trichoharzin (45) with an alkylated trans-decalin skeleton was found.47 Phomodiol (46), an antifungal polyketide, has been found in Phomopsis sp.48 In a screening of soil microorganisms for new antibiotics, aldecalmycin (47) with high efficacy against Gram-positive bacteria was obtained from a culture broth of Streptomyces sp.49 Australifungin (48), structurally related to 37, was isolated from a fungus Sporormiella australis and exhibited strong antifungal activity against a panel of clinically relevant Aspergillus, Candida, and Cryptococcus strains with minimum inhibitory concentration (MIC) values ranging from 0.015 to 1.0 μg mL−1.50 It was also the first reported non-sphingosine-based inhibitor of sphingolipid biosynthesis. Another polyketide, deoxynortrichoharzin (49) was obtained from the saltwater culture of Paecilomyces cf. javanica isolated from the marine sponge Jaspis cf. coriacea, which did not show any cytotoxic activity against solid tumor cells in culture.51 Two betaenone derivatives, 10-hydroxy-18-methoxybetaenone (50) and 10-hydroxy-18-N-2-naphtyl-N-phenylaminobetaenone (51) were produced by an undescribed fungus of the genus Microsphaeropsis isolated from the Mediterranean sponge Aplysina aerophoba.52 Derivative 50 showed inhibitory activity against PKC-ε, CDK4, and EGF receptor tyrosine kinases, with IC50 values of 36.0, 11.5, and 10.5 μM, respectively, whereas 51 did not. Decumbenones A (52) and B (53), together with versiol (54) were isolated from the fungus, Penicillium decumbens.53 Only one of them, compound 52, inhibited melanization in Magnaporthe grisea, the rice blast pathogen, suggesting the importance of the structural units of the diene and COCH2CH2OH for its inhibition efficacy.
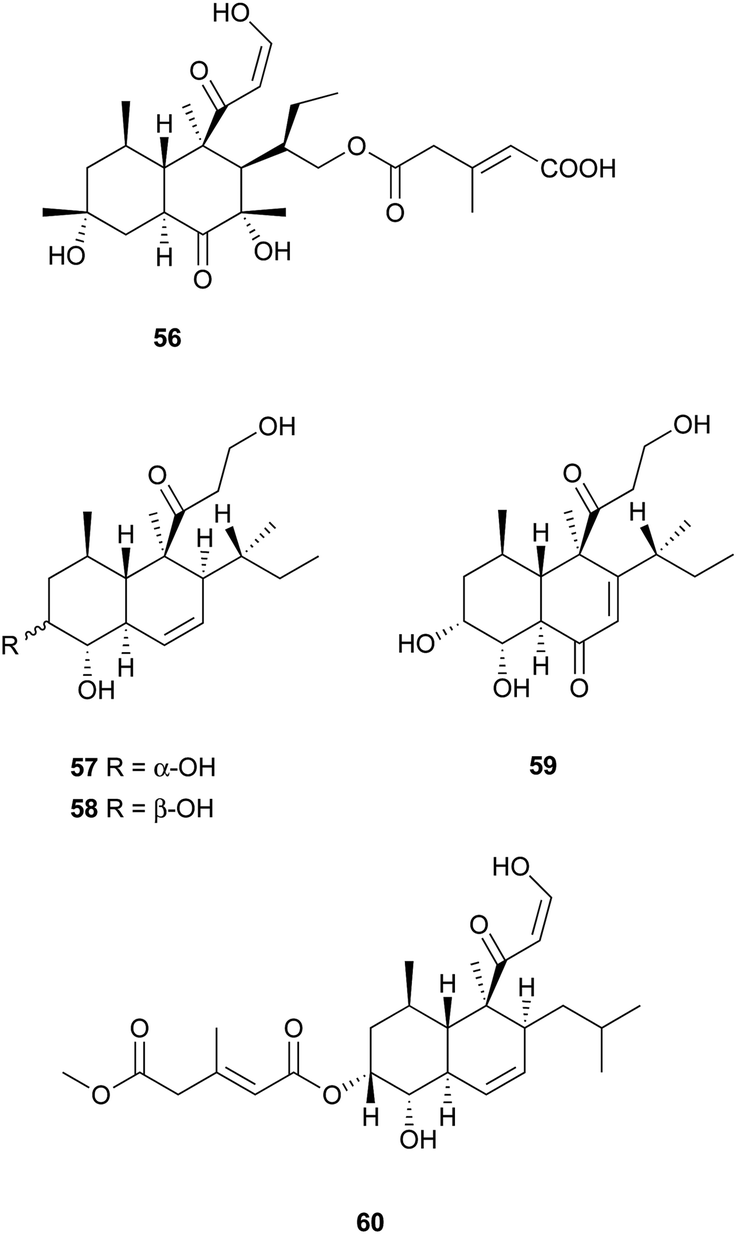
Aspermytin A (55), a heptaketide with a trans-decalin framework, was reported from a cultured marine fungus, Aspergillus sp., inhabiting the mussel Mytilus edulis.54 It induced significant neurite outgrowth in rat pheochromocytoma (PC-12) cells at concentration of 50 μM. FR225654 (56), a novel gluconeogenesis inhibitor was isolated from the culture of Phoma sp.55 Eujavanicols A–C (57–59) were purified from an antifungal-active fraction of E. javanicum, but showed no antifungal activity.56 More recently, Tandyukisin (60), a novel decalin derivative with an enolic β-ketoaldehyde, was produced by a marine sponge-derived fungus, T. harzianum.57 It exhibited moderate cytotoxic activity against the murine P388 leukemia, the human HL-60 leukemia, and the murine L1210 leukemia cell lines with IC50 values ranging from 41 to 55 mM.
3.4 Side chains with a pentanedienoic acid
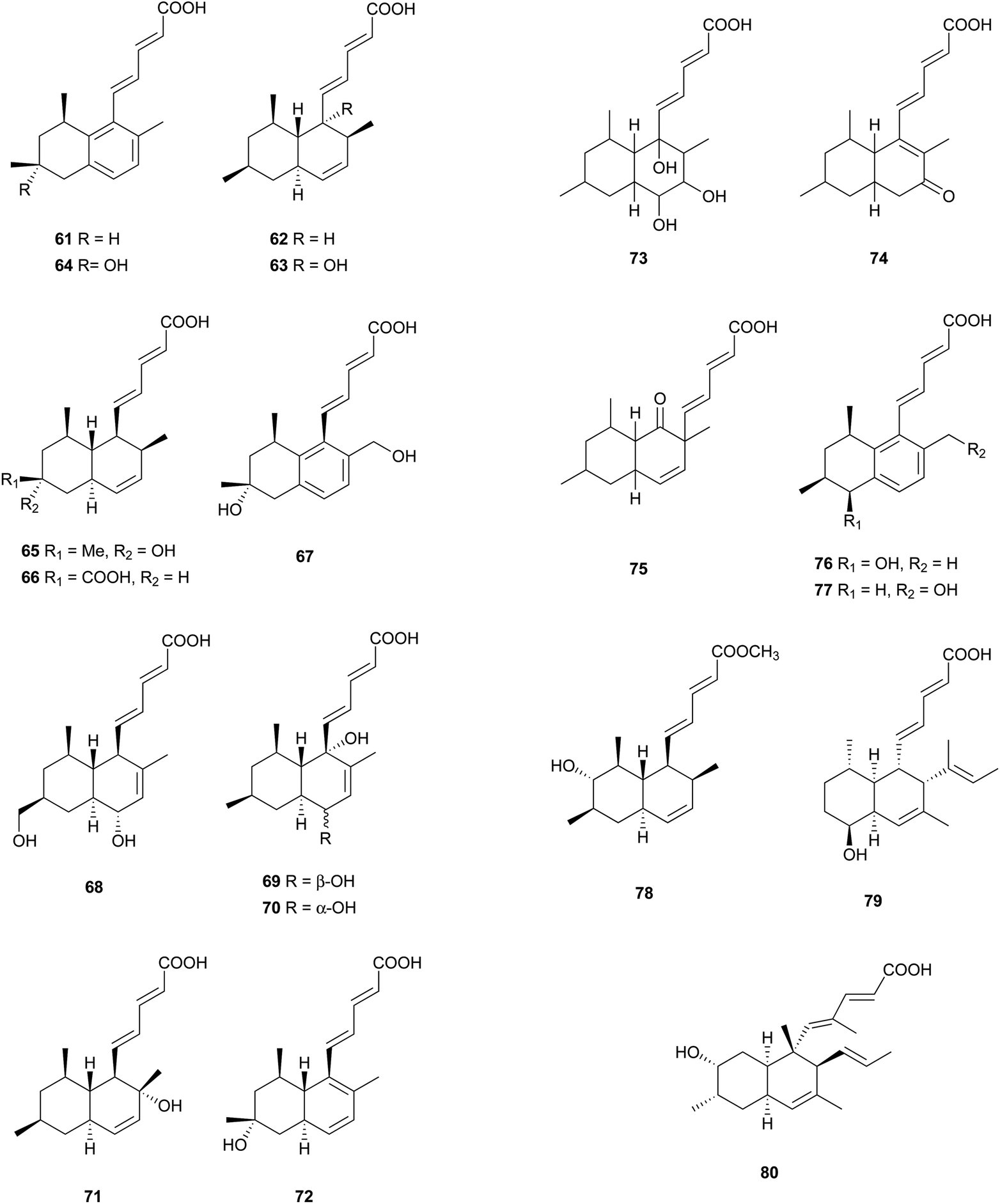
Most of these carboxylic acids have been detected in the cultures of Penicillium species, and possess a penta-2,4-dienoic acid unit connected to a trans-decalin, showing a plethora of interesting activities. A few cis-decalin skeletons can also be found, which seems to play an important role for their relevant biological activities.Tanzawaic acids A–D (61–64) from Penicillium citrinum,58 tanzawaic acids E (65) and F (66) from a marine-derived strain of Penicillium steckii,59 tanzawaic acids G (67) and H (68) from an endophytic fungus P. citrinum,60 and tanzawaic acids I–L (69–72) from a soil derived Penicillium sp.61 are representative trans-decalin pentanedienoic acids. Among metabolites 61–64, only 62 significantly inhibited superoxide anion production in human neutrophils (IC50 = 26 μg mL−1).58 Acids 67 and 68 exhibited no cytotoxicity on the growth of the L5178Y mouse lymphoma cell line (IC50 > 10 μM) and no antimicrobial activity against Staphylococcus aureus, Streptococcus pneumoniae, and Escherichia coli at a concentration of 64 μg mL−1.60 Compounds 61, 65, and 71 showed inhibition of the conidial germination in the rice blast fungus Magnaporthe oryzae at concentrations of around 25 μg mL−1.61 Ōmura and co-workers reported five anticoccidial agents: hynapenes A–C (73–75), from a soil-inhabiting Penicillium sp. that were effective against monensin-resistant Eimeria tenella,62 and arohynapenes A (76) and B (77) from a water-inhabiting Penicillium sp.63 Bioassay-guided fractionation led to the isolation of coprophilin (78), a trans-decalinpentanoic acid methyl ester from an unidentified fungus.6478, as a anticoccidial agent, inhibited the growth of E. tenella with an MIC value of 1.5 μM, compared to that of 123 μM for 73, 34.7 μM for 74 and 75, 35 μM for 76, and 7.0 μM for 77.62,63 Phomopsidin (79) with a cis-decalin core, was obtained from Phomopsis sp., and is a new inhibitor of the assembly of microtubule proteins at an IC50 of 5.7 μM.65 Using 13C-labeled precursors, 79 was proposed to be a trimethylated nonaketide (Fig. 2).66 Biological tests for many structurally related compounds 61–64 suggested that the cis-decalin structure of 79 is important for the anti-microtubule activity.66 In antisense-based screening strategies, pannomycin (80) was isolated from Geomyces pannorum.67 It exhibited weak antibacterial activity.
 | ||
| Fig. 2 Biosynthetic origins of carbons in 79. | ||
3.5 Decalins with oxygenated diene/triene/tetraene side chains
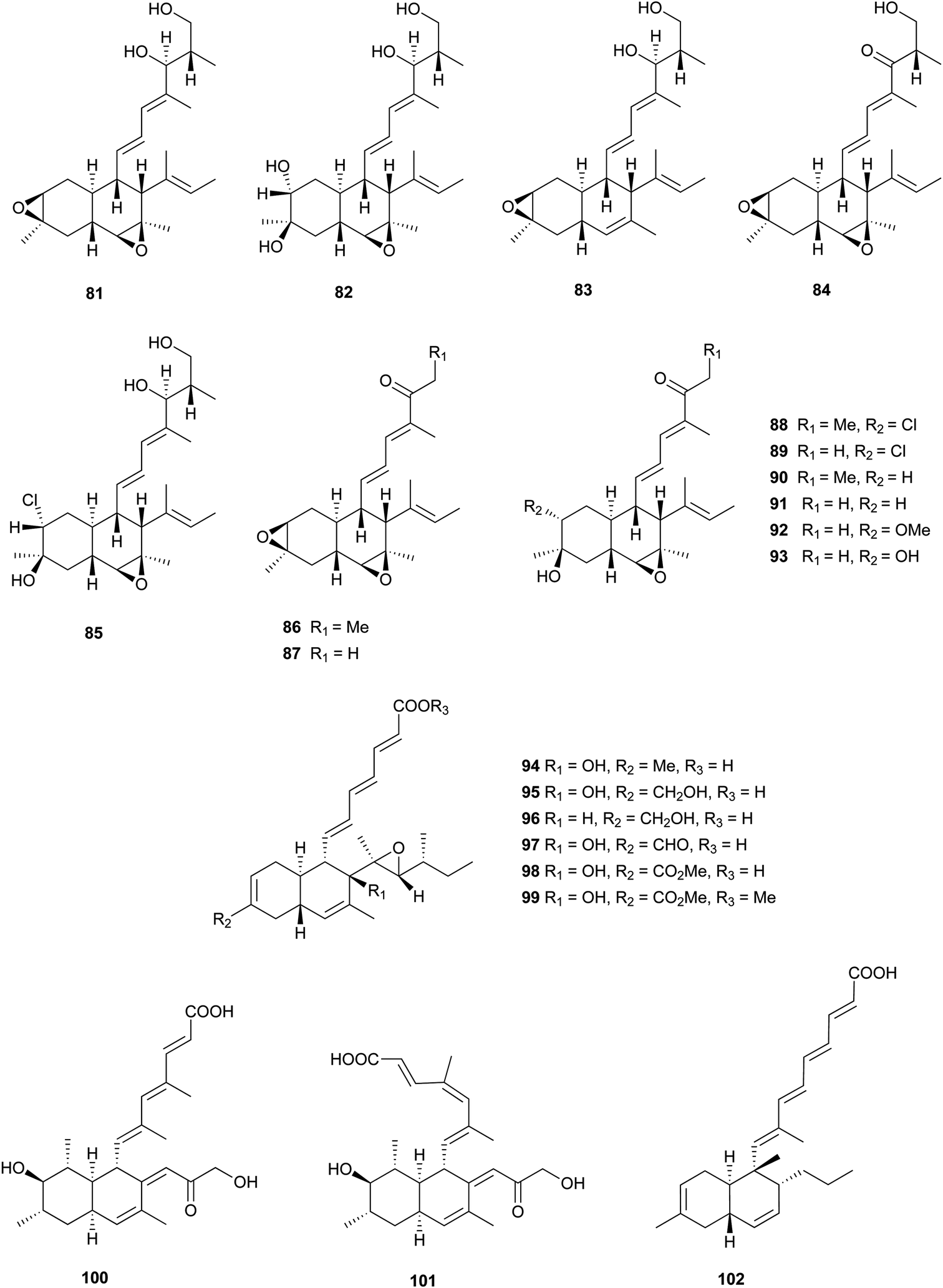
These kinds of decalin derivatives are highly methylated and oxygenated polyketides, and typically have a 6-, 7-, or 9-membered side chain similar to pentanedienoic acids.A weakly antifungal polyketide, fusarielin A (81), and three biosynthetically related fusarielins B–D (82–84), were isolated from Fusarium sp.68 Dehydroxychlorofusarielin B (85) is a mild antibacterial compound isolated from the marine-derived fungus Aspergillus sp.69 In addition, 81 and 82 were also isolated from this fungus and demonstrated weak antibacterial activity. ICM0301A-H (86–93) were inhibitors of angiogenesis in human umbilical vein endothelial cells (HUVECs) with IC50 values of 2.2–9.3 μg mL−1.70 Six fungal-derived polyketides, cladobotric acids A–F (94–99), were isolated from a Cladobotryum species from New Zealand.71 All of them showed modest growth inhibition against the murine P388 leukemia cell line with IC50 values of 6.6, 27.8, 19.4, 24.9, 1.4, and 15.6 μM, respectively, and were even active against Bacillus subtilis and Candida albicans. Furthermore, compound 98 was active against Trichophyton mentagrophytes and Cladosporium resinae. Feeding experiments with 13C-labeled precursors disclosed their polyketide biosynthesis from 11 intact C2 units (Fig. 3).69,71 Antifungal bioassay-guided isolation yielded two new cis-decalin derivatives, 100 and 101, from an endophytic Penicillium sp. isolated from the inner bark of the Pacific yew tree, Taxus brevifolia.72 Both were selectively active against the plant pathogen Sclerotinia sclerotiorum (17 mm zone at 1.1 × 10−4 μmol/disk for 100; 16 mm zone at 3.4 × 10−4 μmol/disk for 101). In a screening program for potent compounds inducing osteoblast differentiation in C3H10T1/2 cells, decalpenic acid (102) bearing a tetraenoic acid side chain was isolated from Penicillium verruculosum.73 Through activation of retinoic acid receptor γ, it induces early osteoblastic markers in pluripotent mesenchymal cells.74
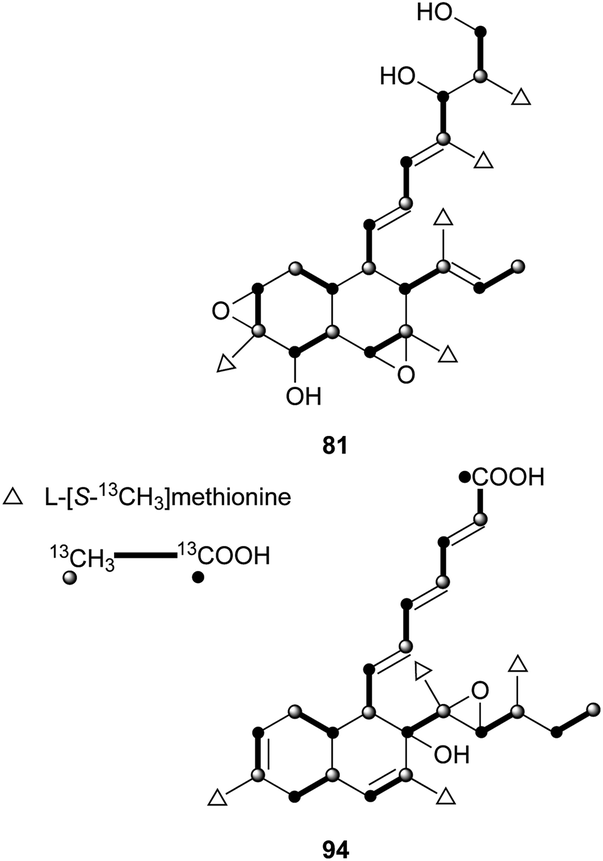 | ||
| Fig. 3 Biosynthesis of 81 and 94 on the basis of the data from feeding experiments. | ||
3.6 Pyrone derivatives
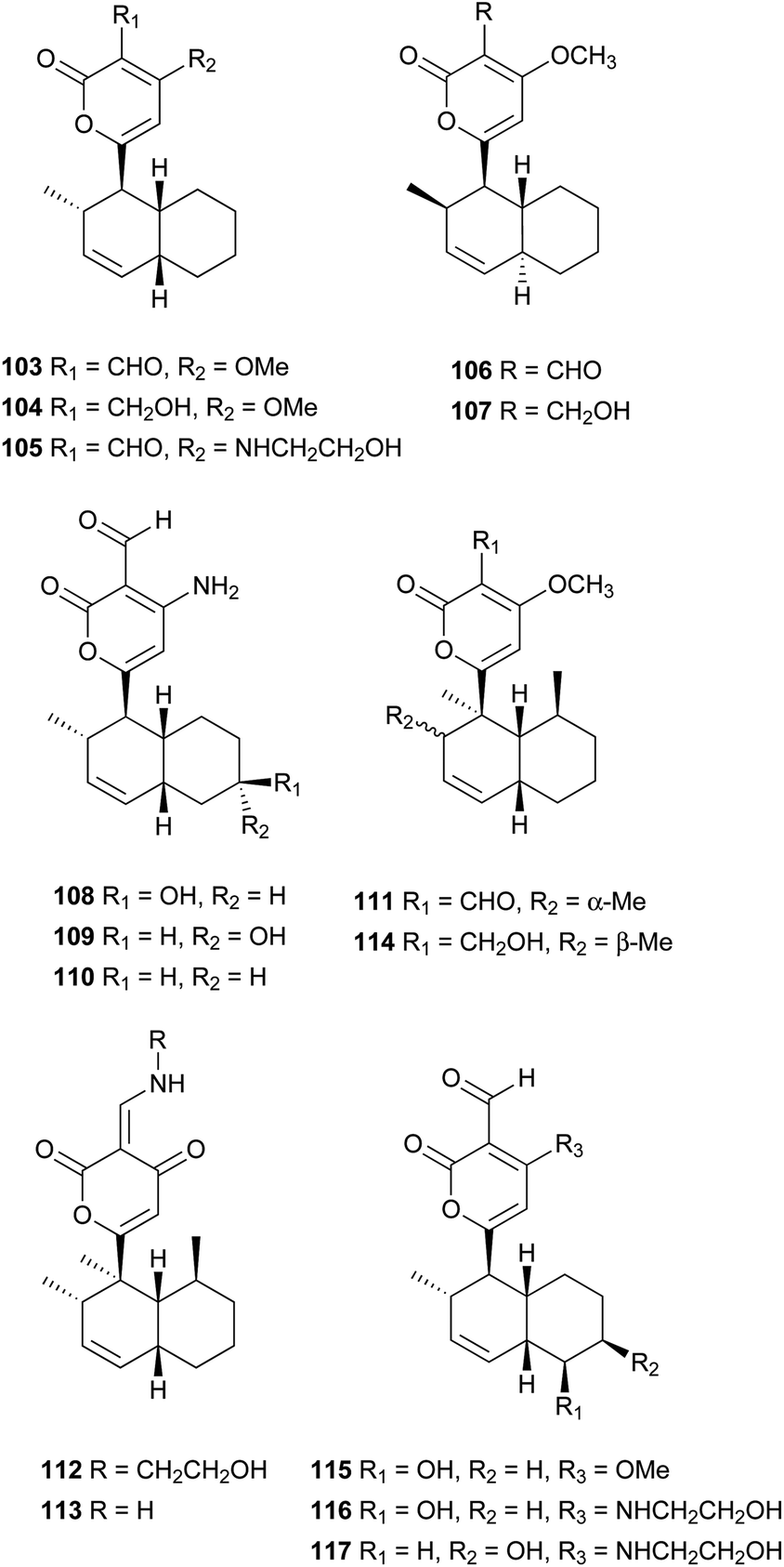
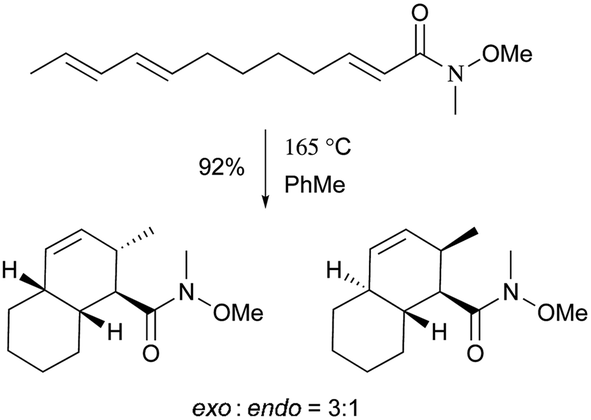
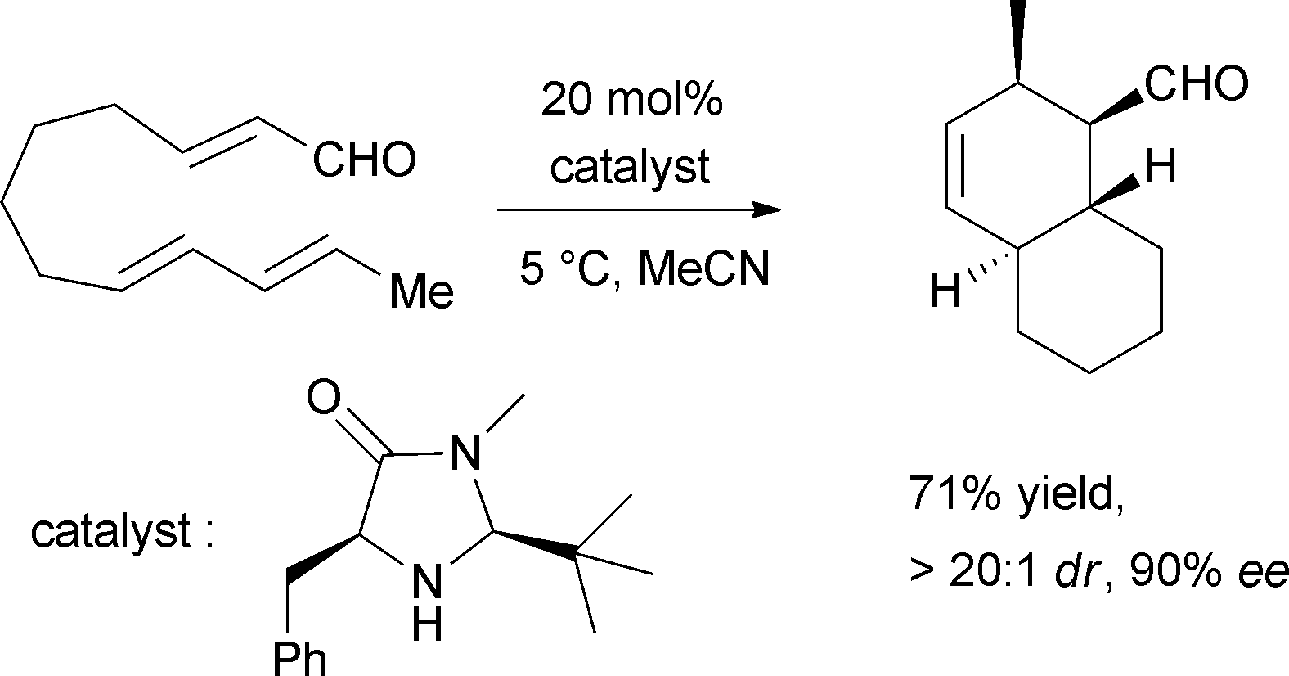
Pyrone derivatives are formed when the pyrone ring is substituted by the methoxyl, aldehyde, hydroxymethyl, amino or ethanolamide group and connected to the decalin framework directly via carbon–carbon bond. Most of these decalin derivatives possess the cis-decalin system. The modified decalin motif with two types of relative configurations has been proposed to be formed via a biological intramolecular Diels–Alder reaction to give exo or endo products.75,76Solanapyrones A–E (103–107), the phytotoxins, were isolated from the fungus Alternaria solani which causes early blight disease in tomato and potato plants.77–79 Phytotoxin 103 was reported to be an inhibitor of DNA polymerase β and γ, with IC50 values of 30 and 37 μM, respectively.78 The biosynthetic pathway for 103 was investigated by feeding experiments and gene expression studies (Scheme 4).10,76,79 The decalin scaffold of 106 and 107 was first synthesized by a domino Michael reaction (Scheme 5).80 In order to complete the total synthesis of 103 and 104, Lygo et al. reported an optimized IMDA cycloaddition to the decalin fragment (Scheme 6).81 MacMillan and co-workers developed a new powerful organocatalytic IMDA reaction protocol to achieve the asymmetric synthesis of 106 (Scheme 7).82 Chemical investigation of an unidentified filamentous marine fungus led to the isolation of new phytotoxic compounds, solanapyrones E–G (108–110) and the known compound 105.83 Compound 108, with a cis-decalin unit, has the same compound name as the trans-decalin derivative 107. Solanapyrones J–M (111–114), new solanapyrone analogues with modest antifungal and antibacterial activities, were obtained from an unidentified fungicolous fungus.84 Nigrosporapyrones A–C (115–117) were isolated from the marine-derived fungus Nigrospora.85 Compound 115 exhibited a weak antibacterial activity with an MIC value of 128 μg mL−1.
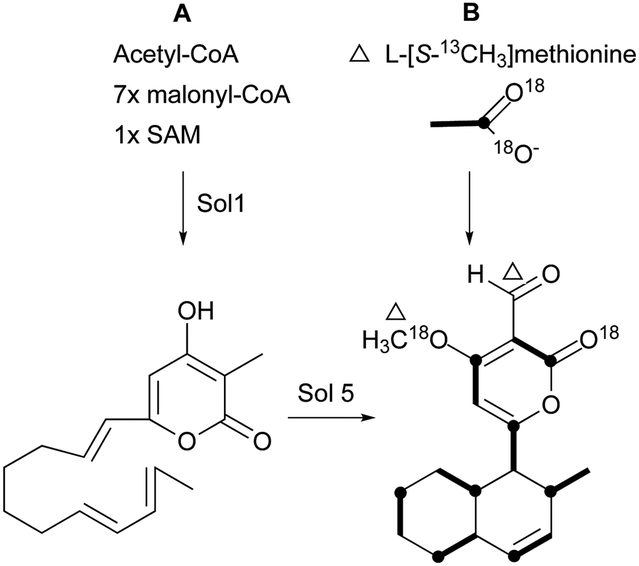 | ||
| Scheme 4 (A) The prosolanapyrone synthase (PSS) encoded by Sol1 catalyzes octaketide prosolanpyrone formation and then a flavin-dependent oxidase (solanapyrone synthase, SPS) encoded by Sol5 catalyzes Diels–Alder cyclization to form 103. (B) Incorporation of labeled acetate and methionine into 103. | ||
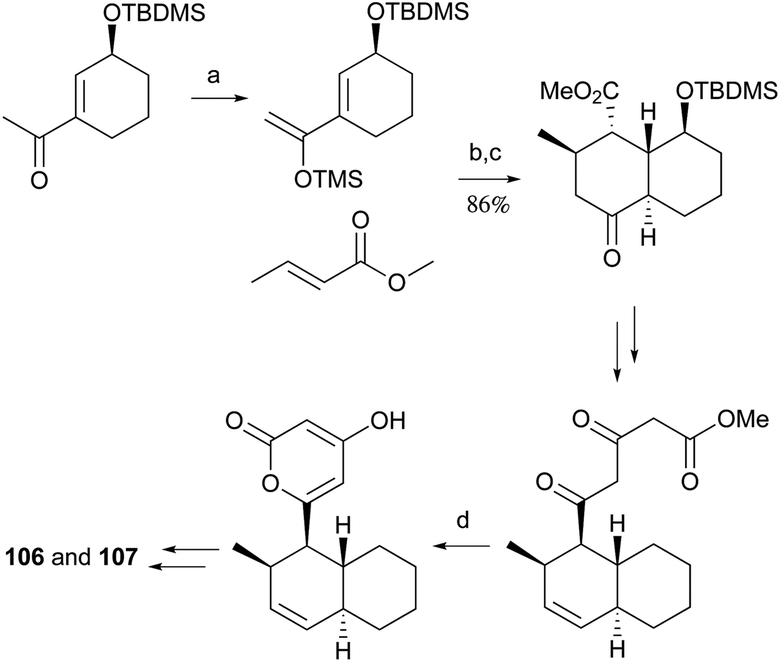 | ||
| Scheme 5 The first total syntheses of 106 and 107: the domino Michael reaction to decalone and the general biosynthetic strategy of pyrone. (a) LDA, TMSCl, 93%; (b) MeLi, methyl crotonate, HMPA, THF; (c) MeONa, MeOH; (d) DBU, benzene, 93%. | ||
 | ||
| Scheme 6 The exo-selective thermal Diels–Alder reaction used for the total syntheses of 103 and 104. | ||
 | ||
| Scheme 7 MacMillan's effective organocatalytic IMDA reaction to the decalin scaffold in the total synthesis of 106. | ||
3.7 Macrolides
Nodusmicin (118) and nargenicin (119) represent a class of macrolide antibiotics that were isolated from Saccharopolyspora hirsuta by Whaley et al. in 198086 and from Nocardia argentinensis by Celmer et al. in the same year, respectively.87 They possess a ten-membered lactone ring fused to an oxygen-bridged cis-decalin system. The acetylated products, 18-O-acetylnodusmicin (120) and 18-O-acetylnargenicin (121), were also isolated and identified from N. argentinensis.88 A more complex analog, coloradocin (122) (luminamicin) with an additional 14-membered macrolactone containing an enol ether in conjugation with an unsaturated cyclic anhydride functionality, was found from the actinomycete strains, Actinoplanes coloradoensis and Nocardioides sp.89 Compound 122 showed selective activity against anaerobic and microaerophilic bacteria whereas compounds 118 and 119 inhibited some aerobic bacteria, thereby suggesting the importance of the additive macrocyclic moiety.90 The biosynthetic origin of this family was investigated through feeding experiments, as shown for compound 122 in Fig. 4.88,91 Compared with the assembly of a polyketide chain in compounds 118–121, 122 also involves an acetate and a succinate unit incorporated into the additional 14-membered macrolactone.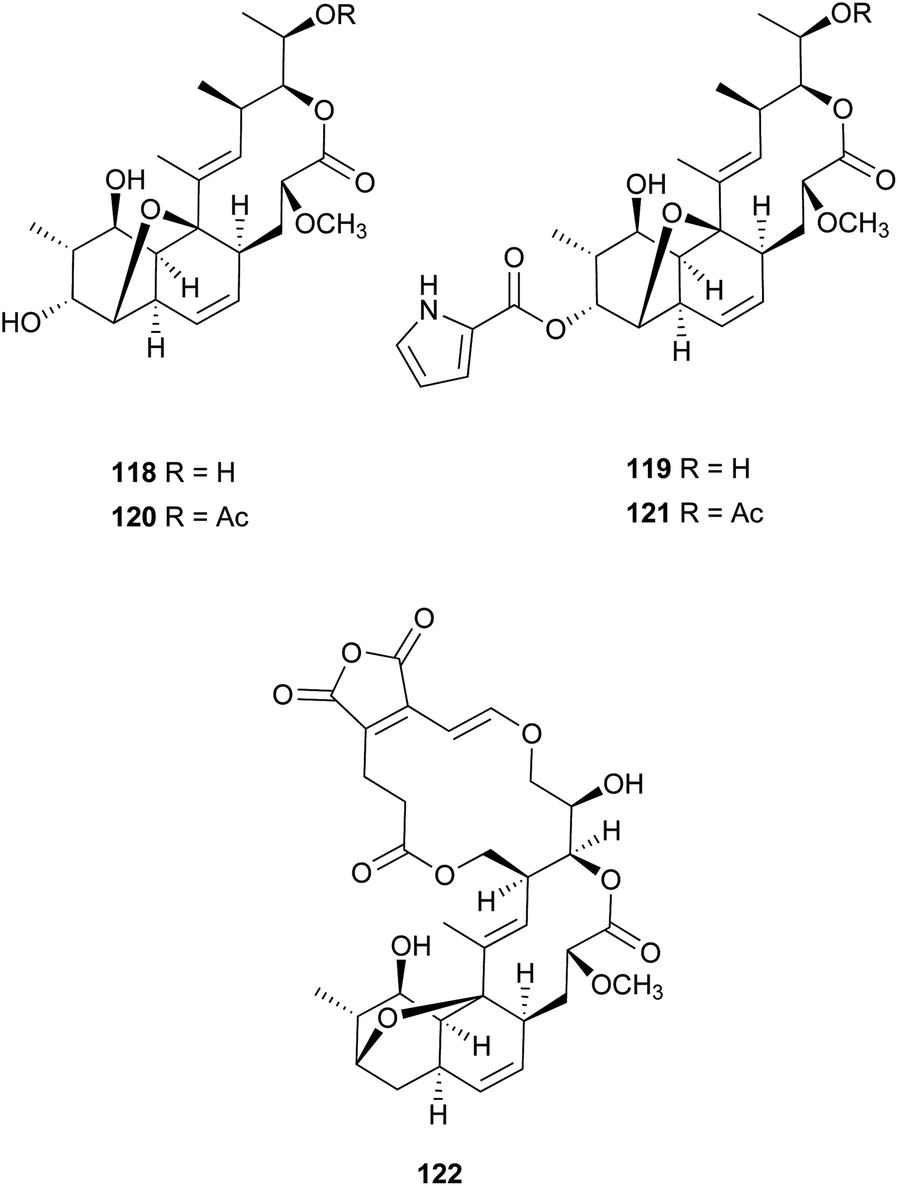
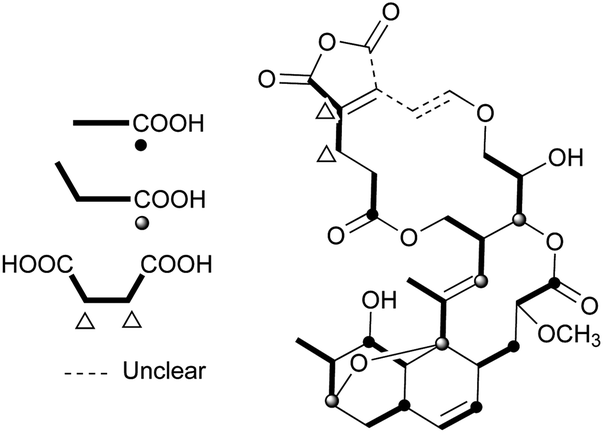 | ||
| Fig. 4 Biosynthetic origin of 122 on the basis of the data from feeding experiments. | ||
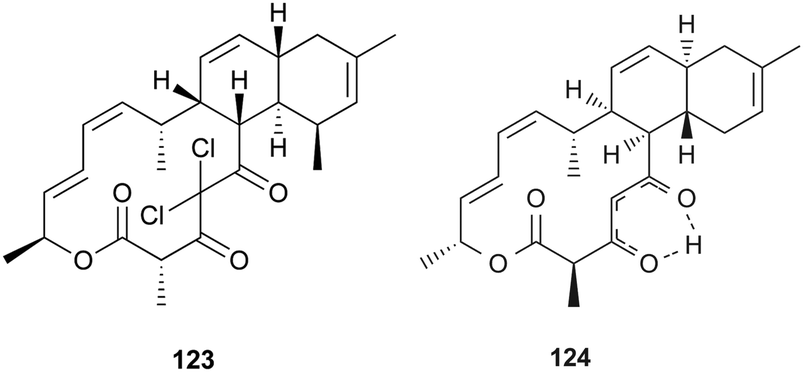
In a broad screening program for active secondary metabolites produced by myxobacteria, Sorangium cellulosum was found to produce a novel polyketide, chlorotonil A (123), with a unique gem-dichloro-1,3-dione functionality in a 14-membered macrolide ring fused to an unsaturated trans-decalin.92 The total synthesis of 123 was reported by Rahn and Kalesse in the same year.93 The decalin system was prepared by a highly stereoselective halogen (bromine)-directed Diels–Alder reaction over standard Diels–Alder routes (Scheme 8). More recently, antibacterial activity-guided fractionation yielded the antibiotic anthracimycin (124) from a Streptomyces species isolated from near-shore marine sediments by the group of Fenical.94 It exhibited significant inhibition of Gram-positive pathogens such as Bacillus anthracis and clinically-relevant methicillin-resistant S. aureus (MRSA) with MIC values of 0.031 and 0.06 μg mL−1, respectively. The semisynthetic derivative, dichloro-anthracimycin has similar activity against Gram-positive pathogenic bacteria as 124, but is active against Gram-negative pathogens. However, 124 lacked activity against Gram-negative pathogens or was weakly active against them. The biosynthesis of compounds 123 and 124 could be proposed via a polyketide pathway with 11 acetate units.
 | ||
| Scheme 8 Synthesis of the decalin system in compound 123. A) Highly stereoselective halogen (bromine)-directed Diels–Alder reaction. B) The same reaction condition without bromine substituent. | ||
3.8 Pyrrolidine-2-one
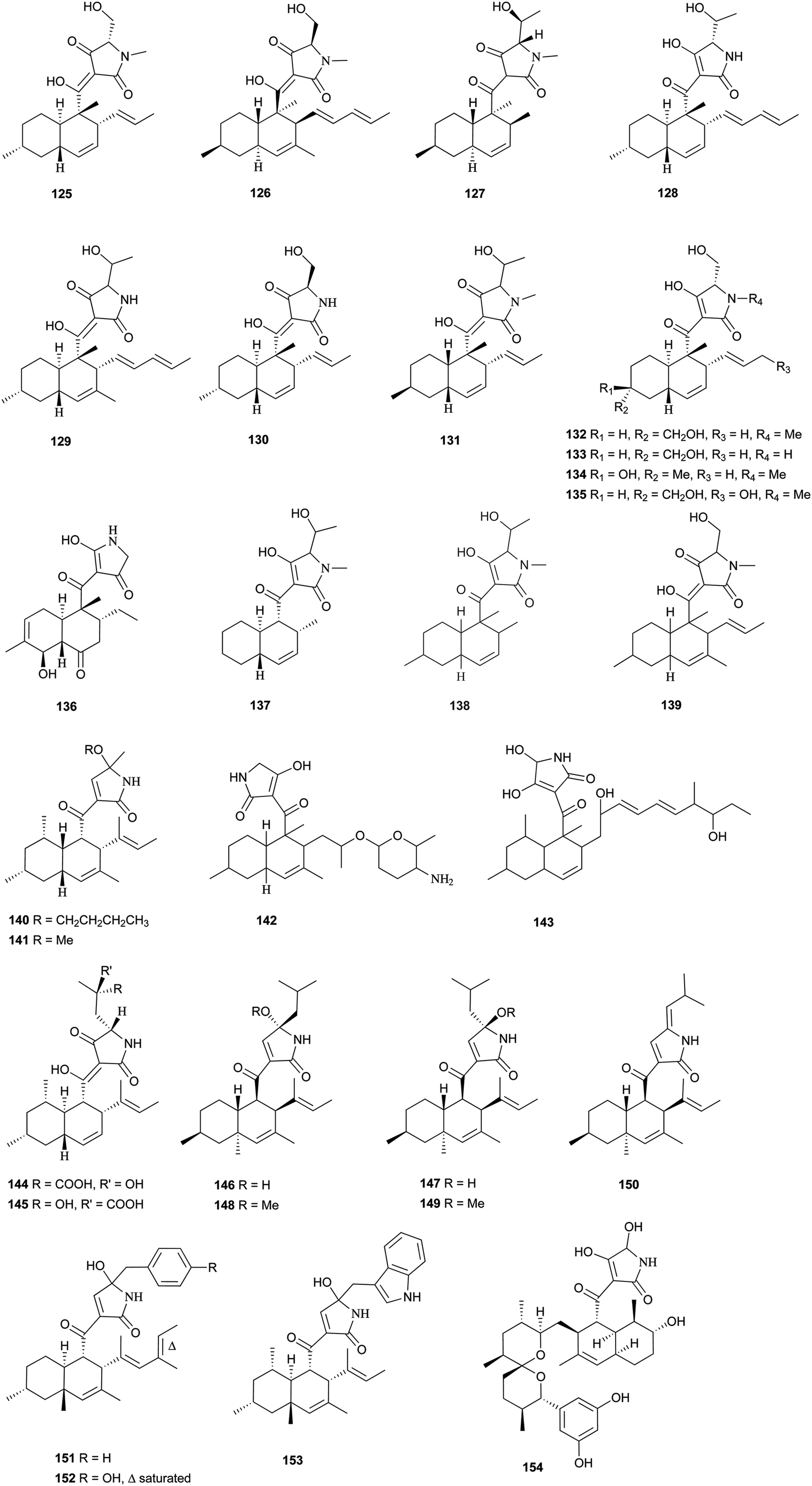
Pyrrolidine-2-one, an important biologically active moiety, is often found in fungal metabolites arising from the mixed PKS-nonribosomal peptide synthetase (NRPS) pathway and has been reported in several reviews.95–97 However, to the best of our knowledge, there is a dearth of information on pyrrolidine-2-one metabolites. Here, we highlight all those compounds that preferentially cooperate with a decalin system. Decalin-type pyrrolidine-2-one derivatives are comprised of two important biosynthetic units. The first one is a linear C14, C16, C18 or more Cn-fragment(s) with several methyl substituents. This linear polyketide unit is then cyclized by a biological intramolecular Diels–Alder cycloaddition to form the decalin scaffold. The second one is comprised of amino acids serine, threonine, leucine, phenylalanine, tyrosine or tryptophan-derived heterocyclic ring (pyrrolidine-2-one) joined to the decalin system. Typically, as a character of this family, this ring is tetramic acid that may also undergo a series of tautomeric shifts to form its derivatives, for example, having a reduced carbonyl carbon. Unfortunately, the exact ‘timing’ of N-methylation for this kind of compound remains unclear. These compounds have significant inhibitory activities against Gram-positive bacteria.Equisetin (125), showing considerable biological activity in an array of assays and inhibiting HIV-1 integrase,96 was first isolated in 1974 from Fusarium equiseti by Burmeister et al.98,99 It comprises a substituted decalin system bearing a quaternary stereogenic center and an N-methylserine-derived heterocycle, tetramic acid. The proposed biosynthesis of 125 catalyzed by a PKS/NRPS hybrid is shown in Scheme 9.100–102 The total synthesis has already been reported by the groups of Danishefsky,103 Shishido,104 and Dixon.105 More recently, Gao and co-workers reported synthetic studies of 125 and the biosynthetically related (+)-fusarisetin A based on the proposed biosynthetic hypothesis (Scheme 10).106 A cyclization sequence involving an intermolecular Diels–Alder reaction followed by a Dieckmann cyclization yielded 125 (Scheme 11). Compound 125 and a new enantiomer, phomasetin (126), were isolated from Fusarium heterosporum and Phoma sp., respectively.107 They are almost equally active in inhibiting recombinant integrase enzyme in vitro with IC50 values of 7–20 μM. Cryptocin (127), showing activity against a wide variety of plant pathogenic fungi (MIC of 0.78–1.56 μg mL−1) but not against human pathogenic fungi, was isolated from an endophytic fungus Cryptosporiopsis cf. quercina arising from the stems of Triptergyium wilfordii.108 Two endophytic Alternaria species produce another antibiotic called altersetin (128).109 It significantly inhibited several pathogenic Gram-positive bacteria with MIC values of no more than 1 μg mL−1, but has no or much less effect on Gram-negative bacteria and pathogenic yeast. Similarly, Coniochaeta ellipsoidea produces a tetramic acid, coniosetin (129), with strong efficacy against Gram-positive bacteria and sensitive against resistant microbial pathogens, especially the multi-drug-resistant S. aureus at a concentration of 0.3 μg mL−1.110 From the co-culture of T. harzianum and Catharanthus roseus, an equisetin derivative without the N-methyl, named trichosetin (130), was isolated.111 Interestingly, this compound was not detected in the individual cultures. Cissetin (131) with a cis-decalin ring fusion exhibited similar antibiotic activities as those of compounds 125 and 130 containing a different trans-decalin system, suggesting the possible biological function of tetramic acid.112 Ophiosetin (132) was isolated from the mycopathogenic fungus Elaphocordyceps ophioglossoides.113 Co-culture of the fungus Fusarium pallidoroseum with the bacterium Saccharopolyspora erythraea afforded N-demethylophiosetin (133) and pallidorosetins A and B (134, 135), along with compounds 125 and 132.114 Compound 125 exhibited a GI50 of 144 nM against the leukemia cell line CCRF-CEM. Following a histone deacetylase (HDAC)-based yeast screening method, streptosetin A (136) was purified and found to show weak inhibitory activity against yeast Sir2p and human SIRT1 and SIRT2.115 The structure of compound 136 was confirmed by X-ray crystal structure analysis and CD theoretical calculation. In an antibiotic screening program, methiosetin (137) was discovered as a modest antibacterial agent against S. aureus and Haemophilus influenzae.116 CJ-17,572 (138) and CJ-21,058 (139) were isolated from Pezicula sp. and an unidentified fungus, respectively.117,118 Two structurally unusual tetramic acids, ascosalipyrrolidinones A (140) and B (141), were obtained from an endophytic fungus, Ascochyta salicorniae isolated from the green alga Ulva sp.119 The antimicrobial, anti-algal, nematicidal, antiplasmodial, anti-trypanosomal and cytotoxic properties as well as brine shrimp lethality of 141 were assessed. BU-4514N (142) with significant NGF-mimic activity and antibacterial efficacy against Gram-positive bacteria, and delaminomycin A (143), a novel extracellular matrix receptor antagonist, have been isolated from the fermentation broth of Microtetraspora sp.120 and Streptomyces albulus,121 respectively. Sch 210971 (144) and its epimer, Sch 210972 (145), showed potent inhibitory activity (IC50 of 1.2 μM and 79 nM, respectively) in the chemokine receptor CCR-5 in vitro binding assay, indicating an interesting structure–activity relationship of the relative configuration at tetramic acid.122 Myceliothermophins A–E (146–150) (Fig. 5) (see Supporting Information of ref. 123) are the decalin polyketides containing tetramic acid moieties which were isolated from a fungus Myceliophthora thermophila.123 Only compounds 146, 148, and 150 exhibited inhibitions against four cancer cell lines, A549, Hep3B, MCF-7, and HepG2, indicating the importance of the relative configuration of tetramic acids. Oteromycin (151)124 and ZG-1494α (152),125 two phenylalanine-derived tetramic acids, were isolated from an unidentified fungus and Penicillium rubrum, respectively. Compound 151 was reported to be a novel antagonist of endothelin receptor while 152 was an inhibitor of platelet-activating factor acetyltransferase. An endophytic fungus, Codinaeopsis gonytrichoides, produced an antimalarial compound that is active against Plasmodium falciparum with an IC50 of 4.7 μM.126 This metabolite, with its unusual heterocyclic unit linking indole and decalin fragments, was named codinaeopsin (153) and was proposed to be biosynthesized by the PKS-NRPS hybrid involving an IMDA-like addition (Scheme 12).126 A hexacyclic secondary metabolite, integramycin (154) isolated from Actinoplanes sp., exhibited an IC50 value of 4 μM against HIV-1 integrase.127
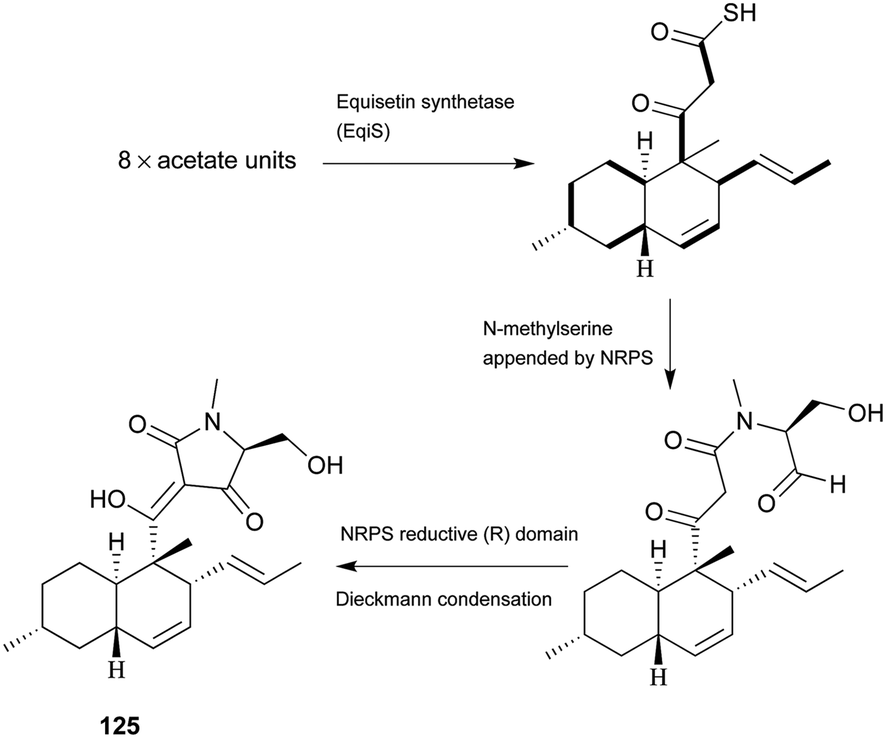 | ||
| Scheme 9 The biosynthetic origin of 125. Bold bonds in the polyketide portion represent 13C-labelled acetate units. The timing of N-methylation is unclear. | ||
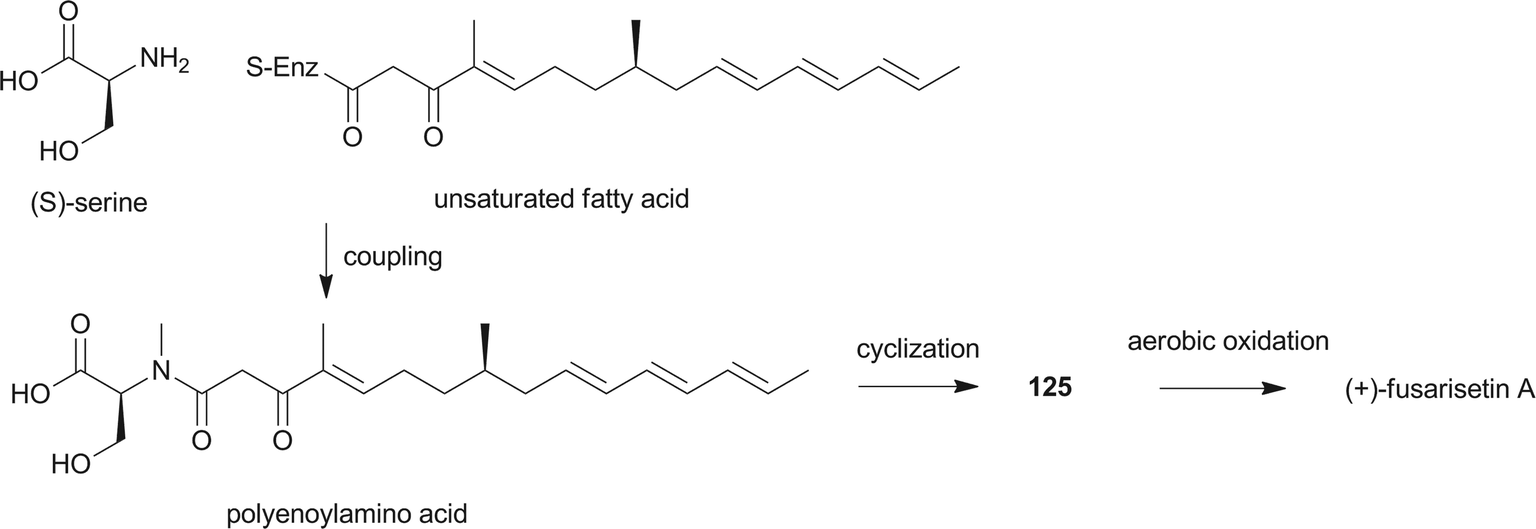 | ||
| Scheme 10 Biosynthetic hypothesis for the total syntheses of 125 and (+)-fusarisetin A. | ||
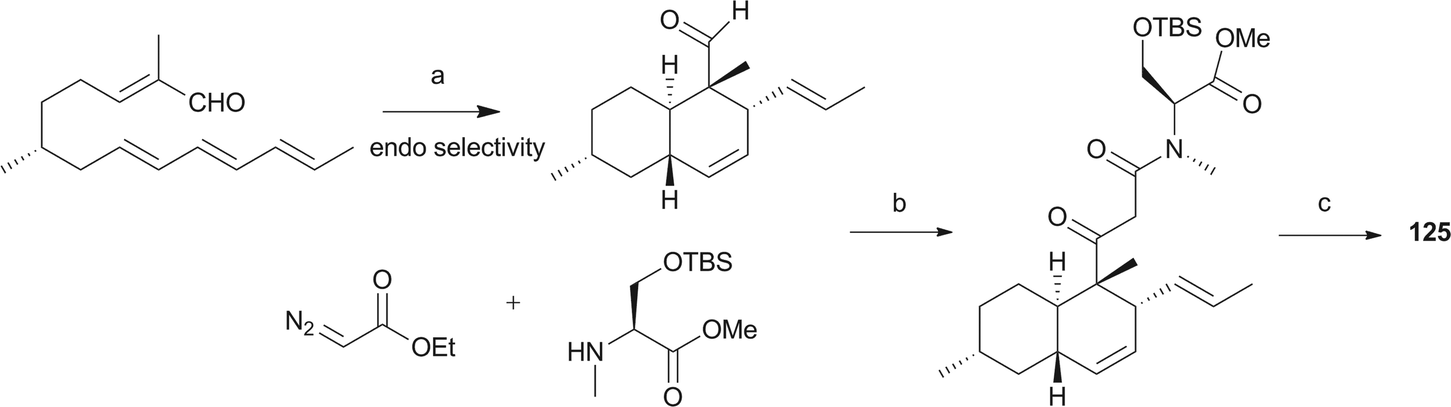 | ||
Scheme 11 Key construction of decalin ring system and the preparation of 125. (a) BF3·Et2O, −78 °C, CH2Cl2, 20 min; (b) 0 °C, 1.5 h; then (S)-serine derivative, DMAP, toluene, 118 °C, 18 h; (c) NaOMe, MeOH, 25 °C, 2 h, 72%, d.r. = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (at C3); then HF, CH3CN, 25 °C, 2 h, 95%. :1 (at C3); then HF, CH3CN, 25 °C, 2 h, 95%. | ||
 | ||
| Fig. 5 Biosynthetic origin of myceliothermophins A-D (146–149). | ||
 | ||
| Scheme 12 Proposed biosynthesis of 153 and the labeling patterns observed in feeding experiments. | ||
Lydicamycin (155) with a long side chain proved to be a potent antibiotic against Gram-positive bacteria such as B. subtilis and S. aureus with MIC values of 1.5 and 6.2 μg mL−1, respectively.128,129 It was isolated from the culture broth of an actinomycete, Streptomyces lydicus. From another actinomycete, Streptomyces platensis, four novel congeners, TPU-0037-A–D (156–159), were obtained.130 They also exhibited inhibition of some Gram-positive bacteria with MIC values of 1.56–12.5 μg mL−1.
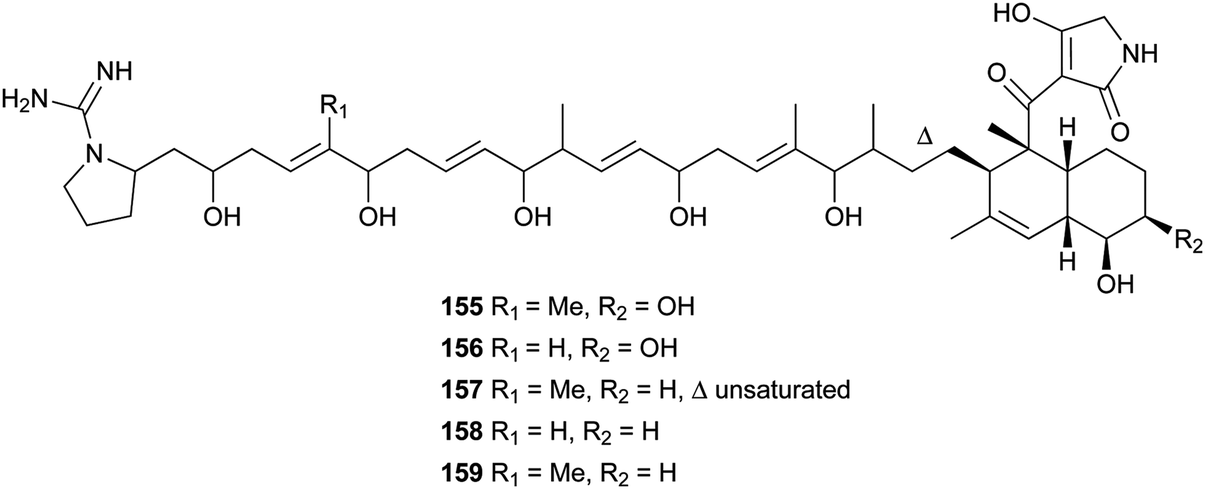
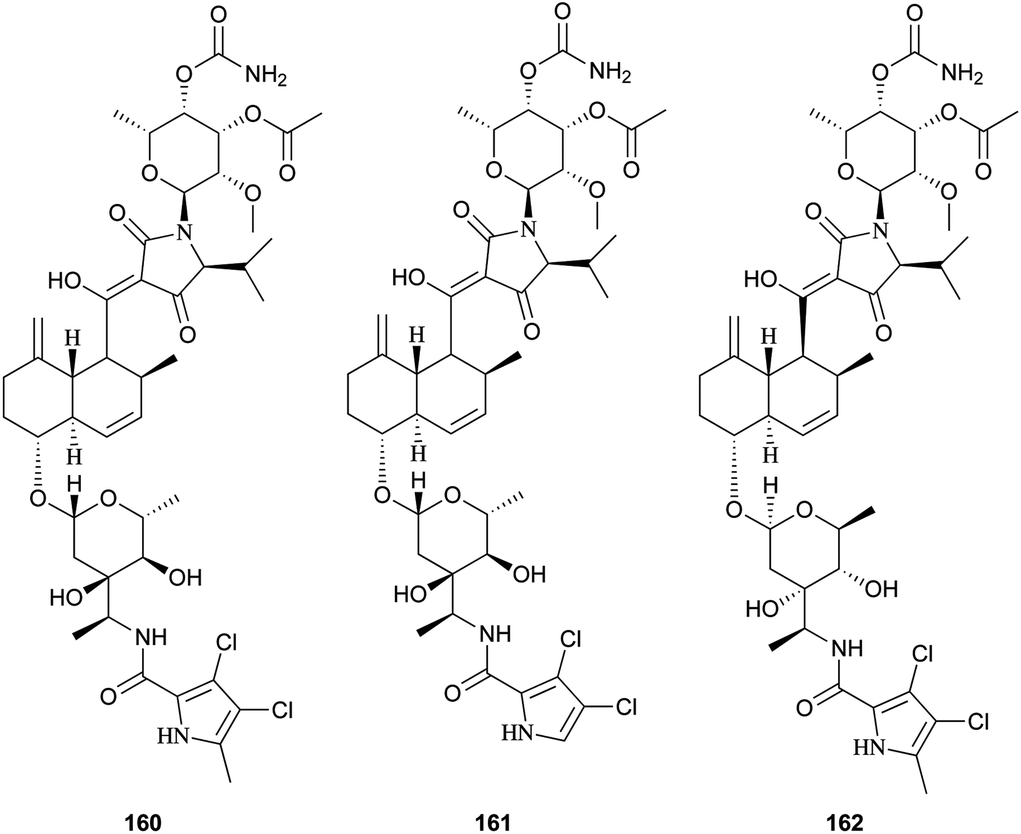
Recently, three novel broad-spectrum antibiotics (160–162) active against Gram-positive bacteria by inhibiting DNA gyrase and bacterial topoisomerase IV were reported.131–133 They consist of a trans-decalin, tetramic acid, two unusual sugars, and dichloropyrrole carboxylic acid. Kibdelomycin (160) and its demethylated congener kibdelomycin A (161) were isolated from a previously undescribed strain of Kibdelosporangium by Singh and co-workers.131 Compound 160 also demonstrated potent and selective activity against toxigenic Clostridium difficile.132 Amycolamicin (162) (AMM) was obtained from the culture broth of the soil actinomycete, Amycolatopsis sp.133
3.9 4-Hydroxy-2-pyridone alkaloids
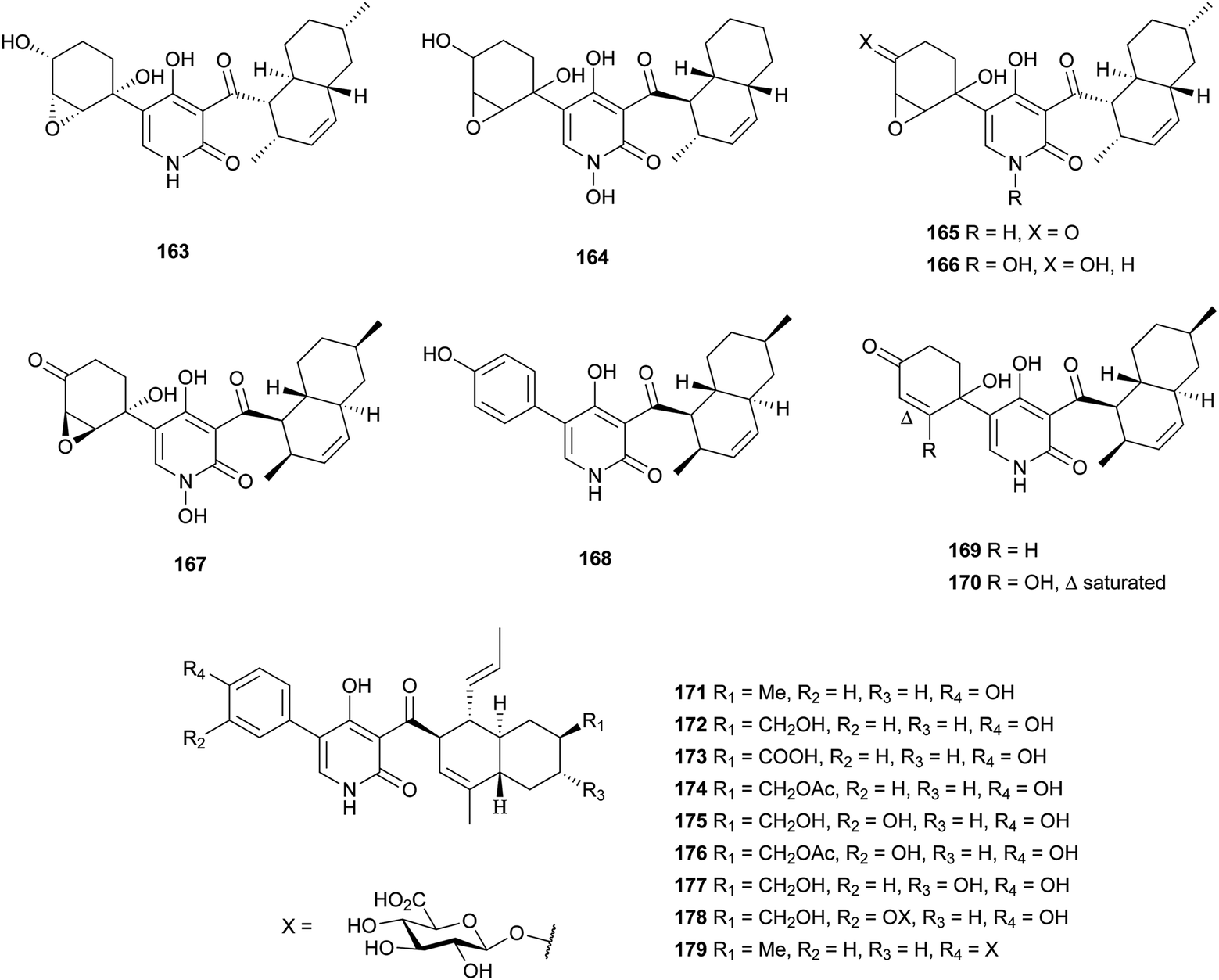
4-Hydroxy-2-pyridone alkaloids have attracted much attention in the scientific community owing to their diverse biological activities.134 A review focusing only on their structures and synthetic approaches has been published.134 Here, we mainly pay attention to the biological activities and biosynthesis of 4-hydroxy-2-pyridone alkaloids with a decalin scaffold, and include some recent discoveries. Their biosynthesis is related to that of polyketide tetramic acid, phenylalanine-derived heterocyclic ring (vide supra, Section 3.8). In their biosynthesis, the key rearrangement of the tetramic acid ring eventually results in the construction of the 4-hydroxy-2-pyridone.Chemical investigation of an EtOAc extract from the coprophilous fungus Apiospora montagnei led to the isolation of an antifungal metabolite named apiosporamide (163).135 Fischerin (164) was isolated from an ascomycete, Neosartorya fischeri. It was toxic to mice, causing lethal peritonitis.136 The biogenetic pathway of 164 related to tetramic acid has been studied (Scheme 13). This kind of biosynthesis related to tetramic acid derivatives was also supported by the group of Hertweck in their discovery of a silent PKS-NRPS hybrid gene cluster and the resulting new pyridine metabolites.137 YM-215343 (165), isolated from Phoma sp., showed antifungal activities against the pathogenic fungi C. albicans, Cryptococcus neoformans, and Aspergillus fumigatus (MIC values of 2–16 μg mL−1) and was cytotoxic against HeLa S3 cells (IC50 of 3.4 μg mL−1).138 An insect-associated fungus tentatively identified as Cytospora, was found to produce a cholesteryl ester transfer protein inhibitor 166 (IC50 of 40 μM).139 More recently, four new members of this family, didymellamides A–D (167–170) were isolated from the marine-derived fungus Stagonosporopsis cucurbitacearum growing on the surface of an unidentified sponge.140 Compound 167 showed antifungal activity against azole-resistant C. albicans with an MIC value of 3.1 μg mL−1.140 The antifungal antibiotic, ilicicolin H (171), was first isolated in 1971 from the imperfect fungus Cylindrocladium ilicicola harboring the dead leaf of beech (Fagus sp.).141 It was found to inhibit the yeast cytochrome bc1 complex by interacting with the Qn site of the complex.142 Using feeding experiments conducted with different labeled acetates and phenylalanine, biosynthesis of 171 was investigated and found to be similar to 164.143 Singh et al. worked on the biotransformation of compound 171 using Actinoplanes sp. and Streptomyces sp., and found eight new oxidized products 172–179.144 The predominant modification was selective oxidation of the methyl group on the decalin ring. Only compound 171 had significant antifungal activity against C. albicans with an MIC value of 8 ng mL−1. The total syntheses of compounds 163, 165, and 171 have been summarized in a recent review.134
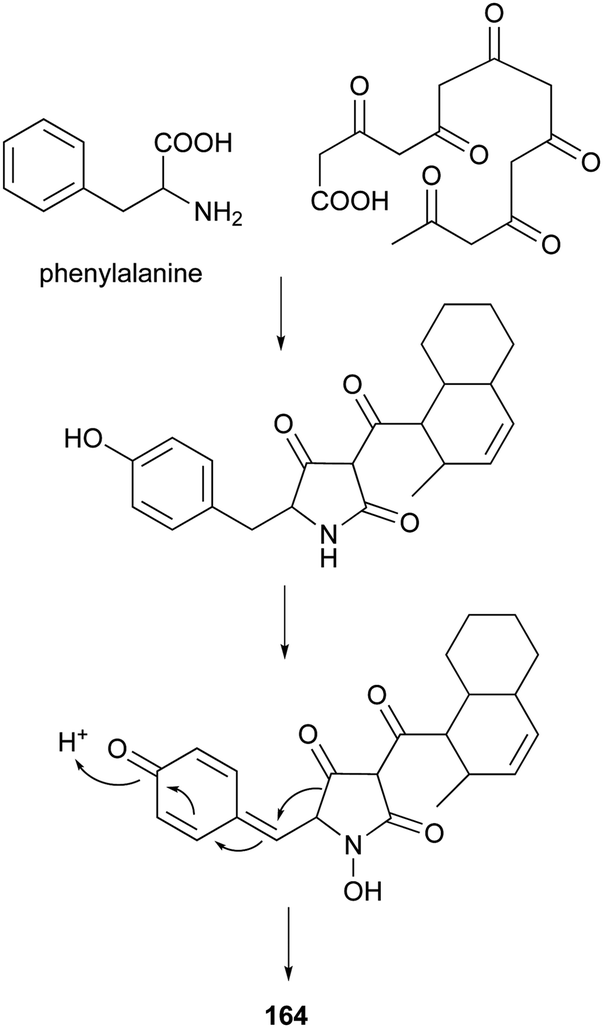 | ||
| Scheme 13 The proposed biogenesis of 164. | ||
3.10 Spirotetronates
Spirotetronates, the macrolide natural products consist of a trans-decalin system, a spiro ring between a cyclohexene and a tetronic acid, several similar or dissimilar deoxysugars and/or a multi-substituted benzene or pyrrole.Chlorothricin (180), the first reported member of spirotetronate family, was isolated from an actinomycete Streptomyces antibioticus in 1969 by Keller-Schierlein et al.145 Tetrocarcin A (181) was isolated in 1979 from the bacterium Micromonospora chalcea by Tomita et al.146 and was found to be identical to antlermicin A (also isolated from M. chalcea).147 Waitz et al. isolated kijanimicin (182) (or Sch 25663) in 1981 from a complex of antibiotics produced by a previously undescribed species of Actinomadura, A. kijaniata.148 Its structure was investigated and confirmed by Mallams et al. in the same year.149,150
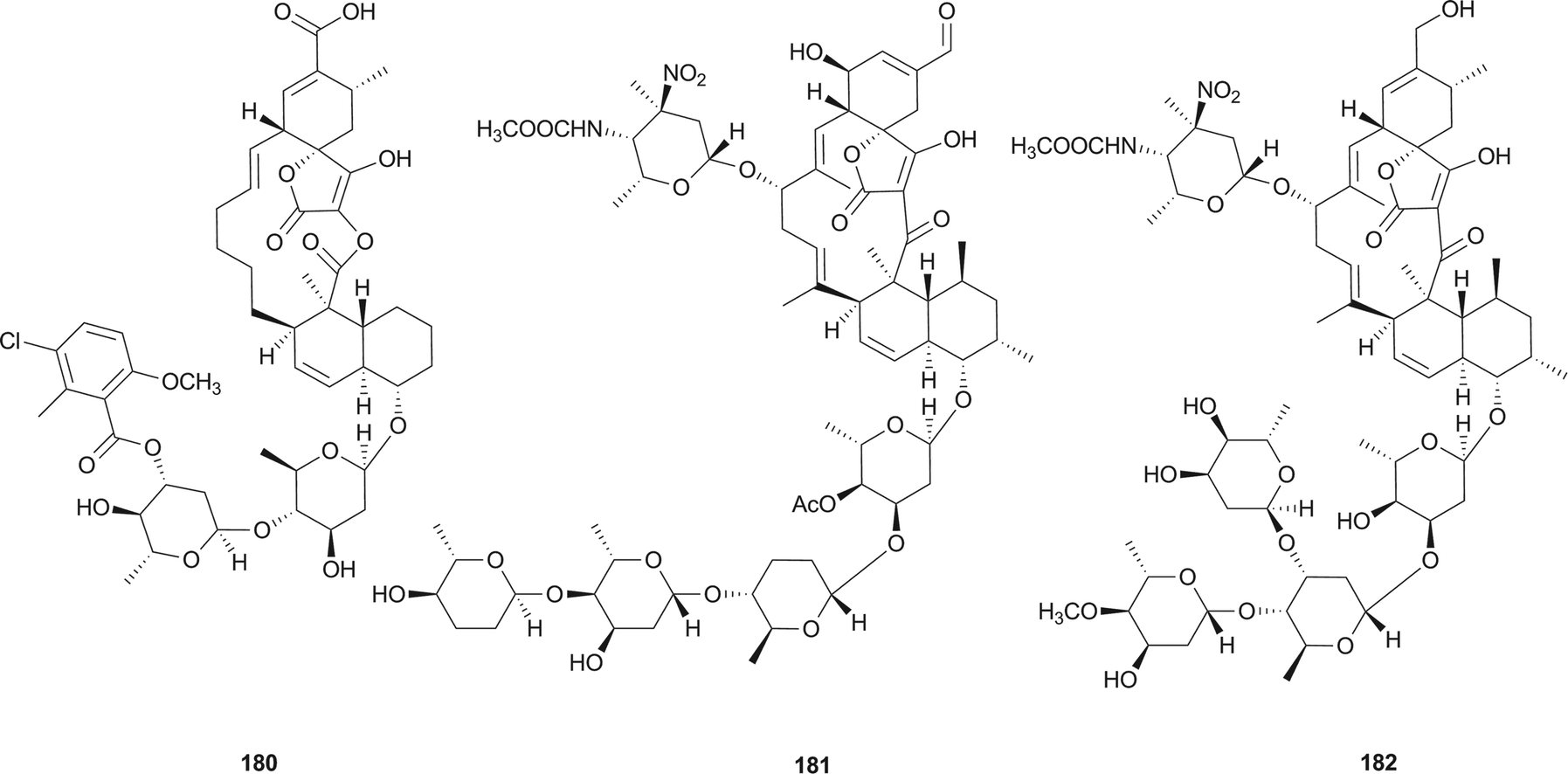
Compounds 180–182 are well-known members of more than 60 spirotetronate-type compounds, and exhibit antibacterial activities against Gram-positive bacteria and show selective antitumor activities.151 Other biological activities are increasingly being revealed. Compound 180 has been shown to inhibit the biosynthesis of cholesterol from mevalonate with an IC50 value of 0.1 mM,152 and further inhibited pyruvate carboxylases purified from vertebrate sources not owing to the occupancy of the acyl-CoA site.153 Metabolite 181 has been proven to be an efficient inducer of apoptosis,154 showing selective inhibition against the mitochondrial functions of Bcl-2 to suppress its anti-apoptotic function in Bcl-2-overexpressing cells,155 mediating apoptosis via endoplasmic reticulum stress preferentially in B-chronic lymphocytic leukemia cells,156 and inactivating the PI3-kinase pathway to directly induce apoptosis of human breast cancer cells.157
The biosynthetic gene clusters for compounds 180–182 were reported in 2006,158 2008,159 and 2007,151 respectively. During the same period, the proposed biosynthetic pathways for the representative 180 (Scheme 14), 181, and 182 were also reported. It is notable that their biosynthesis involves two [4 + 2] Diels–Alder cycloadditions resulting in the formation of the trans-decalin and the spiro-fusion ring systems. However, it is still unclear whether these two Diels–Alder reactions are performed enzymatically or non-enzymatically. The above results indicate the common biosynthetic route for spirotetronate antibiotics to include: (1) formation of the polyketide linear chain; (2) incorporation of a glycerol-derived three-carbon unit;160,161 (3) involvement of two [4 + 2] Diels–Alder cycloadditions resulting from either enzymatic or non-enzymatic processes; and (4) modification of the aglycone cores by some moieties, such as various deoxysugars.
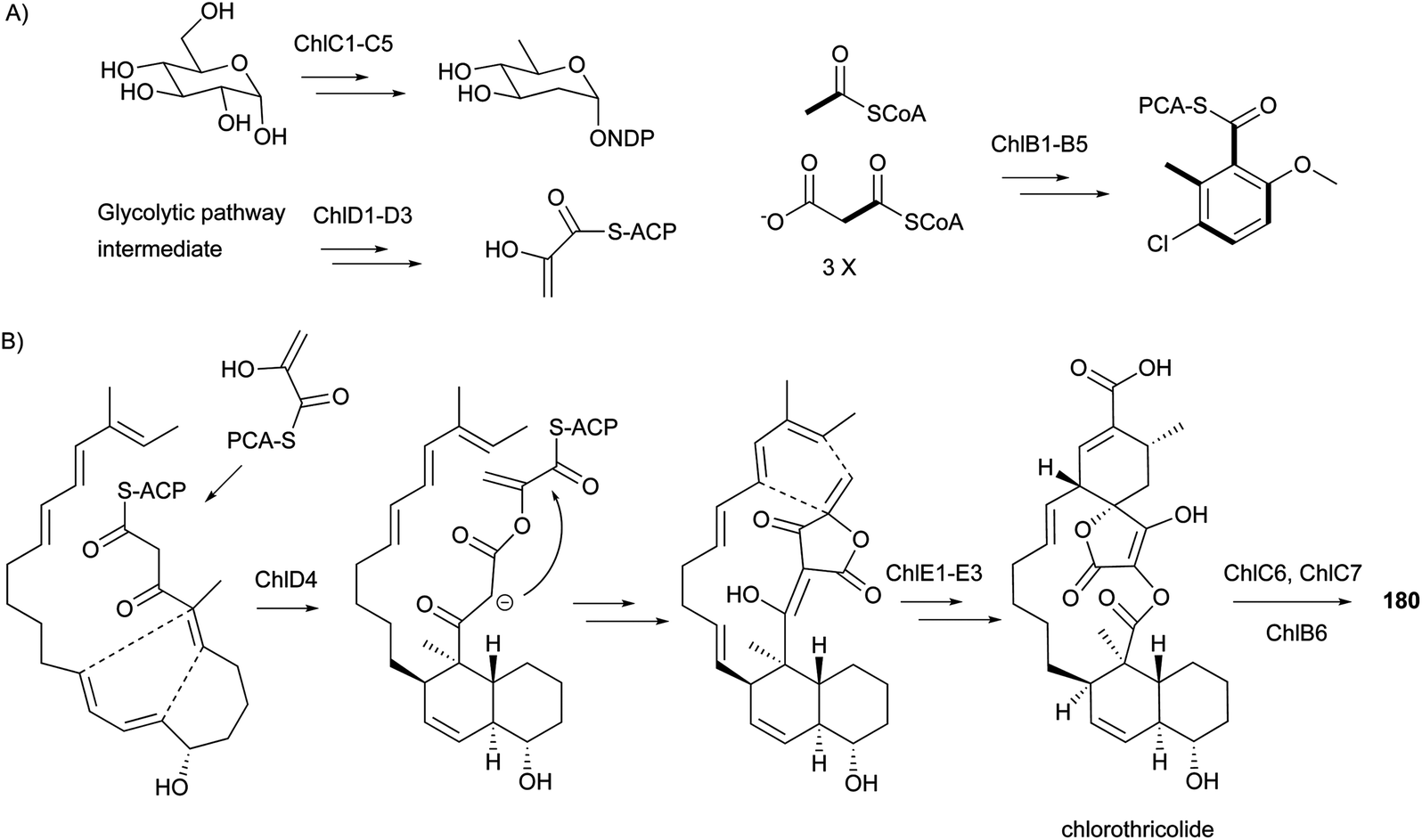 | ||
| Scheme 14 Biosynthetic study on spirotetronate antibiotics. (A) Proposed biosynthesis for deoxysugar olivose, 3-carbon unit enoylpyruvate, and 2-methoxy-5-chloro-6-methylsalicyclic acid. (B) Proposed two [4 + 2] IMDA cycloadditions in the biosynthesis of 180. | ||
To the best of our knowledge, total syntheses of compounds 180–182 have not yet been reported, even though many syntheses for their aglycones have been achieved and many preparations of the functional intermediates have been accomplished.162–168 Enantioselective synthesis of (–)-chlorothricolide, the aglycone of 180, was achieved in 1994 by Roush and Sciotti (Scheme 15).162,163 Prior to this work, the group of Yoshii reported the chemical synthesis of racemic 24-O-methylchlorothricolide (Scheme 16).164 Both syntheses involved Diels–Alder cyclizations to construct the spirotetronate structure and decalin motif of the aglycones. The aglycon of compound 181, tetronolide, was first synthesized by the group of Yoshii in 1991.165,166 Two key steps, the aldol coupling of spirotetronate and a Diels–Alder product decalin system and the internal cyclization, were involved in the construction of the macro-ring as shown in Scheme 17.165,166 A tandem ketene-trapping [4 + 2] cycloaddition strategy for the total synthesis of (+)-tetronolide was also described.167 The synthetic methods of the functional fragments of spirotetronates involving the intermolecular Diels–Alder reaction can be increasingly found in many reports.168,169
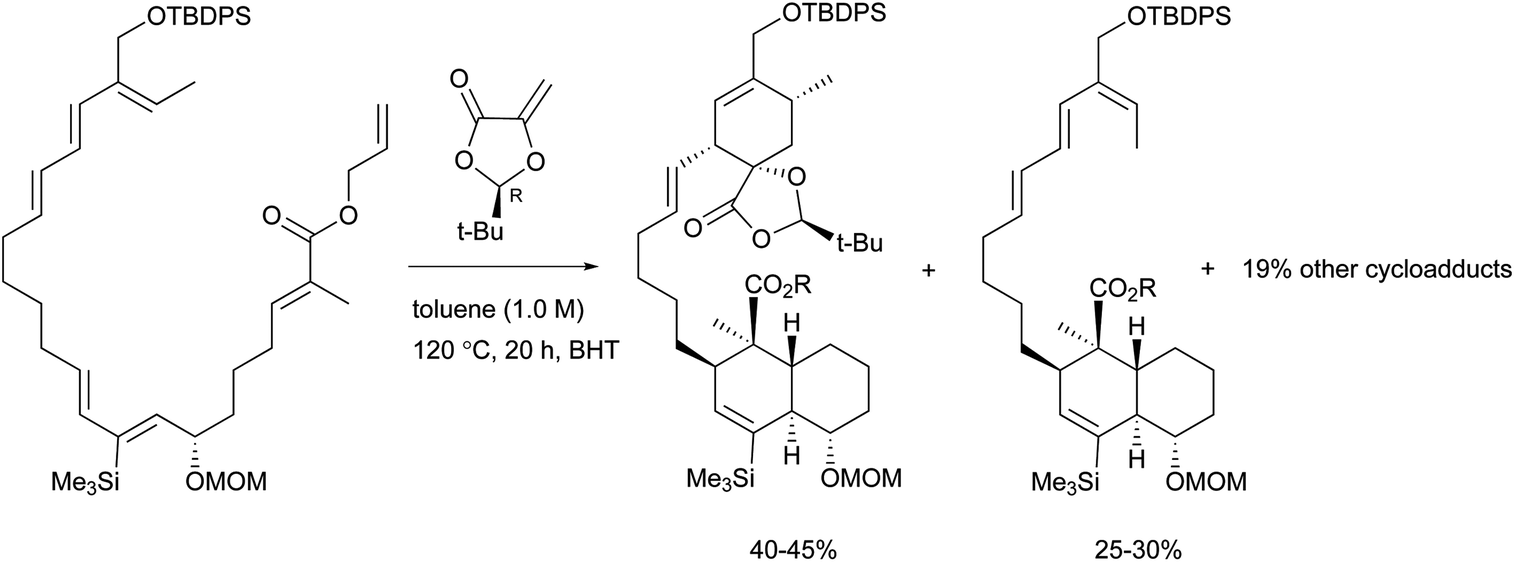 | ||
| Scheme 15 Roush's key Diels–Alder reactions in the enantioselective total synthesis of (-)-chlorothricolide. | ||
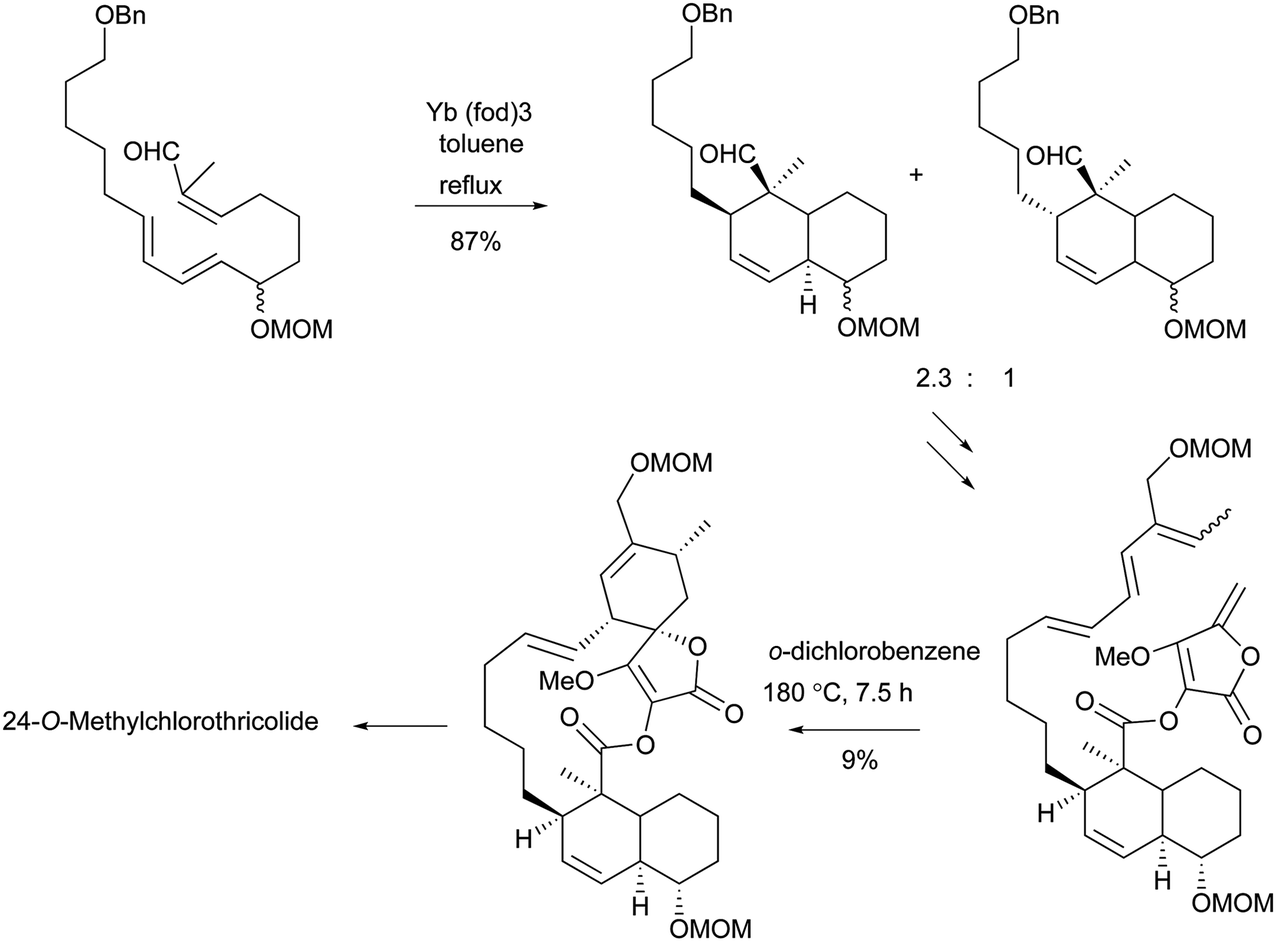 | ||
| Scheme 16 Yoshii's IMDA approach to the synthesis of racemic 24-O-methylchlorothricolide. | ||
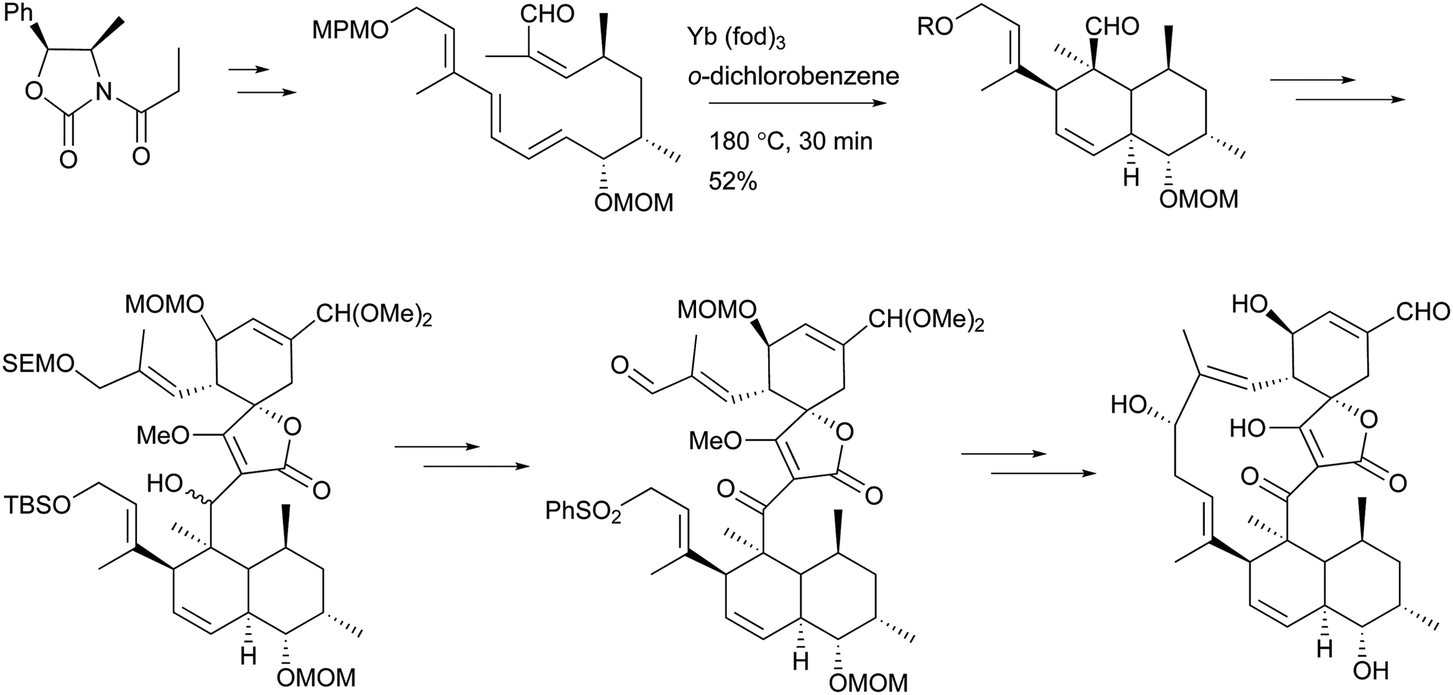 | ||
| Scheme 17 Yoshii's synthesis of the aglycone of 181, tetronolide. | ||
3.11 Pyrrolizidines
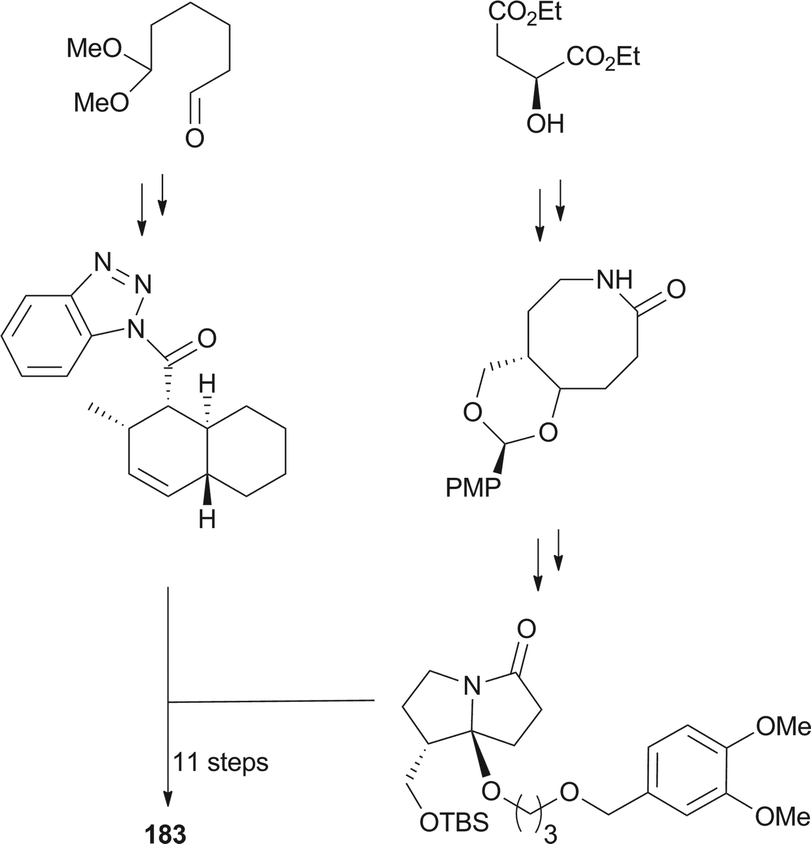
UCS1025-A (183) and -B (184), possessing the unprecedented furopyrrolizin-2,6-dione system, were isolated in 2000 from the broth of the fungus Acremonium sp.170 Their structural elucidations were achieved by spectral data and X-ray crystallographic analysis,171 indicating two more tautomeric isomers in UCS1025A. Compared with compound 184, 183 exhibited significant inhibitory activities against the Gram-positive bacteria S. aureus, B. subtilis and Enterococcus hirae, and Gram-negative bacterium Proteus vulgaris with the MIC values ranging from 1.3 to 5.2 μg mL−1. Moreover, they possessed weak antiproliferative activities against human tumor cell lines ACHN, A431, MCF-7, and T24.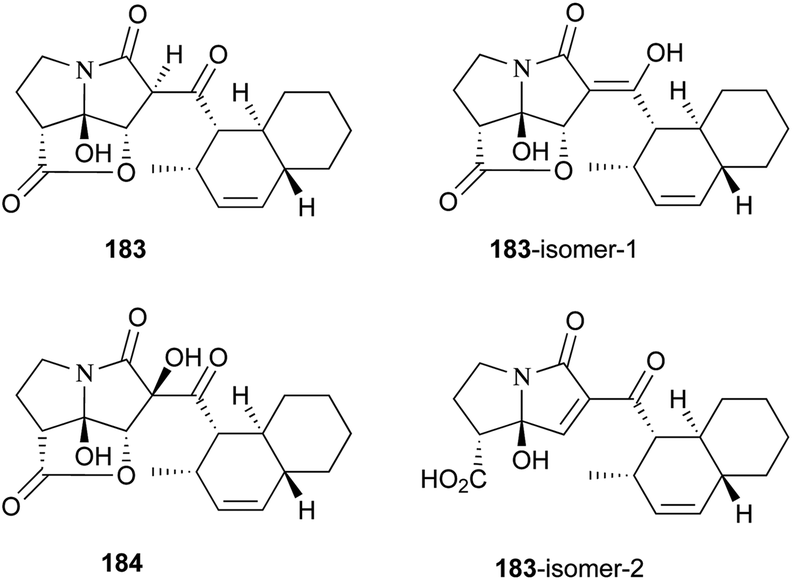
The total synthesis of compound 183 has been accomplished by four groups worldwide.2 The first synthesis of 183 was reported by Lambert and Danishefsky,172 who achieved asymmetric access to the furopyrrolizidine fragment and used a powerful enantioselective organocatalytic intramolecular Diels–Alder reaction to obtain the required decalin.82 The remarkable BEt3-mediated reaction allowed direct coupling of the furopyrrolizidine fragment and the decalin aldehyde to provide the full skeleton of 183 (Scheme 18).172
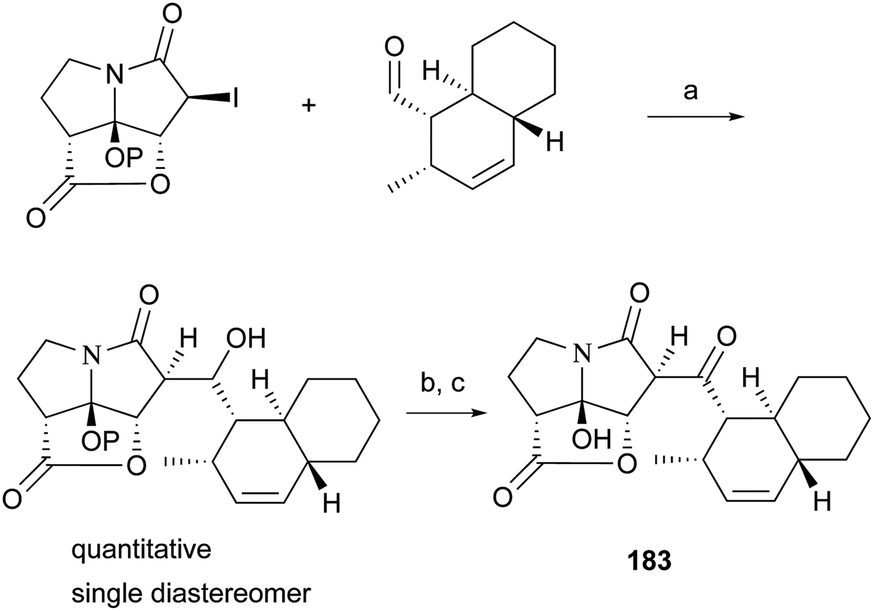 | ||
| Scheme 18 Danishefsky's coupling protocol for the synthesis of 183. (a) BEt3, toluene, −78 °C, P = TBS; (b) TBAF, THF, 85%; (c) Dess–Martin periodinane, CH2Cl2, 84%. | ||
A biomimetic total synthesis of (±)-UCS1025A involving seven linear steps was accomplished by Hoye et al. (Scheme 19).11 The interesting automatic cyclization of a long chain in phosphate buffer (pH 7.2, D2O) to the decalin system indicated a possible non-enzymatic biosynthetic event in vivo. Furthermore, an effective trialkylsilyl triflate (TMSOTf)-mediated cyclization of ester-imide to pyrrolizidine was reported by Hoye et al. (Scheme 20).173
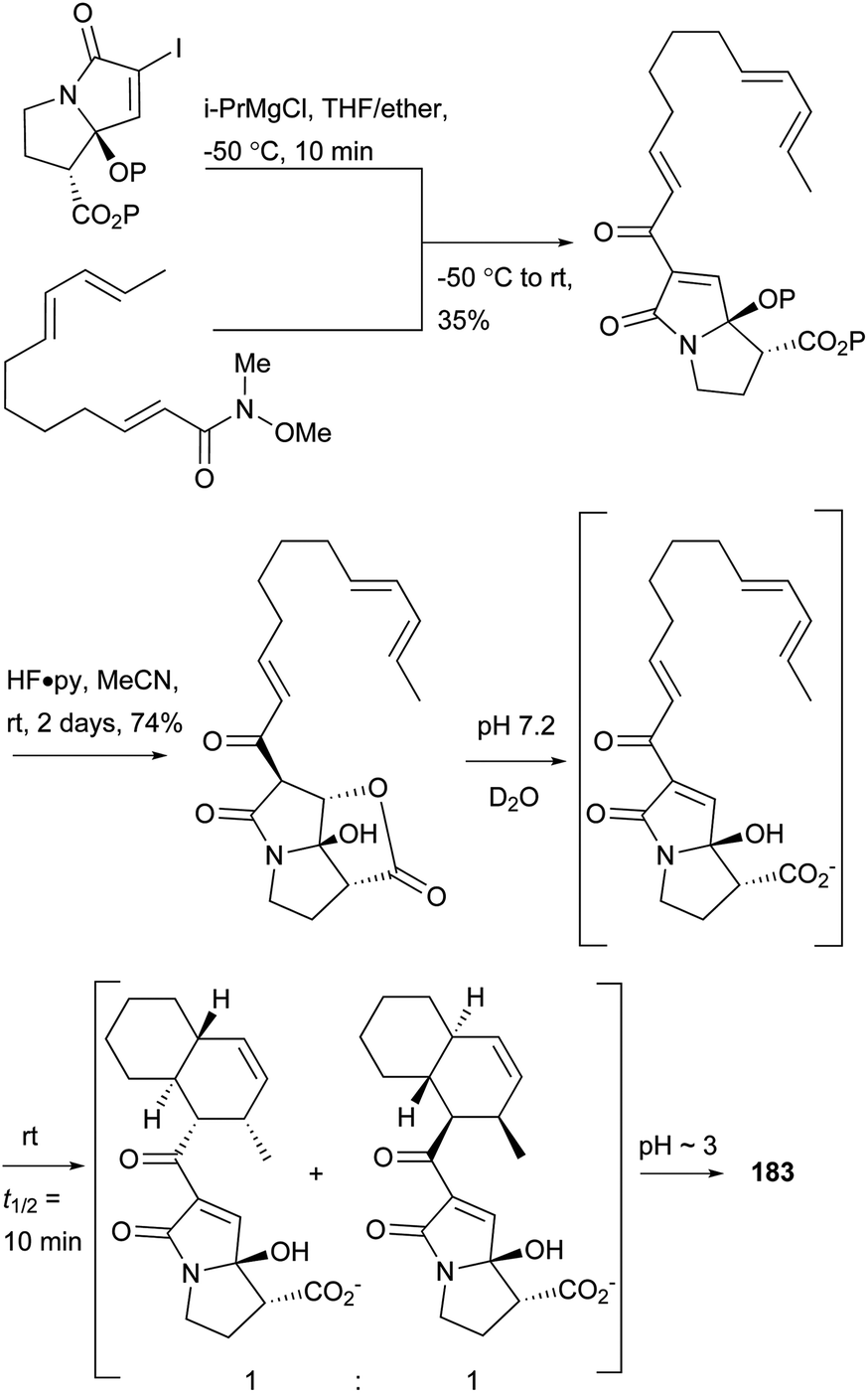 | ||
| Scheme 19 Hoye's biomimetic total synthesis of 183. | ||
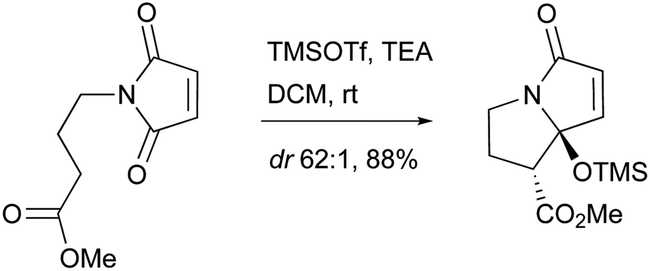 | ||
| Scheme 20 Hoye's silylative Dieckmann-like cyclization to pyrrolizidine fragment. | ||
The group of Christmann synthesized a simplified analogue of compound 183 using an aldol coupling approach (Scheme 21).174 They performed an improved MacMillan organocatalytic Diels–Alder reaction to obtain the concise trans-decalin moiety with an enantiomeric excess (ee) of 99%. For the pyrrolizidine fragment of compound 183, compared to Danishefsky's synthesis in nine steps,172 the group of Christmann showed its two-step enantioselective synthesis (Scheme 22).175 The conditions can also be applied to the commercially available maleimide and some substituted maleimidic acids. An enantioselective lactonization and the trituration enrichment at a high eutectic ee for the pyrrolizidine fragment were also reported.
 | ||
| Scheme 21 Christmann’s aldol coupling involved in the synthesis of an analogue of 183. Condition: NaHMDS, THF, 0 °C, then decalin substance, −78 °C, 3.5 h, 55%. | ||
 | ||
| Scheme 22 Christmann’s two-step synthesis of the pyrrolizidine fragment. | ||
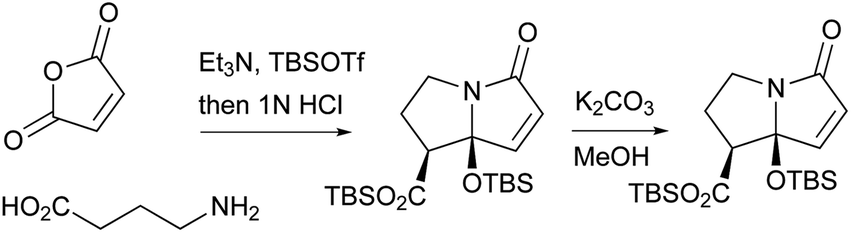
A stereoselective total synthesis of compound 183 was performed by Kan and co-workers, involving an intramolecular Diels–Alder reaction to the decalin skeleton, an intramolecular Staudinger/aza-Wittig reaction to the eight-membered lactam, the stereoselective construction of a labile hemiaminal moiety to assess the pyrrolizidinone skeleton, and the condensation of decalin and the pyrrolizidinone system (Scheme 23).2
 | ||
| Scheme 23 Kan’s stereo-controlled total synthetic method of 183. | ||
Two new pyrrolizidinone antibiotics closely related to 183 and 184, CJ-16,264 (185) and CJ-16,367 (186), were isolated from an unidentified soil fungus CL39457.176 Recently, a novel proteasome inhibitor, pyrrolizilactone (187), was discovered from an uncharacterized fungus,177 and inhibited the trypsin-linked activity of the proteasome.178 All these pyrrolizidine antibiotics could be biogenetically related to tetramic acid derivatives. Specifically, apart from a common polyketide precursor and an amino acid (glycine) as suggested in tetramic acid biosynthesis, an unknown C4 unit could be involved in the construction of these pyrrolizidine antibiotics.171

3.12 Others
Two unprecedented polycyclic polyketides, alchivemycins A (188) and B (189), were isolated from a plant-derived actinomycete Streptomyces sp.179,180 Their structures were confirmed by X-ray crystal structure analysis along with chemical and spectroscopic methods. They showed a selective antimicrobial activity against Micrococcus luteus with MIC values of 0.03 and 0.004 μg mL−1, respectively without inhibitory effects on B. subtilis, E. coli, or C. albicans. Furthermore, they exhibited potency in inhibiting murine colon carcinoma 26-L5 cell invasion (IC50 of 0.34 and 1.9 μM, respectively) without showing any cytotoxic effects. The unprecedented heterocyclic ring system, 2H-tetrahydro-4,6-dioxo-1,2-oxazine, was proposed to be biosynthesized via a similar PKS-NRPS pathway as that of tetramic acid.180 This hypothesis was further investigated by feeding 13C-labeled precursors (Fig. 6). Apiosporic acid (190) is a polyketide-derived compound from a marine fungus A. montagnei isolated from the inner tissue of the North Sea alga Polysiphonia violacea.181 Nahuoic acid A (191), the first known selective SAM-competitive inhibitor of SETD8 (IC50 of 6.5 ± 0.5 μM), was produced by a Streptomyces sp. isolated from a tropical marine sediment.182 The actinomycete Kitasatospora griseola was known to produce two unprecedented glucosylated polyketides, satosporins A 192 and B 193.183 Their absolute configurations were confirmed using TDDFT/CD calculations and chemical derivatization methods. Two novel antimicrobial agents, tetrodecamycin (194) and dihydrotetrodecamycin (195) were obtained from the broth of Streptomyces nashvillensis.184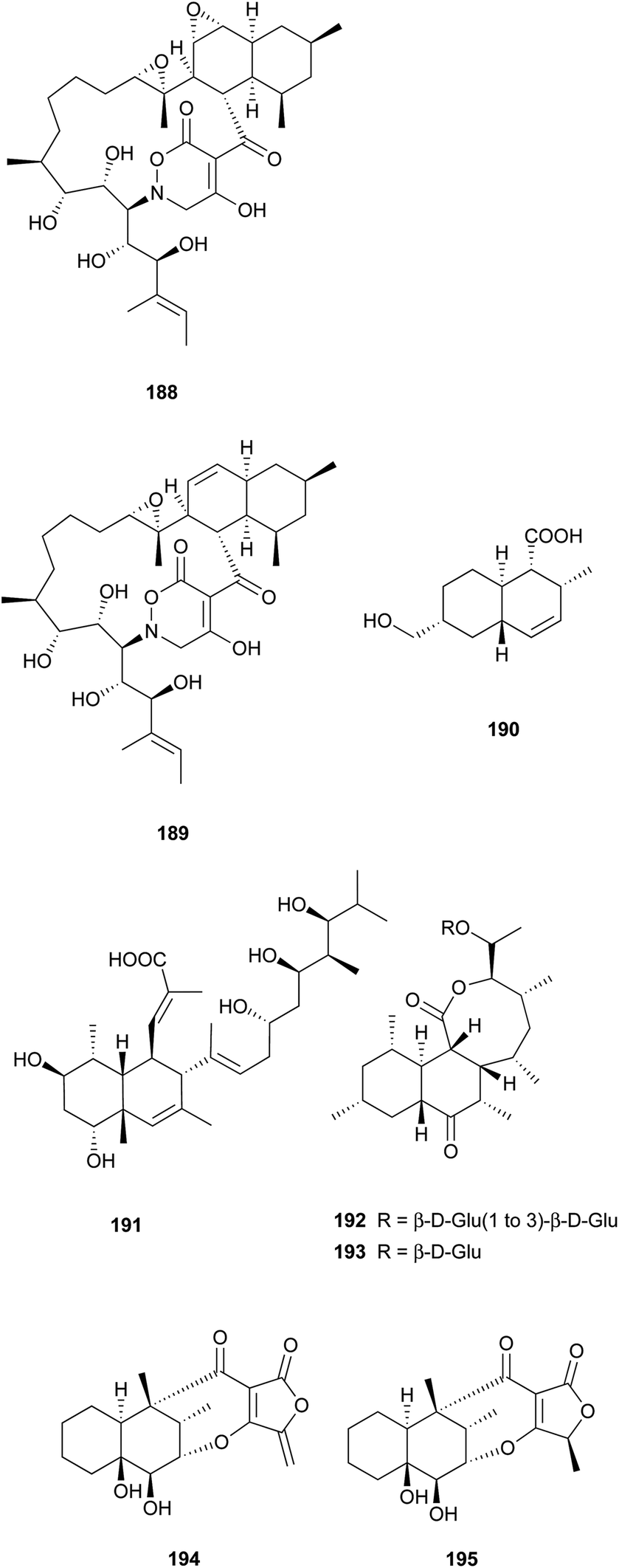
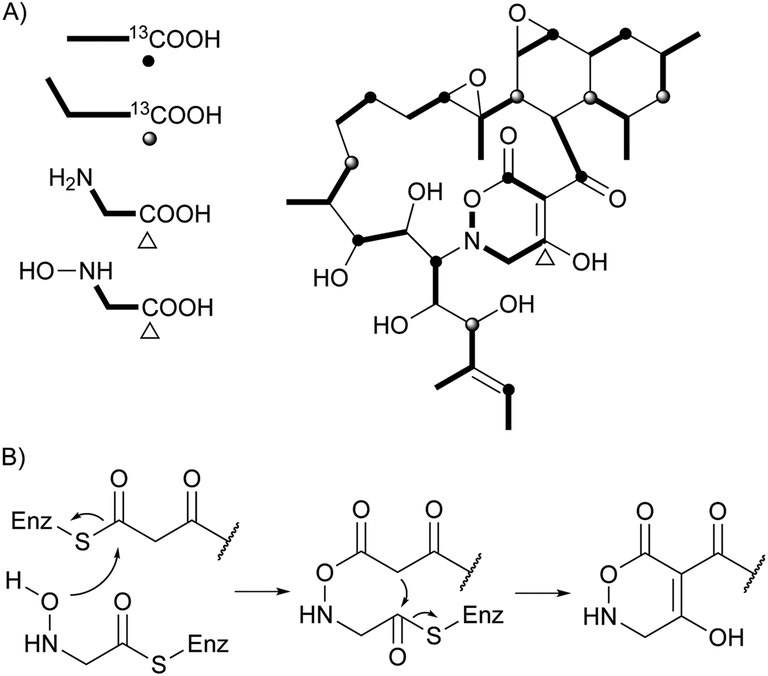 | ||
| Fig. 6 Biosynthetic investigation for 188. (A) Incorporation of 13C-labeled precursors into 188. (B) Proposed biosynthetic pathway for tetrahydrooxazine ring. | ||
4 Isoprenoid decalin
The isolation and structures of natural sesquiterpenoids, diterpenoids, or marine natural products containing isoprenoid-derived decalin have been covered in a series of reports.4–6 Therefore, in this manuscript, we only highlight the important features of isoprenoid-derived decalin secondary metabolites isolated from microorganisms (mainly fungi). Almost always, these isoprenoid decalin-derived compounds of microbial origin belong to sesquiterpenes mainly including cadinane-, eremophilane- and bicyclofarnesane-type sesquiterpenes according to the classification in earlier reviews.4–6 Cadinane sesquiterpenes have demonstrated highly selective and notable nematicidal activities, but have remained largely inactive in antimicrobial and cytotoxic assays.185–189 Eremophilane sesquiterpenes, typically isolated from fungi of the genera Xylaria and Penicillium exhibit diverse biological properties, such as cytotoxic,190,191 antimicrobial,192 and inhibition of HIV integrase.193 More importantly, most of the bicyclofarnesane-type sesquiterpenes are composed of an isoprenoid-derived decalin ring system linked to a polyketide ring unit with some substituted functionalized groups such as an amino acid. The typical diverse structures of this family are the compounds 196,194197,195198,195199,196200,197201,198202,198203,199 and 204.200 The proposed biosynthesis of compound 196 is shown in Scheme 24 (see Supporting Information of ref. 194). Similarly, a coupling between a polyketide-derived pyranone or pyrone and a diterpene (decalin part) contribute to the structural diversity of the diterpenes 205201 and 206.202,203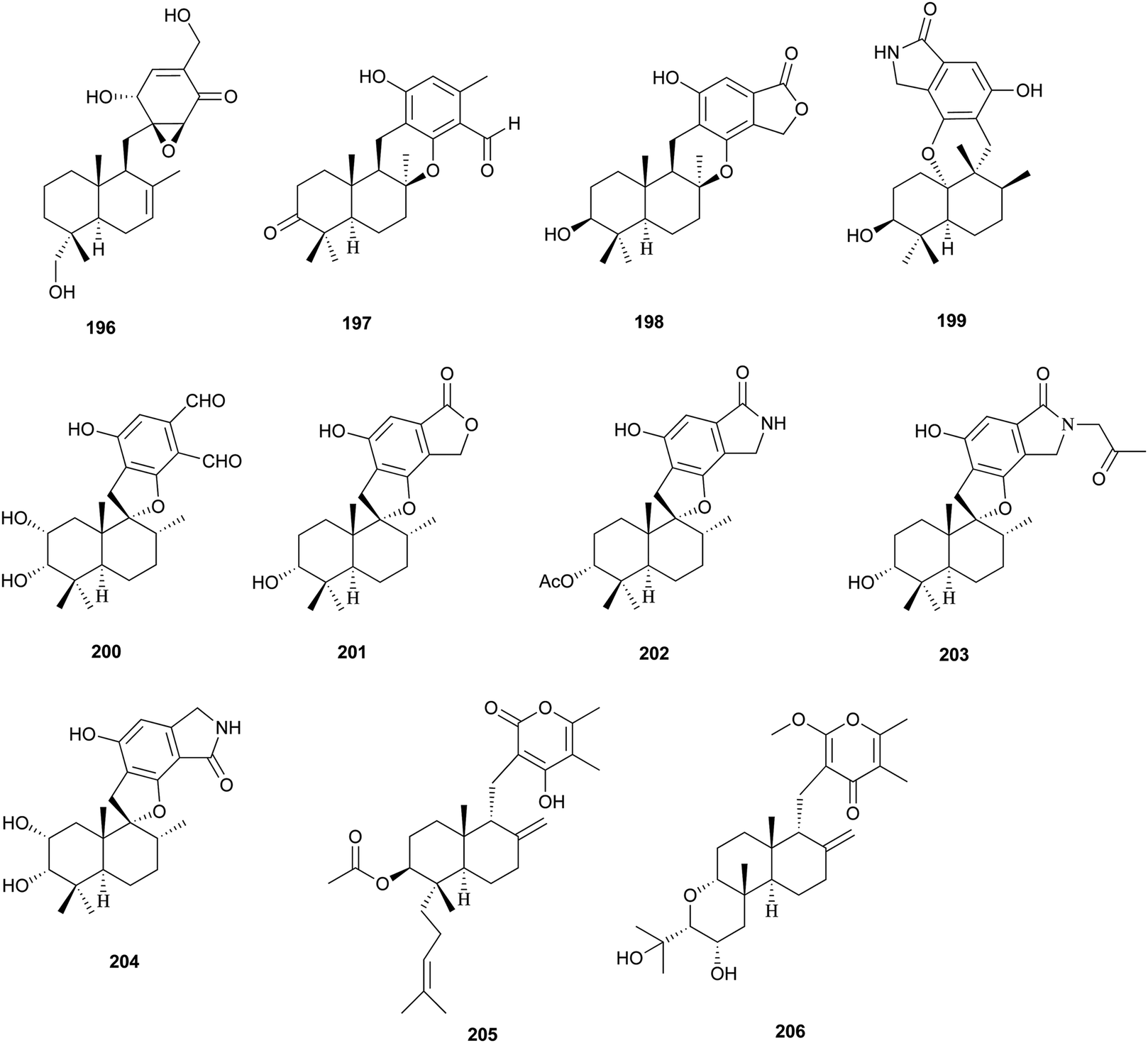
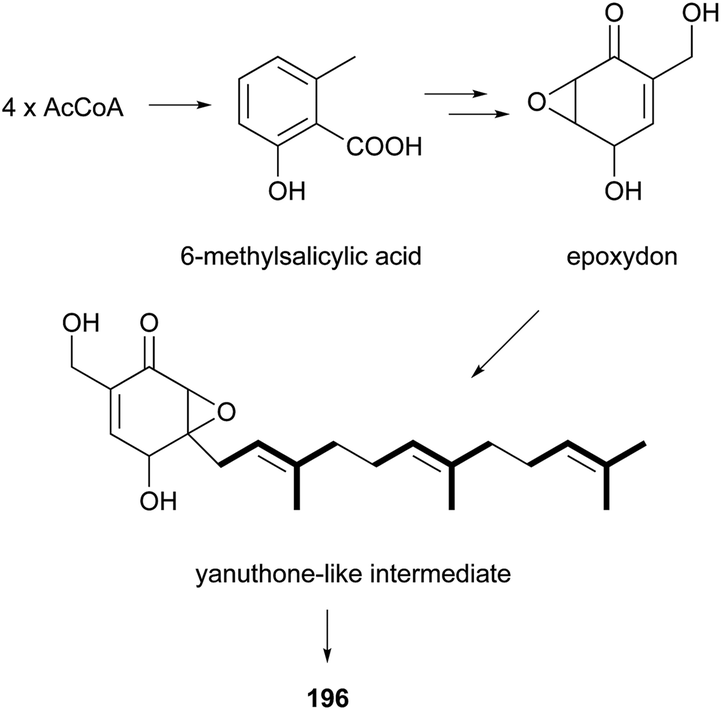 | ||
| Scheme 24 The proposed biosynthesis for 196. | ||
5 Conclusions
A number of bioactive microbial secondary metabolites with the decalin scaffold are increasingly being discovered. Herein, we present a comprehensive review on nearly 200 polyketide decalin secondary metabolites and 11 representative sesquiterpenoids and diterpenoids with the decalin system. The structural diversity of decalin-derived microbial natural products is a consequence of the large manifold of fungal or bacterial species, and also a result of the biosynthetic capability of these fascinating producers to assemble decalin compounds in single or mixed biosynthetic pathways. Furthermore, they display diverse and remarkable biological activities.More importantly, nearly all the polyketide decalin-derived compounds we have discussed in this manuscript have a double bond between C3 and C4 (sometimes oxygenated), suggesting an enzymatic or non-enzymatic IMDA cycloaddition to form the decalin scaffold. Moreover, all the decalin-containing secondary metabolites presented here seem to be assembled by a linear polyketide or isoprenoid unit, which then cyclize to the decalin scaffold followed by incorporation of functionalized substituted groups. This suggests that the biosynthetic pathways of many of these compounds are extremely related, as exemplified in this review. Specifically, the decalin cyclization type endo-trans-fused one (C or D) is the most common, even though two other fused classes are dominant in some classifications, such as the pyrone derivatives (exo-cis-fused, A) and macrolides (exo-cis-fused, B). This phenomenon may be the result of a steric or enzymatic effect. Lastly, compounds with the decalin system conjoining different functionalized moieties such as lactones in monacolins and tetramic acids often have different biological activities. Therefore, these intriguing similarities or differences prove helpful in elucidating their biosynthesis and might provide a scientific handle for aiding biomimetic total synthesis. Finally, the decalin scaffold is more likely to serve as the rigid or basic template for construction of the whole structure.
6 Acknowledgements
G.L. gratefully acknowledges the China Scholarship Council (CSC) for a doctoral fellowship. S.K. was a Visiting Researcher at the Department of Plant Sciences, University of Oxford, South Parks Road, Oxford OX1 3RB, UK, during preparation of the manuscript (Apr 2013 to Mar 2014). S.K. gratefully acknowledges Dr Gail M. Preston for hosting and TU Dortmund for supporting his stay at the University of Oxford. The authors are grateful to Prof. Michael Müller of the University of Freiburg, Germany for valuable comments and suggestions.7 References
- E. M. Stocking and R. M. Williams, Angew. Chem., Int. Ed., 2003, 42, 3078–3115 CrossRef CAS PubMed.
- K. Uchida, T. Ogawa, Y. Yasuda, H. Mimura, T. Fujimoto, T. Fukuyama, T. Wakimoto, T. Asakawa, Y. Hamashima and T. Kan, Angew. Chem., Int. Ed., 2012, 51, 12850–12853 CrossRef CAS PubMed.
- V. Singh, S. R. Iyer and S. Pal, Tetrahedron, 2005, 61, 9197–9231 CrossRef CAS PubMed.
- B. M. Fraga, Nat. Prod. Rep., 2013, 30, 1226–1264 RSC.
- J. R. Hanson, Nat. Prod. Rep., 2009, 26, 1156–1171 RSC.
- J. R. Hanson, Nat. Prod. Rep., 2013, 30, 1346–1356 RSC.
- W. L. Kelly, Org. Biomol. Chem., 2008, 6, 4483–4493 CAS.
- H. J. Kim, M. W. Ruszczycky, S.-h. Choi, Y.-n. Liu and H.-w. Liu, Nature, 2011, 473, 109–112 CrossRef CAS PubMed.
- K. Auclair, A. Sutherland, J. Kennedy, D. J. Witter, J. P. Van den Heever, C. R. Hutchinson and J. C. Vederas, J. Am. Chem. Soc., 2000, 122, 11519–11520 CrossRef CAS.
- K. Kasahara, T. Miyamoto, T. Fujimoto, H. Oguri, T. Tokiwano, H. Oikawa, Y. Ebizuka and I. Fujii, ChemBioChem, 2010, 11, 1245–1252 CrossRef CAS PubMed.
- T. R. Hoye and V. Dvornikovs, J. Am. Chem. Soc., 2006, 128, 2550–2551 CrossRef CAS PubMed.
- M. A. Varner and R. B. Grossman, Tetrahedron, 1999, 55, 13867–13886 CrossRef CAS.
- T. Tokoroyama, Synthesis, 2000, 611–633 CrossRef CAS PubMed.
- H. Akita, Heterocycles, 2013, 87, 1625–1658 CrossRef CAS PubMed.
- E. S. Istvan and J. Deisenhofer, Science, 2001, 292, 1160–1164 CrossRef CAS PubMed.
- W. Xu, Y.-H. Chooi, J. W. Choi, S. Li, J. C. Vederas, N. A. Da Silva and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 6472–6475 CrossRef CAS PubMed.
- A. Endo, J. Antibiot., 1979, 32, 852–854 CrossRef CAS.
- A. Endo, J. Antibiot., 1980, 33, 334–336 CrossRef CAS.
- A. W. Alberts, J. Chen, G. Kuron, V. Hunt, J. Huff, C. Hoffman, J. Rothrock, M. Lopez, H. Joshua, E. Harris, A. Patchett, R. Monaghan, S. Currie, E. Stapley, G. Albers-Schonberg, O. Hensens, J. Hirshfield, K. Hoogsteen, J. Liesch and J. Springer, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 3957–3961 CrossRef CAS.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS.
- X. Xie, M. J. Meehan, W. Xu, P. C. Dorrestein and Y. Tang, J. Am. Chem. Soc., 2009, 131, 8388–8389 CrossRef CAS PubMed.
- J. Barriuso, D. T. Nguyen, J. W.-H. Li, J. N. Roberts, G. MacNevin, J. L. Chaytor, S. L. Marcus, J. C. Vederas and D.-K. Ro, J. Am. Chem. Soc., 2011, 133, 8078–8081 CrossRef CAS PubMed.
- B. D. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. W.-H. Li, J. C. Vederas, Y. Tang and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11144–11149 CrossRef CAS PubMed.
- G. Albers-Schönberg, H. Joshua, M. B. Lopez, O. D. Hensens, J. P. Springer, J. Chen, S. Ostrove, C. H. Hoffman, A. W. Alberts and A. A. Patchett, J. Antibiot., 1981, 34, 507–512 CrossRef.
- A. Endo, K. Hasumi and S. Negishi, J. Antibiot., 1985, 38, 420–422 CrossRef CAS.
- A. Endo, K. Hasumi, T. Nakamura, M. Kunishima and M. Masuda, J. Antibiot., 1985, 38, 321–327 CrossRef CAS.
- A. Endo, D. Komagata and H. Shimada, J. Antibiot., 1986, 39, 1670–1673 CrossRef CAS.
- L. R. Treiber, R. A. Reamer, C. S. Rooney and H. G. Ramjit, J. Antibiot., 1989, 42, 30–36 CrossRef CAS.
- A. G. Brown, T. C. Smale, T. J. King, R. Hasenkamp and R. H. Thompson, J. Chem. Soc., Perkin Trans. 1, 1976, 1165–1170 RSC.
- A. Endo, M. Kuroda and Y. Tsujita, J. Antibiot., 1976, 29, 1346–1348 CrossRef CAS.
- Y. K. Tony Lam, V. P. Gullo, R. T. Goegelman, D. Jorn, L. Huang, C. DeRiso, R. L. Monaghan and I. Putter, J. Antibiot., 1981, 34, 614–616 CrossRef.
- S. Murakawa, K. Sakai and A. Endo, J. Antibiot., 1994, 47, 108–109 CrossRef CAS.
- S. Okamoto, T. Hosoe, T. Itabashi, K. Nozawa, K. Okada, G. M.de C. Takaki, M. Chikamori, T. Yaguchi, K. Fukushima, M. Miyaji and K. Kawai, J. Nat. Prod., 2004, 67, 1580–1583 CrossRef CAS PubMed.
- M.-T. Liu, J.-J. Li, X.-Y. Shang, S. Li, L.-L. Li, N. Luan and Z.-L. Jin, Magn. Reson. Chem., 2011, 49, 129–131 CrossRef CAS PubMed.
- M.-T. Liu, N. Luan, J.-J. Li, X. Huang, Y.-F. Wang, A.-L. Wang and X.-Y. Shang, Magn. Reson. Chem., 2012, 50, 709–712 CrossRef CAS PubMed.
- L. Zhu, L.-F. Yau, J.-G. Lu, G.-Y. Zhu, J.-R. Wang, Q.-B. Han, W.-L. Hsiao and Z.-H. Jiang, J. Agric. Food Chem., 2012, 60, 934–939 CrossRef CAS PubMed.
- L. Zhu, J.-G. Lu, T. Li, G.-Y. Zhu, Q.-B. Han, W.-L. Hsiao, L. Liu and Z.-H. Jiang, J. Nat. Prod., 2012, 75, 567–571 CrossRef CAS PubMed.
- Y.-T. Zhang, Y. Wang, X.-T. Zhang, D.-L. Wu, X.-Q. Zhang and W.-C. Ye, J. Asian Nat. Prod. Res., 2009, 11, 792–795 CrossRef CAS PubMed.
- I. Barash, G. Pupkin, D. Netzer and Y. Kashman, Plant Physiol., 1982, 69, 23–27 CrossRef CAS PubMed.
- I. Barash, S. Manulis, Y. Kashman, J. P. Springer, M. H. M. Chen, J. Clardy and G. A. Strobel, Science, 1983, 220, 1065–1066 CAS.
- S. Manulis, Y. Kashman, D. Netzert and I. Barash, Phytochemistry, 1984, 23, 2193–2198 CrossRef CAS.
- I. Barash and S. Manulis, in Iron, Siderophores, and Plant Diseases, Springer 1986, 117, pp. 273–281 Search PubMed.
- A. Ichihara, H. Oikawa, K. Hayashi, S. Sakamura, A. Furusaki and T. Matsumoto, J. Am. Chem. Soc., 1983, 105, 2907–2908 CrossRef CAS.
- A. Ichihara, H. Oikawa, M. Hashimoto, S. Sakamura, T. Haraguchi and H. Nagano, Agric. Biol. Chem., 1983, 47, 2965–2967 CrossRef CAS.
- H. Oikawa, A. Ichihara and S. Sakamura, J. Chem. Soc., Chem. Commun., 1984, 814–815 RSC.
- H. Oikawa, A. Ichihara and S. Sakamura, J. Chem. Soc., Chem. Commun., 1988, 600–602 RSC.
- M. Kobayashi, H. Uehara, K. Matsunami, S. Aoki and I. Kitagawa, Tetrahedron Lett., 1993, 34, 7925–7928 CrossRef CAS.
- W. S. Horn, R. E. Schwartz, M. S. J. Simmonds and W. M. Blaney, Tetrahedron Lett., 1994, 35, 6037–6040 CrossRef CAS.
- R. Sawa, Y. Takahashi, S. Itoh, K. Shimanaka, N. Kinoshita, Y. Homma, M. Hamada, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1994, 47, 1266–1272 CrossRef CAS.
- O. D. Hensens, G. L. Helms, E. T. T. Jones and G. H. Harris, J. Org. Chem., 1995, 60, 1772–1776 CrossRef CAS.
- L. Rahbæk, S. Sperry, J. E. Piper and P. Crews, J. Nat. Prod., 1998, 61, 1571–1573 CrossRef PubMed.
- G. Brauers, R. A. Edrada, R. Ebel, P. Proksch, V. Wray, A. Berg, U. Gräfe, C. Schächtele, F. Totzke, G. Finkenzeller, D. Marme, J. Kraus, M. Münchbach, M. Michel, G. Bringmann and K. Schaumann, J. Nat. Prod., 2000, 63, 739–745 CrossRef CAS PubMed.
- Y. Fujii, M. Asahara, M. Ichinoe and H. Nakajima, Phytochemistry, 2002, 60, 703–708 CrossRef CAS.
- S. Tsukamoto, S. Miura, Y. Yamashita and T. Ohta, Bioorg. Med. Chem. Lett., 2004, 14, 417–420 CrossRef CAS PubMed.
- Y. Ohtsu, S. Yoshimura, T. Kinoshita, S. Takase and H. Nakajima, J. Antibiot., 2005, 58, 479–482 CrossRef CAS PubMed.
- S. Nakadate, K. Nozawa, H. Horie, Y. Fujii, M. Nagai, T. Hosoe, K. Kawai, T. Yaguchi and K. Fukushima, J. Nat. Prod., 2007, 70, 1510–1512 CrossRef CAS PubMed.
- T. Yamada, Y. Mizutani, Y. Umebayashi, N. Inno, M. Kawashima, T. Kikuchi and R. Tanaka, Tetrahedron Lett., 2014, 55, 662–664 CrossRef CAS PubMed.
- M. Kuramoto, K. Yamada, M. Shikano, K. Yazawa, H. Arimoto, T. Okamura and D. Uemura, Chem. Lett., 1997, 885–886 CrossRef CAS.
- J. Malmstrøm, C. Christophersen and J. C. Frisvad, Phytochemistry, 2000, 54, 301–309 CrossRef.
- M. El-Neketi, W. Ebrahim, W. Lin, S. Gedara, F. Badria, H.-E. A. Saad, D. Lai and P. Proksch, J. Nat. Prod., 2013, 76, 1099–1104 CrossRef CAS PubMed.
- L. P. Sandjo, E. Thines, T. Opatz and A. Schüffler, Beilstein J. Org. Chem., 2014, 10, 251–258 CrossRef CAS PubMed.
- N. Tabata, H. Tomoda, R. Masuma, K. Haneda, Y. Iwai and S. Ōmura, J. Antibiot., 1993, 46, 1849–1853 CrossRef CAS.
- R. Masuma, N. Tabata, H. Tomoda, K. Haneda, Y. Iwai and S. Ōmura, J. Antibiot., 1994, 47, 46–53 CrossRef CAS.
- J. G. Ondeyka, R. A. Giacobbe, G. F. Bills, C. Cuadrillero, D. Schmatz, M. A. Goetz, D. L. Zink and S. B. Singh, Bioorg. Med. Chem. Lett., 1998, 8, 3439–3442 CrossRef CAS.
- M. Namikoshi, H. Kobayashi, T. Yoshimoto and T. Hosoya, J. Antibiot., 1997, 50, 890–892 CrossRef CAS.
- H. Kobayashi, S. Meguro, T. Yoshimoto and M. Namikoshi, Tetrahedron, 2003, 59, 455–459 CrossRef CAS.
- C. A. Parish, M. de la Cruz, S. K. Smith, D. Zink, J. Baxter, S. Tucker-Samaras, J. Collado, G. Platas, G. Bills, M. T. Díez, F. Vicente, F. Peláez and K. Wilson, J. Nat. Prod., 2009, 72, 59–62 CrossRef CAS PubMed.
- H. Kobayashi, R. Sunaga, K. Furihata, N. Morisaki and S. Iwasaki, J. Antibiot., 1995, 48, 42–52 CrossRef CAS.
- H. P. Nguyen, D. Zhang, U. Lee, J. S. Kang, H. D. Choi and B. W. Son, J. Nat. Prod., 2007, 70, 1188–1190 CrossRef CAS PubMed.
- H. Kumagai, T. Someno, K. Dobashi, K. Isshiki, M. Ishizuka and D. Ikeda, J. Antibiot., 2004, 57, 97–103 CrossRef CAS.
- M. I. Mitova, G. Lang, J. W. Blunt, N. J. Cummings, A. L. J. Cole, W. T. Robinson and M. H. G. Munro, J. Org. Chem., 2006, 71, 492–497 CrossRef CAS PubMed.
- D. B. Stierle, A. A. Stierle and B. K. Ganser, J. Nat. Prod., 1999, 62, 1147–1150 CrossRef CAS PubMed.
- S. Sakamoto, F. Kojima, M. Igarashi, R. Sawa, M. Umekita, Y. Kubota, K. Nakae, S. Yamaguchi, H. Adachi, Y. Nishimura and Y. Akamatsu, J. Antibiot., 2010, 63, 703–708 CrossRef CAS PubMed.
- S. Sakamoto, F. Kojima, I. Momose, M. Kawada, H. Adachi and Y. Nishimura, Biochem. Biophys. Res. Commun., 2012, 422, 751–757 CrossRef CAS PubMed.
- H. Oikawa, T. Yokota, A. Ichihara and S. Sakamura, J. Chem. Soc., Chem. Commun., 1989, 1284–1285 RSC.
- H. Oikawa, Y. Suzuki, A. Naya, K. Katayama and A. Ichihara, J. Am. Chem. Soc., 1994, 116, 3605–3606 CrossRef CAS.
- A. Ichihara, H. Tazaki and S. Sakamura, Tetrahedron Lett., 1983, 24, 5373–5376 CrossRef CAS.
- Y. Mizushina, S. Kamisuki, N. Kasai, N. Shimazaki, M. Takemura, H. Asahara, S. Linn, S. Yoshida, A. Matsukage, O. Koiwai, F. Sugawara, H. Yoshida and K. Sakaguchi, J. Biol. Chem., 2002, 277, 630–638 CrossRef CAS PubMed.
- H. Oikawa, T. Yokota, C. Sakano, Y. Suzuki, A. Naya and A. Ichihara, Biosci., Biotechnol., Biochem., 1998, 62, 2016–2022 CrossRef CAS.
- H. Hagiwara, K. Kobayashi, S. Miya, T. Hoshi, T. Suzuki, M. Ando, T. Okamoto, M. Kobayashi, I. Yamamoto, S. Ohtsubo, M. Kato and H. Uda, J. Org. Chem., 2002, 67, 5969–5976 CrossRef CAS PubMed.
- B. Lygo, M. Bhatia, J. W. B. Cooke and D. J. Hirst, Tetrahedron Lett., 2003, 44, 2529–2532 CrossRef CAS.
- R. M. Wilson, W. S. Jen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 11616–11617 CrossRef CAS PubMed.
- K. M. Jenkins, S. G. Toske, P. R. Jensen and W. Fenical, Phytochemistry, 1998, 49, 2299–2304 CrossRef CAS.
- L. E. Schmidt, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2007, 70, 1317–1320 CrossRef CAS PubMed.
- K. Trisuwan, V. Rukachaisirikul, Y. Sukpondma, S. Preedanon, S. Phongpaichit and J. Sakayaroj, Phytochemistry, 2009, 70, 554–557 CrossRef CAS PubMed.
- H. A. Whaley, C. G. Chidester, S. A. Mizsak and R. J. Wnuk, Tetrahedron Lett., 1980, 21, 3659–3662 CrossRef CAS.
- W. D. Celmer, G. N. Chmurny, C. E. Moppett, R. S. Ware, P. C. Watts and E. B. Whipple, J. Am. Chem. Soc., 1980, 102, 4203–4209 CrossRef CAS.
- W. C. Snyder and K. L. Rinehart, Jr, J. Am. Chem. Soc., 1984, 106, 787–789 CrossRef CAS.
- R. R. Rasmussen, M. H. Scherr, D. N. Whittern, A. M. Buko and J. B. McAlpine, J. Antibiot., 1987, 40, 1383–1393 CrossRef CAS.
- H. Gouda, T. Sunazuka, H. Ui, M. Handa, Y. Sakoh, Y. Iwai, S. Hirono and S. Ōmura, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18286–18291 CrossRef CAS PubMed.
- J. B. McAlpine, L. A. Mitscher, M. Jackson, R. R. Rasmussen, D. V. Velde and E. Veliz, Tetrahedron, 1996, 52, 10327–10334 CrossRef CAS.
- K. Gerth, H. Steinmetz, G. Höfle and R. Jansen, Angew. Chem., Int. Ed., 2008, 47, 600–602 CrossRef CAS PubMed.
- N. Rahn and M. Kalesse, Angew. Chem., Int. Ed., 2008, 47, 597–599 CrossRef CAS PubMed.
- K. H. Jang, S.-J. Nam, J. B. Locke, C. A. Kauffman, D. S. Beatty, L. A. Paul and W. Fenical, Angew. Chem., Int. Ed., 2013, 52, 7822–7824 CrossRef CAS PubMed.
- B. J. Royles, Chem. Rev., 1995, 95, 1981–2001 CrossRef CAS.
- R. Schobert and A. Schlenk, Bioorg. Med. Chem., 2008, 16, 4203–4221 CrossRef CAS PubMed.
- B. Nay, N. Riache and L. Evanno, Nat. Prod. Rep., 2009, 26, 1044–1062 RSC.
- H. R. Burmeister, G. A. Bennett, R. F. Vesonder and C. W. Hesseltine, Antimicrob. Agents Chemother., 1974, 5, 634–639 CrossRef CAS.
- R. F. Vesonder, L. W. Tjarks, W. K. Rohwedder, H. R. Burmeister and J. A. Laugal, J. Antibiot., 1979, 32, 759–761 CrossRef CAS.
- E. C. Marfori, T. Bamba, S. Kajiyama, E. Fukusaki and A. Kobayashi, Tetrahedron, 2002, 58, 6655–6658 CrossRef CAS.
- D. Boettger and C. Hertweck, ChemBioChem, 2013, 14, 28–42 CrossRef CAS PubMed.
- J. W. Sims and E. W. Schmidt, J. Am. Chem. Soc., 2008, 130, 11149–11155 CrossRef CAS PubMed.
- E. Turos, J. E. Audia and S. J. Danishefsky, J. Am. Chem. Soc., 1989, 111, 8231–8236 CrossRef CAS.
- K. Yuki, M. Shindo and K. Shishido, Tetrahedron Lett., 2001, 42, 2517–2519 CrossRef CAS.
- L. T. Burke, D. J. Dixon, S. V. Ley and F. Rodríguez, Org. Biomol. Chem., 2005, 3, 274–280 CAS.
- J. Yin, L. Kong, C. Wang, Y. Shi, S. Cai and S. Gao, Chem.-Eur. J., 2013, 19, 13040–13046 CrossRef CAS PubMed.
- S. B. Singh, D. L. Zink, M. A. Goetz, A. W. Dombrowski, J. D. Polishook and D. J. Hazuda, Tetrahedron Lett., 1998, 39, 2243–2246 CrossRef CAS.
- J. Y. Li, G. Strobel, J. Harper, E. Lobkovsky and J. Clardy, Org. Lett., 2000, 2, 767–770 CrossRef CAS.
- V. Hellwig, T. Grothe, A. Mayer-Bartschmid, R. Endermann, F.-U. Geschke, T. Henkel and M. Stadler, J. Antibiot., 2002, 55, 881–892 CrossRef CAS.
- M. P. Segeth, A. Bonnefoy, M. Brönstrup, M. Knauf, D. Schummer, L. Toti, L. Vértesy, M.-C. Wetzel-Raynal, J. Wink and G. Seibert, J. Antibiot., 2003, 56, 114–122 CrossRef CAS.
- E. C. Marfori, S. Kajiyama, E. Fukusaki and A. Kobayashi, Z. Naturforsch., 2002, 57c, 465–470 Search PubMed.
- C. Boros, A. Dix, B. Katz, Y. Vasina and C. Pearce, J. Antibiot., 2003, 56, 862–865 CrossRef CAS.
- S. P. Putri, H. Kinoshita, F. Ihara, Y. Igarashi and T. Nihira, J. Antibiot., 2010, 63, 195–198 CrossRef CAS PubMed.
- J. Whitt, S. M. Shipley, D. J. Newman and K. M. Zuck, J. Nat. Prod., 2014, 77, 173–177 CrossRef CAS PubMed.
- T. Amagata, J. Xiao, Y.-P. Chen, N. Holsopple, A. G. Oliver, T. Gokey, A. B. Guliaev and K. Minoura, J. Nat. Prod., 2012, 75, 2193–2199 CrossRef CAS PubMed.
- K. Herath, H. Jayasuriya, D. L. Zink, J. Sigmund, F. Vicente, M. de la Cruz, A. Basilio, G. F. Bills, J. D. Polishook, R. Donald, J. Phillips, M. Goetz and S. B. Singh, J. Nat. Prod., 2012, 75, 420–424 CrossRef CAS PubMed.
- Y. Sugie, K. A. Dekker, T. Inagaki, Y.-J. Kim, T. Sakakibara, S. Sakemi, A. Sugiura, L. Brennan, J. Duignan, J. A. Sutcliffe and Y. Kojima, J. Antibiot., 2002, 55, 19–24 CrossRef CAS.
- Y. Sugie, S. Inagaki, Y. Kato, H. Nishida, C.-H. Pang, T. Saito, S. Sakemi, F. Dib-Hajj, J. P. Mueller, J. Sutcliffe and Y. Kojima, J. Antibiot., 2002, 55, 25–29 CrossRef CAS.
- C. Osterhage, R. Kaminsky, G. M. König and A. D. Wright, J. Org. Chem., 2000, 65, 6412–6417 CrossRef CAS.
- S. Toda, S. Yamamoto, O. Tenmyo, T. Tsuno, T. Hasegawa, M. Rosser, M. Oka, Y. Sawada, M. Konishi and T. Oki, J. Antibiot., 1993, 46, 875–883 CrossRef CAS.
- M. Ueno, T. Someno, R. Sawa, H. Iinuma, H. Naganawa, M. Ishizuka and T. Takeuchi, J. Antibiot., 1993, 46, 979–984 CrossRef CAS.
- S.-W. Yang, R. Mierzwa, J. Terracciano, M. Patel, V. Gullo, N. Wagner, B. Baroudy, M. Puar, T.-M. Chan, A. T. McPhail and M. Chu, J. Nat. Prod., 2006, 69, 1025–1028 CrossRef CAS PubMed.
- Y.-L. Yang, C.-P. Lu, M.-Y. Chen, K.-Y. Chen, Y.-C. Wu and S.-H. Wu, Chem.-Eur. J., 2007, 13, 6985–6991 CrossRef CAS PubMed.
- S. B. Singh, M. A. Goetz, E. T. Jones, G. F. Bills, R. A. Giacobbe, L. Herranz, S. Stevens-Miles and D. L. Williams, Jr, J. Org. Chem., 1995, 60, 7040–7042 CrossRef CAS.
- R. R. West, J. Van Ness, A.-M. Varming, B. Rassing, S. Biggs, S. Gasper, P. A. Mckernan and J. Piggott, J. Antibiot., 1996, 49, 967–973 CrossRef CAS.
- R. Kontnik and J. Clardy, Org. Lett., 2008, 10, 4149–4151 CrossRef CAS PubMed.
- S. B. Singh, D. L. Zink, B. Heimbach, O. Genilloud, A. Teran, K. C. Silverman, R. B. Lingham, P. Felock and D. J. Hazuda, Org. Lett., 2002, 4, 1123–1126 CrossRef CAS PubMed.
- Y. Hayakawa, N. Kanamaru, A. Shimazu and H. Seto, J. Antibiot., 1991, 44, 282–287 CrossRef CAS.
- Y. Hayakawa, N. Kanamaru, N. Morisaki, H. Seto and K. Furihata, Tetrahedron Lett., 1991, 32, 213–216 CrossRef CAS.
- T. Furumai, K. Eto, T. Sasaki, H. Higuchi, H. Onaka, N. Saito, T. Fujita, H. Naoki and Y. Igarashi, J. Antibiot., 2002, 55, 873–880 CrossRef CAS.
- J. W. Phillips, M. A. Goetz, S. K. Smith, D. L. Zink, J. Polishook, R. Onishi, S. Salowe, J. Wiltsie, J. Allocco, J. Sigmund, K. Dorso, S. Lee, S. Skwish, M. de la Cruz, J. Martín, F. Vicente, O. Genilloud, J. Lu, R. E. Painter, K. Young, K. Overbye, R. G. K. Donald and S. B. Singh, Chem. Biol., 2011, 18, 955–965 CrossRef CAS PubMed.
- S. B. Singh, M. A. Goetz, S. K. Smith, D. L. Zink, J. Polishook, R. Onishi, S. Salowe, J. Wiltsie, J. Allocco, J. Sigmund, K. Dorso, M. de la Cruz, J. Martín, F. Vicente, O. Genilloud, R. G. K. Donald and J. W. Phillips, Bioorg. Med. Chem. Lett., 2012, 22, 7127–7130 CrossRef CAS PubMed.
- R. Sawa, Y. Takahashi, H. Hashizume, K. Sasaki, Y. Ishizaki, M. Umekita, M. Hatano, H. Abe, T. Watanabe, N. Kinoshita, Y. Homma, C. Hayashi, K. Inoue, S. Ohba, T. Masuda, M. Arakawa, Y. Kobayashi, M. Hamada, M. Igarashi, H. Adachi, Y. Nishimura and Y. Akamatsu, Chem.-Eur. J., 2012, 18, 15772–15781 CrossRef CAS PubMed.
- H. J. Jessen and K. Gademann, Nat. Prod. Rep., 2010, 27, 1168–1185 RSC.
- A. A. Alfatafta, J. B. Gloer, J. A. Scott and D. Malloch, J. Nat. Prod., 1994, 57, 1696–1702 CrossRef CAS.
- H. Fujimoto, M. Ikeda, K. Yamamoto and M. Yamazaki, J. Nat. Prod., 1993, 56, 1268–1275 CrossRef CAS.
- S. Bergmann, J. Schümann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS PubMed.
- M. Shibazaki, M. Taniguchi, T. Yokoi, K. Nagai, M. Watanabe, K. Suzuki and T. Yamamoto, J. Antibiot., 2004, 57, 379–382 CrossRef CAS.
- J. C. Lee, S. J. Coval and J. Clardy, J. Antibiot., 1996, 49, 693–696 CrossRef CAS.
- A. Haga, H. Tamoto, M. Ishino, E. Kimura, T. Sugita, K. Kinoshita, K. Takahashi, M. Shiro and K. Koyama, J. Nat. Prod., 2013, 76, 750–754 CrossRef CAS PubMed.
- S. Hayakawa, H. Minato and K. Katagiri, J. Antibiot., 1971, 24, 653–654 CrossRef CAS.
- E. B. Gutierrez-Cirlos, T. Merbitz-Zahradnik and B. L. Trumpower, J. Biol. Chem., 2004, 279, 8708–8714 CrossRef CAS PubMed.
- M. Tanabe and S. Urano, Tetrahedron, 1983, 39, 3569–3574 CrossRef CAS.
- S. B. Singh, X. Li and T. Chen, Tetrahedron Lett., 2011, 52, 6190–6191 CrossRef CAS PubMed.
- W. Keller-Schierlein, R. Muntwyler, W. Pache and H. Zähner, Helv. Chim. Acta, 1969, 52, 127–142 CrossRef CAS.
- F. Tomita, T. Tamaoki, K. Shirahata, M. Kasai, M. Morimoto, S. Ohkubo, K. Mineura and S. Ishii, J. Antibiot., 1980, 33, 668–670 CrossRef CAS.
- K. Kobinata, M. Uramoto, T. Mizuno and K. Isono, J. Antibiot., 1980, 33, 244–246 CrossRef CAS.
- J. A. Waitz, A. Horan, M. Kalyanpur, B. K. Lee, D. Loebenberg, J. A. Marquez, G. Miller and M. G. Patel, J. Antibiot., 1981, 34, 1101–1106 CrossRef CAS.
- A. K. Mallams, M. S. Puar and R. R. Rossman, J. Am. Chem. Soc., 1981, 103, 3938–3940 CrossRef CAS.
- A. K. Mallams, M. S. Puar, R. R. Rossman, A. T. McPhail and R. D. Macfarlane, J. Am. Chem. Soc., 1981, 103, 3940–3943 CrossRef CAS.
- H. Zhang, J. A. White-Phillip, C. E. Melançon, H.-j. Kwon, W.-l. Yu and H.-w. Liu, J. Am. Chem. Soc., 2007, 129, 14670–14683 CrossRef CAS PubMed.
- A. Kawashima, Y. Nakamura, Y. Ohta, T. Akama, M. Yamagishi and K. Hanada, J. Antibiot., 1992, 45, 207–212 CrossRef CAS.
- P. W. Schindler and M. C. Scrutton, Eur. J. Biochem., 1975, 55, 543–553 CrossRef CAS PubMed.
- I. Tinhofer, G. Anether, M. Senfter, K. Pfaller, D. Bernhard, M. Hara and R. Greil, FASEB J., 2002, 16, 1295–1297 CAS.
- T. Nakashima, M. Miura and M. Hara, Cancer Res., 2000, 60, 1229–1235 CAS.
- G. Anether, I. Tinhofer, M. Senfter and R. Greil, Blood, 2003, 101, 4561–4568 CrossRef CAS PubMed.
- H. Nakajima, K. Sakaguchi, I. Fujiwara, M. Mizuta, M. Tsuruga, J. Magae and N. Mizuta, Biochem. Biophys. Res. Commun., 2007, 356, 260–265 CrossRef CAS PubMed.
- X.-Y. Jia, Z.-H. Tian, L. Shao, X.-D. Qu, Q.-F. Zhao, J. Tang, G.-L. Tang and W. Liu, Chem. Biol., 2006, 13, 575–585 CrossRef CAS PubMed.
- J. Fang, Y. Zhang, L. Huang, X. Jia, Q. Zhang, X. Zhang, G. Tang and W. Liu, J. Bacteriol., 2008, 190, 6014–6025 CrossRef CAS PubMed.
- J. J. Lee, J. P. Lee, P. J. Keller, C. E. Cottrell, C.-J. Chang, H. Zähner and H. G. Floss, J. Antibiot., 1986, 39, 1123–1134 CrossRef CAS.
- Y. Sun, H. Hong, F. Gillies, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 150–156 CrossRef CAS PubMed.
- W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1994, 116, 6457–6458 CrossRef CAS.
- W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1998, 120, 7411–7419 CrossRef CAS.
- K. Takeda, Y. Igarashi, K. Okazaki, E. Yoshii and K. Yamaguchi, J. Org. Chem., 1990, 55, 3431–3434 CrossRef CAS.
- K. Takeda, H. Kato, H. Sasahara and E. Yoshii, J. Chem. Soc., Chem. Commun., 1986, 1197–1198 RSC.
- K. Takeda, E. Kawanishi, H. Nakamura and E. Yoshii, Tetrahedron Lett., 1991, 32, 4925–4928 CrossRef CAS.
- R. K. Boeckman, Jr., P. Shao, S. T. Wrobleski, D. J. Boehmler, G. R. Heintzelman and A. J. Barbosa, J. Am. Chem. Soc., 2006, 128, 10572–10588 CrossRef PubMed.
- W. R. Roush, C. Limberakis, R. K. Kunz and D. A. Barda, Org. Lett., 2002, 4, 1543–1546 CrossRef CAS PubMed.
- D. Niu and T. R. Hoye, Org. Lett., 2012, 14, 828–831 CrossRef CAS PubMed.
- R. Nakai, H. Ogawa, A. Asai, K. Ando, T. Agatsuma, S. Matsumiya, S. Akinaga, Y. Yamashita and T. Mizukami, J. Antibiot., 2000, 53, 294–296 CrossRef CAS.
- T. Agatsuma, T. Akama, S. Nara, S. Matsumiya, R. Nakai, H. Ogawa, S. Otaki, S. Ikeda, Y. Saitoh and Y. Kanda, Org. Lett., 2002, 4, 4387–4390 CrossRef CAS PubMed.
- T. H. Lambert and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 426–427 CrossRef CAS PubMed.
- T. R. Hoye, V. Dvornikovs and E. Sizova, Org. Lett., 2006, 8, 5191–5194 CrossRef CAS PubMed.
- R. M. de Figueiredo, M. Voith, R. Fröhlich and M. Christmann, Synlett., 2007, 3, 391–394 Search PubMed.
- R. M. de Figueiredo, R. Fröhlich and M. Christmann, Angew. Chem., Int. Ed., 2007, 46, 2883–2886 CrossRef CAS PubMed.
- Y. Sugie, H. Hirai, H. Kachi-Tonai, Y.-J. Kim, Y. Kojima, Y. Shiomi, A. Sugiura, Y. Suzuki, N. Yoshikawa, L. Brennan, J. Duignan, L. H. Huang, J. Sutcliffe and N. Kojima, J. Antibiot., 2001, 54, 917–925 CrossRef CAS.
- T. Nogawa, M. Kawatani, M. Uramoto, A. Okano, H. Aono, Y. Futamura, H. Koshino, S. Takahashi and H. Osada, J. Antibiot., 2013, 66, 621–623 CrossRef CAS PubMed.
- Y. Futamura, M. Kawatani, M. Muroi, H. Aono, T. Nogawa and H. Osada, ChemBioChem, 2013, 14, 2456–2463 CrossRef CAS PubMed.
- Y. Igarashi, Y. Kim, Y. In, T. Ishida, Y. Kan, T. Fujita, T. Iwashita, H. Tabata, H. Onaka and T. Furumai, Org. Lett., 2010, 12, 3402–3405 CrossRef CAS PubMed.
- Y. Kim, Y. In, T. Ishida, H. Onaka and Y. Igarashi, Org. Lett., 2013, 15, 3514–3517 CrossRef CAS PubMed.
- C. Klemke, S. Kehraus, A. D. Wright and G. M. König, J. Nat. Prod., 2004, 67, 1058–1063 CrossRef CAS PubMed.
- D. E. Williams, D. S. Dalisay, F. Li, J. Amphlett, W. Maneerat, M. A. G. Chavez, Y. A. Wang, T. Matainaho, W. Yu, P. J. Brown, C. H. Arrowsmith, M. Vedadi and R. J. Andersen, Org. Lett., 2013, 15, 414–417 CrossRef CAS PubMed.
- J. C. Arens, F. Berrué, J. K. Pearson and R. G. Kerr, Org. Lett., 2013, 15, 3864–3867 CrossRef CAS PubMed.
- T. Tsuchida, H. Iinuma, C. Nishida, N. Kinoshita, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1995, 48, 1104–1109 CrossRef CAS.
- F. Hiramatsu, T. Murayama, T. Koseki, K. Okada and Y. Shiono, Helv. Chim. Acta, 2008, 91, 1595–1603 CrossRef CAS.
- F. Hiramatsu, T. Murayama, T. Koseki and Y. Shiono, Phytochemistry, 2007, 68, 1267–1271 CrossRef CAS PubMed.
- M. Clericuzio, R. Negri, M. Cossi, G. Gilardoni, D. Gozzini and G. Vidari, Phytochemistry, 2013, 93, 192–198 CrossRef CAS PubMed.
- Y. Shiono, F. Hiramatsu, T. Murayama, T. Koseki, T. Funakoshi, K. Ueda and H. Yasuda, Z. Naturforsch, 2007, 62b, 1585–1589 Search PubMed.
- G.-H. Li, M. Duan, Z.-F. Yu, L. Li, J.-Y. Dong, X.-B. Wang, J.-W. Guo, R. Huang, M. Wang and K.-Q. Zhang, Phytochemistry, 2008, 69, 1439–1445 CrossRef CAS PubMed.
- G. Wu, A. Lin, Q. Gu, T. Zhu and D. Li, Mar. Drugs, 2013, 11, 1399–1408 CrossRef CAS PubMed.
- H. Oh, P. R. Jensen, B. T. Murphy, C. Fiorilla, J. F. Sullivan, T. Ramsey and W. Fenical, J. Nat. Prod., 2010, 73, 998–1001 CrossRef CAS PubMed.
- M. Isaka, P. Chinthanom, T. Boonruangprapa, N. Rungjindamai and U. Pinruan, J. Nat. Prod., 2010, 73, 683–687 CrossRef CAS PubMed.
- S. B. Singh, D. Zink, J. Polishook, D. Valentino, A. Shafiee, K. Silverman, P. Felock, A. Teran, D. Vilella, D. J. Hazuda and R. B. Lingham, Tetrahedron Lett., 1999, 40, 8775–8779 CrossRef CAS.
- I. E. Mohamed, H. Gross, A. Pontius, S. Kehraus, A. Krick, G. Kelter, A. Maier, H.-H. Fiebig and G. M. König, Org. Lett., 2009, 11, 5014–5017 CrossRef CAS PubMed.
- C. Hemtasin, S. Kanokmedhakul, K. Kanokmedhakul, C. Hahnvajanawong, K. Soytong, S. Prabpai and P. Kongsaeree, J. Nat. Prod., 2011, 74, 609–613 CrossRef CAS PubMed.
- K. Minagawa, S. Kouzuki, J. Yoshimoto, Y. Kawamura, H. Tani, T. Iwata, Y. Terui, H. Nakai, S. Yagi, N. Hattori, T. Fujiwara and T. Kamigauchi, J. Antibiot., 2002, 55, 155–164 CrossRef CAS.
- H. Kaise, M. Shinohara, W. Miyazaki, T. Izawa, Y. Nakano, M. Sugawara, K. Sugiura and K. Sasaki, J. Chem. Soc., Chem. Commun., 1979, 726–727 RSC.
- B. B. Jarvis, J. Salemme and A. Morais, Nat. Toxins, 1995, 3, 10–16 CrossRef CAS.
- X. Ma, L. Li, T. Zhu, M. Ba, G. Li, Q. Gu, Y. Guo and D. Li, J. Nat. Prod., 2013, 76, 2298–2306 CrossRef CAS PubMed.
- K. Sakai, K. Watanabe, K. Masuda, M. Tsuji, K. Hasumi and A. Endo, J. Antibiot., 1995, 48, 447–456 CrossRef CAS.
- R. Uchida, R. Imasato, Y. Yamaguchi, R. Masuma, K. Shiomi, H. Tomoda and S. Ōmura, J. Antibiot., 2005, 58, 397–404 CrossRef CAS PubMed.
- S. B. Singh, D. L. Zink, A. W. Dombrowski, G. Dezeny, G. F. Bills, J. P. Felix, R. S. Slaughter and M. A. Goetz, Org. Lett., 2001, 3, 247–250 CrossRef CAS.
- W. Wilk, H. Waldmann and M. Kaiser, Bioorg. Med. Chem., 2009, 17, 2304–2309 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |