Different strategies to enhance the activity of lipase catalysts
Marzia
Marciello†‡
,
Marco
Filice‡
and
Jose M.
Palomo
*
Departamento de Biocatálisis, Instituto de Catálisis (CSIC), c/Marie Curie 2, Cantoblanco Campus UAM, 28049, Madrid, Spain. E-mail: josempalomo@icp.csic.es; Fax: +34-91-585-4760; Tel: +34-91-585-4809
First published on 22nd June 2012
Abstract
The design of new strategies for improving the catalytic activity of lipases in their application on non-natural substrates is in great demand due to their biotechnological applications. This perspective illustrates the power of technologies, such as the immobilization of enzymes on nanomaterials and the site-specific chemical modification of enzyme surfaces, to prepare highly active semisynthetic lipases. These techniques have produced optimal biocatalysts for different important bioprocesses such as biodiesel production, regioselective deprotection of carbohydrates and kinetic resolution of different drug precursors.
 Marzia Marciello | Marzia Marciello obtained her BS in pharmaceutical chemistry and technology from the University of Pavia, Italy, in 2003, and her PhD degree (summa cum laude) from the Pharmaceutical Chemistry Department of the Parma University, Italy, in 2008. During her postdoctoral research she worked for three years in the Biocatalysis Department in Institute of Catalysis (ICP, CSIC) in Madrid. She currently works as an Associate Research Scientist (European Project) in the Material Science Institute of Madrid (ICMM, CSIC). Her current research interests include the synthesis and functionalization with biomolecules of magnetic nanoparticles for cancer imaging and treatment. |
 Marco Filice | Marco Filice obtained his BS in pharmaceutical chemistry and technology at the University of Pavia, Italy, in 2003. He received his PhD degree (summa cum laude) in 2007 at the Pharmaceutical Chemistry Department in the University of Pavia. During his postdoctoral research he joined the Biocatalysis Department in the Institute of Catalysis (IPC, CSIC) in Madrid in 2008 where currently he works as an Associate Research Scientist with a JAE-DOC contract. His current research interests include carbohydrate and nucleoside chemistry with enzymes, applied biocatalysis, site-directed chemical modification of enzymes and asymmetric biotransformations in fine and pharmaceutical chemistry. |
 Jose M. Palomo | Jose M. Palomo received his PhD degree (summa cum laude) in 2003 at the Autonoma University of Madrid working at the Biocatalysis Department in the Institute of Catalysis (IPC, CSIC) in Madrid. During his postdoctoral research he joined the Chemical Biology Department of the Max Planck Institute in Dortmund in 2004. In 2006, he began his appointment as an Associate Research Scientist at the Biocatalysis Department in ICP-CSIC. From 2009, he is a Tenured Scientist (Associate Professor) in ICP-CSIC. His current research interests include immobilization of biomolecules, design of semisynthetic enzymes, asymmetric biotransformations and carbohydrate chemistry with enzymes. |
1. Introduction
Natural enzymes are capable of catalyzing reactions with up to 1017-fold rate accelerations1 and with exquisite control of the regio- and stereochemistry. Such control makes the use of enzymes very attractive as alternatives to traditional catalysts in the synthesis of complex and high added value molecules, especially where chemical routes are difficult to implement.2,3 Enzymes have evolved over billions of years to operate most effectively under physiological conditions on a narrow range of natural substrates and usually at concentrations in the low mM range. In contrast, an efficient industrial synthetic process may require a biocatalytic enzyme to operate on non-natural substrates and under conditions in which the enzyme shows very low or even no activity.4,5 As a result of these requirements, the majority of naturally occurring enzymes are not suitable for industrial-scale biocatalytic processes without further modification of the enzyme itself.One of the most representative examples are lipases (acyl-hydrolases EC 3.1.1.3). In nature they catalyze the hydrolysis of the ester bond in tri-, di- and mono-glycerides of long-chain fatty acids into fatty acids and glycerol. They differ from esterase (EC 3.1.1.1) due to their ability to hydrolyze triglyceride at the lipid–water interface.6 Nevertheless, this aspect, which involves a complex catalytic mechanism, shows the power of these enzymes as biocatalysts.7
Lipases show a very high versatility, recognizing a broad range of different substrates.8–19 They catalyze the hydrolysis, transesterification and synthesis of esters and exhibit enantioselective properties.18–20 The ability of lipases to perform very specific chemical transformations (biotransformations) has made them increasingly popular in the food, detergent and chemical industries.18–21 In particular, these enzymes are quite interesting in the pharmaceutical industry and in fine organic chemistry for the production of chiral reagents by the resolution of racemic mixtures, desymmetrization reactions or regioselective protection and deprotection of functional groups.18,19 Lipases have emerged as one of the leading biocatalysts with a proven potential towards contributing to the multibillion dollar and underexploited bioindustry.22,23
The main aspect of catalysis by lipases is based on the movement of an oligopeptide chain (called a “lid”) on the protein structure which blocks the active site in a closed conformation state, making the enzyme inactive. However, in homogeneous aqueous media, lipases exist in an equilibrium between this closed conformation and another, where the lid is shifted making the active site accessible to the substrate (an open conformation). This equilibrium is mainly shifted towards the closed form under these conditions.7,24–26 However, in nature lipases are adsorbed onto the oil–water interface in order to be active, which means that this equilibrium is completely shifted to this open form when a hydrophobic interface is present, thus obtaining the maximum activity.27–30 Therefore, the development of strategies to shift this equilibrium to the open and active form seems to represent a way to achieve highly active lipase catalysts in a different set of reactions.
Chemical modification of proteins should be considered as an interesting tool to manipulate and/or improve the enzyme properties, together with immobilization on solid supports13 and molecular biology.9 Despite the fact that the latter, nowadays, is becoming an almost standard laboratory technique, chemical modification follows due to its straightforward role. In fact, for many years, chemical manipulation of the protein surface was hampered by the low control of the modification reaction, resulting in one of the biggest drawbacks related to this technique. In the last few years, this handicap has been overcome thanks to the wide range of novel site-directed bio-orthogonal transformation techniques permitting the fine control of the installation and manipulation of various chemical groups on the protein surface.31 Furthermore, chemical modification also shows some advantages compared to genetic manipulation (side-directed mutagenesis or directed evolution).32 For example, theoretically, there are no limits to the nature of the groups installed on the enzyme,33 chemical modification is performed on an already folded protein (differently to that required by molecular biology where the modification can cause incorrect folding) and the knowledge of the crystal structure of the enzyme is not mandatory. Finally, a great advantage of chemical modification is that it is faster than genetic modification and it permits the generation of a large combinatorial library of different chemically-modified enzymes.
Another approach, which has been extensively used for the improvement of lipase activity, has been the application of immobilization techniques.34–38 Different strategies considering the nature of the support (e.g., polysaccharides, epoxyacrilic resins, silica and glass slides), the pore size of the support or the binding methodology (ionic exchange, hydrophobic adsorption, entrapment, crosslinking, etc.) have been critical for a particular set of enzyme properties.34–38
Recently a growing interest towards the use of nanomaterials (especially nanoparticles and nanofibers) as carriers for enzyme immobilization has been observed.39 Different approaches (i.e. enzyme adsorption, covalent attachment or enzyme encapsulation) with the aim to improve the stability, the activity and the reusability of the biocatalyst have been performed.40 The advantages of using this kind of support for enzyme immobilization are characterized by minimum diffusion limitations. The reduction in the size of the enzyme-carrier materials can also generally improve the efficiency of immobilized enzymes. In fact, smaller particles can provide a larger surface area for the attachment of enzymes, leading to higher enzyme loading per unit mass of particles.41 Keeping in mind these considerations, nanomaterials provide an ideal solution to the usually contradictory issues encountered in the optimization of immobilized enzymes: minimum diffusional limitation, maximum surface area per unit mass and high enzyme loading.
In this perspective, we have summarized the major avenues of progress regarding the improvement of the catalytic activity of lipases by chemical modification protocols or directed immobilization on nanostructures.
2. Improved lipase activity by chemical modification
The site-directed chemical modification of a protein represents a versatile method towards modulating the catalytic properties of an enzyme in industrial processes. The conjugation of synthetic polymers to proteins by covalent attachment has significantly improved different properties such as stability, activity, biocompatibility, etc.42Recently, a thermophilic lipase from Bacillus thermocatenulatus (BTL2) was modified via a site-directed method on a solid phase at a free cysteine residue (Cys64) using the tailor-made polymers, pyridyldisulfide poly-aminated-dextrans (NH2-dextran) or monocarboxylated-polyethyleneglycol (PEG-COOH) (Fig. 1).43 Different parameters, such as the chemical nature of the polymer, polymer size or enzyme immobilization protocol, were evaluated. This fine-tuned strategy enhanced the catalytic activity of this lipase towards several substrates. For example, the modification of immobilized glyoxyl-BTL2 with PEG-COOH increased the enzyme activity 5-fold towards the hydrolysis of 2-O-butyryl-2-phenylacetic acid and 3-fold in the hydrolysis of ethyl butyrate (Table 1). The NH2-dextran modification also improved the enzyme activity 2-fold towards p-nitrophenyl butyrate and 1.5-fold towards 2-O-butyryl-2-phenylacetic acid.
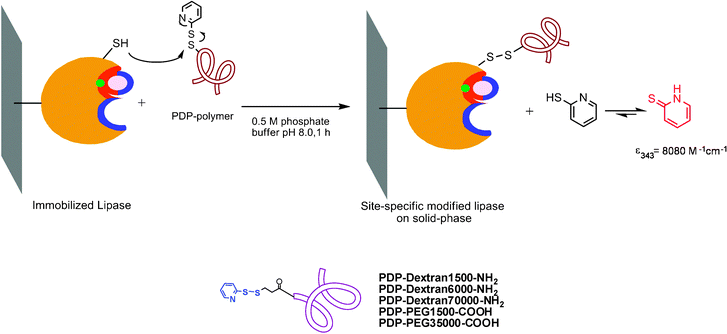 | ||
| Fig. 1 Site-directed chemical modification of immobilized BTL2 with tailor-made polymers.60 Reproduced by permission of Elsevier. | ||
| Biocatalyst | Specific activity | ||
|---|---|---|---|
| 1 a | 2 a | 3 b | |
| a μmol min−1 mglip−1; b nmol h−1 mglip−1. 1: p-nitrophenyl butyrate; 2: ethyl butyrate; 3: 2-O-butyryl-2 phenylacetic acid. | |||
| Glyoxyl-BTL2 | 10.15 | 4.72 | 0.74 |
| NH2-dextran-glyoxyl-BTL2 | 22.4 | 2 | 1.04 |
| COOH-PEG-glyoxyl-BTL2 | 12.25 | 14.28 | 3.62 |
Another example of site-specific chemical modification of lipases for creating artificial glycoproteins has been recently reported.44 The introduction of a sugar tag on a protein represents an interesting advantage in protein chemistry. In fact, in addition to improving the functional diversity of proteins45 (e.g., protein–protein bioconjugations with lectins46 or covalent attachment of multifluorescent labels), a chemical glycosylation could be useful especially for the alteration of the catalytic properties of the enzymes. Thus, site-specific incorporation of dextran polymers on the N-terminus of the Candida antarctica lipase (fraction B) (CAL-B) by reductive amination has been performed. The oligosaccharide incorporation was performed on a solid phase (Fig. 2) in order to achieve a rapid and efficient strategy to obtain a complete enzyme transformation. The chemical modification of CAL-B with dextran of 1500 kDa was the best alternative. The effect of the incorporation of this low-molecular weight dextran onto the CAL-B surface was subsequently evaluated in different reactions. The activity of the modified enzyme improved more than 2-fold in the hydrolysis of p-nitrophenylbutyrate (p-NPB) and (±)-methyl mandelate. The highest synthetic enzyme activity in the transesterification reaction between methyl butyrate and glycerol in aqueous media was achieved after this glycosylation step (80% yield in the monoglyceryl ester).
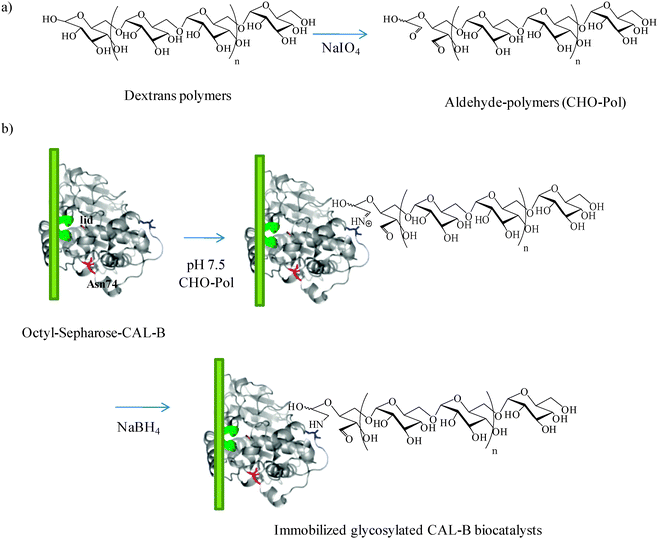 | ||
| Fig. 2 Site-directed chemical modification of immobilized CAL-B with dextran.44 Reproduced by permission of Wiley. | ||
The multifunctionality of biomacromolecules causes a major and continuing demand for the development of new chemical technologies which provide alternatives to the methods mentioned above. The required chemistry must be compatible with the functional groups found in proteins and proceed chemoselectively under mild conditions and in aqueous solution, preferably in the absence of any potentially denaturating co-solvent. Thus, the chemical incorporation of a diene group on a lipase represents an interesting modification of proteins for the site-specific incorporation of different molecules (fluorescent labels, polymers and peptides) by a chemoselective Diels–Alder cycloaddition reaction.
The chemical modification of the Candida antarctica (fraction B) lipase (CAL-B) by a trans–trans hexadiene derivative has also been applied for selective enzyme immobilization on ionic exchange matrices and for the direct or subsequent chemical incorporation of fluorescent labels.
The introduction of this dienyl anchor point was an obligatory element required for the dienophile fluorescent labelling (Fig. 3).47 Surprisingly, this specific modification on CAL-B permitted the full immobilization of this enzyme on Q Sepharose by ionic exchange at pH 7 (Fig. 4), whereas the unmodified enzyme was not immobilized under any experimental conditions. Furthermore, the activity of this CAL-B-diene variant increased 2-fold for the first time in the hydrolysis of p-NPB after the immobilization in an ionic exchange interaction (Fig. 4) (a characteristic only observed for this enzyme on the hydrophobic interfaces). Therefore, this site-specific modification permitted the enhancement of the lipase activity and was successfully used for the incorporation of fluorescent molecules by a Diels–Alder cycloaddition.47
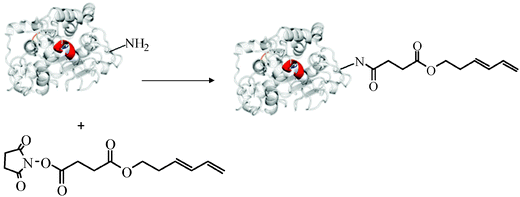 | ||
| Fig. 3 Site-directed anchoring of a trans–trans hexadiene moiety to the N-terminus of CAL-B.47 Reproduced by permission of The Royal Society of Chemistry. | ||
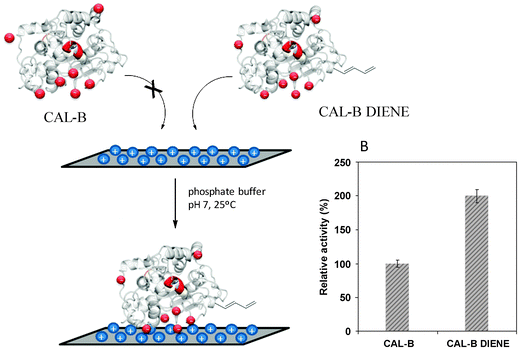 | ||
| Fig. 4 Immobilization of CAL-B on Q-Sepharose supports. (A) Immobilization scheme. (B) Relative specific activity of unmodified CAL-B and the CAL-B diene.47 Reproduced by permission of The Royal Society of Chemistry. | ||
Another example is based on the surface derivatization of lipase microcrystals with polyanionic anhydrides.48 Gupta and co-workers developed a useful methodology to obtain hyperactivated and modified lipase biocatalysts. A chemical modification of the lysine groups of the Pseudomonas cepacia lipase with pyromellitic dianhydride (modifying 72% of the total amino groups) and a further optimized precipitation of the modified biocatalyst was performed (Fig. 5).49 PCMC (protein coated microcrystals), CLPCMC (crosslinked protein coated microcrystals), EPROS (enzyme precipitated and rinsed with organic solvents) and pH tuned preparations of the modified and unmodified lipase were prepared. These biocatalysts were tested in the transesterification reaction of tributyrin using n-octane and DMF as the reaction mediums.49 CLPCMC lipase preparation was the best catalyst, with an activity almost 16-fold higher than the unmodified pH tuned lipase (Table 2). This biocatalyst remained the best at the different conditions tested (e.g. a 1% and 5% (v/v) water concentration) whereas the unmodified pH tuned lipase showed no activity at all. For example, the initial activity rate of the CLPCMC lipase preparation was 36-fold higher than the PCMC lipase derivative with a 1% (v/v) in water content. This enhancement was even higher at a 5% (v/v) water content (360-fold) (Table 3). Therefore this type of modification represents an interesting method towards producing highly active lipase catalysts in organic solvents.49
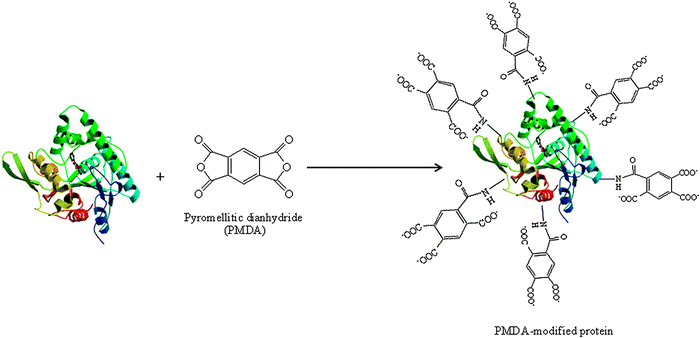 | ||
| Fig. 5 Chemical modification of the protein with pyromellitic dianhydride (PMDA). | ||
| Lipase formulation | Initial rates (nmol min−1 mg−1)a |
|---|---|
| a The initial rates were calculated from aliquots taken within 0.25–1 h. The percentage conversions during these time periods were in the range of 1–25% and were in a linear range. The enzyme used was a biocatalyst formulation made from 2 mg of the solid enzyme (in a 2 ml reaction volume) in each case. | |
| pH tuned (unmodified) | 128 |
| EPROS of unmodified lipase | 240 |
| PCMC of unmodified lipase | 620 |
| CLPCMC of unmodified lipase | 974 |
| pH tuned (modified) | 513 |
| EPROS of PMDA modified lipase | 472 |
| PCMC of PMDA modified lipase | 1150 |
| CLPCMC of PMDA modified lipase | 1970 |
| Biocatalyst preparation | Initial rates (nmol min−1 mg−1)a | |
|---|---|---|
| 1% (v/v) water | 5% (v/v) water | |
| a Initial rates were calculated from aliquots taken within 1–5 h. The percentage conversions during these time periods were in the range of 1–20% and were in a linear range. The enzyme used was a biocatalyst preparation made from 2 mg of the solid enzyme (in a 2 ml reaction volume) in each case. | ||
| pH tuned | 0 | 0 |
| EPROS of unmodified lipase | <0.001 | 0.19 |
| EPROS of modified lipase | <0.001 | 0.42 |
| PCMC of unmodified lipase | 0.01 | <0.001 |
| PCMC of modified lipase | 0.02 | <0.001 |
| CLPCMC of unmodified lipase | 0.05 | 2.5 |
| CLPCMC of modified lipase | 0.72 | 7.2 |
3. Improved lipase activity by immobilization on nanostructures
Among the most recent examples of nanomaterial applications in biocatalysis, Sörensen et al. have reported the immobilization of the lipase from Thermomyces lanuginosus (TLL) within mesoporous spherical particles, produced by a new emulsion and solvent evaporation method (ESE). In this research paper, the increase in the enzyme specific activity after immobilization on hydrophilic and hydrophobic mesoporous particles versus the free enzyme solution was compared.50Hydrophobic silica particles were prepared by silylation of the pore walls of the hydrophilic particles with trimethylchlorosilane, previously prepared by templating with Pluronic F127 and poly (propylene glycol).The lipase was labelled with a fluorophore (Alexa Fluor® 488) to facilitate confocal laser scanning microscopy (CLSM) studies.
Mesoporous particles were selected as the support for (i) a large pore size (improving the enzyme stability) and (ii) spherically-shaped particles (increasing the superficial area of the support). They present a large particle size distribution from 2 to 40 μm (Fig. 6).
 | ||
| Fig. 6 SEM photograph of mesoporous silica spheres.50 Reproduced by permission of The Royal Society of Chemistry. | ||
Two different hydrophilic mesoporous silica particles were studied, with hydrophilic or hydrophobic pore walls. The hydrophobization of the pore walls resulted in a slight decrease in the pore size with respect to the hydrophilic ones (from 67/39 to 65/36 Å). A decrease in the loading efficiency of the lipase immobilization into the hydrophobic spheres was observed, probably due to the small reduction in pore size (similar to the enzyme size) after silylation (46 Å). The specific activity of the lipase was improved after immobilization on the hydrophobic carriers, determined by CLSM studies in combination with UV-Vis measurements. CLSM was used to evaluate the lipase distribution in the mesoporous spheres and the bond strength. The confocal photographs of the hydrophilic and hydrophobic particles show the complete penetration of the enzyme into the core of the particles (Fig. 7). CLSM analysis also showed an insignificant leakage of the immobilized TLL for the hydrophobic material while a higher release rate and amount (fluorescence decrease of a 25% in 2 h) was observed for the hydrophilic support. UV-Vis spectroscopy measurements display the trend in the enzymatic activity. The decreased activity for the hydrophilic material with respect to the hydrophobic material was confirmed by CLSM experiments. In particular, the specific activity values were increased 2.5-fold when the lipase was immobilized on the hydrophobic particles. The activity of the lipase immobilized on the hydrophilic material was comparable with the free enzyme.
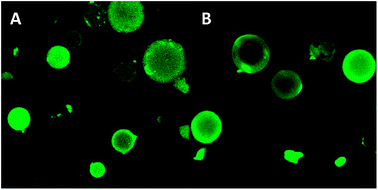 | ||
| Fig. 7 CLSM microphographs of the (A) hydrophilic and (B) hydrophobic mesoporous spheres with the immobilized fluorescent-labelled lipase.50 Reproduced by permission of The Royal Society of Chemistry. | ||
Chronopoulou et al. have reported the ability of nanostructured poly-DL-lactic acid (PDLLA)-based carriers to enhance the activity and stability of the Candida rugosa lipase (CRL) (Fig. 8).51
 | ||
| Fig. 8 SEM micrograph of the PDLLA nanoparticles (A) and EF-TEM images of immunolabeled CRL in the presence (B) and absence (C) of the PDLLA nanoparticles.51 Reproduced by permission of The Royal Society of Chemistry. | ||
PDLLA spherical nanoparticles, with an average diameter of 220 nm, were prepared using an innovative method based on an osmosis process. CRL was immobilized on PDLLA nanoparticles by physical adsorption and the protein loading was quite high (59 mg/100 mg of the polymer).
The protein was marked by gold and analyzed by transmission electron microscopy (TEM). Practically all the biocatalyst was located inside the PDLLA nanoparticles. This result indicates that the technique used to remove the non-immobilized enzyme, based on filtration through a cellulose nitrate filter membrane with a pore diameter of 0.025 μm, was effective.
Enzymatic activity of free and immobilized CRL on the PDLLA nanoparticles was evaluated in aqueous and organic mediums. In the aqueous medium, the lipolytic activity of CRL was strongly increased by immobilization on the PDLLA nanoparticles, almost 3-fold higher than the soluble enzyme activity (Fig. 9).
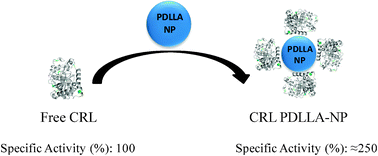 | ||
| Fig. 9 Hyperactivating immobilization of CRL onto the PDLLA-NP surface. | ||
Lipases are used as biocatalysts in synthetic reactions because they can catalyze esterifications and trans-esterifications in non-aqueous media. For this reason the activity of a lipase in different organic solvents was evaluated.
While in polar solvents, such as acetonitrile, both the free and immobilized CRL conjugates lost almost all their activity, whereas incubation in more hydrophobic solvents (i.e. n-hexane or tert-butyl-methyl ether) led to higher activity values. In particular, it was shown that after 6 h of incubation at 40 °C, the relative activity of the CRL/PDLLA bioconjugates increased 3.6 times with respect to the soluble enzyme.
Soluble and immobilized lipase was used to catalyze the transesterification of sulcatol with vinyl acetate in n-hexane. The relative activity of the lipase immobilized on the nanoparticles was 1.4 times higher than the free lipase (Fig. 10).
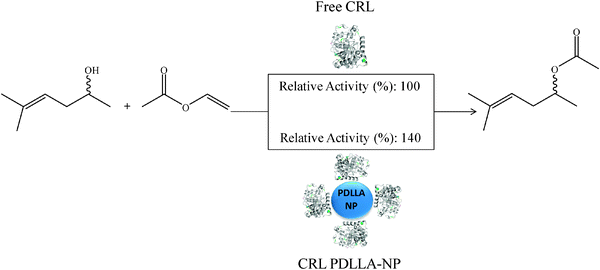 | ||
| Fig. 10 Sulcatol transesterification with vinyl acetate, catalyzed by free and immobilized CRL. | ||
The immobilization of the lipase CRL on the PDLLA nanoparticles was interestingly revealed to enhance its activity. In particular the CRL/PDLLA bioconjugates showed a strong lipolytic activity with respect to the free enzyme.
A study conducted by Chen and coworkers demonstrated that zirconia nanoparticles, modified by a carboxylic surfactant with a long alkyl chain, can significantly enhance the activity of the immobilized lipase in an organic-phase synthesis.52 Some advantages resulting from the use of an inorganic support with respect to a polymeric material are a higher mechanical strength, higher chemical stability in an organic media and lower cost.
Zirconia nanoparticles with a diameter of 20 nm were prepared and coated with two surfactants in its carboxylate anionic form (Tween 85 and Erucic acid) in order to obtain an inorganic support but with a hydrophobic surface (Fig. 11).
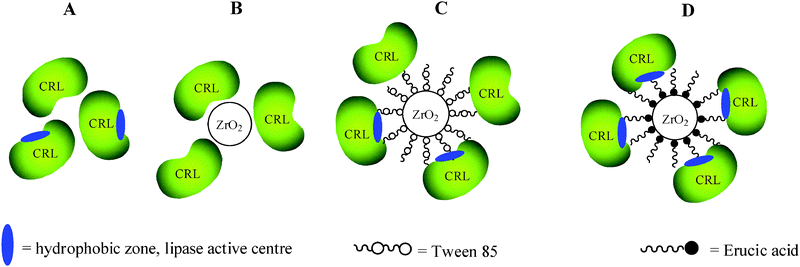 | ||
| Fig. 11 Schematic representation of (A) a crude CRL powder, (B) CRL immobilized on unmodified zirconia nanoparticles, (C) CRL immobilized on Tween modified nanoparticles and (D) CRL immobilized on Erucic zirconia nanoparticles. | ||
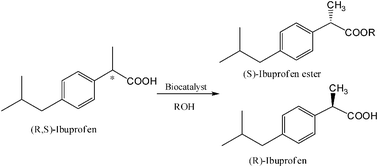
Subsequently, lipases from Candida rugosa (CRL) and Pseudomonas cepacia (PCL) were immobilized on the modified zirconia nanoparticles by hydrophobic physical interactions. These biocatalysts were used in the kinetic resolution of (R,S)-ibuprofen and (R,S)-1-phenylethanol through esterification and acylation, respectively, in an organic solvent (isooctane) (Tables 4 and 5). Both the lipases immobilized on the zirconia nanoparticles modified with erucic acid exhibited much higher activity and enantioselectivity than the soluble form. Table 4 shows the values of the loading protein capacity and relative activity of the immobilized CRL compared with the crude powder. It was possible to immobilize the lipase on modified and unmodified nanoparticles. In particular, the CRL amount loaded onto both the erucic and Tween nanoparticles was higher than the unmodified ones. The lipase activity was very high when immobilized on the erucic nanoparticles (with a relative activity of 214%), probably due to the major hydrophobicity of its structure. The selectivity of CRL also increased when it was immobilized on the erucic nanocomposite and the enantioselectivity (E) value of the nanobioconjugate was almost 2 times higher than that of the crude CRL powder. The interaction of the lipase with the hydrophobic surface of modified zirconia nanoparticles permits the stabilization of the open form of the enzyme,24 exposing the active site to the reaction medium and substrates (Fig. 11), making it more active than the free lipase. The enhancement in the lipase activity with the erucic acid modification instead of with Tween was a question of hydrophobicity (Table 4).
| Biocatalyst | Protein loading (mg g−1) | Relative activity (%) | ee (%) |
|---|---|---|---|
| Time = 4.5 h. | |||
| Crude PCL powder | — | 100 | 31.2 |
| 1.2-PCL-ZrO2 | 43.6 | 110 | 10.2 |
| 1.2-PCL-erucic-ZrO2 | 46.2 | 231 | 95.5 |
| 0.8-PCL-erucic-ZrO2 | 28.7 | 211 | 91.6 |
For this reason erucic-ZrO2 was chosen for immobilization of the Pseudomonas cepacia lipase (PCL). The lipase activity was increased by more than 2 times in the kinetic resolution of (R,S)-1-phenylethanol after immobilization when compared to the soluble lipase activity (Table 5). This study demonstrated that inorganic zirconia nanoparticles, characterized by a hydrophilic surface, can be easily modified in hydrophobic supports (using surfactants with a long alkyl chain) and significantly increases the lipase activity. The CRL-erucic-ZrO2 preparation worked well in the enantioselective reactions in organic media with good recycling parameters, making it a promising biocatalyst for industrial applications.
Another interesting example has been described by Andrade et al., regarding an excellent improvement in the Burkholderia cepacia lipase catalytic performance when immobilized on supermagnetic nanoparticles.53 Three methodologies to immobilize the lipase were compared: adsorption, chemisorption with carboxibenzaldehyde and chemisorption with glutaraldehyde. Furthermore, the catalytic activity was studied in an enantioselective acetylation reaction.
Magnetite (Fe3O4) nanoparticles, with a diameter of 10 nm, were synthesized using a co-precipitation method.54 Fe3+ and Fe2+ ions were mixed in an alkaline solution of NaOH under an inert atmosphere to prevent oxidation by oxygen. For the same reason the magnetic nanoparticles were then sylanized with γ-aminopropyltriethoxysilane (APTS), leaving the γ-aminopropyl groups free to react (Fig. 12A). Furthermore, this process stabilizes the silicate coating by forming strong bonds between the iron and silica. The first immobilization methodology consisted of a simple adsorption of the lipase onto the γ-aminopropyl groups of the magnetic nanoparticle through ionic interactions between the positively charged nanoparticles (APTS-MagNP) and negatively charged groups of the lipase. The amount of protein loaded was 0.21 mg into 20 mg of the APTS-magnetic nanoparticles.
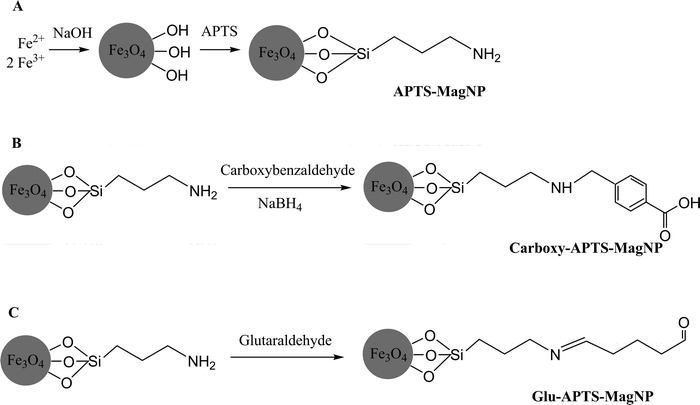 | ||
| Fig. 12 Nanoparticles sylanized with γ-aminopropyltriethoxysilane (APTS-MagNP, (A), modified with 4-carboxybenzaldehyde (Carboxy-APTS-MagNP), (B), modified with glutaraldehyde (Glu-APTS-MagNP), (C). | ||
The second immobilization protocol consisted of covalent binding between the carboxyl group of the magnetic nanoparticles after modification of the γ-aminopropyl groups with 4-carboxybenzaldehyde and the amino groups of lipase via carbodiimide activation (Carboxy-APTS-MagNP) (Fig. 12B). Using this method, it was possible to load 0.25 mg of the protein into 20 mg of the nanoparticles. The third immobilization procedure consisted of a covalent link between the nanoparticles modified by glutaraldehyde (Fig. 12C) and the lipase, binding 0.28 mg of the lipase to 20 mg of the nanoparticles (Glu-APTS-MagNP).
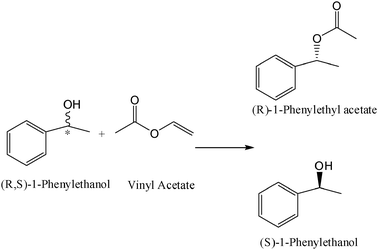
The lipase activity of the different preparations was evaluated in the kinetic resolution of (R,S)-2-bromo-1-(phenyl) ethanol (a drug precursor) by enantioselective acetylation (Table 6). This reaction was carried out at different temperatures. The selectivity of lipase was enhanced after immobilization, with excellent enantiomeric values (E >200) when compared to the soluble enzyme (E <30) (Table 6). The lipase immobilized on Glu-APTS-MagNP showed the best catalytic properties at different temperatures, with a relative activity of 150% with respect to the free lipase. Furthermore, this lipase immobilization preparation resulted quite thermostable. These excellent properties permitted the kinetic resolution at 52 °C (the temperature at which the free lipase was inactive), resulting in a more effective transesterification reaction than the free lipase.
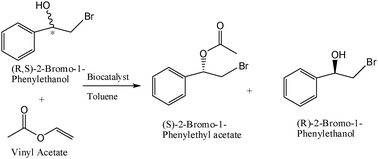
| Biocatalyst | T (°C) | Conversion (%) | ee (%)c | E(S/R)d |
|---|---|---|---|---|
| a Time = 24 h. b Time = 48 h. c Enantiomeric excess of the product. d Enantioselectivity value. | ||||
| Free lipasea | 32 | 20 | 78 | <30 |
| APTS-MagNPa | 32 | 13 | >99 | >200 |
| Carboxy-APTS-MagNPa | 32 | 10 | >99 | >200 |
| Glu-APTS-MagNPa | 32 | 18 | >99 | >200 |
| APTS-MagNPb | 42 | 14 | >99 | >200 |
| Carboxy-APTS-MagNPb | 42 | 24 | >99 | >200 |
| Glu-APTS-MagNPb | 42 | 26 | >99 | >200 |
| APTS-MagNPb | 52 | 9 | >99 | >200 |
| Carboxy-APTS-MagNPb | 52 | 21 | >99 | >200 |
| Glu-APTS-MagNPb | 52 | 24 | >99 | >200 |
Among these emerging nanomaterials, the use of carbon nanotubes (CNTs) as scaffolds in the immobilization of biocatalysts is growing more important in the techniques for enhancing the properties. Ji and co-workers, for example, describe the preparation of a highly active (and enantioselective and stable) biocatalyst using lipase from Rhizopus arrhizus (RAL) immobilized onto a multi-walled carbon nanotube (MWCNT) surface.55
After acidic treatment of the MWCNTs, RAL was covalently linked to the nanotube surface via a carbodiimide amide formation (Fig. 13) and was physically characterized by UV, CD and FTIR studies which confirmed the successful immobilization. Finally, the catalytic properties of this new derivative were tested in an ester-synthesis mediated resolution of the model molecule (R,S)-1-phenyl ethanol (Table 7). The lipase immobilized on the MWCNTs' surface was almost 3-fold more active when compared to the free enzyme and maintained excellent enzyme enantioselectivity (ee > 99%, E > 100 toward the (R)-enantiomer). Furthermore, a lower temperature-dependence of the MWCNT-lipase derivative was observed (a 45–50% conversion range of the racemate at 35–60 °C) versus a 25–35% conversion by the free lipase in the same conditions. This improvement in catalysis efficiency may be attributed to the physical properties of the CNTs that possess an efficient conductivity for heat.56 This intrinsic property of the nanocarrier can improve the heat transfer efficiency between the reaction mixture solvent and the lipase, hence permitting the acceleration of the lipase catalysis.
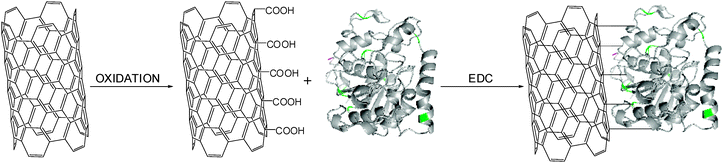 | ||
| Fig. 13 Covalent immobilization of lipase on the carbon nanotube surface. | ||
Using this method, the carbon nanotube assumes a critical role, becoming a crucial parameter in the modulation of the lipase properties and conferring an additional advantage to the nanomaterials in the issue of carrier selection for the immobilization of proteins.
Recently, an interesting property-enhancing immobilization protocol, characterized by the precipitation of proteins onto the surface of micron-sized salt crystals (protein-coated micro-crystals (PCMCs)), and its subsequent cross-linking with a homobifunctional agent, for example linear bialdehydes, (crosslinked protein-coated microcrystals CLPCMC) has been developed (Fig. 14).57
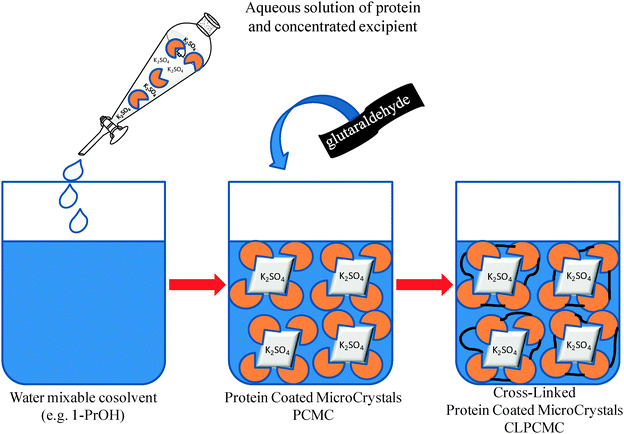 | ||
| Fig. 14 Schematic procedure for the obtainment of protein microcrystals. | ||
For example, Yan et al. have applied this technique (PCMC and CLPCMC) on the Geotricum sp. lipase (GSL), highly improving almost all the catalytic properties (enzyme activity, thermostability, organic solvent tolerance, pH stability and operational stability). The biocatalysts were tested in a biodiesel production process from waste oil.58
Both the PCMC-GSL and CLPCMC-GSL preparations exhibited higher yields in the reaction, 72% and 69% respectively, when compared to the free lipase (29%). Therefore, the new biocatalysts showed a 2.5-fold increased activity with respect to the soluble lipase.
The activity of the new enzyme derivatives was even improved in both polar and non-polar organic solvents. CLPCMC-GSL incubated in polar organic solvents (propanol and acetone) retained more than 80% of its relative biodiesel production, whilst PCMC-GSL and the free lipase were recovered in a 60% and 30% yield, respectively, in the biodiesel production.
Furthermore, CLPCMC-GSL exhibited the highest activity in suitable acidic environments (in a pH range of 4–6) and with higher temperatures (in the range of 45–50 °C) than the corresponding PCMC preparation and free lipase. In this last case, for example, CLPCMC-GSL increased its activity 1.2-fold when compared to PCMC-GSL and 1.5-fold when compared to free lipase.
4. Improved lipase activity by additive addition
Another strategy to increase the lipase activity is represented by the addition of additives with different structures (i.e. organic solvents, detergents, etc.) into the reaction medium during the biocatalytic process.59 Following this approach, we recently described how the presence of very small amounts of ionic liquids in the reaction medium can greatly alter the activity of differently immobilized Rhizomucor miehei lipase preparations in the hydrolysis of peracetylated lactal (Fig. 15).60 The enzymatic activity of the different RML preparations varied depending on the relationship between the nature of the immobilized enzyme and the chemical structure of the ionic liquid (Fig. 16).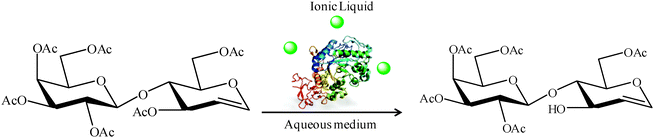 | ||
| Fig. 15 Regioselective deprotection of peracetylated lactal in the presence of small amounts of an ionic liquid. | ||
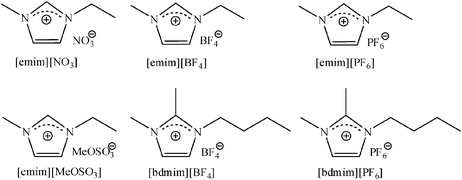 | ||
| Fig. 16 The chemical structure of different ionic liquids.60 Reproduced by permission of The Royal Society of Chemistry. | ||
In fact, when the lipase is immobilized on octyl-Sepharose, it exists in a fixed open conformation with a high hydrophobic pocket surrounding the active site. For this lipase immobilized preparation, the best results were achieved using the most hydrophobic ionic liquids (i.e. [bdmim][PF6] increased the activity 6-fold), probably because they can easily penetrate and alter the active site environment, favoring the substrate (hydrophobic) access and avoiding the undesired bihydrolysis of the monodeprotected product (more hydrophilic than the substrate). The study of the kinetic parameters demonstrated that the affinity of the substrate was better (a lower Km) and the turnover number (Kcat) was increased. On the other hand, when the lipase was immobilized on an ionic exchanger (Q-Sepharose), considering the most hydrophilic area was around the active site, the most hydrophilic ionic liquids gave the best results. In fact, using [emim][MeOSO3] as an additive, the activity of Q-Sepharose-RML increased by 2-fold (Table 8).
| Support | ILs | Time (h) | Activitya |
|---|---|---|---|
| a The initial rate in mmol mg prot−1 h−1. It was calculated at 10–20% yield. An ionic liquid without an enzyme was tested and no conversion was detected. | |||
| CNBr | — | 144 | 0.038±0.001 |
| [emim][BF4] | 144 | 0.052±0.002 | |
| [bdmim][PF6] | 144 | 0.064±0.002 | |
| Octyl | — | 24 | 0.58±0.01 |
| [emim][BF4] | 8 | 2.40±0.02 | |
| [bdmim][PF6] | 7 | 3.30±0.02 | |
| Q-Sepharose | — | 48 | 0.36±0.005 |
| [emim][NO3] | 72 | 0.10±0.002 | |
| [emim][MeOSO3] | 26 | 0.77±0.003 | |
Another interesting example is based on the activity improvement from differently immobilized preparations of a thermophilic lipase from Bacillus thermocatelunatus with additive additions (Fig. 17). Using this method, a straightforward synergistic hyperactivating effect was obtained.61
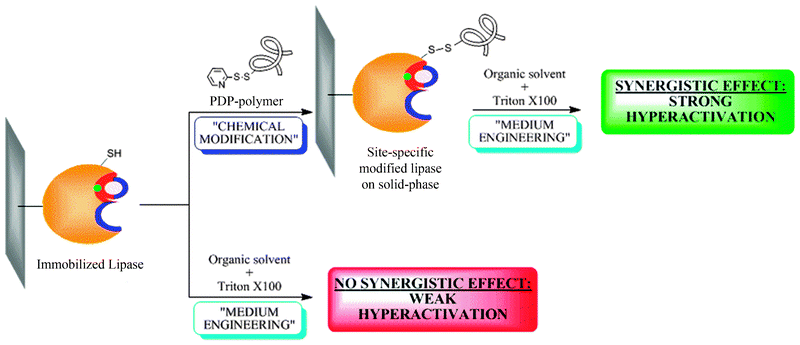 | ||
| Fig. 17 Synergistic effect of medium-engineering on site-directed chemically modified BTL2. | ||
The hyperactivation profiles of the lipase from the addition of additives depended on the immobilization protocol (one-point vs. a multipuntual covalent linkage), the site-directed chemical modification (tailor-made polymers) and the additive, co-solvent, detergent (Triton X-100) or a combination of both (Table 9). Among all the studies performed, the best result was achieved by combining the dextran-1500-NH2 CNBr-BTL2 preparation (described in Section 2) with both chemical additives: a co-solvent (dioxane) and detergent (Triton X-100), obtaining 11-fold higher activity when compared with the CNBr-BTL2 preparation used without any additive.
| Biocatalyst | Relative activity (%) | ||
|---|---|---|---|
| Triton X-100 | Dioxane (20%) | detergent + co-solvent | |
| CNBr-BTL2 | (0.01%) 275 | 330 | 320 |
| NH2-dextran-CNBr-BTL2 | (0.01%) 188 | 257 | 500 |
| COOH-PEG-CNBr-BTL2 | (0.01%) 290 | 312 | 1100 |
| Glyoxyl-BTL2 | (0.1%) 290 | 150 | 150 |
| NH2-dextran-glyoxyl-BTL2 | (0.1%) 100 | 279 | 350 |
| COOH-PEG-glyoxyl-BTL2 | (0.1%) 100 | 300 | 300 |
5. Conclusions
The examples covered in this perspective illustrate the power of new technologies, such as using nanomaterials for the immobilization of enzymes and site-specific chemical modification protocols to obtain semisynthetic lipases with a highly improved activity towards different kinds of unnatural substrates. For example, by employing the new biocatalysts produced with the techniques described herein, it has been possible to improve the outgoing of some important bioprocesses, such as the regioselective deprotection of peracetylated carbohydrates,60 biodiesel production,58 the enantiomeric resolution of a NSAID drug (ibuprofen),51 the kinetic resolution of natural products by transesterification in an organic medium 50 or an important chiral drug precursor.54These kinds of methodologies could be compatible with other techniques, e.g. molecular biology (site-directed mutagenesis or directed evolution) for the possible design of a second generation of lipase catalysts with improved catalytic properties (activity, stability and even selectivity) or with novel activities in organic chemical reactions (Diels–Alder cycloadditions, Mannich reactions, or ring-closing metathesis), which are not found in nature.
Acknowledgements
The authors are grateful for the help and comments from Dr Ángel Berenguer (Instituto de Materiales, Universidad de Alicante).References
- A. Radzicka and R. Wolfenden, Science, 1995, 267, 90 CAS.
- H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694 CrossRef CAS.
- A. J. Straathof, S. Panke and A. Schmid, Curr. Opin. Biotechnol., 2002, 13, 548 CrossRef CAS.
- R. N. Patel, Curr. Org. Chem., 2006, 10, 1289 CrossRef CAS.
- H. Fukuda, S. Hama, S. Tamalampudi and H. Noda, Trends Biotechnol., 2008, 26, 668 CrossRef CAS.
- R. Verger, Trends Biotechnol., 1997, 15, 32 CrossRef CAS.
- L. Sarda and P. Desnuelle, Biochim. Biophys. Acta, 1958, 30, 513 CrossRef CAS.
- E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, 110 Search PubMed.
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138 CrossRef CAS.
- R. Wohlgemuth, Curr. Opin. Microbiol., 2010, 13, 283 CrossRef CAS.
- E. Marqués-López, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC.
- G. Volpato, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Med. Chem., 2010, 17, 3855 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451 CrossRef CAS.
- P. Domínguez de Maria, C. A. García-Burgos, G. Bargeman and R. Van Germert, Synthesis, 2007, 10, 1439 CrossRef.
- D. Gocke, C. L. Nguyen, M. Pohl, T. Stillger, L. Walter and M. Muller, Adv. Synth. Catal., 2007, 349, 1425 CrossRef CAS.
- R. Sangeetha, I. Arulpandi and A. Geetha, Res. J. Microbiol., 2011, 6, 1 CrossRef CAS.
- K. Faber, Biotransformations in Organic Chemistry, Springer-Verlag, Berlin, 5th edn, 2004 Search PubMed.
- J. M. Palomo, Curr. Bioact. Compd., 2008, 4, 126 CrossRef CAS.
- J. M. Palomo, Curr. Org. Synth., 2009, 6, 1 CrossRef CAS.
- N. Gupta, V. Shai and R. Gupta, Process Biochem. (Amsterdam, Neth.), 2007, 42, 518 CAS.
- L. P. G. Franken, N. S. Marcon, H. Treichel, D. Oliveira, D. M. G. Freire and C. Dariva, et al. , Food Bioprocess Technol., 2009, 3, 511 CrossRef.
- B. Joseph, P. W. Ramteke and G. Thomas, Biotechnol. Adv., 2008, 26, 457 CrossRef CAS.
- A. Pandey, S. Benjamin, C. R. Soccol, P. Nigam, N. Krieger and U. T. Soccol, Biotechnol. Appl. Biochem., 1999, 29, 119 CAS.
- A. M. Brzozowski, U. Derewenda, Z. S. Derewenda, G. G. Dodson, D. M. Lawson, J. P. Turkenburg, T. Bjorkling, B. Huge-Jensen, S. S. Patkar and L. Thim, Nature, 1991, 351, 491 CrossRef CAS.
- U. Derewenda, A. M. Brzozowski, D. M. Lawson and Z. S. Derewenda, Biochemistry, 1992, 31, 1532 CrossRef CAS.
- A. Lowrier, G. J. J. Drtina and A. M. Klibanov, Biotechnol. Bioeng., 1996, 50, 1 CrossRef.
- J. M. Palomo, G. Muñoz, G. Fernández-Lorente, C. Mateo, R. Fernández-Lafuente and J. M. Guisán, J. Mol. Catal. B: Enzym., 2002, 19–20, 279 CrossRef CAS.
- G. Fernández-Lorente, Z. Cabrera, C. Godoy, R. Fernandez-Lafuente, J. M. Palomo and J. M. Guisan, Process Biochem. (Amsterdam, Neth.), 2008, 43, 1061 Search PubMed.
- J. M. Palomo, M. Fuentes, G. Fernández-Lorente, C. Mateo, J. M Guisan and R. Fernández-Lafuente, Biomacromolecules, 2003, 4, 1 CrossRef CAS.
- G. Fernandez-Lorente, J. M. Palomo, Z. Cabrera, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 41, 565 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS.
- E. Basl, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213 CrossRef.
- B. G. Davis, Curr. Opin. Biotechnol., 2003, 14, 379 CrossRef CAS.
- Z. Cabrera, G. Fernández-Lorente, R. Fernandez-Lafuente, J. M. Palomo and J. M. Guisan, J. Mol. Catal. B: Enzym., 2009, 57, 171 CrossRef CAS.
- J. M. Palomo, R. L. Segura, G. Fernández-Lorente, M. Pernas, M. L. Rua, J. M. Guisán and R. Fernández-Lafuente, Biotechnol. Prog., 2004, 20, 630 CrossRef CAS.
- G. Fernández-Lorente, C. Ortiz, R. L. Segura, R. Fernández-Lafuente, J. M. Guisán and J. M. Palomo, Biotechnol. Bioeng., 2005, 92, 773 CrossRef.
- M. L. Foresti, G. A. Alimenti and M. L. Ferreira, Enzyme Microb. Technol., 2005, 36, 338 CrossRef CAS.
- L. Wilson, J. M. Palomo, G. Fernández-Lorente, A. Illanes, J. M. Guisán and R. Fernández-Lafuente, Enzyme Microb. Technol., 2006, 38, 975 CrossRef CAS.
- F. Caruso and C. Schuler, Langmuir, 2000, 16, 9595 CrossRef CAS.
- J. Kim, J. W. Grate and P. Wang, Chem. Eng. Sci., 2006, 61, 1017 CrossRef CAS.
- H. Jia, G. Zhu and P. Wang, Biotechnol. Bioeng., 2003, 84, 406 CrossRef CAS.
- (a) G. G. Kochendoerfer, Curr. Opin. Chem. Biol., 2005, 9, 555 CrossRef CAS; (b) K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45 RSC.
- C. A. Godoy, B. de las Rivas, M. Filice, G. Fernandez-Lorente, J. M. Guisan and J. M. Palomo, Process Biochem., 2010, 45, 534 CrossRef CAS.
- M. L. E. Gutarra, O. Romero, O. Abian, F. A. G. Torres, D. M. G. Freire, A. M. Castro, J. M. Guisan and J. M. Palomo, ChemCatChem, 2011, 3, 1902 CrossRef CAS.
- M. L. E. Gutarra, C. Mateo, D. M. G. Freire, F. A. G. Torres, A. M. Castro, J. M. Guisan and J. M. Palomo, Catal. Sci. Technol., 2011, 1, 260 CAS.
- M. D. Leipold, I. Herrera, O. Ornatsky, V. Baranov and M. Nitz, J. Proteome Res., 2009, 8, 443 CrossRef CAS.
- M. Filice, O. Romero, J. M. Guisan and J. M. Palomo, Org. Biomol. Chem., 2011, 9, 5535 CAS.
- D. G. Rees, I. I. Gerashchenko, E. V. Kudryashova, V. V. Mozhaev and P. J. Halling, Biocatal. Biotransform., 2002, 20, 161 CrossRef CAS.
- K. Solanki and M. N. Gupta, Bioorg. Med. Chem. Lett., 2011, 21, 2934 CrossRef CAS.
- M. H. Sörensen, J. B. S. Ng, L. Bergström and P. C. A. Alberius, J. Colloid Interface Sci., 2010, 343, 359 CrossRef.
- L. Chronopoulou, G. Kamel, C. Sparag, F. Bordi, S. Lupi, M. Diociaiuti and C. Palocci, Soft Matter, 2011, 7, 2653 RSC.
- Y. Z. Chen, C. T. Yang, C. B. Ching and R. Xu, Langmuir, 2008, 24, 8877 CrossRef CAS.
- L. H. Andrade, L. P. Rebelo, C. G. C. M. Netto and H. E. Toma, J. Mol. Catal. B: Enzym., 2010, 66, 55 CrossRef CAS.
- S.-J. Lee, J.-R. Jeong, S.-C. Shin, J.-C. Kim and J.-D. Kim, J. Magn. Magn. Mater., 2004, 282, 147 CrossRef CAS.
- P. Ji, H. Tan, X. Xu and W. Feng, AIChE J., 2010, 56(11), 3005 CrossRef CAS.
- L. X. Zheng, M. J. O’Connell, S. K. Doorn, X. Z. Liao, Y. H. Zhao, E. A. Akhadov, M. A. Hoffbauer, B. J. Roop, Q. X. Jia, R. C. Dye, D. E. Peterson, S. M. Huang, J. Liu and Y. T Zhu, Nat. Mater., 2004, 3, 673 CrossRef CAS.
- (a) M. Kreiner, B. D. Moore and M. C. Parker, Chem. Commun., 2001, 1096 RSC; (b) M. Kreiner and M. C. Parker, Biotechnol. Lett., 2005, 27, 1571 CrossRef CAS; (c) A. Khosravani, M. C. Parker, R. Parton and J. Coote, Vaccine, 2007, 25, 4361 CrossRef CAS.
- J. Yan, Y. Yan, S. Liu, J. Hu and G. Wang, Bioresour. Technol., 2011, 102, 4755 CrossRef CAS.
- G. Fernández-Lorente, J. M. Palomo, Z. Cabrera, R. Fernández-Lafuente and J. M. Guisán, Biotechnol. Bioeng., 2007, 97, 242 CrossRef.
- M. Filice, J. M. Guisan and J. M. Palomo, Green Chem., 2010, 12, 1365 RSC.
- C. A. Godoy, G. Fernandez-Lorente, B. de las Rivas, M. Filice, J. M. Guisan and J. M. Palomo, J. Mol. Catal. B: Enzym., 2011, 70, 144 CrossRef CAS.
Footnotes |
| † Current address: Departamento de Biomateriales y Materiales Bioinspirados. Instituto de Ciencia de Materiales (CSIC), Madrid, Spain |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |