
Microwave-pulse synthesis of tunable 2D porous nickel-enriched LaMnxNi1−xO3 solid solution for efficient electrocatalytic urea oxidation†
Zhiao
Wu‡
,
Jinglin
Xian‡
,
Jiao
Dai‡
,
Guangyu
Fang
,
Miao
Fan
,
Haoran
Tian
,
Jiayue
Guo
,
Zhenhui
Huang
,
Huiyu
Jiang
,
Weilin
Xu
and
Jun
Wan
 *
*
State Key Laboratory of New Textile Materials and Advanced Processing Technologies, Hubei Key Laboratory of Biomass Fibers and Eco-Dyeing & Finishing, Wuhan Textile University, Wuhan 430200, China. E-mail: wanj@wtu.edu.cn
First published on 15th February 2024
Abstract
Amidst the pressing demand for carbon neutrality and clean energy policies, the electrocatalytic urea oxidation reaction (UOR) has gained attention as an efficient and environmentally friendly energy conversion pathway. High-valence/content two-dimensional (2D) nickel-based ABO3 perovskite oxides, particularly LaNiO3, are highlighted for their elevated intrinsic catalytic activity and rich electronic configuration advantages. To enhance the intrinsically limited conductivity of LaNiO3, a modified strategy involving B-site substitution can be used to construct a solid solution structure, significantly improving the electronic configuration. However, as Ni is the primary active site for the UOR, substituting the B-site will create a trade-off between enhancing electron migration and decreasing active site content. Specifically, conventional synthesis methods with a slow-entropy-change pose challenges in precise control over atomic ratios and 2D structure design, hindering elucidation of the impact mechanism of the electronic configuration on UOR performance. Herein, we utilized the microwave-pulse method for rapid synthesis of highly tunable 2D porous nickel-enriched LaMnxNi1−xO3 perovskite. Leveraging transient high-temperature and high-energy conditions, we achieved rapid one-step equilibrium between 2D porous structure design and precise control of solid solution atomic ratios. This strategy allows for the controlled synthesis of 2D nickel-enriched solid solutions with excellent conductivity and UOR activity, while effectively avoiding the generation of by-products, providing a detailed analysis of the UOR activity mechanism dependent on distinct electronic configurations. The synthesized LaMn0.2Ni0.8O3 structure exhibits optimal UOR performance. This strategy provides a new avenue for the design of more unique structured nickel-based solid solutions and the investigation of their structure–performance relationships in the electrocatalytic UOR.
Introduction
Efficient and green electrocatalytic UOR, as a means of energy conversion with a more oxidative anodic reaction replacing the oxygen evolution reaction (OER) in water splitting, has emerged as a promising carbon-neutral strategy with practical development potential.1–6 Compared to a theoretical decomposition voltage of 1.23 V for the OER, the UOR exhibits a lower voltage of 0.37 V,7–10 attributed to its overall reaction process based on a six-electron transfer mechanism: CO(NH2)2 + H2O → N2 + 3H2 + CO2. High-valence/content nickel-based ABO3 perovskite oxides, particularly easily synthesized LaNiO3, have garnered increasing attention due to their high intrinsic catalytic activity and rich electronic configuration advantages (Fig. 1a).11 Unfortunately, the inherent limited conductivity of oxides typically significantly impacts their bulk/interface intrinsic catalytic activity. In recent years, as 2D materials have demonstrated unique physicochemical advantages at the atomic-level interface,12,13 the preparation of 2D LaNiO3 has proven effective in both enhancing electronic mobility and improving interface ion transport and reactions.14 In addition to the unique nanostructure design, introducing a foreign element M through the B-site substitution modification strategy for LaNiO3 effectively forms solid solution structure LaMnxNi1−xO3, leading to significant improvement in its electronic configuration (Fig. 1b).15 For instance, the introduction of Co into LaNiO3 by Yun et al. can significantly alter its band structure, resulting in a substantial enhancement of its conductivity.16 Similarly, the incorporation of an appropriate amount of Fe into LaNiO3 by Fu et al. forms a homogeneous solid solution, leading to a significant improvement in its electronic configuration and enhanced interface catalytic activity.17 However, unlike other types of electrocatalysis where active sites are typically formed by the coupling of multiple metal centers, the primary active site for the UOR has been confirmed to be the Ni element.18–23 In strongly alkaline media, nickel readily transforms into nickel-based oxide or hydroxide nanostructures, leading to redox reactions occurring cyclically between NiOOH, accompanied by the oxidation and decomposition of urea.24–27 This indicates that the B-site substitution of the Ni element will lead to a trade-off between enhanced electron mobility and decreased active site content. Therefore, achieving a delicate balance between the one-step design of the 2D structure of LaMnxNi1−xO3 and precise control of the B-site electronic configuration for optimal UOR activity, while elucidating the electron configuration-dependent mechanism of UOR activity, is of utmost importance and a challenge. | ||
| Fig. 1 (a) The process of a nickel-based catalyst reaction in the UOR. (b) LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8) crystal structure diagram. | ||
To successfully engineer a 2D structure for LaNiO3 (LNO) while elucidating the impact mechanism of Ni content on its UOR activity through a clear understanding of the optimal electronic configuration in B-site substitution, poses a challenge of balancing the thermodynamics of 2D oriented growth with the kinetics of transiently controlling the Ni atomic ratio. Actually, there are currently various methods available for the successful preparation of nickel-based oxide solid solutions, which represent a type of alloy phase material capable of effectively integrating the target elements with the matrix elements, maintaining the homogeneous crystal structure characteristics of the matrix elements. This is a strategy capable of utilizing a targeted atomic ratio to influence the electronic configuration of the parent material, aiding in deciphering its corresponding impact mechanism on electrocatalytic activity.28 For example, Rao et al. synthesized Mn0.1Ni0.9O nanoparticles by solution combustion after high temperature calcination for half an hour, which significantly improved their electrocatalytic activity in the OER.29,30 Additionally, Wang et al. successfully synthesized stable multi-metal NiCoFe-O@NF nanoparticles using a prolonged hydrothermal method, inducing enriched Co2+ and Ni3+ configurations, thereby adjusting the electronic structure and demonstrating exceptional dual-catalytic activity for both the hydrogen evolution reaction and OER under alkaline conditions.31 Although conventional synthesis methods involving slow-entropy-change processes can effectively enhance the bulk/interface catalytic activity of nickel-based oxides through solid solution design, they often exhibit typical nanoparticle structures and relatively arbitrary atomic ratios, making it challenging to simultaneously control 2D selective oriented growth and the specific atomic ratio of the target elements. Moreover, undesired by-products and impurities may also be generated due to inappropriate precursor ratios. Thus, it is imperative to seek a method for precise design of the Ni atomic ratio in 2D LaMnxNi1−xO3 solid solution through B-site substitution.
Over the past decade, advanced non-liquid-phase microwave technology has been able to exploit mechanisms such as dipolar loss, conduction loss, and magnetic loss to rapidly construct numerous unique nanostructures from both thermodynamic and kinetic perspectives. For instance, Sun et al. achieved rapid synthesis of high-quality graphene within 200 seconds using the microwave method,32 while Wan et al. demonstrated the 2D structural evolution of TMCs over 3 times of microwave pulse approach.33 Subsequently, precise control over metal element doping, especially the successful introduction of high atomic ratios, was accomplished using the microwave discharge method.34 Considering the involvement of Ni as the primary active site for the UOR, this suggests a conceptual and feasible approach to construct nickel-rich 2D LaMnxNi1−xO3 solid solutions using microwave technology.
Herein, taking manganese (Mn) as an example with rich oxidation states and abundant sources, we successfully prepared highly tunable 2D porous LaMnxNi1−xO3 perovskite using a microwave-pulse method. By leveraging the thermodynamic and kinetic advantages of microwave transient high temperature and high energy, a rapid one-step achievement of balanced design for 2D porous structures and atomic ratio precision control can be realized. This strategy can effectively avoid the generation of by-products, precisely regulating the Ni–Mn atomic ratio to achieve the optimal coupling of conductivity and UOR catalytic activity. Subsequently, the mechanisms underlying the electronic configuration-dependent UOR activity are further elucidated. The synthesized LaMn0.2Ni0.8O3 structure exhibits an optimal electrocatalytic UOR performance (a low potential of 1.27 V at a current density of 10 mA cm−2 and a low Tafel value of 44.6 mV dec−1), highlighting the synergistic effect of its conductivity and nickel active site content. This strategy provides a novel approach for the precise design of more 2D nickel-based UOR electrocatalysts and mechanistic studies of structure–performance relationships.
Results and discussion
To investigate the equilibrium between the design of 2D porous structures and the precise control of the atomic ratio in solid solutions, we designed the following experiment. Fig. 2a depicts the process of rapidly preparing porous 2D LNMO perovskites via the microwave pulse method, with partial B-site substitution of Ni. Initially, glucose and ammonium nitrate were added to a specific ratio of manganese nitrate and nickel nitrate, ground uniformly in a mortar, and placed in a quartz crucible filled with carbon nanotubes on the periphery. The carbon nanotubes, having a high dielectric constant, rapidly responded to the microwaves to generate transient high temperatures.35,36 Glucose and nitrate act as templates and foaming agents, collectively promoting precise control of the ratio of solid solutions and the design of 2D porous structures. Microwave pulses induced rapid and uniform temperature modulation, allowing for the one-step controlled and uniform design of nickel-rich LaMnxNi1−xO3 with various atomic ratios without further processing. We prepared a series of LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8), and the atomic ratios determined by ICP-MS closely matched the feed ratios (Fig. S1 and Table S1†), demonstrating the precise control of the atomic ratio in solid solutions by the microwave pulse method. Due to the contribution of the transient heating and cooling characteristics of microwave pulse heating, which can maintain the crystal structure and morphology to the greatest extent at specific time points, we carried out precise non-in situ characterization of the reaction process. The infrared spectra and X-ray diffraction (XRD) data of the microwave pulses at 1 min, 1.5 min, and 10 min (referred to as LNMO-T1, LNMO-T1.5, and LNMO-T10) clearly showed the weakening of carbon peaks due to melting of glucose as a template after experiencing high-energy microwave pulses.37 This indicates that as the duration of the high-temperature shock increases, the surface carbon layer gradually diminishes, exposing the underlying 2D porous structure. We recorded the entire reaction process using 3D microscope photographs and physical optical photographs, observing that the reactants bubble and burst, forming a 2D porous structure (Fig. S2†). According to the temperature records of a specially designed microwave infrared thermometer, a single microwave shock has three stages: microwave initiation, high-temperature pulse, and rapid self-quenching.38,39 (1) During the microwave initiation stage, the temperature in the reaction area rapidly increased under microwave irradiation (increasing to ∼500 °C, lasting for about 10 s), causing a violent redox reaction of glucose and nitrate ions under the action of a large amount of gas. This led to rapid internal expansion of the polymer, resulting in the formation of ultra-thin 2D porous perovskites embedded with carbon nanosheets. (2) The high-temperature shock stage involved continuously heating the CNTs to a peak (∼738 °C) via microwaves, creating a stable high-temperature environment for carbon decomposition and leaving behind 2D porous perovskites. (3) During the rapid self-quenching stage, the rapid cooling rate was conducive to kinetic control over the thermodynamic mixing mechanism, forming a 2D porous morphology of the solid solution.40 Prolonged high-temperature reactions tend to cause aggregation or sintering of materials, which is detrimental to the formation of 2D structures. Employing a method involving short-duration, multiple pulses enables a process of transient heating and self-quenching. This approach not only prevents the occurrence of side reactions but also aids in maintaining the structural integrity and dimensional characteristics of the materials, thereby addressing the challenges associated with high-temperature processes. To verify the 2D porous structure of LNMO, the morphology of the materials was characterized using scanning electron microscopy (SEM). The resulting LNMO-0.2 porous nanosheets demonstrated flexibility and curling, as shown in Fig. 2b, indicating the exceptional uniformity of the prepared perovskite nanosheets. The post-reaction 2D porous LNMO-x nanosheets exhibited volumetric expansion, approximately three times the volume of the precursor mixture of glucose and nitrate salts (Fig. S3†), due to the accumulation of fluffy 2D nanosheets. Transmission electron microscope (TEM) images revealed that the flexible, ultra-thin LNMO-0.2 nanosheets were uniformly covered with porous structures (Fig. 2c). It is widely believed that such porous structures can expose more active sites and provide advanced electrocatalytic activity. SEM and TEM images of LNO in Fig. S4 and S5† also display similar 2D porous structures. Moreover, the thickness of the LNMO-0.2 porous nanosheets was measured using atomic force microscopy (AFM), revealing a thickness of approximately 1.5 nm (Fig. 2d), confirming the ultra-thin nature of the nanosheets. Subsequently, the high-resolution TEM (HRTEM) image in Fig. 2e clearly displayed the lattice fringes of LNMO-0.2, with a lattice spacing of 0.272 nm, corresponding to the (110) diffraction plane.41 Notably, due to crystal distortion, the introduction of Mn widened the lattice spacing of LNO (Fig. S6†). Additionally, electron diffraction (Fig. 2f) showed polycrystalline rings, indicating that the nanosheets were formed from interconnected single-crystal particles. Energy-dispersive X-ray spectroscopy (EDS) characterization (Fig. 2g) indicated a uniform distribution of La, Mn, and Ni elements throughout the entire LNMO-0.2 sample. | ||
| Fig. 2 (a) Schematic diagram of the synthesis of 2D LNMO-x by the microwave shock method; FTIR spectra and XRD patterns of LNMO within different synthesis times; physical photos of the sample during the reaction process; real-time temperature changes of samples heated by microwave pulses. (b) SEM images of LNMO-0.2. (c) TEM images of LNMO-0.2. (d) AFM image of LNMO-0.2. (e) HRTEM image of LNMO-0.2. (f) The corresponding SAED patterns of LNMO-0.2. (g) The EDS elemental mapping scanning from TEM of LNMO-0.2. | ||
The electronic configurations of Ni and Mn, which differ in their d orbital electron arrangements, are [Ar]3d84s2 for Ni and [Ar]3d54s2 for Mn. The substitution of Ni cations at the B-site with Mn leads to interactions between their electrons, potentially enhancing the structure and properties (Fig. 3a). To investigate the impact of Mn substitution at the B-site on the crystal structure of 2D porous LNO perovskites, XRD analysis was conducted on LNO, LNMO-0.2, LNMO-0.4, LNMO-0.6, and LNMO-0.8 samples to examine their phases and crystal structures. Rietveld refinement using GSAS software and the EXPGUI interface further determined the space groups and lattice parameters of these samples (Fig. 3b), revealing that LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8) possesses a rhombohedral perovskite structure (space group: R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c). Due to the larger ionic radius of Mn3+ compared to Ni3+, the lattice parameters of LNMO changed. From Fig. S7†, it is evident that the metal–oxygen bonding stretching vibration occurs around a wavenumber of 600 nm−1. A slight shift of the main diffraction peak at 32.9° for the LNO (110) plane towards lower angles indicated an expansion of the lattice spacing (Fig. 3c). This expansion can be attributed to the changes in lattice parameters due to the variation in ionic radii between Mn3+ (0.66 Å) and Ni3+ (0.63 Å) cations. Fig. 3d shows that the Ni–O bond length is increasing, from 1.97 Å to 2.04 Å. Similarly, the Mn–O bond length increases from 1.98 Å to 2.04 Å. Fig. 3e presents the changes in bond angles; the angle between O–Mn1–O and O–Ni2–O increases from 90.04° to 93.34°. Meanwhile, the angle between O–Mn2–O and O–Ni1–O decreases from 88.45° to 86.66°. This further confirms the substitution of Mn for Ni at the B-site. Fig. 3f shows the refined error values for different samples, which can be considered as strong evidence of the successful preparation of a series of LNMO samples and the significant substitution of Mn for Ni at the B-site.
c). Due to the larger ionic radius of Mn3+ compared to Ni3+, the lattice parameters of LNMO changed. From Fig. S7†, it is evident that the metal–oxygen bonding stretching vibration occurs around a wavenumber of 600 nm−1. A slight shift of the main diffraction peak at 32.9° for the LNO (110) plane towards lower angles indicated an expansion of the lattice spacing (Fig. 3c). This expansion can be attributed to the changes in lattice parameters due to the variation in ionic radii between Mn3+ (0.66 Å) and Ni3+ (0.63 Å) cations. Fig. 3d shows that the Ni–O bond length is increasing, from 1.97 Å to 2.04 Å. Similarly, the Mn–O bond length increases from 1.98 Å to 2.04 Å. Fig. 3e presents the changes in bond angles; the angle between O–Mn1–O and O–Ni2–O increases from 90.04° to 93.34°. Meanwhile, the angle between O–Mn2–O and O–Ni1–O decreases from 88.45° to 86.66°. This further confirms the substitution of Mn for Ni at the B-site. Fig. 3f shows the refined error values for different samples, which can be considered as strong evidence of the successful preparation of a series of LNMO samples and the significant substitution of Mn for Ni at the B-site.
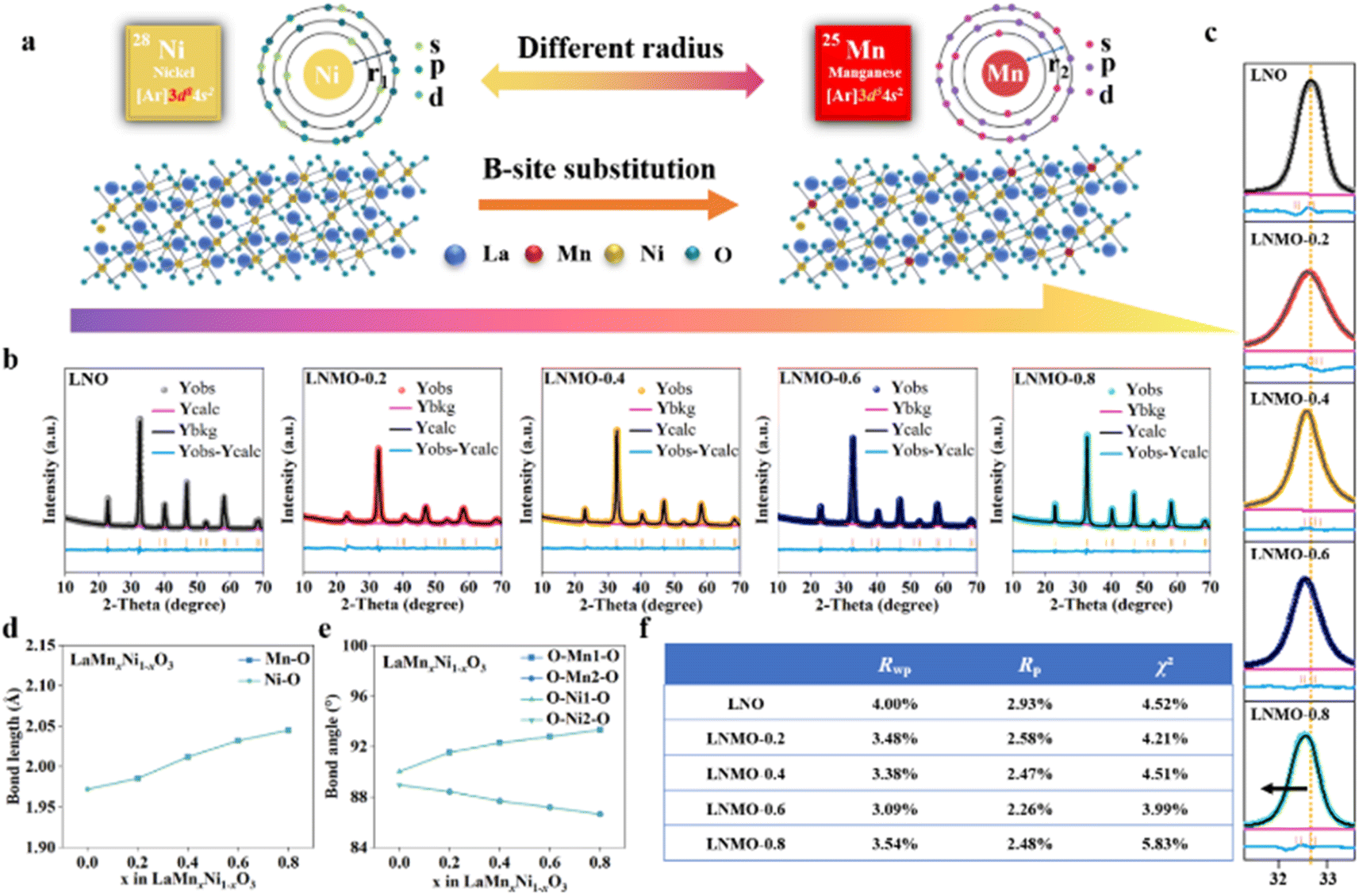 | ||
| Fig. 3 (a) B-site substitution diagram of LNMO-x solid solution. (b) XRD patterns of LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8) with their Rietveld refinement results. (c) Partial enlargement of the XRD pattern. (d) Change in bond length of LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8). (e) Change in bond angle of LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8). (f) Error analysis values of Rietveld data for the XRD pattern. | ||
To better understand the electronic structure of solid solutions with varying Mn/Ni ratios, X-ray photoelectron spectroscopy (XPS) was used to characterize the valence states of the constituent elements in the LNMO-x samples. The full XPS spectrum of LNMO-x revealed the presence of La, Mn, Ni, and O elements (Fig. S8†). The Ni 2p orbitals partially overlapped with the La 3d orbitals, with the characteristic peak at 851 eV attributed to La 3d3/2. The Ni 2p3/2 orbitals were divided into Ni3+ and Ni2+, with the peak at 852.8 eV corresponding to the satellite peak of Ni 2p3/2. The Mn 2p orbital data showed that 642.7 eV and 654.4 eV correspond to Mn 2p3/2 and Mn 2p1/2, respectively. The Mn 2p3/2 orbitals were divided into Mn4+ and Mn3+ peaks, resulting in a strong Mn–O bond.42 In the rate-determining step of the OER, a stronger Mn–O bond can also promote charge transfer between surface Mn ions and oxygen intermediates (such as O22− and O2−) (Fig. S9†).43 If the formation potential of the active phase is too high, the OER might compete with the UOR, thereby reducing the selectivity of the UOR.44,45 In alkaline media, surface Ni2+ ions adsorb hydroxide ions to form Ni(OH)2, which can be oxidized to NiOOH at higher potentials. NiOOH is commonly considered as the active site for the UOR, making the intrinsic content of Ni3+ a reasonable indicator of UOR activity.46 Therefore, maintaining a high concentration of Ni3+ is crucial for UOR activity. We further analyzed the content ratio of Ni and Mn elements. As shown in Fig. 4a, we found that the content of Ni3+ in LNMO-0.2 was 65.03%, and that of Ni was 34.97%, which was an increase compared to 58.8% and 41.2% in LNO. However, with further introduction of Mn, the Ni3+/Ni2+ ratio decreased. Similarly, Fig. 4b shows the trend of Mn4+/Mn3+ content variation. As shown by the characteristic peaks of the O 1s orbital, the peak at 529.1 eV corresponds to lattice oxygen, 531.9 eV to surface adsorbed oxygen and hydroxyl groups, and 534.4 eV to surface adsorbed water molecules.47 The strong peak of surface adsorbed oxygen and hydroxyl groups is likely related to the generation of oxygen vacancies (Fig. S10†). To more accurately characterize the variation in oxygen vacancy (Ov) content, electron paramagnetic resonance (EPR) analysis was performed on LNO and LNMO-0.2. As shown in Fig. S11†, under the condition of g = 2.002, the EPR signal intensity of LNMO-0.2 was stronger than that of LNO (indicating a higher concentration of unpaired electrons/Ov), suggesting that the introduction of Mn effectively increases the Ov concentration.48Fig. 4c displays the relationship between Ni3+/Ni2+ and oxygen vacancies, revealing a positive correlation between the concentration of oxygen vacancies and the Ni3+/Ni2+ ratio. According to the principle proposed by Shao-Horn, an optimized eg filling near 1.2 should be an effective strategy for developing transition metal oxides as efficient catalysts, as an eg electron ratio close to 1.2 enhances the hybridization between the 3d–eg orbitals of the transition metal and the 2p orbitals of oxygen.49–51Based on this, the eg orbital filling numbers for LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8) were calculated to be 0.597, 1.150, 1.002, 0.850, and 0.696, respectively (Fig. 4d). In nickel ions (Ni2+), the high occupancy of the eg orbitals, as manifested in the t2g6eg2 configuration, results in a strong binding affinity with reaction intermediates, thereby impeding the desorption of gaseous products. However, an eg electron filling number approaching 1.2 can significantly enhance the adsorption and desorption processes of these intermediates, which contributes to improving the catalysis of the UOR.9 Increased oxygen vacancies often lead to enhanced electrical conductivity, and variations in the bandgap width typically indicate changes in conductivity. The UV-vis absorption spectra of LNMO-x showed that the measured bandgap width for LNMO-0.2 was 1.83 eV, a decrease of 0.42 eV compared to the bandgap width of LNO (2.25 eV) (Fig. 4e). Bandgap information for other samples is presented in Fig. S12.†Fig. 4f provides a schematic representation of the changes in the bandgap width. The observed reduction in the bandgap width suggests enhanced conductivity, which is beneficial for rapid electron transfer in materials such as electrocatalysts. These results indicate that the partial substitution of Mn at the B-site in LNO affects its electron transfer properties, enhancing its conductivity. Introducing more oxygen defects in the structure affects the proportion of Ni3+ at the B-site, thereby increasing the number of reactive sites. Overall, the LNMO-0.2 structure, characterized by a significant presence of oxygen vacancies and a high Ni3+ content, is the result of a balanced approach to 2D porous structure design and precise control of the atomic ratio in solid solutions. Therefore, LNMO-0.2 holds promise as a highly active material for electrocatalytic UOR applications.
 | ||
| Fig. 4 (a) Statistics of Ni 2p3/2 on chemical structure analysis of surfaces using XPS. (b) Statistics of Mn 2p3/2 on chemical structure analysis of surfaces using XPS. (c) The Ni3+/Ni2+ ratio of LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8). (d) The number of electrons LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8) fills in the eg orbital. (e) The band gap of LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8). (f) Schematic representation of the variation of the band gap. | ||
To assess the impact of partial Mn substitution at the B-site on the UOR activity of LNO, comprehensive electrochemical characterization of the synthesized electrocatalysts was conducted using a standard three-electrode setup in a 1 M KOH + 0.33 M urea electrolyte. Fig. 5a presents the linear sweep voltammetry (LSV) polarization curves for LNO and LNMO-0.6, and it was observed that the UOR possesses a lower thermodynamic potential compared to the OER, indicating more rapid reaction kinetics for the UOR. However, beyond a potential of 1.4 V, a competitive relationship between the UOR and OER may emerge. Consequently, with the further incorporation of Mn, there is a likelihood of a decrease in the catalytic activity of the UOR. Fig. 5b shows the LSV polarization curves for LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8). Notably, LNMO-0.2 exhibited a lower potential at a current density of 10 mA cm−2 (1.275 V) compared to LNO (1.434 V), LNMO-0.4 (1.326 V), LNMO-0.6 (1.374 V), and LNMO-0.8 (1.413 V), indicating the highest UOR activity among the perovskite oxide electrocatalysts studied. Moreover, the Tafel slope values of the samples showed significant differences (Fig. 5c). The Tafel slope of the LNMO-0.2 sample was significantly lower at 44.6 mV dec−1 compared to that of LNO (98.1 mV dec−1), LNMO-0.4 (58.0 mV dec−1), LNMO-0.6 (66.7 mV dec−1), and LNMO-0.8 (89.2 mV dec−1). This observation strongly suggests enhanced reaction kinetics in the UOR process for the LNMO-0.2 sample, indicating its potential as a superior electrocatalyst with improved electrocatalytic performance. A significant decrease in the Tafel slope indicates accelerated reaction kinetics, which may imply a change in the rate-determining step. The observed reduction in the Tafel slope can be attributed to the favourable adsorption of intermediates, resulting from electronic modulation caused by the presence of Mn4+. The overpotentials and Tafel values of all samples are presented in Fig. 5d. LNMO-0.2 showed the lowest potential and Tafel slope, indicating excellent UOR activity.
 | ||
| Fig. 5 (a) LSV curves of LNMO-0.2 in an N2-saturated 1 M KOH + 0.33 M urea solution and without urea. (b) LSV curves of a LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8) based catalyst supported on a glass carbon electrode in an N2-saturated 1 M KOH and 0.33 M urea solution. (c) Tafel plots extracted from LSV curves. (d) Voltage and Tafel data of LaMnxNi1−xO3 (x = 0, 0.2, 0.4, 0.6, 0.8) at 10 mA cm−2. (e) Plots of current density against the scan rate. (f) Nyquist plots of the as-prepared catalysts. (g) The UOR performance comparison of different catalysts. (h) The E–t plot of LNMO-0.2 at a current density of 50 mA cm−2. | ||
To gain further insight into the intrinsic activity of the catalysts, the electrochemical surface area (ECSA) was evaluated by measuring the electrochemical double layer capacitance (Cdl) within a scan range of 1.13–1.23 V (Fig. S13†). As shown in Fig. 5e, the calculated Cdl values for LNO, LNMO-0.2, LNMO-0.4, LNMO-0.6, and LNMO-0.8 were estimated to be 5.48, 9.16, 8.59, 7.56, and 6.78 mF cm−2, respectively. These results indicate that the high UOR current density obtained with the LNMO-0.2 electrode is the result of a balance between the precise introduction of Mn and the construction of 2D structure. To evaluate the UOR kinetics of the perovskite oxide electrocatalysts, electrochemical impedance spectroscopy (EIS) analysis was performed on all samples at an open-circuit potential with an AC amplitude of 10 mV, in a frequency range of 100 kHz to 0.01 Hz. Nyquist plots are shown in Fig. 5f. Compared to other electrocatalysts, LNMO-0.2 exhibited the smallest radius, indicating the lowest charge transfer resistance (Rct). The Rct values for LNO, LNMO-0.2, LNMO-0.4, LNMO-0.6, and LNMO-0.8 were 8.53, 5.98, 7.44, 8.03, and 8.19 Ω, respectively. The excellent characteristics of LNMO-0.2, with fast charge transfer and low resistance, are well-aligned with the Tafel slope results, further supporting its potential as a highly effective electrocatalyst for the UOR. The response in the low-frequency region of the Nyquist plots for LNMO-0.4, LNMO-0.6, and LNMO-0.8 showed differences, with the diffusion process in LNMO electrodes evidently accelerating. These results suggest that the substitution of Mn significantly enhances the reaction kinetics, thereby improving the UOR performance. This collectively confirms that substituting Mn at the B-site in LNO perovskite oxides effectively improves the conductivity during the OER process and enhances charge transfer capabilities. The exceptional UOR electrocatalytic activity of LNMO-0.2 surpasses that of many catalysts (Fig. 5g and Table S2†).2,52–65 Electrocatalysis requires the stability of electrodes, as commercial electrodes must maintain a stable potential over extended periods. As shown in Fig. 5h, we assessed the long-term electrochemical stability of LNMO-0.2 using chronoamperometry tests at 50 mA cm−2. The durability assessment was conducted at a constant current density of 50 mA cm−2. The results demonstrated significant stability of the potential, with negligible decay even after approximately 30 hours of continuous operation under alkaline conditions. In the accelerated durability assessment (Fig. S14†), LNMO-0.2 consistently exhibited outstanding UOR performance, even after enduring a 30 hour durability test at a scan rate of 10 mV s−1. Notably, the powder XRD spectrum of LNMO-0.2 remained largely unchanged after the stability test (Fig. S15†). Post-durability test SEM images of LNMO-0.2 showed that the 2D porous morphology was maintained with no significant aggregation observed (Fig. S16†). We employ EDS to analyze the elemental composition of electrode materials after testing. This will help us confirm that the distribution and concentration of La, Mn, and O elements in the material remain essentially unchanged over prolonged operation, while the Ni content decreases. This demonstrates the role of Ni as an active site in the catalyst (Fig. S17†).
HRTEM will allow us to observe the microstructure of the catalyst at high resolution after stability testing. We found that the crystal structure of the catalyst remains stable throughout long-term electrochemical reactions, with no particle agglomeration or morphological changes (Fig. S18†).
XPS analysis will be utilized to assess changes in the surface chemical state of the catalyst, especially the chemical environment and valence changes of Ni active sites. The observed decrease in signal intensity of Ni, with the ratio remaining fundamentally unchanged, indicates that the redox reactions occurring during the electrochemical process are reversible (Fig. S19†). This indicates negligible changes in the surface composition of the sample after the durability test. This 2D porous LNMO-0.2, with partial Mn substitution at the B-site, achieves a balance between 2D porous structure design and precise control of the atomic ratio in solid solutions. It facilitates electron transfer in electrocatalytic processes and exhibits stable UOR performance.
In the field of electrocatalysis, the d band center theory is one of the most representative descriptors. We have utilized descriptors derived from density functional theory (DFT) and molecular orbital theory to interpret the performance of a series of LNMO catalysts. To investigate the impact of Ni introduction on the electronic configuration of LMO, DFT calculations were employed to study the electronic configurations of LMO, LNMO-0.2, LNMO-0.4, LNMO-0.6, and LNMO-0.8. Their projected density of states (PDOS) is displayed in Fig. 6a. The valence band states below the Fermi level (Ef) are primarily derived from Mn 3d and O 2p orbitals. As the concentration of Ni increases, the overlap between the partial DOS of occupied and unoccupied 2p orbitals and the partial DOS of Mn 3d promotes the hybridization of Ni 3d with O 2p. This hybridization can lower the charge transfer barrier between O 2p and Ni 3d, thereby enhancing the kinetics of oxygen electrode reactions.66
 | ||
| Fig. 6 (a) Comprehensive PDOS for various elements in LaMnxNi1−xO3. (b) PDOS of Mn 3d orbitals in LaMnxNi1−xO3. (c) PDOS of Ni 3d orbitals in LaMnxNi1−xO3. (d) Schematic diagram illustrating the shift of the d band center. (e) Trend analysis of the d band center variations for Mn and Ni. | ||
As shown in Fig. 6b and c, the d band center of Mn is far from the Fermi level, while the d band center of Ni shows a shifting process relative to the Fermi level. This shift in the d band center of Ni, as a result of varying Ni concentrations, indicates changes in the electronic structure that can significantly affect the catalytic activity. The proximity of the d-band center to the Fermi level is a key factor in determining the reactivity of transition metal catalysts, as it influences the extent of electronic interaction between the catalyst and reactants.67,68 In summary, the DFT analysis suggests that the introduction of Mn into the LNO structure modifies the electronic interactions, particularly through the hybridization of Ni 3d and O 2p orbitals, which is conducive to improved catalytic activity for the UOR. This finding is consistent with the observed enhanced UOR activity in LNMO catalysts, particularly LNMO-0.2, which displayed the highest activity among the samples studied. From the perspective of molecular orbital theory, when adsorbate molecules approach the metal surface, their orbitals interact with the metal's s and d orbitals, leading to energy level splitting. The resulting bonding orbitals are lower in energy and more stable than the original orbitals, while antibonding orbitals are higher in energy and less stable. The more electrons that fill the antibonding orbitals, the less stable the structure becomes, weakening the bond strength of the intermediates (Fig. 6d).69,70 Upon the interaction between the adsorption energy levels and the continuous energy levels, its energy between bonding orbitals and antibonding orbitals undergoes a certain degree of broadening. The width of this broadening represents the interaction strength between the metal's d orbitals and the adsorbate's p orbitals; the stronger the interaction, the wider the d band broadening. When the broadening narrows while the Fermi level remains constant, the d band center rises. Consequently, the fewer the electrons in the antibonding orbitals, the greater the adsorption energy.71 To further clarify the key factors of the d-band in LNMO, we calculated the d-band centers of Ni and Mn for all samples. In the UOR, the strong binding of Ni3+ active sites with *COO intermediates can lead to poisoning effects. The thermodynamic energy of this reaction is substantial, resulting in lower catalyst activity. Therefore, we aim to reduce the adsorption energy at the Ni active sites. As shown in Fig. 6e, the d band center of Ni exhibits a volcanic shape, decreasing from −1.69 eV to −2.70 eV and then increasing to −1.94 eV. This indicates a weakening of adsorption, which is favorable for the occurrence of the UOR. This also explains why LNMO-0.2 exhibits the best UOR activity. Since the OER competes with the UOR at high potentials, we aim to suppress the occurrence of the OER. According to Sabatier's principle, the binding between the catalyst and substrate should not be too strong or too weak, as this is detrimental to the catalytic reaction. Hence, we observe that Mn's d band center continuously rises, indicating stronger adsorption energy. This is increasingly favorable for the OER, where the rate-determining step is the initial adsorption of oxygen intermediates, inadvertently suppressing reactions. Therefore, by combining theoretical calculations, we have achieved a balance between the design of 2D porous structures and the precise control of the atomic ratio in solid solutions.
Conclusions
In summary, we propose a method for the one-step synthesis of highly tunable 2D porous nickel-enriched LaMnxNi1−xO3 perovskite utilizing microwave pulses. This strategy leverages the transient high-temperature and high-energy advantages of microwaves, achieving a rapid one-step equilibrium between the design of 2D porous structures and precise control over solid solution atomic ratios. Compared to traditional slow-entropy-change strategies, it enables the effective avoidance of byproduct generation, yielding a 2D homogeneous solid solution rich in nickel with controlled conductivity and excellent UOR catalytic activity, facilitating a comprehensive understanding of the UOR activity mechanism dependent on distinct electronic configurations. Through the coordinated interaction of conductivity and nickel active site content, the synthesized LaMn0.2Ni0.8O3 structure exhibits optimal UOR performance with a low potential of 1.27 V at a current density of 10 mA cm−2 and a low Tafel value of 44.6 mV dec−1. We anticipate that this strategy will pave the way for the design of more 2D nickel-based solid solutions as efficient electrocatalysts for the UOR, offering new insights for carbon neutrality and clean energy development.Experimental
Chemicals
In this study, analytical grade chemicals were employed, with specifics as follows: lanthanum nitrate hexahydrate (La(NO3)3·6H2O), manganese nitrate tetrahydrate (Mn(NO3)2·4H2O), nickel nitrate hexahydrate (Ni(NO3)2·6H2O), ammonium nitrate (NH4NO3), and glucose, all of which were procured from Sinopharm Chemical Reagent Co., Ltd, China. Additionally, commercial carbon nanotubes (sourced from Suzhou Tanfeng Graphene Technology Co., Ltd, China) and quartz reaction apparatus (obtained from Lianyungang Zhongqi Quartz Products Co., Ltd, China) were utilized in the experiments.Method and synthesis
A series of nickel-rich LaMnxNi1−xO3 solid solution materials were synthesized using the microwave-assisted method. The substitution of Ni atoms with Mn at the B-site was employed to achieve varying atomic ratios in these nickel-rich solid solutions. The atomic ratios of La, Mn, and Ni were set as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:1, 1:0.2:0.8, 1:0.4:0.6, 1:0.6:0.4, and 1:0.8:0.2, denoted as LNO, LNMO-0.2, LNMO-0.4, LNMO-0.6, and LNMO-0.8, respectively. These samples were subsequently utilized as catalysts in the UOR.
:0:1, 1:0.2:0.8, 1:0.4:0.6, 1:0.6:0.4, and 1:0.8:0.2, denoted as LNO, LNMO-0.2, LNMO-0.4, LNMO-0.6, and LNMO-0.8, respectively. These samples were subsequently utilized as catalysts in the UOR.
In the synthesis of 2D LNMO-x, 0.4 g glucose, 0.433 g La(NO3)3·6H2O, 0.291 g of Ni(NO3)2·6H2O, 0.251 g of Mn(NO3)2·4H2O, and 0.5 g NH4NO3 were ground thoroughly in a mortar. Then the mixture was transferred into a custom-made quartz tube (inner diameter of 12 mm, wall thickness of 2 mm) inside a microwave reaction unit. 2D porous LNMO-x can be obtained after heating for 15 min (heating with four pulses of microwaves) with a power of 900 W without any purification process. The samples synthesized by using microwave shock times of 1 min, 1.5 min and 10 min were named LNMO, LNMO-1.5 and LNMO-10, respectively.
Characterization
The compositional and structural analyses of the synthesized materials were conducted using X-ray diffraction (XRD, Empyrean, Netherlands) and X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA). The morphological characteristics of the samples were examined using a high-resolution field emission scanning electron microscope (FESEM, JSM7800 F, Japan) and a transmission electron microscope (TEM, Tecnai G2 F30, USA). The thickness of the lanthanum oxide (LO) was measured using an atomic force microscope (AFM, Bruker Dimension Icon, Germany). Fourier-transform infrared spectroscopy (FTIR) characterization studies were performed using a Nicolet iS50R spectrometer (USA). A Raytek M13 infrared detector (USA) was employed for real-time temperature monitoring during the reactions.Electrochemical measurements
All electrochemical measurements were conducted on a rotating disk electrode (RDE, PINE Instrument, AFMSRCE model, USA). The electrochemical properties were characterized using a CHI electrochemical workstation (Model CHI660E, China) in a conventional three-electrode system. The working electrode was prepared by dispersing 7 mg of catalyst powder and 7 mg of conductive carbon black in a mixture of 0.7 mL deionized water, 0.3 mL isopropanol, and 20 μL of Nafion solution (5% by weight).After ultrasonication for one hour in an ice bath, the mixture formed a homogeneous ink. Subsequently, 5 μL of this catalyst ink was deposited onto a mirror-polished glassy carbon electrode. The mass loading of each electrode was precisely controlled at 1 mg cm−2. The reference electrode used was Hg/HgO (in 1 M KOH + 0.33 M urea), and the counter electrode was a platinum plate. Calibration of the reference electrode was performed according to: E(RHE) = E(Hg/HgO) + E(Hg/HgO vs. RHE) + 0.0592 pH. All scans were performed under an argon atmosphere.
Polarization curves were obtained at room temperature with a scan rate of 5 mV s−1. The electrochemical surface area (ECSA) was estimated from the double-layer capacitance (Cdl) of the samples. Cdl was determined by measuring the corresponding cyclic voltammetry (CV) curves within a specific potential window at different scan rates ranging from 2 to 20 mV s−1. The absolute sum of cathodic and anodic currents at 0.18 V vs. RHE at different scan rates was plotted, and half of the obtained slope was taken as the Cdl. Electrochemical impedance spectroscopy (EIS) was then conducted at various overpotentials, in a frequency range of 0.01 to 105 Hz and at an amplitude of 10 mV. The stability tests were conducted on an electrochemical workstation, utilizing a three-electrode system. Our two-dimensional porous, nickel-rich LaMnxNi1−xO3 solid solution material served as the working electrode, with an Hg/HgO electrode as the reference electrode and a platinum wire as the counter electrode. Tests were performed at room temperature, using a 1 M KOH + 0.33 M urea solution as the electrolyte.
For the stability tests, we employed constant potential (CP) electrolysis to assess the catalyst's stability. For CV tests, the scan rate was set at 50 mV s−1, with a scanning range from 1.13–1.23 V (vs. RHE). A total of 1000 cycles were conducted to evaluate the electrochemical stability of the material throughout the cycling process. For CP tests, electrolysis was performed at the material's optimized activity potential for at least 30 hours to assess its long-term stability.
Author contributions
Contributed equally to this work and conceptualization: Wu Z., Xian J., Dai J. Data curation and writing – original draft: Wu Z., Xian J. Formal analysis: Wu Z., Dai J., Fang G. Software: Fan M., Tian H., Guo J., Huang Z. Funding acquisition: Wan J., Xu W., Jiang H. Supervision, validation and writing – review & editing: Wan J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (52203070) and the Open Fund of Hubei Key Laboratory of Biomass Fiber and Ecological Dyeing and Finishing (STRZ202203). J. Wan expresses gratitude for the financial support provided by the China Scholarship Council (CSC) Visiting Scholar Program.Notes and references
- J. Li, S. Wang, S. Sun, X. Wu, B. Zhang and L. Feng, J. Mater. Chem. A, 2022, 10, 9308–9326 RSC
.
- R.-Q. Li, Q. Liu and Y. Zhou,
et al.
, J. Mater. Chem. A, 2021, 9, 4159–4166 RSC
- Z.-Y. Yu, C.-C. Lang and M.-R. Gao,
et al.
, Energy Environ. Sci., 2018, 11, 1890–1897 RSC
- Z. Cao, T. Zhou and X. Ma,
et al.
, ACS Sustain. Chem. Eng., 2020, 8, 11007–11015 CAS
- S.-K. Geng, Y. Zheng and S.-Q. Li,
et al.
, Nat. Energy, 2021, 6, 904–912 CrossRef CAS
- H. Yu, J. Wan, M. Goodsite and H. Jin, One Earth, 2023, 6, 267–277 CrossRef
- M. He, C. Feng, T. Liao, S. Hu, H. Wu and Z. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 2225–2233 CrossRef CAS PubMed
- D. Li, W. Wan and Z. Wang,
et al.
, Adv. Energy Mater., 2022, 12, 2201913 CrossRef CAS
- X. Xu, H. Ullah and M. Humayun,
et al.
, Adv. Funct. Mater., 2023, 33(44), 2303986 CrossRef CAS
- Y. Tao, Z. Ma and W. Wang,
et al.
, Adv. Funct. Mater., 2023, 33, 2211169 CrossRef CAS
- D. Zhu, H. Zhang and J. Miao,
et al.
, J. Mater. Chem. A, 2022, 10, 3296–3313 RSC
- H. Jin, Q. Gu and B. Chen,
et al.
, Chem, 2020, 6, 2382–2394 CAS
- J. Kundu, T. Kwon, K. Lee and S. I. Choi, Exploration, 2024, 20220174 CrossRef
- E. F. Rodriguez, F. Xia, D. Chen, A. F. Hollenkamp and R. A. Caruso, J. Mater. Chem. A, 2016, 4, 7772–7780 RSC
- C.-J. Huang, H.-M. Xu, T.-Y. Shuai, Q.-N. Zhan, Z.-J. Zhang and G.-R. Li, Small, 2023, 19(45), 2301130 CrossRef CAS PubMed
- T. G. Yun, Y. Heo, H. Bin Bae and S.-Y. Chung, Nat. Commun., 2021, 12, 824 CrossRef CAS PubMed
- G. Fu, W. Li and J.-Y. Zhang,
et al.
, Small, 2021, 17, 2006930 CrossRef CAS PubMed
- W. Chen, L. Xu and X. Zhu,
et al.
, Angew. Chem., Int. Ed., 2021, 60, 7297–7307 CrossRef CAS PubMed
- W.-K. Han, J.-X. Wei and K. Xiao,
et al.
, Angew. Chem., Int. Ed., 2022, 61, e202206050 CrossRef CAS PubMed
- W. Yan, D. Wang and G. G. Botte, Appl. Catal., B, 2012, 127, 221–226 CrossRef CAS
- W. Zhang, Y. Tang, L. Yu and X.-Y. Yu, Appl. Catal., B, 2020, 260, 118154 CrossRef CAS
- N. K. Shrestha, S. A. Patil, A. S. Salunke, A. I. Inamdar and H. Im, J. Mater. Chem. A, 2023, 11, 14870–14877 RSC
- Y. Zhang, H. Guo, M. Song, L. Sun and R. Song, J. Mater. Chem. A, 2023, 11, 3584–3593 RSC
- L. Wang, Y. Zhu and Y. Wen,
et al.
, Angew.
Chem., Int. Ed., 2021, 60, 10577–10582 CrossRef CAS PubMed
- Y. Zhu, C. Liu and S. Cui,
et al.
, Adv. Mater., 2023, 35, 2301549 CrossRef CAS PubMed
- L. Zhang, L. Wang and H. Lin,
et al.
, Angew. Chem., Int. Ed., 2019, 58, 16820–16825 CrossRef CAS PubMed
- Y. Liu, Z. Yang, Y. Zou, S. Wang and J. He, Energy, Environ. Mater., 2023, e12576 CrossRef
- E. Pastor, M. Sachs, S. Selim, J. R. Durrant, A. A. Bakulin and A. Walsh, Nat. Rev. Mater., 2022, 7, 503–521 CrossRef CAS
- R. R. Rao, S. Corby and A. Bucci,
et al.
, J. Am. Chem. Soc., 2022, 144, 7622–7633 CrossRef CAS PubMed
- Y. Sun, S. Xu, C. A. Ortiz-Ledon, J. Zhu, S. Chen and J. Duan, Exploration, 2021, 1, 20210021 CrossRef PubMed
- Z. Wang, P. Guo and S. Cao,
et al.
, Appl. Catal., B, 2021, 284, 119725 CrossRef CAS
- Y. Sun, L. Yang and K. Xia,
et al.
, Adv. Mater., 2018, 30, 1803189 CrossRef PubMed
- R. Hu, H. Jiang, J. Xian, S. Mi, L. Wei, G. Fang, J. Guo, S. Xu, Z. Liu, H. Jin, W. Xu and J. Wan, Appl. Catal., B, 2022, 317, 121728 CrossRef CAS
- H. Jiang, J. Xian and R. Hu,
et al.
, Chem. Eng. J., 2023, 455, 140804 CrossRef CAS
- G. Fang, K. Liu and M. Fan,
et al.
, Carbon Neutralization, 2023, 2, 1–12 CrossRef
- H. Jiang, J. Li and Z. Xiao,
et al.
, Nanoscale, 2020, 12, 16245–16252 RSC
- R. Hu, H. Jiang and J. Xian,
et al.
, Appl. Catal., B, 2022, 317, 121728 CrossRef CAS
- J. Xian, H. Jiang and Z. Wu,
et al.
, J. Energy Chem., 2024, 88, 232–241 CrossRef CAS
- K. Liu and H. Jin,
et al.
, Sci. Adv., 2022, 8, eabn2030 CrossRef CAS PubMed
- J. Wan, Z. Wu and G. Fang,
et al.
, J. Energy Chem., 2024, 91, 226–235 CrossRef CAS
- M. Qu, X. Ding and Z. Shen,
et al.
, Chem. Mater., 2021, 33, 2062–2071 CrossRef CAS
- T. X. Nguyen, Y. C. Liao, C. C. Lin, Y. H. Su and J. M. Ting, Adv. Funct. Mater., 2021, 31, 2101632 CrossRef CAS
- M. A. Alkhalifah, B. Howchen, J. Staddon, V. Celorrio, D. Tiwari and D. J. Fermin, J. Am. Chem. Soc., 2022, 144, 4439–4447 CrossRef CAS PubMed
- X. Gao, X. Bai and P. Wang,
et al.
, Nat. Commun., 2023, 14, 5842 CrossRef CAS PubMed
- V. M. Zemtsova, A. G. Oshchepkov and E. R. Savinova, ACS Catal., 2023, 13, 13466–13473 CrossRef CAS
- D. Li, X. Zhou and Q. Ruan,
et al.
, Adv. Funct. Mater., 2023, 2313680 CrossRef
- J. Sun, L. Du and B. Sun,
et al.
, ACS Appl. Mater. Interfaces, 2020, 12, 24717–24725 CrossRef CAS PubMed
- Y. Tong, P. Chen and M. Zhang,
et al.
, ACS Catal., 2017, 8, 1–7 Search PubMed
- A. Sheelam, S. Balu and A. Muneeb,
et al.
, ACS Appl. Energy Mater., 2022, 5, 343–354 CrossRef CAS
- J. Luo, X. Wang and Y. Gu,
et al.
, Chem. Eng. J., 2023, 472, 144839 CrossRef CAS
- J. Suntivich, K. J. May and H. A. Gasteiger,
et al.
, Nat. Chem., 2011, 334, 1383–1385 CAS
- H. Choi, S. Surendran and D. Kim,
et al.
, Environ. Sci.: Nano, 2021, 8, 3110–3121 RSC
- L. Yang, R. He and X. Wang,
et al.
, Nano Energy, 2023, 115, 108714 CrossRef CAS
- S. Lu, M. Hummel and Z. Gu,
et al.
, ACS Sustain. Chem. Eng., 2021, 9, 1703–1713 CrossRef CAS
- M. Pan, G. Qian and T. Yu,
et al.
, Chem. Eng. J., 2022, 435, 134986 CrossRef CAS
- S. Li, J. Fan and S. Li,
et al.
, J. Nanostructure Chem., 2021, 11, 735–749 CrossRef CAS
- S. Ligani Fereja, P. Li and Z. Zhang,
et al.
, Chem. Eng. J., 2022, 432, 134274 CrossRef CAS
- G. Qian, W. Chen and J. Chen,
et al.
, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.04.006
- D. C. Nguyen, T. L. L. Doan, S. Prabhakaran, D. H. Kim, N. H. Kim and J. H. Lee, Appl. Catal., B, 2022, 313, 121430 CrossRef CAS
- X. Gu, D. Yang, Z. Liu, S. Wang and L. Feng, Electrochim. Acta, 2020, 353, 136516 CrossRef CAS
- Y. Wang, Y. Liu, M. Zhang, B. Liu, Z. Zhao and K. Yan, Sci. China Mater., 2022, 65, 1805–1813 CrossRef CAS
- Q. Zhang, F. M. D. Kazim and S. Ma,
et al.
, Appl. Catal., B, 2021, 280, 119436 CrossRef CAS
- B. Zhang, S. Wang, Z. Ma and Y. Qiu, Appl. Surf. Sci., 2019, 496, 143710 CrossRef CAS
- X. Ao, Y. Gu and C. Li,
et al.
, Appl. Catal., B, 2022, 315, 121586 CrossRef CAS
- X. Jia, H. Kang and X. Yang,
et al.
, Appl. Catal., B, 2022, 312, 121389 CrossRef CAS
- G. Fu, W. Li and J. Y. Zhang,
et al.
, Small, 2021, 17, e2006930 CrossRef PubMed
- H. Huang, H. Jia and Z. Liu,
et al.
, Angew Chem. Int. Ed. Engl., 2017, 56, 3594–3598 CrossRef CAS PubMed
- Z. Lu, K. Jiang, G. Chen, H. Wang and Y. Cui, Adv. Mater., 2018, 30, e1800978 CrossRef PubMed
- D. A. Kuznetsov, B. Han and Y. Yu,
et al.
, Joule, 2018, 2, 225–244 CrossRef CAS
- Y. Liu, P. Vijayakumar, Q. Liu, T. Sakthivel, F. Chen and Z. Dai, Nanomicro Lett., 2022, 14, 43 CrossRef CAS PubMed
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00235k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |