
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile fabrication of binary copper–palladium alloy thin film catalysts for exceptional hydrogen evolution performance†
Muhammad Ali
Ehsan
 *a,
Akilarasan
Muthumariappan
b,
Muhammad
Ali
a,
Abbas Saeed
Hakeem
a and
Wasif
Farooq
bc
*a,
Akilarasan
Muthumariappan
b,
Muhammad
Ali
a,
Abbas Saeed
Hakeem
a and
Wasif
Farooq
bc
aInterdisciplinary Research Center for Hydrogen Technologies and Carbon Management (IRC-HTCM), King Fahd University of Petroleum & Minerals, Box 5040, Dhahran 31261, Saudi Arabia. E-mail: meali@kfupm.edu.sa; Tel: +966138607511
bInterdisciplinary Research Centre for Refining and Advanced Chemicals (IRC-RAC), King Fahd University of Petroleum & Minerals, Dhahran 31261, Kingdom of Saudi Arabia
cDepartment of Chemical Engineering, King Fahd University of Petroleum and Minerals (KFUPM), Dhahran, 31261, Saudi Arabia
First published on 4th September 2024
Abstract
The hydrogen evolution reaction (HER) plays a crucial role in realizing the ambitious objectives of renewable hydrogen (H2) production and CO2 neutrality. The efficacy of the HER process mainly relies on the electrocatalysts that are highly active, stable, and cost-effective, ensuring efficient and sustainable H2 generation. In this study, binary copper–palladium (CuPd) alloy thin film catalysts directly grown on graphite sheets via aerosol-assisted chemical vapor deposition were employed for the HER in 0.5 M H2SO4. A unique array of tower-like microstructures were fabricated by varying the deposition time from 1 to 2 hours, demonstrating excellent HER activity by achieving high current densities of 100 and 1000 mA cm−2 at low overpotentials of 64 and 137 mV, respectively. It also exhibited favorable Tafel kinetics (28 mV dec−1), a high electrochemical surface area (3046 cm2), and reasonable stability over 24 hours, surpassing the benchmark Pt, Pd, and other reputed noble metal-based catalysts. The synergy between transition and noble metals (Cu–Pd) and the array of tower structure in the alloy has been shown to enhance conductivity and offer abundant active sites, resulting in its superior performance in the HER. Furthermore, density functional theory simulations indicated a decrease in the Gibbs free energy value for the binary CuPd alloy (−0.12 eV) compared to that of metallic Pd and Cu in the HER process, thereby validating the experimental observations. This study presents a straightforward deposition technique to design robust and efficient thin film electrocatalysts and optimize electrochemically active sites to achieve faster HER rates with low overpotential.
1. Introduction
The development of efficient and sustainable energy solutions is imperative in addressing the escalating challenges posed by climate change, dwindling fossil fuel reserves, and economic instability. There has been widespread interest in hydrogen (H2) as a renewable energy source due to its high calorific value and environmentally benign properties, making it a potential substitute for fossil fuels.1,2 The natural reserves of pure H2 are nearly non-existent on the Earth, necessitating its extraction from fossil fuels through reforming and gasification processes. However, these methods contribute significantly to CO2 emissions, undermining the renewable nature of H2.3 Alternatively, electrochemical water splitting has received more attention, as it involves electrochemical reaction known as the hydrogen evolution reaction (HER), which can produce pure H2 abundantly using renewable energy sources.4–7 Nevertheless, to ensure the efficient implementation of the HER for sustainable H2 production, it is crucial to design highly active, durable, and economically viable catalysts.8–10The noble metal platinum (Pt) stands out as the most favorable catalyst for the electrochemical HER, but its high cost and susceptibility to corrosion during catalysis impose limitations on its large-scale application.11,12 Therefore, the pursuit of non-Pt catalysts with exceptional catalytic activity, high electrical conductivity, and outstanding stability remains a formidable endeavor within the field of water splitting research.13–15 Elements like Pd, Rh, Re, and Ir, akin to Pt, have been the focal points of research in HER catalysis.16,17 Particularly, Pd-based nanomaterials are widely recognized for their catalytic ability and have become essential in various electrochemical processes, primarily due to their involvement in hydrogen-related reactions.18–20 A famous strategy for developing Pd-based catalysts involves alloying Pd with 3d transition metals like Ni, Co, Cu, etc.21–24 This approach not only reduces the usage of noble metals but also fosters a synergistic interplay between alloying metals, redistributing charge density, and adjusting surface and electronic properties, thereby enhancing the electrocatalytic properties of the alloy catalyst.25,26 Consequently, Pd alloys with binary and ternary compositions display an increased affinity for adsorbing substantial quantities of hydrogen from electrolytes, consistently demonstrating outstanding performance in the HER and achieving high current densities.27,28
For instance, a PdCo alloy encapsulated in N-doped carbon (PdCo@CN) was produced by calcining Pd-doped MOFs under a N2 atmosphere. The PdCo@CN catalyst exhibited promising HER activity, a current density of 10 mA cm−2 at only 80 mV, a small Tafel slope of 31 mV dec−1, and long-term stability in an acidic solution.29 Similarly, PdNi alloy films fabricated via a CVD approach have demonstrated high efficiency in HER catalysis. This alloy electrocatalyst achieved a benchmark current density at an overpotential of just 20 mV, with a Tafel slope value of 50.2 mV dec−1, and exhibited good chemical and mechanical stability.30 Y. Jia et al. recently developed a Pd–Cu hydride (PdCu0.2H0.43) catalyst, combining the benefits of both Pd–M and PdHx structures, resulting in significantly enhanced HER activity and durability.31 The PdCu0.2H0.43 catalyst demonstrated remarkable HER activity including a mere overpotential of 28 mV at a minimal Tafel slope of 23 mV dec−1 and exceptional durability in the HER, attributed to its optimal hydrogen adsorption free energy and reduced metal dissolution rate.31
Despite these notable studies, there is still a need for improving the design of Pd–M alloy catalysts with unique phases and distinct morphologies at scalable levels to further address the activity and durability issues of these catalysts. In this regard, fabricating thin film electrocatalysts on a conductive support through appropriate deposition techniques has the potential to greatly expedite the HER kinetics.32 Previous studies reveal that alloy catalysts are typically synthesized as powders and then converted into electrodes using chemical binders and reagents to ensure mechanical strength and adhesion to the support surface.33 The addition of binders sometimes masks the active sites of the catalyst and slows down the rate of the electrochemical reaction and hence the HER activity.34,35 Moreover, under high applied voltage, there is a risk of catalyst layer defoliation, rendering it unsuitable for commercial application. Considering these significant challenges, our main focus is on fabricating binder-free thin films of Pd alloy catalysts using a modified CVD approach known as aerosol-assisted chemical vapor deposition (AACVD).36
Thus, in this study, we present deposition of a binary copper–palladium (CuPd) alloy catalyst through a one-step AACVD and investigate its potential for hydrogen production through HER catalysis in an acidic medium. Our synthesis method involves using commercial Cu and Pd acetylacetonate precursors, which are dissolved in a methanol–chloroform solvent mixture. Upon conversion into gaseous vapors, they undergo decomposition and reaction at a high temperature of 475 °C, resulting in the formation of a thin film of CuPd alloy on a graphite sheet under an inert gas flow. Deposition times of 1 and 2 hours are used to produce thin films with varying morphologies while maintaining the CuPd composition at a constant 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. Moreover, the films are deposited directly onto a conductive substrate, making them readily available for use as electrodes without any modifications. Consequently, different morphologies of CuPd alloy films are employed in the acidic HER to optimize their catalytic performance. Particularly, the CuPd alloy grown with tower-like morphology after 2 h of deposition exhibits a low overpotential of 64 mV at 100 mA cm−2. Even a large current density of 1000 mA cm−2 is attained at 137 mV, demonstrating excellent stability for 24 hours. Density functional theory (DFT) measurements corroborated the experimental findings, offering precise quantification of the Gibbs free energy, thus affirming the superior catalytic performance of the CuPd alloy.
:1 molar ratio. Moreover, the films are deposited directly onto a conductive substrate, making them readily available for use as electrodes without any modifications. Consequently, different morphologies of CuPd alloy films are employed in the acidic HER to optimize their catalytic performance. Particularly, the CuPd alloy grown with tower-like morphology after 2 h of deposition exhibits a low overpotential of 64 mV at 100 mA cm−2. Even a large current density of 1000 mA cm−2 is attained at 137 mV, demonstrating excellent stability for 24 hours. Density functional theory (DFT) measurements corroborated the experimental findings, offering precise quantification of the Gibbs free energy, thus affirming the superior catalytic performance of the CuPd alloy.
2. Experimental section
2.1. Materials and methods
Copper(II) acetylacetonate (Cu(C5H7O2)2, 97%), palladium(II) acetylacetonate (Pd(C5H7O2)2, 99%) and solvents (methanol and chloroform) were purchased from Sigma-Aldrich. All chemicals and solvents were used directly in the experiments without purification.2.2. Deposition of binary CuPd alloy and monometallic Cu and Pd thin films
A custom-designed AACVD system was utilized for the deposition of a binary CuPd alloy and monometallic Cu and Pd thin films. Before starting the deposition procedure, graphite strips with dimensions of 1 × 2 cm2 were cleaned using ethanol and acetone solvents. The precursor solution was formulated by dissolving Cu (acac)2 (50 mg, 191 mmol) and Pd (acac)2 (58 mg, 191 mmol) in a solvent mixture of toluene/chloroform at a 70:30 volume ratio. The resulting blue color solution was utilized in the AACVD and the deposition was performed for 1 and 2 hours to fabricate binary CuPd alloy thin films with different microstructural properties. For deposition of monometallic Cu and Pd films, the individual precursor of Cu/Pd (acac)2 solution was utilized in AACVD at a similar temperature of 475 °C.
A schematic illustration of the thin film fabrication process via AACVD is shown in Fig. 1. A typical AACVD experiment begins with converting a clear binary precursor solution into an aerosol mist using an ultrasonic humidifier. This mist is then directed towards a horizontal tube furnace set at 475 °C, containing a quartz tube lined with graphite strips. As the mist enters the heated region, the solvent evaporates and the gaseous precursor decomposes, releasing products that settle on the graphite surface. Here, nucleation and growth occur, forming a film layer of the desired material. The exhaust released is directed into a water trap for disposal. Throughout this process, mist transfer and decomposition occur in the presence of a carrier gas containing 5% H2 balanced with N2 (99.999% purity) flowing at a rate of 100 cm3 min−1. Deposition is stopped after either 1 or 2 hours, with the aerosol supply and furnace shut off subsequently. The furnace is then cooled to room temperature under the flow of an inert gas. The CuPd alloy exhibits a dull grey hue, with films uniformly and securely adhered to the graphite sheet, showing no signs of damage or cracking.
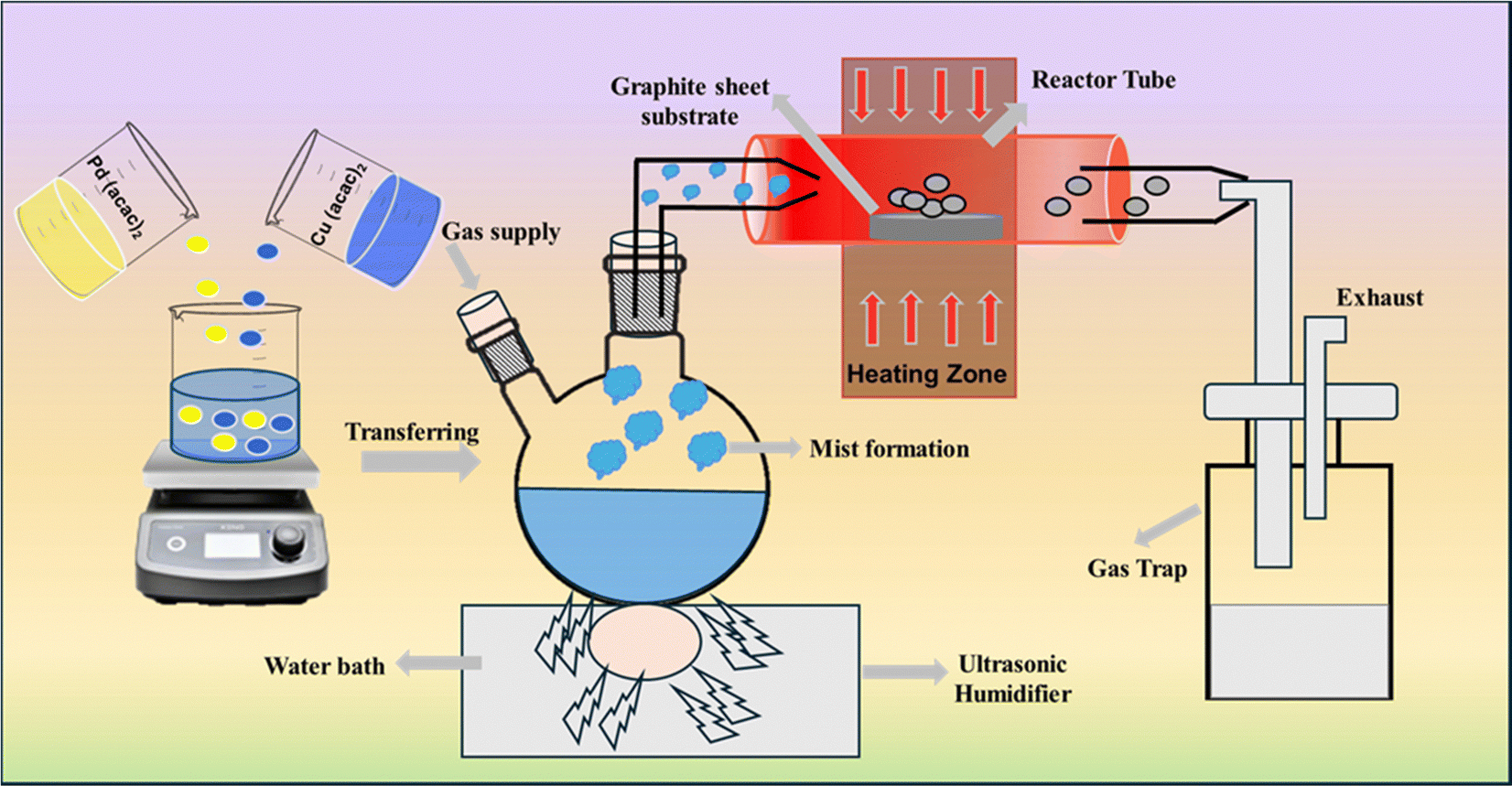 | ||
| Fig. 1 Schematic illustration of the AACVD process for depositing the binary CuPd alloy and monometallic Cu and Pd thin films at 475 °C. | ||
2.3. Structural characterization
The X-ray diffraction (XRD) patterns of the fabricated thin films were measured in the 2θ range of 20°–90° using an X-ray diffractometer (MiniFlex, Rigaku, Japan). Microstructure and morphological investigations of the fabricated thin films were conducted with a scanning electron microscope (MIRA3, TESCAN). The elemental composition of the thin films was determined using an energy-dispersive X-ray spectrophotometer (INCA Energy 200, Oxford Instruments, UK). The chemical states and electronic structure of the film elements were investigated with an X-ray photoelectron spectroscope (Thermo Scientific ESCALAB 250Xi, USA) with an Al Kα (1486.6 eV) source.2.4. Electrochemical measurements
All electrochemical experiments were conducted using a Gamry potentiostat (model: INTERFACE 1010 E). We employed a standard three-electrode system for detailed measurements following the procedures outlined in our prior work, which are illustrated in the ESI.† The electrochemical data were recorded using the iR compensation option in the Gamry workstation program. The resulting data were then plotted and are presented in the manuscript without further iR correction.2.5. Computational methodology
The Vienna ab initio simulation package (VASP) was utilized for first-principles calculations to investigate the structural, electronic and catalytic properties of Cu, Pd and the binary CuPd alloy using the projector-augmented wave (PAW) technique.37 The RPBE exchange–correlation functional was employed at the generalized gradient approximation (GGA) level for all the calculations on periodically repeated metal slabs.38 The valence electron wave functions were expanded using a plane wave basis set that had a cutoff kinetic energy of 400 eV. A conjugate-gradient technique was utilized to optimize the lattice constant as well as the atomic coordinates of all structures. The relaxation process continued until the Hellmann–Feynman forces acting on all atoms were below 0.01 eV Å−1 and the total energy was less than 10−5 eV. The two-dimensional Brillouin zone was sampled using the Monkhorst–Pack method with 3 × 3 × 1 k-points.39 In order to prevent any interaction between periodic images, a vacuum layer with a thickness of 15 Å was added in the z-direction of all structures. The catalytic activity of Pd, Cu and the PdCu alloy was assessed using the Gibbs free energy change, which serves as a typical descriptor for the rate of the overall process. The Gibbs free energy of hydrogen on a catalyst can be expressed as| ΔGH* = ΔEH + ΔEZPE − TΔSH | (1) |
3. Results and discussion
3.1. Structural analysis
The crystalline structure of monometallic Cu, Pd and binary CuPd alloy thin films was analyzed using XRD. The highly crystalline peaks of the graphite substrate were eliminated to ensure an accurate phase analysis of the crystalline products. Fig. 2 shows the comparative XRD patterns of pure metals and their binary alloy samples deposited for time periods of 1 and 2 h. Pure Pd exhibits four crystalline peaks at 2θ = 40.0°, 46.5°, 68.2°, and 82.5°, which can be indexed to the (111), (200), (220), and (311) lattice planes of metallic Pd (International Centre for Diffraction Data (ICDD) No. 01–088–2335).16 While the XRD pattern of metallic Cu explicitly exhibits three crystalline peaks at 2θ = 44°, 51.3°, and 75.5°, corresponding to the (111), (200), and (220) planes, which indicate the formation of a cubic crystal system (ICDD No. 01-071-7832), the XRD patterns of binary alloy samples show peaks at 2θ = 43.2°, 62.7°, 71.2°, and 70°, which were attributed to the (110), (111), (200), and (211) lattice planes of the single-phase Cu0.6Pd0.4 alloy crystallized in the cubic phase (ICDD No. 01-078-4406).40 The XRD patterns clearly demonstrate that the high intensity (100%) crystalline peak of the bimetallic alloy emerged at a larger 2θ value (43.2°) compared to pure Pd (40°), which suggests the incorporation of Cu atoms into the Pd crystal to generate a cubic Cu0.6Pd0.4 alloy structure. The crystalline peaks of both alloy samples appeared at the same 2θ values, indicating the formation of a similar Cu0.6Pd0.4 phase. No other crystalline peaks related to any other possible phase of the CuPd alloy, metallic Pd, Co, or any of their oxide species were observed, confirming the formation of single-phase Cu0.6Pd0.4 alloys.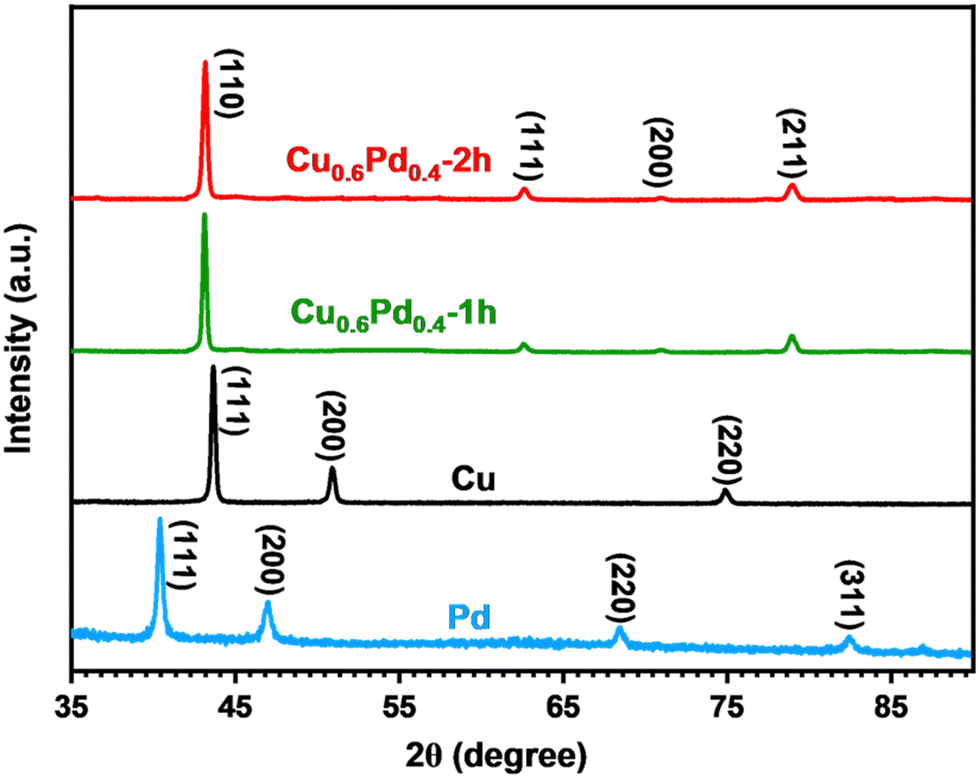 | ||
| Fig. 2 Overlaid XRD patterns of monometallic Pd, Cu and binary Cu0.6Pd0.4 alloy films prepared via the AACVD process. | ||
Fig. 3 illustrates the scanning electron microscopy (SEM) images of the binary CuPd alloy and monometallic Cu and Pd grown on a graphite substrate at a constant temperature of 475 °C. Low-magnification SEM images (Fig. 3(a)–(d)) showed the uniformly grown material covering a large substrate area without any breaks or voids. However, at this magnification level, the morphological features remain unrecognizable. The high-resolution images further elucidate the thin film microstructure and the developed morphological patterns. The binary CuPd alloy deposited for 1 h exhibits vertically oriented structures that extend upward from the plane of the graphite substrate (Fig. 3(a′)). Upon further zooming in on a particular object, one can discern the emergence of tower-like structures standing tall in a vertical orientation, decorated with continuous buds reminiscent of a Christmas tree (Fig. 3(a′′)). As the deposition time extends to 2 hours, there is a noticeable enhancement in the growth of these vertical structures. This leads to the appearance of clusters of tower-like objects, resembling a skyline seen from a bird's-eye view, with numerous skyscraper-like buildings (Fig. 3(b′)). From a magnified view of an individual tower object, it becomes evident that its length exceeds 5 μm, with scales heavily grown on its surface, displaying a lateral perspective of a pine tree. In contrast, pure metallic Cu and Pd show nanoparticle-like morphology. The magnified image shows that the interconnected nanoparticles are undergoing agglomeration, resulting in the formation of large particles. The SEM results demonstrate that vapors of Cu and Pd precursors underwent decomposition and mutual reaction at a high temperature of 475 °C, resulting in the formation of a distinctive tower-like microstructure. Such a morphology is rarely observed in materials synthesized using other solution-based techniques. A key advantage of the AACVD approach is its ability to grow novel features directly on the substrate surface, even without the use of structural directing reagents or templates, all within a remarkably short processing time. While developing catalysts for water splitting, the emergence of novel morphological patterns is highly desirable. This facilitates the creation of a high density of reaction sites, all uniformly exposed to enhance the rates of the HER.
 | ||
| Fig. 3 SEM images captured at different resolutions; binary CuPd alloy deposited for 1 h (a) and 2 h (b). Monometallic Cu (c) and Pd (d) grown on a graphite substrate. | ||
The elemental composition of the fabricated thin films was established through energy dispersive X-ray (EDX) analysis (Fig. S1, ESI†). The monometallic films contained Cu and Pd elements in their individual samples; however, the binary alloy displayed the presence of both elements. The CuPd alloy sample deposited for 1 h consists of 39.69% Pd and 60.31% Cu, while the alloy sample deposited for 2 h comprises 41.78% of Pd and 58.22% of Cu. Both alloy samples roughly show a metallic ratio of Pd to Cu at ∼1:1.5, consistent with the chemical formula Cu0.6Pd0.4 identified from XRD analysis. In the EDX spectra, there is the absence of an oxygen peak, suggesting the synthesis of pure monometallic and alloy materials. Moreover, the EDX mapping of alloy samples confirmed the homogeneous distribution of Pd and Cu elements within the film matrix (Fig. S2, ESI†).
X-ray photoelectron spectroscopy (XPS) analysis was used to study the chemical states and electronic structures of the key elements involved in Cu0.6Pd0.4 alloy fabricated for 2 h. The presence of essential elements of Cu and Pd was recognized with survey scan XPS (Fig. S3, ESI†). The high resolution deconvoluted spectrum of Pd 3d showed peaks at binding energies of 335.8 (3d5/2) and 341.1 eV (3d3/2), indicating the presence of Pd metal in its zero oxidation (0) state (Fig. 4(a)).41 The relatively small peaks at 337.4 and 341.9 eV indicate the existence of Pd2+ species due to the inevitable surface oxidation when the sample was exposed to air.42 Analogously, the Cu 2p XPS spectrum (Fig. 4(b)) of the Cu0.6Pd0.4 alloy involves two fitted peaks at binding energies of 932.23 and 952.08 eV, which correspond to the Cu 2p3/2 and Cu 2p1/2 of metallic Cu (0).43 The peaks at 933.08 and 953.58 eV belong to the Cu 2p3/2 and Cu 2p1/2 of Cu (2+), while the peak at 945.28 eV is a satellite peak.44
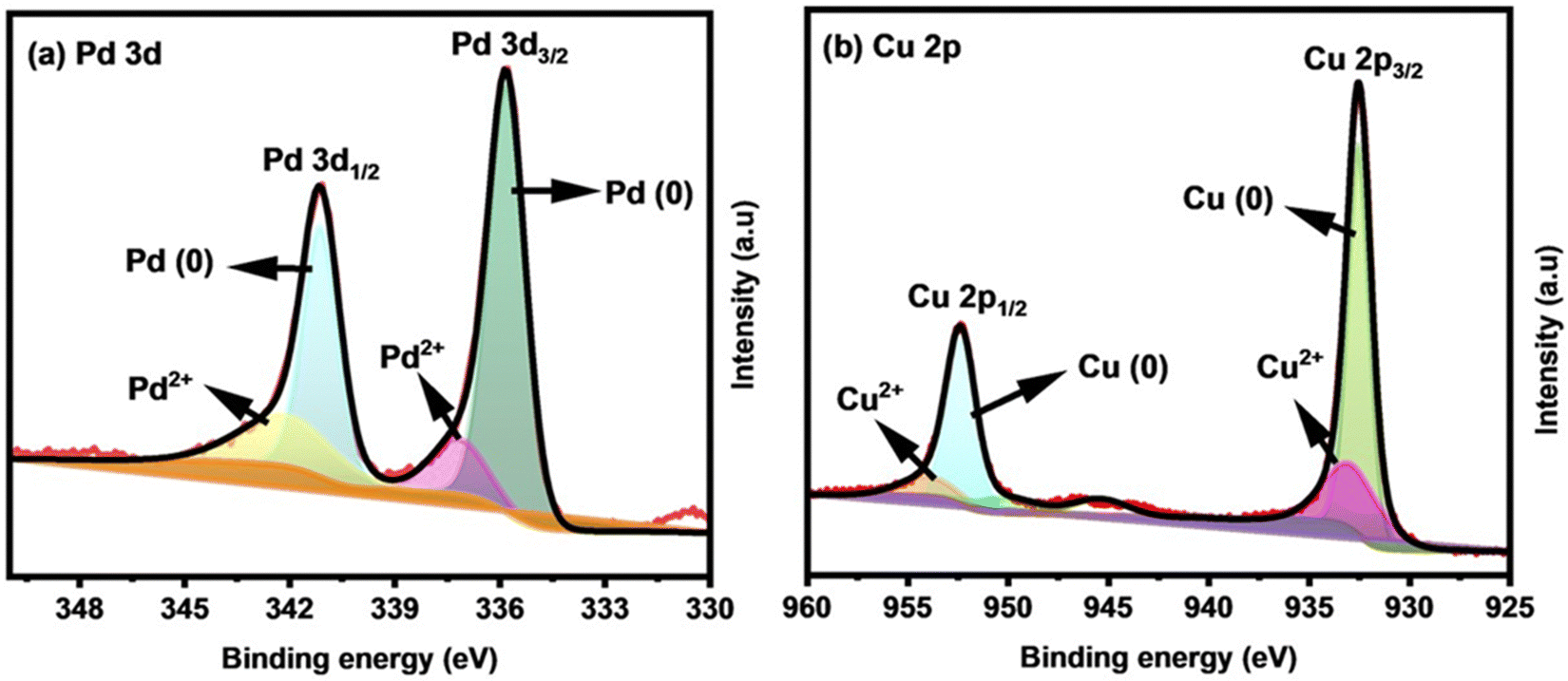 | ||
| Fig. 4 High resolution XPS spectra of the CuPd alloy: (a) Pd 3d and (b) Cu 2p. | ||
3.2. Electrochemical HER studies
The HER performance of thin film electrocatalysts was assessed in a standard three-electrode setup using 0.5 M H2SO4 electrolyte solution. Before measuring the HER activity, the binary CuPd electrocatalyst was activated through consecutive cyclic voltammetry (CV) scans for 150 cycles until a stable and uniform CV signal was obtained.45,46Fig. 5(a) and (b) displays the CV results of activated electrocatalysts, with the first and 150th CV curves overlaid to notice the changes occurring throughout the CV process. For both alloy samples, the 150th CV curve exhibits a noticeable shift towards lower potentials, suggesting that alloy surfaces underwent activation during CV cycling.46 A comparison of the final CV curves (150th) reveals that the CuPd deposited for 2 h has undergone greater activation, as evidenced by a more pronounced shift towards lower potentials compared to the 1 h deposited catalyst (Fig. 5c). This suggests that the distinctive tower-like array developed in the CuPd-2 h catalyst offers more catalytically active sites, consequently accelerating the HER rate, as demonstrated by the high peak current density achieved at low overpotentials. Moreover, CV curves show an oxidation peak around 0 V (vs. reversible hydrogen electrode (RHE)), referring to the adsorption or desorption of intermediate species on the electrode surface.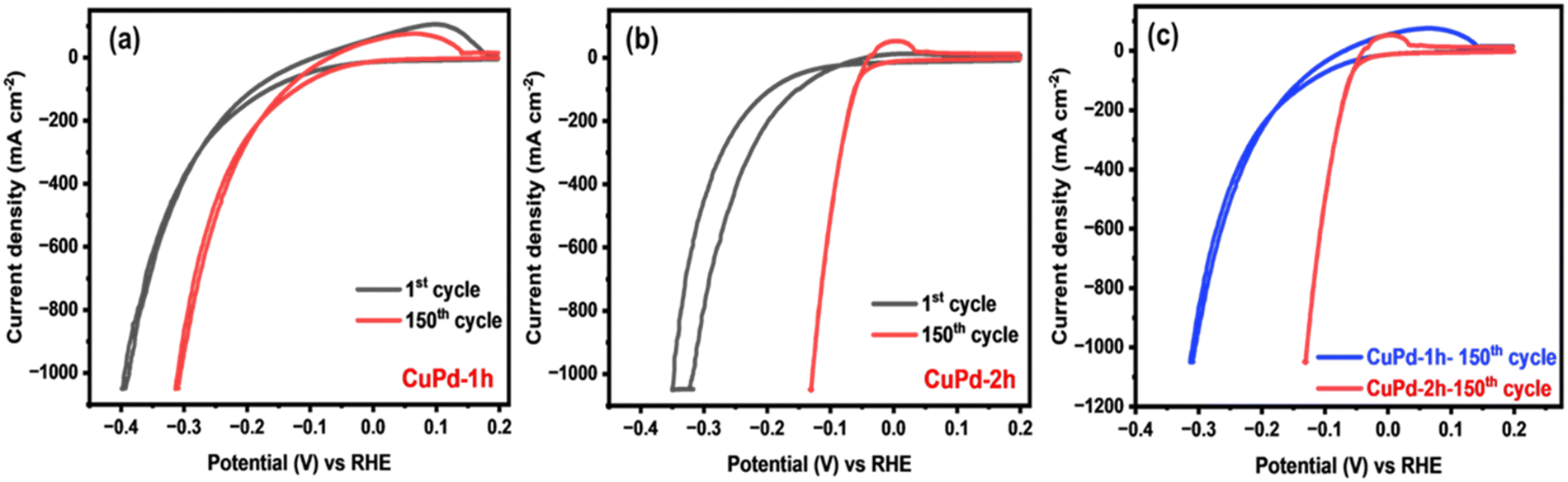 | ||
| Fig. 5 CV analysis of binary CuPd alloy electrocatalysts performed at a scan speed of 50 mV s−1 in 0.5 M H2SO4 electrolyte deposited for 1 h (a) and 2 h (b) and comparison of the 150th CV curves of both alloy samples (c). | ||
Following the CV test, linear sweep voltammetry (LSV) measurements were conducted at a low scan rate of 2 mV s−1 to emphasize the HER performances of the investigated electrocatalysts. Fig. 6(a) clearly reveals that the LSV curves of pure Pd and the binary CuPd alloy reach a significant current density of 1000 mA cm−2 at varying overpotentials, except that of monometallic Cu, which shows poor performance under the given electrochemical conditions. An enlarged view, as shown in Fig. 6(b), provides further insight into the distinctions between the LSV profiles. CuPd-2 h shows HER behavior similar to pure Pd, while CuPd-1 h slightly underperforms in comparison. In the case of the pure Pd catalyst, a typical Pd–H bond peak is observed around ∼0 V (vs. RHE),16 which is absent in both CuPd alloys, indicating structural modulation in pure Pd when interacted with Cu, ultimately leading to the formation of the Cu–Pd alloy. Fig. 6(c) illustrates the comparison of overpotentials at different current densities. For instance, at 100 mA cm−2 the overpotentials increased in the following order: CuPd-2 h (64 mV) < pure Pd (80 mV) < CuPd-1 h (148 mV) < Cu (255 mV). Meanwhile, at a large current density of 1000 mA cm−2, the CuPd-2 h catalyst required the lowest overpotential of 137 mV and clearly outperforms pure Pd (190 mV), while CuPd-1 h still requires a higher overpotential of 311 mV. It is worth noting that the catalytic activity of pure Pd closely resembles that of the benchmark Pt in the HER, despite both Pt and Pd being expensive metals. However, by alloying Pd with the more cost-effective Cu, it becomes possible to readily modulate the electronic structure within the resulting Cu–Pd alloy and achieve better catalytic activity than benchmark catalysts.47 Moreover, the tower-like microstructure engineered in the CuPd-2 h alloy offers abundant active sites conducive to enhancing the kinetics of the HER. The synergistic interplay between the Cu and Pd metals and the distinctive microstructures not only lowers the required overpotential but also augments the HER activity as evidenced by the observed LSV results.
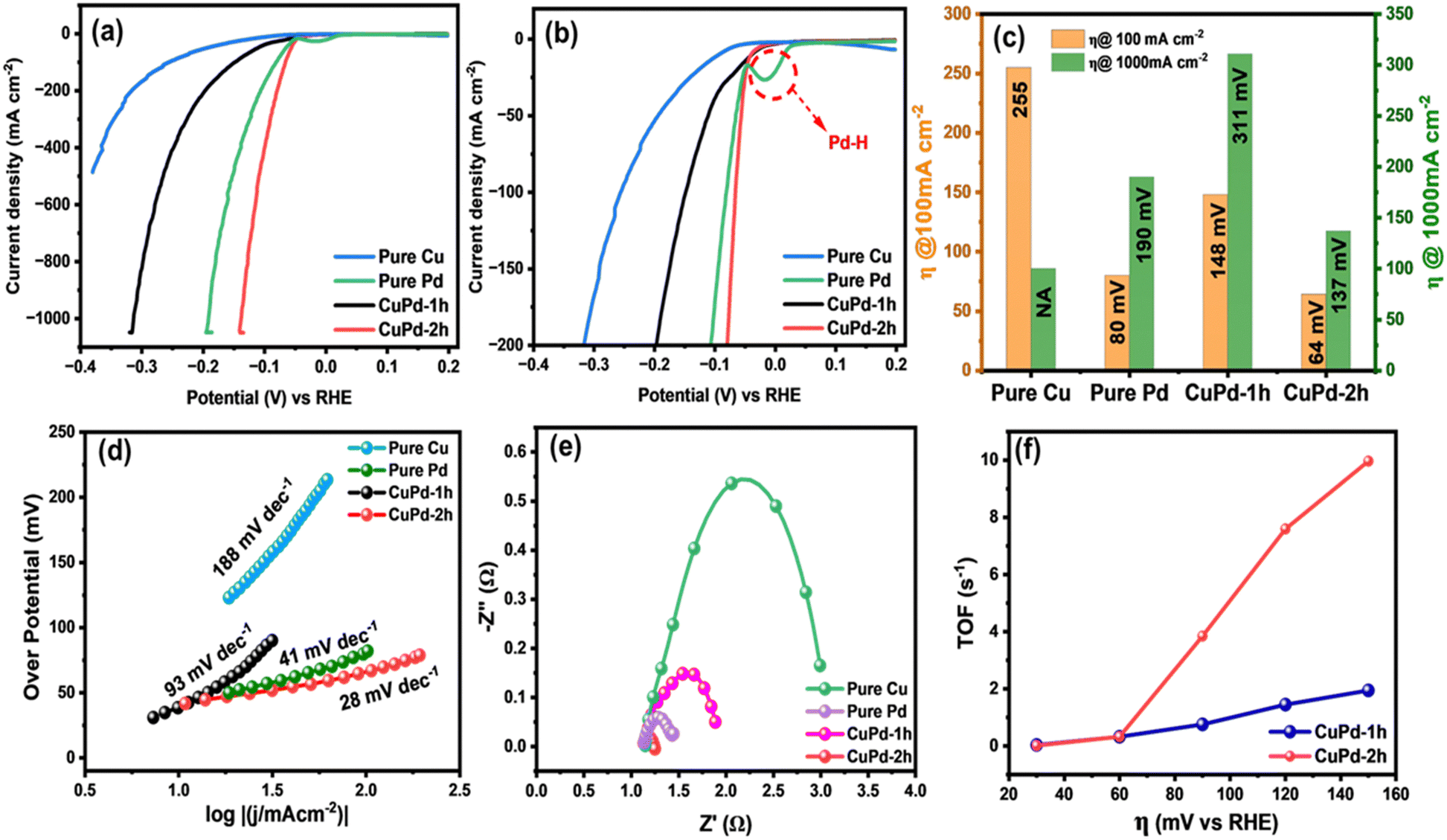 | ||
| Fig. 6 Electrochemical HER measurements of different electrocatalysts in 0.5 M H2SO4 solution. LSV curves (a), enlarged LSV curves (b), overpotential (η) comparison at different current densities of 100 and 1000 mAcm−2 (c), Tafel slopes derived from the corresponding LSV curves (d), EIS Nyquist plots (e), and turnover frequency (TOF) curves (f). | ||
The catalytic attributes of an electrocatalyst can be assessed through its Tafel slope, where a smaller slope generally indicates quicker reaction kinetics.48 To further explore the HER catalytic mechanism, the Tafel slopes were obtained by plotting overpotential (η) against the logarithm of the current density (j). Interestingly, the estimated Tafel slope for CuPd-2 h (28 mV dec−1) was smaller than those of pure Pd (41 mV dec−1), CuPd-1 h (93 mV dec−1) and pure Cu (188 mV dec−1), comparable to or even better than those of the catalysts previously reported and described in Table S1 (ESI†). The lower Tafel slope of the CuPd-2 h catalyst indicates that the rate-limiting step is the charge-transfer (Volmer) process, and the Volmer–Heyrovsky reaction is the pathway for the HER.49
Further, electrochemical impedance spectroscopy (EIS) measurements were conducted to compare the electrical conductivity of different catalysts.50Fig. 6(d) presents a comparative Nyquist plot of all investigated catalysts. The charge-transfer resistance (Rct) was measured to be 0.11, 0.76, 0.32, and 1.87 Ω cm2 for CuPd-2 h, pure Pd, CuPd-1 h and pure Cu, respectively. The far smaller Rct and minute arc size of the CuPd-2 h catalyst compared with those of pure Pd and the other control catalysts suggest faster charge transfer in the reaction at the catalyst–electrolyte interface. Thus, CuPd-2 h exhibited the highest electrical conductivity, which can translate into faster electronic communication and higher HER performance on the electrode surface.
TOF is a valuable indicator for characterizing the intrinsic catalytic activity of different catalysts.51 The TOF values of CuPd alloys measured at different overpotentials (at η = 20, 40, 60, 80, 100, and 120 mV) are shown in Fig. 6(f). At an overpotential of 120 mV, the TOFs of CuPd-2 h and CuPd-1 h were 7.58 and 1.75 s−1, respectively. The remarkably higher TOF of CuPd-2 h indicates a faster reaction on the catalyst surface under the electrochemical conditions employed.
Moreover, electrochemically active surface area (ECSA) serves as a crucial parameter for identifying a catalyst with distinguished catalytic activity.52,53 To determine the ECSA, it is essential to ascertain the value of the double-layer capacitance (Cdl) through simultaneous CV measurements in the non-faradaic region at various scan rates ranging from 10 to 60 mV (vs. RHE). Fig. 7(a) and (c) shows the resultant CV curves of binary CuPd alloy catalysts. The Cdl value of each catalyst was measured by plotting anodic current density vs. the scan rate as shown in Fig. 7(b) and (d). According to the calculations, the CuPd-2 h alloy delivered a higher Cdl (106 mF cm−2) than CuPd-1 h (77 mF cm−2). The ECSA of the CuPd-2 h catalyst, which was as high as 3046 cm2, indicated that more accessible active sites were formed on its surface compared with the CuPd-1 h (2200 cm2) catalyst.
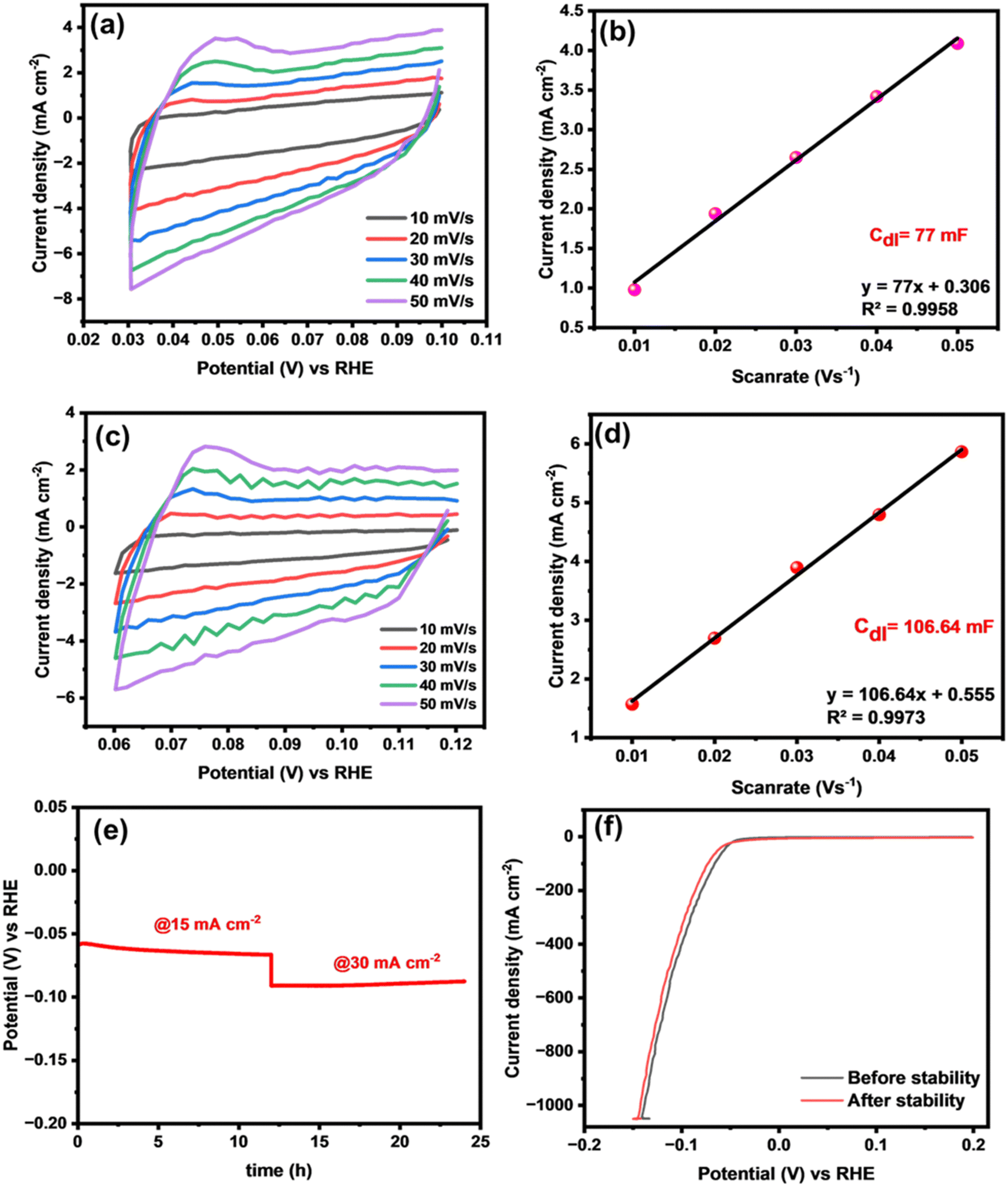 | ||
| Fig. 7 ECSA measurements involving CVs recorded at different scan rates in the non-Faradic region: CuPd-1 h (a) and CuPd-2 h (b). The capacitive current densities of CuPd-1 h (c) and CuPd-2 h (d) catalysts plotted against the scan rate. Chronopotentiometric stability test of the binary CuPd-2 h catalyst (e) and comparison of polarization curves before and after the 24-h stability test (f). | ||
In addition to demonstrating outstanding catalytic activity, it is imperative for catalysts to exhibit robust electrochemical stability, to verify their suitability for large-scale applications. Therefore, a chronopotentiometric test was conducted using the optimized CuPd-2 h catalyst to evaluate its long-term stability for HER catalysis in a 0.5 M H2SO4 solution. Fig. 7(e) demonstrates the efficient performance of the catalyst under two different applied current densities, 15 and 30 mA cm−2, over a continuous 24-hour period, showing no notable decay in potential signals, thus indicating the durability of the system for the HER under the employed electrochemical conditions. After the stability test, the HER activity was again measured using LSV polarization curves. Fig. 7(f) shows comparable LSV responses before and after the 24-h stability test, indicating the sustainable HER performance of the alloy catalyst.
Subsequent to the chronopotentiometric test, the surface of the CuPd-2 h binary catalyst was reassessed using SEM and EDX to ascertain any potential morphological and compositional modifications (Fig. S4, ESI†). The tower shape is slightly crumbled due to the influence of acidic electrolyte throughout continuous HER measurements. Additionally, through EDX analysis (Fig. S4(c), ESI†), the elemental concentrations of Pd and Cu were determined to be 46.9% and 53.1%, respectively, resulting in an empirical molar ratio of 1:1.5. This ratio precisely corresponds to that of the originally synthesized Cu0.6Pd0.4-2 h catalyst. Furthermore, EDX mapping (Fig. S4(d), ESI†) demonstrated that the catalyst retained the elemental uniformity between Cu and Pd elements, even following a 24-hour stability test conducted in acidic electrolyte (Fig. S4(d), ESI†). Overall, the post-characterization findings suggest that the binary catalyst effectively preserved its structural and compositional integrity throughout the electrochemical HER investigations conducted in a 0.5 M H2SO4 electrolyte. A distinctive feature of this study is the synthesis of the CuPd alloy catalyst in a thin film format using a single-step AACVD method, which requires only a brief processing time of 2 hours. The performance of this catalyst is on par with that of the advanced Pd/Cu–Pt nanoring catalyst, which was produced through a more complex two-step solution process that includes extensive washing, separation, and annealing stages.54
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and an optimized lattice constant of 2.98 Å. Therefore, we cleaved the bulk structure into a (110) surface to conduct HER calculations. It is widely recognized that for an efficient HER catalyst, the Gibbs free energy change should be nearly zero.55 DFT calculations revealed a clear difference in the hydrogen adsorption energies between the CuPd alloy (−0.36 eV) and monometallic Pd (−0.38 eV) and Cu (−0.05 eV). On the surfaces of Pd and Cu crystals, the Gibbs free energy of a hydrogen atom is calculated to be −0.14 eV and 0.19 eV, respectively. However, the Gibbs free energy for the CuPd surface is −0.12 eV, which is closer to zero than that of Pd or Cu, as shown in Fig. 8(a). The calculated Gibbs free energy suggests that the adsorbed hydrogen does not exhibit excessively weak or strong binding to the CuPd surface. This characteristic boosts the activity of the surface in the HER. During the process of optimizing the geometry of hydrogen adsorbed on the Pd (Cu) surface, it was seen that the hydrogen atom formed bonds with three neighboring Pd (Cu) atoms on the surface. Similarly, hydrogen adsorbed on the CuPd surface formed bonds with one Pd and two neighboring Cu atoms as shown in Fig. 8(b) and (c). The bond lengths of Pd–H and Cu–H are measured to be 1.72 Å and 1.79 Å, respectively, suggesting a more active interaction of H with Pd compared to that of Cu. Previous studies have demonstrated comparable occurrences of bridging bonding in the context of hydrogen adsorption on other materials.56–58 The Bader charge calculations demonstrated that the addition of Cu to the Pd structure caused a redistribution of charges between the Pd and Cu atoms in the alloy due to the difference in their electronegativities.59,60 This redistribution of charges contributed to a better understanding of the catalytic activity of Cu and Pd in the binary CuPd alloy. The charge redistribution in CuPd enhances the catalytic activity of Pd in the alloy by reducing the Gibbs free energy from −0.14 to −0.12 eV, in comparison to the intrinsic Pd surface. Barder charge analysis indicates that Pd acquired a 0.2e from Cu, leading to an increase in the catalytic activity of Pd in the alloy. The combination of experimental data and DFT calculations conclusively shows that the formation of the CuPd alloy greatly improves the HER compared to its constituent materials.
m and an optimized lattice constant of 2.98 Å. Therefore, we cleaved the bulk structure into a (110) surface to conduct HER calculations. It is widely recognized that for an efficient HER catalyst, the Gibbs free energy change should be nearly zero.55 DFT calculations revealed a clear difference in the hydrogen adsorption energies between the CuPd alloy (−0.36 eV) and monometallic Pd (−0.38 eV) and Cu (−0.05 eV). On the surfaces of Pd and Cu crystals, the Gibbs free energy of a hydrogen atom is calculated to be −0.14 eV and 0.19 eV, respectively. However, the Gibbs free energy for the CuPd surface is −0.12 eV, which is closer to zero than that of Pd or Cu, as shown in Fig. 8(a). The calculated Gibbs free energy suggests that the adsorbed hydrogen does not exhibit excessively weak or strong binding to the CuPd surface. This characteristic boosts the activity of the surface in the HER. During the process of optimizing the geometry of hydrogen adsorbed on the Pd (Cu) surface, it was seen that the hydrogen atom formed bonds with three neighboring Pd (Cu) atoms on the surface. Similarly, hydrogen adsorbed on the CuPd surface formed bonds with one Pd and two neighboring Cu atoms as shown in Fig. 8(b) and (c). The bond lengths of Pd–H and Cu–H are measured to be 1.72 Å and 1.79 Å, respectively, suggesting a more active interaction of H with Pd compared to that of Cu. Previous studies have demonstrated comparable occurrences of bridging bonding in the context of hydrogen adsorption on other materials.56–58 The Bader charge calculations demonstrated that the addition of Cu to the Pd structure caused a redistribution of charges between the Pd and Cu atoms in the alloy due to the difference in their electronegativities.59,60 This redistribution of charges contributed to a better understanding of the catalytic activity of Cu and Pd in the binary CuPd alloy. The charge redistribution in CuPd enhances the catalytic activity of Pd in the alloy by reducing the Gibbs free energy from −0.14 to −0.12 eV, in comparison to the intrinsic Pd surface. Barder charge analysis indicates that Pd acquired a 0.2e from Cu, leading to an increase in the catalytic activity of Pd in the alloy. The combination of experimental data and DFT calculations conclusively shows that the formation of the CuPd alloy greatly improves the HER compared to its constituent materials.
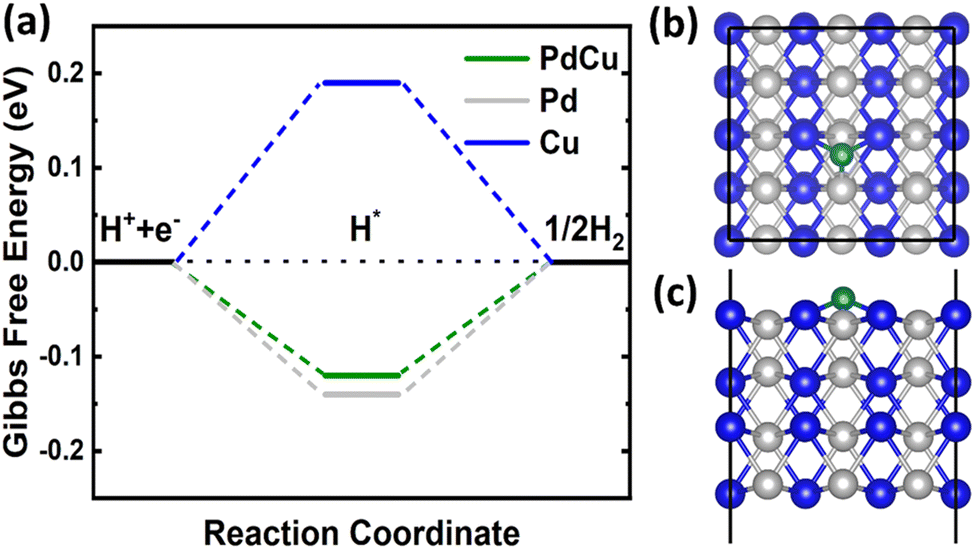 | ||
| Fig. 8 Free-energy diagram for hydrogen evolution on Pd, Cu and PdCu surfaces (a). Top (b) and side (c) views of hydrogen adsorption on the PdCu surface. Grey, blue, and green balls represent Pd, Cu, and H atoms, respectively. | ||
4. Conclusions
A straightforward and efficient AACVD process has been successfully employed to produce thin films of binary Cu–Pd and monometallic Cu and Pd on graphite sheets using readily available acetylacetonate compounds of Cu and Pd. These fabricated film electrocatalysts have been assessed for their efficacy in promoting the HER under acidic conditions. The optimized binary Cu0.6Pd0.4 electrocatalyst, deposited for 2 hours, exhibited a distinctive microstructure resembling a cluster of towers and demonstrated remarkable performance, surpassing even benchmark catalysts such as Pt and Pd, thereby substantially reducing the cost associated with noble metal catalysts. Detailed electrochemical analyses unveiled that the binary alloy catalyst required minimal overpotentials of 64 and 137 mV at current densities of 100 and 1000 mA cm−2, respectively, along with faster Tafel kinetics (28 mV dec−1) and sufficient catalytic stability over 24 hours. The enhanced catalytic activity is attributed to the facilitated synergistic interactions between the noble-transition metal (Cu–Pd) and the abundance of active sites provided by the thin film structure. Both of these factors significantly contribute to the improved catalytic activity and stability of the binary alloy catalysts. These experimental results have been verified by DFT calculations, revealing the enhanced catalytic efficiency of the binary CuPd alloy (−0.12 eV) compared to monometallic Pd (−0.14 eV) and Cu (0.19 eV).Author contributions
Muhammad Ali Ehsan: writing – review & editing, writing – original draft, supervision, project administration, methodology, formal analysis, data curation, conceptualization. Akilarasan Muthumariappan: visualization, validation, software, formal analysis. Muhammad Ali: writing – review & editing, writing – original draft, software, investigation. Abbas Saeed Hakeem: writing – review & editing, writing – original draft, investigation, formal analysis. Wasif Farooq: visualization, validation, resources, formal analysis.Data availability
This is to confirm that the data presented in this manuscript have already been included in both the manuscript and the ESI.†Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
Muhammad Ali Ehsan and Abbas Saeed Hakeem would like to acknowledge the Interdisciplinary Research Center for Hydrogen Technologies and Carbon Management (IRC-HTCM) and Deanship of Research Oversight and Coordination (DROC), King Fahd University of Petroleum and Minerals (KFUPM), Dhahran 31261, for supporting this research work.References
- A. Kovač, M. Paranos and D. Marciuš, Int. J. Hydrogen Energy, 2021, 46, 10016–10035 CrossRef.
- H. Song, S. Luo, H. Huang, B. Deng and J. Ye, ACS Energy Lett., 2022, 7, 1043–1065 CrossRef CAS.
- R. W. Howarth and M. Z. Jacobson, Energy Sci. Eng., 2021, 9, 1676–1687 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- J. Wang, Y. Gao, H. Kong, J. Kim, S. Choi, F. Ciucci, Y. Hao, S. Yang, Z. Shao and J. Lim, Chem. Soc. Rev., 2020, 49, 9154–9196 RSC.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- J. N. Tiwari, A. N. Singh, S. Sultan and K. S. Kim, Adv. Energy Mater., 2020, 10, 2000280 CrossRef CAS.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- L. Tian, Z. Li, X. Xu and C. Zhang, J. Mater. Chem. A, 2021, 9, 13459–13470 RSC.
- M. Wang, L. Zhang, Y. He and H. Zhu, J. Mater. Chem. A, 2021, 9, 5320–5363 RSC.
- M. A. Ehsan, M. H. Suliman, A. Rehman, A. S. Hakeem, A. Al Ghanim and M. Qamar, Int. J. Hydrogen Energy, 2020, 45, 15076–15085 CrossRef CAS.
- T.-H. Yang, J. Ahn, S. Shi, P. Wang, R. Gao and D. Qin, Chem. Rev., 2020, 121, 796–833 CrossRef PubMed.
- B. Owens-Baird, Y. V. Kolen'ko and K. Kovnir, Chem. – Eur. J., 2018, 24, 7298–7311 CrossRef CAS PubMed.
- M. A. Ehsan, A. Adam, A. Rehman, A. S. Hakeem, A. A. Isab and M. Qamar, Sustainable Energy Fuels, 2021, 5, 459–468 RSC.
- J. N. Tiwari, A. M. Harzandi, M. Ha, S. Sultan, C. W. Myung, H. J. Park, D. Y. Kim, P. Thangavel, A. N. Singh and P. Sharma, Adv. Energy Mater., 2019, 9, 1900931 CrossRef.
- M. A. Ehsan, M. H. Suliman, A. Rehman, A. S. Hakeem, Z. H. Yamani and M. Qamar, New J. Chem., 2020, 44, 7795–7801 RSC.
- S. Sarkar and S. C. Peter, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC.
- N.-U.-A. Babar, A. Khan, M. H. Suliman, M. A. Marwat, M. Nawaz Tahir and M. A. Ehsan, Energy Fuels, 2022, 36, 5910–5919 CrossRef CAS.
- H. Li, A. Caravella and H. Xu, J. Mater. Chem. A, 2016, 4, 14069–14094 RSC.
- H. Li and G. Li, J. Mater. Chem. A, 2023, 11, 9383–9400 RSC.
- M. M. Flores Espinosa, T. Cheng, M. Xu, L. Abatemarco, C. Choi, X. Pan, W. A. Goddard III, Z. Zhao and Y. Huang, ACS Energy Lett., 2020, 5, 3672–3680 CrossRef CAS.
- J. Bao, W. Liu, Y. Zhou, T. Li, Y. Wang, S. Liang, Y. Xue, C. Guo, Y. Zhang and Y. Hu, ACS Appl. Mater. Interfaces, 2019, 12, 2243–2251 CrossRef.
- S. Yang, Z. Si, G. Li, P. Zhan, C. Liu, L. Lu, B. Han, H. Xie and P. Qin, Small, 2023, 19, 2207651 CrossRef CAS PubMed.
- Y. Yao, D. S. He, Y. Lin, X. Feng, X. Wang, P. Yin, X. Hong, G. Zhou, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 5501–5505 CrossRef CAS PubMed.
- W. Li, Y. Zhao, Y. Liu, M. Sun, G. I. Waterhouse, B. Huang, K. Zhang, T. Zhang and S. Lu, Angew. Chem., 2021, 133, 3327–3335 CrossRef.
- Q. Wu, M. Luo, J. Han, W. Peng, Y. Zhao, D. Chen, M. Peng, J. Liu, F. M. De Groot and Y. Tan, ACS Energy Lett., 2019, 5, 192–199 CrossRef.
- B. Patella, C. Zanca, F. Ganci, S. Carbone, F. Bonafede, G. Aiello, R. Miceli, F. Pellitteri, P. Mandin and R. Inguanta, Materials, 2023, 16, 474 CrossRef CAS PubMed.
- M. Luo, P. Lu, W. Yao, C. Huang, Q. Xu, Q. Wu, Y. Kuwahara and H. Yamashita, ACS Appl. Mater. Interfaces, 2016, 8, 20667–20674 CrossRef CAS PubMed.
- J. Chen, G. Xia, P. Jiang, Y. Yang, R. Li, R. Shi, J. Su and Q. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 13378–13383 CrossRef CAS.
- N.-U.-A. Babar, A. Khan, M. H. Suliman, M. A. Marwat, M. Nawaz Tahir and M. A. Ehsan, Energy Fuels, 2022, 36, 5910–5919 CrossRef CAS.
- Y. Jia, T.-H. Huang, S. Lin, L. Guo, Y.-M. Yu, J.-H. Wang, K.-W. Wang and S. Dai, Nano Lett., 2022, 22, 1391–1397 CrossRef CAS PubMed.
- N. Nalajala, K. K. Patra, P. A. Bharad and C. S. Gopinath, RSC Adv., 2019, 9, 6094–6100 RSC.
- I. A. Cechanaviciute, R. P. Antony, O. A. Krysiak, T. Quast, S. Dieckhöfer, S. Saddeler, P. Telaar, Y. T. Chen, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2023, 62, e202218493 CrossRef CAS.
- Y. Y. How, A. Numan, M. N. Mustafa, R. Walvekar, M. Khalid and N. M. Mubarak, J. Energy Storage, 2022, 51, 104324 CrossRef.
- O. Gerard, A. Numan, S. Krishnan, M. Khalid, R. Subramaniam and R. Kasi, J. Energy Storage, 2022, 50, 104283 CrossRef.
- M. A. Ehsan, S. S. Shah, S. I. Basha, A. S. Hakeem and M. A. Aziz, Chem. Rec., 2022, 22, e202100278 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- H. Peng and J. P. Perdew, Phys. Rev. B, 2017, 95, 081105 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- M. Friedrich and M. Armbrüster, Chem. Mater., 2009, 21, 5886–5891 CrossRef CAS.
- A. Khan, A. S. Hakeem, H. D. Mohamed, M. H. A. Al-Saeed and M. A. Ehsan, J. Environ. Chem. Eng., 2022, 10, 107959 CrossRef.
- H.-Y. Chen, H.-J. Niu, X. Ma, J.-J. Feng, X. Weng, H. Huang and A.-J. Wang, J. Colloid Interface Sci., 2020, 561, 372–378 CrossRef CAS PubMed.
- Y. Ren, Y. Bai, G. Wang, Y. Liu, C. Mou, J. Chen, B. Wei, H. Wang and Y. Sun, Energy Fuels, 2023, 37, 9289–9296 CrossRef CAS.
- C. Wu, J. Zhu, H. Wang, G. Wang, T. Chen and Y. Tan, ACS Catal., 2019, 10, 721–735 CrossRef.
- P. F. Liu, H. Yin, H. Q. Fu, M. Y. Zu, H. G. Yang and H. Zhao, J. Mater. Chem. A, 2020, 8, 10096–10129 RSC.
- H. Schäfer, S. Sadaf, L. Walder, K. Kuepper, S. Dinklage, J. Wollschläger, L. Schneider, M. Steinhart, J. Hardege and D. Daum, Energy Environ. Sci., 2015, 8, 2685–2697 RSC.
- C. Sarkar, P. Koley, I. Shown, J. Lee, Y.-F. Liao, K. An, J. Tardio, L. Nakka, K.-H. Chen and J. Mondal, ACS Sustainable Chem. Eng., 2019, 7, 10349–10362 CrossRef CAS.
- K. Xu, H. Cheng, H. Lv, J. Wang, L. Liu, S. Liu, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1703322 CrossRef PubMed.
- Y.-H. Fang, G.-F. Wei and Z.-P. Liu, J. Phys. Chem. C, 2013, 117, 7669–7680 CrossRef CAS.
- K. S. Joya, M. A. Ehsan, M. Sohail and Z. H. Yamani, J. Mater. Chem. A, 2019, 7, 9137–9144 RSC.
- Y. Ao, S. Chen, C. Wang and X. Lu, J. Colloid Interface Sci., 2021, 601, 495–504 CrossRef CAS PubMed.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- G. Li, L. Anderson, Y. Chen, M. Pan and P.-Y. A. Chuang, Sustainable Energy Fuels, 2018, 2, 237–251 RSC.
- T. Chao, X. Luo, W. Chen, B. Jiang, J. Ge, Y. Lin, G. Wu, X. Wang, Y. Hu and Z. Zhuang, Angew. Chem., Int. Ed., 2017, 56, 16047–16051 CrossRef CAS PubMed.
- X. Zhao, Z. H. Levell, S. Yu and Y. Liu, Chem. Rev., 2022, 122, 10675–10709 CrossRef CAS PubMed.
- M. Ali, N. Tit and Z. H. Yamani, Int. J. Energy Res., 2020, 44, 10926–10936 CrossRef CAS.
- R. Yao, K. Sun, K. Zhang, Y. Wu, Y. Du, Q. Zhao, G. Liu, C. Chen, Y. Sun and J. Li, Nat. Commun., 2024, 15, 2218 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- R. F. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XPS survey spectra, post-HER SEM images, EDX characterization, and a table showing a comparison of the HER performance of Pd-based electrocatalysts. See DOI: https://doi.org/10.1039/d4ma00410h |
| This journal is © The Royal Society of Chemistry 2024 |