Synthesis of a high-density jet fuel with creosol and formaldehyde†
Received
17th January 2023
, Accepted 18th March 2023
First published on 20th March 2023
Abstract
A new two-step process was developed for the synthesis of jet fuel range high-density bicycloalkanes with creosol and formaldehyde, two platform compounds that can be derived from lignocellulose. In the first step, C17 oxygenate was obtained by the phenolic condensation of creosol and formaldehyde. Among the investigated catalysts, 40% HPW/SiO2 (a SiO2-loaded phosphotungstic acid catalyst with a phosphotungstic acid weight percentage of 40%) exhibited the highest activity for this reaction. Over it, a high yield (64%) and good selectivity (92%) of C17 oxygenate were achieved under mild reaction conditions (323 K, 6 h). In the second step, the C17 oxygenate was hydrodeoxygenated to C14 and C15 bicycloalkanes over Ru-based bifunctional catalysts. Under the optimized conditions, a high total yield of C14 and C15 bicycloalkanes (89%) was achieved. According to our measurements, the C14 and C15 bicycloalkane mixture as obtained had a high density (0.90 g mL−1), low freezing point (246.5 K), and high heat value (46.2 MJ kg−1 or 41.6 MJ L−1). It has potential to be used as a fuel additive to improve the volumetric heat value of current bio-jet fuels.
Introduction
To fulfil the need for sustainable development and tackle environmental problems, the substitution of fossil energy with renewable and carbon dioxide neutral biomass to fuels1 and chemicals2 has drawn considerable attention. Jet fuel is a highly demanded transportation fuel for aviation. Lignocellulose is the main component of agriculture waste and forest residues. Compared with other biomass, lignocellulose has some evident advantages, such as low cost and high availability. Following the pioneering work of Dumesic et al.,3 great efforts have been devoted to the synthesis of jet fuel range hydrocarbons by the C–C coupling reactions of lignocellulosic platform compounds, followed by hydrodeoxygenation.
Creosol is a model compound that can be easily obtained from lignin.4 Likewise, formaldehyde can also be obtained from lignocellulose or CO2 by several routes. First, formaldehyde can be produced from the partial oxidization of methane5 from the biological fermentation or the hydrogenolysis of lignocellulose.6 Second, formaldehyde can be manufactured with CO2 by hydrogenation, photochemical, electrochemical, and photoelectrochemical methods.7 Third, formaldehyde can also be obtained from the acid hydrolysis of lignin.8 In the previous work by our group,9 it was found that jet fuel range polycycloalkanes could be obtained from the hydrodeoxygenation of bisphenols that may be produced with phenol, a model compound that can be obtained from the hydrogenolysis of lignin. To the best of our knowledge, there is no report about the synthesis of polycycloalkane from lignin-derived phenols that were connected with other functional groups (such as –OCH3 group and alkyl group). In this work, it is reported for the first time that jet fuel range bicycloalkanes can be selectively produced by the phenolic condensation reaction of creosol and formaldehyde, followed by hydrodeoxygenation over a Ru-based bifunctional catalyst. The strategy for this process is illustrated in Scheme 1.
 |
| | Scheme 1 Strategy for the synthesis of jet fuel range C14–C15 high-density bicycloalkanes with creosol and formaldehyde. | |
Experimental
Materials
All of the chemicals used in this work were purchased from Energy Chemicals® and directly used as received without any further purification. The SiO2-supported heteropolyacid catalysts were prepared by an impregnation method. Typically, 1.0 g SiO2 support (supplied by Aladdin Chemistry Co. Ltd.) was added to an aqueous solution of heteropolyacids, with the appropriate concentration (see Table 1). The mixture was stirred vigorously for 4 h at room temperature. The water was removed by rotary evaporation. Subsequently, the sample was dried at 383 K overnight in an oven and calcined at 573 K in a muffle furnace. The nomenclature of the SiO2-supported heteropolyacid catalysts and the heteropolyacids used in the preparation of the catalysts is shown in Table 1.
Table 1 Nomenclature of the SiO2-supported heteropolyacid catalysts and the heteropolyacids used in the preparation of the catalysts
| Catalyst |
Heteropolyacid used in the preparation of the catalysts |
Theoretical content of heteropolyacid (wt%) |
| 30% HSiMo/SiO2 |
Silicomolybdic acid |
30 |
| 30% HPMo/SiO2 |
Phosphomolybdic acid |
30 |
| 30% HSiW/SiO2 |
Silicotungstic acid |
30 |
| 30% HPW/SiO2 |
Phosphotungstic acid |
30 |
| 20% HPW/SiO2 |
Phosphotungstic acid |
20 |
| 40% HPW/SiO2 |
Phosphotungstic acid |
40 |
| 50% HPW/SiO2 |
Phosphotungstic acid |
50 |
The Ru catalysts used in the hydrodeoxygenation process were prepared by the impregnation of zeolites (H-ZSM-5, H-β and H-Y, which were purchased from Nankai University) with aqueous solutions of RuCl3. According to the information from the supplier, the SiO2/Al2O3 molar ratios of the H-ZSM-5, H-β, and H-Y zeolites were 50, 25, and 5.4, respectively. The products were dried at 383 K for 10 h and then reduced by hydrogen at 573 K for 3 h. For comparison, the concentration of Ru in the catalysts was controlled as 5% by weight.
Characterization
The N2-physisorption measurements of the catalysts were performed on a Micromeritics ASAP 2010 apparatus. Before the measurements, the samples were evacuated at 573 K for 6 h. The CO-chemisorption, NH3-chemisorption, and NH3-temperature-programmed desorption (NH3-TPD) tests of the samples were conducted using a Micromeritics AutoChem II 2920 Characterization System. Before the NH3-TPD tests, the samples were pretreated by a He flow at 573 K for 0.5 h (as we did for the activity tests) and cooled down to 373 K in the He flow. After the saturated adsorption of NH3 at 373 K, the desorption of NH3 was carried out in He flow from 373 K to 1173 K (at a rate of 10 K min−1). The desorbed NH3 was monitored by an OmniStar mass spectrometer. The X-ray diffraction (XRD) patterns of the Ru-based HDO catalysts were collected using a PW3040/60X′ Pert PRO (PANAlytical) diffractometer. The TEM images of the Ru-based HDO catalysts were obtained by a JEM-2100F scanning transmission electron microscopy (STEM) system. The metal dispersions on the surfaces of the Ru-based catalysts were measured with a Micromeritics AutoChem II 2920 Automated Catalyst Characterization System by CO chemisorption. These values corresponded to the ratio of surface metal atoms to total metal atoms assuming that the stoichiometry of the adsorbed CO to the surface metal atom was one. Before the tests, the samples were dried in a He flow at 393 K for 0.5 h and cooled down in a He flow to 323 K. After the stabilization of the baseline, the CO adsorption was carried out at 323 K by the pulse adsorption of 10% CO in He.
Activity tests
Phenolic condensation.
The phenolic condensation reaction of creosol and 37 wt% formaldehyde aqueous solution was carried out in a 35 mL glass batch reactor. The temperature of the reactor was controlled using a water bath. Typically, 25 mmol creosol, 10 mmol formaldehyde, and 0.1 g SiO2-supported heteropolyacid catalyst were stirred at 333 K for 2 h. The reactions were stopped by rapidly quenching the glass batch reactor in cool water. Subsequently, tridecane was added into the reaction system as an internal standard. The mixture was diluted by CH2Cl2 and analyzed by an Agilent 7890A gas chromatography (GC) instrument equipped with an HP-5 capillary column (30 m, 0.32 mm ID, 0.5 mm film) and a flame ionization detector (FID). Formaldehyde is highly reactive. As a result, we could not detect it by GC. Due to this reason, we used creosol conversion in the activity tests. The conversion of creosol and yield of 1A were calculated by using the following equations:
| Conversion of creosol (%) = (mol of creosol in the feedstock − mol of unreacted creosol in the product)/(mol of creosol in the feedstock) × 100% |
| Yield of 1A (%) = 2 × (mol of 1A in the product)/(mol of creosol in the feedstock) × 100% |
Hydrodeoxygenation (HDO).
The HDO process of the condensation product was carried out in a stainless-steel batch reactor. Typically, 0.2 g substrate, 0.1 g Ru-based catalyst, and 40 mL cyclohexane (used as a solvent) were employed for each test. The reaction was conducted under 4 MPa H2 at 473 K for 5 h and terminated by putting the reactor in cool water. After releasing unreacted hydrogen, dodecane (used as an internal standard) was added into the reaction system. The mixture was filtered and analyzed by GC and GC-MS systems equipped with HP-INNOWAX capillary columns (30 m, 0.25 mm ID, 0.5 μm film thickness). The yields of dicycloalkanes were calculated by the following equation:
| Yield of specific dicycloalkane (%) = (mol of specific dicycloalkane in the product)/(mol of 1A used in the feedstock) × 100% |
The density and freezing point of the polycycloalkane mixture were measured by a DMA 4500 M digital densitometer and a SETARAM DSC141 system, respectively.
Results and discussion
Phenolic condensation
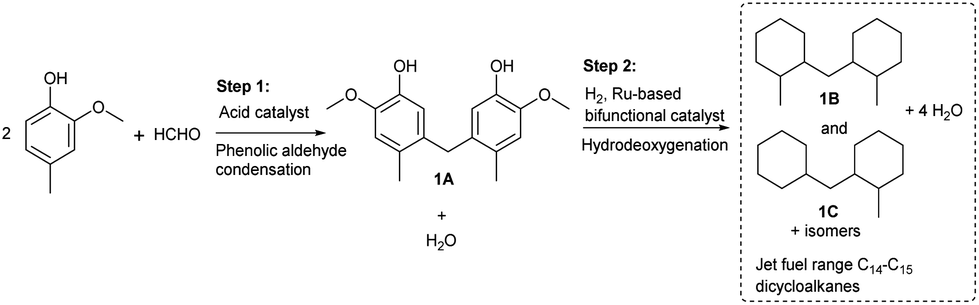
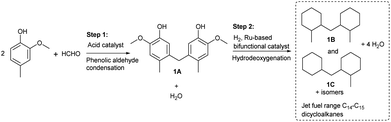
In this work, a series of SiO2-loaded heteropolyacids were first found to be effective catalysts for the phenolic condensation of creosol and formaldehyde. From the analysis of the GC and NMR spectra (see Fig. S1–S3 in the ESI†), 5,5′-methylenebis(2-methoxy-4-methylphenol) from the phenolic condensation of formaldehyde with creosol (i.e.1A in Scheme 1) was obtained as the main product. Besides 1A, small amounts of its isomers were also detected in the product (see Fig. S1 in the ESI†). Based on its bicyclic carbon chain structure, the 1A as obtained could be used as a precursor for the production of jet fuel range high-density bicycloalkane. Among the investigated catalysts, the 30% HPW/SiO2 exhibited the highest activity for the phenolic condensation of creosol and formaldehyde (see Fig. 1). Over it, a good 1A yield (37.8%) was achieved after the reaction was carried out at 333 K for 2 h. The activity of the SiO2-loaded heteropolyacid catalysts decreased in the order of HPW/SiO2 > HPMo/SiO2 > HPSiW/SiO2 > HSiMo/SiO2, which was consistent with the amounts of acid sites on the surfaces of these catalysts (see Table 2). From the NH3-TPD results (see Fig. S4 in the ESI†), no clear relationship was observed between the acid strength of the SiO2-loaded heteropolyacids and their activity for phenolic condensation.
 |
| | Fig. 1 Creosol conversions and the yields of 1A over the different heteropolyacid catalysts. Reaction conditions: 333 K, 2 h; 25 mmol creosol, 10 mmol formaldehyde, and 0.1 g catalyst were used in each test. | |
Table 2 Specific BET surface areas (SBET) and the amounts of acid sites of the different solid acid catalysts
| Catalyst |
S
BET
(m2 g−1) |
Amount of acid sitesb (μmol g−1) |
|
Calculated from the N2-physisorption results.
Calculated from the NH3-chemisorption results.
|
| 30% HSiMo/SiO2 |
204 |
377 |
| 30% HSiW/SiO2 |
166 |
465 |
| 30% HPMo/SiO2 |
197 |
521 |
| 30% HPW/SiO2 |
233 |
824 |
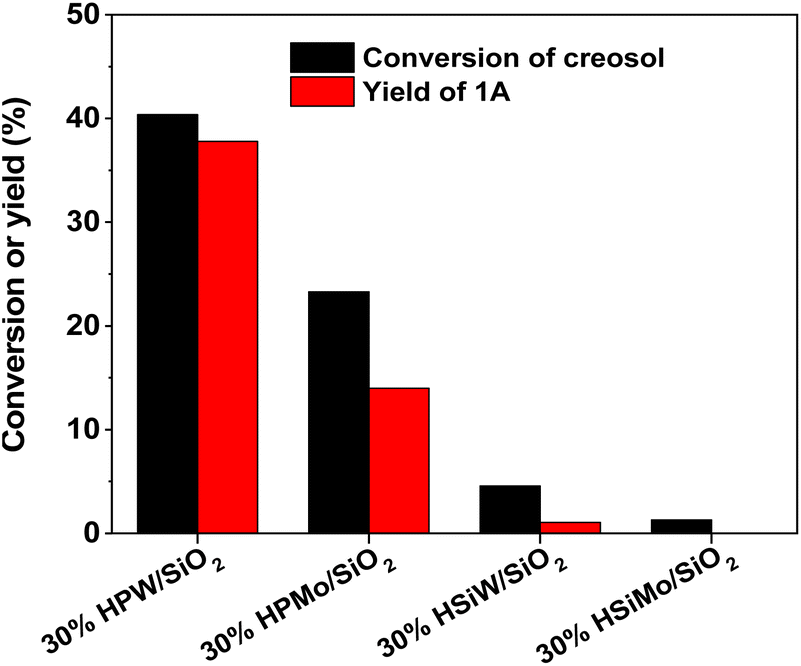
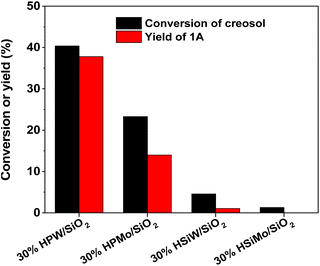
The effect of the HPW content on the catalytic performance of HPW/SiO2 was investigated as well. As can be seen from Fig. 2, the activity of the HPW/SiO2 catalyst increased with the content of phosphotungstic acid, reaching a maximum when the content of phosphotungstic acid was about 40 wt%, but then decreased with the further increment of the phosphotungstic acid content.
 |
| | Fig. 2 Creosol conversions and the yields of 1A over the HPW/SiO2 catalysts. Reaction conditions: 333 K, 2 h; 25 mmol creosol, 10 mmol formaldehyde and 0.1 g catalyst were used in each test. | |
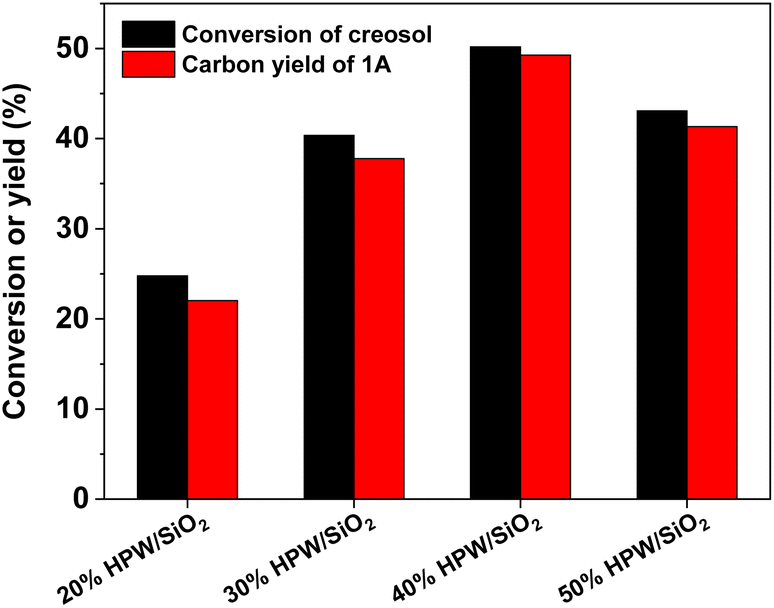
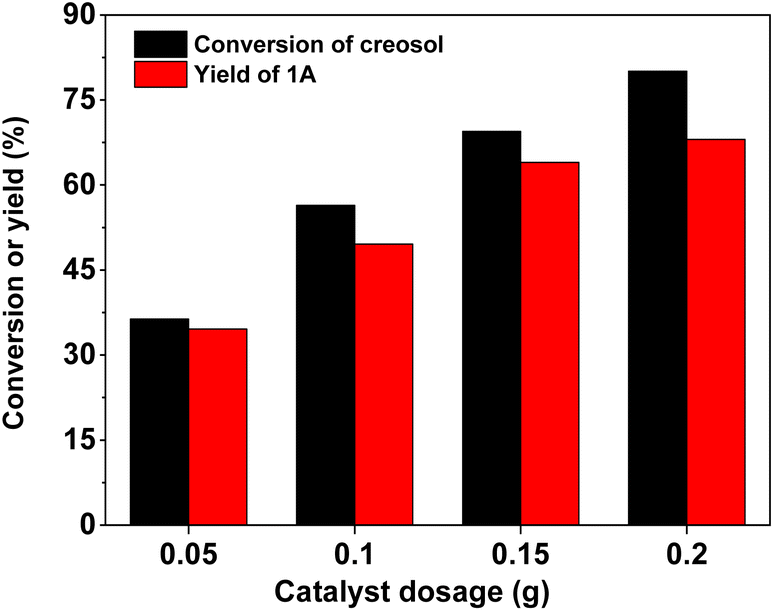
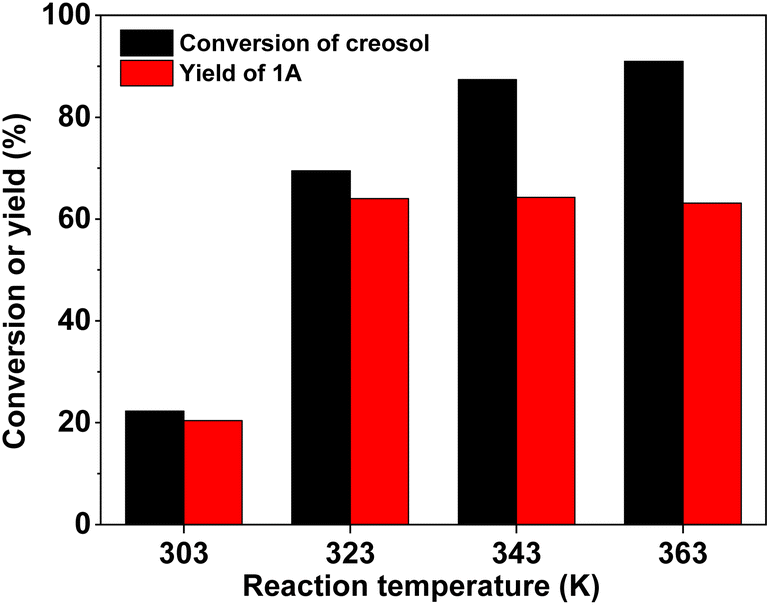
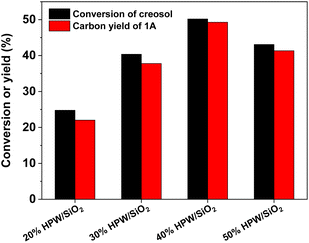
With the further optimization of the reaction time, the catalyst dosage and reaction temperature (see Fig. 3–5), a good 1A yield (64%) was achieved when the reaction was carried out over 0.15 g 40% HPW/SiO2 catalyst at 323 K for 6 h. Under such reaction conditions, a high 1A selectivity (92%) was achieved. In real application, the 1A yield can be further improved by recycling the unreacted creosol, which is a commonly used method in the modern chemical industry for similar reactions.
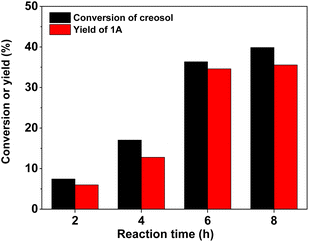 |
| | Fig. 3 Creosol conversions and the yields of 1A over the 40% HPW/SiO2 catalyst as a function of the reaction time. Reaction conditions: 323 K; 20 mmol creosol, 10 mmol formaldehyde, and 0.05 g catalyst were used in each test. | |
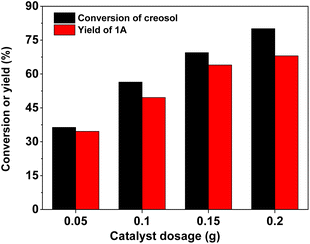 |
| | Fig. 4 Creosol conversions and the yields of 1A over the 40% HPW/SiO2 catalyst as a function of the catalyst dosage. Reaction conditions: 323 K, 6 h; 20 mmol creosol and 10 mmol formaldehyde were used in each test. | |
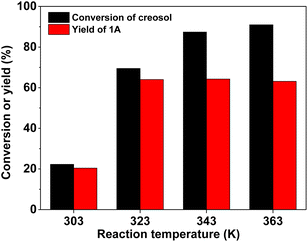 |
| | Fig. 5 Creosol conversion and the yield of 1A over the 40% HPW/SiO2 catalyst as a function of the reaction temperature. Reaction conditions: 6 h; 20 mmol creosol, 10 mmol formaldehyde, and 0.15 g catalyst were used in each test. | |
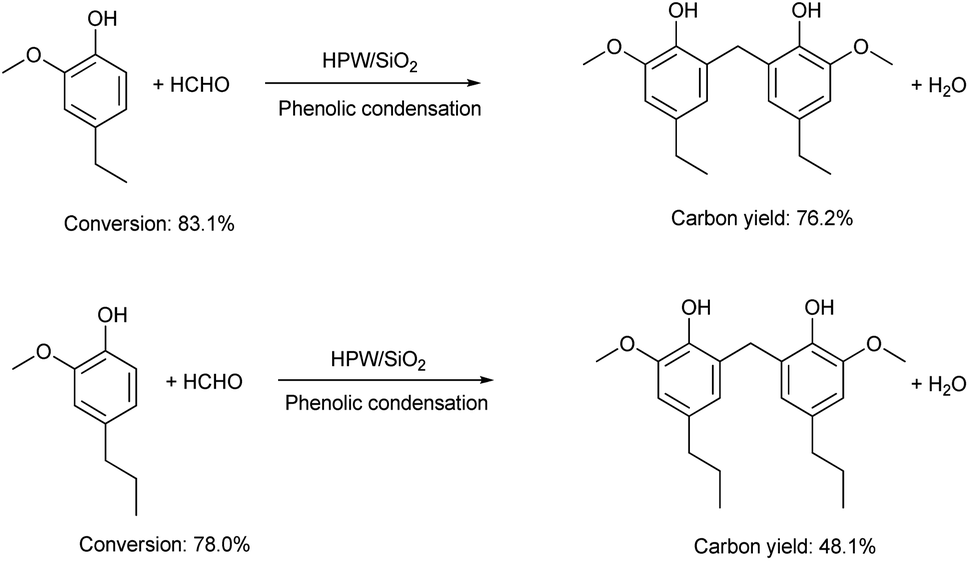
The application of the HPW/SiO2 catalyst for the production of jet fuel precursors with other lignin-derived phenols was also explored. According to the results in Fig. 6 and S5–S8 in the ESI,† the HPW/SiO2 catalyst was also effective for the phenolic condensation of formaldehyde and 4-ethyl guaiacol or 4-propyl guaiacol, which is advantageous in real application.
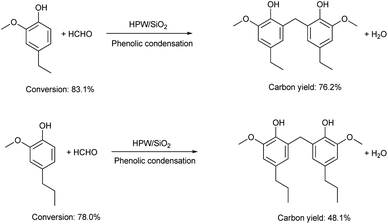 |
| | Fig. 6 Conversions of lignin-derived phenols and the carbon yields of different bisphenols over the 40% HPW/SiO2 catalyst. Reaction conditions: 373 K, 6 h; 20 mmol phenol, 10 mmol formaldehyde and 0.15 g 40% HPW/SiO2 catalyst were used in the test. | |
The reusability of the HPW/SiO2 catalyst was also checked. According to the results illustrated in Fig. S9 of the ESI,† the activity of the 40% HPW/SiO2 catalyst evidently decreased after it was used for the activity test. Based on the ICP analysis of the liquid product, such a deactivation could be rationalized by the leaching of phosphotungstic acid during the reaction. As a solution to this problem, a more water-tolerant catalyst should be developed in the future research.
Hydrodeoxygenation
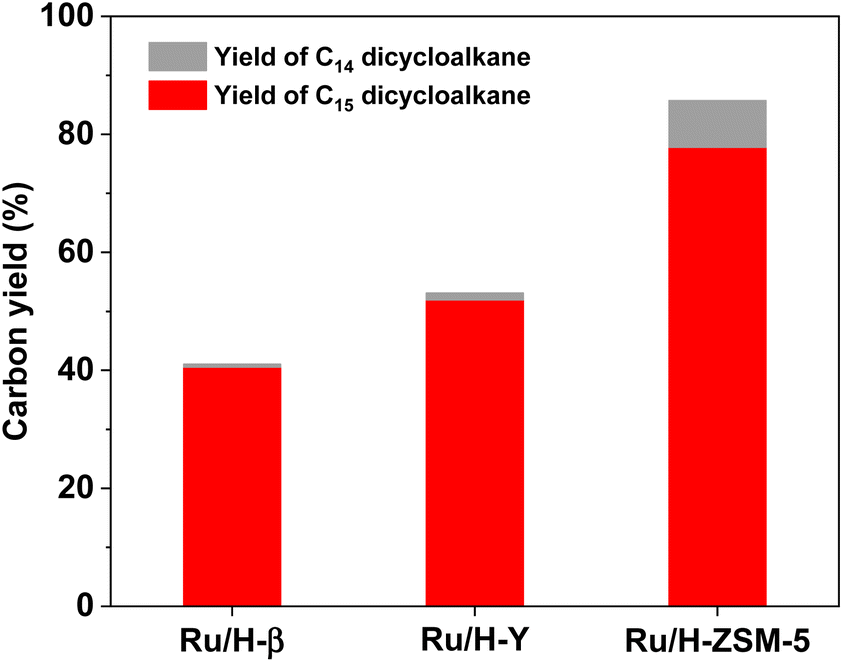
As the final aim of this work, we also explored the synthesis of jet fuel range dicycloalkanes by the hydrodeoxygenation (HDO) of 1A purified from the phenolic condensation of formaldehyde and creosol. Based on the GC-MS analysis (see Fig. S10 and S11 in the ESI†), 1A was completely converted (i.e. 100% conversion), and bis(2-methylcyclohexyl)methane (i.e.1B in Scheme 1) was obtained as the main product over a series of Ru-loaded zeolite catalysts. Besides 1B, small amounts of 1-(cyclohexylmethyl)-3-methylcyclohexane (i.e.1C in Scheme 1) were also identified in the HDO products. According to Scheme 1, 1C may be generated from the C–C cleavage (or hydrocracking) of 1B. Among the investigated Ru-based catalysts, the Ru/H-ZSM-5 catalyst exhibited the best performance (see Fig. 7). Over it, a high total carbon yield (85.7%) of C14 and C15 dicycloalkanes (i.e.1B and 1C in Scheme 1) was achieved after the HDO reaction was carried out at 473 K and 4 MPa H2 for 5 h.
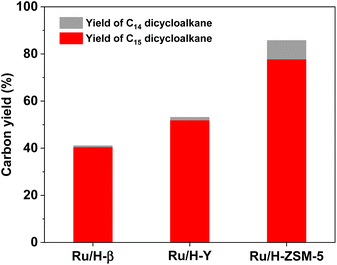 |
| | Fig. 7 Yields of different dicycloalkanes from the HDO of 1A over the Ru-based catalyst. Reaction conditions: 473 K, 4 MPa H2, 5 h; 0.2 g 1A, 0.1 g catalyst; 40 mL solvent was used for each test. | |
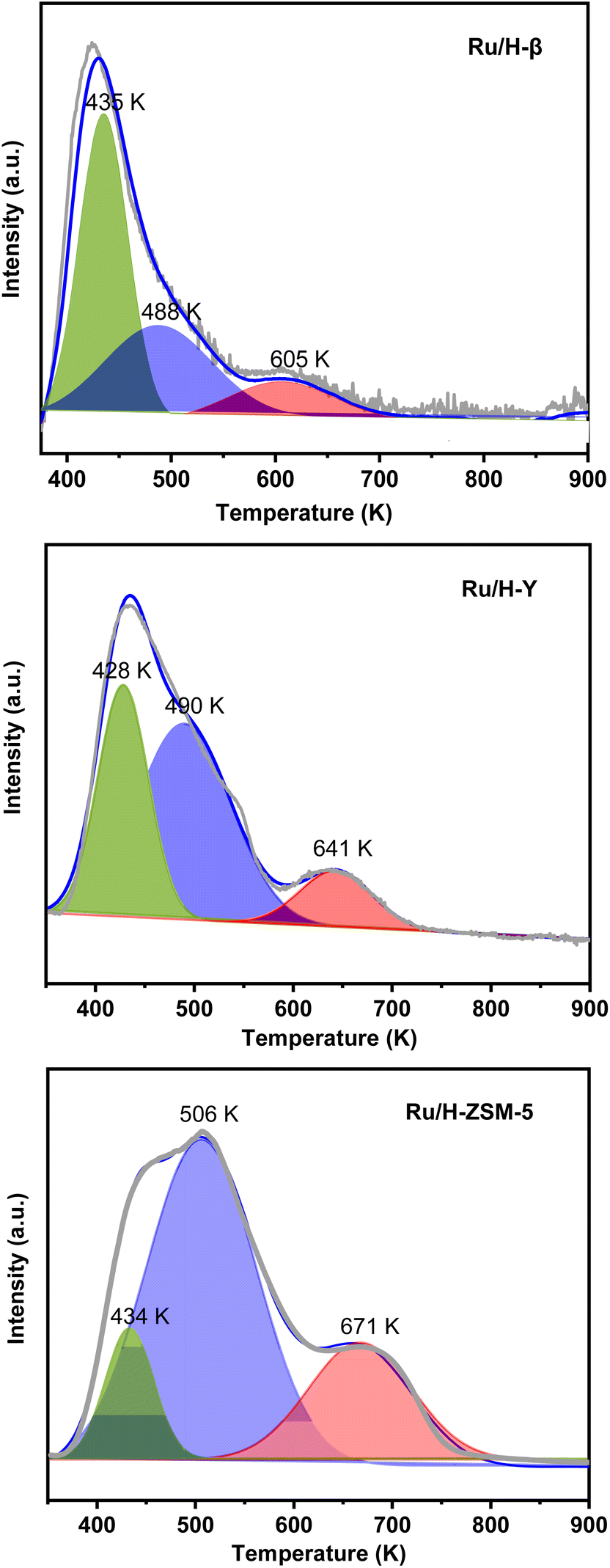
To figure out the reason for the good HDO performance of Ru/H-ZSM-5, the Ru-based catalysts were characterized by different technologies. Based on the results of the N2-physisorption tests, XRD, TEM, CO-chemisorption, and the pore sizes of the zeolite support (see Table 2 and Fig. S12, S13 of the ESI†), no clear relationship was noticed between the pore sizes of the zeolite support, the specific BET surface areas, or the Ru dispersions (or the average Ru particle sizes) of the Ru-based catalysts and their catalytic performances in the HDO process. From the results of the NH3-chemisorption and NH3-TPD tests (see Table 3 and Fig. 8), it was noticed that the Ru/H-ZSM-5 catalyst had a higher acid strength and higher amount of acid sites than the Ru/H-Y and Ru/H-β catalysts. In the previous work of Huber et al.,11 it was suggested that acid-catalyzed dehydration followed by hydrogenation is the main pathway for the HDO of biomass-derived oxygenates. In our recent work,12 it was also found that dehydration is a reaction that is sensitive to the acid strength of the catalyst. By comparison, a strong acid is more active than a weak acid for the dehydration. From the points of view of the acid strength and the amount of acid sites, we believe that the higher acidity of Ru/H-ZSM-5 may be the reason for its higher HDO activity.
Table 3 Specific BET surface areas (SBET) and the amounts of acid sites of the different catalysts
| Catalyst |
S
BET
(m2 g−1) |
Amount of acid sitesb (mmol g−1) |
Ru dispersionc (%) |
Pore sized (nm) |
|
Calculated from the N2-physisorption results.
Calculated from the NH3-chemisorption results.
Calculated from the CO-chemisorption results.
The values reported in the literature.10
|
| Ru/H-β |
420 |
1.25 |
25.8 |
5.1 × 5.5 |
| 5.3 × 5.6 |
| Ru/H–Y |
546 |
1.0 |
21.8 |
7.4 × 7.4 |
| Ru/H-ZSM-5 |
313 |
1.7 |
3.0 |
5.1 × 5.5 |
| 5.3 × 5.6 |
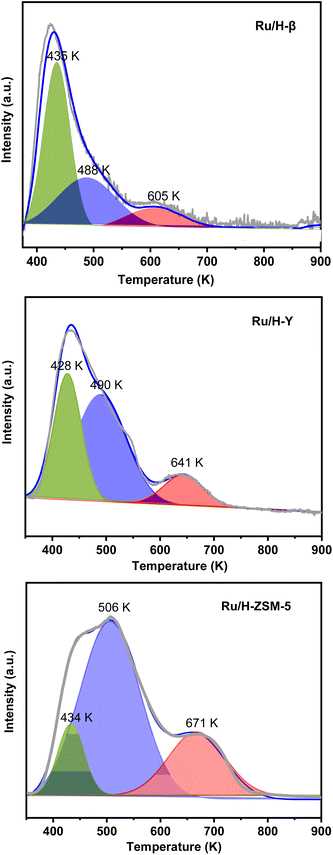 |
| | Fig. 8 NH3-TPD profiles of the different Ru-based catalysts. | |
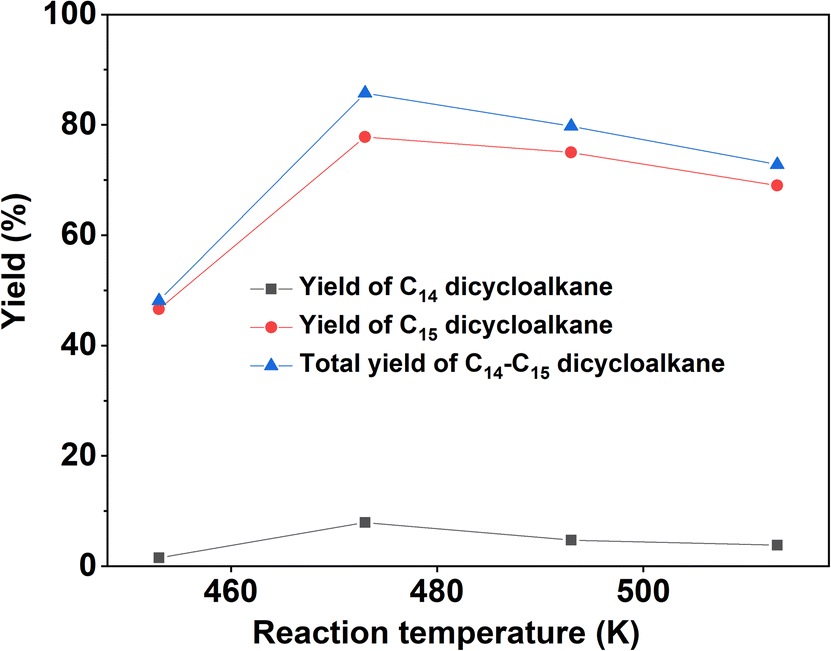
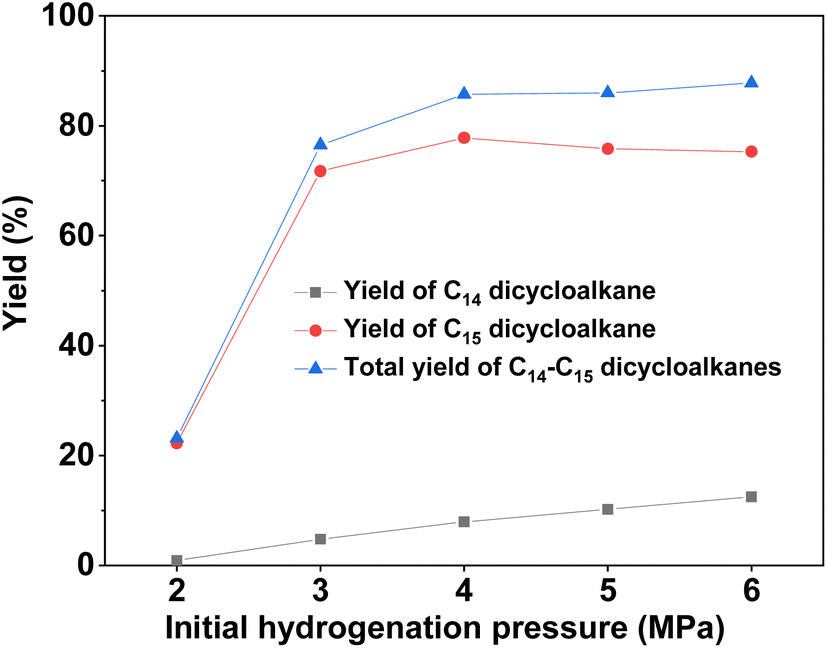
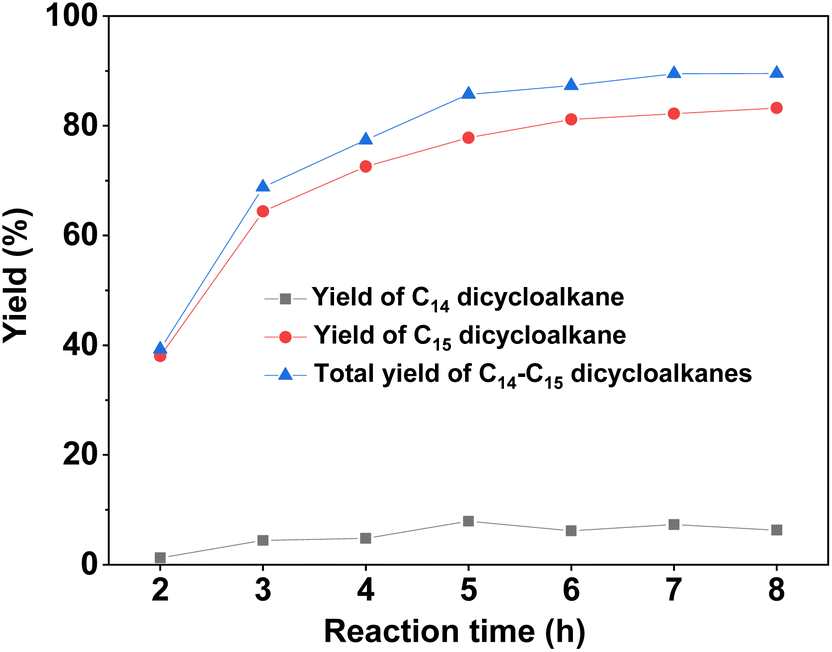
With the further optimization of the reaction temperature, initial hydrogen pressure, and reaction time (see Fig. 9–11), a high total yield (89%) of 1B and 1C was achieved when the reaction was carried out over Ru/H-ZSM-5 at 473 K for 8 h. According to our measurements, the mixture of C14 and C15 dicycloalkanes obtained under the optimum conditions had a high density (0.90 g mL−1), low freezing point (246.5 K), and high heat value (46.2 MJ kg−1 or 41.6 MJ L−1). As a potential application, it can be used as an additive to improve the volumetric heat value of current bio-jet fuels.
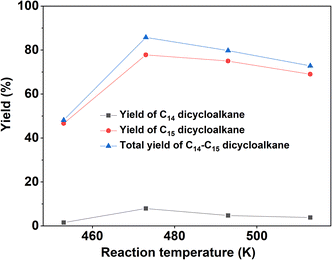 |
| | Fig. 9 Yields of different dicycloalkanes from the HDO of 1A over the Ru/H-ZSM-5 catalyst as a function of the reaction temperature. Reaction conditions: 4 MPa H2, 5 h; 0.2 g 1A, 0.1 g catalyst; 40 mL cyclohexane (used as solvent) was used for each test. | |
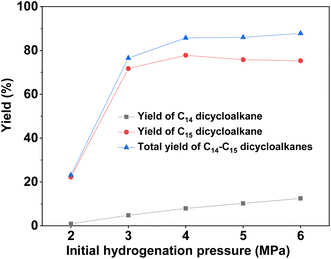 |
| | Fig. 10 Yields of different dicycloalkanes from the HDO of 1A over the Ru/H-ZSM-5 catalyst as a function of the initial hydrogen pressure at room temperature. Reaction conditions: 5 h, 473 K; 0.2 g 1A, 0.1 g catalyst; 40 mL cyclohexane was used for each test. | |
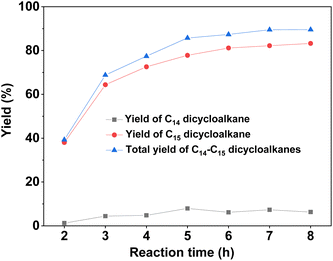 |
| | Fig. 11 Yields of different dicycloalkanes from the HDO of 1A over the Ru/H-ZSM-5 catalyst as a function of the initial hydrogen pressure at room temperature. Reaction conditions: 4 MPa H2, 473 K; 0.2 g 1A, 0.1 g catalyst; 40 mL cyclohexane was used for each test. | |
Conclusions
Jet fuel range high-density C14–C15 dicycloalkanes were synthesized by the phenolic condensation of creosol and formaldehyde. Among the SiO2-loaded heteropolyacid catalysts tested, HPW/SiO2 exhibited the highest activity for the phenolic condensation of creosol and formaldehyde, which could be rationalized by its higher amount of acid sites. After being hydrodeoxygenated over the acidic zeolite-loaded Ru catalysts, the phenolic condensation product of creosol and formaldehyde was converted to a mixture of C14 and C15 bicycloalkanes with a high density (0.90 g mL−1), low freezing point (246.5 K), and high heat value (46.2 MJ kg−1 or 41.6 MJ L−1). Among the investigated Ru-based catalysts, Ru/H-ZSM-5 exhibited the best HDO performance, which could be understood as due to the higher acidity of the H-ZSM-5 support. As a potential application, the mixture of C14 and C15 bicycloalkanes as obtained can be used as a fuel additive to improve the volumetric heat value of current bio-jet fuels.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China (no. 2022YFB4201802), National Natural Science Foundation of China (no. 22178335; 21721004; 22078318), Joint Fund of the Yulin University and the Dalian National Laboratory for Clean Energy (Grant. YLU-DNL Fund 2021020).
Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed; H. Li, A. Riisager, S. Saravanamurugan, A. Pandey, R. S. Sangwan, S. Yang and R. Luque, ACS Catal., 2018, 8, 148–187 CrossRef; C. Zhao, Y. Kou, A. A. Lemonidou, X. B. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef PubMed; B. G. Harvey and R. L. Quintana, Energy Environ. Sci., 2010, 3, 352–357 RSC; A. Corma, O. de la Torre, M. Renz and N. Villandier, Angew. Chem., Int. Ed., 2011, 50, 2375–2378 CrossRef PubMed; T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef PubMed; P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235–239 CrossRef PubMed; A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328–6344 RSC; M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854–1857 CrossRef PubMed; Q.-N. Xia, Q. Cuan, X.-H. Liu, X.-Q. Gong, G.-Z. Lu and Y.-Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 9755–9760 CrossRef PubMed; Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 11162 CrossRef PubMed; Z. Zhao, H. Shi, C. Wan, M. Y. Hu, Y. Liu, D. Mei, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 9178–9185 CrossRef; J. A. Muldoon and B. G. Harvey, ChemSusChem, 2020, 13, 5777–5807 CrossRef PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed; M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef PubMed; C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef; Y. Liu, C. Luo and H. C. Liu, Angew. Chem., Int. Ed., 2012, 51, 3249–3253 CrossRef; Y. L. Wang, W. P. Deng, B. J. Wang, Q. H. Zhang, X. Y. Wan, Z. C. Tang, Y. Wang, C. Zhu, Z. X. Cao, G. C. Wang and H. L. Wan, Nat. Commun., 2013, 4, 2141 CrossRef PubMed; T. P. Vispute, H. Y. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef PubMed; Y. T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 1387–1390 CrossRef PubMed; Y. T. Cheng, Z. P. Wang, C. J. Gilbert, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 11097–11100 CrossRef PubMed; Y. Wang, M. Peng, J. Zhang, Z. Zhang, J. An, S. Du, H. An, F. Fan, X. Liu, P. Zhai, D. Ma and F. Wang, Nat. Commun., 2018, 9, 5183 CrossRef PubMed; Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385 CrossRef PubMed; W. Deng, L. Yan, B. Wang, Q. Zhang, H. Song, S. Wang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 4712–4719 CrossRef PubMed; S. Xiang, L. Dong, Z.-Q. Wang, X. Han, L. L. Daemen, J. Li, Y. Cheng, Y. Guo, X. Liu, Y. Hu, A. J. Ramirez-Cuesta, S. Yang, X.-Q. Gong and Y. Wang, Nat. Commun., 2022, 13, 3657 CrossRef.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed; E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef PubMed; J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed; H. A. Meylemans, T. J. Groshens and B. G. Harvey, ChemSusChem, 2012, 5, 206–210 CrossRef PubMed; S. F. Koelewijn, S. Van den Bosch, T. Renders, W. Schutyser, B. Lagrain, M. Smet, J. Thomas, W. Dehaen, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2017, 19, 2561–2570 RSC; S.-F. Koelewijn, D. Ruijten, L. Trullemans, T. Renders, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2019, 21, 6622–6633 RSC.
- B. Kunkel and S. Wohlrab, Catal. Commun., 2021, 155, 106317 CrossRef CAS.
- H. Zhou, M. Wang and F. Wang, Joule, 2021, 5, 3031–3044 CrossRef CAS; Y. Wang, Chem Catalysis, 2021, 1, 765–767 CrossRef; X. Si, R. Lu, Z. Zhao, X. Yang, F. Wang, H. Jiang, X. Luo, A. Wang, Z. Feng, J. Xu and F. Lu, Nat. Commun., 2022, 13, 258 CrossRef PubMed; Z. Ren, X. Si, J. Chen, X. Li and F. Lu, ACS Catal., 2022, 12, 5549–5558 CrossRef.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS; S. Zhao, H.-Q. Liang, X.-M. Hu, S. Li and K. Daasbjerg, Angew. Chem., Int. Ed., 2022, 61, e202204008 Search PubMed.
- M. Tasooji and C. E. Frazier, ACS Sustainable Chem. Eng., 2021, 9, 207–215 CrossRef CAS; G. Wan and C. E. Frazier, ACS Sustainable Chem. Eng., 2017, 5, 4830–4836 CrossRef.
- H. Tang, Y. Hu, G. Li, A. Wang, G. Xu, C. Yu, X. Wang, T. Zhang and N. Li, Green Chem., 2019, 21, 3789–3795 RSC.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- N. Li and G. W. Huber, J. Catal., 2010, 270, 48–59 CrossRef CAS.
- F. Chen, N. Li, S. Li, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Green Chem., 2016, 18, 5751–5755 RSC; Y. Hu, N. Li, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, ChemSusChem, 2017, 10, 2880–2885 CrossRef CAS PubMed; H. Tang, N. Li, G. Li, W. Wang, A. Wang, Y. Cong and X. Wang, ACS Sustainable Chem. Eng., 2018, 6, 5645–5652 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ac,
Yu
Cong
a and
Ning
Li
ac,
Yu
Cong
a and
Ning
Li