
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ investigation of the mechanochemically promoted Pd–Ce interaction under stoichiometric methane oxidation conditions†
Maila
Danielis
 a,
Núria J.
Divins
bc,
Jordi
Llorca
bc,
Lluís
Soler
bc,
Xènia
Garcia
bc,
Isabel
Serrano
b,
Luis E.
Betancourt
d,
Wenqian
Xu
e,
José A.
Rodríguez
d,
Sanjaya D.
Senanayake
d,
Sara
Colussi
a and
Alessandro
Trovarelli
*a
a,
Núria J.
Divins
bc,
Jordi
Llorca
bc,
Lluís
Soler
bc,
Xènia
Garcia
bc,
Isabel
Serrano
b,
Luis E.
Betancourt
d,
Wenqian
Xu
e,
José A.
Rodríguez
d,
Sanjaya D.
Senanayake
d,
Sara
Colussi
a and
Alessandro
Trovarelli
*a
aDipartimento Politecnico, Università di Udine, and INSTM, via del Cotonificio 108, 33100 Udine, Italy. E-mail: alessandro.trovarelli@uniud.it
bInstitute of Energy Technologies and Department of Chemical Engineering, Universitat Politècnica de Catalunya, EEBE, Eduard Maristany 10-14, 08019 Barcelona, Spain
cBarcelona Research Center in Multiscale Science and Engineering, Universitat Politècnica de Catalunya, EEBE, Eduard Maristany 10-14, 08019 Barcelona, Spain
dChemistry Division, Brookhaven National Laboratory, Upton, NY 11973, USA
eX-Ray Science Division, Advanced Photon Source, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 8th December 2022
Abstract
The optimization of the supported Pd phase for CH4 activation on Pd/CeO2 catalysts has been a matter of great interest in the recent literature, aiming at the design of efficient methane abatement catalysts for Natural Gas fueled Vehicles (NGVs). Under lean conditions, a mixed Pd0/PdO combination has been indicated as exhibiting the best performance, while controversial results have been reported under stoichiometric conditions depending on the support oxide, where either Al2O3 or zeolite-based supports are usually considered. Here, by means of synchrotron-based in situ NAP-XPS and XRD measurements, we follow the evolution of Pd species on Pd/CeO2 samples prepared by dry mechanochemical synthesis (M) under stoichiometric CH4 oxidation feed, unravelling a stable Pd0/Pd2+ arrangement in a close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio as the most active palladium state for CH4 activation when excess oxygen is not available, in contrast to what was reported for Pd/alumina materials, where metallic Pd0 nanoparticles showed the highest activity. The combination of NAP-XPS analysis and activity test results highlights the promotional effect of the Pd–Ce interaction, resulting in enhanced oxygen transfer and improved activity and stability of the Pd/CeO2 catalyst prepared by a novel mechanochemical approach even under low O2 content, large excess of water vapor (10 vol%) and high temperature exposure (>700 °C).
:1 ratio as the most active palladium state for CH4 activation when excess oxygen is not available, in contrast to what was reported for Pd/alumina materials, where metallic Pd0 nanoparticles showed the highest activity. The combination of NAP-XPS analysis and activity test results highlights the promotional effect of the Pd–Ce interaction, resulting in enhanced oxygen transfer and improved activity and stability of the Pd/CeO2 catalyst prepared by a novel mechanochemical approach even under low O2 content, large excess of water vapor (10 vol%) and high temperature exposure (>700 °C).
Broader contextThe increasing global energy demand coupled with the imperative need to decrease greenhouse gas (GHG) emissions has strongly pushed in recent years towards the research and development of more sustainable energy sources. Amidst the various possible alternatives to liquid fossil fuels, natural gas (NG) has proved to be a feasible transition fuel while hydrogen- and electric-technologies become socially and commercially available. Increasing NG use would also serve as a cleaner alternative to flaring, which is especially common near oil-drilling sources. However, methane itself is a strong greenhouse gas and its utilization should be strictly controlled. For this reason, catalytic materials have to be optimized to ensure full methane conversion at low temperature, whether for abatement applications or for CH4 transformation into value-added chemicals. Here, we investigate in detail the surface interaction generated by dry milling between cerium oxide and palladium, specifically for enhanced activation of methane under stoichiometric conditions for unburned CH4 emission control. The insights gained regarding the metal–support interactions and their interplay for promoted CH4 conversion can be of general interest for the design of tailored methane valorization catalysts, reducing the amount of noble metal and relying on a more sustainable synthesis method such as mechano-chemistry. |
Introduction
Considering the significant contribution of the mobility sector to the global emission of greenhouse gases (GHGs), the need for sustainable alternative fuels and efficient after-treatment systems is of utmost importance in order to reach the decarbonization goals set forth by the EU and UN for the next few decades1,2 and ensure improved air quality.3 Among others, the substitution of traditional fossil fuels by natural gas appears to be a promising, ready-to-go option in the transition towards fully electric or H2-fueled fleets.4–6 The low C:H ratio of methane, the main component of natural gas, ensures lower production of CO2, NOx, and soot particulates in the engine compared to other fossil fuels,5,7,8 and its facile substitution by biomethane coupled with the well-developed natural gas distribution grids has already pushed towards a significant diffusion of natural gas fueled vehicles (NGVs) in recent decades.9
However, unburned CH4 is a strong greenhouse gas and its leakage needs to be carefully controlled.6,10,11 In the engine, the combustion technology of choice (stoichiometric vs. lean) depends on the desired power and torque requirements of the vehicle,12,13 and subsequently affects the catalytic after-treatment conditions.14,15 Platinum Group Metal (PGM)-based catalysts have been implemented to burn residual methane in the exhaust gases of NGVs to ensure abatement within imposed legislation limits,11 as either oxidative catalysts (OCs) for lean burn engine technologies or as three-way catalysts (TWCs) in stoichiometric engines.16,17 Stoichiometric methane oxidation represents a different system compared to excess O2 conditions,13,15 presenting several challenges in terms of oxidation states and stability of PdO species and the possible occurrence of other predominant reactions (partial methane oxidation, steam reforming).18 The stoichiometric point, indeed, often represents the most difficult lambda range for three-way catalyst development.11
In the latter context, Pd-based catalysts are currently reported as the most active for CH4 abatement;17,19–21 however, they are also characterized by low resistance to deactivation in the presence of steam under an O2-poor atmosphere and are susceptible to thermal aging.14,22–24 Thus, most recent efforts are aimed at improving the performances of Pd catalysts in these aspects. One widely employed solution is the addition of Pt to Pd-based catalysts,25–28 yet the optimization of the Pd phase is also highly desirable to improve catalytic activity and stability without increasing the noble metal loading and overall cost of the after-treatment system.29–31 In this regard, many recent studies have focused on the effect of optimal PdO particle size and of the oxidation state transitions in Pd/Al2O3-based samples during lean/rich switches and under isothermal conditions (<773 K).32–36 On these samples, CeO2 addition was observed to stabilize the oxidized PdO species thanks to its oxygen storage capacity,36–39 while Pd0 species led to higher activity on pure Pd/Al2O3 materials.33,34 Similar findings were reported for zeolite-supported Pd catalysts, where the stabilization of Pd2+ ions in the SSZ-13 structure resulted in decreased catalytic performance.40 Nevertheless, to the best of our knowledge a comprehensive study on Pd/CeO2 catalysts under stoichiometric methane oxidation after-treatment conditions has not yet been reported, including the effects of the co-presence of H2O and high temperature exposure, which are necessary to evaluate the catalyst's resistance to thermal aging.41
We have recently reported the development of a Pd/CeO2 catalytic system obtained by solvent-free mechanochemical synthesis which yields a combination of undercoordinated Pd0/Pd2+ sites in close contact with the surface ceria lattice, thus leading to enhanced methane conversion performance in lean conditions42,43 and under industrial bench test conditions,44 which range from lean oxidation to steam reforming in rich CH4 feed. Here, we investigate the evolution of the supported Pd species under a stoichiometric feed (1:2 CH4:O2), followed by means of in situ XRD and NAP-XPS techniques. The effect of the unique Pd–Ce interaction on the catalytic activity of Pd/CeO2 samples in dry and wet conditions was thoroughly investigated, aiming at filling the gap on the most active Pd species for methane conversion under stoichiometric CH4:O2 pressure.
Experimental
Catalyst synthesis
4 wt% Pd/CeO2 samples were prepared on commercial ceria (Rhodia), and calcined at 1173 K in static air for 3 hours before noble metal loading (surface area: 25 m2 g−1). Pd was dispersed on the support by dry mechano-chemical synthesis (M) and incipient wetness impregnation (IW) following established synthesis routes reported elsewhere.42 Briefly, for the milled sample (4PdAcCeM), 84.4 mg of palladium acetate (Sigma-Aldrich, 99.9%) and 960 mg of CeO2 were milled for 20 minutes in a 15 mL ZrO2 milling jar loaded with one ZrO2 grinding ball (15 mm diameter, ca. 10 g weight) using a Pulverisette 23 mini-mill operating at 15 Hz. The reference 4PdCeIW sample was prepared by incipient wetness impregnation, wetting 1 g of CeO2 powder with an appropriate amount of commercial palladium nitrate solution (Sigma-Aldrich, 99.999%) to reach a final Pd loading of 4 wt%. After complete wetting, the powders were dried at 373 K overnight and subsequently calcined at 1173 K for 3 h in static air. A high metal loading (4 wt%) was chosen to obtain a stronger signal and a higher definition from AP-XPS and XRD techniques, where the surface and catalytic properties can be considered representative also of lower Pd loadings.42Ex situ catalyst characterization
The surface area and pore volume of the prepared samples were investigated by analysis of N2 adsorption isotherms at 77 K in a Micromeritics Tristar 3000 analyzer using BET and BJH methods, respectively. ICP-MS analysis, performed by Mikroanalytisches Labor Pascher (Remagen, Germany), was used to measure the amount of loaded Pd on the prepared samples. Ex situ Raman spectroscopy was used to probe the surface Pd and CeO2 states before and after the reaction. Raman spectra were collected using an Xplora Plus Micro-Raman system (Horiba, Kyoto, Japan) using a 532 nm laser source. The spectra were acquired at room temperature with a resolution of 1 cm−1 and 5 accumulations of 60 s with a 50× LWD objective. Temperature Programmed Oxidation (TPO) analysis was carried out to probe the decomposition-reoxidation behavior of the supported Pd samples. For TPO tests, 150 mg of catalyst powder were placed in a quartz reactor supported on a quartz wool bed. The sample was then exposed to a flowing mixture (60 mL min−1) containing 2 vol% O2 in N2, and the oxygen release and consumption was monitored with an ABB Magnos 106 paramagnetic analyzer during 3 heating/cooling cycles (from RT up to 1273 K, at a heating/cooling ramp of 10 K min−1).In situ catalyst characterization
Time-resolved XRD measurements were performed at beamline 17 BM (λ = 0.24169 Å) at the Advanced Photon Source (APS) in Argonne, IL, using a Clausen cell flow reactor.45 A 30 mL min−1 flow rate of a gas mixture containing 0.3% CH4, 0.6% O2 and balance He was introduced, and then the sample was heated to 773 K at a 10 °C min−1 ramping rate; two heating/cooling cycles were performed to reach stable behavior. An in-line residual gas analyzer was used to track the evolution of the gaseous species right after the flow cell. A Si flat detector (PerkinElmer) was used to collect two-dimensional XRD images every 60 s throughout the reaction processes. The XRD data were subsequently processed with GSAS-II software.46Information on the surface electronic state during the reaction was gathered by means of Ambient-Pressure X-ray Photoelectron Spectroscopy (AP-XPS) at the CIRCE beamline of the ALBA Synchrotron Light Source, following the surface chemical composition under operando conditions at 2 mbar and different temperatures. Different excitation photon energies were used to obtain XP spectra of Ce 3d, O 1s, Pd 3d, and C 1s (acquiring photoelectrons with photon energy 485 eV (kinetic energy – KE – 150 eV), corresponding approximately to 0.5 nm inelastic mean free path (IMFP), photon energy 682 eV (KE 350 eV, IMFP = 0.9 nm) and photon energy 1032 eV (KE 700 eV, IMFP = 1.4 nm)). Catalyst powders were pressed with a gold mesh with a 0.25 mm nominal aperture (99.9%, GoodFellow) to form a compact disk and to minimize charging effects. The AP-XPS spectra were measured with a commercial PHOIBOS 150 NAP energy analyzer, equipped with four differentially pumped stages. Methane (0.6 mbar) and oxygen (1.4 mbar) were dosed into the analysis chamber by high-precision leak valves. The evolution of products was followed by a quadrupole mass spectrometer Microvision 2 Residual Gas Analyzer (MKS Instruments). Samples were heated using an infrared laser (λ = 808 nm) focused on a stainless-steel plate on top of which the samples were mounted. The temperature was monitored with a K-type thermocouple in contact with the samples. Due to the upper temperature limits of the XPS chamber, samples were pretreated ex situ by running a complete methane oxidation test up to 1173 K before analysis. Data processing was performed with the CasaXPS program (Casa Software Ltd, UK). To compensate the charging effects, spectra were calibrated with respect to the U′′′ Ce 3d peak at 916.8 eV.47,48 Atomic fractions were calculated using peak areas normalized on the basis of acquisition parameters after background subtraction. The relative sensitivity factors were calculated taking into account the ionization cross-section of each spectral region,49 the photon flux corresponding to the photon energy used for each measurement, and the transmission function of the analyzer. Cerium 3d spectra were deconvoluted using six peaks for Ce4+ (V, V′′, V′′′, U, U′′ and U′′′), which correspond to three pairs of spin–orbit doublets, and four peaks for Ce3+ (V0, V′, U0 and U′), corresponding to two doublets, based on the peak positions reported by Mullins et al.,47 where U and V refer to the 3d3/2 and 3d5/2 spin–orbit components, respectively.
Catalytic activity tests
Methane oxidation tests were carried out on 30 mg of catalyst powder, loaded on a quartz wool bed in a quartz reactor (ID 6 mm) and placed inside of a tubular furnace. For standard Temperature Programmed Combustion (TPC) tests, the sample was exposed to 90 mL min−1 of a high precision 0.5%CH4/1%O2/He gas mixture (SIAD) and heated up to 1173 K at a 10 K min−1 heating rate; two heating/cooling cycles were performed, and the second cycle is reported as representative of catalytic activity unless otherwise stated. To evaluate the effect of excess steam, TPC experiments in “wet” conditions were performed by adding 10 vol% water vapor to the stoichiometric gas feed, introducing 0.007 mL min−1 of liquid water through heated lines using a high-performance liquid chromatography (HPLC) pump. Time-on-stream (TOS) stability tests, in both dry and wet conditions, were performed holding the sample at 723 K for 24 hours; one oxidation cycle up to 1173 K in dry conditions was always performed as pretreatment.Reaction rates were calculated using methane conversion values below 5% to ensure the kinetic regime; conversion values were measured during the 2nd light off heating ramp of TPC experiments at 563 K and 613 K for dry and wet gas feed, respectively.
Results and discussion
The textural and morphological properties of the 4 wt%Pd/CeO2 samples obtained by milling (4PdAcCeM) and by the incipient wetness impregnation method (4PdCeIW) were studied by means of BET and XRD analysis, and the actual Pd loading was measured by ICP-MS analysis. The measured surface area and Pd loading are summarized in Table 1.| Sample | Calcination temperature (K) | BET S.A. (m2 g−1) | Pore volume (cm3 g−1) | Measured Pd loading (wt%) |
|---|---|---|---|---|
| 4PdCeIW | 1173 | 16 | 0.09 | 3.94 |
| 4PdAcCeM | 1173 | 22 | 0.17 | 3.92 |
Temperature programmed oxidation (TPO) experiments were performed to evaluate the redox behavior of the supported Pd species and to assess the amount of loaded palladium undergoing the characteristic PdO–Pd–PdO decomposition–reoxidation hysteresis on both samples. TPO tests were carried out under 1 vol% O2 and 2 vol% O2 in N2, representing the experimental conditions for stoichiometric and lean CH4 oxidation, respectively, with respect to 0.5 vol% CH4 in the feed.
Qualitative analysis of the high temperature decomposition profiles, reported in Fig. S1 (ESI†), shows that the lower availability of O2 has a negligible effect on the palladium species stabilized on the IW and M samples, with the latter maintaining the high temperature O2 release contribution corresponding to PdOx species in strong interaction with the ceria surface.50 Only a slight shift to lower temperature is observed, as expected by the decrease in O2 partial pressure.50 In addition, quantitative analysis of the recorded oxygen uptake and release profiles, reported in Table S1 (ESI†), confirms that palladium undergoes the typical PdO ↔ Pd cycle on both samples in the same amount observed under excess oxygen conditions (2 vol%).
To investigate in detail the bulk and surface structure of the Pd species and their evolution under the reaction conditions, the structural characteristics of the investigated samples were followed by in situ XRD under dry stoichiometric methane oxidation gas feed (0.3% CH4, 0.6% O2, He to balance) up to a temperature of 773 K. Full time-resolved patterns (Fig. S2, ESI†) show that, even under low O2 partial pressure, the CeO2 lattice structure remains unaltered during the reaction and the palladium species are well dispersed. No additional strains in the CeO2 lattice due to Pd incorporation could be detected, suggesting that no diffusion of oxidic Pd species occurs within the ceria lattice. The time-resolved patterns focused on the PdO–Pd 2θ range (5.2–6.8°) are reported in Fig. 1 to highlight the evolution of the supported Pd species. In situ XRD measurements highlight PdO nanoparticles as the predominant species on both samples, with a minor appearance of metallic Pd throughout the reaction. On the IW sample the higher intensity of the reflections corresponding to the (101) and (110) planes of palladium oxide, at 2θ = 5.21° and 6.42°, respectively, indicate larger PdO particles,42 as observed under lean oxidation conditions42 and corroborated by ex situ Raman spectra collected on the samples, reported in Fig. S3 (ESI†). Accurate Scherrer or Rietveld calculation of the average PdO particle size is hindered by the vicinity of the CeO2 (111) peak, and hence only a semi-quantitative estimate can be inferred from the XRD profiles. Semi-quantitative Rietveld analysis, performed on the secondary (110) PdO peak at 2θ = 6.42°, results in an estimated particle size of ca. 20 nm on 4PdCeIW compared to ca. 15 nm on 4PdAcCeM. The large particle size might be due to the elevated temperature of the pretreatment (1173 K). Nevertheless, apart from the slight difference in the calculated PdO particle size, the XRD profiles show negligible differences between the bulk structures of the milled and IW samples.
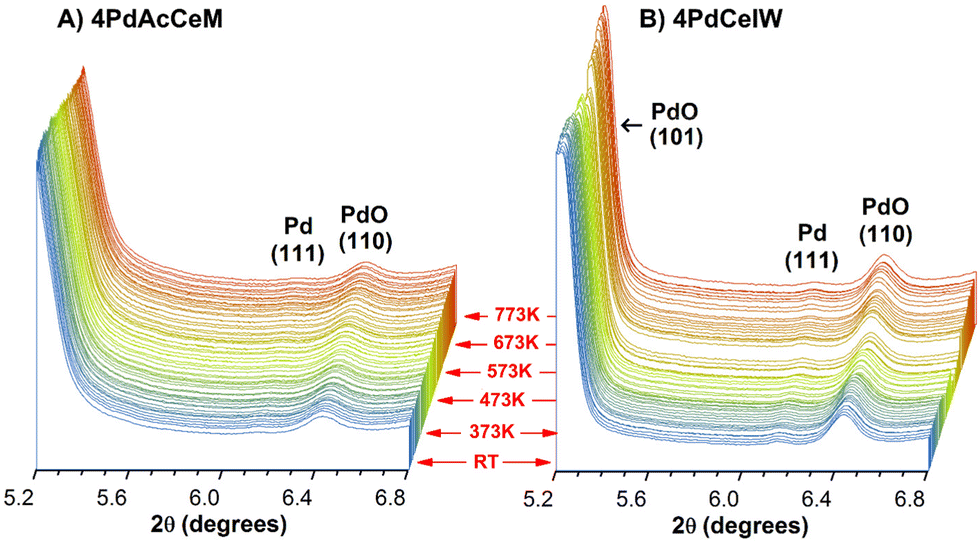 | ||
| Fig. 1 In situ XRD diffraction patterns collected under stoichiometric methane oxidation conditions (0.3% CH4, 0.6% O2 in He) on (A) 4PdAcCeM and (B) 4PdCeIW; samples were heated from RT to 773 K at a 10 K min−1 heating rate, and scans were collected every 60s. | ||
Consequently, to better understand the reaction mechanisms and the evolution of the catalyst surface states, the oxidation state of palladium and ceria during stoichiometric methane combustion at 673–873 K was followed by Near Ambient Pressure X-ray Photoelectron Spectroscopy (NAP-XPS) experiments. For consistency, both samples were pretreated at 1173 K under the reaction conditions before analysis. As shown in Fig. 2, there are important differences between the surfaces of samples 4PdCeIW and 4PdAcCeM (Fig. 2(A–C) and (D–F), respectively). On the IW sample, only Pd2+ and Pd0 species are detected with no appearance of Pd4+, contrary to what was observed under lean conditions,42 with the amount of metallic Pd slightly increasing from 14% to 19% with increasing temperature (Fig. 2A). In parallel, a negligible growth of Ce3+ is observed (Fig. 2B and Table S2, ESI†). In Fig. 2C, scans in the O 1s range highlight the presence of reaction products (CO2 and H2O); however, the surface of the catalyst remains predominantly covered by OH− species accumulated during the reaction. The milled sample presents a larger amount of Pd in the metallic state, with a remarkable increase in temperature from 22% to 40% (Fig. 2D) and a slightly more significant degree of Ce3+ formation (Fig. 2E), suggesting a higher rate of oxygen being removed to react with the CH4 in the gas feed. Nonetheless, Ce4+ is maintained as the predominant species on the 4PdAcCeM sample.
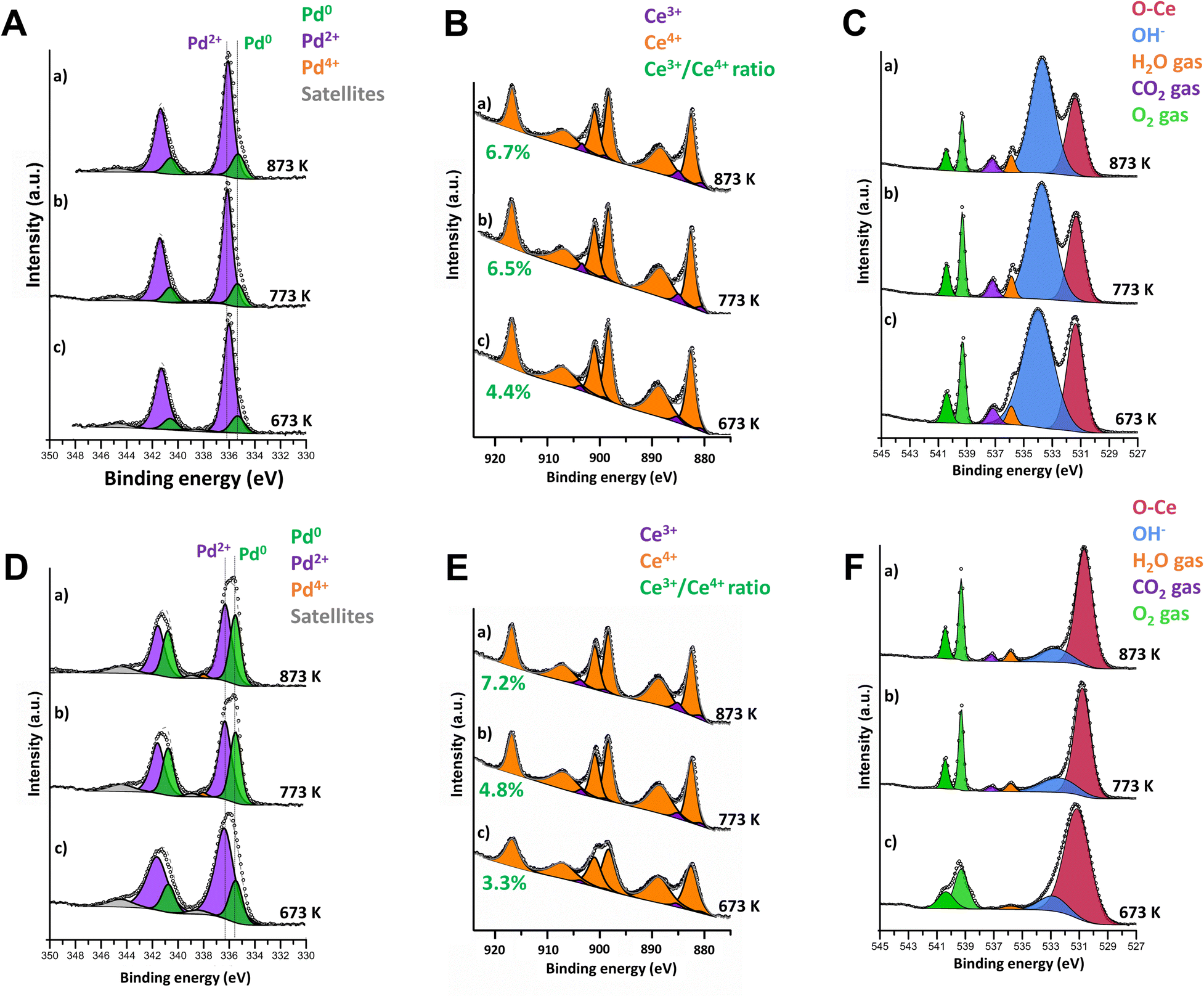 | ||
| Fig. 2 NAP-XPS spectra collected on 4PdCeIW (A–C) and 4PdAcCeM (D–F) in the Pd 3d (A and D), Ce 3d (B and E) and O 1s (C and F) regions under 0.6 mbar CH4 and 1.4 mbar O2 at increasing temperatures: (c) 673 K, (b) 773 K, (a) 873 K. | ||
In contrast with what was observed on the IW sample, the M catalyst surface is devoid of hydroxyl groups, and only CO2 and H2O products are detected (Fig. 2F). In addition, from the quantitative analysis of the O 1s spectra, reported in Table S2 (ESI†), on the M sample ca. 90% of the surface oxygen is found as lattice O–Ce, suggesting that a much higher amount of mobile lattice oxygen is available during reaction to be exchanged to the partially oxidized PdOx particles. On the other hand, on the IW sample the contribution of the OH− band accounts for more than 65% of the surface oxygen species. Likely, the surface of the IW sample is more prone to OH formation and stabilization, hence resulting in the blockage of active undercoordinated Pd sites and in slower CH4 conversion.51
Since the surface vs. bulk structures of supported PdO particles have been reported as playing a key role in the observed methane dissociation energy,52,53 NAP-XPS spectra at 873 K were also acquired with different photon energies, i.e., varying the penetration depth, to further investigate the surface, subsurface, and core oxidation states of the supported palladium particles on 4PdCeIW and 4PdAcCeM. Their comparison is reported in Fig. 3, with the corresponding XPS spectra and quantitative analysis reported in full in the ESI† (Fig. S5 and Table S2). Remarkably, and differently than other supported Pd systems reported in the literature,32,54 on both samples, the distribution of metallic Pd appears almost uniform throughout the supported PdOx particles, with a slightly larger amount of oxidized Pd2+ on the outermost layers compared to the inner core. The analysis thus confirms that on the milled sample, palladium nanoparticles present a higher contribution of metallic Pd with respect to the impregnated one, irrespective of the probing depth.
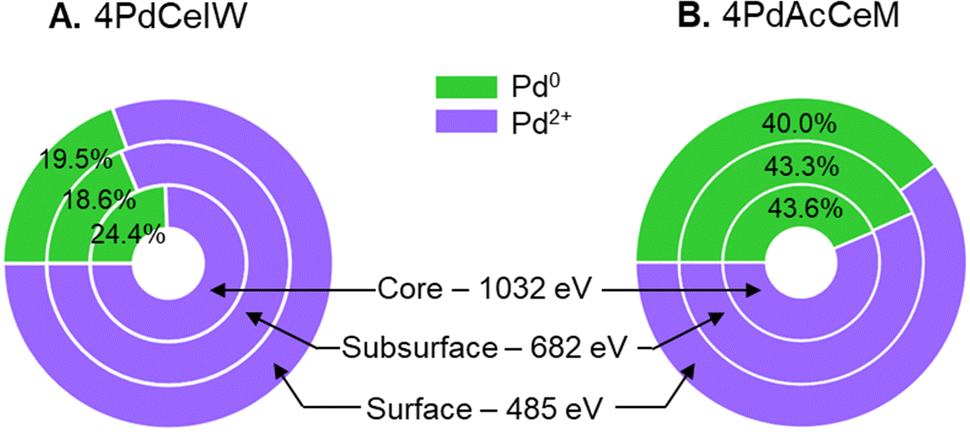 | ||
| Fig. 3 Comparison of the measured Pd0/Pd2+ amount at different penetration depths on (A) 4PdCeIW and (B) 4PdAcCeM under 0.6 mbar CH4 and 1.4 mbar O2 at 873 K. | ||
The methane oxidation performance of the prepared samples was evaluated in the absence and in the presence of excess water vapor (10 vol%), both in transient and stationary conditions. Fig. 4 reports the light-off curves for methane oxidation tests carried out under dry and wet stoichiometric conditions up to 1173 K; the temperatures corresponding to 10% and 50% CH4 conversion, as well as the calculated reaction rates, are reported in Table 2. Under stoichiometric conditions, the milled sample shows better performance compared to the conventional IW catalyst, with the latter suffering from more significant activity loss due to the PdO-to-Pd decomposition both during the heating and the cooling ramp. The same trend was observed on 1 wt%Pd/CeO2 samples (Fig. S6, ESI†), with the samples prepared by milling both outperforming the corresponding IW catalyst. In addition, the improved catalytic activity of 4PdAcCeM is maintained also in the presence of excess steam (10 vol%).
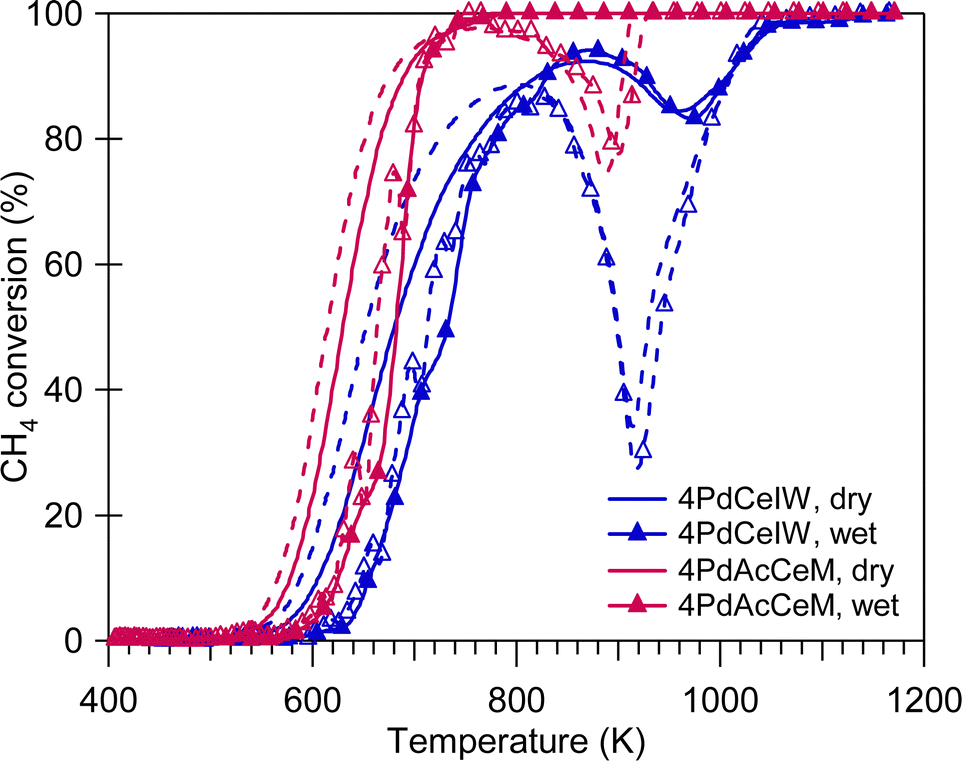 | ||
| Fig. 4 Methane oxidation activity of 4PdAcCeM and 4PdCeIW under stoichiometric methane oxidation conditions, in a dry atmosphere and with 10 vol% H2O added to the feed (wet). Solid line: heating; dashed line: cooling. | ||
000 h−1) at temperatures ensuring XCH4 < 5%, i.e. kinetic regime
| Sample | Pd atomica (%) | Measured Pd loading (wt%) | Feed | T 10 (K) | T 50 (K) | Rate (μmolCH4 gPd−1 s−1) |
|---|---|---|---|---|---|---|
| a Measured at 673 K in AP-XPS chamber. b Measured at 563 K. c Measured at 613 K. | ||||||
| 4PdCeIW | 14.2 | 3.94 | Dry | 607 | 681 | 4.8b |
| Wet (10%H2O) | 657 | 733 | 4.2c | |||
| 4PdAcCeM | 24.3 | 3.92 | Dry | 578 | 630 | 13.5b |
| Wet (10%H2O) | 626 | 683 | 14.9c | |||
The quantitative results obtained by TPO experiments (Table S1, ESI†) exclude an effect of less palladium available for reaction on the 4PdCeIW sample, since previous results from our group showed that a lower amount of noble metal actively participating in the reaction would be linked to a minor amount of exchanged oxygen observed by TPO analysis.42,55 Additionally, the difference in catalytic performance at high temperature can hardly be attributed to alternative reaction mechanisms occurring on the two samples. Partial oxidation and steam reforming of methane are reported as the main CH4 conversion mechanisms for fuel rich mixtures at high temperatures.18 However, in our previous work44 these catalysts were observed to possess comparable steam reforming activity while here, under both dry and wet stoichiometric oxidation conditions, no CO formation is detected, suggesting that no partial oxidation is occurring; therefore, on the investigated Pd/CeO2 samples full methane oxidation is still the driving conversion mechanism. Indeed, on all samples, irrespective of the gas feed conditions, the selectivity to CO2 is equal to 100%. Consequently, having excluded the effect of a minor amount of palladium available for reaction and the occurrence of side reactions, the enhanced catalytic performance of the 4PdAcCeM milled sample can be primarily ascribed to the peculiar surface arrangement identified by NAP-XPS.
The improved steam deactivation resistance observed under transient wet oxidation conditions (Fig. 4) is maintained under prolonged exposure at lower temperature, as illustrated in Fig. 5 where the methane oxidation activity of 4PdCeIW and 4PdAcCeM held for 24 hours at 723 K in dry and wet conditions is reported. Below 773 K, the competitive adsorption of O2 and H2O on the catalyst surface and the accumulation of OH− groups strongly hinder the oxygen transfer from the oxide support to the palladium species, and thus are indicated in the literature as the main steam inhibition mechanisms.51,56 These effects can be mitigated by using redox active supports compared to Al2O3,57,58 aiming at improving lattice oxygen transfer and maintaining the supported palladium species in an oxidized state. Here, the larger availability of ceria lattice oxygen species, observed by NAP-XPS under dry conditions on the milled sample, is able to efficiently inhibit the H2O-induced deactivation, allowing the 4PdAcCeM catalyst to retain a higher conversion throughout the 24-hour isothermal period under a large excess of steam such as the one present in real NGV exhaust gases (above 10 vol%). Conversely, the higher tendency of the conventional 4PdCeIW catalyst to retain OH− groups on the surface (Fig. 2C) results in a more pronounced loss of catalytic activity over the time-on-stream test (Fig. 5), which is further exacerbated by the addition of extra water vapor in the reaction feed.
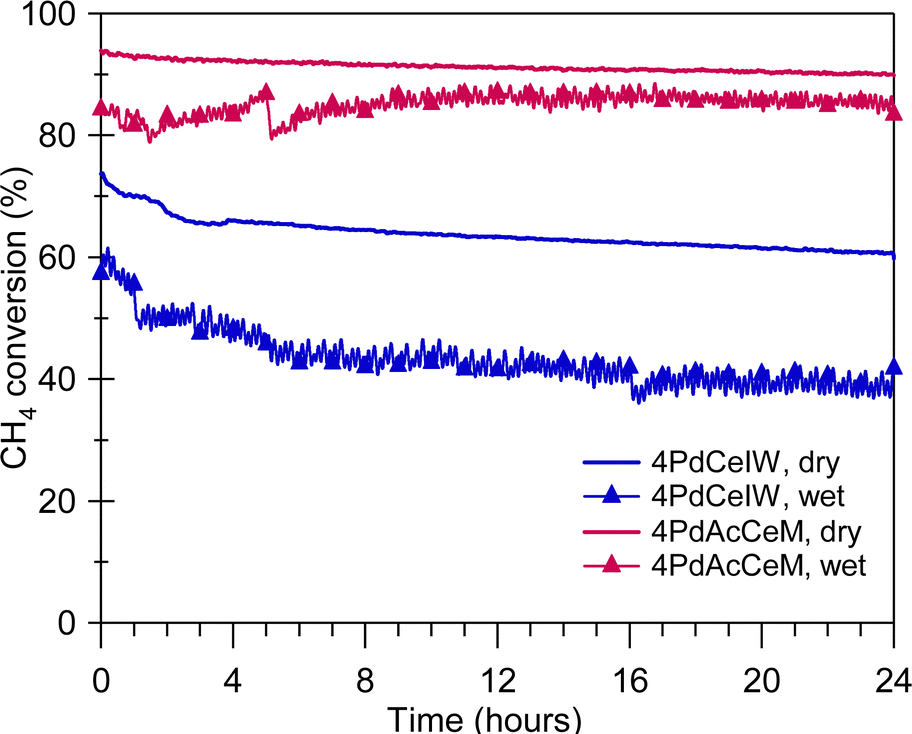 | ||
| Fig. 5 Methane oxidation activity of 4PdAcCeM and 4PdCeIW under stoichiometric methane oxidation conditions held at 723 K for 24 hours; in a dry atmosphere (solid line) and with 10 vol% H2O added to the feed (trace with symbols). Measured CO2 selectivity on all samples: 100%. | ||
In order to explain the observed differences in catalytic performance, XRD analysis appears to exclude a strong effect of the Pd particle size, which was instead linked to promoted reducibility to Pd0 and reported as a key element in alumina-supported Pd catalysts.33,59 However, the presence of surface amorphous phases, such as those observed on a similar milled sample under lean O2 methane oxidation conditions,42 would escape XRD detection, hindering a straightforward comparison between the two samples. Conversely, the combination of in situ XRD and NAP-XPS characterization (Fig. 1 and 2) presents additional insights into the surface environment of the investigated catalysts and suggests a predominant effect of the surface electronic state of palladium on CeO2. On both samples, differently from what was observed under lean conditions, a combination of metallic and oxidized Pd species is observed, with the 4PdCeIW sample exhibiting predominant Pd2+ features both on the surface and in the bulk of the PdO particles, while a Pd0/Pd2+ ratio closer to 0.5 is detected on the 4PdAcCeM sample (Fig. 3). In general, an undercoordinated PdOx on Pd or PdO configuration was reported by both theoretical39,53,60 and experimental works42,54,61 as very active for the low temperature activation of methane under lean conditions, where the close interaction between neighboring Pd0 and Pd–O species would lead to lower CH4 activation energy.52,53 In addition, a PdO-rich configuration was indicated as a key point for improved stability of Pd/CeO2/Al2O3 catalysts under lean/rich oscillating NGV abatement conditions.32,35
Here, under stoichiometric conditions, we further reveal that on CeO2 a less oxidized PdOx state, with a Pd0/Pd2+ ratio around 0.4–0.5, appears to be more favorable for CH4 activation, as demonstrated by the performance of the 4PdAcCeM milled sample. This is in line with previous literature results, where a Pd core–PdOx shell structure with a Pd0/Pd2+ ratio close to 0.5 was reported as most favorable for CH4 oxidation on Pd/CeO254 and, similarly, as a strong promoter to reduce steam-induced deactivation on Pd/Al2O3.32 However, both studies were carried out in the presence of excess O2. Moreover, on the alumina support this structure had to be formed and maintained via repeated reducing pulses during reaction, while here we show that the CeO2 support and the mechano-chemical synthesis method allow for the stabilization of such a structure throughout the reaction in a constant stoichiometric feed. NAP-XPS analysis with varying probing depths (Fig. 3) shows that no core–shell structure is formed in the Pd/CeO2 system, thus likely contributing to the stability of the supported PdOx particles. These results further suggest that the amount of PdO available for the reaction is important, but the source of O atoms plays a role as well: i.e., mobile oxygen atoms supplied through ceria to undercoordinated PdOx particles are likely more active than PdO nanoparticles well supported on Al2O3, suggesting that the Pd0–PdO–CeOx interface is responsible for enhanced methane oxidation activity under stoichiometric conditions. In addition, the NAP-XPS results in the O 1s range show that the peculiar configuration not only results in enhanced CH4 activation performance but also in improved activity and stability in the presence of large quantities of H2O, which are reported as strong competitors for surface O2 adsorption sites24,56 and thus lead to the formation of inactive palladium species.62
Conclusions
The catalytic abatement performance of Pd/CeO2 catalysts prepared by solvent-free milling and incipient wetness impregnation methods was evaluated under stoichiometric reaction feed and followed in situ to reveal the Pd species involved in the CH4 conversion under low O2 pressure. Transient and stationary reaction tests show that the 4PdAcCeM milled sample displays higher methane oxidation activity and stability compared to 4PdCeIW, also in the presence of excess water in the feed. The in situ studies carried out during the reaction point at different Pd oxidation states involved under a 1:2 CH4:O2 gas feed, with a higher amount of metallic Pd species and significant mobility of the lattice Ce–O leading to higher activity and stability on the sample obtained by milling. Indeed, under a stoichiometric Air-to-Fuel (A/F) ratio, the optimal configuration for PdOx particles supported on CeO2 appears to be uniform palladium nanoparticles with a balanced ratio of Pd0 and Pd2+ species (ca. 0.5), as exhibited by the milled sample. In addition, NAP-XPS results indicate that the enhanced lattice oxygen availability of the 4PdAcCeM catalyst, coupled with the optimal Pd0/Pd2+ ratio around 0.5, are the main factors contributing to the improved activity and stability of the Pd/CeO2 milled samples under O2-deficient gas feed compositions. Conversely, on the reference sample prepared by the conventional IW technique, the PdOx particles are maintained in a higher oxidation state, which proves to be less effective for CH4 activation and more prone to steam-induced deactivation when no excess O2 is available.
The differences observed between the Pd/CeO2 system herein studied and the previous Pd/Al2O3 works reported in the literature, where nanosized Pd0 particles displayed the highest activity and stability, highlight a much stronger role of CeO2 as a support oxide. In addition, here we further show how the possibility to control the active Pd sites by a simple, one-pot mechanochemical process, leading to a Pd–Ce nanoscale arrangement that proves to be extremely active for CH4 oxidation both in lean, stoichiometric and slightly rich atmospheres, can be highly interesting for industrial applications of the proposed synthesis process.
Author contributions
Maila Danielis: investigation, methodology, formal analysis, data curation, conceptualization, validation, writing – original draft. Núria J. Divins: investigation, methodology, data curation, formal analysis, writing – review & editing. Jordi Llorca: investigation, supervision, funding acquisition. Lluís Soler: investigation, formal analysis, data curation. Xènia Garcia: investigation, formal analysis. Isabel Serrano: investigation, formal analysis. Luis E. Betancourt: investigation, methodology, data curation. Wenqian Xu: resources, data curation. José A. Rodríguez: supervision, resources, writing – review & editing. Sanjaya D. Senanayake: supervision, resources, funding acquisition. Sara Colussi: conceptualization, supervision, writing – review & editing. Alessandro Trovarelli: conceptualization, supervision, funding acquisition, writing – review & editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. D. and S. C. are grateful to Fondazione CRUI for Go for it Project CUP G29C20000830001. N. J. D. acknowledges the funding received from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No. 897197. L. S. is grateful to MICINN Ramon y Cajal program for an individual fellowship grant agreement RYC2019-026704-I. J. L. is a Serra Húnter Fellow and is grateful to the ICREA Academia program and projects MICINN/FEDER PID2021-124572OB-C31 and GC 2017 SGR 128. NAP-XPS experiments were performed at the CIRCE beamline at the ALBA Synchrotron with the collaboration of ALBA staff. The work carried out at Brookhaven National Laboratory was supported by the US Department of Energy, Chemical Sciences Division (DE-SC0012704). S. D. S. is supported by a US Department of Energy Early Career Award. The XRD experiments used resources of the Advanced Photon Source Beamline 17-BM (XRD) at Argonne National Laboratory, which is an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science and was supported by the U.S. DOE under contract no. DE-AC02-06CH11357. This research used resources of the Advanced Light Source, a U.S. DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.References
- T. Haasz, J. J. Gómez Vilchez, R. Kunze, P. Deane, D. Fraboulet, U. Fahl and E. Mulholland, Energy Strategy Rev., 2018, 20, 124–132 CrossRef.
- Intergovernmental Panel on Climate Change, Climate change 2014: mitigation of climate change: Working Group III contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. O. Edenhofer, Cambridge University Press, New York, NY, 2014 Search PubMed.
- A. K. Datye and M. Votsmeier, Nat. Mater., 2021, 20, 1049–1059 CrossRef CAS.
- M. Gustafsson, N. Svensson, M. Eklund and B. Fredriksson Möller, Transportation Research Part D: Transport and Environment, 2021, 97, 102911 CrossRef.
- M. Clairotte, R. Suarez-Bertoa, A. A. Zardini, B. Giechaskiel, J. Pavlovic, V. Valverde, B. Ciuffo and C. Astorga, Environ. Sci. Eur., 2020, 32, 125 CrossRef CAS.
- H. Chen, J. He and X. Zhong, J. Energy Inst., 2019, 92, 1123–1136 CrossRef CAS.
- M. Hajbabaei, G. Karavalakis, K. C. Johnson, L. Lee and T. D. Durbin, Energy, 2013, 62, 425–434 CrossRef CAS.
- A. M. Pourkhesalian, A. H. Shamekhi and F. Salimi, Fuel, 2010, 89, 1056–1063 CrossRef CAS.
- H. Engerer and M. Horn, Energy Policy, 2010, 38, 1017–1029 CrossRef.
- C. Huang, W. Shan, Z. Lian, Y. Zhang and H. He, Catal. Sci. Technol., 2020, 10, 6407–6419 RSC.
- A. Raj, Johnson Matthey Technol. Rev., 2016, 60, 228–235 CrossRef CAS.
- H. M. Cho and B.-Q. He, Energy Convers. Manage., 2007, 48, 608–618 CrossRef CAS.
- P. Einewall, P. Tunestål and B. Johansson, Lean Burn Natural Gas Opearations vs. Stoichiometric Operation with EGR and a Three Way Catalyst, SAE Special Publications, 2005, vol. 2005, pp.343–362 Search PubMed.
- P. Lott and O. Deutschmann, Emiss. Control Sci. Technol., 2021, 7, 1–6 CrossRef CAS.
- D. Lou, Y. Ren, X. Li, Y. Zhang and X. Sun, Energies, 2020, 13, 4905 CrossRef CAS.
- X. Liu, J. Chen, G. Zhang, Y. Wu, P. Shen, L. Zhong and Y. Chen, J. Environ. Chem. Eng., 2021, 9, 105570 CrossRef CAS.
- D. Jiang, K. Khivantsev and Y. Wang, ACS Catal., 2020, 10, 14304–14314 CrossRef CAS.
- H. Stotz, L. Maier and O. Deutschmann, Top. Catal., 2017, 60, 83–109 CrossRef CAS.
- M. Monai, T. Montini, R. J. Gorte and P. Fornasiero, Eur. J. Inorg. Chem., 2018, 2884–2893 CrossRef CAS.
- R. J. Farrauto, Science, 2012, 337, 659–660 CrossRef CAS PubMed.
- M. Cargnello, J. J. D. Jaen, J. C. H. Garrido, K. Bakhmutsky, T. Montini, J. J. C. Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS.
- R. Sajiv Kumar, R. E. Hayes and N. Semagina, Catal. Today, 2021, 382, 82–95 CrossRef CAS.
- P. Granger, Catal. Sci. Technol., 2017, 7, 5195–5211 RSC.
- O. Mihai, G. Smedler, U. Nylén, M. Olofsson and L. Olsson, Catal. Sci. Technol., 2017, 7, 3084–3096 RSC.
- N. J. Divins, A. Braga, X. Vendrell, I. Serrano, X. Garcia, L. Soler, I. Lucentini, M. Danielis, A. Mussio, S. Colussi, I. J. Villar-Garcia, C. Escudero, A. Trovarelli and J. Llorca, Nat. Commun., 2022, 13, 5080 CrossRef CAS PubMed.
- A. Mussio, M. Danielis, N. J. Divins, J. Llorca, S. Colussi and A. Trovarelli, ACS Appl. Mater. Interfaces, 2021, 13, 31614–31623 CrossRef CAS PubMed.
- P. H. Ho, J.-W. Woo, R. Feizie Ilmasani, J. Han and L. Olsson, Ind. Eng. Chem. Res., 2021, 60, 6596–6612 CrossRef CAS.
- K. A. Karinshak, P. Lott, M. P. Harold and O. Deutschmann, ChemCatChem, 2020, 12, 3712–3720 CrossRef CAS.
- A. E. Hughes, N. Haque, S. A. Northey and S. Giddey, Resources, 2021, 10, 93 CrossRef.
- H. Xiong, D. Kunwar, D. Jiang, C. E. García-Vargas, H. Li, C. Du, G. Canning, X. I. Pereira-Hernandez, Q. Wan, S. Lin, S. C. Purdy, J. T. Miller, K. Leung, S. S. Chou, H. H. Brongersma, R. ter Veen, J. Huang, H. Guo, Y. Wang and A. K. Datye, Nat. Catal., 2021, 4, 830–839 CrossRef CAS.
- S. Colussi, P. Fornasiero and A. Trovarelli, Chin. J. Catal., 2020, 41, 938–950 CrossRef CAS.
- T. Franken, M. Roger, A. W. Petrov, A. H. Clark, M. Agote-Arán, F. Krumeich, O. Kröcher and D. Ferri, ACS Catal., 2021, 11, 4870–4879 CrossRef CAS.
- J. Chen, J. Zhong, Y. Wu, W. Hu, P. Qu, X. Xiao, G. Zhang, X. Liu, Y. Jiao, L. Zhong and Y. Chen, ACS Catal., 2020, 10, 10339–10349 CrossRef CAS.
- J. Chen, Y. Wu, W. Hu, P. Qu, G. Zhang, P. Granger, L. Zhong and Y. Chen, Appl. Catal., B, 2020, 264, 118475 CrossRef.
- J. Nilsson, P.-A. Carlsson, N. M. Martin, E. C. Adams, G. Agostini, H. Grönbeck and M. Skoglundh, J. Catal., 2017, 356, 237–245 CrossRef CAS.
- D. Ferri, M. Elsener and O. Kröcher, Appl. Catal., B, 2018, 220, 67–77 CrossRef CAS.
- F. Huang, J. Chen, W. Hu, G. Li, Y. Wu, S. Yuan, L. Zhong and Y. Chen, Appl. Catal., B, 2017, 219, 73–81 CrossRef CAS.
- S. K. Matam, G. L. Chiarello, Y. Lu, A. Weidenkaff and D. Ferri, Top. Catal., 2013, 56, 239–242 CrossRef CAS.
- A. Hellman, A. Resta, N. M. Martin, J. Gustafson, A. Trinchero, P.-A. Carlsson, O. Balmes, R. Felici, R. van Rijn, J. W. M. Frenken, J. N. Andersen, E. Lundgren and H. Grönbeck, J. Phys. Chem. Lett., 2012, 3, 678–682 CrossRef CAS PubMed.
- I. Friberg, A. H. Clark, P. H. Ho, N. Sadokhina, G. J. Smales, J. Woo, X. Auvray, D. Ferri, M. Nachtegaal, O. Kröcher and L. Olsson, Catal. Today, 2021, 382, 3–12 CrossRef CAS.
- K. G. Rappé, C. DiMaggio, J. A. Pihl, J. R. Theis, S. H. Oh, G. B. Fisher, J. Parks, V. G. Easterling, M. Yang, M. L. Stewart and K. C. Howden, Emiss. Control Sci. Technol., 2019, 5, 183–214 CrossRef.
- M. Danielis, L. E. Betancourt, I. Orozco, N. J. Divins, J. Llorca, J. A. Rodríguez, S. D. Senanayake, S. Colussi and A. Trovarelli, Appl. Catal., B, 2021, 282, 119567 CrossRef CAS.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Angew. Chem., Int. Ed., 2018, 57, 10212–10216 CrossRef CAS PubMed.
- M. Danielis, S. Colussi, J. Llorca, R. H. Dolan, G. Cavataio and A. Trovarelli, Ind. Eng. Chem. Res., 2021, 60, 6435–6445 CrossRef CAS.
- P. J. Chupas, K. W. Chapman, C. Kurtz, J. C. Hanson, P. L. Lee and C. P. Grey, J. Appl. Crystallogr., 2008, 41, 822–824 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- D. R. Mullins, S. H. Overbury and D. R. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- M. M. Natile and A. Glisenti, Surf. Sci. Spectra, 2021, 13, 17 CrossRef.
- R. Pugliese and G. Paolucci, Atomic calculation of photoionization cross-sections and asymmetry parameters, (accessed September 7, 2022), https://vuo.elettra.eu/services/elements/WebElements.html.
- S. Colussi, A. Trovarelli, E. Vesselli, A. Baraldi, G. Comelli, G. Groppi and J. Llorca, Appl. Catal., A, 2010, 390, 1–10 CrossRef CAS.
- X. Li, X. Wang, K. Roy, J. A. van Bokhoven and L. Artiglia, ACS Catal., 2020, 10, 5783–5792 CrossRef CAS.
- R. J. Bunting, X. Cheng, J. Thompson and P. Hu, ACS Catal., 2019, 9, 10317–10323 CrossRef CAS.
- N. M. Kinnunen, J. T. Hirvi, M. Suvanto and T. A. Pakkanen, J. Phys. Chem. C, 2011, 115, 19197–19202 CrossRef CAS.
- K. Murata, D. Kosuge, J. Ohyama, Y. Mahara, Y. Yamamoto, S. Arai and A. Satsuma, ACS Catal., 2020, 10, 1381–1387 CrossRef CAS.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Catal. Sci. Technol., 2019, 9, 4232–4238 RSC.
- R. Gholami, M. Alyani and K. Smith, Catalysts, 2015, 5, 561–594 CrossRef CAS.
- W. Barrett, J. Shen, Y. Hu, R. E. Hayes, R. W. J. Scott and N. Semagina, ChemCatChem, 2020, 12, 944–952 CrossRef CAS.
- M. Monai, T. Montini, C. Chen, E. Fonda, R. J. Gorte and P. Fornasiero, ChemCatChem, 2015, 7, 2038–2046 CrossRef CAS.
- J. J. Willis, A. Gallo, D. Sokaras, H. Aljama, S. H. Nowak, E. D. Goodman, L. Wu, C. J. Tassone, T. F. Jaramillo, F. Abild-Pedersen and M. Cargnello, ACS Catal., 2017, 7, 7810–7821 CrossRef CAS.
- N. M. Martin, M. Van den Bossche, A. Hellman, H. Grönbeck, C. Hakanoglu, J. Gustafson, S. Blomberg, N. Johansson, Z. Liu, S. Axnanda, J. F. Weaver and E. Lundgren, ACS Catal., 2014, 4, 3330–3334 CrossRef CAS.
- Y. Mahara, K. Murata, K. Ueda, J. Ohyama, K. Kato and A. Satsuma, ChemCatChem, 2018, 10, 3384–3387 CrossRef CAS.
- P. Velin, F. Hemmingsson, A. Schaefer, M. Skoglundh, K. A. Lomachenko, A. Raj, D. Thompsett, G. Smedler and P. Carlsson, ChemCatChem, 2021, 13, 3765–3771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00067a |
| This journal is © The Royal Society of Chemistry 2023 |