
Oxidative instability of ionomers in hydroxide-exchange-membrane water electrolyzers†
Grace A.
Lindquist
 a,
Jamie C.
Gaitor
b,
Willow L.
Thompson
a,
Valerie
Brogden
a,
Kevin J. T.
Noonan
b and
Shannon W.
Boettcher
*a
a,
Jamie C.
Gaitor
b,
Willow L.
Thompson
a,
Valerie
Brogden
a,
Kevin J. T.
Noonan
b and
Shannon W.
Boettcher
*a
aDepartment of Chemistry and Biochemistry and the Oregon Center for Electrochemistry University of Oregon Eugene, Oregon 97403, USA. E-mail: swb@uoregon.edu
bDepartment of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213-2617, USA. E-mail: noonan@andrew.cmu.edu
First published on 14th August 2023
Abstract
Hydroxide-exchange membrane (HEM) electrolyzers can produce green H2 with only earth-abundant catalysts and electrolyte-free (nominally pure) water feed, significantly decreasing system cost and complexity. However, HEM technology suffers from short lifetimes, attributed in part to poor stability of anion-exchange polymers used in the membrane and catalyst layers. We use electrochemical analysis and ex situ characterization techniques to study anion-exchange-polymer degradation in electrolyzers. Using multiple ionomers, catalyst-layer additives, and electrolyte feed, we show how anode-ionomer oxidation is the dominant degradation mechanism for all HEM-based electrolyzer cells tested. We find improved device stability using oxidation-resistant catalyst-layer binders and propose new design strategies for advanced ionomer and catalyst-layer development.
Broader contextLow-temperature membrane electrolyzers are primed for dramatic growth in scale for high-purity, green H2 production. Hydroxide-exchange-membrane (HEM) electrolyzers are a new technology that allow for significant cost reductions by using all inexpensive materials, but progress has been limited by short lifetimes in the absence of corrosive liquid electrolytes. We studied underlying degradation processes in state-of-the-art HEM electrolyzers under device-relevant conditions where interfacial interactions, local conditions, and transport phenomena modulate reaction pathways. We discovered dominant oxidation-driven degradation mechanisms specific to operation in electrolyte-free water across diverse polymer structures and provide catalyst-layer design strategies to overcome this critical issue limiting otherwise promising HEM electrolyzer technology. |
Introduction
H2 production has seen substantial investment as political and environmental factors drive a demand for increased renewable energy adoption.1–3 Renewable energy technology typically yields electrical power, which can then be stored or converted to energy carriers such as renewable/green H2 with possible longer-duration and increased scale of energy storage compared to batteries.4 Green H2 also enables the difficult decarbonization of manufacturing and industry sectors like chemical, metal, and fertilizer production.5Of the H2 production technologies, low-temperature membrane electrolysis is attractive as it operates using electrolyte-free water – as opposed to KOH electrolytes which complicates balance-of-plant components – and produces high-purity (>99.9%) H2. Proton-exchange-membrane (PEM) electrolysis is an established technology at megawatt scale6 with gigawatt-scale pilot projects deployed.7–9 PEM electrolyzers use perfluorinated sulfonic acid polymers, like Nafion®, a cation-exchange ionomer with high H+ conductivity.10,11 PEM electrolyzers achieve ∼80% voltage efficiency at 1 A cm−2 or higher12 and steady-state degradation rates well below 20 μV h−1.13 However, the locally acidic environment necessitates the use of expensive precious-metal catalysts, in particular Ir, which limit device scale-up and deployment.14 Substantial catalyst loading reductions are needed for PEM to achieve H2 cost and scale targets.2
Hydroxide-exchange-membrane (HEM) electrolyzers are a developing technology that might address the limitations of PEM electrolyzers (Fig. 1). They operate using an anion-selective membrane, which creates a locally alkaline environment enabling the use of inexpensive, earth-abundant catalyst and cell materials. However, the mobility of OH− is half that of H+ (in dilute aqueous solution), and therefore HEMs must have a higher charge-carrying capacity than PEMs to achieve comparable conductivity. For example, Nafion membranes have an ion-exchange capacity (IEC) of ∼1 meq g−1 giving a H+ conductivity of ∼70–100 mS cm−1 at 80 °C.15,16 HEMs have required IEC values of >2 meq g−1 to achieve comparable OH− conductivity.17–19 These transport limitations, combined with the poor stability of anion-exchange polymers, has prevented HEM electrolyzers from reaching maturity and substantial commercial penetration.20
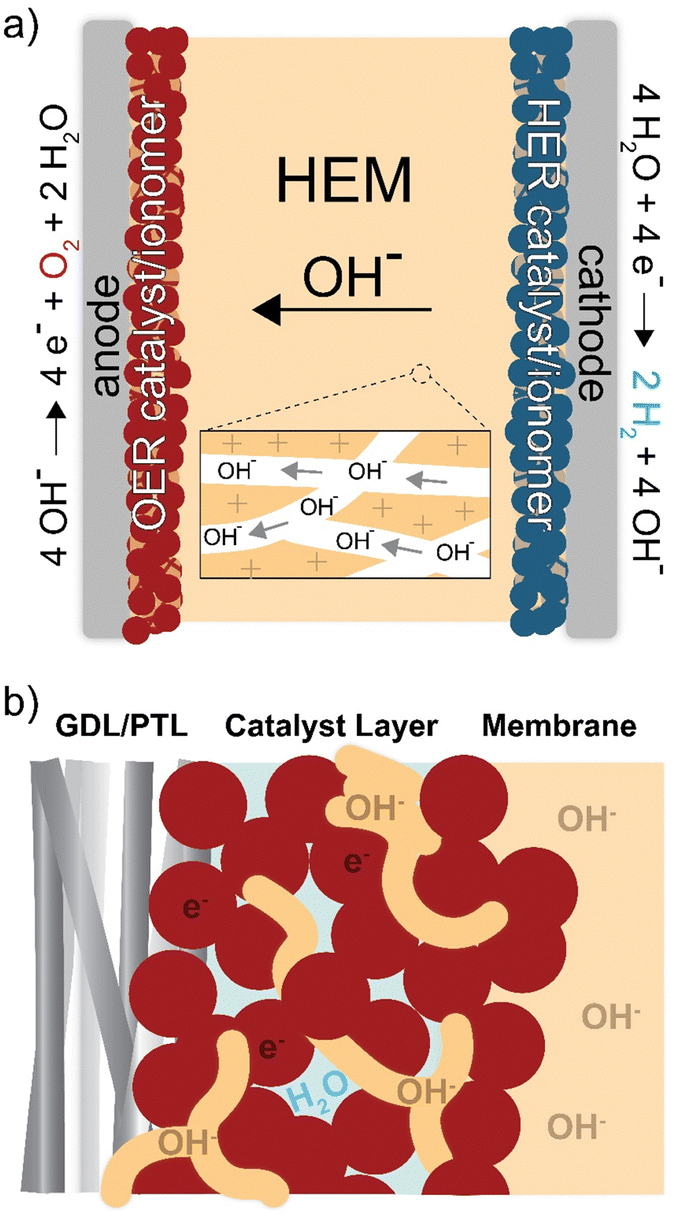 | ||
| Fig. 1 Ion transport and reactivity in HEM electrolyzers. Schematics of (a) electrolyzer cell and (b) anode-catalyst layer. The OH− anions conduct through the ionomer, and electrons conduct through the catalyst. Oxygen evolution occurs at the ionomer/catalyst interface. | ||
In addition to the ionomer (ion-exchange polymer) playing an essential role as the membrane, it is also used in the catalyst layer (Fig. 1(b)). The catalyst layer is deposited on the membrane or electrode porous-transport layer (PTL) using an ink. The ink is comprised of the catalyst, solvent, and dissolved ionomer that acts as a catalyst binder and enables ion transport to the active catalyst surface.21 When the ink is deposited, the solvent evaporates and creates a porous layer of ionomer, catalyst, and void space for liquid/gas transport to/from the catalyst. The interactions between materials in this region directly impact device performance and durability,22–24 due to the coupled effects of electric, ionic, and reactant/product transport impedance and catalyst-surface reaction kinetics.25
Anion-exchange ionomers have historically limited the performance and durability of HEM electrolyzer devices.20,25–29 Common cation-exchange ionomers are perfluorinated-sulfonic-acid-(PFSA)-type materials with high chemical stability.16 Most anion-exchange ionomers are hydrocarbons (e.g. polybenzimidazoles, polyethers, polyphenylenes, etc.) which are more-susceptible to chemical degradation by nucleophilic OH− in the strong alkaline conditions. While substantial polymer development in the past decade has improved electrolyte-free HEM electrolyzer performance,19,30–35 further improvements are needed. Most efforts at improving the stability of HEM polymers have focused on alkaline chemical stability, including adding protecting groups near electrophilic sites,36–38 partial fluorination,39 and tuning side-chain length40 or cation identity.41,42Ex situ chemical stability, however, is not necessarily reflective of device conditions nor indicative of how a polymer will perform in a membrane-electrode assembly (MEA), in particular during operation with electrolyte-free (nominally pure) water feed. In the MEA, the polymer may degrade by other chemical and electrochemical means besides OH− attack. At the anode the ionomer is held at a strongly oxidizing potential and exposed to possible oxidizing species/intermediates produced during the oxygen-evolution reaction (OER). The oxidative and radical stability of anion-exchange ionomers has been investigated to various extents,43–47 but few studies are conducted under device-relevant operating configurations and conditions.
Here we report a comprehensive ionomer failure analysis of HEM electrolyzer MEAs operating with electrolyte-free water and uncover common oxidative processes that must be solved for commercialization. Using integrated reference electrodes and impedance analysis on the full MEA electrolyzer, post-mortem surface analysis, and cross-sectional electron-microscopy and chemical imaging, we find ionomer oxidation as a dominant degradation mechanism across all ionomer chemistries studied in locally alkaline conditions, even those with all sp3 carbon and PFSAs. We further investigate the effect of additives and alternative feed modes on the extent of oxidation, showing pure-water operation degrading <0.5 mV h−1 over 100 h using an oxidatively stable anode-catalyst layer. Lastly, we introduce catalyst-layer design strategies for next-generation HEM electrolyzers.
Results and discussion
Degradation of TP-85 anion exchange ionomer
HEM electrolyzers with an active area of 1 cm2 were prepared and assembled as described in the methods section and elsewhere (Fig. S11a, ESI†).24,31 IrOx on platinized Ti and Pt black on Toray carbon-paper were used as the anode and cathode porous-transport electrodes (PTEs), respectively. Both electrodes were prepared with PiperION-A5 ionomer dispersions and devices were assembled with a 40 μm-thick PiperION TP-85 membrane. For this study, the membrane and cathode remained constant for all experiments and only the anode PTE, including anode ionomer, was varied.Understanding electrolyzer device degradation during operation is challenging and usually limited to information gained from two-electrode studies. To better understand the contribution of individual components to the total cell voltage, a reference electrode was integrated into the MEA.50 A strip of membrane is attached to the edge of the membrane in the MEA and extended outside the cell hardware, where a reference electrode is attached to the membrane strip and used to measure the anode and cathode components to the total cell voltage (Fig. 2). A high conductivity HEM is used as the reference strip to minimize reference-electrode-potential errors from non-symmetric current distributions.48,49
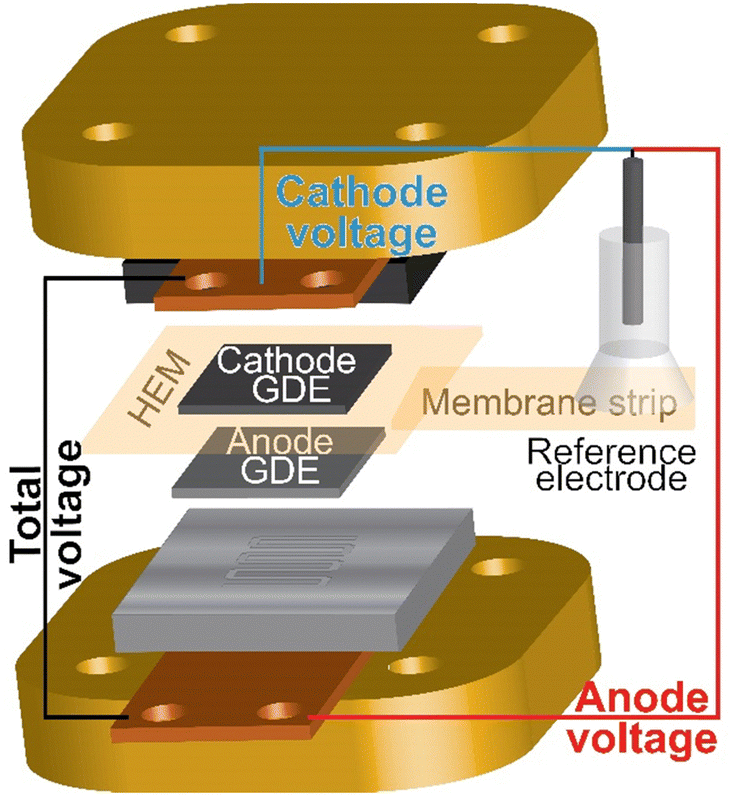 | ||
| Fig. 2 Schematic of the reference electrode integrated in the electrolyzer MEA and hardware. A reference electrode is placed on a strip of membrane attached to the HEM in the MEA. The voltage is measured from the cathode end plate or anode end plate versus the reference. Precision gasketing and a high-conductivity HEM reference strip are used to minimize misalignment reference-potential errors.48,49 | ||
HEM electrolyzers were operated with electrolyte-free (nominally pure) water. The initial performance was 2.1 V at 1 A cm−2 at 70 °C (Fig. 3(a)). The polarization curve is recorded following a ∼20 min break-in period, during which some degradation occurs contributing to the high voltage performance relative to PEM benchmark systems. During operation at 1 A cm−2, the total cell voltage decreases for a short period before rapidly degrading at 22 mV h−1 from 1–10 h, then stabilizing to 4 mV h−1 from 10–20 h (Fig. 3(b)). The reference electrode shows this voltage degradation occurs at the anode. The cathode degradation was constant throughout the run at ∼1 mV h−1, which may be due to non-optimal water management that could be solved with better ionomer and cathode-electrode design that is not the focus of this work.
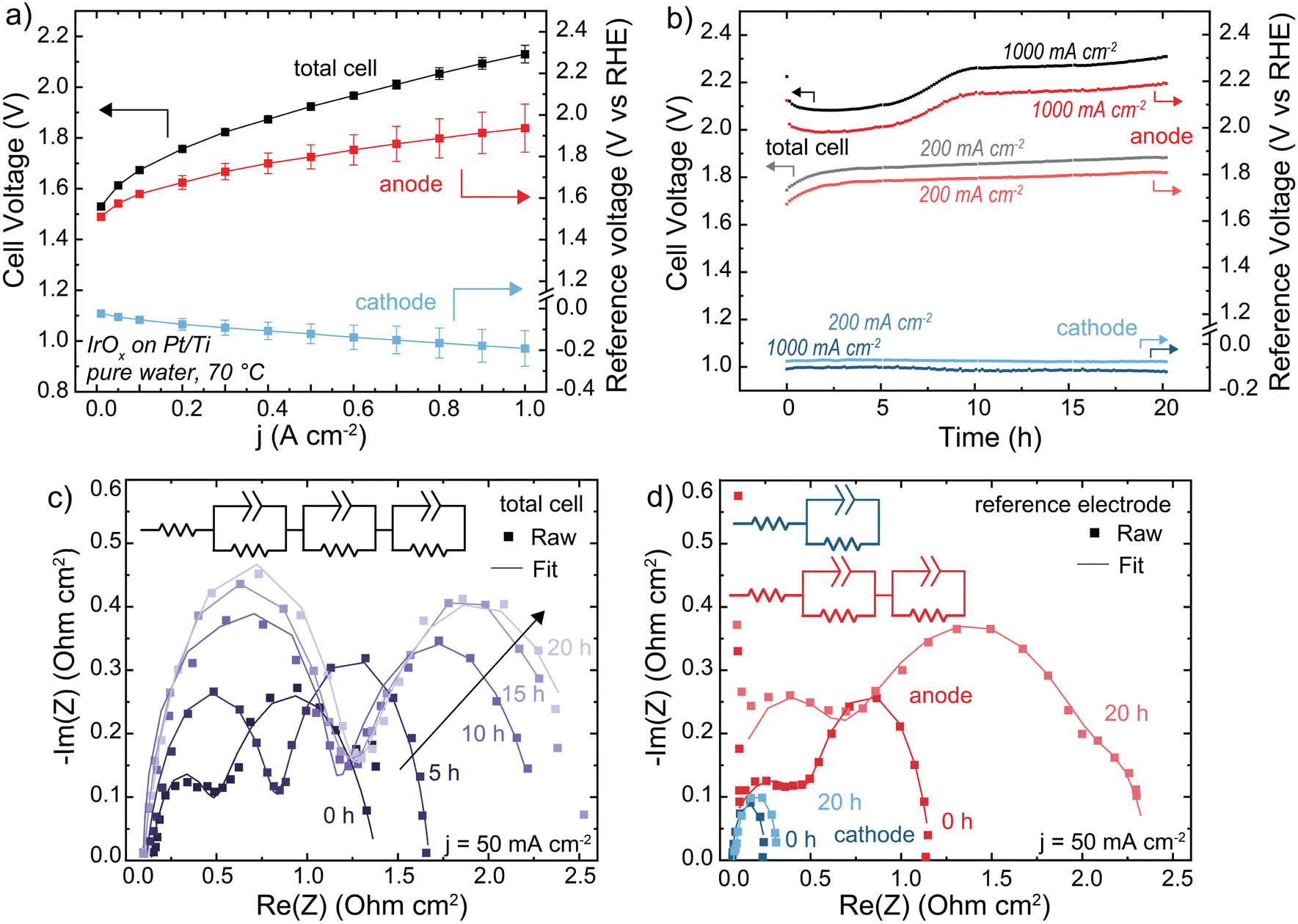 | ||
| Fig. 3 Performance and stability of a HEM electrolyzer. Cells were operated with an IrOx/20% TP-85 catalyst layer on Pt/Ti PTL (anode), Pt-black/10% TP-85 catalyst layer on Toray carbon-paper PTL (cathode), and TP-85 membrane at 70 °C. (a) Polarization curve showing anode and cathode contribution to total cell voltage. Reported data is the average of three polarization curves and the error bars are one standard deviation. (b) Cell durability at 200 mA cm−2 (gray total cell, light red anode and light blue cathode) and 1 A cm−2 (black total cell, dark red anode and dark blue cathode). (c) Nyquist plots of total cell impedance collected at 50 mA cm−2 every 5 h of the 1 A cm−2 test. Cell current was decreased to 50 mA cm−2 for impedance testing, then increased to 1 A cm−2 to continue durability testing. (d) Nyquist plots of the anode and cathode impedance measured at 50 mA cm−2 before and after 20 h operation at 1 A cm−2. Nyquist plots were fit to the inset equivalent circuit. All fit parameters are shown in Tables S2–S4 (ESI†). Degradation is primarily evident in the anode impedance. | ||
The Nyquist plot of the full cell shows two semi-circles (Fig. 3(c)). During operation, the low frequency resistances of both semicircles increase with time. If these two processes were assumed to be the anode and cathode, this would suggest an increase in both OER and HER charge-transfer resistance. However, the anode and cathode Nyquist plots using the reference electrode show two semicircles for the anode, and one at the cathode (Fig. 3(d)). The two semicircles in the total cell cannot be assigned to each electrode – one semicircle in the total-cell impedance represents a combination of both cathodic and anodic processes. The second RC component shown in the anode-reference experiment may be due to a variety of processes, including ionomer oxidation reactions, a slow corrosion or dissolution mechanism, or water-dissociation reactions from OER occurring in lower pH regions in the catalyst layer. Previous measurements found OER faradaic efficiency of >∼98% for the same MEA system.31 Impedance analysis of electrochemical devices is complicated51 and more work is needed to assign mechanisms to each semicircle at the (degrading) anode. After operation at 1 A cm−2, both charge-transfer resistances at the anode increased significantly while the cathode increased only slightly, consistent with our conclusion that cell degradation occurs primarily at the anode. For EIS analysis of electrochemical devices with complex porous electrodes, it is not uncommon for multiple distinct processes to contribute to one resolvable semi-circle.52 The fit parameters for all EIS are shown in Tables S2–S4 (ESI†). The fit parameters obtained from anode and cathode contributions (three RC components) can be used to simulate the observed (two semi-circle) total cell data (Fig. S13, ESI†). Quantifying and separating the three separate electrode-response impedance arcs would not have been possible by fitting the full-cell impedance alone, further demonstrating the value of reference-electrode measurements.
When the operating current is lowered to 200 mA cm−2 the total-cell voltage-degradation rate decreases to 7 mV h−1 from 0–10 h, and 3 mV h−1 from 10–20 h (Fig. 3(a)). This voltage degradation is at the anode; the average cathode degradation rate was 50 μV h−1. Interestingly, at 200 mA cm−2 the initial large decrease in voltage was not observed. A decrease in voltage can still indicate cell degradation, for example membrane thinning which decreases cell resistance, and is not necessarily representative of cell performance improving.20 The ionomer loading in the PTE is 20 wt% relative to catalyst mass, which was chosen to ensure sufficient ionomer sample signal for XPS and resolution for SEM cross-section analysis (discussed later). A high ionomer loading can contribute additional iR or mass-transport losses to the cell voltage. This initial voltage decrease is less severe when the ionomer content is decreased to 10 wt% (Fig. S14, ESI†), and thus we interpret the initial voltage decrease at 1 A cm−2 to be due to various coupled catalyst-layer reorganization and degradation processes, which initially decrease catalyst-layer resistance and appear as an improvement in cell voltage.
X-ray photoelectron spectroscopy (XPS) analysis of the PTE surface was conducted to understand the anode-degradation mechanism (Fig. 4). The pristine anode PTE shows the expected spectra for the undamaged ionomer. No Ir XPS peak is observed (Fig. S15, ESI†), as the PTE surface is sprayed with a top layer of ionomer and XPS only penetrates a few nanometers into the surface of the material. After operating at 1 A cm−2 for 20 h, the C 1s spectra from the cathode catalyst layer remains unchanged, while the anode C 1s spectra shows a new higher-binding-energy peak between 288 and 289 eV, consistent with carbonyl and/or ester group formation,24,31,44,53 and a loss of C–N content. This is accompanied by a loss of N 1s and F 1s peaks (Fig. S16, ESI†), demonstrating both polymer backbone and cation groups have degraded and dissolved, at least from the surface layer analyzed. XP spectra of the anode and cathode face of the HEM match the PTE spectra (Fig. S17, ESI†). The surface of the anode PTE operated at 200 mA cm−2 for 20 h shows no obvious signal from ionomer oxidation, consistent with the more-stable operating voltage and slower oxidation kinetics at lower anode potentials.
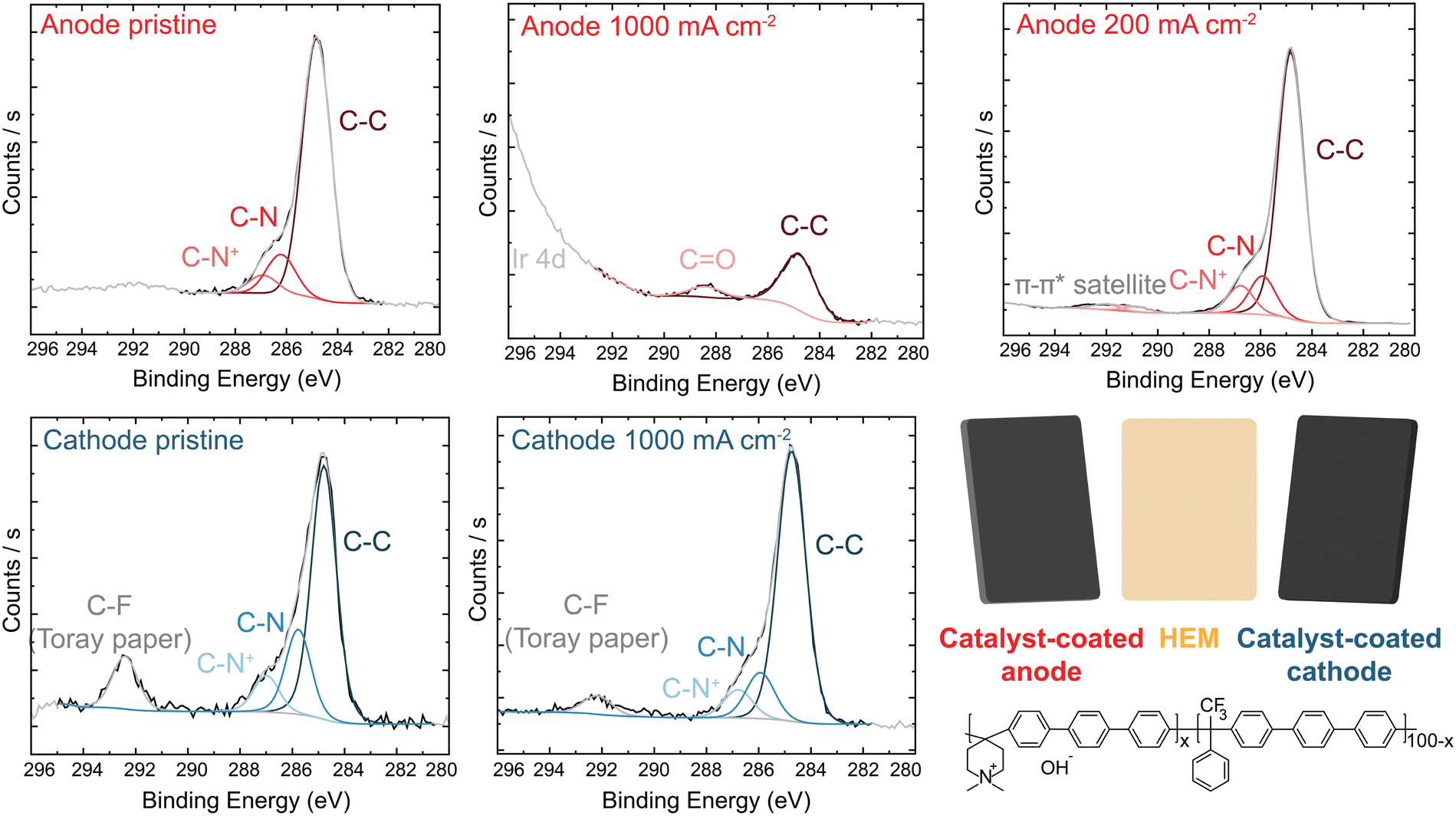 | ||
| Fig. 4 XPS analysis of IrOx/20% TP-85 anode and Pt-black/10% TP-85 cathode. C 1s spectra of (a) anode and (b) cathode PTE. Inset shows the chemical structure of the TP-85 ionomer. Ionomer degradation is only observed on the anode PTE operating at high current. | ||
We note that XPS signal from carbonate or bicarbonate may also appear in the binding-energy range where oxidized-carbon species were found. All electrodes are quenched in 3 M NaCl before drying and thus any carbonate from operation should be exchanged to Cl− before analysis. We have shown this procedure is sufficient to exchange carbonate to chloride after testing of ionomer films in supporting carbonate electrolyte.44 To confirm this assumption here, an MEA was prepared and pure water was flowed through the device at 70 °C without applied current. The PTE was ion exchanged and dried following the same procedure as the operated samples. XPS analysis shows only pristine polymer with no higher-binding-energy carbon peak (Fig. S18, ESI†), indicating complete exchange of any absorbed carbonate. Therefore, growth of the high-binding-energy peak in the C region, combined with a loss of N and F signal, demonstrates severe ionomer oxidation in the anode catalyst layer. This oxidation is likely leading to reduced ionic transport to the catalyst, decreased electrochemical active area due to detachment of catalyst particles, and thus the increase in the anode charge-transfer resistance measured by impedance.
PTEs were then cross-sectioned using a plasma-focused-ion-beam (PFIB) and imaged with a SEM. The pristine PTE has ionomer uniformly dispersed throughout the catalyst layer (Fig. 5(a)). After operation, no ionomer is observed, and only large aggregates of IrOx remain (Fig. 5(b)). Elemental counts are shown in Fig. S19 (ESI†). The cross section of the control MEA with only water flow shows no ionomer degradation or loss from the catalyst layer (Fig. S20, ESI†). The observed ionomer loss is thus a coupled chemical/electrochemical/mechanical process, and not purely detachment due to poor catalyst-layer adhesion. It is noted the control test did not account for possible detachment from the produced gases. Gas production is not believed to influence the ionomer degradation, as gas is also evolving at the cathode (at twice the rate, due to stoichiometry of water electrolysis) where the catalyst layer remains intact. However, after oxidation, the evolving gas likely contributes to the removal of the degraded ionomer from the catalyst layer.
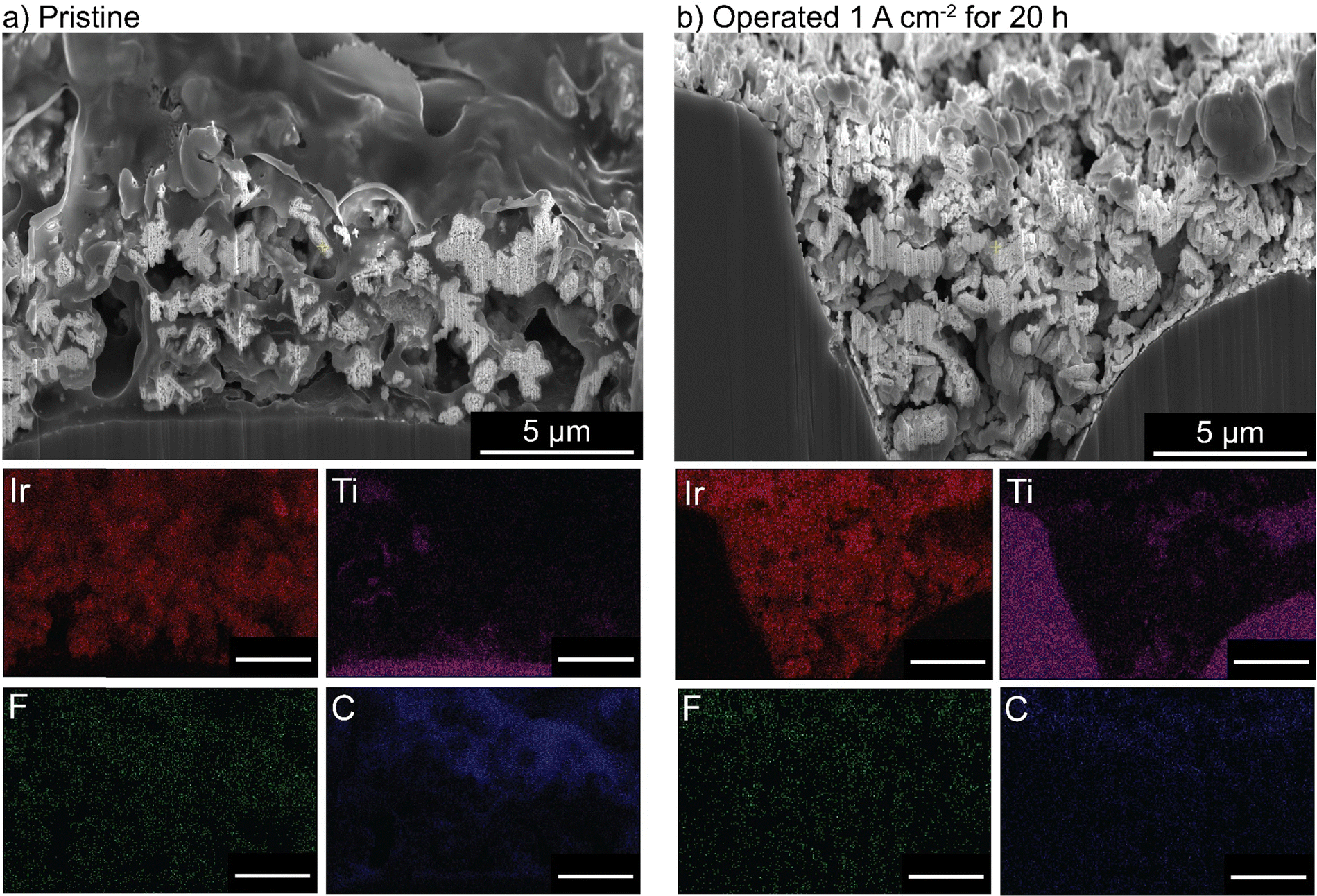 | ||
| Fig. 5 Cross-sectional imaging and EDX of an IrOx/20% TP-85 anode PTE. (a) Before and (b) after 20 h operation at 1 A cm−2. All scale bars are 5 μm. No ionomer is observed in the catalyst layer after operation. | ||
As the membrane is made of the same polymer as the ionomer in the catalyst layers, the anode face of the HEM is also susceptible to oxidation and shows oxidative damage by XPS (Fig. S17, ESI†). The difference in degradation rate at 1 A cm−2 observed in Fig. 3 may be attributed to a difference in membrane versus ionomer oxidation kinetics due in part to proximity with reactive OER catalyst.
For HEM electrolyzers multiple degradation mitigation strategies have been pursued. These generally fall into three categories; improved ionomer design, introducing stable catalyst layer additives, and operation with supporting electrolyte – each of which is explored and discussed in the next section.
All-sp3 norbornene-backbone ionomers
Many polymer design strategies have been pursued to improve alkaline ionomer durability. The polynorbornene (PNB) backbone is of particular interest. The aromatic regions of HEM backbones are likely weak sites for oxidation,43,44,47 therefore a fully sp3-hybridized backbone should be more resistant to oxidative damage. These and related polymers have shown promising performance and durability in HEM fuel cells54,55 and electrolyzers.56–58Anode PTEs were prepared using Co3O4 catalyst and the PNB ionomer on a woven stainless-steel substrate. MEAs were prepared with the same TP-85 membrane and Pt-black/TP-85 cathode PTE as previously. The PNB ionomers show nearly identical polarization performance as the equivalent TP-85 electrode (Fig. 6(a) inset). When operating at 500 mA cm−2 the PNB ionomer appears more stable, with a more-linear voltage degradation as opposed to the rapid onset and stabilization behavior observed for the TP-85 (Fig. 6(a), light red and light orange). However, substantial oxidation is still observed by XPS in conjugation with a loss of ammonium cation and appearance of uncharged nitrogen species (Fig. 6(b)). At 1 A cm−2, both electrodes show similar voltage degradation (Fig. 6(a), dark red and dark orange) and structural oxidation by XPS (Fig. 6(b)). The extent of oxidation appears less compared to the IrOx OER catalysts, which we have found previously to be due to the lower electrical conductivity of Co3O4 that limits the reaction zone to near the PTL and thus less ionomer oxidation is observed on the top (opposite side) of the deconstructed post-mortem catalyst layer.31 Different catalysts are also expected to differently interact chemically/electrostatically with ionomer which may lead to different adsorption and oxidation behavior. In any case, the results here show the PNB ionomer oxidation is comparable to the TP-85 under the same operating conditions, despite having all single bonds.
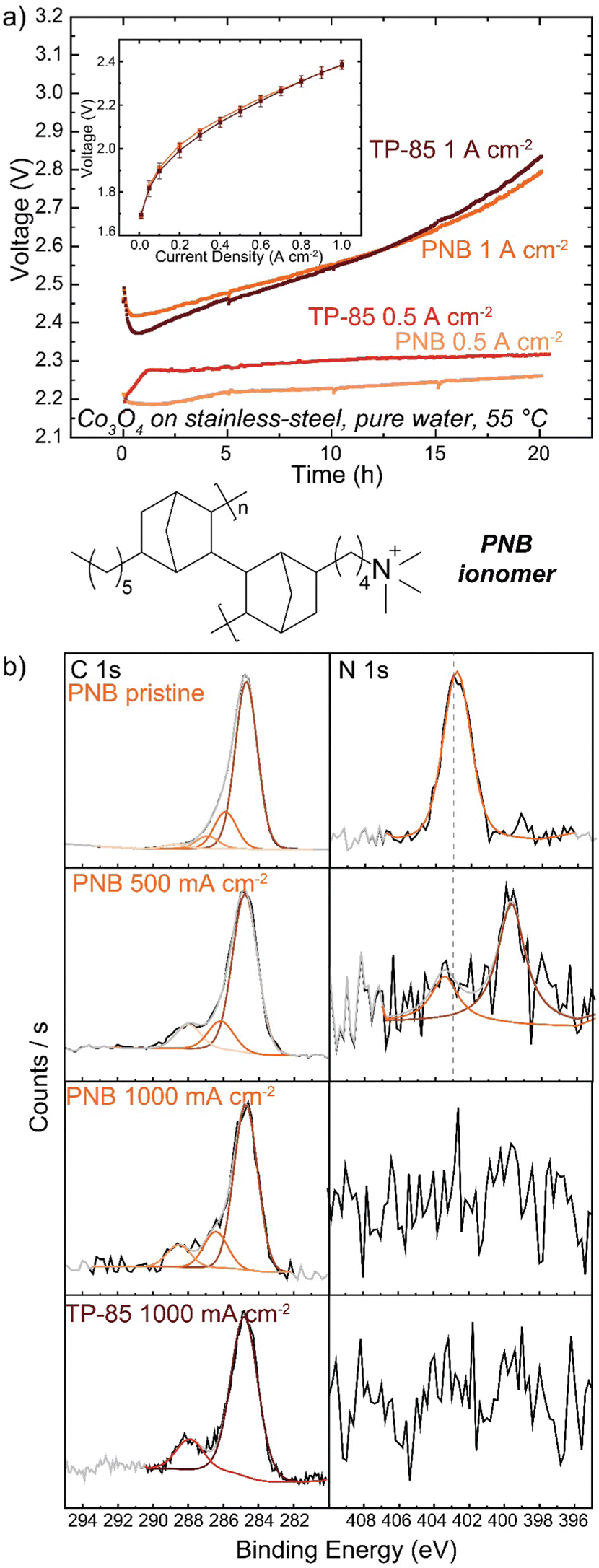 | ||
| Fig. 6 PNB ionomer performance and durability. Cells were operated with a Co3O4/10% PNB or Co3O4/10% TP-85 catalyst on stainless-steel anode, TP-85 membrane, and Pt-black/10% TP-85 cathode at 55 °C. (a) Cell durability at 500 mA cm−2 (light red TP-85 and light orange PNB) and 1 A cm−2 (dark red TP-85 and dark orange PNB). Inset shows polarization performance (dark red TP-85 and dark orange PNB). (b) XPS comparison of pristine and operated anode PTEs. Both systems show similar voltage degradation and anode ionomer oxidation, despite differences in ionomer chemistry. | ||
Other reports of PNB-based ionomers have shown stable voltage performance up to 1 A cm−2 during electrolyte-free water-electrolysis operation.56–58 Other polymer characteristics can impact the extent of oxidation independent of polymer structure. Here, the ion-exchange capacity of the ionomer was tuned to result in the same voltage-polarization performance as the TP-85 system, but the two polymers possess different water uptake and OH− conductivity properties (Table S1, ESI†). Ionomer water uptake can affect device stability, and a low water uptake material at the anode was shown to improve stability despite the performance losses from low ionic conductivity.57 Water uptake will also affect how chemical OH− and radical oxygen species access and interact with the ionomer, and thus ionomers with lower water uptake may not degrade as rapidly. The most-stable electrodes appear to be prepared with ground ionomer-resin particles as opposed to the conventional dissolved/dispersed ionomer solution in ink as used here. Such electrodes often include PTFE additives. This observation presents an interesting question as to the effect of catalyst layer geometry and morphology on ionomer electrochemical degradation and the role of non-ion-conducting additives as stabilizers in the catalyst layer.
Degradation in electrodes with stabilizing additives
Despite Nafion being a cation-conducting polymer, some HEM studies have pursued it as a binder to improve system lifetimes (because PEM electrolyzers show far-superior stability). A Nafion anode PTE was prepared with IrOx catalyst on platinized Ti and operated with a TP-85 membrane and Pt-black/TP-85 cathode PTE. The performance and durability were compared to a conventional PEM baseline with a Nafion membrane and Pt-black/Nafion cathode. The PEM electrolyzer reached 2 V at 2.5 A cm−2 (Fig. 7(a)). The PEM cells were operated at 1 A cm−2 and 2.5 A cm−2 to compare equivalent current and voltage conditions to the performance of the TP-85 HEM and ionomer system. Both PEM systems showed degradation rates below 1 mV h−1 (Fig. 7(b)). There was some initial voltage degradation, which is common for the startup of PEM systems as cell break-in/conditioning is longer, often many hours, during which ion transport channels are established. Further, most PEM electrolyzers use catalyst-coated membranes, while the PEM devices prepared here were catalyst-coated PTEs for direct comparison to the HEM electrolyzers. The initial degradation may be attributed to interfacial optimization between the catalyst layer and membrane. The degradation rate stabilizes to near-expected PEM rates. The degradation rate of the Nafion PTE operated with a HEM was an order of magnitude larger than that of the PEM at the equivalent operating current. XPS analysis of the surface of the PEM-operated Nafion anodes shows no obvious oxidation of the pristine material (Fig. 7(c), yellow and green). However, the surface of the HEM-operated Nafion shows the growth of C–C and oxidized carbon species, a decrease in the higher C–F contribution, and a loss of higher-binding-energy F, consistent with a loss of CF3 content in the polymer. The sulfonate group is still resolvable, but with higher signal-to-noise than the PEM system. This suggests a side-chain scission or loss mechanism, which is believed to be a dominant degradation pathway for Nafion polymer.16,59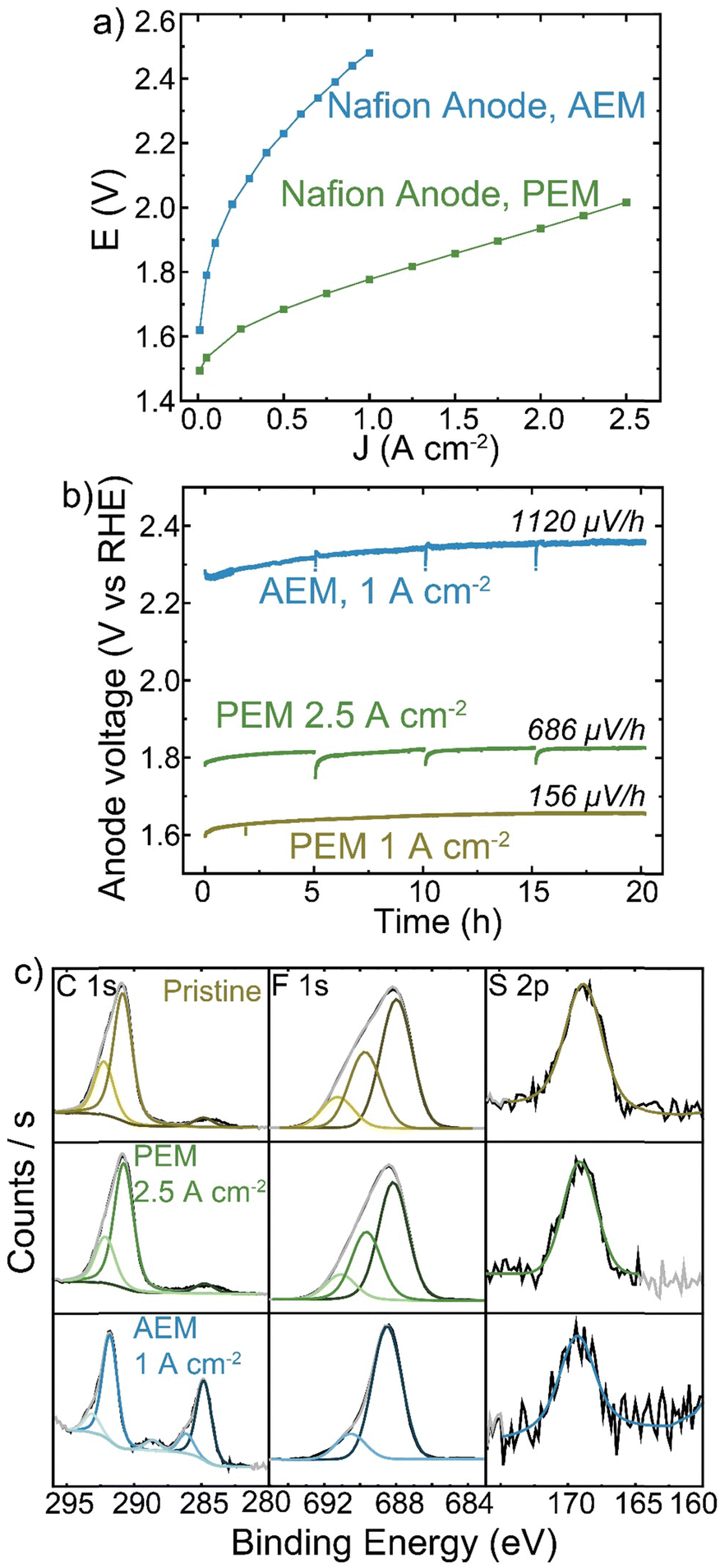 | ||
| Fig. 7 Nafion PTE operation in different membrane-pH environments. All cells were operated with an IrOx/5% Nafion catalyst on Pt/Ti anode, TP-85 or Nafion membrane, and Pt-black/10% TP-85 or Pt-black/5% Nafion cathode (matching the membrane) and at 70 °C. (a) polarization comparison of Nafion PTE operating in a PEM MEA (green) and HEM MEA (blue). (b) Nafion anode durability when operating in a PEM MEA at 1 A cm−2 (yellow) and 2.5 A cm−2 (green) and HEM MEA at 1 A cm−2 (blue). (c) XPS of Nafion PTEs before operation (yellow), after operation with a PEM MEA at 2.5 A cm−2 (green) and after operation with a HEM MEA at 1 A cm−2 (blue). Degradation at the Nafion anode PTE is only observed when operating with a HEM. | ||
When operated with a HEM, the Nafion anode could create a bipolar interface between the anode and membrane, with proton transport through the anode and hydroxide transport from the cathode through the HEM recombining to form water at the anode/membrane interface. However, studies using mixed cation-ionomer/alkaline-membrane devices suggest the membrane pH environment will dictate the pH at the electrode/membrane interface more than ion transport in the ionomer of the catalyst layer.60,61 The degradation of Nafion by XPS analysis with the HEM but not PEM observed here support the existence of a high pH environment at the catalyst layer/HEM interface. SEM cross-section analysis did not yield conclusive results regarding bulk catalyst-layer degradation, as the Nafion content was only 5 wt% and no significant difference in ionomer environment is observed between the pristine Nafion PTE and the electrode operated with a HEM (Fig. S21, ESI†).
Nafion is known to be stable across a wide pH range and as a membrane is chemically stable in many acid and alkaline electrochemical device applications. However, as a binder in a HEM catalyst layer it experiences high-surface-area contact with the oxidizing anode catalyst and may be exposed to a high concentration of radical oxygen species from intermediates. Combined with the high-pH environment, even Nafion thus suffers oxidative damage.
One mechanistic explanation for this broad instability is that the ionomer near the catalyst is polarized in the strong double-layer electric field, leading to increased susceptibility to nucleophilic attack by OH−, whereas in acidic systems no equivalent strong nucleophile exists.
Alkaline oxidative environments are common in organic cleaning solutions used in semiconductor processing, for example RCA2 cleaning solutions.62
PTFE is also used as a non-conductive stabilizing additive. Anode electrodes were prepared with IrOx catalyst on platinized Ti and either TP-85, PTFE, or a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 wt% mixture of the two in the catalyst ink. The mass of total binder and ionomer relative to catalyst in the ink was kept constant for all electrodes. The reference electrode technique was used to determine changes to the cathode and anode as components of the total-cell voltage. Interestingly, the replacement of half the mass of ionomer with PTFE did not affect cell performance (Fig. 8(a)). When only PTFE is present in the anode catalyst layer the voltage performance is very poor, as there is little-to-no ionic conductivity in the anode catalyst layer and thus only OER catalyst in direct contact with the HEM is active. The voltage-degradation profiles are quite different for the three electrodes (Fig. 8(b)). Both electrodes containing TP-85 ionomer show a rapid degradation onset before stabilizing. The degradation profile of the electrode that contains a PTFE/TP-85 mixture matches that of the electrode operated with 10% TP-85 (the equivalent mass of just TP-85 in the electrode) and reaches a steady-state degradation rate of 5.4 mV h−1 for the last 10 h, comparable to the TP-85 system. The rapid degradation onset is not observed for the PTFE-only system. The steady-state degradation rate is 7 mV h−1 over the entire run.
:50 wt% mixture of the two in the catalyst ink. The mass of total binder and ionomer relative to catalyst in the ink was kept constant for all electrodes. The reference electrode technique was used to determine changes to the cathode and anode as components of the total-cell voltage. Interestingly, the replacement of half the mass of ionomer with PTFE did not affect cell performance (Fig. 8(a)). When only PTFE is present in the anode catalyst layer the voltage performance is very poor, as there is little-to-no ionic conductivity in the anode catalyst layer and thus only OER catalyst in direct contact with the HEM is active. The voltage-degradation profiles are quite different for the three electrodes (Fig. 8(b)). Both electrodes containing TP-85 ionomer show a rapid degradation onset before stabilizing. The degradation profile of the electrode that contains a PTFE/TP-85 mixture matches that of the electrode operated with 10% TP-85 (the equivalent mass of just TP-85 in the electrode) and reaches a steady-state degradation rate of 5.4 mV h−1 for the last 10 h, comparable to the TP-85 system. The rapid degradation onset is not observed for the PTFE-only system. The steady-state degradation rate is 7 mV h−1 over the entire run.
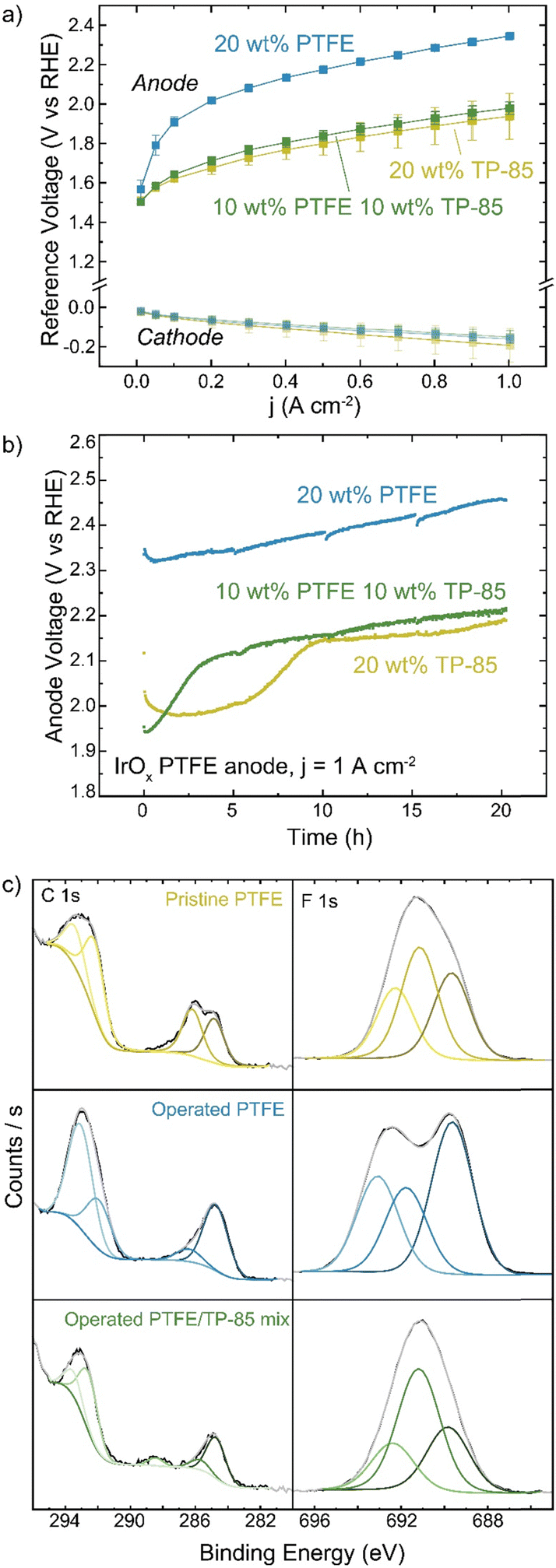 | ||
| Fig. 8 Effect of PTFE on electrolyzer performance. Cells were operated with an IrOx catalyst on Pt/Ti anode with the indicated ionomer and/or binder, TP-85 membrane, and Pt-black/10% TP-85 cathode at 70 °C. (a) polarization curves of the anode and cathode potentials. Data shown is the average of three replicate measurements and error bars are one standard deviation. (b) anode voltage during durability testing at 1 A cm−2 and (c) XPS of the pristine PTFE anode (yellow), operated PTFE anode (blue), and operated mixed PTFE/TP-85 anode (green). No oxidation is observed for the PTFE-only system. | ||
XPS of the mixed-polymer PTE shows C–F contribution from the PTFE and oxidized carbon (Fig. 8). XPS of the PTFE-only system shows no oxidized carbon by XPS (Fig. 8(c)). Interestingly, the operated PTFE shows a change in the F 1s region. This suggests a defluorination degradation mechanism, however, this is not consistent with the lack of changes in the C–F content in the C 1s region. The change in the F 1s region is thus attributed to a polymer/sample inhomogeneity or sample-charging artifact.
Cross-sectioning of the pristine mixed PTFE/TP-85 electrode shows the distribution of binder was not homogenous through the catalyst layer (Fig. 9). The polymers are indistinguishable by EDX, as they both contain C and F content, but they show distinctly different texture/morphology. Some regions show the binder has a smooth texture, the same as what is observed for the TP-85-only catalyst layers and is thus assigned to the TP-85 ionomer (Fig. 9(b)). Other regions show a porous binder environment (Fig. 9(a)), which is assigned as PTFE binder. After operation, the PTFE/TP-85 electrode shows regions of the porous-textured binder (Fig. 9(c)) and large catalyst aggregations with no binder (Fig. 9(d)), which were likely regions where TP-85 was degraded and flushed from the system.
 | ||
| Fig. 9 Cross-sectional analysis of IrOx/mixed binder anode PTE. SEM images of different regions of the pristine catalyst layer (a) and (b) and SEM with EDX maps of two operated regions (c) and (d). Only PTFE binder is observed in the catalyst layer after operation. | ||
Despite different degrees of oxidative damage, all systems show similar voltage degradation from 10–20 h. All three electrolyzers were operated with a TP-85 membrane that is susceptible to oxidation at the anode PTE surface. The similar steady-state degradation rate after 10 h may be due to catalyst oxidizing the membrane surface, increasing ionic resistance between membrane and catalyst layer. The PTFE-only system shows a slightly higher steady-state degradation rate, which is likely due to catalyst detachment or non-uniformities in the catalyst layer. PTFE contains no ionic components and does not interact strongly in the ink solution with the solvent or catalyst, creating a poor catalyst dispersion. The quality of the ink and catalyst layer deposited can impact device performance independent of the properties of the individual components.24 Further, PTFE on its own is a poor catalyst binder, and the catalyst was observed to detach from the electrode during MEA preparation. SEM imaging shows the distribution of PTFE in the pristine catalyst layer was also non-uniform with large agglomerates of PTFE. The PTFE electrode charged too rapidly under the electron beam to obtain usable images (but a video is available as Video S1, ESI†). The PTFE catalyst layer after operation shows large areas of the bare exposed Ti support and no large PTFE agglomerates, suggesting some PTFE washed away during operation.
As PTFE is the only polymer to show no oxidative damage during pure-water operation in our study here, longer-duration testing was conducted. When operating with Co3O4 catalyst at 500 mA cm−2, the cell operated for 100 h with a degradation rate of 0.3 mV h−1 (Fig. 10(a)). No changes to polymer structure were observed by XPS after operation (Fig. 10(b)). While cell voltage was too high for commercial applications, advanced electrode designs that use stable binders or additives, but maintain ionic conductivity in the catalyst layer, may be a viable solution for pure-water HEM operation.
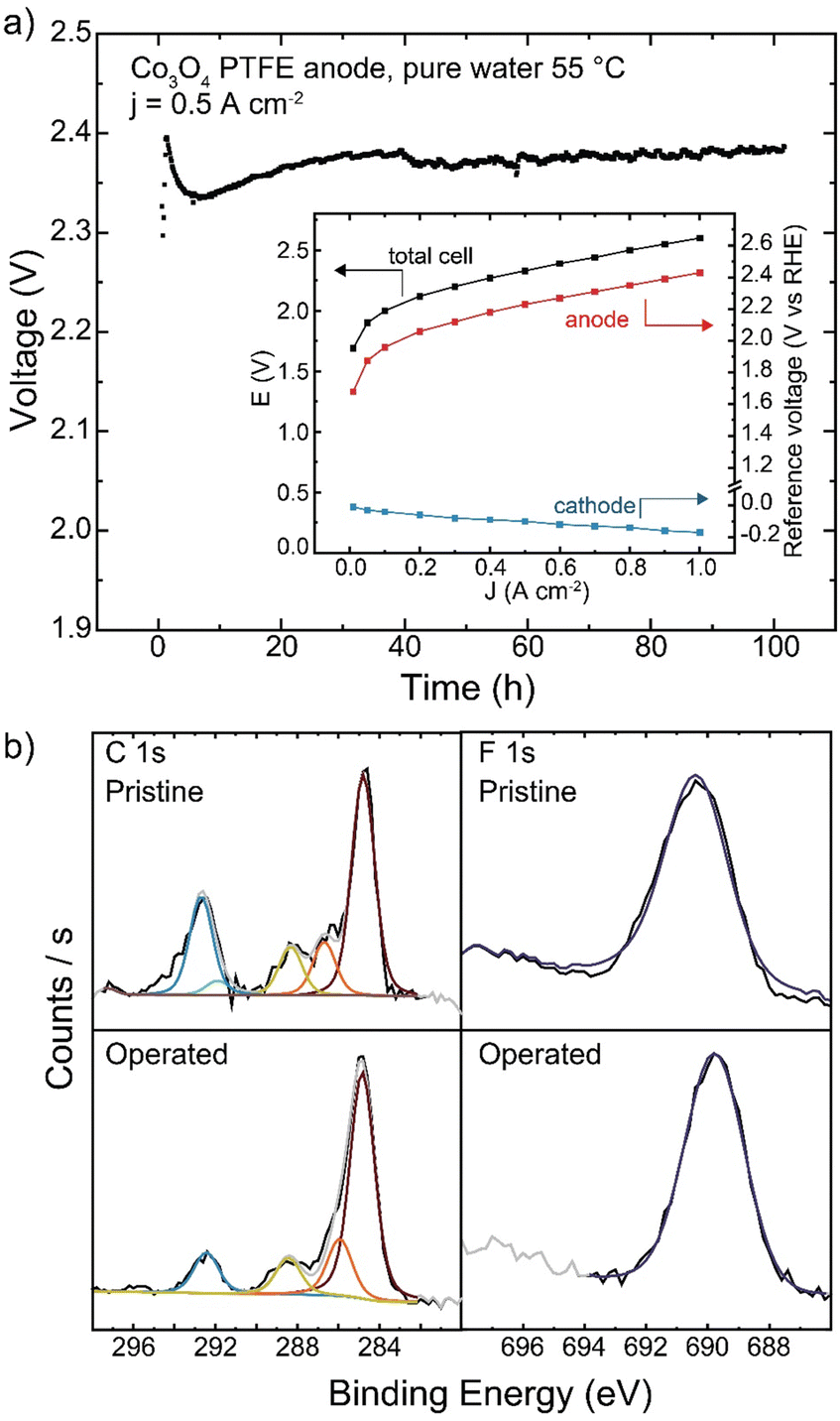 | ||
| Fig. 10 Long-term operation of a PTFE-containing anode. (a) HEM electrolyzer operation with a Co3O4/20% PTFE-coated stainless-steel anode PTE, TP-85 membrane, and Pt-black/10% TP-85 cathode for 100 h at 55 °C. Inset shows cathode and anode contributions to total cell voltage. (b) XPS of the anode catalyst layer before and after 100 h operation. | ||
Effect of supporting hydroxide electrolyte
HEM electrolyzers have improved performance and stability when operating in KOH electrolyte.19,56 A Co3O4 TP-85 anode on a Ni–alloy substrate (to prevent corrosion in KOH on the PTE) was operated in 0.1 M KOH and also in electrolyte-free water (Fig. 11). When fed with KOH, the catalyst layer is saturated with electrolyte and all catalyst sites are exposed to OH−, as opposed to electrolyte-free water where only catalyst sites in contact with the ionomer are active. The addition of electrolyte is expected to increase catalyst electrochemically active surface area, increase ionic conductivity of the catalyst layer, and decrease transport losses. This is reflected in the polarization data, as improvements in 0.1 M KOH are observed in the kinetic, ohmic, and mass-transport regions of the polarization curve (Fig. 11(a)). The cells were then operated at 1 A cm−2 for 20 h (Fig. 11(b)), and a smaller degradation rate <1 mV h−1 was found, compared to the rapid degradation of the electrolyte-free water cell at 17 mV h−1. A third cell was then operated with 0.1 M KOH at 3 A cm−2 so the starting operating voltage was ∼2 V, creating anode potentials where ionomer oxidation is known to occur in the electrolyte-free water system. The voltage was also more stable at a higher operating current, with a cell degradation of 2.4 mV h−1. The anodes operated in 0.1 M KOH also show no obvious evidence of oxidized carbon, even after operating at voltages comparable to the electrolyte-free-water devices (Fig. 11(c)). Only the electrolyte-free cell shows a growth of oxidized carbon in conjunction with a loss of N and F signal.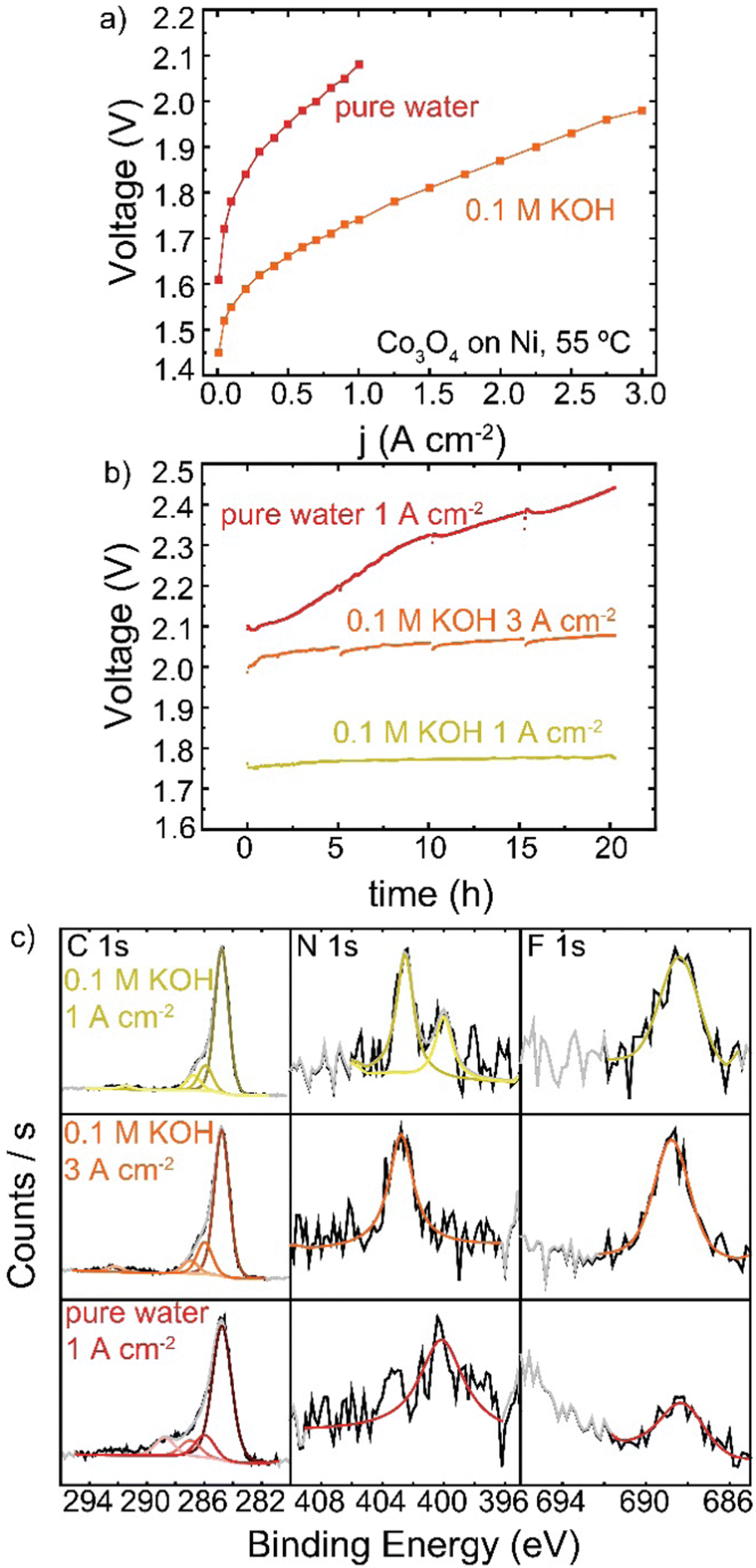 | ||
| Fig. 11 HEM electrolyzer operation in 0.1 M KOH versus pure-water feed. All cells were operated with a Co3O4/20% TP-85-coated Ni PTE, TP-85 membrane, and Pt-black/10% TP-85 cathode at 55 °C. (a) polarization curves of electrolyte-free water (red), and 0.1 M KOH (orange) water feed, (b) durability of electrolyte-free water (red) and 0.1 M KOH (yellow) feed at 1 A cm−2 and 0.1 M KOH at 3 A cm−2 (orange; for operation at a comparable starting voltage to the electrolyte-free water system). Even under high voltages/currents, the presence of 0.1 M KOH dramatically suppresses oxidation, perhaps due to differences in double-layer structure. | ||
In electrolyte-free water, the weakest, most-easily oxidized polymer sites degrade first. This leads to an increase in cell voltage, which may induce a larger driving force at the remaining catalyst/ionomer interface for oxidation, driving cascading degradation until substantial ionomer is oxidized. In KOH, the degradation of ionically conductive polymer is compensated by the presence of supporting OH−. Local oxidation may lead to some catalyst detachment and loss of binder but does not appear to dramatically increase cell voltage and therefore does not increase the driving potential for oxidation.
The potential distribution and structure of the electrical double layer at the catalyst surface may be quite different in electrolyte-free water versus in supporting electrolyte. In alkaline conditions, metal-oxide surfaces are likely negatively charged (due to deprotonation) leading to absorption of cationic or polymer backbone groups from the ionomer, as has been invoked earlier for other reasons.38,41,63–68 Without supporting electrolytes, ionomer therefore must play a fundamental role in the formation of the double layer requiring it to be in close vicinity to the polarized catalyst. The presence of soluble mobile ions in supporting electrolyte likely leads to the displacement of ionomer from direct involvement in the double layer, and this may be responsible for substantially reducing the degradation rate compared to pure water. Alternative strategies to exclude ionomer from the double-layer region therefore might be expected to also reduce oxidation rates.
While these results show operation in supporting electrolyte suppresses ionomer oxidation improving cell stability, the durability testing here was relatively short. The ionomer and membrane may continue degrading at longer operating time, particularly over months or years, leading to slow catalyst detachment or membrane thinning as is observed on a shorter time-scales in the pure water tests.
Conclusion
We compared HEM electrolyzer operation with chemically varied ionomers, catalyst layer additives, and feed modes to understand the extent of ionomer oxidation and its impact on cell performance. Anode ionomer oxidation is the dominant degradation mechanism for all HEM-based devices operating in nominally pure water. All hydrocarbon-based anion-exchange ionomers oxidize rapidly, losing both backbone and cationic side-chain groups. Nafion oxidizes in a HEM electrolyzer, but not PEM electrolyzer, indicating the high rate of degradation can be in part attributed to the combination of the strongly oxidizing environment and high pH. No oxidative damage was observed when using PTFE, showing the promise of stable additives in improving HEM electrolyzer operation.Regarding the future of HEM electrolyzer technology, the most-immediate route to commercialization appears to be operation with supporting OH− electrolyte. KOH-fed HEM electrolyzers present possible advantages over conventional liquid–alkaline electrolyzers, including higher-efficiency/current operation with lower-concentration KOH, perhaps reducing balance-of-plant costs and enabling the use of stainless-steel as opposed to more-expensive pure Ni components. The membrane, as opposed to porous separator, also enables electrochemical compression of the output gases. However, the introduction of supporting electrolyte, even to just the anode, may be accompanied by shunt-current losses and reverse-current degradation upon cell shutdown, making intermittent operation probably more-challenging than for competing PEM electrolyzers. Substantial cell and stack design engineering appears required to address these issues, and the extent to which these limitations affect low-concentration KOH-fed HEMWEs is not understood. Further, the study here only investigated the short-term durability of KOH-fed cells. KOH systems may suffer from similar degradation pathways at longer time periods.
The ideal path for HEM electrolyzer development is electrolyte-free operation. This work reveals the significant challenge facing nominally-pure-water HEM electrolyzer commercialization, but these challenges can likely be overcome. Thus far, HEM electrolyzer advancement has looked to PEM technology for innovation and development insight. For nominally pure-water operation, next-generation HEM cells need new design strategies to address HEM-specific problems. Advanced anode ionomers designed to resist oxidative degradation are necessary, but are not likely to fully resolve degradation limitations, as we found no organic ion-conducting material was oxidatively stable under electrolyte-free operating conditions in this study. While PTFE showed stable operation, the high ion-transport resistance in the catalyst layer results in low voltage efficiencies and is therefore alone not a promising route to commercialization. Improved catalyst-layer interfacial design should minimize ionomer contact, and therefore degradation, but maintain OH− conductivity. These could enable competitive performance and lifetimes for commercialized pure-water HEM electrolyzer devices at dramatically lower materials costs than current membrane electrolyzer technologies.
Materials and methods
Polynorbornene ionomer synthesis
000 Å; Polymer Standard Services). The eluent tetrahydrofuran (THF) was doped with 10 mM lithium bis(trifluoromethanesulfonyl)imide (flow rate of 1 mL min−1, 40 °C). A nine-point calibration based on polystyrene standards (Polystyrene, ReadyCal Kit, Polymer Standard Services) was applied for determination of molecular weight.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :40 NB-5-Hex-co-NB-5-BuBr copolymer.
In a N2 glovebox, tetrakis(pentafluorophenyl)boron lithium ethyl etherate (0.0264 mmol), tri-tert-butylphosphine palladium(II) methyl chloride (0.0264 mmol), and dry CH2Cl2 (6.6 mL) were added to an oven-dried Schlenk flask equipped with a stir bar. 5-n-hexyl-2-norbornene (7.92 mmol) and 5-(4-bromobutyl)-2-norbornene (5.28 mmol) were added to a separate vessel and dissolved in dry CH2Cl2 (59.1 mL). Both solutions were brought to a benchtop stir plate, and the Pd catalyst reaction mixture was stirred for 15 min at 22 °C to ensure formation of the active cationic Pd catalyst. The solution of 5-n-hexyl-2-norbornene and 5-(4-bromobutyl)-2-norbornene in CH2Cl2 was then injected into the activated catalyst solution. The polymerization reaction was stirred, and a 0.05 mL aliquot was removed at various timepoints for crude analysis by 1H NMR spectroscopy to ensure complete consumption of the two monomers (disappearance of the vinyl protons). Polymers were precipitated into a large excess of methanol, which yielded an off-white stringy polymer that was filtered and dried in vacuo for 17 h at 22 °C (2.51 g, 96% yield) (Scheme 1). 1H NMR (500 MHz, CDCl3) δ ppm: 3.4 (br s, 2H), 2.6–0.96 (br, all other protons except for hexyl norbornene –CH3), 0.88 (br s, 4.5 H) (Fig. S2, ESI†).
:40 NB-5-Hex-co-NB-5-BuBr copolymer.
In a N2 glovebox, tetrakis(pentafluorophenyl)boron lithium ethyl etherate (0.0264 mmol), tri-tert-butylphosphine palladium(II) methyl chloride (0.0264 mmol), and dry CH2Cl2 (6.6 mL) were added to an oven-dried Schlenk flask equipped with a stir bar. 5-n-hexyl-2-norbornene (7.92 mmol) and 5-(4-bromobutyl)-2-norbornene (5.28 mmol) were added to a separate vessel and dissolved in dry CH2Cl2 (59.1 mL). Both solutions were brought to a benchtop stir plate, and the Pd catalyst reaction mixture was stirred for 15 min at 22 °C to ensure formation of the active cationic Pd catalyst. The solution of 5-n-hexyl-2-norbornene and 5-(4-bromobutyl)-2-norbornene in CH2Cl2 was then injected into the activated catalyst solution. The polymerization reaction was stirred, and a 0.05 mL aliquot was removed at various timepoints for crude analysis by 1H NMR spectroscopy to ensure complete consumption of the two monomers (disappearance of the vinyl protons). Polymers were precipitated into a large excess of methanol, which yielded an off-white stringy polymer that was filtered and dried in vacuo for 17 h at 22 °C (2.51 g, 96% yield) (Scheme 1). 1H NMR (500 MHz, CDCl3) δ ppm: 3.4 (br s, 2H), 2.6–0.96 (br, all other protons except for hexyl norbornene –CH3), 0.88 (br s, 4.5 H) (Fig. S2, ESI†).
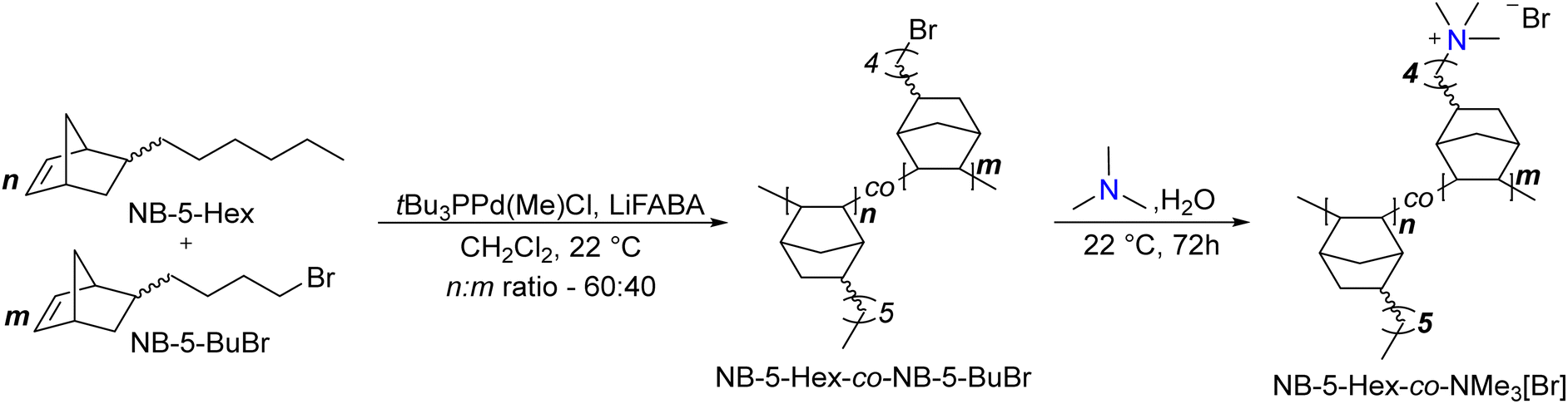 | ||
| Scheme 1 General synthesis of 60:40 NB-5-Hex-co-NMe3 HEM. | ||
:40 copolymer, a 2:4.5 ratio should be observed for the –CH2Br signal on the NB-5-BuBr the terminal methyl group from the hexyl chain of the NB-5-Hex, so the reported assignments for the terminal methyl group are set to 4.5.
:40 NB-5-Hex-co-NB-5-BuBr copolymers.
150 mg of polymer was dissolved in 3 mL of CHCl3. Upon complete dissolution, the solution was filtered through a 0.22 μm syringe filter onto a stainless-steel dish (diameter – 5 cm). The CHCl3 evaporated over an hour to afford a clear freestanding film, which was removed from the dish by immersion in deionized water. The polymer was then dried in vacuo to remove water and any other residual solvents.
:40 NB-5-Hex-co-NMe3 polymer.
The dried NB-5-Hex-co-NB-5-BuBr polymer film was immersed in an aqueous solution of 25% (w/v) trimethylamine for 48 h at room temperature. The solution was then replaced with fresh aqueous trimethylamine and the films were immersed for an additional 24 h. The films were removed and immersed in 3 × 100 mL portions of deionized water for 1 h each. The films were then dried in vacuo to afford the trimethylammonium-functionalized polymers. Accurate 1H integrals were difficult to obtain due to overlapping solvent and signal broadness. Spectral data: 1H NMR (CDCl3:CD3OD 1:1) δ ppm: 3.0 (br s, 9H, NMe3 –CH3), 2.5–0.76 (br, all other protons except for hexyl norbornene –CH3), 0.56 (br s, 6H) (Fig. S2, ESI†).
Author contributions
G. A. L. and S. W. B. designed the study, analyzed the data, and wrote the manuscript. G. A. L. collected and analyzed all electrolyzer, SEM, and XPS data, with electrolyzer support from W. L. T. and SEM support from V. B. The PNB ionomer system was designed by J. C. G. and K. J. T., with synthesis and characterization conducted by J. C. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Fuel Cell Technologies Office (FCTO) under award DE-EE0008841 and by De Nora Tech, LLC. J. C. G. acknowledges support from the Center for Alkaline-Based Energy Solutions (CABES), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences (BES) under Award DE-SC0019445. G. A. L. acknowledges additional support from the University of Oregon through the John Keana Graduate Fellowship. We acknowledge the use of shared instrumentation in the Center for Advanced Materials Characterization in Oregon (CAMCOR). The authors are grateful to Nel Hydrogen for providing the platinized PTL material, Dr Stephen Golledge for XPS analysis guidance, L. Chen for assistance with the integrated reference electrode experiments, the Yan lab at University of Delaware for their advice regarding KOH electrolyzer testing, and Cy Fujimoto and Nick D’Antona for insightful discussion.References
- G. Bender, Scalable Electrolytic Systems for Renewable Hydrogen Production: Cooperative Research and Development Final Report, CRADA Number CRD-18-747, Report NREL/TP-5900-76136 United States 10.2172/1601587 NREL English, National Renewable Energy Lab. (NREL), Golden, CO (United States), 2020.
- IRENA, Green Hydrogen Cost Reduction: Scaling up Electrolysers to Meet the 1.5 °C Climate Goal, International Renewable Energy Agency, 2020.
- Hydrogen Council, Path to hydrogen competitiveness: A cost perspecitve, 2020.
- IEA, Technology Roadmap - Hydrogen and Fuel Cells, International Energy Alliance, Paris, 2015 Search PubMed.
- B. Pivovar, N. Rustagi and S. Satyapal, Electrochem. Soc. Interface, 2018, 27, 47–52 CrossRef CAS.
- Nel Hydrogen, M Series Proton PEM Electrolyzer, https://nelhydrogen.com/product/m-series-3/, (accessed April 2, 2020).
- Plug Power, Plug lands 1 GW electrolyzer order with H2 Energy Europe, https://www.ir.plugpower.com/press-releases/news-details/2022/Plug-Lands-1-GW-Electrolyzer-Order-with-H2-Energy-Europe/default.aspx, (accessed March 29, 2023).
- Nel Hydrogen, Nel ASA: Official opening of the Herøya facility, https://nelhydrogen.com/press-release/nel-asa-official-opening-of-the-heroya-facility/, (accessed March 29, 2023).
- Cummins Inc., Cummins drives domestic green hydrogen economy forward with first U.S. electrolyzer manufacturing facility, https://www.cummins.com/news/releases/2022/10/10/cummins-drives-domestic-green-hydrogen-economy-forward-first-us, (accessed March 29, 2023).
- J. J. Sumner, S. E. Creager, J. J. Ma and D. D. DesMarteau, J. Electrochem. Soc., 1998, 145, 107 CrossRef CAS.
- Y. Sone, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254 CrossRef CAS.
- P. Millet, R. Ngameni, S. A. Grigoriev and V. N. Fateev, Int. J. Hydrogen Energy, 2011, 36, 4156–4163 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- J. Hemauer, S. Rehfeldt, H. Klein and A. Peschel, Int. J. Hydrogen Energy, 2023, 48(66), 25619–25634 CrossRef.
- J. Peron, A. Mani, X. Zhao, D. Edwards, M. Adachi, T. Soboleva, Z. Shi, Z. Xie, T. Navessin and S. Holdcroft, J. Membr. Sci., 2010, 356, 44–51 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- Ionomr, Ionomer Hydrogen Info Sheet, https://ionomr.com/wp-content/uploads/2018/12/Ionomr-Hydrogen-Info-Sheet.pdf, (accessed September 15, 2020).
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- D. G. Li, E. J. Park, W. L. Zhu, Q. R. Shi, Y. Zhou, H. Y. Tian, Y. H. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Energy Environ. Sci., 2021, 14, 3393–3419 RSC.
- Y. Chen, D. Su, Y. Chen, Z. Zhu and W. Li, Cell Rep. Phys. Sci., 2021, 2, 100602 CrossRef CAS.
- A. Y. Faid, L. Xie, A. O. Barnett, F. Seland, D. Kirk and S. Sunde, Int. J. Hydrogen Energy, 2020, 45, 28272–28284 CrossRef CAS.
- S. M. Alia, K. S. Reeves, J. S. Baxter and D. A. Cullen, J. Electrochem. Soc., 2020, 167, 144512 CrossRef CAS.
- G. A. Lindquist, S. Z. Oener, R. Krivina, A. R. Motz, A. Keane, C. Capuano, K. E. Ayers and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2021, 13, 51917–51924 CrossRef CAS PubMed.
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef CAS PubMed.
- F. P. Lohmann-Richters, S. Renz, W. Lehnert, M. Müller and M. Carmo, J. Electrochem. Soc., 2021, 168, 114501 CrossRef CAS.
- K. F. L. Hagesteijn, S. Jiang and B. P. Ladewig, J. Mater. Sci., 2018, 53, 11131–11150 CrossRef CAS.
- T. A. M. Suter, K. Smith, J. Hack, L. Rasha, Z. Rana, G. M. A. Angel, P. R. Shearing, T. S. Miller and D. J. L. Brett, Adv. Energy Mater., 2021, 11, 2101025 CrossRef CAS.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- L. Wan, J. Liu, Z. Xu, Q. Xu, M. Pang, P. Wang and B. Wang, Small, 2022, 18, 2200380 CrossRef CAS PubMed.
- R. A. Krivina, G. A. Lindquist, S. R. Beaudoin, T. N. Stovall, W. L. Thompson, L. P. Twight, D. Marsh, J. Grzyb, K. Fabrizio, J. E. Hutchison and S. W. Boettcher, Adv. Mater., 2022, 34, 2203033 CrossRef CAS PubMed.
- F. Razmjooei, T. Morawietz, E. Taghizadeh, E. Hadjixenophontos, L. Mues, M. Gerle, B. D. Wood, C. Harms, A. S. Gago, S. A. Ansar and K. A. Friedrich, Joule, 2021, 5, 1776–1799 CrossRef CAS.
- N. Chen, S. Y. Paek, J. Y. Lee, J. H. Park, S. Y. Lee and Y. M. Lee, Energy Environ. Sci., 2021, 14, 6338–6348 RSC.
- J. Xiao, A. M. Oliveira, L. Wang, Y. Zhao, T. Wang, J. Wang, B. P. Setzler and Y. Yan, ACS Catal., 2021, 11, 264–270 CrossRef CAS.
- R. Soni, S. Miyanishi, H. Kuroki and T. Yamaguchi, ACS Appl. Energy Mater., 2020, 4, 1053–1058 CrossRef.
- J. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
- J. T. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6, 1089–1093 CrossRef CAS PubMed.
- K. M. Hugar, H. A. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed.
- A. M. Park, Z. R. Owczarczyk, L. E. Garner, A. C. Yang-Neyerlin, H. Long, C. M. Antunes, M. R. Sturgeon, M. J. Lindell, S. J. Hamrock, M. Yandrasits and B. S. Pivovar, ECS Trans., 2017, 80, 957 CrossRef CAS.
- E. J. Park, S. Maurya, M. R. Hibbs, C. H. Fujimoto, K.-D. Kreuer and Y. S. Kim, Macromolecules, 2019, 52, 5419–5428 CrossRef CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed.
- W. E. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805–2838 RSC.
- D. Li, I. Matanovic, A. S. Lee, E. J. Park, C. Fujimoto, H. T. Chung and Y. S. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 9696–9701 CrossRef CAS PubMed.
- R. A. Krivina, G. A. Lindquist, M. C. Yang, A. K. Cook, C. H. Hendon, A. R. Motz, C. Capuano, K. E. Ayers, J. E. Hutchison and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2022, 14, 18261–18274 CrossRef CAS PubMed.
- T. Nemeth, T. Nauser and L. Gubler, ChemSusChem, 2022, 15, e202201571 CrossRef CAS PubMed.
- Y. Z. Zhang, J. Parrondo, S. Sankarasubramanian and V. Ramani, ChemSusChem, 2017, 10, 3056–3062 CrossRef CAS PubMed.
- S. Maurya, A. S. Lee, D. Li, E. J. Park, D. P. Leonard, S. Noh, C. Bae and Y. S. Kim, J. Power Sources, 2019, 436, 226866 CrossRef CAS.
- S. B. Adler, J. Electrochem. Soc., 2002, 149, E166 CrossRef CAS.
- R. Zeng, R. C. T. Slade and J. R. Varcoe, Electrochim. Acta, 2010, 56, 607–619 CrossRef CAS.
- Q. Xu, S. Z. Oener, G. Lindquist, H. Jiang, C. Li and S. W. Boettcher, ACS Energy Lett., 2020, 6, 305–312 CrossRef.
- V. Vivier and M. E. Orazem, Chem. Rev., 2022, 122, 11131–11168 CrossRef CAS PubMed.
- A. C. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3(3), 162–193 CrossRef CAS PubMed.
- Thermo Fisher Scientific, X-ray photoelectron spectroscopy of atomic elements, https://www.thermofisher.com/us/en/home/materials-science/learning-center/periodic-table.html, (accessed April 7, 2023).
- D. P. Leonard, M. Lehmann, J. M. Klein, I. Matanovic, C. Fujimoto, T. Saito and Y. S. Kim, Adv. Energy Mater., 2023, 13, 2203488 CrossRef CAS.
- X. Wu, N. Chen, C. Hu, H.-A. Klok, Y. M. Lee and X. Hu, Adv. Mater., 2023, 35(26), 2210432 CrossRef CAS PubMed.
- N. Hassan, Y. Zheng, P. Kohl and W. E. Mustain, J. Electrochem. Soc., 2022, 169, 044526 CrossRef CAS.
- G. Huang, M. Mandal, N. U. Hassan, K. Groenhout, A. Dobbs, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2020, 167, 164514 CrossRef CAS.
- G. Huang, M. Mandal, N. U. Hassan, K. Groenhout, A. Dobbs, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2021, 168, 024503 CrossRef CAS.
- L. Ghassemzadeh and S. Holdcroft, J. Am. Chem. Soc., 2013, 135, 8181–8184 CrossRef CAS PubMed.
- B. Mayerhöfer, D. McLaughlin, T. Böhm, M. Hegelheimer, D. Seeberger and S. Thiele, ACS Appl. Energy Mater., 2020, 3, 9635–9644 CrossRef.
- B. Mayerhöfer, K. Ehelebe, F. D. Speck, M. Bierling, J. Bender, J. A. Kerres, K. J. J. Mayrhofer, S. Cherevko, R. Peach and S. Thiele, J. Mater. Chem. A, 2021, 9, 14285–14295 RSC.
- W. Kern, J. Electrochem. Soc., 1990, 137, 1887 CrossRef CAS.
- S. Maurya, J. H. Dumont, C. N. Villarrubia, I. Matanovic, D. Li, Y. S. Kim, S. Noh, J. Han, C. Bae, H. A. Miller, C. H. Fujimoto and D. R. Dekel, ACS Catal., 2018, 8, 9429–9439 CrossRef CAS.
- I. Matanovic, S. Maurya, E. J. Park, J. Y. Jeon, C. Bae and Y. S. Kim, Chem. Mater., 2019, 31, 4195–4204 CrossRef CAS.
- A. R. Motz, D. Li, A. Keane, L. D. Manriquez, E. J. Park, S. Maurya, H. Chung, C. Fujimoto, J. Jeon, M. K. Pagels, C. Bae, K. E. Ayers and Y. S. Kim, J. Mater. Chem. A, 2021, 9, 22670–22683 RSC.
- S. Maurya, C. H. Fujimoto, M. R. Hibbs, C. Narvaez Villarrubia and Y. S. Kim, Chem. Mater., 2018, 30, 2188–2192 CrossRef CAS.
- H. T. Chung, U. Martinez, I. Matanovic and Y. S. Kim, J. Phys. Chem. Lett., 2016, 7, 4464–4469 CrossRef CAS PubMed.
- I. T. McCrum, M. A. Hickner and M. J. Janik, J. Electrochem. Soc., 2018, 165, F114 CrossRef CAS.
- M. Yamashita, I. Takamiya, K. Jin and K. Nozaki, Organometallics, 2006, 25, 4588–4595 CrossRef CAS.
- R. Selhorst, J. Gaitor, M. Lee, D. Markovich, Y. Yu, M. Treichel, C. Olavarria Gallegos, T. Kowalewski, L. F. Kourkoutis, R. C. Hayward and K. J. T. Noonan, ACS Appl. Energy Mater., 2021, 4, 10273–10279 CrossRef CAS.
- J. R. Glenn, G. A. Lindquist, G. M. Roberts, S. W. Boettcher and K. E. Ayers, Front. Energy Res., 2022, 10 DOI:10.3389/fenrg.2022.908672.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee01293j |
| This journal is © The Royal Society of Chemistry 2023 |