
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of the preparation method on the catalytic properties of copper-containing zeolite Y applied for NH3-SCR-DeNOx†
Rujito S. R.
Suharbiansah
a,
Muhammad Fernadi
Lukman
 b,
Chiara
Nannuzzi
c,
Anna
Wach
d,
Kinga
Góra-Marek
e,
Michael
Liebau
a,
Ana
Palčić
f,
Andreas
Pöppl
b,
Gloria
Berlier
c,
Silvia
Bordiga
c,
Roger
Gläser
a and
Magdalena
Jabłońska
*a
b,
Chiara
Nannuzzi
c,
Anna
Wach
d,
Kinga
Góra-Marek
e,
Michael
Liebau
a,
Ana
Palčić
f,
Andreas
Pöppl
b,
Gloria
Berlier
c,
Silvia
Bordiga
c,
Roger
Gläser
a and
Magdalena
Jabłońska
*a
aInstitute of Chemical Technology, Universität Leipzig, Linnéstr. 3, 04103 Leipzig, Germany. E-mail: magdalena.jablonska@uni-leipzig.de
bFelix Bloch Institute for Solid State Physics, Universität Leipzig, Linnéstr. 5, 04103, Leipzig, Germany
cDepartment of Chemistry, NIS Center and INSTM Reference Center, University of Turin, Via P. Giuria 7, 10125 Turin, Italy
dPaul Scherrer Institute, 5232 Villigen, Switzerland
eFaculty of Chemistry, Jagiellonian University in Krakow, Gronostajowa 2, 30-387, Krakow, Poland
fLaboratory for the Synthesis of New Materials, Division of Materials Chemistry, Ruđer Bošković Institute, Bijenička 54, 10000 Zagreb, Croatia
First published on 4th May 2023
Abstract
Two series of zeolite Y were prepared with varying particle sizes (estimated to be 100 or 400 nm) by applying different synthesis gel composition ratios as well as different ageing and hydrothermal steps. Copper-containing zeolite Y samples were obtained through ion-exchange, thoroughly characterized and tested for the selective catalytic reduction with NH3 (NH3-SCR-DeNOx). The catalytic activity of the prepared materials varied significantly due to the different nature of copper species. Results show that catalysts CuY-0h-A and CuY-24h-A (approx. particle sizes 2676 and 475 nm, respectively) facilitate a higher NO conversion both in the presence and absence of water vapor, exhibiting however stronger N2O formation above 250 °C. On the other hand, nearly X-ray amorphous Y-0h-B and Y-24h-B after ion-exchange led to decreased activity of CuY-0h-B and CuY-24h-B (approx. particles sizes 345 and 128 nm, respectively). Additional insight into the activity of the catalysts was obtained through in situ DR UV-vis spectroscopy.
1. Introduction
NOx (NO2 and NO) produced by the combustion processes in trucks, cars, stationary combustion equipment, and power plants are detrimental to the environment as well as human health. For instance, they may cause the formation of acid rain and photochemical smog and lead to ozone depletion. Thus, regulations around the world (e.g., the European emission standards) set limits for the emission of NOx.1 The selective catalytic reduction of NOx with ammonia (NH3-SCR-DeNOx) is an efficient method for the reduction of NOx, while copper-containing zeolites have been widely reported as efficient catalysts for this process (e.g., ref. 2). Among them, the catalytic properties of Cu-containing zeolite Y have been less extensively investigated compared to other zeolites, with the presented literature results being ambiguous. For instance, Kwak et al.3 achieved less than 70% NOx conversion for (7.2 wt%) copper-containing zeolite Y above 300 °C (Na–Y, Zeolyst, CBV 100, n(Si)/n(Al) = 2.6, ion-exchanged with an aqueous solution of copper(II) nitrate, GHSV 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1). In contrast to these results, our latest study4 showed that a catalyst based on commercial zeolite Y (Zeolyst, CBV 100, n(Si)/n(Al) = 2.6), i.e., (6.5 wt%)Cu–Y presents a NO conversion higher than 80% between 125 and 450 °C. Furthermore, we found4 that different synthesis routes lead to the formation of zeolite Y with variations in particle size which affect the efficiency of the ion-exchange process as the specific surface area available for direct contact with the solution is determined by the size of the particles.5 The synthesis temperature also influences the properties of the final zeolite product and thus the nature of copper species in Cu-exchanged materials.6 Differences in the ion-exchange conditions (salt, concentration, time and temperature) also afforded the divergence in copper site speciation and therefore the catalyst activity and selectivity. Being able to prepare catalysts with predictable physico-chemical properties represents a breakthrough and is of great significance for their commercial application. Thus, in the present study, we applied different synthesis routes for the preparation of zeolite Y, using different initial gel compositions as well as different ageing periods (0 or 24 h in air) and hydrothermal (at 70 °C or 100 °C for 21 h) treatments. The effect of the structural and morphological properties of zeolite Y on the nature and distribution of copper species was investigated by applying XRD, SEM, TEM, N2 sorption, ICP-OES, DR UV-vis, TPR-H2, CW-EPR, and XANES techniques. Subsequently, the associated catalytic activity and selectivity were investigated over copper-containing zeolite Y samples for NH3-SCR-DeNOx. Furthermore, by applying in situ DR UV-vis spectroscopy to NH3-SCR-DeNOx, we examined the Cu dynamics under operating conditions.
000 h−1). In contrast to these results, our latest study4 showed that a catalyst based on commercial zeolite Y (Zeolyst, CBV 100, n(Si)/n(Al) = 2.6), i.e., (6.5 wt%)Cu–Y presents a NO conversion higher than 80% between 125 and 450 °C. Furthermore, we found4 that different synthesis routes lead to the formation of zeolite Y with variations in particle size which affect the efficiency of the ion-exchange process as the specific surface area available for direct contact with the solution is determined by the size of the particles.5 The synthesis temperature also influences the properties of the final zeolite product and thus the nature of copper species in Cu-exchanged materials.6 Differences in the ion-exchange conditions (salt, concentration, time and temperature) also afforded the divergence in copper site speciation and therefore the catalyst activity and selectivity. Being able to prepare catalysts with predictable physico-chemical properties represents a breakthrough and is of great significance for their commercial application. Thus, in the present study, we applied different synthesis routes for the preparation of zeolite Y, using different initial gel compositions as well as different ageing periods (0 or 24 h in air) and hydrothermal (at 70 °C or 100 °C for 21 h) treatments. The effect of the structural and morphological properties of zeolite Y on the nature and distribution of copper species was investigated by applying XRD, SEM, TEM, N2 sorption, ICP-OES, DR UV-vis, TPR-H2, CW-EPR, and XANES techniques. Subsequently, the associated catalytic activity and selectivity were investigated over copper-containing zeolite Y samples for NH3-SCR-DeNOx. Furthermore, by applying in situ DR UV-vis spectroscopy to NH3-SCR-DeNOx, we examined the Cu dynamics under operating conditions.
2. Experimental
2.1 Preparation of zeolite Y
The first set of zeolite Y samples was prepared by a dense gel method using an initial molar gel composition of 8Na2O:1Al2O3:10SiO2:410H2O according to the modified procedure by Dabbawala et al.7 Firstly, 94.5 g of water was poured into a polypropylene (PP) bottle, followed by the addition of 8.4 g of NaOH (≥98 wt%, Sigma-Aldrich). This mixture was manually shaken until a clear solution was formed and then 2.88 g of NaAlO2 (Sigma-Aldrich, Al2O3 (50–56 wt%), Na2O (37–45 wt%)) was added to it. This solution was stirred for 15 min, after which 22.5 g of an aqueous colloidal silica sol (LUDOX-HS40®, 40 wt%, Sigma-Aldrich) was added dropwise.
In this series, two samples of zeolite Y were prepared, namely, ZY-0h-A and ZY-24h-A, where 0 h or 24 h indicates the aging period of the initial hydrogel at room temperature (ca. 25 °C) before subjecting it to the same hydrothermal treatment at 100 °C for 21 h. After the 21 h long hydrothermal treatment, the samples were filtered and washed until pH 7 was reached and dried under ambient conditions. Finally, ZY-0h-A and ZY-24h-A were calcined at 550 °C for 4 h in static air (with a heating rate of 1 °C min−1).
In the second set of zeolite Y, a hydrogel with an initial molar composition of 12Na2O:1Al2O3:14SiO2:270H2O was prepared. Alumina and silica solutions were prepared separately. The alumina solution was prepared by adding 9 g of NaOH (≥98 wt%, Sigma-Aldrich) and 2.1 g of NaAlO2 (Sigma-Aldrich, Al2O3 (50–56 wt%) and Na2O (37–45 wt%)) to 15.0 g of water, and the resulting solution was homogenized by manual shaking. The silica solution was prepared by adding 22.5 g of an aqueous colloidal silica sol (LUDOX-HS40®, 40 wt%, Sigma-Aldrich) to 21.0 g of water while stirring. Both the alumina and silica solutions were placed in an ice bath for 0.5 h. Afterwards, the alumina solution was transferred dropwise to the silica solution under stirring.
Additionally, in this series two zeolite Y samples were also prepared, namely, ZY-0h-B and ZY-24h-B. ZY-0h-B refers to the material that was immediately subjected to hydrothermal treatment in an oven at 70 °C for 21 h, while ZY-24h-B was aged for 24 h at room temperature (ca. 25 °C) before it was subjected to the same hydrothermal treatment as ZY-0h-B. After the hydrothermal treatment, both samples were filtered and washed until pH 7 was reached. The samples were dried at room temperature and subsequently calcined at 550 °C for 4 h in static air (with a heating rate of 1 °C min−1).
2.2 Copper-containing zeolite Y
All the prepared zeolite Y samples (ZY-0h-A, ZY-24h-A, ZY-0h-B, and ZY-24h-B) were ion-exchanged using a 0.05 M aqueous solution of copper(II) acetate (≥98 wt%, Alfa Aesar) at room temperature (ca. 25 °C) for 24 h (1 g of zeolite per 100 ml of copper solution). After 24 h, the copper-containing zeolite Y samples were filtered and washed until pH 7 was reached, dried and calcined at 550 °C for 4 h in static air (with a heating rate of 1 °C min−1).The zeolite Y samples as well as their copper-containing forms were characterized in terms of their structure, morphology and chemical composition (XRD, SEM, TEM, and ICP-OES), texture (N2 sorption), the nature of copper species (DR UV-vis, TEM, TPR-H2, X-band CW-EPR, and XANES techniques), and catalytic activity and selectivity (NH3-SCR-DeNOx, including in situ DR UV-vis spectroscopy). The details of the experimental procedures can be found in the ESI.†
3. Results and discussion
3.1 Structural, morphological and textural properties
Fig. 1 shows the XRD patterns obtained for the prepared zeolite Y and copper-containing zeolite Y samples. The characteristic diffraction peaks of zeolite Y (Fig. 1a) can be seen at 2θ = 6.3°, 10.3°, 12.2°, 16°, 19.1°, 20.7°, 23.3°, 24.1°, 27.6°, etc., which can be assigned to the (111), (220), (311), (331), (511) (440), (533) and (642) crystal planes of zeolite Y with a cubic structure.8 The distinct peak shape indicates the high crystallinity of the materials. Loading the Cu species in the series of samples studied affected their crystal structure (Fig. 1b). In the A series of samples this effect is minor as the intensity ratio of the peak at 6.3° with respect to the rest of the pattern only slightly decreased. On the other hand, the samples in the B series underwent crystallinity degradation as evidenced by a significant decrease in the relative peak intensities and the broad maximum in the range of 17–33° typical of the amorphous phase. What's more, the peaks have almost completely disappeared in the sample Cu-Y-24h-B. We observed even stronger degradation of the structure after copper-exchange for the EMT type zeolite (i.e., the hexagonal counterpart of the cubic zeolite Y)9 in our previous separate studies. No characteristic diffraction peaks attributed to CuOx species were observed even in the samples with a copper species loading of 12.9–13.1 wt% (Table 1). This can be either related to the high distribution of CuOx species on the supports or the particle size of copper species that are too small to exceed the XRD detection limit.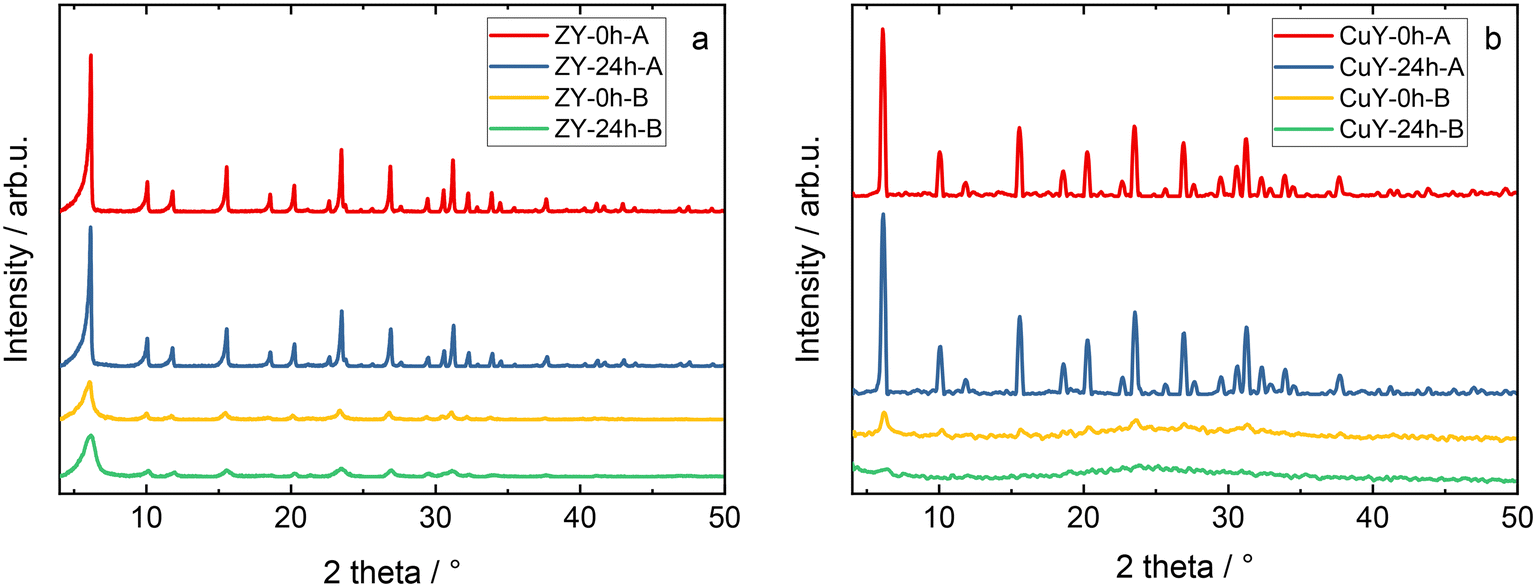 | ||
| Fig. 1 XRD patterns of a) zeolite Y samples and b) its copper-containing form. The sample labels are the same as in Table 1. | ||
| Sample | Average particle size/nm | ω Al/wt% | ω Si/wt% | ω Na/wt% | ω Cu/wt% | n(Si)/n(Al) | n(Cu)/n(Al) |
|---|---|---|---|---|---|---|---|
| ZY-0h-A | 2676 | 13.9 | 22.9 | 11.1 | — | 1.58 | — |
| ZY-24h-A | 475 | 15.2 | 20.7 | 12.2 | — | 1.31 | — |
| ZY-0h-B | 345 | 14.4 | 20.0 | 11.8 | — | 1.33 | — |
| ZY-24h-B | 128 | 13.2 | 20.8 | 11.1 | — | 1.51 | — |
| CuY-0h-A | — | 10.0 | 18.8 | 3.3 | 8.7 | 1.81 | 0.37 |
| CuY-24h-A | — | 10.2 | 20.8 | 2.7 | 9.9 | 1.96 | 0.41 |
| CuY-0h-B | — | 13.0 | 19.7 | 2.8 | 12.9 | 1.46 | 0.42 |
| CuY-24h-B | — | 10.0 | 19.7 | 3.1 | 13.1 | 1.89 | 0.56 |
The particle size distributions of the crystals were determined based on SEM micrographs (Fig. 2). They are influenced by the applied preparation methods, in particular the aging step at room temperature. The average particle sizes (Table 1) of the zeolite Y samples that were subjected to hydrothermal treatment without aging (i.e., ZY-0h-A and ZY-0h-B) are larger than the particle sizes of the samples aged at room temperature. These results are in agreement with the study of Ginter et al.,10 which showed that increasing the aging time leads to a decrease in the average size of the particles of zeolite Y. According to the study conducted by Dabbawala et al.,7 the average particle size of zeolite Y prepared from an initial molar gel composition of 8Na2O:1Al2O3:10SiO2:410H2O is ca. 400 nm. Meanwhile, materials with a particle size of ca. 100 nm were obtained from an initial molar gel composition of 12Na2O:1Al2O3:14SiO2:270H2O. In both cases, the gels were aged for 24 h at room temperature. In addition, in our work, for both preparation methods, we prepared the materials without the aging step at room temperature. The zeolite Y prepared without the aging step (ZY-0h-A) has an average particle size of 2676 nm, while for ZY-24h-A, we obtained an average particle size of 475 nm, meeting the assumption that the aging process leads to a decrease in the average particle size of the synthesized zeolite Y. ZY-0h-B, prepared without aging, has an average particle size of 345 nm in contrast to ZY-24h-B with an average crystal size of 128 nm (Table 1) as predicted from the research of Dabbawala et al.7 (i.e., the increase in particle size is due to the increase in water content in the precursor suspension).
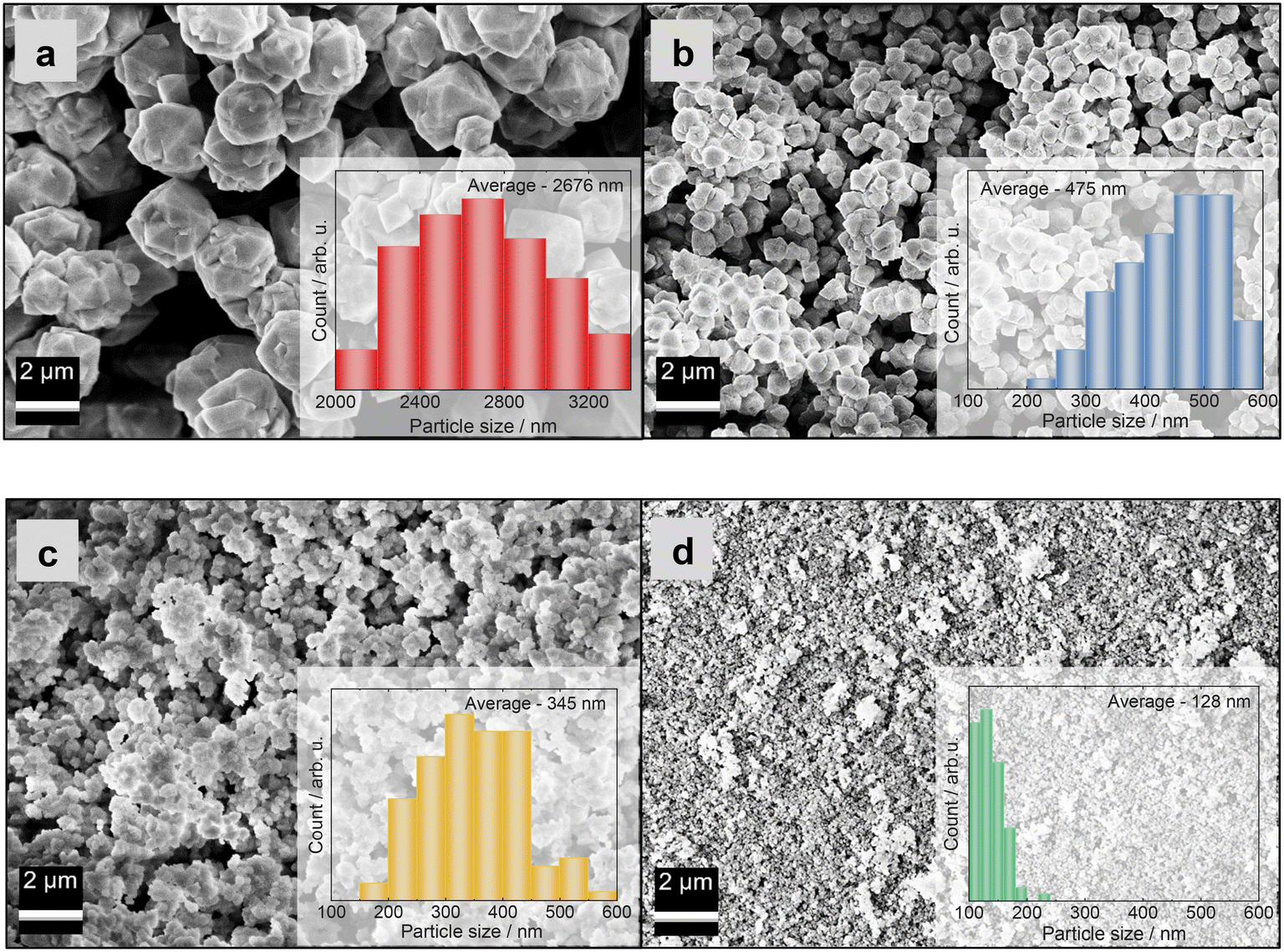 | ||
| Fig. 2 SEM images of zeolite Y samples: a) ZY-0h-A, b) ZY-24h-A, c) ZY-0h-B and d) ZY-24h-B. The particle size distributions (inlays in a–d) were obtained by measuring 100 particles from SEM images. | ||
Fig. 2 and 3 show the surface morphology of the prepared zeolite Y and copper-containing zeolite Y samples investigated by applying SEM and TEM analyses. As shown in Fig. 2a and b, a certain degree of intergrowth of the crystals is observed in the ZY-0h-A and ZY-24h-A samples. Still, most of the particles exhibit discernible crystal faces and well-defined edges; meanwhile in Fig. 2c and d and SI1† aggregations are visible in the ZY-0h-B and ZY-24h-B samples. The aged ZY-24h-A and ZY-24h-B have particle size distributions in the range of 150–600 nm and 80–200 nm, respectively, while ZY-0h-A and ZY-0h-B (which were not aged at room temperature) have particle size distributions in the range of 2000–3400 nm and 150–500 nm, respectively. The particle size distributions were derived from ImageJ software and are shown in Fig. 2(a–d). We also performed STEM analysis in both series of samples A and B (Fig. SI2†). The distribution and dispersion of copper species within the zeolite Y particles are clearly shown. The TEM image of CuY-0h-B (Fig. 3a) revealed faujasite (FAU) particles with a visible porous network. In addition, intergrown planes corresponding to the boundaries between intergrown crystalline domains can be distinguished. Depending on the particle orientation, the two-dimensional projection of the crystal on the TEM micrograph is observed as bi-pyramidal, isometric or even plate-like (Fig. 3b and c). The edges of these particles seem blurry and without any particular ordering. However, in the interior are visible evenly distributed voids having dimensions that coincide with the diameter of the pore opening of the FAU-type zeolite framework supercage. The arrangement of these voids corresponds to the ABC stacking of the FAU-sheets confirming the X-ray findings that some fraction of the zeolite Y crystalline structure is preserved and the material is partially amorphous. This observation indicates that the amorphization of the CuY-0h-B sample occurs at the external surface of the zeolite Y particles, yet the interior remains rather highly ordered.
 | ||
| Fig. 3 Magnified TEM images of CuY-0h-B: a) 100.000× magnification, b) 125.000× magnification, and c) 250.000× magnification. | ||
Table 2 shows the results of the analysis of the chemical composition of the prepared zeolite Y and copper-containing zeolite Y samples measured with ICP-OES for the elements Al, Si, Na, and Cu. The n(Si)/n(Al) ratio obtained for Na-containing zeolites varies in the range of 1.31–1.58 without any clear trend between the applied preparation methods. A relatively high amount of Na ions was also reported in our previous studies over Cu-containing zeolite Y samples.4 CuY-0h-A and CuY-24h-A have higher ratios of 1.81 and 1.96, respectively, compared to CuY-0h-B and CuY-24h-B (n(Si)/n(Al) of 1.46–1.89). In both series of samples, there is an increase of n(Si)/n(Al) upon ion-exchange with Cu species, documenting partial dealumination during Cu-modification. Interestingly, the dealumination process is more pronounced for the samples of series A than that for B as evidenced by the degree of change in the n(Si)/n(Al) ratio. In accordance with their higher Al content, samples CuY-0h-B and CuY-24h-B have higher copper species loadings. An additional reason for this could be the smaller particle size for the B-series of samples facilitating the ion-exchange process by enhancing the contact surface. Moreover, the partial amorphization of the B-materials during the process of exchange with the Cu precursor leading to the production of a high number of silanol sites on the external surface results in retaining of Cu ions on the catalyst's surface. Fig. SI3† demonstrates the correlation between the amount of amorphous material and the silanol groups. Specifically, according to XRD findings, sample CuY-24h-A has a significantly higher ordering degree than CuY-24h-B. The OH group band regions in the respective IR spectra depict several well-defined bands in CuY-24h-A, such as isolated internal silanols (3730 cm−1), Si–(OH)–Al groups located in supercages (3635 cm−1) and hexagonal prisms at a lower frequency mixed with silanols nests. Furthermore, the relatively intense band of Cu2+–OH groups (3680 cm−1) indicates that the process of ion exchange (introduction of isolated exchangeable Cu2+ cations) was also accompanied by the process of hydrolysis. CuY-24h-B presents a band at 3745 cm−1 (attributed to external silanols) and a broad band in the range 3700–3250 cm−1 having a maximum at 3550 cm−1 (attributed to silanols nests). This intense and broad absorption overshadows the expected contributions of residual Si(OH)Al groups and presumably OH groups associated with copper ions. Furthermore, the materials prepared with an aging step at room temperature reveal a higher copper species content than the zeolite Y materials harvested from the freshly treated hydrogel. This could also occur due to smaller particles of the end product in the aged systems.
| Sample | A S(BET)/m2 g−1 | V (TOT)/cm3 g−1 | V (MIC)/cm3 g−1 | V (MES)/cm3 g−1 |
|---|---|---|---|---|
| ZY-0h-A | 854 | 0.32 | 0.32 | 0.00 |
| ZY-24h-A | 824 | 0.31 | 0.30 | 0.01 |
| ZY-0h-B | 680 | 0.34 | 0.28 | 0.06 |
| ZY-24h-B | 685 | 0.36 | 0.35 | 0.01 |
| CuY-0h-A | 714 | 0.28 | 0.28 | 0.00 |
| CuY-24h-A | 741 | 0.30 | 0.28 | 0.02 |
| CuY-0h-B | 276 | 0.20 | 0.12 | 0.08 |
| CuY-24h-B | 174 | 0.26 | 0.17 | 0.09 |
Fig. 4 shows the N2 sorption isotherms and pore width distributions of zeolite Y samples and its copper-containing forms. The Na-containing zeolite Y samples exhibit isotherm type I, indicating the presence of micropores, or a combination of isotherm types I and IV, typical of co-present mesopores and micropores.11 Analogously, the respective Cu-containing forms show type I or a combination of types I and IV isotherms. However, after ion-exchange with copper species, less N2 is adsorbed in copper-containing zeolite Y samples. Usually, this suggests pore blockage by extra-framework species and/or structure degradation (amorphization) as evidenced by XRD. In particular, for CuY-24h-B, the isotherm shape is very different form that of the pristine material. An increase of the adsorbed volume at higher relative pressures is clearly visible. It should be noted that this is probably due to complicated partly constricted pore networks with pore blocking and/or cavitation.12 A significantly lower specific surface area (Table 2) was obtained for the materials prepared in the second set of Na-containing zeolite Y samples (i.e., from an initial composition of 12Na2O:1Al2O3:14SiO2:270H2O). A decrease in the total pore volume for all the copper-containing zeolite Y samples also appears to arise due to pore blockage and/or amorphization after modification with copper species.4,13,14
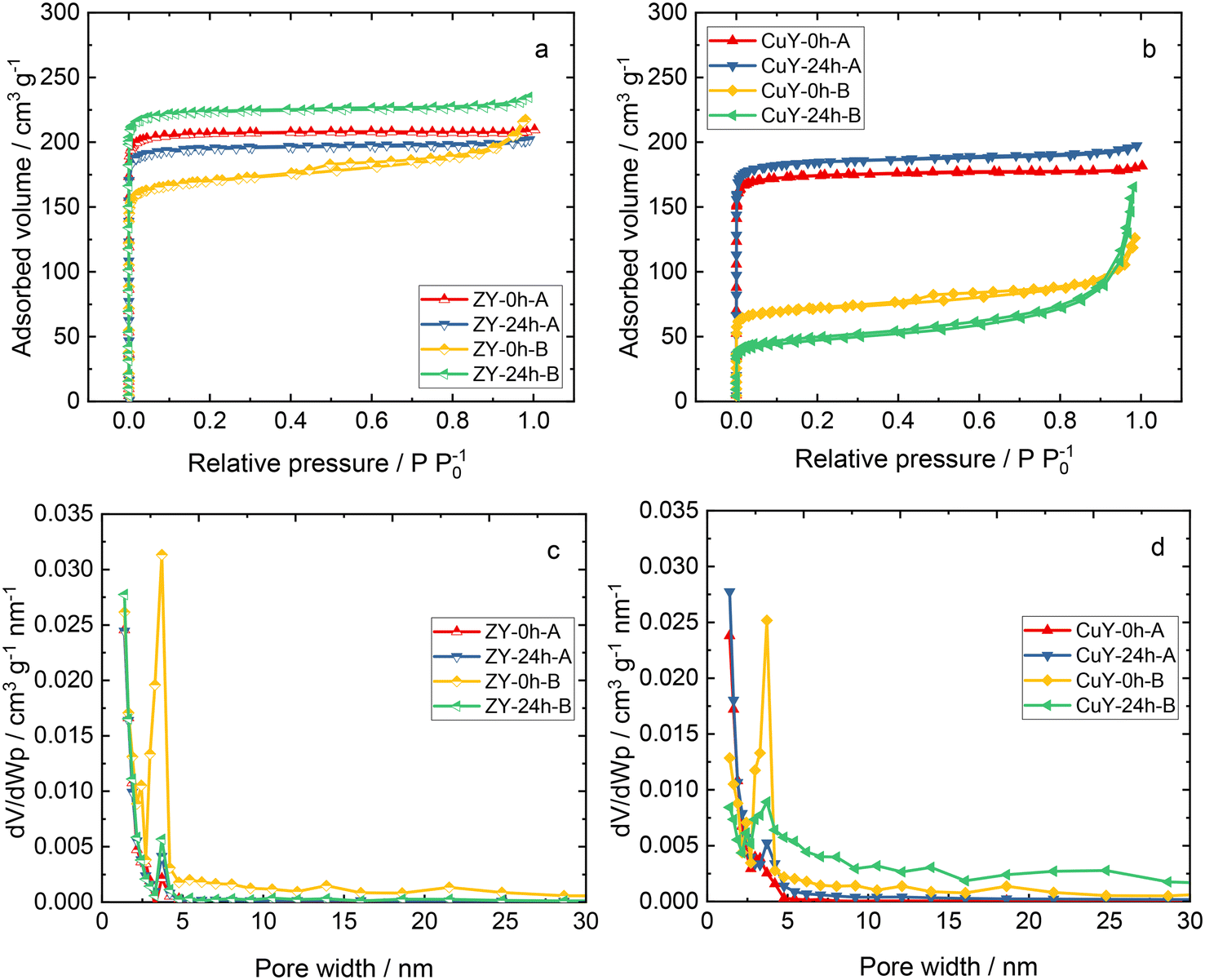 | ||
| Fig. 4 a and b) N2 sorption isotherms and c and d) pore width distributions of zeolite Y samples and its copper-containing forms. | ||
3.2 Nature of copper species
Fig. 5a shows the DR UV-vis spectra of copper-containing zeolite Y samples (calcined at 550 °C for 4 h in static air). The absorption bands at high energy are due to oxygen-to-metal charge transfer related to Cu2+ ions stabilized in the zeolite framework, while the d–d transitions related to hydrated Cu2+ ions are characterized by a broad band below 20000 cm−1.15–17 The reducibility of the copper-containing zeolite Y samples is evaluated via TPR-H2. According to the literature,18,19 Cu species in zeolites are reduced via two main reduction steps, the reduction of Cu2+ to Cu+ and the reduction of Cu+ to Cu0. For instance, Wang et al.16 reported a reduction of isolated Cu2+ to Cu+ below 300 °C, and the reduction of CuOx to Cu0 and Cu+ ions to metallic Cu above 300 °C over copper-containing zeolite Y samples. The reduction of dimeric [Cu–O–Cu]2+ species is controversially discussed in the literature, while it is also believed to be reduced below 300 °C.4 Besides information on reducibility, some authors ascribed the reduction properties of copper species to their position in the zeolite. For instance, the reduction peaks below 300 °C are attributed to the reduction of copper species located in the super-cage of the FAU-type framework. The reduction peaks above 300 °C are assigned to the reduction of copper species in sodalite cavities of zeolite Y (e.g., ref. 20 and 21). In our case in Fig. 5b, the reduction peaks vary in shape and reduction temperatures. CuY-0h-A and CuY-24h-A exhibit reduction peaks centered at about 220 and 320 °C, respectively, while in the case of other materials, i.e., CuY-0h-B and CuY-24h-B, the reduction peaks are shifted to higher temperatures (centered at ca. 290 °C and 420 °C, respectively). Indeed, CuY-0h-B and CuY-24h-B contain higher amounts of copper species (based on ICP-OES analysis), thus, higher amounts of aggregated species (i.e., revealing lower reducibility) are expected. This suggests that the preparation method of zeolite Y, resulting in different numbers of framework Al atoms and silanol groups, influences the nature of the Cu species incorporated into the zeolite structure.
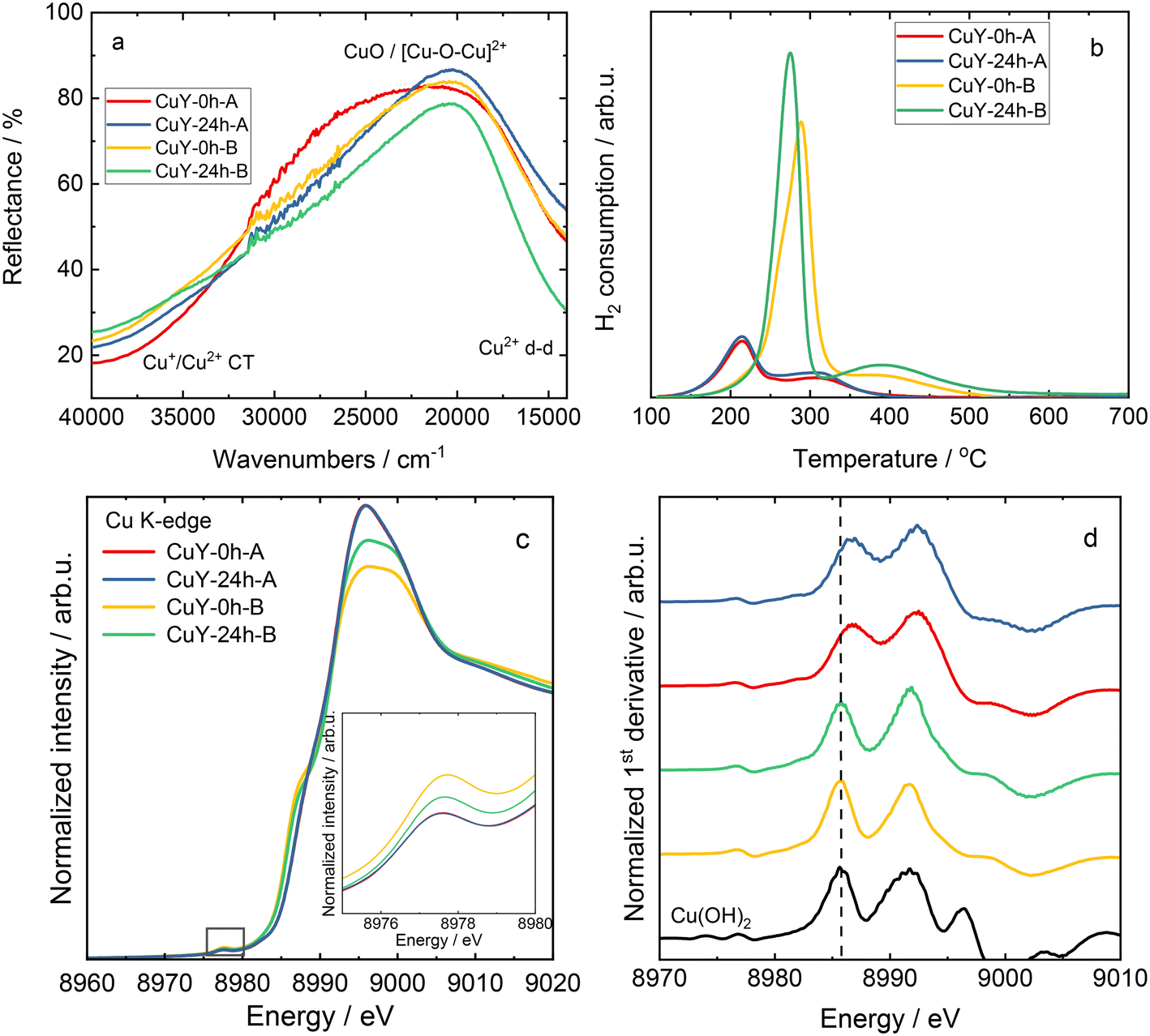 | ||
| Fig. 5 a) DR UV-vis spectra and b) TPR-H2 profiles for copper-containing zeolite Y samples, as well as c) Cu K-edge XANES spectra of copper-containing zeolite Y samples, with the pre-edge region emphasized in the inset, and d) Cu K-edge first derivative spectra; the sample labels in c) and d) are the same. | ||
To further confirm the nature of copper species, X-ray absorption near-edge structure (XANES) spectra analysis was performed. The normalized copper K-edge XANES spectra collected for Cu reference compounds (Cu foil, Cu2O, CuO and Cu(OH)2) are shown in Fig. SI4.† In all the samples, the rising edge feature located in the range of 8979–8987 eV is assigned to dipole-allowed transitions of an electron from the 1s orbital to a 4p orbital (1s → 4p transitions).22 The absorption maxima above the edge are due to multiple scattering, and their intensity and position depend on the local geometry of the material. Typically, the rising-edge energy positions of transition metal K-edges are used as signatures of the oxidation state of the absorbing metal center.23 The energy position of the absorption edge of the XANES spectra collected for reference materials was determined as the maximum of the first derivative. As shown in Fig. SI4b,† the edge positions exhibit the expected increasing trend with the copper oxidation state. However, Cu reference compounds with a formal 2+ oxidation state display different absorption edge positions depending on the ligand environment (coordination number and symmetry). In particular, Cu(II) species in an octahedral symmetry, that is in Cu(OH)2, exhibits larger edge energy than CuO with a tetragonal symmetry. In addition, a weak pre-edge feature (see the inset in Fig. SI4a†) is observed at 8978.2 and 8977.8 eV for CuO and Cu(OH)2, respectively. This pre-edge feature is attributed to the 1s → 3d quadrupole transition. Although these transitions are forbidden by dipole selection rules, the pre-edge peak is observed due to 3d–4p mixing and direct quadrupolar coupling.23 The pre-edge feature is indicative of Cu(II) species and is not observed in Cu2O since the 1s → 3d transition does not occur for the fully occupied d orbitals (d10) in Cu(I) species.
The X-ray absorption near edge structure is sensitive to the chemical state and thus was used to further verify the nature of the Cu species in zeolite Y samples. It should be noted that due to the heterogeneity of Cu species as well as the presence of nano-sized particles, standard reference materials (Cu, Cu2O, CuO and Cu(OH)2) were used as a qualitative approximation for Cu speciation in copper-containing zeolite Y samples in the XAS analysis. Fig. 5c and d show the normalized XANES spectra, along with the corresponding first derivative curves, collected for the materials. In the case of samples prepared with the same method, the collected XANES spectra display virtually identical spectral profiles. Nevertheless, differences in the spectral shapes and intensities are observed between the zeolites prepared with various treatments. All samples exhibit a distinctive 1s → 3d pre-edge peak at 8977.6–8977.7 eV, which can be used as a fingerprint of the electronic structure of 3d9 Cu(II) species formed during the ion-exchange process. Moreover, the collected results, i.e., the absence of an intense feature at ca. 8982 eV and the position of the absorption edge (determined from the maximum of the first derivative), implied that no detectable Cu(I) species existed in the prepared samples. As depicted in Fig. 5c, the XANES spectra collected for copper-containing zeolite Y samples from the A-series show characteristic spectral features of hydrated Cu2+ ions. In particular, a weak pre-edge feature (8977.6 eV) and a high-intensity white line (ca. 8995.9 eV) are typical of Cu2+ species in a highly coordinated form, with a combination of water, framework oxygen and OH groups in a coordination sphere. This is in accordance with the literature data for copper-containing zeolites prepared by an aqueous ion-exchange method (e.g., ref. 22 and 24–26). On the other hand, for the B-series of materials, a prominent rising edge feature is observed at ca. 8987.6 eV. This is accompanied by a lower intensity of the white line and slight enhancement of the pre-edge feature compared to the spectra of A-series of samples. This might be explained by small changes in covalency or lowering of the coordination symmetry of the copper sites due to the dehydration and formation of oxygenated species.27,28
Continuous-wave electron paramagnetic resonance (CW-EPR) spectroscopy at the X-band frequency (9.4 GHz) has been used extensively to provide information about the oxidation state, structure, and quantity of isolated Cu2+ paramagnetic centers in copper-containing zeolites (e.g., X, Y, ZSM-5 and SAPO-34).29–32 In the case of the hydrated (calcined at 550 °C for 4 h in static air) CuY-0h-A and CuY-24h-A samples, only a broad isotropic signal with unresolved hyperfine splitting, a linewidth of about 23 mT, and an isotropic g-value of 2.164 (termed as species B) is observed at room temperature (Fig. 6a). At this temperature, the Cu2+ aqueous complexes are reported to rotate inside the cavity of the zeolite framework leading to a fully motional averaged spectrum with a single isotropic line (species B).29,32 However it is transformed into an anisotropic species A* at −196 °C with spin Hamiltonian parameters gzz = 2.390, gxx,yy = 2.074, and Azz = 410 MHz. Spectral simulation and weighing contributions of suggested species A, A* and B for all hydrated samples at two different temperatures (25 and −196 °C) are given in Fig. SI5 and listed in Table SI1.† Larsen et al.33 suggested that species A* as a [Cu(H2O)6]2+ complex located close to the channel intersection (site II). For samples CuY-0h-B and CuY-24h-B we see relatively more complex spectra at room temperature (Fig. 6a) that indicates a superposition of a poorly resolved Cu2+ powder pattern (simulated as species A with gzz = 2.352, gxx,yy = 2.074, and Azz = 404 MHz) with a similar isotropic signal to those observed for CuY-0h-A and CuY-24h-A. Then at a lower temperature (−196 °C), specifically for CuY-24h-B, the spectrum transforms into species A* whereas there is no species transformation in the case of CuY-0h-B which stays satisfactorily described using species A. There is some residual contribution from species B with its isotropic signal to the total Cu2+ EPR spectrum at −196 °C. We assign this species as cupric ion species in high local concentration which are subjected to electron spin exchange processes, leading to the formation of an isotropic signal at the mean g-value of 2.16.
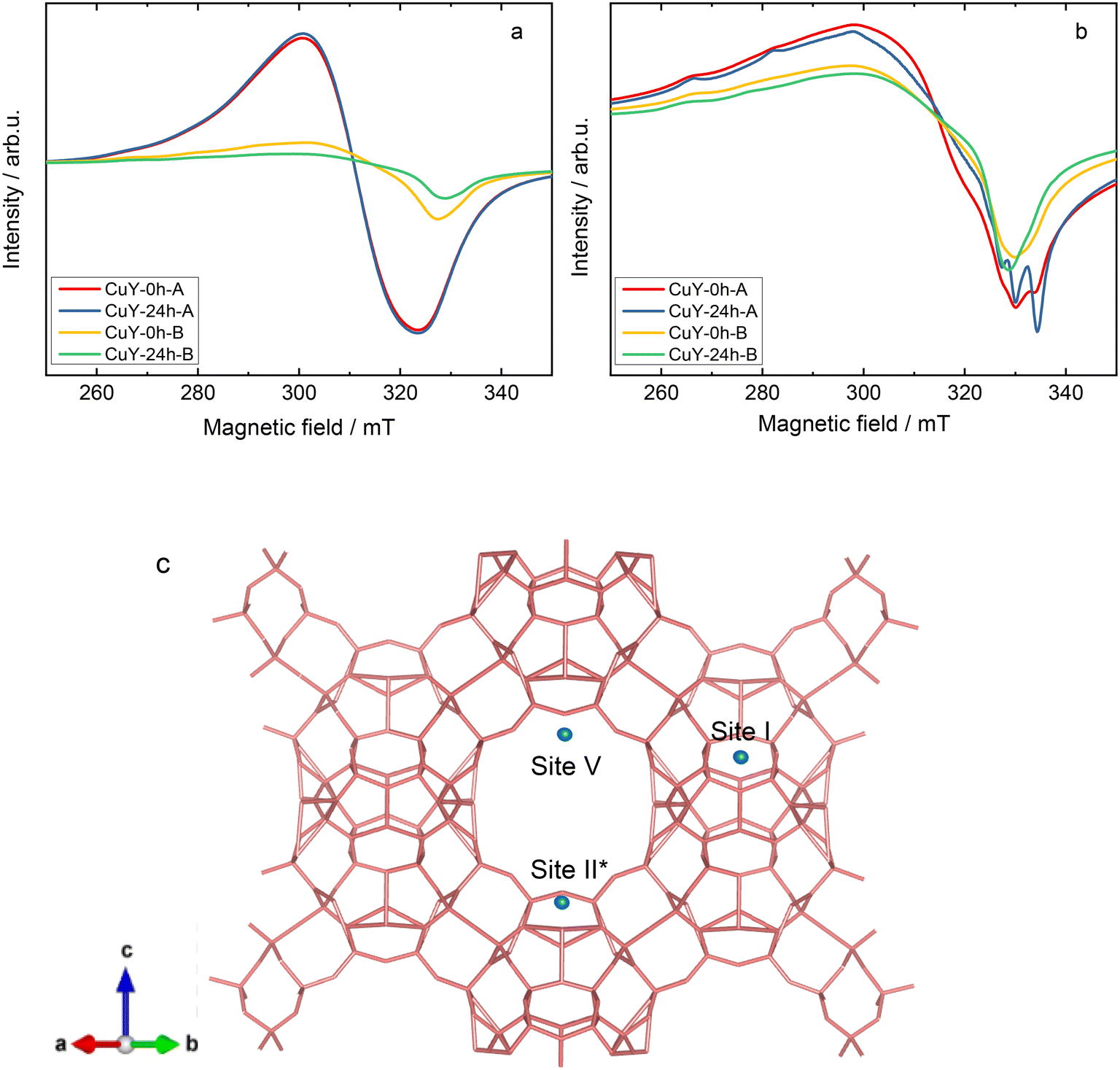 | ||
| Fig. 6 a) EPR spectra of hydrated (calcined at 550 °C for 4 h in static air) copper-containing zeolite Y samples recorded at room temperature (ca. 25 °C), b) spectra of dehydrated (activated at 200 °C for 2 h under vacuum) copper-containing zeolite Y samples recorded at −196 °C, and c) schematic structure of zeolite Y with the possible Cu cation positions shown by the site number based on EPR interpretations. | ||
Comparatively, both hydrated CuY-0h-B and CuY-24h-B have less intensities with a tendency towards anisotropic features due to a more restricted movement of Cu2+ centers. This is probably due to a clustering of Cu2+ on the pores hence reducing the amount of isolated Cu2+ that are EPR active even though their Cu2+ content is slightly higher compared to CuY-0h-A and CuY-24h-A (according to the ICP-OES analysis). The clustering effect may be explained by the fact that if the electron spins from the two Cu2+ centers are very close together, they will commonly couple antiferromagnetically and then give a diamagnetic singlet ground state.31 This result is also in line with N2 sorption analysis (Fig. 4a and b) and TEM images (Fig. SI2c and d†) that support a strong indication of copper aggregates in the hydrated CuY-0h-B and CuY-24h-B.
Upon dehydration of copper-containing zeolite Y samples at 200 °C under vacuum, it can be seen that an isotropic signal was observed when the EPR spectra were recorded at a lower temperature (−196 °C) due to some loss of motional freedom. However, typical Cu2+ hyperfine coupling still can only be partly resolved due to a relatively high concentration of copper species (i.e., >8 wt% obtained from ICP-OES analysis) in these samples hence increasing the line broadening. Spectral simulations of the spectra of the dehydrated samples exhibit the superposition of up to four distinct Cu2+ species in the dehydrated samples. The complete spin Hamiltonian parameters of these four species A, B, C, and D as deduced from the experimental spectra can be found in Table 3. Here it needs to be clarified that the uncertainty of spin Hamiltonian determination is quite high since the spectra suffered from considerable homogeneous line broadening as well as significant g and A strain effects. According to previous EPR studies on copper-containing zeolite Y samples at −196 °C,29,34 species A with gzz = 2.352 and Azz = 404 MHz is assigned in the current study to moieties which correspond to Cu2+ fully coordinated by six water molecules [Cu(H2O)6]2+ while species C with gzz = 2.345 and Azz = 430 MHz can be assigned to Cu2+ complexes coordinated by three water molecules and three oxygen molecules from the zeolite framework [Cu(H2O)3(Of)3]2+. Species B is added into the spectral simulation again as exchanged narrowed Cu2+ species in a high local concentration (ca. giso = 2.16). Species D (i.e., gzz = 2.324 and Azz = 523 MHz) is associated with Cu2+ species that coordinate to six oxygen molecules from zeolitic framework [Cu(Of)6]2+. The spectral simulation for the activated CuY-0h-A and CuY-24h-A samples is shown in Fig. 7, whereas the simulation for CuY-0h-B and CuY-24h-B is provided in Fig. SI6.† Note that all the spectral simulations take into account the contribution of the four different species described in Table 4. According to the weighing contribution for each species in the spectral simulation, species D is the major component for all activated CuY samples while species A and B appear as the minor components. In addition, a contribution of the exchanged narrowed species C was applied to CuY-0h-B sample to improve the spectral simulation. The most probable location of species A in the copper-containing zeolite Y structure is at the super-cage (site V – copper location near the surface of twelve-membered ring channels) since it allows large complexes such as cupric hexahydrates to move freely with less steric hindrance. In the case of species C, a study based on pulse EPR at −196 °C (ref. 34) assigned this [Cu(H2O)3(Of)3]2+ complex at site II* (site II – which is displaced towards the α cage along an axis perpendicular to the hexagonal window in the sodalite cage). Lastly, species D ([Cu(Of)6]2+) is assigned to the center of the hexagonal prism site (site I – copper ions located in the cage units). A schematic representation of cupric sites in Y zeolites can be found in Fig. 6c. Our studies remain in agreement with the data reported by Yu and Kevan34 which provided details about Cu(II) speciation on zeolite Y. Also, Wang et al.16 reported about EPR analysis over copper-containing zeolite samples. Two of the three species (i.e., Cu(II)(H2O)6 and [Cu(H2O)3(Of)3]2+) in our analysis are within the range reported by the authors with consistent assignment of an octahedral configuration. Please note, however, that our current EPR data only focus on Cu species displaced after activation and not interaction with reaction molecules; e.g., the migration of Cu species to super-cages after coming into contact with NH3 was broadly investigated via IR spectroscopy (e.g., ref. 35).
| Samples | Relative EPR intensity | A/% | B/% | C/% | D/% |
|---|---|---|---|---|---|
| CuY-0h-A | 2.8 | 20 | 20 | 0 | 60 |
| CuY-24h-A | 3.1 | 20 | 20 | 0 | 60 |
| CuY-0h-B | 1.3 | 30 | 10 | 20 | 40 |
| CuY-24h-B | 1.0 | 20 | 30 | 0 | 50 |
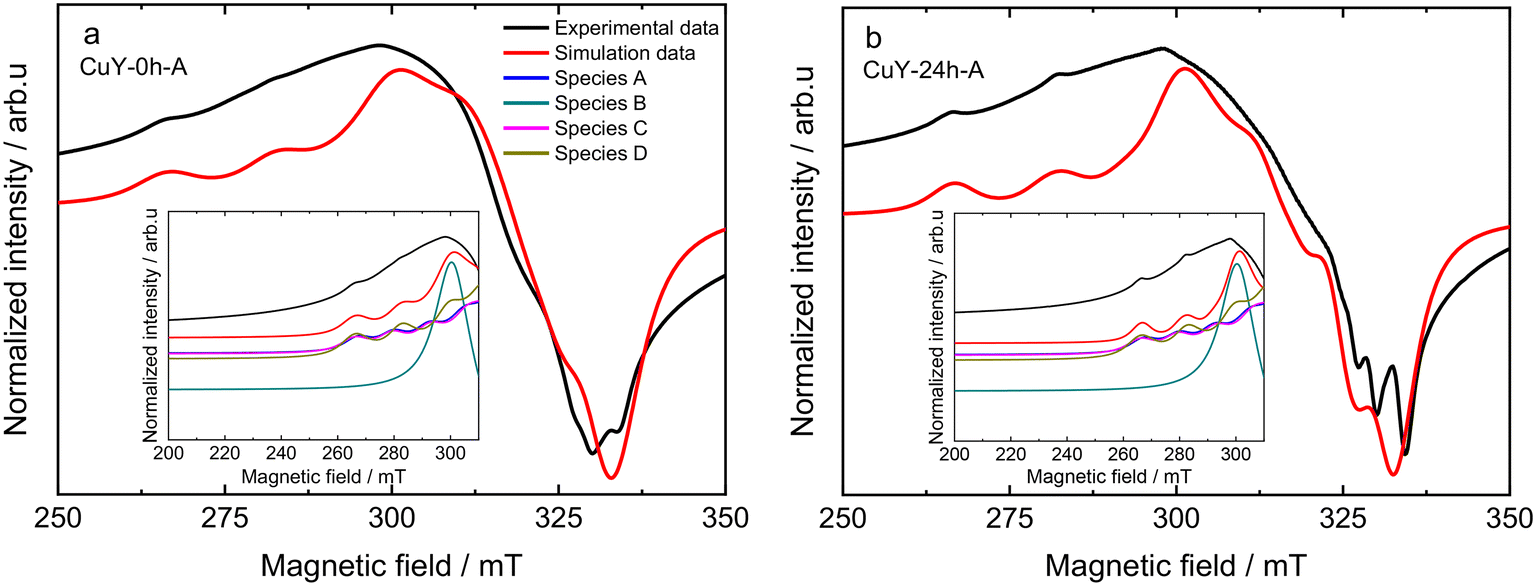 | ||
| Fig. 7 Spectral simulation of activated a) CuY-0h-A and b) CuY-24h-A samples recorded at −196 °C (samples were activated at 200 °C for 2 h under vacuum). The insets show the contributions of different copper species to the simulations. | ||
| Cu species | g xx,yy | g zz | A xx,yy/MHz | A zz /MHz | Lwpp/mT | Assignment |
|---|---|---|---|---|---|---|
| a g iso is an isotropic g-value. | ||||||
| A | 2.074 ± 0.005 | 2.352 ± 0.005 | 30 ± 10 | 404 ± 5 | 5.0 ± 0.3 | Cu(II)(H2O)6 site V |
| Aa | 2.070 ± 0.005 | 2.390 ± 0.005 | 30 ± 10 | 410 ± 5 | 5.0 ± 0.3 | Cu(II)(H2O)6 site II |
| B | 2.16a | — | — | — | — | Cu(II)(H2O)6 site V |
| C | 2.074 ± 0.005 | 2.357 ± 0.005 | 30 ± 10 | 430 ± 5 | 5.0 ± 0.3 | [Cu(H2O)3(Of)3]2+ site IIa |
| D | 2.074 ± 0.005 | 2.352 ± 0.005 | 30 ± 10 | 523 ± 5 | 5.0 ± 0.3 | ([Cu(Of)6]2+) site I |
TPD-NOx is beneficial to investigate the dynamics of the surface during the NO interaction with copper-containing zeolite Y samples (Fig. 8). Qualitative and quantitative analyses of the surface intermediates upon adsorption and interaction of the reagents with the catalysts were performed. However, the profiles varied significantly depending on the desorption conditions, i.e., desorption in 4 vol% O2/He or pure He for both A- and B-series of samples. According to literature reports36,37 as well as our recent studies over copper-containing zeolite Y samples,4 the desorption peak below 250–300 °C corresponds to NO molecules that desorb from isolated Cu+/Cu2+. The peak arising between 300–425 °C is attributed to the thermal decomposition of nitrate and/or nitrite species generated by NO adsorption on dimeric [Cu–O–Cu]2+: 2NO + [Cu–O–Cu]2+ → Cu+–NO + [Cu–NO2]+. The peak centered at around 460–480 °C can be assigned to the desorption of NO2 formed in the reaction between NO and the dimeric [Cu–O–Cu]2+ active sites: NO + [Cu–O–Cu]2+ → NO2 + [Cu–□–Cu]2+ (□ represents the surface oxygen vacancy).4 Concerning the investigated copper-containing zeolite Y samples, however, the oxidation of NO to NO2 can be excluded, since the TPD-NOx results show desorption of NO and NOx in approximately equimolar amounts. Ruggeri et al.38 pointed out that the evolution of NO and NO2 refers to the thermal decomposition of nitrites up to 250 °C, while NO2 decomposition to NO and O2 appears above 370 °C.
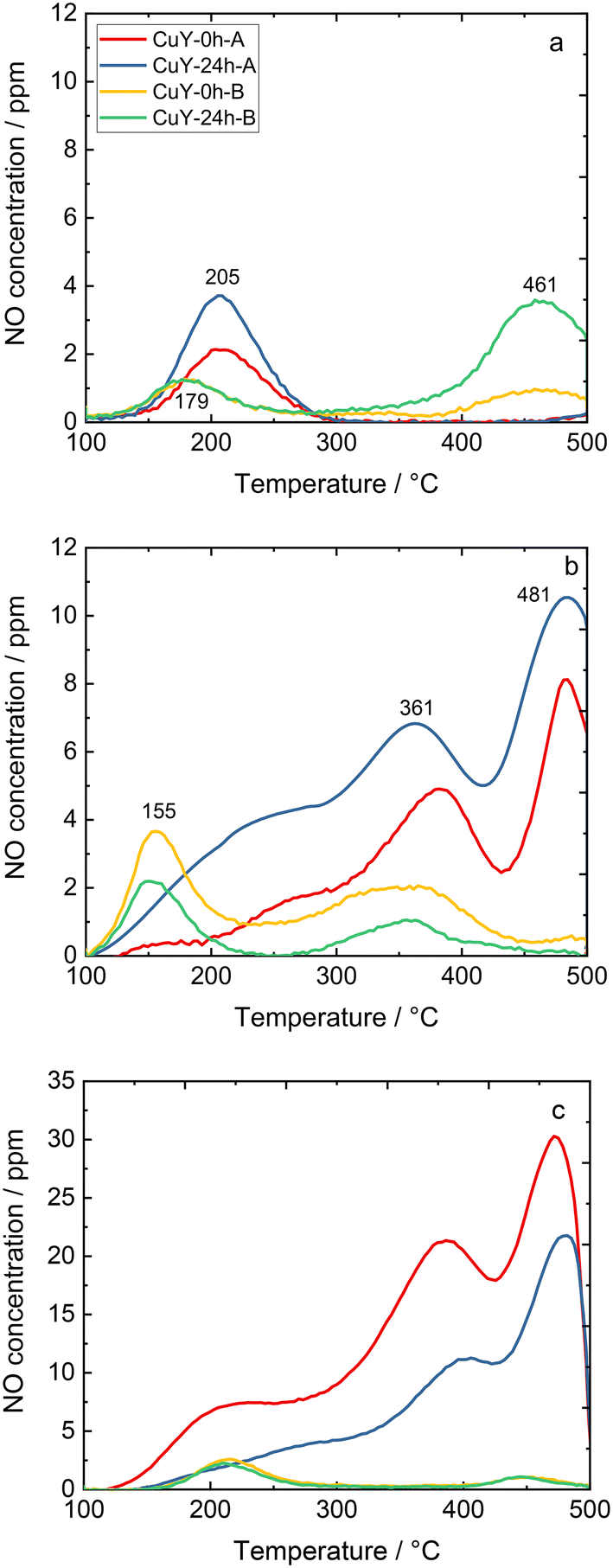 | ||
| Fig. 8 TPD-NOx profiles of copper-containing zeolite Y samples: a) NOx desorption in He (pre-treatment in He), b) NOx desorption in a 4 vol% O2/He mixture (pre-treatment in He), and c) NOx desorption in He (pre-treatment in 5 vol% O2/He); the sample labels are the same for a)–c). | ||
On the other hand, the profile depends strongly on the applied conditions. For example, for CuY-A materials one main NO desorption peak is visible after pre-treatment in He (Fig. 8a), while three distinct peaks can be found after pre-treatment in He and desorption in O2/He (two main peaks can be distinguished with the broad band above 250 °C, Fig. 8b). By quantification, the amount of NO that adsorbed in CuY-A samples was relatively higher than in CuY-B samples (16–29 μmol g−1versus 1–5 μmol g−1), when O2/He pre-treatment was applied first (Fig. 8c). This measurement proves that the higher amount of copper species accommodated in the B-series of zeolite Y does not directly imply a greater number of active sites for NOx adsorption, probably in relation to the formation of aggregates. Indeed, besides the Cu content, the amorphization degree of the zeolite Y framework (after ion-exchange with Cu species) influences the adsorption capacity of NOx molecules. A significantly higher amount of desorbed NO is found for the materials treated under oxidizing conditions, i.e., O2/He, than under reducing conditions, i.e., pure He. To identify the nature of surface species formed under adsorption conditions, FT-IR spectroscopy was carried out. Fig. 9 shows the development of stable adducts of NO with Cu2+ and Cu+ cations within the temperature increase. All the zeolites show bands located at ca. 1945–1890 cm−1 arising from the interaction of NO and exchangeable bare Cu2+ cations, i.e., Cu2+(NO) mononitrosyl complexes.15 From the highest intensity of Cu2+(NO) mononitrosyl bands in vacuum-treated CuY-24h-A, we can conclude that it has the highest population of isolated copper(II) cations while in CuY-24h-B the population of Cu2+ sites is considerably lower (five-fold). The appearance of nitrate and nitrite species (NOx−) in CuY-24h-A and CuY-24h-B results from the interaction of NO with oxo- and hydroxo-copper(II) sites thus the intensities of the bands in the 1700–1300 cm−1 region can be used for the estimation of the relative abundance of isolated Cu2+ and Cu2+oxo sites. Even taking into account the possibility of the participation of a water molecule in the 1645–1630 cm−1 band, the participation of copper(II) oxo-forms in Cu-Y-24h-B is clearly visible. Thus, in Cu-Y-24h-A the dominant speciation form of copper is isolated Cu2+ cations. NOx− appeared however in pure O2-pretreated Cu-Y-24h-A which was accompanied by a significantly decreased intensity of the Cu2+(NO) bands. The lower Cu2+(NO) abundance in Cu-Y-24h-A can be ascribed to evolution upon oxygen treatment of copper-oxo forms that easily react with NO producing NOx−. The latter species are stable up to 350 °C and then decompose, as documented by their bands shrinking at a temperature higher than 350 °C. Consequently, in Cu-Y-24h-A the low-temperature desorption peak in TPD-NOx can be ascribed to desorption of NO from mononitrosyl Cu2+(NO) while the high-temperature peak can be ascribed to NO from the decomposition of NOx− species. In the Cu-Y-24h-B sample the number of Cu2+ accessible to NO increases upon O2 pre-treatment; this is manifested as an upsurge intensity of Cu2+(NO) bands. Mononitrosyls are more resistant to both desorption and transformation to NOx−; some Cu2+(NO) species were preserved even at 250 °C while the NOx− species became more homogenous and now are characterized by the only band at 1374 cm−1 typical of chelating and bridging bidentate nitrito species or bidentate nitrito species.39,40 Similarly, mononitrosyl and NOx− species were recognized as species boosting the activity in NH3-SCR-DeNOx over Cu-containing ZSM-5.41
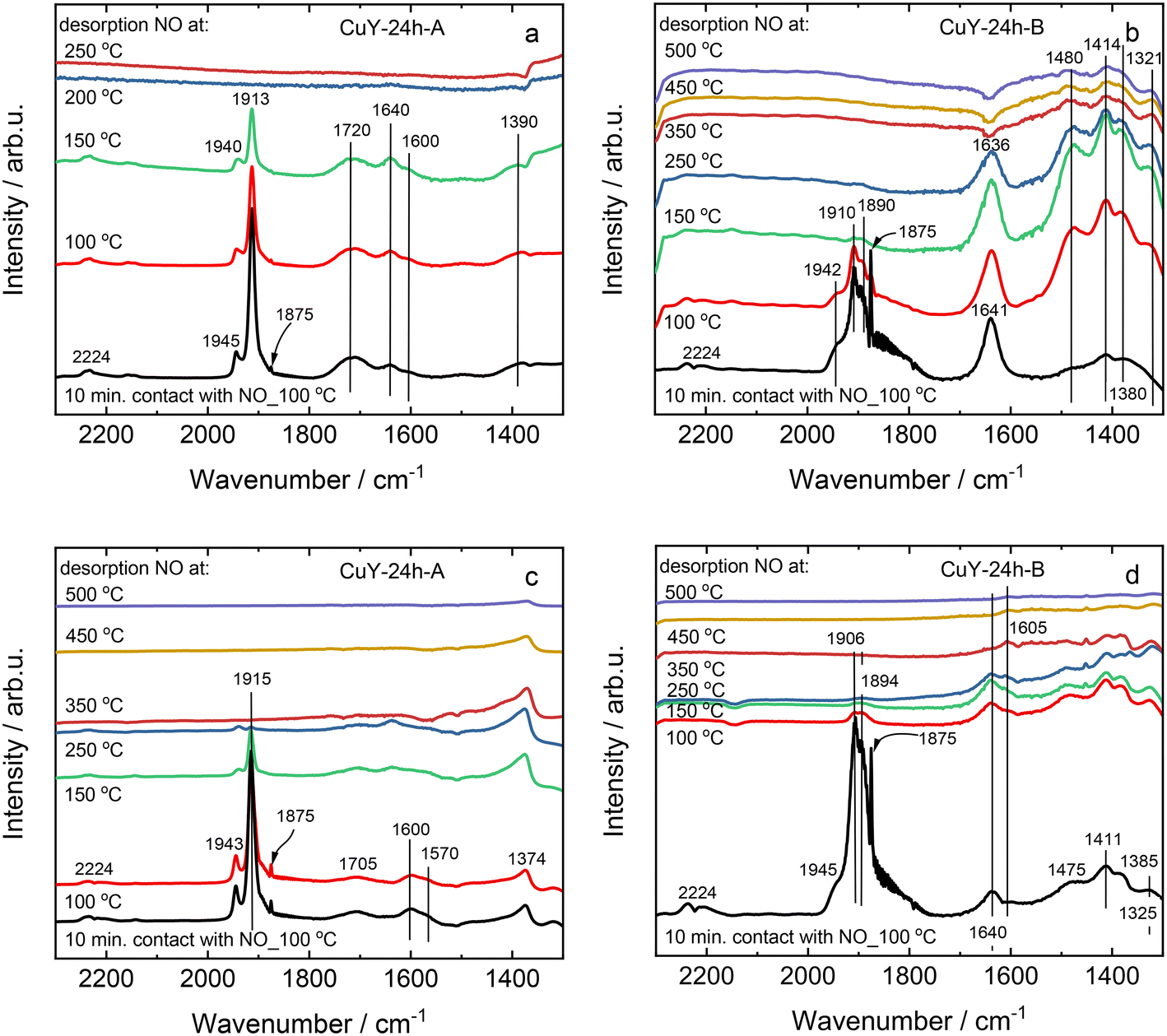 | ||
| Fig. 9 The IR spectra of copper-containing zeolite Y samples under pre-treatment in a and b) vacuum conditions, or c and d) pure O2, after coming into contact with pure NO at 100 °C followed by its desorption at 100–500 °C. | ||
3.3 Catalytic investigation over copper-containing zeolite Y
Fig. 10 shows the catalytic activity experiments over the copper-containing zeolite Y samples in NH3-SCR-DeNOx. The CuY-0h-A and CuY-24h-A catalysts reveal higher NO conversion both without H2O and in the presence of H2O compared to the CuY-0h-B and CuY-24h-B catalysts (Fig. 10a and b). The results of the catalytic studies fully agree with our previous investigations where Cu-24h-A (named as Cu-Y) was already introduced and compared with other Cu-containing zeolite Y catalysts reported in the literature.4 The CuY-24h-A catalyst remains active in five consecutive runs without H2O in the feed (Fig. SI7a†) as well as during five consecutive time-on-stream tests carried out for 24 h at 200 °C in the presence of H2O (Fig. 10e). The structure of zeolite Y is preserved after the five consecutive runs both without H2O and in its presence (Fig. SI8†). For all the materials, the NO conversion decreases above 350 °C due to the competitive NH3 oxidation, which is more pronounced for the CuY-0h-B and CuY-24h-B catalysts. Meanwhile, CuY-0h-A and CuY-24h-A reveal significantly higher N2O yields (without and with H2O in the feed) compared to CuY-0h-B and CuY-24h-B. The N2O yield below 200 °C was <6% for all the samples. The NH3 oxidation results revealed the oxidation of ammonia to N2O over the A-series of zeolite Y-based catalysts (Fig. SI7b†). Fig. 10c and d show the N2O formation in the presence and absence of water vapor for Cu-containing Y catalysts. A significant effect of H2O is observed, especially for the CuY-A samples. Interestingly, different effects are seen at two N2O maxima in the low (ca. 100–300 °C) and high-temperature regions (ca. 300–450 °C). As a comparison, Han et al.42 found that water promotes N2 selectivity via ammonium nitrate decomposition into NH3 and HNO3 forming NO2 instead of N2O. Thus, they observed less N2O formation under NH3-SCR-DeNOx under wet conditions. In our case, the lower N2O formation in the presence of H2O arises due to the competitive adsorption between water vapor and ammonia for the active sites. Nevertheless, a significant amount of N2O is also detected. As the n(Si)/n(Al) ratio governs the copper site speciation further optimization of the catalysts to reduce N2O production will be investigated in the following studies. A significantly higher NO conversion is revealed for the sample pre-treated under oxidizing conditions (Fig. 10f). This result remains in contrast to the data obtained before for Cu-containing SAPO-34, SSZ-13 and ZSM-5 where a higher NO conversion was achieved over Ar-activated samples.43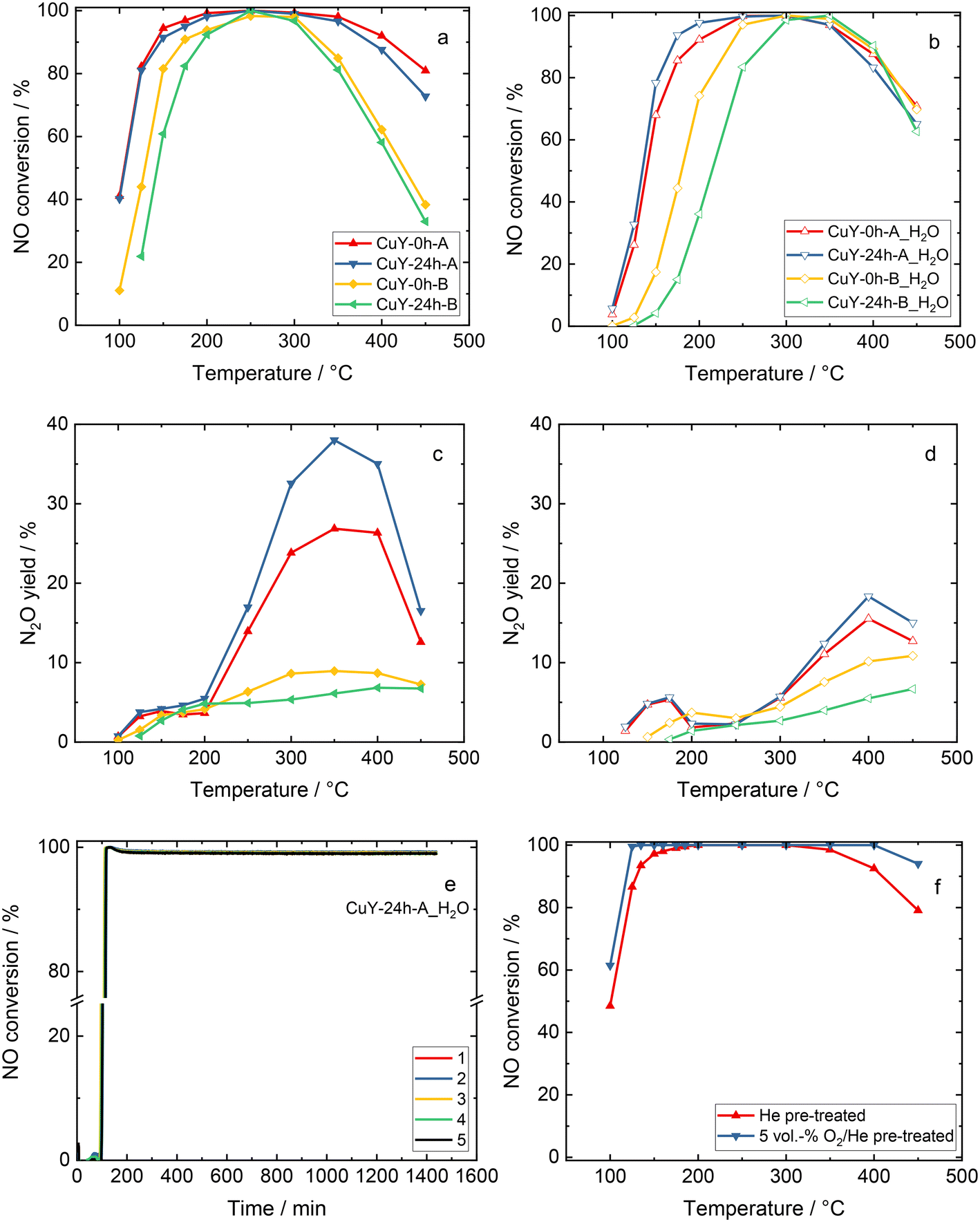 | ||
| Fig. 10 a and b) NO conversion and c and d) N2O yield during NH3-SCR-DeNOx over copper-containing zeolite Y samples. Reaction conditions: mK = 0.2 g; GHSV = 30000 h−1, c(NO) = 500 ppm, c(NH3) = 575 ppm, c(O2) = 4 vol%, (c(H2O) = 5 vol% when used), He balance, FTOT = 120 ml min−1, e) NO conversion over CuY-24h-A for five consecutive runs during the time-on-stream stability tests in the presence of water vapor at 200 °C, and f) NO conversion over CuY-24h-A after pre-treatment in He or 5 vol% O2/He. | ||
The effect of different zeolite particle sizes on the catalytic activity in NH3-SCR-DeNOx was broadly investigated in the case of Cu-CHA. For instance, Gao et al.44 synthesized three types of Cu-SSZ-13 catalysts with different particle sizes (i.e., 0.26 μm, 0.45 μm and 1.3 μm), however, the activities in NH3-SCR-DeNOx over these three catalysts were quite similar to each other. This phenomenon was also attested by Wang et al.45 who showed that the particle size has no effects on NH3-SCR-DeNOx, i.e., similar Cu2+ amounts and distributions in samples with varying particle sizes (0.4–2 μm) should have similar activities as reported for Cu-containing SSZ-13. Similarly, also in our case, the considerable difference in particle size between the CuY-A samples (ca. 2676 versus 475 nm) did not enhance the NO reduction capabilities.
Fig. 11 shows the in situ DR UV-vis spectra measured on sample CuY-24h-A in the NH3-SCR-DeNOx mixture, after pre-treatment in pure He. The spectrum measured at 100 °C before sending the NH3-SCR-DeNOx mixture is characterized by a relatively intense band at ca. 12000 cm−1 related to the d–d transitions of Cu2+ ions, with the corresponding intense oxygen-to-metal charge transfer absorption at higher energy. A broad and very weak absorption is observed on the tail of the oxygen-to-metal charge transfer, between 27000 and 20000 cm−1, which could be related to a very small amount of mono or dinuclear CuxOy complexes as observed in other copper-containing zeolites (e.g., ZSM-5, mordenite and chabazite).17,28,46 Exposure of the NH3-SCR-DeNOx mixture at 100 °C causes a progressive increase and change of shape of the d–d band (now centered at 12500 cm−1 with an evident component at 15600 cm−1), and an important change above 20000 cm−1. Specifically, the oxygen-to-metal charge transfer absorption shifts to higher energy and an evident component is formed with the maximum at around 26600 cm−1. A similar component was interpreted by Oda et al.46 on Cu-CHA in terms of amino solvated (μ-η2,η2-peroxo) dicopper(II) complexes proposed as active species in the low temperature NH3-SCR-DeNOx.47–49 Interestingly, this feature is gradually consumed during the NH3-SCR-DeNOx at 200 °C, when the NO conversion of the catalyst passes from 40% to almost 100% (Fig. 10), suggesting an involvement of related Cu sites in the low temperature reactivity. The changes observed by increasing the temperature to 300 °C are mainly found in the d–d region, where the d–d band decreases in intensity and moves back to ca. 11600 cm−1. More evident changes are seen on heating the sample in the NH3-SCR-DeNOx mixture at 400 and 500 °C, where the catalyst shows a decrease in NO conversion and an important increase in N2O production. This would imply the involvement in the NH3 oxidation reaction of different CuxOy sites, tentatively ascribed to the large absorption centred at around 22500 cm−1. The NO conversion measured in the in situ DR UV-vis set-up (Fig. SI9†) is in good agreement with the data measured in the catalytic set-up (Fig. 10), except for a lower conversion at 100 °C.
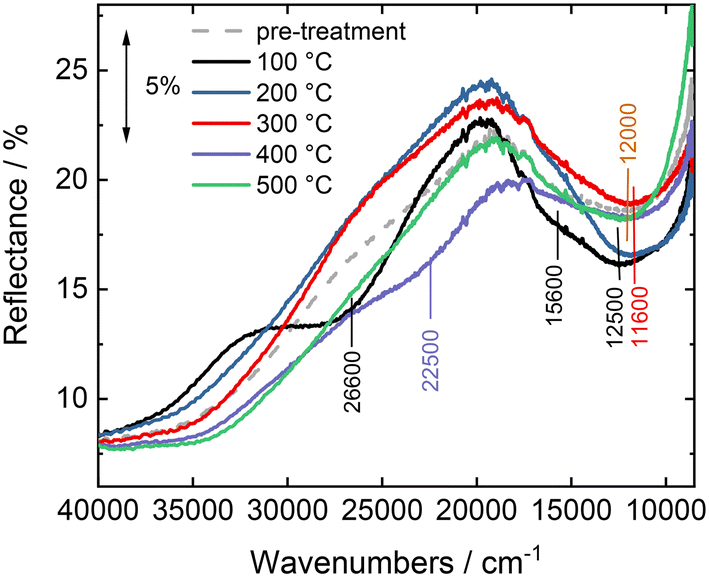 | ||
| Fig. 11
In situ DR UV-vis spectra of CuY-24h-A studied during NH3-SCR-DeNOx at increasing temperature. The spectra were obtained after 30 min of contact with the NH3-SCR-DeNOx at the set temperature. Reaction conditions: mK = 0.1 g; GHSV = 30000 h−1, c(NO) = 500 ppm, c(NH3) = 575 ppm, c(O2) = 4 vol%, He balance, and FTOT = 120 ml min−1. | ||
4. Conclusions
The effect of the preparation method on the catalytic properties of copper-containing zeolite Y samples applied for NH3-SCR-DeNOx was investigated. As a result, materials with varied particle sizes (approx. 128 or 2676 nm) and thus different copper species were obtained. However, even the considerable difference in particle size between the A-series of samples (ca. 2676 for Cu-Y-24h-A versus 475 nm for Cu-Y-0h-A) did not enhance the NO reduction capabilities. In the A-series of samples, the dominant speciation form of copper is isolated Cu2+ cations, while the presence of copper(II) oxo-forms in the B-series of samples is clearly visible. The CuY-0h-A and CuY-24h-A catalysts showed a higher NO conversion both in the presence and absence of water, but they also produced more N2O at temperatures above 250 °C. The enhanced activity of Cu-containing zeolite Y in NH3-SCR-DeNOx is correlated with the formation of mononitrosyl and NOx− species appearing in the SCR reduction half-cycle as well as the μ-η2,η2-peroxo dicopper(II) complexes in the SCR oxidation half-cycle.Author contributions
Rujito S. R. Suharbiansah: conceptualization, methodology, investigation, data curation, writing – original draft, and writing – review & editing. Muhammad Fernadi Lukman: investigation, data curation, and writing – review & editing. Chiara Nannuzzi: investigation and data curation. Anna Wach: investigation, data curation, and writing – review & editing. Kinga Góra-Marek: investigation, data curation, and writing – review & editing. Michael Liebau: investigation and data curation. Ana Palčić: writing – review & editing. Gloria Berlier: review. Silvia Bordiga: review. Andreas Pöppl: review. Roger Gläser: review. Magdalena Jabłońska: conceptualization, methodology, investigation, data curation, writing – original draft, writing – review & editing, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. R. S. acknowledges the DAAD scholarship programme. M. J. acknowledges a DFG Research Grant (JA 2998/2-1). A. P. acknowledges the funding from the Croatian Science Foundation under the project UIP-2019-04-4977. M. F. L. acknowledges funding from the DFG (Project ID 443871192, GRK 2721: Hydrogen Isotopes 1,2,3). H. K. G.-M. acknowledges Grant 2021/41/B/ST4/00048 from the National Science Centre, Poland. The authors would like to thank the Paul Scherrer Institut, Viligen, Switzerland for the provision of the synchrotron radiation facility at the SuperXAS beamline of the SLS.References
- Regulation (EC) No 715/2007 of the European Parliament and of the Council, Available online: https://eur-lex.europa.eu/eli/reg/2007/715/oj (accessed on 20 December 2022) Search PubMed.
- M. Jabłońska, RSC Adv., 2022, 12, 25240–25261 RSC.
- J. H. Kwak, D. Tran, S. D. Burton, J. Szanyi, J. H. Lee and C. H. Peden, J. Catal., 2012, 287, 203–209 CrossRef CAS.
- R. S. Suharbiansah, K. Pyra, M. Liebau, D. Poppitz, K. Góra-Marek, R. Gläser and M. Jabłońska, Microporous Mesoporous Mater., 2022, 334, 111793 CrossRef CAS.
- A. Palčić, P. C. Bruzzese, K. Pyra, M. Bertmer, K. Góra-Marek, D. Poppitz, A. Pöppl, R. Gläser and M. Jabłońska, Catalysts, 2020, 10, 506 CrossRef.
- M. Jabłońska, K. Góra-Marek, M. Grilc, P. C. Bruzzese, D. Poppitz, K. Pyra, M. Liebau, A. Pöppl, B. Likozar and R. Gläser, Catalysts, 2021, 11, 843 CrossRef.
- A. A. Dabbawala, V. Tzitzios, K. Sunny, K. Polychronopoulou, G. Basina, I. Ismail, V. Pillai, A. Tharalekshmy, S. Stephen and S. M. Alhassan, Surf. Coat. Technol., 2018, 350, 369–375 CrossRef CAS.
- J. M. Ortigosa, C. W. Lopes, G. Agostini, A. E. Palomares, T. Blasco and F. Rey, Microporous Mesoporous Mater., 2021, 323, 111230 CrossRef.
- V. Georgieva, A. Vicente, C. Fernandez, R. Retoux, A. Palčić, V. Valtchev and S. Mintova, Cryst. Growth Des., 2015, 15, 1898–1906 CrossRef CAS.
- D. M. Ginter, A. T. Bell and C. J. Radke, Zeolites, 1992, 12, 742–749 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- P. Sharma, J. S. Song, M. H. Han and C. H. Cho, Sci. Rep., 2016, 6, 22734 CrossRef CAS PubMed.
- R. A. Shutilov, G. A. Zenkovets, E. A. Paukshtis and V. Y. Gavrilov, Kinet. Catal., 2014, 55, 243–251 CrossRef CAS.
- J. Chen, G. Peng, T. Liang, W. Zhang, W. Zheng, H. Zhao, L. Guo and X. Wu, Nanomaterials, 2020, 10(11), 2170 CrossRef CAS PubMed.
- K. A. Tarach, M. Jabłońska, K. Pyra, M. Liebau, B. Reiprich, R. Gläser and K. Góra-Marek, Appl. Catal., B, 2021, 284, 119752 CrossRef CAS.
- H. Wang, R. Xu, Y. Jin and R. Zhang, Catal. Today, 2019, 327, 295–307 CrossRef CAS.
- C. Negri, M. Signorile, N. G. Porcaro, E. Borfecchia, G. Berlier, T. V. Janssens and S. Bordiga, Appl. Catal., A, 2019, 578, 1–9 CrossRef CAS.
- Y. Wang, Z. Liu, C. Tan, H. Sun and Z. Li, RSC Adv., 2020, 10, 3293–3300 RSC.
- L. Yan, T. Fu, J. Wang, N. Narkhede and Z. Li, J. Chem. Res., 2020, 44, 710–720 CrossRef CAS.
- S. Kieger, G. Delahay, B. Coq and B. Neveu, J. Catal., 1999, 183, 267–280 CrossRef CAS.
- J. Wan, H. Yang, Y. Shi, Y. Liu, J. Zhang, J. Zhang, G. Wu and R. Zhou, J. Environ. Sci., 2022, 126, 445–458 CrossRef PubMed.
- E. M. C. Alayon, M. Nachtegaal, E. Kleymenov and J. A. van Bokhoven, Microporous Mesoporous Mater., 2013, 166, 131–136 CrossRef CAS.
- M. L. Baker, M. W. Mara, J. J. Yan, K. O. Hodgson, B. Hedman and E. I. Solomon, Coord. Chem. Rev., 2017, 345, 182–208 CrossRef CAS PubMed.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16–22 CrossRef CAS.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563 RSC.
- M. Sano, S. Komorita and H. Yamatera, Inorg. Chem., 1992, 31, 459–463 CrossRef CAS.
- M. H. Groothaert, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, J. Am. Chem. Soc., 2003, 125, 7629–7640 CrossRef CAS.
- P. J. Carl and S. C. Larsen, J. Phys. Chem. B, 2000, 104, 6568–6575 CrossRef CAS.
- F. Gao, E. D. Walter, N. M. Washton, J. Szanyi and C. H. F. Peden, ACS Catal., 2013, 3, 2083–2093 CrossRef CAS.
- A. Godiksen, F. N. Stappen, P. N. R. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS.
- A. Godiksen, P. N. R. Vennestrøm, S. B. Rasmussen and S. Mossin, Top. Catal., 2017, 60, 13–29 CrossRef CAS.
- S. C. Larsen, A. Aylor, A. T. Bell and J. A. Reimer, J. Phys. Chem., 1994, 98, 11533–11540 CrossRef CAS.
- J. S. Yu and L. Kevan, J. Phys. Chem., 1990, 94, 7612–7620 CrossRef CAS.
- K. Góra-Marek, Vib. Spectrosc., 2010, 52, 31–38 CrossRef.
- D. K. Lee, Korean J. Chem. Eng., 2004, 21, 611–620 CrossRef CAS.
- M. Jabłońska, K. Góra-Marek, M. Grilc, P. C. Bruzzese, D. Poppitz, K. Pyra, M. Liebau, A. Pöppl, B. Likozar and R. Gläser, Catalysts, 2021, 11, 843 CrossRef.
- M. P. Ruggeri, T. Selleri, M. Colombo, I. Nova and E. Tronconi, J. Catal., 2015, 328, 258–269 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev.: Sci. Eng., 2000, 42, 71–144 CrossRef CAS.
- T. Tanaka, T. Okuhara and M. Misono, Appl. Catal., B, 1994, 4, L1–L9 CrossRef CAS.
- M. Jabłońska, K. Góra-Marek, P. C. Bruzzese, A. Palčić, K. Pyra, K. Tarach, M. Bertmer, D. Poppitz, A. Pöppl and R. Gläser, ChemCatChem, 2022, 14, 1–14 CrossRef.
- J. Han, A. Wang, G. Isapour, H. Härelind, M. Skoglundh, D. Creaser and L. Olsson, Ind. Eng. Chem. Res., 2021, 60, 17826–17839 CrossRef CAS.
- D. Chen, Y. Yan, A. Guo, V. Rizzotto, H. Lei, Z. Qiao, H. Liang, M. Jabłońska, X. Jiang, J. Jiang, R. Palkovits, P. Chen, D. Ye and U. Simon, Appl. Catal., B, 2023, 322, 122118 CrossRef CAS.
- S. Prodinger, M. A. Derewinski, Y. Wang, N. M. Washton, E. D. Walter, J. Szanyi, F. Gao, Y. Wang and C. H. Peden, Appl. Catal., B, 2017, 201, 461–469 CrossRef CAS.
- J. Wang, L. Shao, C. Wang, J. Wang, M. Shen and W. Li, J. Catal., 2018, 367, 221–228 CrossRef CAS.
- A. Oda, H. Shionoya, Y. Hotta, T. Takewaki, K. Sawabe and A. Satsuma, ACS Catal., 2020, 10, 12333–12339 CrossRef CAS.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero and A. Yezerets, Science, 2017, 357, 898–903 CrossRef CAS PubMed.
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. W. Janssens, M. Cutini, S. Bordiga and G. Berlier, J. Am. Chem. Soc., 2020, 15884–15896 CrossRef CAS PubMed.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, J. Am. Chem. Soc., 2017, 4935–4942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00269a |
| This journal is © The Royal Society of Chemistry 2023 |