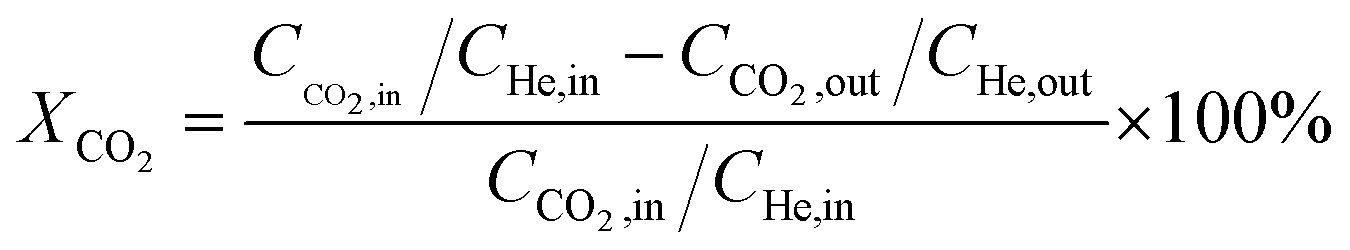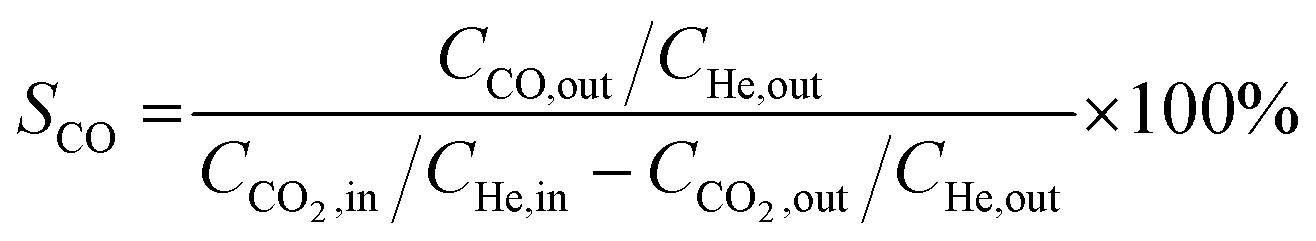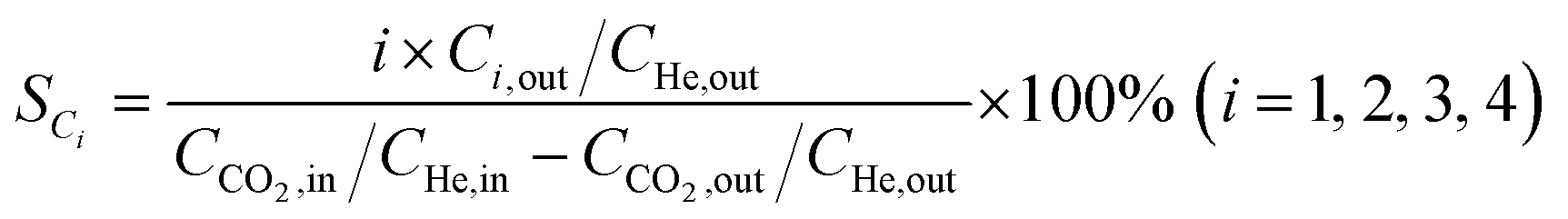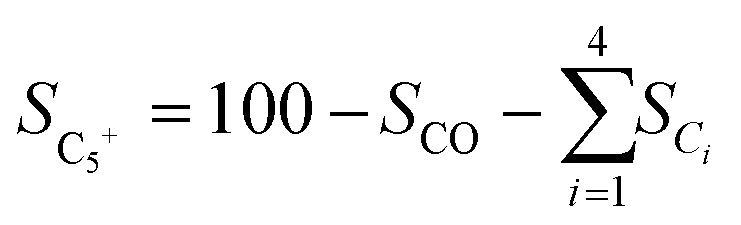Bimetallic Fe–Co catalysts for the one step selective hydrogenation of CO2 to liquid hydrocarbons†
Received
1st November 2022
, Accepted 25th January 2023
First published on 25th January 2023
Abstract
The direct conversion of CO2 to value-added products has received considerable attention as it can effectively mitigate CO2 emission and alleviate over-reliance on fossil fuels. We report the synthesis of a series of K-promoted Fe–Co bimetallic catalysts along with their performance for the selective hydrogenation of CO2 to liquid hydrocarbons (mostly linear α-olefins (LOAs)). High dispersion of K and Co on the catalysts was achieved through a modified one pot sol–gel approach. Both K and Co significantly influence catalyst activity and selectivity. By systematically studying the adsorption energies of key reactants on modeled iron oxide and carbide surfaces by density functional theory, we demonstrate that addition of K increases the affinity of the catalyst towards the adsorbed species. On the other hand, the presence of Co facilitates the spontaneous dissociation of H2. As a result of the high dispersion of components achieved through the one pot synthesis, at 300 °C, 20 bar (H2/CO2 = 2) and 7200 mL gcat.−1 h−1, the optimized catalyst exhibits a C5+ space time yield of 15.8 mmol gcat.−1 h−1 along with a C5+ selectivity of 51% at a CO2 conversion of 35%.
1. Introduction
Directly converting CO2 to higher hydrocarbons (light olefins (C2–C4), gasoline (C5–C12) or jet fuel (C8–C16))1–3 and oxygenated chemicals (methanol, dimethyl ether (DME), ethanol)4–6 by heterogeneous catalytic CO2 hydrogenation has gained a great deal of attention in the past few decades. In a future power-to-liquids (PtL) scenario with large integration of renewable energy (RE), H2 will be primarily obtained by water electrolysis with renewable energy sources (RES, e.g., solar, wind), while CO2 could be provided by carbon capture from point sources or even the atmosphere.7–9 Therefore, the transformation of CO2 to liquid fuels may be considered as one of the main avenues to achieve carbon neutrality.| | | CO2 + H2 → CO + H2O ΔH298 = +41 kJ mol−1 | (1) |
| | | CO + 2H2 → (–CH2–) + H2O ΔH298 = −152 kJ mol−1 | (2) |
| | | CO2 + 4H2 → CH4 + 2H2O ΔH298 = −165 kJ mol−1 | (3) |
The direct selective hydrogenation of CO2 to liquid hydrocarbons is generally described as the combination of the reverse water gas shift (RWGS) reaction (eqn (1)) with the successive hydrogenation of in situ produced CO to hydrocarbons through conventional Fischer–Tropsch synthesis (FTS, eqn (2)).3,10,11 Due to the thermodynamic stability and chemical inertness of CO2, efficient catalysts are required to decrease the activation energy of this process.9–11 Among the various options, cobalt and iron-based catalysts are the most widely studied, as they are commercially used for traditional CO-FTS.10–12 However, different from CO-FTS, more H2 is required for CO2-FTS and the concentration of CO is usually lower, leading to a higher H/C ratio on the surface of the catalyst, consequently favoring the formation of CH4 (methanation reaction, eqn (3)).13 Moreover, the amount of water in the reactor is higher and this may lead to catalyst oxidation under reaction conditions.3 Cobalt is substantially inactive for the RWGS reaction, therefore, when replacing CO with CO2 in the feedstock, cobalt-based catalysts display limited activities toward long-chain hydrocarbons, with CH4 as the main product.8,11,14 In contrast, iron-based catalysts are not only active for the RWGS reaction but also for CO-FTS when adequately promoted.10–13
Indeed, a large bulk of the literature focuses on the modification of iron-based catalysts with alkali metals, e.g., K and Na, to increase hydrocarbon selectivity.8,11–13,15 It has been widely reported that alkali metals could markedly alter the surface basicity of the catalyst, transferring electrons to metal sites, and thus enhance the adsorption of CO2 and reduce its dissociation barrier. This contributes to the formation and stabilization of metal carbides, which are believed to be the active phase for FTS.11,15–17 Enhanced CO2 adsorption, in contrast, impedes H2 adsorption on the surface of the catalyst, consequently suppressing, to a certain extent, CH4 formation.11,16–18 Last but not least, lower hydrogenation activity results in higher alkene selectivity.16,17 In addition to alkali metals, transition metals, e.g., Cu, Zn, and Mn, have also been explored to enhance or modulate the catalytic performance and hydrocarbon selectivity.10–14 These can improve the reducibility of the catalyst, influencing the carburization and hydrogenation performance.8,10,11,15 Cobalt has also been combined with Fe, but surprisingly focusing mostly on the production of light olefins.19–21 Recently, Jiang et al. have studied K-promoted cobalt doped Fe-based catalysts for CO2 hydrogenation to C2+ hydrocarbons.22 The remarkably enhanced catalytic performance by cobalt addition was mainly attributed to the increased conversion of CO intermediates via CO-FTS. In addition, they revealed that a suitable affinity between iron and cobalt was crucial. Meanwhile, Kim et al. found that Co–Fe alloy carbides ((Fe1−xCox)5C2, x ≤ 0.2) could be formed under reaction conditions, accounting for the outstanding performance of their Na–CoFe2O4/CNT catalyst.23 Interestingly, Hwang and co-workers reported an alloyed Fe–Co catalyst (FeK/Co-NC) with high activity for the direct conversion of CO2 to long-chain hydrocarbons with a C5+ selectivity up to 42%. They attributed the superior performance to the formation of Fe–Co alloys, which was stable in both carburized and oxide forms. DFT calculations suggested that Fe–Co mixed carbides could promote chain growth and suppress CH4 formation. However, the CH4 selectivity was still too high (21.6%).24
In this contribution, with the objective of strongly decreasing methane selectivity while increasing C5+ selectivity, we explored the use of the sol gel method for the synthesis of highly dispersed Fe–Co bimetallic catalysts.25,26 The performance of these catalysts in the direct hydrogenation of CO2 to high value-added liquid hydrocarbons has been studied in detail, including full characterization of the liquid product. The optimized catalyst exhibits a high activity with a C5+ space time yield (STY) of 15.8 mmol gcat.−1 h−1 and a notable C5+ selectivity of 51% under demanding reaction conditions. To our delight, liquid phase analysis demonstrated that most liquid products were highly valuable linear α-olefins (LOAs) in the C10 to C20 range, further highlighting the potential of CO2 as a valuable chemical feedstock.
2. Experimental
2.1 Catalyst preparation
All the catalysts were prepared by a modified sol–gel method with citric acid (≥99.5%, Sigma-Aldrich) as a chelating agent and metal nitrates as metal precursors (iron(III) nitrate nonahydrate, ≥98%, Sigma-Aldrich; potassium nitrate, ≥99%, Sigma-Aldrich; cobalt(II) nitrate hexahydrate, 98.0–102.0%, Alfa Aesar). Typically, a certain amount of citric acid was dissolved in distilled water forming solution A. Solution B with a desired amount of metal nitrates (in a molar ratio of (Fe + Co)/citric acid = 2, and Fe/Co ranging from 3 to 15, Fe/K varying from 2 to 8) was added into the above solution dropwise under stirring. The obtained mixture solution C was heated up to 80 °C using a water bath under stirring for about 1–2 h to obtain a dark brown slurry. This gel was transferred to a pre-heated muffle furnace (Nabertherm S.A.S, connected to the vent system) at 120 °C for 2 h. Then the obtained pre-carbonized foam structure was ground and calcined in air at 350 °C for 4 h (as illustrated in Fig. S1†). The final products were named as FexCoKy, where x represents the Fe/Co molar ratio and y refers to the Fe/K molar ratio (as listed in Table S1†).
2.2 Catalyst characterization
Powder X-ray diffraction (XRD) was performed on a Bruker D8 Advance X-ray deflector equipment equipped with a Cu Kα (λ = 1.5418 Å) radiation source at 40 kV and 40 mA. The typical 2θ scanning range was from 10° to 80° with a scan step size of 0.02° in continuous mode. The crystalline phase was identified by comparison with the powder diffraction file (PDF) from the International Center for Diffraction Data (ICDD); thermogravimetric analysis (TGA) was conducted in the 30–800 °C temperature range with a heating rate of 10 °C min−1 using a TGA/DSC 1 STARe system operating under an air flow (20 mL min−1). Temperature-programmed desorption of carbon dioxide (CO2-TPD) was carried out to examine the basicity of the catalyst using a Micromeritics AutoChem 2950 HP chemisorption analyzer. Briefly, 50 mg catalyst was first reduced with H2 at 350 °C for 1 h, and then the system was cooled down to 50 °C, followed by CO2 adsorption at 50 °C for 30 min. After sweeping with He for 1 h to remove the remaining and weakly adsorbed CO2, the system was heated up to 800 °C with a ramp rate of 10 °C min−1 and the desorbed CO2 was detected on-line. Inductively coupled plasma-optical emission spectrometry (ICP-OES) analysis was performed on an Agilent 5110 ICPOES to analyze the chemical composition of the materials. Prior to the analysis, the samples were fully digested with acidic solution (HCL and HNO3) using the Milestone Ethos One Microwave Digestion System. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) coupled with energy dispersive X-ray spectrometry (EDS) observation of the samples was conducted with a Titan Themis-Z microscope (Thermo-Fisher Scientific). The reduced catalyst was obtained by external PID reduction of the fresh calcined sample following the sample reduction procedure.
2.3 Catalytic activity evaluation
Catalytic activity evaluation was conducted using a fixed bed continuous-flow reactor on a 4-channel Florence® platform from Avantium. Typically, 50 mg catalyst was loaded in each stainless steel reactor with one blank as a reference for each test. Prior to the reaction, the catalyst was reduced in situ with pure H2 at 350 °C for 1 h with a total flow of 30 mL min−1. After reduction, the system was cooled down to the reaction temperature, then the mixture of reactant gases with a H2/CO2 (v/v) ratio of 2 (33.33% CO2 and 66.67% H2) was fed into the reactor using He as internal standard reference gas (mixture/He ratio of 5, v/v) to provide a total gas hourly space velocity (GHSV) of 3600 mL gcat.−1 h−1. The system was then pressurized using a membrane based pressure controller. The effluent gases from the reactor were analyzed online using gas chromatography (GC, Agilent Technologies 7890B). CO2, CO, H2, and He were analyzed using a thermal conductivity detector (TCD) equipped with 2 HayeSep pre-column and an MS5A, whereas C1–C8 hydrogenations were detected using a flame ionization detector (FID) equipped with Gaspro and Innowax columns. The liquid products were collected and analyzed using off-line GC and GC-MS. Typical TCD and FID chromatograms are shown in Fig. S2.† The CO2 conversion (XCO2) and product selectivity (SCO and SCi) were calculated on a carbon basis using the following equations:| | 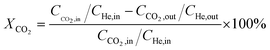 | (4) |
| | 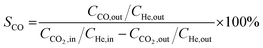 | (5) |
| | 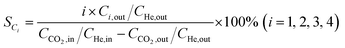 | (6) |
| | 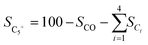 | (7) |
where Ci,in/out refers to the concentration of the i component in the inlet or outlet mixture detected and calibrated by the GC.
3. Results and discussion
3.1 Characterization results
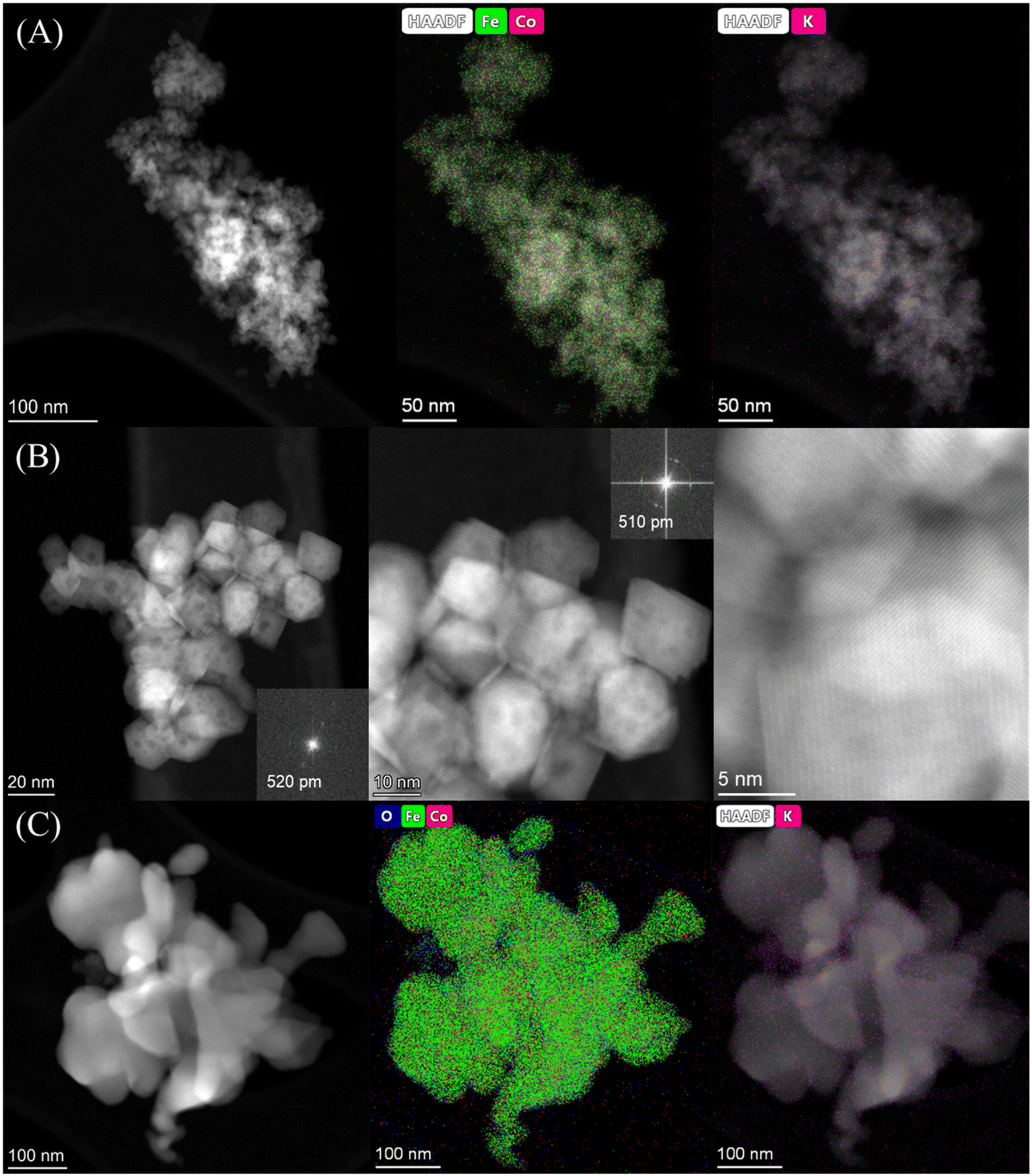
HAADF-STEM images and the corresponding EDS elemental mapping of the as-synthesized catalyst are shown in Fig. 1(A and B) and S3.† According to STEM, cobalt and potassium were uniformly dispersed on the catalyst surface (Fig. 1A). This provided close proximity between iron, cobalt and potassium and could be beneficial in improving the reduction and carburization of the catalyst, and thus the catalytic performance, which have been evidenced in the following analysis.20,24 The obtained fresh Fe9CoK6 metal oxide displayed cubic crystals with an average diameter of 15 nm (Fig. 1B and S3†). No obvious residual carbon was observed for the as-synthesized catalysts, which is in accordance with the TGA analysis (Fig. S4†). The XRD patterns of the as-synthesized catalysts are displayed in Fig. S5.† The main diffraction peaks located at 2θ = 15.0°, 18.4°, 23.8°, 26.1°, 30.2°, 35.6°, 37.3°, 43.3°, 53.7°, 57.3°, 62.9°, 71.4° and 74.5° were assigned to the (110), (111), (210), (211), (220), (311), (222), (400), (422), (511), (440), (620) and (533) crystalline planes of Fe2O3 (PDF: 00-039-1346), respectively. No discernible characteristic diffraction peaks of cobalt oxides were detected over the Fe–Co catalyst in the XRD analysis (Fig. S5A†), suggesting a high dispersion of cobalt species, which is in excellent agreement with the STEM results. The addition of cobalt resulted in a slight shift of the main Fe2O3 diffraction peaks towards lower 2θ values in the presence of potassium (Fig. S5B†), indicating the potential incorporation of cobalt into the lattice structure of iron oxide. This can greatly increase the interaction between cobalt and iron and facilitate the formation of Fe–Co mixed oxides, thus inhibiting the migration and aggregation of metal species during the reaction. CO2-TPD clearly evidenced the modified surface basicity of the catalyst by the introduction of potassium carbonate, which greatly enhanced the adsorption of CO2. This has been widely reported to be beneficial for the catalytic performance (Fig. S6†).14–17 In addition, H2-TPR of the as-synthesized catalysts (Fig. S7A†) revealed that the introduction of Co promoted the reduction of the catalysts as the reduction peak shifted towards a lower temperature.20,27 Moreover, the promotion of K, to some extent, resulted in a slight increase of the reducibility of the catalyst at the moderate temperature range, indicating adjusted interaction of each component.
 |
| | Fig. 1 HAADF-STEM images and corresponding elemental mapping images of the Fe9CoK6 catalyst (A and B) before reduction and (C) after reduction. The insets in B show the FFT patterns. | |
After reduction in H2 (Fig. 1C), the average particle size increased, and the as-synthesized cubic nanoparticles turned into a less defined morphology. However, a good dispersion of cobalt and potassium was well maintained (Fig. S8†), in good agreement with the following XRD results (Fig. S7B†). Some oxygen species have also been detected on the reduced catalyst surface, which should be attributed to the passivation of the reduced metallic phase by air during the sample preparation and also could be contributed by the potassium carbonate as it is stable under the reduction conditions. The XRD profiles of the reduced samples further confirmed the effective transformation of metal oxides into metallic phases. The sharp characteristic reflection peaks centered at 44.7°, 65.1° and 82.4° for all the samples were assigned to the (110), (200), (211) planes of metallic iron (PDF: 04-007-9753), respectively. No distinct peaks were related to metallic cobalt for the Fe–Co bimetallic catalysts. This suggested that cobalt atoms were either highly dispersed or probably involved in the formation of an iron-rich Fe–Co alloy structure partially. It has been reported by Kim and Zhang recently that even the formation of alloys could also benefit the hydrogenation process.23,28 Xu and co-workers revealed that electron-rich iron atoms in the CoFe alloy facilitated the CO intermediate dissociation, and thus substantially promoted the in situ generation of active carbide phases, which consequently improved the hydrocarbon productivity and suppressed the CO2 methanation.27 From the XRD data, the absence of iron oxide phases for cobalt doped catalysts after reduction indicates that cobalt enhances the reducibility of the catalysts, which is in line with the H2-TPR results (Fig. S7A†). This may be attributed to the enhanced reducibility by hydrogen spillover on the surface of cobalt to the intimate reducible iron oxides under the reduction microenvironment.27,29 The obtained metallic phases will be further oxidized and carburized to the corresponding metal oxides and carbides during the hydrogenation and are active for the corresponding RWGS and FTS, which will be fully unveiled in the following analysis.30
The HAADF-STEM images of the spent Fe9CoK6 reveal the structure reconstruction of the metal nanoparticles after around 45 h time on stream (TOS) at 300 °C with a GHSV of 7200 mL gcat.−1 h−1 and a H2/CO2 ratio of 2 (Fig. 2). The particle size ranges from around 90 nm to 150 nm approximately for the spent one with a distinct different morphology compared to the reduced catalyst (Fig. 1C). This is due to the surface reconstruction and phase transformation during the reaction, e.g., the generation of new species (metal carbides and oxides), which have been widely reported in previous studies and demonstrated in the following analysis.30–32 The EDS elemental mapping images of the spent catalyst clearly disclosed the highly dispersed cobalt and potassium on the catalyst surface after 45 h TOS test, indicating excellent stability of the catalyst. This should be related to the close proximity and appropriate interaction between each component. Meanwhile, EDS elemental maps visibly depicted the appearance of carbonous species, which should come from the metal carbide phases, and also probably from the remaining heavy hydrocarbons on the surface of the catalyst (Fig. S9†). Oxygen species (with larger content than that of the fresh reduced one) could be observed clearly on the surface of the spent catalyst, suggesting the oxidation of the reduced catalyst under the test conditions. This is due to the phase transformation during the reaction (formation of iron oxides) and has been evidenced by the XRD analysis. It is generally accepted that the metallic phase could be oxidized by CO2 and be further promoted by the H2O by-product once the RWGS reaction starts.17,22 After 85 h stability test, EDS elemental mapping (Fig. S10†) disclosed a slight aggregation of cobalt species on the surface of iron oxides. Similar to our previous observation, an enrichment of potassium on the catalyst surface after reaction has also been detected. This is due to the high mobility of the potassium ions.33
 |
| | Fig. 2 HAADF-STEM images and corresponding compositional elemental mapping of the spent Fe9CoK6 catalyst after 45 h time on stream at 300 °C with a H2 to CO2 ratio of 2 and a GHSV of 7200 mL gcat.−1 h−1. | |
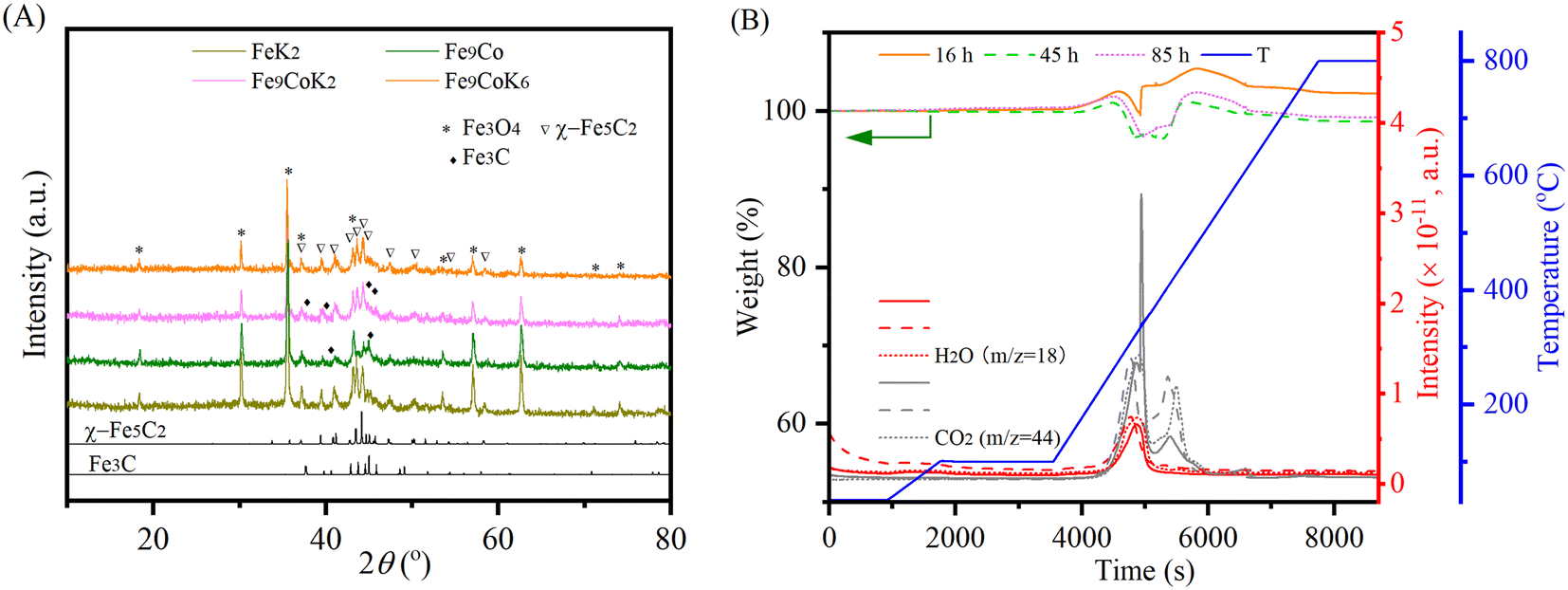
The XRD profiles of the spent catalysts are shown in Fig. 3A and S11A.† The main diffraction peaks were associated with Fe3O4 and iron carbide phases, in good agreement with the literature.30,32 No discrete diffraction peaks were assigned to cobalt species in the spent catalysts. This together with the observed unobtrusive higher angular shift for Fe3O4 diffraction peaks may indicate the inclusion of cobalt into the iron oxides or the iron carbides structures.24 This could decrease the bonding strength between iron and CO and thus promote its dissociation and further hydrogenation to hydrocarbons.23,27 Fe3O4 is generally supposed to be formed by the oxidation of metallic iron or iron carbide species in the reaction environment, while iron carbide phases are generated by the carburization of the metallic iron phase and iron(II) oxide species with carbon species and facilitated by the cobalt and potassium promoter.3,22,30 The oxidation and carburization continue until a dynamic balance state is reached.32 It is well established that in the tandem CO2-FTS mechanism, the RWGS reaction occurs on the surface of Fe3O4, then the CO intermediate will migrate to the adjacent active metal carbide sites, where the CO-FTS reaction is induced.3,22,27 The presence of χ-Fe5C2 and Fe3C species on the spent K modified catalyst indicates that potassium promoted the carburization during the reaction. In contrast, for the unpromoted Fe9Co catalyst, mostly Fe3C species have been identified after the reaction, which are supposed to be less active for long chain growth to higher hydrocarbons than the χ-Fe5C2 phase during FTS.19,27,32 TGA and MS curves of the spent catalysts clearly confirmed the oxidation of the carbonaceous and carbide species, similar to our previous study34 (Fig. 3B and S11B†).
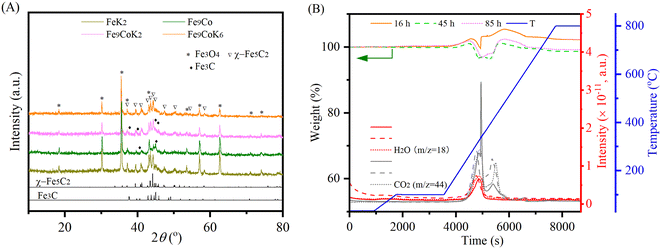 |
| | Fig. 3 (A) XRD patterns of the spent catalysts with 16 h TOS and (B) TGA-MS profiles of the spent Fe9CoK6 catalysts with different TOS at 300 °C with a H2 to CO2 ratio of 2 and GHSVs of (A) 3600 mL gcat.−1 h−1 and (B) 7200 mL gcat.−1 h−1. | |
3.2 Catalytic performance
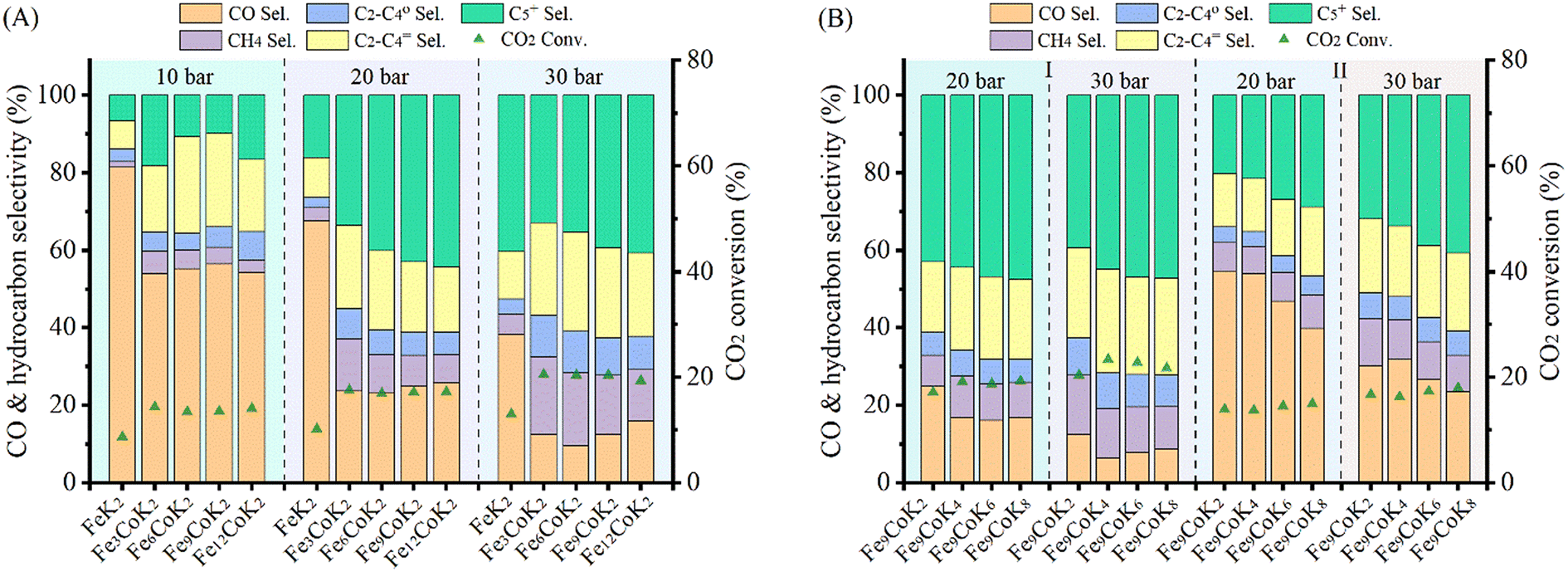
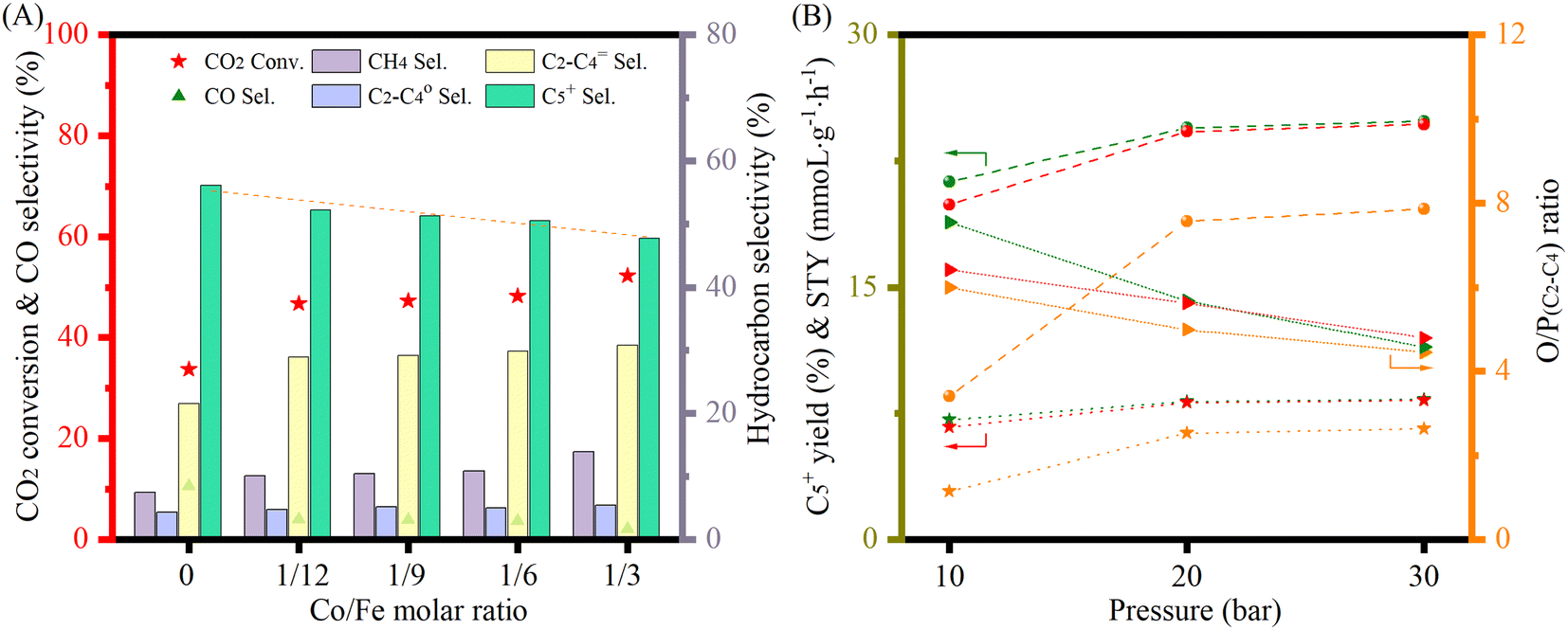
The catalytic performance of the materials was first investigated at 250 °C with an H2 to CO2 molar ratio of 2 under varying pressures from 10 to 30 bar (Fig. 4A). Under the studied reaction conditions, the increase of pressure increased the CO2 conversion from an average 14% at 10 bar to 20% at 30 bar together with an obvious increase of CH4 and C5+ selectivity, whereas the CO selectivity exhibited an opposite trend, decreasing dramatically with increasing pressure. The addition of cobalt into the FeK2 catalyst evidently enhanced catalyst activity (with CO2 conversion increased by a 75%) and significantly reduced CO selectivity (from 67.7% to 23.7%) but was also accompanied by an increase in CH4 selectivity (from 3.3% to 13.4% for FeK2 and Fe3CoK2 at 20 bar, respectively) (as summarized in Table S2,† entries 1 vs. 2). The total yield to higher hydrocarbons could be considerably boosted by addition of small amounts of cobalt. The C5+ yield of the Fe12CoK2 catalyst was about 4 times higher than that of FeK2 (Table S2,† entry 5). It is well known that cobalt has negligible activity in both water gas shift (WGS) and RWGS (eqn (1)), therefore such remarkable enhancement should be ascribed to a higher FTS activity, which goes in agreement with the sharp decrease in CO selectivity once cobalt was introduced. The synergistic promotion of cobalt and iron combination in the presence of potassium greatly forces the reaction towards the formation of hydrocarbons.8,22 Similar results have been reported by Jiang and co-workers.22 In fact, cobalt is a well-known active phase for both low temperature CO-FTS (eqn (2)) and CO2 methanation (eqn (3)).35,36 Highly dispersed cobalt could effectively convert the in situ produced CO formed on the adjoined iron oxides surface into either heavy hydrocarbons by CO-FTS through C–C bond coupling or CH4 through CO methanation. Furthermore, CH4 could also be formed on cobalt species through CO2 methanation (eqn (3)).37,38 These together lead to the increased longer chain hydrocarbons and CH4 selectivity, especially at 20 bar.
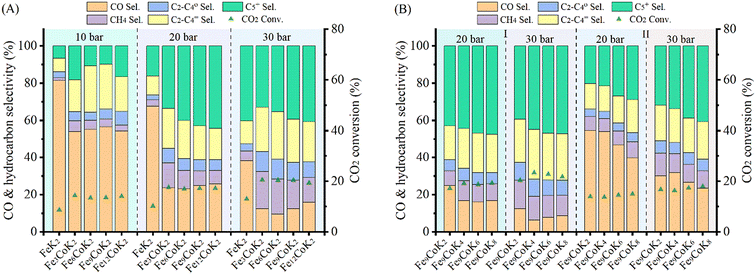 |
| | Fig. 4 Catalytic performance of Fe–Co catalysts under different pressures at 250 °C with a H2 to CO2 ratio of 2 and GHSVs of 3600 mL gcat.−1 h−1 (A and B-zone I) and 7200 mL gcat.−1 h−1 (B-zone II). | |
While in the case of FeK2 higher pressures are favorable for the synthesis of C5+ products (Fig. 4A), a different behavior has been observed for Fe–Co bimetallic catalysts. With pressure increasing from 10 to 20 bar, the bimetallic catalysts exhibited noticeably higher selectivity towards higher hydrocarbons, as the total C2+ selectivity rose from around 18% to an average 45% at 10 bar, and from 32% to 75% at 20 bar for FeK2 and Fe–Co bimetallic catalysts, respectively. Accordingly, the chain growth probability, α, calculated based on C3–C7 products was 0.54 for the former, while it increased to 0.59 for Fe9CoK2 at 20 bar (Fig. S12†). However, for the catalyst containing cobalt, further increasing the pressure over 20 bar has a negative effect (Fig. S11†). It is obvious that the C5+ selectivity progressively decreased with increasing cobalt loading at both 20 and 30 bar. Meanwhile, the methane selectivity and the formation of light hydrocarbons (C2–C4, mainly light olefins) was apparently enhanced by cobalt addition and promoted with the reaction pressure increase. These indicate that, at high pressure, cobalt may primarily limit the carbon chain growth to heavy hydrocarbons by reducing the chance of C–C bond coupling on the contiguous iron carbide phases but rather facilitating the CO2/CO methanation under the test conditions.8 These positive and negative effects seem to be well balanced for catalysts with a low content of cobalt.
The influence of K loading and gas hourly space velocity (GHSV) on the catalytic activity of Fe–Co bimetallic catalysts with an Fe/Co molar ratio of 9 has been studied at 20 and 30 bar (Fig. 4B). The decrease of K loading resulted in a general decrease of CO selectivity and a slight increase of CO2 conversion and C5+ selectivity. A high K loading results in the formation of more potassium carbonate on the surface (Fig. S13†), potentially limiting accessibility to the active sites. This is in agreement with what has been reported by Jiang et al.,22 with the C5+ selectivity being almost stable when the pressure increased from 20 bar to 30 bar with a GHSV of 3600 mL gcat.−1 h−1, suggesting that K loading mostly affects the conversion between CO and C1–C4 hydrocarbons under the experimental conditions (Fig. 4B zone-I). However, when doubling the GHSV, the selectivity to C5+ and C2–C4 olefins considerably decreased along with a significant increase of CO selectivity (Fig. 4B zone-II). This could be attributed to the fact that increasing GHSV reduces the residence time of the reactants and CO intermediates on the active sites, which prevents their subsequent dissociation and hydrogenation to higher hydrocarbons. It is interesting to note that, at this temperature, higher pressure and low K loadings are required to obtain a high yield of heavy hydrocarbons at higher GHSV, but also accompanied with high CO selectivity. In addition, the CO selectivity almost remained constant for the Fe–Co catalysts (Fig. 4A, Table S2†). These phenomena reveal that the reaction under the current test conditions, to a large extent, is limited by CO hydrogenation over the active species, and a higher temperature is probably needed for boosting the catalytic performance.10,27,39 This observation is consistent with the fact that iron-based CO2-FTS catalysts generally work more efficiently at higher reaction temperature.8,10 Such a hypothesis has also been partially confirmed by Khangale and co-workers in their study on the effect of reaction temperature on CO2 conversion to liquid hydrocarbons using a 15% Co–5% K/Al2O3 catalyst.40 Analogous results have been also observed for Fe6CoK catalysts with varying K contents under different conditions (Fig. S14 and S15†).
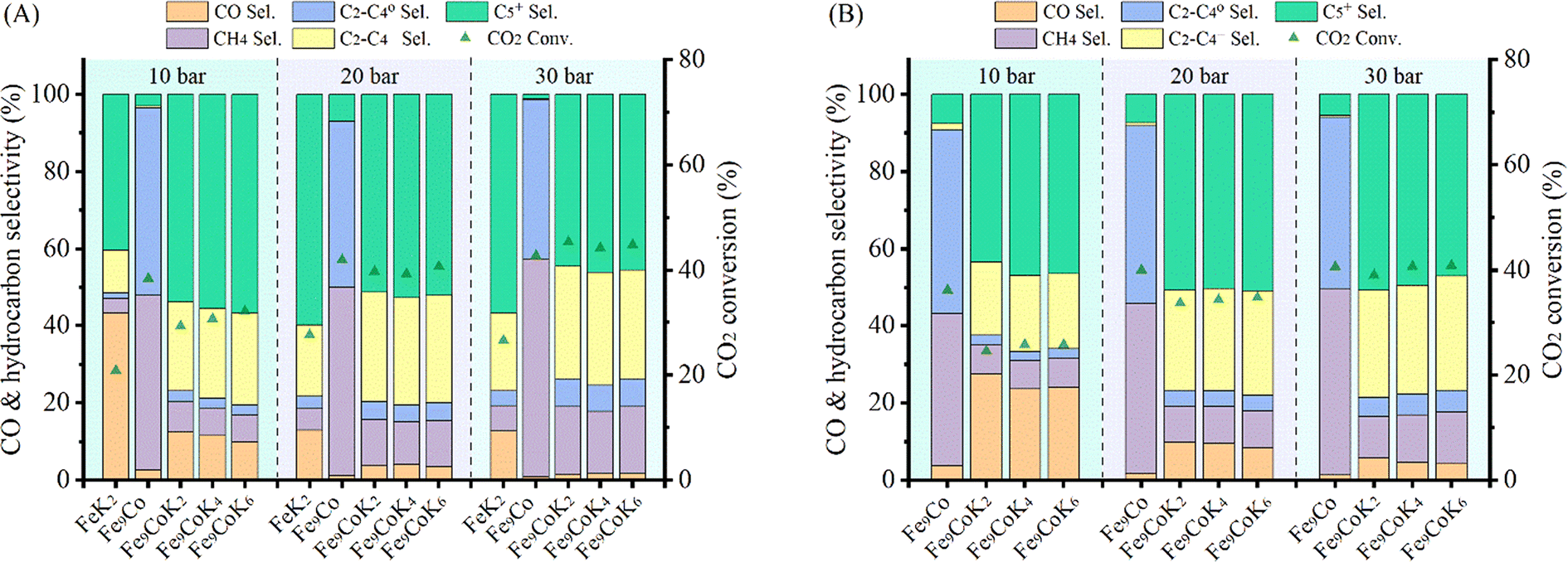
The catalytic performance was further studied at 300 °C with a H2 to CO2 ratio of 2 at different pressures and GHSVs (Fig. 5 and S16†). FeK2 and Fe9Co were studied as reference catalysts (Fig. 5A). It is evident that the temperature increase remarkably increased the CO2 conversion and higher hydrocarbon selectivity while the CO selectivity was sharply decreased. This is probably because the reaction rate of the CO-FTS was significantly increased at this higher temperature, leading to fast and consecutive consumption of the CO intermediate formed by the RWGS reaction, and thus further driving the equilibrium towards the conversion of CO2.8,10,40 The CO2 conversion for K-promoted catalysts doubled when compared to the results obtained at 250 °C, while a very low CO selectivity (<4%) and higher selectivity for long chain hydrocarbons (>50%) for Fe–Co catalysts at a GHSV of 3600 mL gcat.−1 h−1 (Table 1) could be achieved at this temperature. At this point, the selectivity towards CH4 also increased, but to a lesser extent than that for higher hydrocarbons. Accordingly, the yield of heavy hydrocarbons (C5+) was much higher than that at 250 °C. At the same time, the light olefins to paraffins (C2–C4) ratio (O/P) almost doubled at higher reaction temperatures compared with that at 250 °C (Table S2†). Notably, the main products (>90%) over the Fe9Co catalyst were CH4 and C2–C4 paraffins, whereas for the Fe–Co–K catalysts, C2–C4 olefins and C5+ were the dominant ones, highlighting the crucial role of K in: (i) promoting the formation of intermediate CO and (ii) modulating the hydrogenation activity of the catalyst.8,18,22,41 Indeed, the C5+ selectivity and space time yield (STY) with Fe9CoK6 was more than 6 times higher than that obtained with the Fe9Co catalyst along with a methane selectivity only a quarter of the latter (Table 1, entries 4 vs. 2). The amount of K had a smaller influence on the hydrocarbon distribution (Fig. 5A). When combined with the notable promotion effect of cobalt, the CO2 conversion reached up to 45% with a total hydrocarbon carbon selectivity ≥98% in one single-pass at 30 bar. Meanwhile, the carbon chain growth probability α increased to 0.63 for the Fe9CoK2 catalyst compared with that of 0.59 at 250 °C and 20 bar (Fig. S12 and S17†). Similar results have also been found for Fe6CoK catalysts (Fig. S15 and S18†). Interestingly, the FeK2 catalyst exhibited the highest C5+ selectivity under the same test conditions at 20 bar with a GHSV of 3600 mL gcat.−1 h−1, but due to the lower CO2 conversion (27.6%), the overall yield of C5+ over FeK2 (16.5%) is lower, in agreement with the earlier observation by Hwang et al.24 Moreover, doubling the GHSV leads to a visible increase of CO selectivity together with an evident decrease of CO2 conversion (Fig. 5B).
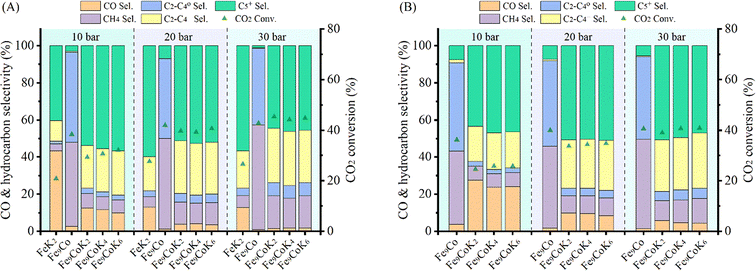 |
| | Fig. 5 Catalytic performance of Fe9CoK catalysts with different K loadings under different pressures at 300 °C with a H2 to CO2 ratio of 2 and GHSVs of (A) 3600 mL gcat.−1 h−1 and (B) 7200 mL gcat.−1 h−1. | |
Table 1 Catalytic performance summary at 300 °C, 20 bar with a H2/CO2 ratio of 2
| Sample |
GHSV (mL gcat.−1 h−1) |
XCO2 (%) |
S
CO (%) |
SCH4 (%) |
SC2–C4 (%) |
SC5+ (%) |
C5+ yield (%) |
O/P (C2–C4) |
C5+ STY (mmol gcat.−1 h−1) |
| FeK2 |
3600 |
27.6 |
13.0 |
5.7 |
21.6 |
59.7 |
16.5 |
6.4 |
7.4 |
| Fe9Co |
41.9 |
1.3 |
48.9 |
43.1 |
6.8 |
2.8 |
0.0 |
1.3 |
| Fe9CoK2 |
39.7 |
3.8 |
11.9 |
33.1 |
51.2 |
20.3 |
6.0 |
9.1 |
| Fe9CoK6 |
40.6 |
3.4 |
12.0 |
32.5 |
52.1 |
21.2 |
6.0 |
9.4 |
| Fe9CoK2 |
7200 |
33.7 |
9.9 |
9.4 |
30.0 |
50.7 |
17.1 |
6.6 |
15.3 |
| Fe9CoK6 |
34.8 |
8.4 |
9.6 |
31.1 |
50.8 |
17.7 |
6.6 |
15.8 |
The stability under stream is shown in Fig. S19.† Over the whole period, the catalyst did not show any sign of deactivation. Finally, for better comparison with the literature (Table 2), the catalytic performance was further explored at 300 °C with an H2 to CO2 ratio of 3 (Fig. 6(A)). The increase of H2/CO2 ratio obviously increased the CO2 conversion, but slightly decreased the C5+ selectivity. Consequently, the yield per pass to C5+ was marginally increased (Table S3†). Low Co doping (≈4 wt%) not only could keep the necessary high CO2 conversion (≈46%), but also could decrease the CO selectivity (from 12% to 4%) and maintain a low CH4 selectivity (<11%). Similar results have also been reported by Satthawong et al. in their study on γ-Al2O3 supported Fe–Co catalysts for CO2 hydrogenation to higher hydrocarbons.51 It is worth noting that catalysts with high cobalt loadings also exhibited high activity (Fig. 6(B)), even though the C5+ selectivity decreased due to the excessive formation of CH4. With the pressure increase, the O/P (C2–C4) ratio decreased. However, the O/P (C2–C4) ratio was high, (e.g., above 5 at 20 bar, Table S3†), indicating that olefins dominated the low hydrocarbons. Even though they are not the target products, they may play a vital role in carbon chain propagation during the reaction and are also important chemical building blocks in the petrochemical industry.
Table 2 Catalytic performance comparison with different Fe-based catalysts for the direct conversion of CO2 to hydrocarbons
| Catalyst |
GHSV mL gcat.−1 h−1 |
H2/CO2 (v/v) |
P (MPa) |
T (°C) |
XCO2 (%) |
S
CO (%) |
SCH4 (%) |
SC2–C4 (%) |
SC5+ (%) |
O/P C2–C4 |
Ref. |
|
Reported as SC2–C3 and SC4+.
|
| Fe9CoK6 |
3600 |
2 |
3.0 |
250 |
22.8 |
7.8 |
11.8 |
33.6 |
46.9 |
3.0 |
This work |
| Fe9CoK6 |
3600 |
2 |
2.0 |
300 |
40.6 |
3.4 |
12.0 |
32.5 |
52.1 |
6.0 |
| Fe9CoK6 |
7200 |
2 |
2.0 |
300 |
34.8 |
8.4 |
9.9 |
31.5 |
50.2 |
6.7 |
| Na–CoFe2O4 |
7200 |
3 |
3.0 |
320 |
41.8 |
9.7 |
20.0 |
44.1 |
26.2 |
5.4 |
20
|
| 10Fe0.8 K0.53Co |
560 |
3 |
2.5 |
300 |
54.6 |
2.0 |
18.9 |
32.1 |
47.0 |
2.4 |
22
|
| Na–CoFe2O4/CNT |
3600 |
3 |
1.0 |
340 |
34.4 |
18.6 |
12.0 |
36.1 |
33.3 |
7.1 |
23
|
| FeK/Co-NC |
2000 |
3 |
2.5 |
300 |
54.6 |
3.4 |
21.6 |
32.6 |
42.4 |
— |
24
|
| ZnCo0.5Fe1.5O4 |
4800 |
3 |
2.5 |
320 |
49.6 |
5.8 |
17.8 |
39.8 |
36.6 |
5.8 |
27
|
| CoFe–0.82Na–U |
5500 |
3 |
3.0 |
240 |
11.0 |
5.4 |
21.5 |
8.6 |
64.5 |
— |
28
|
| 15% Co–6% K/Al2O3 |
1200 |
3 |
0.5 |
300 |
42.3 |
8.2 |
67.6 |
22.3 |
1.9 |
— |
40
|
| Na–Fe3O4 |
4000 |
3 |
3.0 |
320 |
34.0 |
14.3 |
9.6 |
39.5 |
32.6 |
— |
42
|
| FeNa(1.18) |
2000 |
3 |
3.0 |
320 |
40.5 |
13.5 |
13.7 |
46.8 |
26.0 |
6.2 |
43
|
| Fe–Co–K(0.1) |
3600 |
3 |
1.1 |
300 |
23.9 |
31.0 |
23.0 |
11.0 |
35.0 |
0.20 |
44
|
| Fe–Cu–K(0.1)–La(0.1) |
3600 |
3 |
1.1 |
300 |
23.1 |
33.0 |
13.0 |
9.0 |
45.0 |
1 |
44
|
| 10Mn–Na/Fe |
2040 |
3 |
3.0 |
320 |
37.7 |
12.9 |
14.0 |
34.1 |
38.9 |
6.0 |
45
|
| Fe/C–K2CO3 |
2400 |
3 |
1.0 |
320 |
32.4 |
21.4 |
10.0 |
20.8a |
47.8a |
— |
46
|
| Fe/C–KHCO3 |
2400 |
3 |
1.0 |
320 |
33.0 |
20.8 |
10.0 |
21.6a |
47.6a |
— |
46
|
| Fe–Co/K–Al2O3 |
9000 |
3 |
2.0 |
340 |
40.0 |
12.2 |
21.8 |
47.4 |
18.6 |
5.9 |
47
|
| Fe5C2–10K/α-Al2O3 |
3600 |
3 |
3.0 |
320 |
31.5 |
18.6 |
12.1 |
40.2 |
29.1 |
8.1 |
48
|
| FeK/MPC |
2000 |
3 |
2.5 |
300 |
50.6 |
8.2 |
15.4 |
31.9 |
44.5 |
— |
49
|
| FeK/SWCNTs |
9000 |
3 |
2.0 |
340 |
52.7 |
9.6 |
12.2 |
28.1 |
50.1 |
2.6 |
50
|
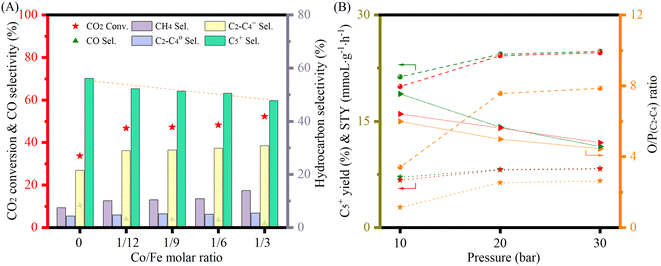 |
| | Fig. 6 (A) Catalytic performance of FeCoK catalysts with varying Co/Fe ratios at 20 bar; (B) C5+ yield (ball) and C5+ STY (star) together with O/P (C2–C4) ratios (triangle) for the FeK2 (orange), Fe3CoK2 (olive) and Fe9CoK2 (red) catalysts at different pressures. Other reaction conditions: 300 °C at a H2 to CO2 ratio of 3 and a GHSV of 3600 mL gcat.−1 h−1. | |
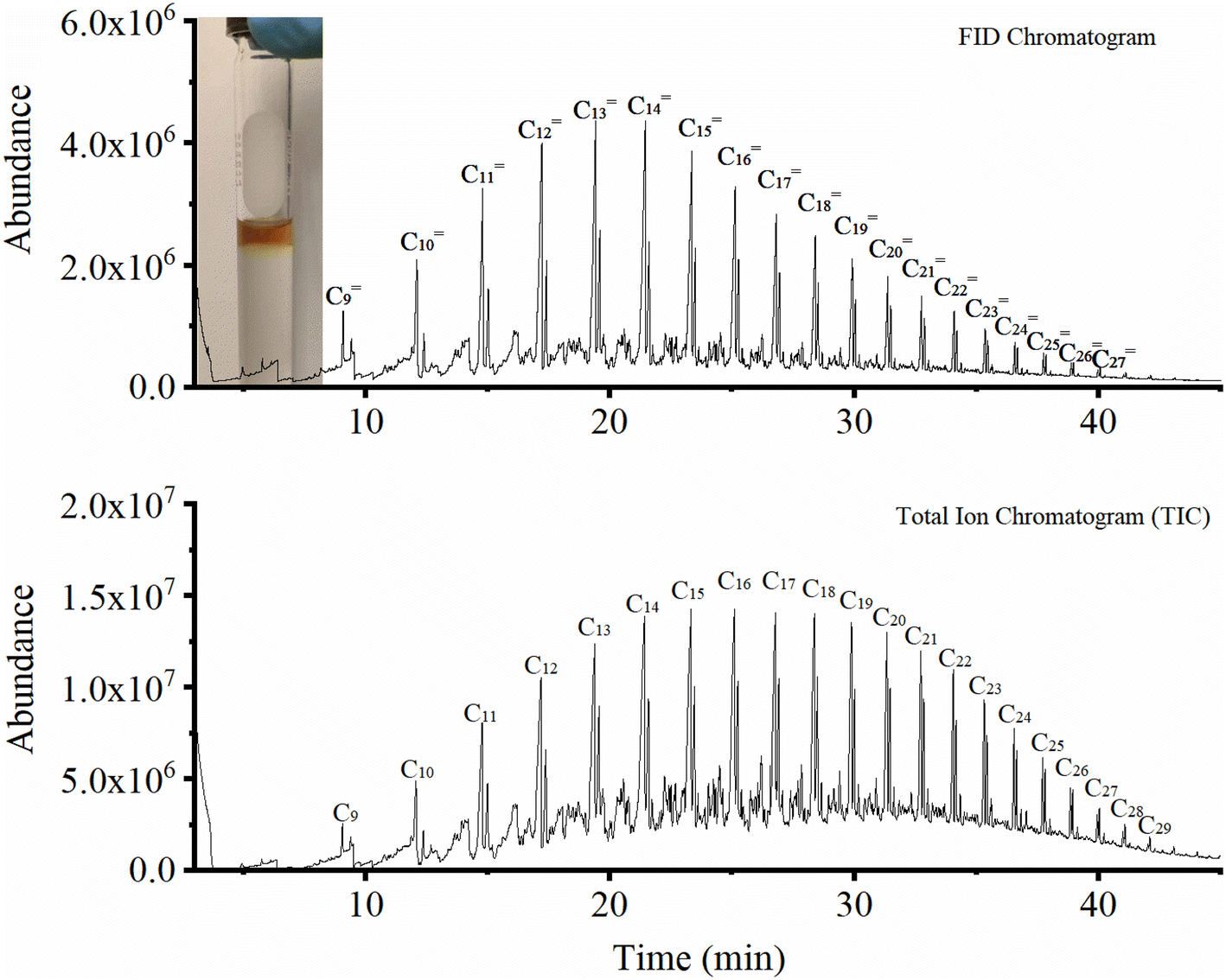
Generally, most studies in the literature report C5+ selectivity based on the fraction of products that cannot be quantified by online gas analysis.23,24,27,40,49,50 Therefore, except for some articles,3,23,24,51 there is very little information about the nature of this important fraction of hydrocarbons. In this work, we collected the liquid phase via condensation in a cold trap and analyzed the product offline. This did not only help close the mass balance but also revealed the nature of the different hydrocarbons formed. As shown in the GC-MS analysis of this fraction (Fig. 7), the obtained liquid over Fe9CoK6 is composed of a hydrocarbon fraction between C8 and C30 along with a small amount of carboxylic acids and alcohols. Among these, linear α-olefins with carbon numbers from C10 to C20 were the main products, in good agreement with ref. 51. With applications ranging from detergents to lubricants, oilfield chemicals and plasticizers, the linear α-olefins market size was over USD 12.5 billion in 2016 and consumption may exceed 7 million tons by 2024, giving an excellent outlook for this catalytic process.52 Another option to valorize the product could be the direct hydrogenation of the liquid phase to linear paraffins that would be in the range of jet fuel.
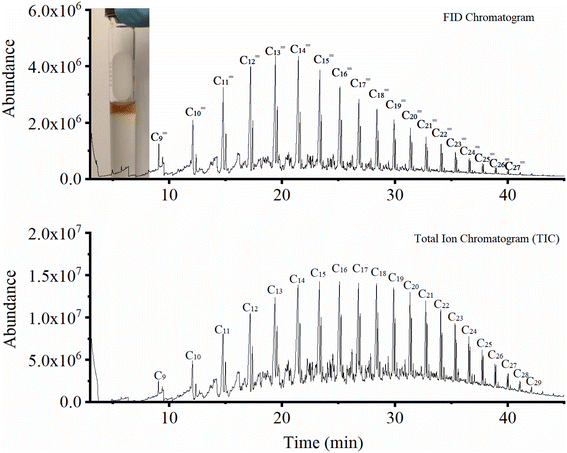 |
| | Fig. 7 GC-MS FID and total iron chromatogram of the liquid products. | |
As discussed above, it has been widely reported that the enhanced CO2 uptake on the catalyst surface through alkali promotion could effectively decrease H2 adsorption, and thus inhibit the over hydrogenation of surface carbonaceous monomers and secondary hydrogenation of produced olefins, resulting in long-chain hydrocarbons and high olefin productivity.8,17 Recently, Guo et al. reported a bio-promoted catalyst (Fe/C-Bio) for directly converting CO2 to linear α-olefins with a selectivity higher than 40% for α-C4–18= and a total olefin selectivity of 72% for C2–18= in hydrocarbons. However, the total CO and CH4 selectivity was still too high (>32%), and LAOs were mainly concentrated on the lower carbon number range of 4 to 9.53 Under reaction conditions, potassium could undergo the transformation between different salts, e.g., potassium carbonate, potassium bicarbonate and potassium formate, and this could contribute to the enhanced activation through the previous proposed “potassium carbonate mechanism” but also the formation of carboxylic acid and alcohols over iron mediated FTS through the CO insertion pathway under the appropriate reaction conditions.33,54–56
To get a glimpse of the reaction mechanism, we systematically studied the adsorption energies of key reactants on modeled iron oxide and carbide surfaces by density functional theory (DFT). Owing to the atomic heterogeneity on iron oxide and carbide surfaces, different types of adsorption sites can be present. Among them, those exhibiting the strongest adsorption energy for each adsorbate were selected and discussed subsequently. The completely optimized geometries of all the discussed modeled systems are shown in Fig. S20 and S21.†
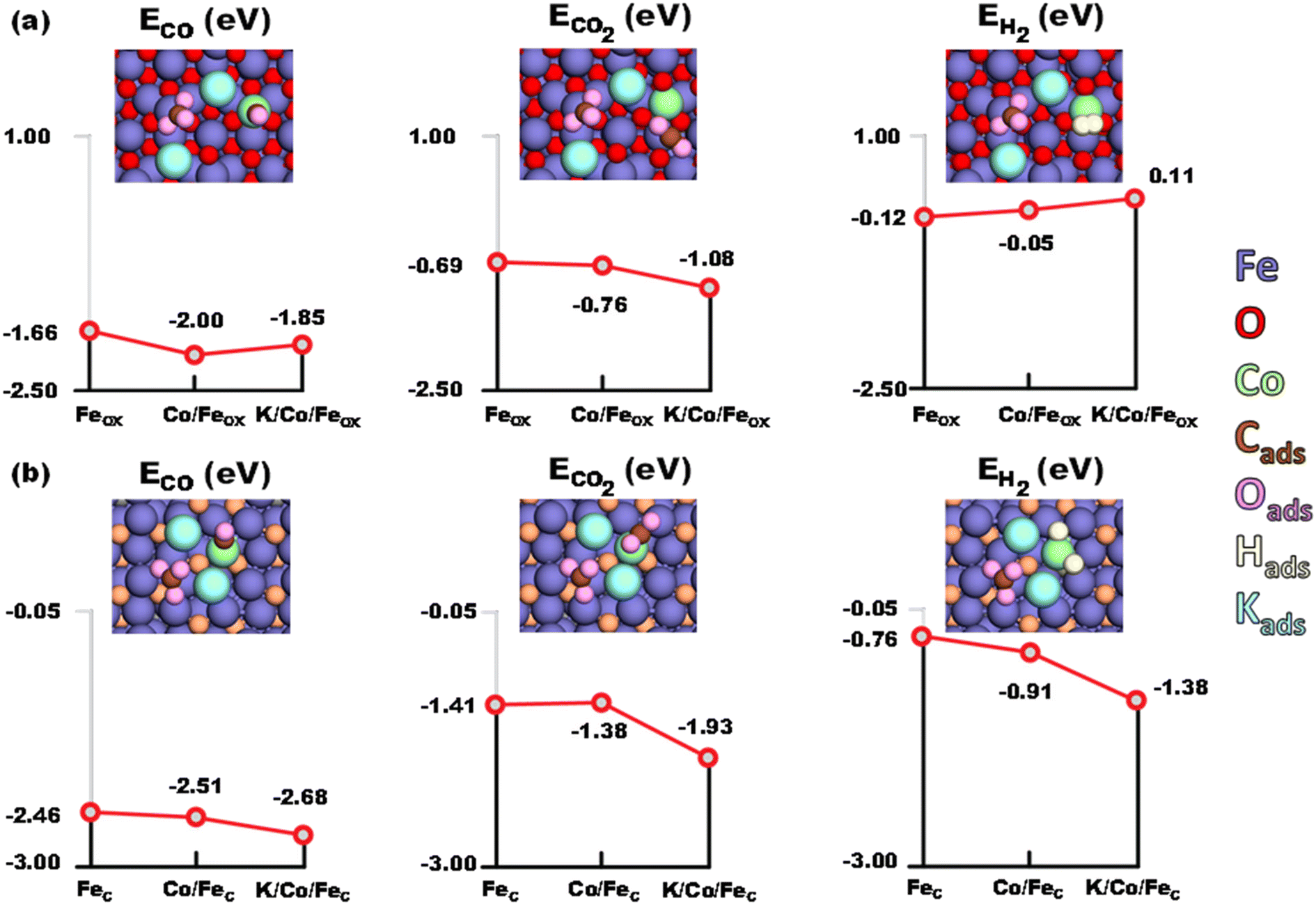
The adsorption of CO, CO2 and H2 on Fe3O4 (Feox hereafter) was first studied. The (111) facet of magnetite was considered as predominantly the most stable one, as reported both experimentally and theoretically.57–60 CO adsorbed moderately on the pristine Feox (111) with an energy gain of −1.66 eV. Intuitively, the adsorption of CO2 (−0.69 eV) was much weaker than that of CO. For H2, the adsorption energy was even weaker, but still exothermic by −0.12 eV. However, different situations have been observed on the cobalt doped Co/Feox surface. It has been revealed that CO prefers binding to the Co site, with an energy gain of 0.34 eV over that binding to the pristine Feox surface. Meanwhile, the adsorption of CO2 on the Co/Feox surface (−0.76 eV) was slightly stronger than that on the Feox surface. Owing to the slight negative charge acquired on Co due to the charge redistribution with the adjacent Fe atoms (−0.2 eV), H2 adsorption on the Co site of the Co/Feox surface was weakened (−0.05 eV) compared to the pristine Feox surface. On the other hand, coordination of a K2CO3 unit on the Co/Feox surface resulted in a small decrease of surface affinity for the adsorbed molecules, except for CO2. Still, CO adsorption on the K/Co/Feox surface was favored (by 0.19 eV) compared to adsorption on the pristine Feox surface. Meanwhile the CO2 adsorption near the K2CO3 unit with an energy of −1.08 eV, which was 0.32 eV stronger compared to the Co/Feox surface, indicates an obviously enhanced adsorption of CO2 by potassium. H2 adsorption was slightly endothermic in the presence of the K2CO3. It is worth mentioning that CO adsorption is in an optimal range to promote RWGS product formation without poisoning the catalyst surface.61,62 This ensures the easy migration of the produced CO from iron oxide to iron carbide for efficient FTS, as revealed below. The optimized geometries of the above discussed model systems are shown in Fig. 8.
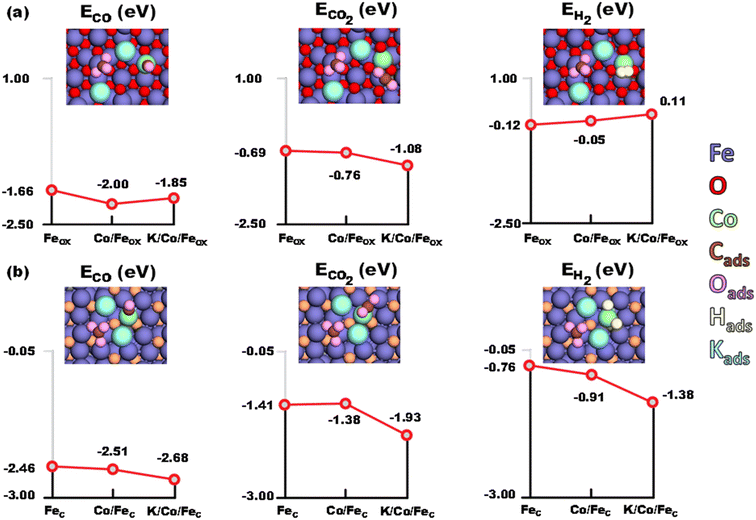 |
| | Fig. 8 Adsorption energies of CO, CO2 and H2 on (a) Fe3O4 and (b) Fe5C2 surfaces, respectively (blue – Fe, red – O, orange – C, aqua – K, lime – Co, brown – C adsorbed, pink – O adsorbed). | |
The adjoining iron carbide phase in the experimental system was modeled as Fe5C2 (Fec hereafter). The (001) face has been selected as it has been reported to be the most stable facet in the earlier studies.63–65 CO was strongly adsorbed (−2.46 eV) on this carbide site, while the CO2 adsorption energy (−1.41 eV) was not as strong as that of CO. H2 has an exothermic adsorption (−0.76 eV), starkly different from that on the Feox surface. The presence of a K2CO3 unit increased the affinity of Co/Fec towards the adsorbed species. The adsorption of CO (−2.68 eV) and CO2 (−1.93 eV) were favored by 0.22 eV and 0.52 eV relative to the pristine Fec, respectively. And the largest variation was observed for H2 adsorption (−1.38 eV), which was enhanced by 0.62 eV. Furthermore, adsorption of H2 on the various Fec based surfaces results in spontaneous H2 dissociation, with the H–H bond length elongating from 2.07 Å in Fec, to 2.10 Å in Co/Fec, and further to 2.68 Å in K/Co/Fec (for comparison, the H–H bond in K/Co/Feox is 0.80 Å). The strong affinity of K/Co/Fec towards CO and H greatly promotes the formation of long chain hydrocarbons through the CO-FTS, which in turn enhanced the RWGS reaction.
4. Conclusions
In summary, K-promoted Fe–Co bimetallic catalysts with varying cobalt and potassium loading have been synthesized following a modified sol–gel approach. When applied in the direct hydrogenation of CO2, these catalysts produce an important amount of C5+ hydrocarbons. Our results demonstrate that both potassium and cobalt play a key role in tuning catalyst selectivity and reactivity. On the one hand, addition of Co strongly increases catalyst activity by facilitating H2 dissociation. This hydrogenation activity needs, however, to be modulated by potassium promotion. Potassium has a dual role: on the one hand, it increases surface basicity and promotes the formation of iron carbides and oxides, responsible for chain growth and the crucial first hydrogenation of CO2 to CO, respectively. On the other hand, potassium strongly modulates the hydrogenation activity of Co, reducing to a large extent the formation of CH4via methanation while facilitating FTS chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge Dr. Omar El Tall, Dr. Jullian Vittenet, Dr. Selvedin Telalovic, Dr. Alla Dikhtiarenko, Dr. Anastasiya Bavykina, Dr. Natalia Morlanes, Dr. Youssef Saih, Dr. Luis Garzon Tovar and Mr. Il Son Khan at KAUST for useful discussions and technical support.
References
- D. Goud, R. Gupta, R. Maligal-Ganesh and S. C. Peter, Review of catalyst design and mechanistic studies for the production of olefins from anthropogenic CO2, ACS Catal., 2020, 10(23), 14258–14282 CrossRef CAS.
- X. Wang, C. Zeng, N. Gong, T. Zhang, Y. Wu, J. Zhang, F. Song, G. Yang and Y. Tan, Effective suppression of CO selectivity for CO2 hydrogenation to high-quality gasoline, ACS Catal., 2021, 11(3), 1528–1547 CrossRef CAS.
- B. Yao, T. Xiao, O. A. Makgae, X. Jie, S. Gonzalez-Cortes, S. Guan, A. I. Kirkland, J. R. Dilworth, H. A. Al-Megren, S. M. Alshihri, P. J. Dobson, G. P. Owen, J. M. Thomas and P. P. Edwards, Transforming carbon dioxide into jet fuel using an organic combustion-synthesized Fe-Mn-K catalyst, Nat. Commun., 2020, 11(1), 1–12 CrossRef PubMed.
- S. Dang, B. Qin, Y. Yang, H. Wang, J. Cai, Y. Han, S. Li, P. Gao and Y. Sun, Rationally designed indium oxide catalysts for CO2 hydrogenation to methanol with high activity and selectivity, Sci. Adv., 2020, 6(25), eaaz2060 CrossRef CAS PubMed.
- K. Stangeland, H. Li and Z. Yu, Thermodynamic analysis of chemical and phase equilibria in CO2 hydrogenation to methanol, dimethyl ether, and higher alcohols, Ind. Eng. Chem. Res., 2018, 57(11), 4081–4094 CrossRef CAS.
- F. Zeng, C. Mebrahtu, X. Xi, L. Liao, J. Ren, J. Xie, H. J. Heeres and R. Palkovits, Catalysts design for higher alcohols synthesis by CO2 hydrogenation: Trends and future perspectives, Appl. Catal., B, 2021, 291, 120073–120100 CrossRef CAS.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Power-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: a review, Energy Environ. Sci., 2020, 13(10), 3207–3252 RSC.
- C. Panzone, R. Philippe, A. Chappaz, P. Fongarland and A. Bengaouer, Power-to-Liquid catalytic CO2 valorization into fuels and chemicals: focus on the Fischer-Tropsch route, J. CO2 Util., 2020, 38, 314–347 CrossRef CAS.
- S. De, A. Dokania, A. Ramirez and J. Gascon, Advances in the design of heterogeneous catalysts and thermocatalytic processes for CO2 utilization, ACS Catal., 2020, 10(23), 14147–14185 CrossRef CAS.
- U. Rodemerck, M. Holeňa, E. Wagner, Q. Smejkal, A. Barkschat and M. Baerns, Catalyst development for CO2 hydrogenation to fuels, ChemCatChem, 2013, 5(7), 1948–1955 CrossRef CAS.
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons, Catal. Sci. Technol., 2017, 7(20), 4580–4598 RSC.
- X. Nie, W. Li, X. Jiang, X. Guo and C. Song, Recent advances in catalytic CO2 hydrogenation to alcohols and hydrocarbons, Adv. Catal., 2019, 65, 121–233 CAS.
- P. Gao, L. Zhang, S. Li, Z. Zhou and Y. Sun, Novel heterogeneous catalysts for CO2 hydrogenation to liquid fuels, ACS Cent. Sci., 2020, 6(10), 1657–1670 CrossRef CAS PubMed.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Heterogeneous catalytic CO2 conversion to value-added hydrocarbons, Energy Environ. Sci., 2010, 3(7), 884–890 RSC.
- T. Numpilai, N. Chanlek, Y. Poo-Arporn, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, M. Chareonpanich, P. Kongkachuichay, N. Yigit, G. Rupprechter and J. Limtrakul, Tuning interactions of surface-adsorbed species over Fe-Co/K-Al2O3 catalyst by different K contents: selective CO2 hydrogenation to light olefins, ChemCatChem, 2020, 12(12), 3306–3320 CrossRef CAS.
- X. Liu, C. Zhang, P. Tian, M. Xu, C. Cao, Z. Yang, M. Zhu and J. Xu, Revealing the Effect of Sodium on Iron-Based Catalysts for CO2 Hydrogenation: Insights from Calculation and Experiment, J. Phys. Chem. C, 2021, 125(14), 7637–7646 CrossRef CAS.
- B. Liang, H. Duan, T. Sun, J. Ma, X. Liu, J. Xu, X. Su, Y. Huang and T. Zhang, Effect of Na promoter on Fe-based catalyst for CO2 hydrogenation to alkenes, ACS Sustainable Chem. Eng., 2019, 7(1), 925–932 CrossRef CAS.
- Z. Shi, H. Yang, P. Gao, X. Chen, H. Liu, L. Zhong, H. Wang, W. Wei and Y. Sun, Effect of alkali metals on the performance of CoCu/TiO2 catalysts for CO2 hydrogenation to long-chain hydrocarbons, Chin. J. Catal., 2018, 39(8), 1294–1302 CrossRef CAS.
- T. Witoon, V. Lapkeatseree, T. Numpilai, C. K. Cheng and J. Limtrakul, CO2 hydrogenation to light olefins over mixed Fe-Co-K-Al oxides catalysts prepared via precipitation and reduction methods, Chem. Eng. J., 2022, 428, 131389–131400 CrossRef CAS.
- F. Yuan, G. Zhang, J. Zhu, F. Ding, A. Zhang, C. Song and X. Guo, Boosting light olefin selectivity in CO2 hydrogenation by adding Co to Fe catalysts within close proximity, Catal. Today, 2021, 371, 142–149 CrossRef CAS.
- T. Numpilai, N. Chanlek, Y. Poo-Arporn, S. Wannapaiboon, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, P. Kongkachuichay, M. Chareonpanich, G. Rupprechter, J. Limtrakul and T. Witoon, Pore size effects on physicochemical properties of Fe-Co/K-Al2O3 catalysts and their catalytic activity in CO2 hydrogenation to light olefins, Appl. Surf. Sci., 2019, 483, 581–592 CrossRef CAS.
- F. Jiang, B. Liu, S. Geng, Y. Xu and X. Liu, Hydrogenation of CO2 into hydrocarbons: enhanced catalytic activity over Fe-based Fischer-Tropsch catalysts, Catal. Sci. Technol., 2018, 8(16), 4097–4107 RSC.
- K. Y. Kim, H. Lee, W. Y. Noh, J. Shin, S. J. Han, S. K. Kim, K. An and J. S. Lee, Cobalt ferrite nanoparticles to form a catalytic Co-Fe alloy carbide phase for selective CO2 hydrogenation to light olefins, ACS Catal., 2020, 10(15), 8660–8671 CrossRef CAS.
- S. M. Hwang, S. J. Han, H. G. Park, H. Lee, K. An, K. W. Jun and S. K. Kim, Atomically Alloyed Fe-Co Catalyst derived from a N-coordinated Co single-atom structure for CO2 hydrogenation, ACS Catal., 2021, 11(4), 2267–2278 CrossRef CAS.
- H. Lu, C. Jiang, Z. Ding, W. Wang, W. Chu and Y. Feng, Effects of ultrasonic impregnation combined with calcination in N2 atmosphere on the property of Co3O4/CeO2 composites for catalytic methane combustion, J. Energy Chem., 2016, 25(3), 387–392 CrossRef.
- K. W. Jeon, J. W. Cho, H. R. Park, H. S. Na, J. O. Shim, W. J. Jang, B. H. Jeon and H. S. Roh, One-pot sol–gel synthesis of a CoMo catalyst for sustainable biofuel production by solvent-and hydrogen-free deoxygenation: effect of the citric acid ratio, Sustainable Energy Fuels, 2020, 4(6), 2841–2849 RSC.
- Q. Xu, X. Xu, G. Fan, L. Yang and F. Li, Unveiling the roles of Fe-Co interactions over ternary spinel-type ZnCoxFe2-xO4 catalysts for highly efficient CO2 hydrogenation to produce light olefins, J. Catal., 2021, 400, 355–366 CrossRef CAS.
- L. Zhang, Y. Dang, X. Zhou, P. Gao, A. P. van Bavel, H. Wang, S. Li, L. Shi, Y. Yang, E. I. Vovk, Y. Gao and Y. Sun, Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts, Innovation, 2021, 2(4), 100170 CAS.
- A. C. Ghogia, S. Cayez, B. F. Machado, A. Nzihou, P. Serp, K. Soulantica and D. Pham Minh, Hydrogen spillover in the Fischer-Tropsch synthesis on carbon-supported cobalt catalysts, ChemCatChem, 2020, 12(4), 1117–1128 CrossRef CAS.
- M. Ding, Y. Yang, B. Wu, Y. Li, T. Wang and L. Ma, Study on reduction and carburization behaviors of iron phases for iron-based Fischer-Tropsch synthesis catalyst, Appl. Energy, 2015, 160, 982–989 CrossRef CAS.
- Y. Zhang, C. Cao, C. Zhang, Z. Zhang, X. Liu, Z. Yang, M. Zhu, B. Meng, J. Xu and Y. F. Han, The study of structure-performance relationship of iron catalyst during a full life cycle for CO2 hydrogenation, J. Catal., 2019, 378, 51–62 CrossRef CAS.
- J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, T. P. Senftle, W. Liu, J. Wang, Y. Wang, A. Zhang, Q. Fu, C. Song and X. Guo, Dynamic structural evolution of iron catalysts involving competitive oxidation and carburization during CO2 hydrogenation, Sci. Adv., 2022, 8(5), eabm3629 CrossRef CAS PubMed.
- A. Ramirez, S. Ould-Chikh, L. Gevers, A. D. Chowdhury, E. Abou-Hamad, A. Aguilar-Tapia, J. L. Hazemann, N. Wehbe, A. J. Al Abdulghani, S. M. Kozlov, L. Cavallo and J. Gascon, Tandem conversion of CO2 to valuable hydrocarbons in highly concentrated potassium iron catalysts, ChemCatChem, 2019, 11(12), 2879–2886 CrossRef CAS.
- A. Russkikh, G. Shterk, B. H. Al-Solami, B. A. Fadhel, A. Ramirez and J. Gascon, Turning Waste into Value: Potassium-Promoted Red Mud as an Effective Catalyst for the Hydrogenation of CO2, ChemSusChem, 2020, 13(11), 2981–2987 CrossRef CAS PubMed.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, New horizon in C1 chemistry: breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels, Chem. Soc. Rev., 2019, 48(12), 3193–3228 RSC.
- J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei and S. Kawi, A review of recent catalyst advances in CO2 methanation processes, Catal. Today, 2020, 356, 471–489 CrossRef CAS.
- W. Chen, R. Pestman, B. Zijlstra, I. A. Filot and E. J. Hensen, Mechanism of cobalt-catalyzed CO hydrogenation: 1. Methanation, ACS Catal., 2017, 7(12), 8050–8060 CrossRef CAS PubMed.
- J. Tu, H. Wu, Q. Qian, S. Han, M. Chu, S. Jia, R. Feng, J. Zhai, M. He and B. Han, Low temperature methanation of CO2 over an amorphous cobalt-based catalyst, Chem. Sci., 2021, 12(11), 3937–3943 RSC.
- Q. Yang, A. Skrypnik, A. Matvienko, H. Lund, M. Holena and E. V. Kondratenko, Revealing property-performance relationships for efficient CO2 hydrogenation to higher hydrocarbons over Fe-based catalysts: Statistical analysis of literature data and its experimental validation, Appl. Catal., B, 2021, 282, 119554–119564 CrossRef CAS.
- P. R. Khangale, R. Meijboom and K. Jalama, CO2 hydrogenation to liquid hydrocarbons via modified Fischer-Tropsch over alumina-supported cobalt catalysts: effect of operating temperature, pressure and potassium loading, J. CO2 Util., 2020, 41, 101268 CrossRef CAS.
- C. G. Visconti, M. Martinelli, L. Falbo, L. Fratalocchi and L. Lietti, CO2 hydrogenation to hydrocarbons over Co and Fe-based Fischer-Tropsch catalysts, Catal. Today, 2016, 277, 161–170 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8(1), 1–9 CrossRef PubMed.
- J. Wei, J. Sun, Z. Wen, C. Fang, Q. Ge and H. Xu, New insights into the effect of sodium on Fe 3 O 4-based nanocatalysts for CO2 hydrogenation to light olefins, Catal. Sci. Technol., 2016, 6(13), 4786–4793 RSC.
- N. Boreriboon, X. Jiang, C. Song and P. Prasassarakich, Fe-based bimetallic catalysts supported on TiO2 for selective CO2 hydrogenation to hydrocarbons, J. CO2 Util., 2018, 25, 330–337 CrossRef CAS.
- B. Liang, T. Sun, J. Ma, H. Duan, L. Li, X. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Mn decorated Na/Fe catalysts for CO2 hydrogenation to light olefins, Catal. Sci. Technol., 2019, 9(2), 456–464 RSC.
- Y. Han, C. Fang, X. Ji, J. Wei, Q. Ge and J. Sun, Interfacing with carbonaceous potassium promoters boosts catalytic CO2 hydrogenation of iron, ACS Catal., 2020, 10(20), 12098–12108 CrossRef CAS.
- N. Chaipraditgul, T. Numpilai, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, C. Wattanakit, J. Limtrakul and T. Witoon, Tuning interaction of surface-adsorbed species over Fe/K-Al2O3 modified with transition metals (Cu, Mn, V, Zn or Co) on light olefins production from CO2 hydrogenation, Fuel, 2021, 283, 119248–119255 CrossRef CAS.
- J. Liu, A. Zhang, X. Jiang, M. Liu, J. Zhu, C. Song and X. Guo, Direct transformation of carbon dioxide to value-added hydrocarbons by physical mixtures of Fe5C2 and K-modified Al2O3, Ind. Eng. Chem. Res., 2018, 57(28), 9120–9126 CrossRef CAS.
- S. M. Hwang, C. Zhang, S. J. Han, H. G. Park, Y. T. Kim, S. Yang, K. W. Jun and S. K. Kim, Mesoporous carbon as an effective support for Fe catalyst for CO2 hydrogenation to liquid hydrocarbons, J. CO2 Util., 2020, 37, 65–73 CrossRef CAS.
- S. Wang, T. Wu, J. Lin, Y. Ji, S. Yan, Y. Pei, S. Xie, B. Zong and M. Qiao, Iron-potassium on single-walled carbon nanotubes as efficient catalyst for CO2 hydrogenation to heavy olefins, ACS Catal., 2020, 10(11), 6389–6401 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Bimetallic Fe-Co catalysts for CO2 hydrogenation to higher hydrocarbons, J. CO2 Util., 2013, 3, 102–106 CrossRef.
- Global Market Insights, Linear Alpha Olefins Market Size By Product, https://www.gminsights.com/industry-analysis/linear-alpha-olefins-market?utm_source=GoogleAds&utm_medium=Adwords&utm_campaign=Chemicals-PPC&gclid=CjwKCAjwur-SBhB6EiwA5sKtjvEAaDw-M4YDulPHwiKoTSAn9tGMwNFcqN-0ew3EXRiEuwlsbkz5-hoCuIsQAvD_BwE.
- L. Guo, J. Sun, X. Ji, J. Wei, Z. Wen, R. Yao, H. Xu and Q. Ge, Directly converting carbon dioxide to linear α-olefins on bio-promoted catalysts, Commun. Chem., 2018, 1(1), 1–8 CrossRef CAS.
- M. K. Gnanamani, H. H. Hamdeh, G. Jacobs, W. D. Shafer, S. D. Hopps, G. A. Thomas and B. H. Davis, Hydrogenation of Carbon Dioxide over K-Promoted FeCo Bimetallic Catalysts Prepared from Mixed Metal Oxalates, ChemCatChem, 2017, 9(7), 1303–1312 CrossRef CAS.
- M. K. Gnanamani, G. Jacobs, H. H. Hamdeh, W. D. Shafer, F. Liu, S. D. Hopps, G. A. Thomas and B. H. Davis, Hydrogenation of carbon dioxide over Co-Fe bimetallic catalysts, ACS Catal., 2016, 6(2), 913–927 CrossRef CAS.
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Advances in higher alcohol synthesis from CO2 hydrogenation, Chem, 2021, 7(4), 849–881 CAS.
- T. Yang, X. D. Wen, C. F. Huo, Y. W. Li, J. Wang and H. Jiao, Structure and energetics of hydrogen adsorption on Fe3O4 (1 1 1), J. Mol. Catal. A: Chem., 2009, 302(1–2), 129–136 CrossRef CAS.
- C. Lemire, R. Meyer, V. E. Henrich, S. Shaikhutdinov and H. J. Freund, The surface structure of Fe3O4 (1 1 1) films as studied by CO adsorption, Surf. Sci., 2004, 572(1), 103–114 CrossRef CAS.
- L. Zhu, K. L. Yao and Z. L. Liu, First-principles study of the polar (111) surface of Fe3O4, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74(3), 035409 CrossRef.
- X. Yu, C. F. Huo, Y. W. Li, J. Wang and H. Jiao, Fe3O4 surface electronic structures and stability from GGA+ U, Surf. Sci., 2012, 606(9–10), 872–879 CrossRef CAS.
- Y. Liu, C. Xu, W. Cen and H. Li, Design strategy of bifunctional catalysts for CO oxidation, Fuel, 2022, 320, 123909 CrossRef CAS.
- L. C. Grabow, B. Hvolbæk and J. K. Nørskov, Understanding trends in catalytic activity: the effect of adsorbate–adsorbate interactions for CO oxidation over transition metals, Top. Catal., 2010, 53(5), 298–310 CrossRef CAS.
- X. Tian, T. Wang, Y. Yang, Y. W. Li, J. Wang and H. Jiao, Surface Morphology of Cu Adsorption on Different Terminations of the Hägg Iron Carbide (χ-Fe5C2) Phase, J. Phys. Chem. C, 2015, 119(13), 7371–7385 CrossRef CAS.
- D. B. Cao, F. Q. Zhang, Y. W. Li and H. Jiao, Density functional theory study of CO adsorption on Fe5C2 (001), -(100), and -(110) surfaces, J. Phys. Chem. B, 2004, 108(26), 9094–9104 CrossRef CAS.
- R. Gao, D. B. Cao, Y. Yang, Y. W. Li, J. Wang and H. Jiao, Adsorption and energetics of H2O molecules and O atoms on the χ-Fe5C2 (111), (-411) and (001) surfaces from DFT, Appl. Catal., A, 2014, 475, 186–194 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Adrian
Ramirez
a,
Genrikh
Shterk
a,
Rafia
Ahmad
a,
Mustafa
Caglayan
a,
Adrian
Ramirez
a,
Genrikh
Shterk
a,
Rafia
Ahmad
a,
Mustafa
Caglayan