
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-chain multicolor-reporter templates for subcellular localization of molecular events in mammalian cells†
Sung-Bae
Kim
 *a,
Ramasamy
Paulmurugan
b,
Nobuo
Kitada
c and
Sojiro A.
Maki
c
*a,
Ramasamy
Paulmurugan
b,
Nobuo
Kitada
c and
Sojiro A.
Maki
c
aEnvironmental Management Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba 305-8569, Japan. E-mail: kimu-sb@aist.go.jp
bMolecular Imaging Program at Stanford, Bio-X Program, Stanford University School of Medicine, Palo Alto, California 94304, USA
cDepartment of Engineering Science, Graduate School of Informatics and Engineering, The University of Electro-Communications, Chofu 182-8585, Japan
First published on 3rd October 2023
Abstract
Single-chain multicolor-reporter imaging templates were developed for the subcellular localization of molecular events in mammalian cells. The templates were constructed by tandem linkage of fluorescent protein variants – fused with luciferases and the subcellular localization signal peptides. The templates simultaneously reported steroid hormonal activities at different optical spectra in the subcellular compartments. The templates contribute to the expansion of a toolbox of optical probes for subcellular localization of molecular events in intact cells.
Introduction
Luciferases emitting bioluminescence (BL) are sensitive optical reporters that can be used to interrogate biological processes in vitro and in vivo. BL is generated by the catalytic oxidation of luciferin with the specific enzyme luciferase in the presence of molecular oxygen (O2), and optionally Mg2+ and adenosine triphosphate (ATP).1,2Bioluminescence imaging (BLI) has clearly contrasted advantages and disadvantages over fluorescence imaging (FLI), which includes: (i) BLI does not require excitation light causing phototoxicity and the difficulty in separating excitation light-associated overlap in the optical signal. Instead, it needs a specific substrate for light emission;3 (ii) the weak BLI signals commonly allow very low background intensities, long linear dynamic ranges, and high signal-to-background (S/B) ratios.4 Because of these features, BLI has been considered a more quantitative imaging modality compared to FLI. The distinctive advantages of BL have been harnessed in the development of various multi-purpose BL imaging probes and which contributed to the expansion of the imaging toolbox.5,6
To date, excellent examples for conventional microscopic imaging have been developed using BL/FL probes: (i) multiplex imaging microscopy via bioluminescent phasor7—this is an efficient technology that helps in unmixing multiple BL signals based on their optical spectra, although it is necessary to have microscopes powered with multiple cameras; (ii) spectral unmixing algorithms are also a practical choice for analysing conventional multiplex imaging systems;8,9 (iii) specific tagging systems, such as HaloTag, to exclude optical contamination between multi-reporters;10 and (iv) multiplex reporter gene assay systems with stringent transcription machineries to minimize optical contamination between reporters, together with optical filters.11 Furthermore, (v) three bioluminescence resonance energy transfer (BRET) configurations were also reported for microscopic imaging, which all use DeepBlueC (DBlueC) as the Renilla luciferase (RLuc) substrate and acceptor fluorescent proteins (FPs) that can be excited at a similar wavelength (∼400 nm) but emit at different colors as a result of distinct Stokes shifts.12
Conventionally, the spatial resolution of BL has been considered inadequate for space-resolved imaging of molecular events in subcellular compartments of live cells, because of the broad spectral bandwidth and poor absolute intensities compared to FPs. This appears as a fundamental demerit of BL, considering that eukaryotic cells are compartmented into various organelles,13 and subcellular imaging probes are important for a deep understanding of life science and become powerful tools for investigating the molecular bases of a broad spectrum of major diseases.14 These clear contrasts of advantages and disadvantages of BLI inspired us to create multiplex imaging templates for determining subcellular molecular events of eukaryotic cells, such as steroid hormone-activated protein–protein interactions (PPIs), with compensating the individual demerits of BL or FL signals.
The present study demonstrates the feasibility of single-chain multicolor-reporter imaging templates in locating subcellular molecular events in living mammalian cells using an optimized optical intensity balance. First, we determined the optical properties of major marine luciferases with respect to the substrate specificity and color variance according to the luciferins. Based on our knowledge, we developed two distinctive single-chain reporter imaging templates, iRFP-R86SG-NLS and A16-mNep-MLS, for locating subcellular molecular events in eukaryotic cells, where iRFP-R86SG-NLS comprises NLS directing the probe to the nucleus, while A16-mNep-MLS contains MLS sequestering it to the endoplasmic reticulum (ER) and cell membrane compartments. The templates facilitate the simultaneous generation of intensity-balanced BL, FL, and BRET signals (meaning that one signal does not overwhelm the other signals) in the intended intracellular compartments. The utilities of the templates were further characterized in various applications, including monitoring of steroid hormonal activities in the subcellular compartments of animal cells. The developed single-chain multicolor reporter templates contribute to the expansion of the toolbox of optical probes for locating subcellular proteins in animal cells.
Results and discussion
Marine luciferases show distinctive substrate specificities to luciferins with unique optical and cellular localization properties in mammalian cells
For studying location specific molecular events in subcellular compartments, we first examined the suitability of various marine luciferases and luciferins for their signal intensities, spectral emission, and subcellular distribution in mammalian cells (Fig. 1[C]). The results show that ALuc16 and ALuc49 are the brightest luciferases upon reacting with nCTZ as a substrate in live MDA-MB231 cells, which is followed by R86SG and ALuc47. On the other hand, R86SG showed an extremely bright signal upon reacting with the substrate BBlue2.3 in the same type of cells.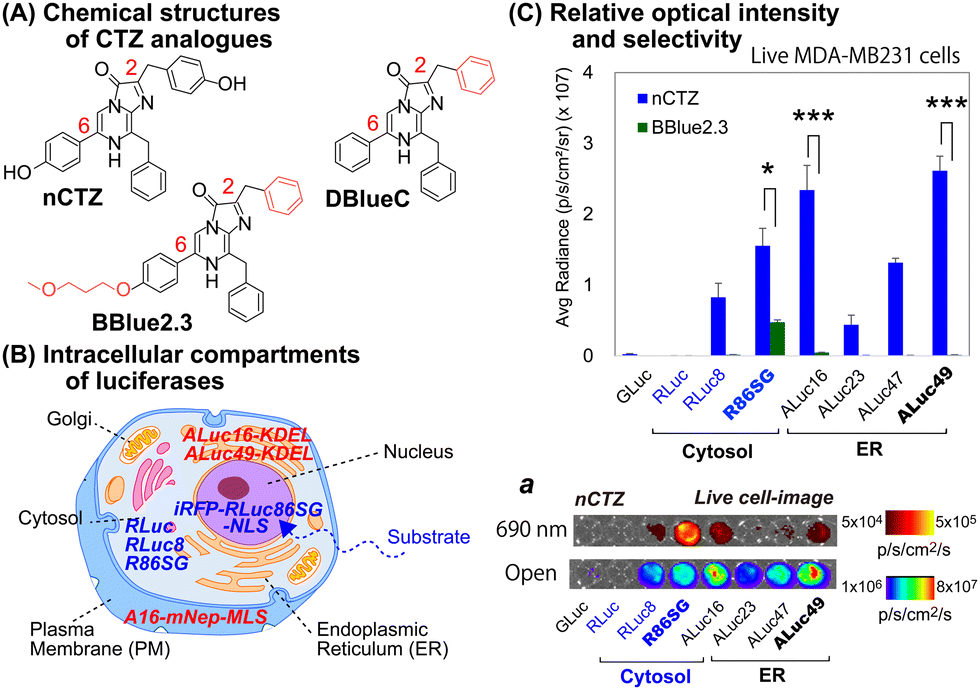 | ||
| Fig. 1 (A) The chemical structures of native coelenterazine (nCTZ) and its analogues (DBlueC and BBlue2.3) used in this study. The CTZ analogues diffuse into the intracellular compartments and react with the expressed marine luciferases. (B) Schematic illustration of a mammalian cell showing the location specific distribution of expressed marine luciferases. Each luciferase is designed to be sequestered into a distinctive cellular compartment, i.e., the cytosol, nucleus, plasma membrane (PM), or endoplasmic reticulum (ER). (C) The quantitative results of live cells that are selectively bright according to the substrates and luciferases used for the study. The absolute BL intensities were independently measured in triplicate from a single experiment (n = 3). The p-values (student t test) are *** ≤ 0.001, **≤ 0.01, and *≤ 0.05. Inset ‘a’ shows the representative BL images of Fig. 1(C), where the BL images were obtained in an open channel or with a 690 nm bandpass filter. | ||
The corresponding optical images in live mammalian cells were acquired using the IVIS Spectrum imaging system with an open or 690 nm bandpass filter (Fig. 1[C], inset ‘a’). The results show that R86SG is advantageous for reporting BL signals in the NIR range, although dominant absolute luciferase signals are observed in an open-filter condition (collecting lights emitted in the entire spectrum).
RLuc and its variants are known to be cytosolic proteins.15 In contrast, the ALuc variants are sequestered into the ER compartment as they contain a natural secretion peptide (SP) at the N-terminus16,17 and an ER retention signal peptide (KDEL) at the C-terminus. We have provided these details in the experimental section. The live cell images shown in Fig. 1(C), inset ‘a’, can be interpreted as the BL intensities of RLucs are localized mostly in the cytosolic compartment of the MDA-MB231 cells. On the other hand, the BL intensities of ALucs are commonly observed in the ER compartment due to the presence of the ER localization signal sequence. This feature is further confirmed by the microscopic images shown in Fig. 3. The subcellular localization and substrate specificity of marine luciferases were briefly summarized in Table 1.
| Substrates | Subcellular localization | ||
|---|---|---|---|
| nCTZ | ER | Cytosol | Nucleus |
| GLuc*, ALuc16-49* | RLuc8, RLuc86SG | iRFP-R86SG-NLS | |
| DBlueC | NLuc | ||
| BBlue2.3 | ALuc16*, ALuc23* | RLuc8, RLuc86SG | iRFP-R86SG-NLS |
The optical filter-based BL spectra reveal that RLucs change their colors based on the substrates used for imaging while ALucs show no variation
The BL spectral properties of RLucs and ALucs were investigated using a spectrophotometer equipped with a full set of bandpass filters covering the visible region in the presence of nCTZ and BBlue2.3 as substrates (Fig. 2). | ||
| Fig. 2 The BL spectra of marine luciferases in transiently transfected COS-7 cells. The upper panel shows the signals observed in live cells, while the lower panel shows the same in cell lysates. (A) and (B) The BL spectra of the live COS-7 cells transfected with different marine luciferases and imaged after the addition of nCTZ or BBlue2.3. (C) and (D) The BL spectra of the COS-7 cells in (A) and (B) in the lysates after the addition of nCTZ or BBlue2.3. | ||
In the context of live MDA-MB231 cells illuminated with the nCTZ substrate, the overall BL intensities of ALucs were superior to those of RLucs and their colors were in the green spectral region with a peak wavelength of ca. 500 nm. However, in the lysates, RLuc8 and R86SG showed enhanced BL intensities of approximately up to ten-fold compared to ALucs, and their peak emission wavelengths were in the green and yellow regions, respectively. When we compared the BL intensities of ALucs and ALuc23, we dramatically lost their BL intensities in the lysates compared to live cells. Although, the variants of ALucs such as ALuc16 and ALuc49 retained their BL intensities both in the lysates and in live cells.
In live MDA-MB231 cells with the BBLue2.3 substrate, the BL spectra of RLuc8 and R86SG were greatly shifted in the blue spectrum, whereas those of ALuc16 and ALuc23 retained their spectra in the green region. In contrast, ALuc47 and ALuc49 did not emit any notable BL signals with the substrate BBLue2.3. The BL intensities of ALuc16 in the lysates were decreased to as low as tenfold compared to the same in live cells, and for ALuc23 the signal intensity was at the background level. In contrast, R86SG retained the BL intensities even in the lysates. The peak wavelength values (λmax) of R86SG in the lysates were dramatically blue shifted in relation to the BBLue2.3 substrate, i.e., the λmax value of R86SG was at 550 nm with nCTZ, whereas the same was found to be at ca. 400 nm with the BBlue2.3 substrate.
All these results may be summarized as follows: (i) in live cells, the greater intensity variance between ALucs and RLucs allows us to ignore the contribution of the weaker reporter side, granting intensity-driven unmixing to multiplex reporter systems; (ii) R86SG and ALuc16 are robust and less influenced by the assay condition, indicating that these luciferases are suitable reporters for designing a wide range of bioassay systems; (iii) the BL emission peak of R86SG is dramatically variable according to the substrates used, whereas ALuc16 variants are substrate independent, and which allows optical filter-driven unmixing to develop multiplex reporter systems; (iv) the BBlue2.3 substrate is exclusively reactive with ALuc16 and ALuc23, but completely unreactive with ALuc47 and ALuc49, implicating the potential of substrate-driven unmixing in developing multiplex reporter systems; and (v) the detergents in the lysis buffer severely suppress the enzymatic activities of some ALucs such as ALuc23, implicating the detergent-driven unmixing to develop multiplex reporter systems.
The bar graphs in Fig. S1(A) and S1(B) (ESI†) exhibit that the blue and green bandpass filters can successfully discriminate the BL intensities from R86SG and ALuc16, respectively. These results feature the optical filter-driven unmixing of BL signals in multiplex reporter systems.
The combination of substrate- and optical filter-driven unmixing strategies of marine luciferases was able to locate cellular compartment specific BL signals in live cells
The substrates- and filter-driven unmixing of the BL signals were determined with the live cells expressing iRFP-R86SG, NLuc, or ALuc16 (Fig. S2[A] and S2[B], ESI†). The strong and linear BL signals were determined according to varying ratios of living COS-7 cells containing each marine luciferase. By increasing the portion of COS-7 cells containing iRFP-R86SG or NLuc, all the BL intensities by the DBlueC substrate showed a linear increase in BL signal, as shown in Fig. S2(A) (ESI†). When we measured the signals using different optical filters, i.e., by open filter, the BL intensities of iRFP-R86SG were not different from those of NLuc. However, through 720 nm and 500 nm bandpass filters, the BL intensities from iRFP-R86SG and NLuc, respectively, were clearly specified. The overall correlation coefficients (R2) observed were found to be between 0.985 and 0.997.The BL signals produced by the BBLue2.3 substrate were also linearly enhanced by increasing the amounts of COS-7 cells as shown in Fig. S2(B) (ESI†), where we measured the BL signals with different bandpass filters, i.e., the BL signals of COS-7 cells containing iRFP-R86SG or ALuc16 were linearly elevated by increasing the amounts of COS-7 cells under the 500 nm and 720 nm bandpass filters. The correlation coefficients were around 0.989 and 0.985.
The A16-mNep-MLS and iRFP-R86SG-NLS imaging probes can localize in the membrane and the nuclear compartments respectively and facilitate sub-cellular imaging applications
The sub-cellular localization properties of the BRET probes, A16-mNep-MLS and iRFP-R86SG-NLS, were confirmed with optical imaging of live cells (Fig. 3[A] and [B]). The pseudo green colors shown in Fig. 3(A) are the images of cells monitored by ER-tracker™ Green (Invitrogen), which highlights the cytosolic and the nuclear compartments of the cells. The pseudo red colors shown in the upper panel of Fig. 3(A) are the signals generated in cells transfected by mNep and iRFP in A16-mNep-MLS and iRFP-R86SG-NLS, respectively. The red-color image shown at the top panel is the signal from A16-mNep-MLS at the ER and the cell membrane compartments. The translocation mechanism is explained as that the secretion peptide (SP) located at the N-terminal end of ALuc16 localizes the luciferase protein to the ER and the membrane compartments, whereas the whole protein is anchored by the fused MLS.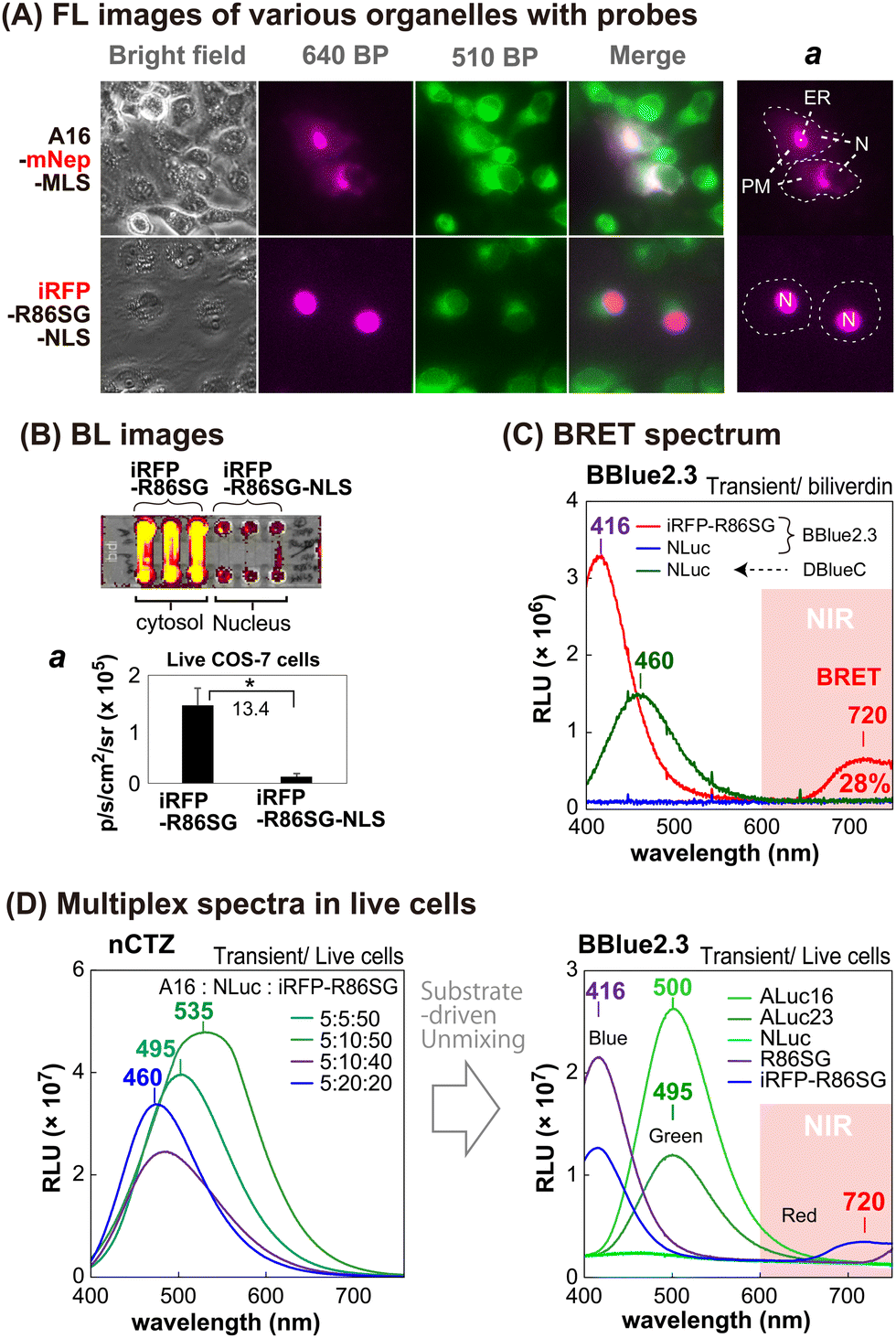 | ||
| Fig. 3 Cellular localization of various luciferases in living mammalian cells. (A) Fluorescence images of the cells showing the subcellular localization of luciferase probes. The red FL images in the top and bottom panels showing the location of A16-mNep-MLS (mostly in the ER and PM) and iRFP-R86SG-NLS (mostly in the nucleus), respectively. Section ‘a’ specifies the location of the two probes. The capital letters, ER, PM, and N, in the image abbreviate the endoplasmic reticulum (ER), the plasma membrane (PM), and the nucleus (N), respectively. (B) NIR BL images of microslides grown with the COS-7 cells expressing iRFP-R86SG and iRFP-R86SG-NLS. Inset ‘a’ showing the variance in the absolute intensities. (C) BRET spectrum of iRFP-R86SG in the presence of BBlue2.3. The spectra of NLuc were referenced. The numbers showing the wavelengths at the peak BL intensities. (D) Separation of the multiplex spectra according to the substrates. The annotated numbers in the left panel showing the mixing ratios of the live cells containing ALuc16, NLuc, and iRFP-R86SG, respectively. The right panel exhibits the substrate-driven unmixing of the BL spectra of various marine luciferases. | ||
On the other hand, in the red-colored images shown in Fig. 3(A), the bottom panel is the BL signal from the iRFP-R86SG-NLS found at the nucleus. The red image is clearly separated from the green image in the live cells. This localization confirms that iRFP-R86SG-NLS is successfully translocated into the nucleus upon expression in cells.
Localization of reporter proteins at the cytosol is advantageous over them localized at the nucleus with respect to their overall BL intensities
We further investigated the advantages of localizing iRFP-R86SG at the cytosol over locating at the nucleus with BL images in live COS-7 cells (Fig. 3[B]). The basic probe design of iRFP-R86SG is originated from our previous study.18The BL images showing the live cells containing iRFP-R86SG (left three columns) are approximately 13.4 times brighter when expressed in the cytosol compared to the same expressed in the nucleus as shown by expressing iRFP-R86SG with and without the NLS signal peptide (right three columns).
The greater BL intensities of iRFP-R86SG compared to iRFP-R86SG-NLS may be explained by the following factors: (i) the substrate is easily accessible to iRFP-R86SG located at the cytosol, compared to iRFP-R86SG-NLS in the nucleus, because the substrate has to be diffused into the nucleus by bypassing two lipophilic membranes to react with iRFP-R86SG-NLS; (ii) the steric hindrance associated with the presence of an additional tag, in this case the NLS, could compromise the protein function and reduce the overall BL signal; and (iii) the overall quantity of proteins that can accumulate in the nucleus could be less compared to the cytosol, due to the compartment size difference.
BRET spectra confirm that the BL signal of iRFP-R86SG can be unmixed from the BL signals of other marine luciferases in the presence of substrate BBlue2.3
We further investigated whether the BL signals of marine luciferases localized in different intracellular compartments can be unmixed to further understand the cellular location-specific activity (Fig. 3[C] and [D]).The BL spectrum shown in Fig. 3(C) revealed two BL peaks (λmax) at 416 nm and 720 nm for iRFP-R86SG, which are interpreted as the peaks of R86SG and iRFP (BRET), respectively. In the presence of substrate BBlue2.3, R86SG emits dominantly blue-shifted BL at 416 nm. The resonance energy is transferred to the adjacent iRFP, which exerts a characteristic emission peak at 720 nm. The portion longer than 650 nm in the spectra occupied approximately 28% of the total BL intensity. This NIR signal greatly contributes to imaging molecular events occurring in the deep tissues of animal models. On the other hand, NLuc showed fairly good BL intensity with a spectrum peaked at 460 nm with substrate DBlueC, but not with BBlue2.3. These results confirm that the substrate BBlue2.3 can be used to unmix the BL signals of iRFP-R86SG from NLuc in a multiplex reporter system.
The blue-shifted emission peak at 416 nm of iRFP-R86SG can be explained as follows: it is well known that substrate nCTZ shows a broad emission spectrum from blue to green (400–535 nm) when reacting with marine luciferases, because it generates four different energy levels of intermediates during the oxidation reaction, i.e., neutral species, amide anion, phenolate anion, and pyrazine anion19,20 In the case of BBLue2.3 substrate, the OH group at C-6 position of nCTZ was substituted with an alkyl ether group. This substitution prevents the formation of the anion intermediates but facilitates the neutral form upon the enzymatic oxidation, which results in an enhanced blue-shifted emission peak at 417 nm.
We further examined whether multiple BL signals collected from a single imaging experiment can be separated by the BBlue2.3 substrate in a multiplex reporter system (Fig. 3[D]). We first made a series of mixtures with live COS-7 cells expressing ALuc16, NLuc, and iRFP-R86SG, where Aluc16 is located in the ER compartment while NLuc and iRFP-R86SG are mostly sequestered in the cytosolic compartment. The mixtures were illuminated with nCTZ or Bblue2.3, and the BL signal collected from the sample was used for spectral unmixing.
In the presence of nCTZ, the spectra of Aluc16, NLuc, and iRFP-R86SG were almost superimposed, where the BL spectra of the mixture simply appeared as a single broad band image, no matter which ratios of the signals were applied. By increasing the portion of the cells containing iRFP-R86SG, the band peaks looked prominent at around 535 nm. In the presence of BBlue2.3, the spectra of R86SG and iRFP-R86SG were strongly blue-shifted and peaked at 416 nm. The spectrum of iRFP-R86SG additionally showed a peak at around 720 nm, which confirms the BRET signal generated by the resonance energy transfer from R86SG to iRFP. On the other hand, the BL spectra of ALuc16 and ALuc23 were independent of the substrates used for the study. NLuc did not luminesce with the BBlue2.3 substrate. The results collectively indicate that the multiple superimposed spectra of various marine luciferases can be unmixed after imaging the BL signals using specific substrates and optical filters. The unmixed signal also locates the molecular events in the intracellular compartments.
The BRET signal of iRFP-R86SG implicates the following facts: R86SG shows blue-shifted BL spectra with a specially designed BBlue2.3 substrate because the emission spectrum is superimposed with the Soret band of the iRFP. The merits of this probe design are that: (i) low background BL intensities are expected in vivo and in vitro because its blue BL emission is attenuated in the tissues and in physiological samples, expecting the minimized background intensities, and (ii) the large blue-to-NIR shifts of the resonance energy allow highly efficient BL imaging in animal models without any overlap from the donor signal.
Simultaneous monitoring of FL and BL signals from the nucleus and the cytosol of living cells and the signal unmixing.
We consecutively monitored the FL and BL signals simultaneously from the cellular compartments of living mammalian cells and unmixed them using selective filters and specific substrates (Fig. S3, ESI†).After transient transfection of COS-7 cells with pcDNA3.1(+) vector encoding A16-mNep-MLS or iRFP-R86SG-NLS, we found that the FL images of A16-mNep-MLS and iRFP-R86SG-NLS were sequestered in the nucleus and the membrane compartments, respectively. The BL images indicate that the BL signals can be unmixed through combining the optical filters and the substrates: i.e., the BL signal from NLuc can be screened using a 500 nm BP filter and DBlueC substrate, whereas the BL signals from A16-mNep-MLS and iRFP-R86SG-NLS can be unmixed from the NLuc signal through combining nCTZ and BBlue2.3 substrates with 640 nm and 720 nm BP filters, respectively. The BL spectra confirmed that the BL signals from A16-mNep-MLS and iRFP-R86SG-NLS luminesce with blue/NIR and green colors, respectively. The BL spectra also showed unique cross-section points at 450 nm and 690 nm that mimic an isosbestic point of the absorbance spectra for these reporters. The ratiometric BL spectra also confirm that the BL signals of the cells are highly quantitative and possible to spectrally unmix using BBlue2.3 as a substrate.
Locating of steroid hormonal activities in the ER and membrane compartments of mammalian cells
We further determined if this unique optical template works as a single-chain imaging probe for locating subcellular molecular events such as protein-folding in mammalian cells (Fig. 4). This feature was determined with a modified version of the template, where the ligand binding domain of human estrogen receptor (ER LBD: 281–549 AA) was placed in between ALuc16 and mNeptune of the template, and finally tagged with MLS (named A16-ERLBD-mNep-MLS).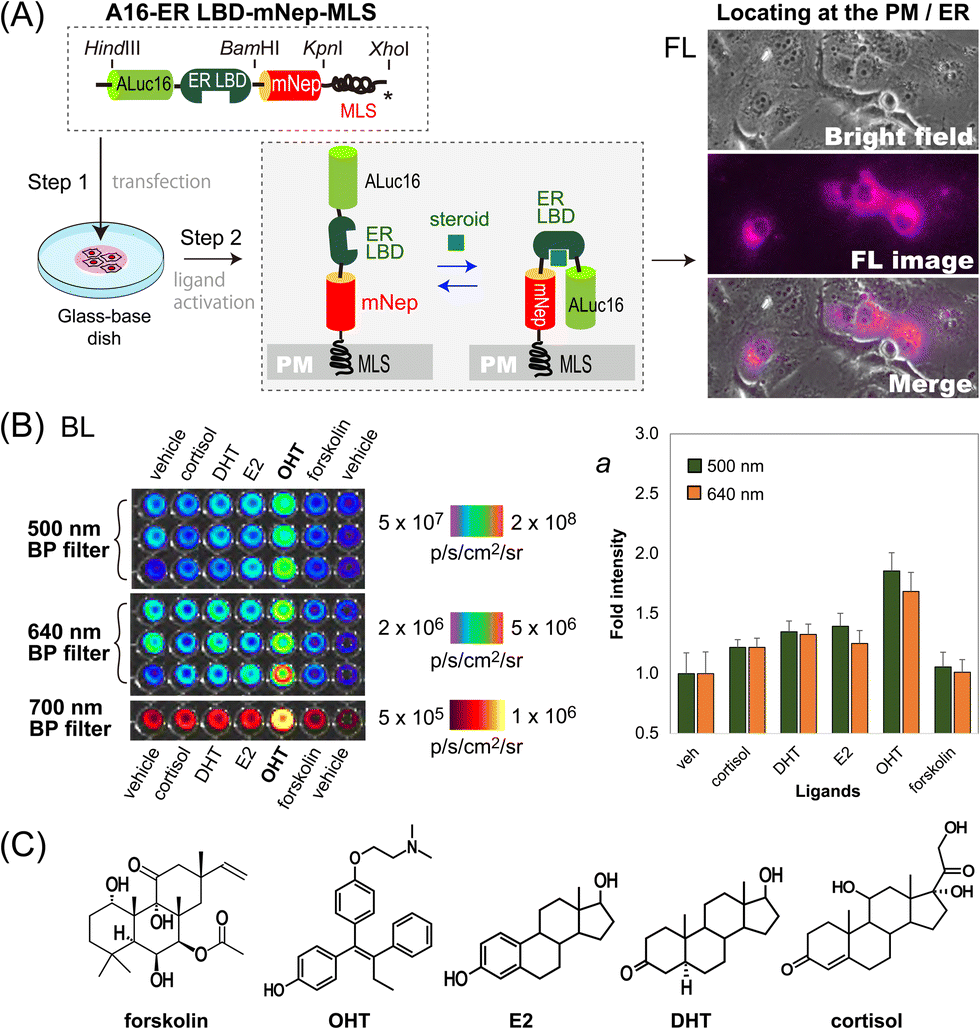 | ||
| Fig. 4 Locating of steroid hormone activities in the subcellular compartments of mammalian cells. (A) Determination of FL images for the sub-cellular localization of A16-ER LBD-mNep-MLS. The probe was designed to transform in response to steroid hormones. (B) Determination of the BL images of the cells after stimulation using different ER ligands. Inset ‘a’ represents the corresponding quantitative values of the BL images. (C) Chemical structures of the ligands used in this study. The ligands include endogenous steroids and its antagonist. | ||
After transient transfection of COS-7 cells using a mammalian expression vector encoding A16-ERLBD-mNep-MLS, we determined the FL images of the live cells. The results in Fig. 4(A) showed that the FL images indeed cover the whole cell boundaries and the ER compartment, meaning that A16-ERLBD-mNep-MLS is properly located in the ER and plasma membrane (PM) compartments.
We further stimulated the COS-7 cells with various ligands: i.e., 17β-estradiol (E2), dihydrotestosterone (DHT), and cortisol as endogenous steroid hormones; 4-hydroxytamoxifen (OHT) as a selective estrogen receptor antagonist; forskolin as an adenylate cyclase activator; and 0.1% (v/v) DMSO as the vehicle negative control. The BL intensities were the highest with the cells stimulated by OHT in both measurements using 500 nm and 640 nm BP filters (Fig. 4[B]). The BL intensities were approximately 1.9- and 1.7-fold higher than those of the vehicle. The cells stimulated by E2, DHT, or cortisol emitted 20–40% elevated BL intensities compared to those by the vehicle. In contrast, the cells activated by forskolin did not enhance the BL intensities, compared with the vehicle.
Overall, the evidence shows that: (i) steroid-activated A16-ERLBD-mNep-MLS enhances the BL intensities in the PM and ER compartments of the animal cells; (ii) moreover, A16-ERLBD-mNep-MLS is selectively bright with the estrogen antagonist OHT, compared with the agonist E2; and (iii) the optical intensities obtained at 640 nm are contributed by the leakage of the ALuc16 signal and the BRET signal from mNeptune, considering that the fluorescent protein mNeptune itself cannot auto-luminesce.
The antagonist specificity corresponds with our previous studies on the ER probes based on protein-fragment complementation, where the antagonist transforms the α-helix12 of the ER LBD in a way to approximate the fragmented luciferases closer compared to the agonists.21,22 Moreover, it is also known that fragmented luciferases in fusion proteins are expressed prematurely and start folding only when they are brought into proximity.23
Considering the above references, the working mechanism of A16-ERLBD-mNep-MLS may be explained as follows: the OHT-activated ER LBD replaces the position of the α-helix12 in a way to approximate the adjacent ALuc16 and mNeptune closer to each other. This association triggers the maturation (folding) of ALuc16 inside the probe, enhancing the BL intensities.
Conclusions
The present study introduces unique multiplex imaging templates for location-specific imaging of molecular events that are occurring in the subcellular compartments of living mammalian cells. We examined many optimal reporters for subcellular imaging and found that the variants of marine luciferases such as R86SG and ALuc49 are useful because they exhibit unique substrate specificity, robust optical brightness, and responsiveness to directed compartment-specific signal peptides by expression in the subcellular compartments. The optical properties were harnessed for imaging subcellular molecular events with two distinctive single-chain multi-reporter imaging templates named iRFP-R86SG-NLS and A16-mNep-MLS. The templates facilitated the simultaneous generation of intensity-balanced BL, FL, and BRET signals (meaning that one signal does not overwhelm the other signals) and consecutive monitoring of a protein of interest in the intended intracellular compartments. We showed that the optical signals can be unmixed based on specific substrates, detergents, or optical filters. We also exemplified the utility of the template with a quantitative imaging of hormonal activities in the PM and ER compartments using a specially designed probe A16-ER LBD-mNep-MLS. Likewise, the templates can be freely modified to locate other molecular events of interest into intracellular compartments. This subcellular imaging modality contributes to the expansion of the toolbox of optical probes that are detectable using standard microscopes and other optical imaging systems for locating subcellular molecular events in living mammalian cells.Author contributions
S.-B.K. contributed conceptualization, methodology, investigation, resources, data curation, writing, and supervision; R.P. contributed validation, formal analysis, and data curation; N.K. contributed methodology, investigation, resources, and data curation; S.A.M. contributed resources, data curation, writing, and supervision; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partly supported by grants from the Japan Society for the Promotion of Science (JSPS Grants 23KK0101, 21H04948, 20K21851, and 17H01215).Notes and references
- O. Shimomura, Bioluminescence, World Scientific Publishing Co. Pte. Ltd, Singapore, 2006 Search PubMed.
- Y. Y. Jiang, X. R. Shi, C. Tang and F. Wang, Coord. Chem. Rev., 2023, 481, 215045 CrossRef CAS.
- T. Ozawa, H. Yoshimura and S. B. Kim, Anal. Chem., 2013, 85, 590–609 CrossRef CAS PubMed.
- S. B. Kim and R. Paulmurugan, Anal. Sci., 2021, 37, 233–247 CrossRef CAS PubMed.
- M. M. Calabretta, D. Gregucci, H. Martinez-Perez-Cejuela and E. Michelini, Biosensors, 2022, 12, 742 CrossRef CAS PubMed.
- Z. Yao, B. S. Zhang and J. A. Prescher, Curr. Opin. Chem. Biol., 2018, 45, 148–156 CrossRef CAS PubMed.
- Z. Yao, C. K. Brennan, L. Scipioni, H. T. Chen, K. K. Ng, G. Tedeschi, K. Parag-Sharma, A. L. Amelio, E. Gratton, M. A. Digman and J. A. Prescher, Nat. Methods, 2022, 19, 893 CrossRef CAS PubMed.
- S. T. M. Gammon, W. M. Leevy, S. Gross, G. W. Gokel and D. Piwnica-Worms, Anal. Chem., 2006, 78, 1520–1527 CrossRef CAS PubMed.
- F. Cutrale, V. Trivedi, L. A. Trinh, C. L. Chiu, J. M. Choi, M. S. Artiga and S. E. Fraser, Nat. Methods, 2017, 14, 149–152 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohane, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- A. Sarrion-Perdigones, L. Chang, Y. Gonzalez, T. Gallego-Flores, D. W. Young and K. J. T. Venken, Nat. Commun., 2019, 10, 5710 CrossRef CAS PubMed.
- B. Breton, E. Sauvageau, J. Zhou, H. Bonin, C. Le Gouill and M. Bouvier, Biophys. J., 2010, 99, 4037–4046 CrossRef CAS PubMed.
- B. Alberts, Molecular biology of the cell, Garland Pub., New York, 3rd edn, 1994 Search PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- A. Kaihara, Y. Kawai, M. Sato, T. Ozawa and Y. Umezawa, Anal. Chem., 2003, 75, 4176–4181 CrossRef CAS PubMed.
- S. B. Kim, M. Torimura and H. Tao, Bioconjugate Chem., 2013, 24, 2067–2075 CrossRef CAS PubMed.
- S. B. Kim, R. Nishihara, D. Citterio and K. Suzuki, ACS Comb. Sci., 2017, 19, 594–599 CrossRef CAS PubMed.
- R. Nishihara, R. Paulmurugan, T. Nakajima, E. Yamamoto, A. Natarajan, R. Afjei, Y. Hiruta, N. Iwasawa, S. Nishiyama, D. Citterio, M. Sato, S. B. Kim and K. Suzuki, Theranostics, 2019, 9, 2646–2661 CrossRef CAS PubMed.
- R. Nishihara, H. Suzuki, E. Hoshino, S. Suganuma, M. Sato, T. Saitoh, S. Nishiyama, N. Iwasawa, D. Citterioa and K. Suzuki, Chem. Commun., 2015, 51, 391–394 RSC.
- O. Shimomura, Biochem. J., 1995, 306, 537–543 CrossRef CAS PubMed.
- R. Paulmurugan and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15883–15888 CrossRef CAS PubMed.
- S. B. Kim, Y. Umezawa, K. A. Kanno and H. Tao, ACS Chem. Biol., 2008, 3, 359–372 CrossRef CAS PubMed.
- S. W. Michnick, Curr. Opin. Struct. Biol., 2001, 11, 472–477 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows the quantitative information on the spectra of Fig. 2; Fig. S2 shows the quantitative relationship between the BL intensities of live cells containing iRFP-R86SG or NLuc. Suppl. Methods comprise all the experimental procedures of this study. See DOI: https://doi.org/10.1039/d3cb00077j |
| This journal is © The Royal Society of Chemistry 2023 |