
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biotransformation-coupled mutasynthesis for the generation of novel pristinamycin derivatives by engineering the phenylglycine residue†
Oliver
Hennrich
a,
Leoni
Weinmann
b,
Andreas
Kulik
 c,
Karen
Harms
d,
Philipp
Klahn
ef,
Jung-Won
Youn
b,
Frank
Surup
d and
Yvonne
Mast
*agh
c,
Karen
Harms
d,
Philipp
Klahn
ef,
Jung-Won
Youn
b,
Frank
Surup
d and
Yvonne
Mast
*agh
aDepartment Bioresources for Bioeconomy and Health Research, Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Inhoffenstraße 7B, 38124 Braunschweig, Germany. E-mail: yvonne.mast@dsmz.de
bInstitute of Microbiology, University Stuttgart, Allmandring 31, D-70569 Stuttgart, Germany
cDepartment Microbial Bioactive Compounds, Interfaculty Institute of Microbiology and Infection Medicine, Faculty of Science, University of Tübingen, Auf der Morgenstelle 28, D-72076 Tübingen, Germany
dMicrobial Drugs Department, Helmholtz-Centre for Infection Research, 38124 Braunschweig, Germany
eDivision of Organic and Medicinal Chemistry, Department of Chemistry and Molecular Biology, University of Gothenburg, Kemigården 4, 412 96 Göteborg, Sweden
fCentre of Antimicrobial Resistance Research in Gothenburg (CARe), Gothenburg, Sweden
gTechnische Universität Braunschweig, Institut für Mikrobiologie, Rebenring 56, 38106 Braunschweig, Germany
hGerman Center for Infection Research (DZIF), Partner Site Tübingen, Tübingen, Germany
First published on 14th October 2023
Abstract
Streptogramins are the last line of defense antimicrobials with pristinamycin as a representative substance used as therapeutics against highly resistant pathogenic bacteria. However, the emergence of (multi)drug-resistant pathogens renders these valuable antibiotics useless; making it necessary to derivatize compounds for new compound characteristics, which is often difficult by chemical de novo synthesis due to the complex nature of the molecules. An alternative to substance derivatization is mutasynthesis. Herein, we report about a mutasynthesis approach, targeting the phenylglycine (Phg) residue for substance derivatization, a pivotal component of streptogramin antibiotics. Mutasynthesis with halogenated Phg(-like) derivatives altogether led to the production of two new derivatized natural compounds, as there are 6-chloropristinamycin I and 6-fluoropristinamycin I based on LC-MS/MS analysis. 6-Chloropristinamycin I and 6-fluoropristinamycin I were isolated by preparative HPLC, structurally confirmed using NMR spectroscopy and tested for antimicrobial bioactivity. In a whole-cell biotransformation approach using an engineered E. coli BL21(DE3) pET28-hmo/pACYC-bcd-gdh strain, Phg derivatives were generated fermentatively. Supplementation with the E. coli biotransformation fermentation broth containing 4-fluorophenylglycine to the pristinamycin mutasynthesis strain resulted in the production of 6-fluoropristinamycin I, demonstrating an advanced level of mutasynthesis.
1. Introduction
Antibiotics are undoubtedly the most important and impactful medical advancement of all time and a cornerstone of modern medicine.1,2 However, the rapid emergence of (multi)drug-resistant pathogens endangers their efficacy and poses a serious threat to global human health.3 According to a recent estimate, more than 1.2 million people died worldwide in 2019 due to drug-resistant bacterial infections.4 Accompanying this development, the pipeline for new antibacterial agents was feared to run empty in recent years with only slightly encouraging rising numbers of drug candidates from 2017 onwards.5,6 Especially, new compound classes are rarely found. The most recent new compound classes that have been introduced to the market are cyclic lipopeptides (daptomycin by Cubist Pharmaceuticals in 2003) and oxazolidinones (linezolid by Pfizer in 2000).7 Thus, discovering and developing new antimicrobials and strategies to combat resistance is a key challenge in the years ahead.The most important sources for many powerful antibiotics are actinomycetes and fungi, which are the origin of up to two-thirds of all antibiotics in clinical use today.8 These involve all different classes of antibiotics, such as aminoglycosides, macrolides, tetracyclines, glycopeptide antibiotics, β-lactams, rifamycins, or streptogramins.8 While the biosynthetic potential of actinomycetes for unknown antibiotics is still considered high,9,10 there are also other approaches that aim to get access to novel bioactive substances, as for instance the structural modification of already existing potent bioactive drugs.11 A good example of modification of a chemical scaffold is the macrolide antibiotic erythromycin, where the natural compound served as a starting point for chemical modification at the C12 position, resulting in a series of novel ketolides.12 All modified compounds showed high activity against Gram-positive bacteria, such as Staphylococcus aureus, Streptococcus pneumoniae, and Streptococcus pyogenes.12 The challenge in modifying such a kind of antibiotics lies in their complex chemical structures, which originate from a multistep biosynthesis. Many compounds are difficult to generate by chemical or semi-synthesis, which limits the ability to optimize compounds by chemical derivatization.13,14 For example, the chemical synthesis of the aglycone of the glycopeptide antibiotic vancomycin involves 19 chemical steps and is associated with several problems, notably the lack of kinetic atroposelectivity in the construction of the tricyclic scaffold, low overall yields, and the difficult synthesis of unnatural amino acids.15,16
Genetic engineering methods are suitable alternatives for the targeted derivatization of complex natural products.17 These methods are based on the biotechnological manipulation of genes involved in antibiotic biosynthesis, which in bacterial antibiotic producers are usually organized in biosynthetic gene clusters (BGCs).18 Mutasynthesis is a method of metabolic engineering that combines aspects of chemical synthesis with genetic engineering techniques (= mutational biosynthesis). The principle of mutasynthesis involves the genetic disruption of one or more gene(s) responsible for the biosynthesis of a key precursor, with subsequent supply of synthetic analogues of the missing precursor, designated as mutasynthons. The incorporation of a mutasynthon into the biosynthetic assembly line of the secondary metabolite then leads to the formation of novel compound derivatives (Fig. 1).19 In this way, mutasynthesis combines genetic manipulation with the principle of precursor-directed biosynthesis20 to exploit substrate promiscuity in the biosynthetic assembly lines of natural product producers.13 This can be used to modify complex natural products that are inaccessible or difficult to access by means of chemical synthesis.13,14,21 The advantage of mutasynthesis over precursor-directed biosynthesis is that by using a precursor block mutant, there is no competition between the natural precursor and the unnatural mutasynthon building block, resulting in the biosynthesis of the desired mutasynthesis product only. A prerequisite for this powerful technology is the knowledge of the biosynthetic pathway, especially that the respective precursor genes are known, the genetic tractability of the antibiotic producer strain, as well as that the mutasynthon is readily taken up by the cell and accepted by the biosynthetic machinery.19,22–24 Mutasynthesis has been successfully applied for the derivatization of various compound classes, such as polyketides,25–27 nonribosomal peptides28 glycopeptide antibiotics,14 aminoglycosides,29 alkaloids,30 aminocoumarins,31 or lincomycins.32 For example, regarding drug optimization, mutasynthesis has successfully been applied to generate derivatives of lincomycin, which showed increased activities against low-level lincosamide resistant staphylococci compared to the original compound.32 Another example includes a series of novel physostigmine alkaloids generated by mutasynthesis, where one of the modified compounds exhibited an improved selectivity and toxicity profile.30
 | ||
| Fig. 1 Schematic presentation of the mutasynthesis principle. Colored hexagons represent the different antibiotic building blocks, which are assembled by the biosynthetic machinery. For mutasynthesis, a block mutant is required where the synthesis of a building block is abolished. Feeding with an unnatural analog (mutasynthon (yellow hexagon)) results in the synthesis of an altered product. The provision of the mutasynthon as a result of the conversion of the precursor (orange hexagon) by the biotransformation step (bacteria symbol) is indicated below. | ||
Streptogramin antibiotics are a substance class that with pristinamycins (Synercid®, Pyostacin®) have found their way into clinical applications. Pristinamycins are used as an antibiotic of last resort to treat multidrug-resistant bacterial infections.33 These antibiotics show good activities against numerous Gram-positive pathogens, including methicillin- and vancomycin-resistant Staphylococcus aureus (MRSA, VRSA) and vancomycin-resistant Enterococcus faecium strains (VREF). Despite the longstanding use of pristinamycin for more than 50 years, especially in French-speaking countries, the frequency of resistance remained low but has increased in recent years,33 which makes it necessary to optimize the substance in terms of broad-spectrum activity and resistance breaking characteristics, which can be achieved by derivatization of the compound by mutasynthesis. However, so far mutasynthesis has been prevented by the lack of information about precursor biosynthesis and its encoding genes. In particular, the genes coding for the biosynthesis of the non-proteinogenic amino acid L-phenyglycine (L-Phg), which is a constituent of the peptide antibiotic pristinamycin I (1), were not known until recently.
L-Phg is essential for the bioactivity of streptogramin antibiotics, such as pristinamycin I (1) or its semi-synthetic derivative Synercid®.34,35 Pristinamycin is a natural compound mixture produced by Streptomyces pristinaespiralis and consists of a mixture of two types of chemically unrelated substances – the macrocyclic polyketide/nonribosomal peptide hybrid compound pristinamycin II (= streptogramin group A (SA)) and the cyclic hexadepsipeptide pristinamycin I (1) (= streptogramin group B (SB)), the latter containing Phg as a structural component (Fig. 2).36 Pristinamycin I and II are co-produced by S. pristinaespiralis in a synergistically active ratio of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70.36 Both compounds inhibit the bacterial protein biosynthesis by binding to the 23S rRNA of the 50S subunit of bacterial ribosomes, whereby pristinamycin II prevents the binding of the aminoacyl-tRNA, while pristinamycin I (1) causes the dissociation of the peptidyl-tRNA from the ribosome.35 Each component alone exhibits moderate bacteriostatic activity, whereas in combination they act synergistically, resulting in a potent bactericidal activity.35 The synergistic mixture is used for clinical application, e.g. as Synercid®, which is a combination of semi-synthetic derivatives of pristinamycin I (quinupristin (2), Fig. 3) and pristinamycin II (dalfopristin).35 Resistances against streptogramin antibiotics include, e.g. the active efflux of the antibiotics or the enzymatic inactivation of either of the two components.36 The most prevalent resistance mechanism against SB antibiotics is the N6 (di)methylation of a specific adenine nucleotide residue (A2058, Escherichia coli numbering) of the ribosomal 23S rRNA.37,38 This modification is mediated by Erm methyltransferases, encoded by plasmid-borne erm genes, which leads to a drastic reduction of affinity for members of the macrolide-lincosamide-streptogramin B (MLSB) group of antibiotics.39 In S. pristinaespiralis, the SB antibiotic 1 is synthesized by the nonribosomal peptide synthetases (NRPSs) SnbA, SnbC, and SnbDE, whereby the latter one incorporates L-Phg as the final amino acid into the growing pristinamycin I peptide chain.36,40L-Phg biosynthesis occurs through a series of enzymatic reactions, catalyzed by the enzymes phenylpyruvate dehydrogenase (PglB/PglC), Phg dehydrogenase (PglA), thioesterase (PglD), and aminotransferase (PglE), which convert phenylpyruvate to L-Phg.18,41,42 The genetic information for Phg biosynthesis is encoded on the S. pristinaespiralis pglA-E operon.41 So far, this is the first reported Phg biosynthetic pathway and has been identified in the frame of characterizing the pristinamycin biosynthetic gene cluster.40,41,43 Crystallographic analysis of a ribosome-bound streptogramin antibiotic revealed the formation of a hydrogen bond between the L-Phg carbonyl oxygen with nucleotide C2586 of the 23S rRNA (E. coli numbering) as part of the interactions involved in the binding of 1 to the ribosome.35 Additionally, the aromatic ring of Phg is involved in establishing hydrophobic interactions with the ribosome34 hinting at the functional importance of the amino acid for bioactivity.
:70.36 Both compounds inhibit the bacterial protein biosynthesis by binding to the 23S rRNA of the 50S subunit of bacterial ribosomes, whereby pristinamycin II prevents the binding of the aminoacyl-tRNA, while pristinamycin I (1) causes the dissociation of the peptidyl-tRNA from the ribosome.35 Each component alone exhibits moderate bacteriostatic activity, whereas in combination they act synergistically, resulting in a potent bactericidal activity.35 The synergistic mixture is used for clinical application, e.g. as Synercid®, which is a combination of semi-synthetic derivatives of pristinamycin I (quinupristin (2), Fig. 3) and pristinamycin II (dalfopristin).35 Resistances against streptogramin antibiotics include, e.g. the active efflux of the antibiotics or the enzymatic inactivation of either of the two components.36 The most prevalent resistance mechanism against SB antibiotics is the N6 (di)methylation of a specific adenine nucleotide residue (A2058, Escherichia coli numbering) of the ribosomal 23S rRNA.37,38 This modification is mediated by Erm methyltransferases, encoded by plasmid-borne erm genes, which leads to a drastic reduction of affinity for members of the macrolide-lincosamide-streptogramin B (MLSB) group of antibiotics.39 In S. pristinaespiralis, the SB antibiotic 1 is synthesized by the nonribosomal peptide synthetases (NRPSs) SnbA, SnbC, and SnbDE, whereby the latter one incorporates L-Phg as the final amino acid into the growing pristinamycin I peptide chain.36,40L-Phg biosynthesis occurs through a series of enzymatic reactions, catalyzed by the enzymes phenylpyruvate dehydrogenase (PglB/PglC), Phg dehydrogenase (PglA), thioesterase (PglD), and aminotransferase (PglE), which convert phenylpyruvate to L-Phg.18,41,42 The genetic information for Phg biosynthesis is encoded on the S. pristinaespiralis pglA-E operon.41 So far, this is the first reported Phg biosynthetic pathway and has been identified in the frame of characterizing the pristinamycin biosynthetic gene cluster.40,41,43 Crystallographic analysis of a ribosome-bound streptogramin antibiotic revealed the formation of a hydrogen bond between the L-Phg carbonyl oxygen with nucleotide C2586 of the 23S rRNA (E. coli numbering) as part of the interactions involved in the binding of 1 to the ribosome.35 Additionally, the aromatic ring of Phg is involved in establishing hydrophobic interactions with the ribosome34 hinting at the functional importance of the amino acid for bioactivity.
 | ||
| Fig. 2 Chemical structure of 1, 3, and 4 with Phg residue indicated in blue. Numbering indicates the order of incorporation into the peptide backbone. | ||
 | ||
| Fig. 3 (A) Structure of quinupristin (2) and illustration of the network of hydrogen bonds and lipophilic interactions with bacterial 23S rRNA bases in the 50S subunit of the bacterial ribosomes (green: E. coli, PBD: 4U26,44 red: Deinococcus radiodurans, PBD: 1SM1,35 Black: Haloarcura marismortui, PBD: 1YJW.34 Illustration of docked pristinamycin (1) into the binding pocket at the 23S rRNA of E. coli (B), H. marismortui (C) and D. radiodurans (D) in comparison to quinupristin (2, depicted in red) prepared with SeeSAR software version 13.0.1 (https://www.biosolveit.de/SeeSAR). Colored spheres around atoms indicate positive (green) or negative (red) contributions to the overall binding affinity based on ligand–target interactions as well as desolvatization energies. | ||
The pristinamycin biosynthetic route is well-described and the producer strain S. pristinaespiralis is genetically tractable,40 making it strain a suitable chassis strain for a streptogramin mutasynthesis approach. In the current study, we report on a mutasynthesis approach for the derivatization of pristinamycin I (1) by targeting the Phg-like residue for modification to generate novel antibiotic derivatives.
2. Results and discussion
2.1 Docking of pristinamycin into the ribosomal binding pocket reveals options for pristinamycin I derivatization
In order to estimate the potential for compound derivatisation, computational docking studies were carried out with known pristinamycin compounds and bacterial ribosomes as drug targets. Quinupristin (2), another clinically applied pristinamycin I derivative bearing an additional (R)-2-(quinuclidin-3-ylthio)ethyl substituent at the 4-oxo-L-pipecolic acid moiety (Fig. 3A), has been co-crystallized with the ribosome of different species including E. coli and the two extremophiles Deinococcus radiodurans and Haloarcura marismortui.34,35,44 The structure analysis of these co-crystals reveals that quinupristin (2) binds the 23S rRNA of the bacterial large ribosomal subunit 50S at the entrance of the ribosomal tunnel through a network of hydrophobic interactions predominantly mediated and hydrogen bonds between the N6 nitrogen of the A2062 nucleotide (E. coli nomenclature in green) and the N4 nitrogen of the C2565 nucleotide (E. coli nomenclature in green) as outlined in Fig. 3A34,35,44 with no direct contact to the ribosome protein backbone. Pristinamycin I (1) is believed to bind in a similar manner to the same binding pocket at 23S rRNA of the bacterial ribosomal 50S subunit. However, as no co-crystal structure of 1 with the bacterial ribosome is available, we have performed a virtual docking of pristinamycin I (1) into the binding pocket of quinupristin (2) at the ribosome based on the three available co-crystal structure with the ribosome of E. coli (PDB: 4U26), D. radiodurans (PDB: 1MS1), and H. marismortui (PBD: 1YJW)34,35,44 using the software SeeSAR (SeeSAR version 13.0.1; BioSolveIT GmbH, Sankt Augustin, Germany, 2023, www.biosolveit.de/SeeSAR). The general overlap of pristinamycin (1) with quinupristin (2) in cases E. coli (Fig. 3B, see PDB file 3B_E. coli in the ESI†) and H. marismortui (Fig. 3C, see PDB file 3C_H. marismorturi in the ESI†) is very high, with only very little deviations especially in the position of the 4-N,N-dimethylamino-L-Phe side chain and the 3-hydroxypipecolinic acid moiety. In the case of D. radiodurans (Fig. 3D, see PDB file 3D_D. radiodurans in ESI†) already 2 is bound in a slightly different orientation compared to E. coli and H. marismortui, in particular, the 3-hydroxypipecolinic acid moiety is oriented to the opposite direction. The docked structure of 1 is then shifted by approximately 2.1 Å into the binding pocket of the 23S rRNA compared to 2. Presumably, this is a consequence of lacking the (R)-2-(quinuclidin-3-ylthio)ethyl substituent of 2 allowing 1 to enter deeper into the pocket. The Phg moiety of 1 points in all cases towards the inside of the 23S rRNA binding pocket. However, unoccupied space between the 23S rRNA nucleotides and the Phg moiety in all docking approaches encouraged us to virtually explore further possible Phg derivatives, which could be accessed through mutasynthesis via an in silico screening of 4-, 3- and 2-substituted Phg derivatives based on the docking of 1 into the binding pocket of E. coli. In general, smaller substituents in the 3- and 4-position seemed to be tolerated for binding to the target. Substitution in the 2-position of the Phg moiety mostly led to derivatives presumably not binding to the target structure anymore.2.2 Construction of a biosynthetic mutant of S. pristinaespiralis for mutasynthesis
To allow for the mutasynthesis of Phg-containing pristinamycin I derivatives, an appropriate biosynthetic mutant deficient in Phg precursor supply had to be generated. In S. pristinaespiralis, pglA is the first gene of the L-Phg biosynthesis encoding operon pglA-E and has been shown to be essential for pristinamycin I production.41 Inactivation of pglA in S. pristinaespiralis (MpglA) resulted in a loss of pristinamycin I production, whereas feeding MpglA with L-Phg restored pristinamycin I production, thus functionally complementing the pglA inactivation.41 The production of the SA antibiotic pristinamycin II is not affected in the MpglA mutant.41 To obtain an S. pristinaespiralis mutasynthesis strain that produces pristinamycin I derivatives exclusively, we aimed to abolish the biosynthesis of the pristinamycin II compound as well. This should allow for PI derivative production in an antibiotic-free background so that the mutasynthesis samples can be directly tested for bioactivity. For this purpose, the gene snaE1, encoding a hybrid PKS/NRPS enzyme essential for pristinamycin II biosynthesis45 was inactivated by insertion of a thiostrepton resistance cassette (thioR) using the mutational plasmid pK18-snaE1tsr, which resulted in the double mutant S. pristinaespiralis ΔpglAΔsnaE1 (Table S1, ESI†). The ΔpglAΔsnaE1 mutant and the S. pristinaespiralis wild-type strain (control) were cultivated in pristinamycin production medium HT7T, respectively. After 72 h of cultivation, the culture extracts were analyzed for pristinamycin production by HPLC. The HPLC spectrum of the S. pristinaespiralis wild-type (WT) extract showed specific peaks at retention times (RT) 8.2 min and 10.35 min (Fig. 4A) with characteristic pristinamycin I and II UV-Vis spectra, respectively (data not shown). In addition, the WT extracts exhibited bioactivity against B. subtilis (Fig. 4B). In contrast, such a HPLC peak pattern was not detected for the S. pristinaespiralis ΔpglAΔsnaE1 extract samples (Fig. 4C) and extracts did not exert antibiotic activity against B. subtilis (Fig. 4D). These results confirmed that the S. pristinaespiralis ΔpglAΔsnaE1 mutant was no longer capable of synthesizing pristinamycin I nor pristinamycin II. Extract samples obtained from S. pristinaespiralis ΔpglAΔsnaE1 cultures, supplemented with L-Phg revealed a single peak at RT 8.2 min in HPLC analysis (Fig. 4E) and exerted moderate bioactivity against B. subtilis (Fig. 4F), which showed that pristinamycin I production was restored exclusively. These results proved the functional correctness of the S. pristinaespiralis ΔpglAΔsnaE1 mutant and enabled its use as a mutasynthesis strain for Phg-directed derivatisation of pristinamycin I.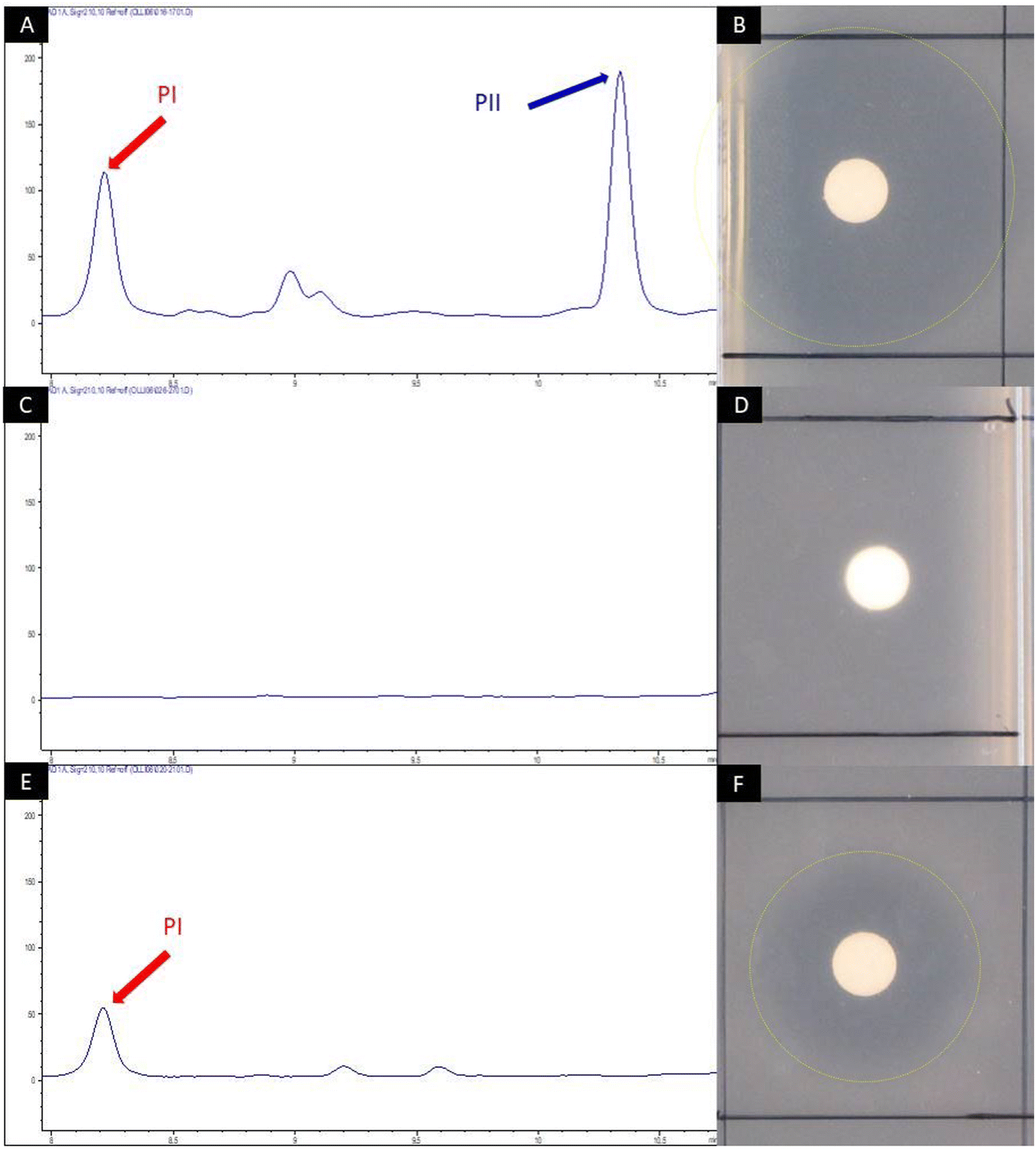 | ||
| Fig. 4 HPLC chromatogram of crude extract samples from S. pristinaespiralis wild-type (A), S. pristinaespiralis ΔpglAΔsnaE1 (C) and ΔpglAΔsnaE1 supplemented with 100 μM L-Phg (E). The chromatogram shows peaks measured at 210 nm. Pristinamycin I (PI) and II (PII)-specific peaks were detected at retention times of 8.2 and 10.35 min and are indicated by red and blue arrows, respectively. Disc diffusion assays with the respective extract samples against B. subtilis (B), (D) and (F). Inhibition zones are marked with yellow circles. | ||
2.3 Generation of novel pristinamycin I derivatives by mutasynthesis
To conduct the mutasynthesis of pristinamycin I, a series of commercially available Phg derivatives was used as mutasynthons for feeding experiments with S. pristinaespiralis ΔpglAΔsnaE1 (Table 1). These included a variety of halogenated and non-halogenated Phgs substituted at different positions of the aromatic residue, as well as enantiomers. The mutasynthons were added to the growing cultures of S. pristinaespiralis ΔpglAΔsnaE1, and ethyl acetate crude extract from S. pristinaespiralis cultures was analyzed for the production of pristinamycin I derivatives by HPLC and HPLC/MS. For S. pristinaespiralis ΔpglAΔsnaE1, feeding with six mutasynthons, namely D-Phg, 2,5-dihydro-D-Phg, 4-fluoro-L-Phg, 2-fluoro-DL-Phg, 4-chloro-DL-Phg, and hydroxy-DL-Phg led to the production of the respective pristinamycin I derivatives, which were identified by HPLC-MS analysis based on their expected calculated masses (Table 1). Thereof, extract samples of S. pristinaespiralis ΔpglAΔsnaE1 cultures supplemented with D-Phg, 4-fluoro-L-Phg, and 4-chloro-DL-Phg showed activity in bioassays against the test organism B. subtilis (Table 1). Because of the comparatively stable and good antibiotic production behavior of the ΔpglAΔsnaE1 mutant when supplemented with 4-fluoro-L-Phg, and 4-chloro-DL-Phg, further efforts were focused on the isolation of the two halogenated pristinamycin I derivatives, which were referred to as 6-fluoropristinamycin I (3) and 6-chloropristinamycin I (4), respectively. Notably, feeding of D-Phg also led to the production of a pristinamycin I derivative as identified by HPLC-MS analysis, although the enantiomeric L-Phg is the natural precursor of pristinamycin I biosynthesis. Here, it remains unclear whether the adenylation domain of SnbE is able to accept D-Phg as a substrate or whether D-Phg was converted to L-Phg prior to incorporation. Since Phgs were solubilized by using sodium hydroxide prior to addition to the culture, chemical racemisation to D/L-Phg mixtures is quite possible. Though, polarimetric analysis of L- and D-Phg under alkaline conditions did not indicate racemisation (data not shown), which however does not exclude racemization in the organism. In a previous study, it has been described that the thioesterase domain of SnbDE can only accept L-Phg but not D-Phg,46 which makes incorporation of D-Phg in PI unlikely. Natural pristinamycin I (produced from S. pristinaespiralis ΔpglAΔsnaE1 supplemented with L-Phg) was used as a control and was referred to as C-PI. Production of 3 and 4 (for structure see Fig. 2) was confirmed using HPLC-MS/MS analysis by comparing the fragmentation pattern of 1 with that of the respective halogenated derivatives (Fig. S1, ESI†). Three prominent, reoccurring mass signals (m/z = 578.3, 663.3, and 839.4) were detected and assigned to fractions of the parental ion pristinamycin I (Fig. S1a, ESI†). These fragments, which were determined to include the L-Phg residue in pristinamycin I, were shifted in masses corresponding to the addition of the respective halogen atom introduced (+34 for the chlorine main isotope, +18 for fluorine; shift is the mass of the atom −1 to account for the mass of the replaced hydrogen (Fig. S1b and c, respectively, ESI†)), suggesting the incorporation of the respective halogenated mutasynthons at the expected position in pristinamycin I. In order to produce sufficient amounts of substance for the structural elucidation of 3 and 4 by NMR analysis, mutasynthesis was carried out on a large scale. In total, 12 l and 6 l cultures were extracted for 3 and 4 isolation, respectively. HPLC fractions were analyzed by HRESIMS analysis to confirm the presence of 3 and 4 (Fig. S2 and S3, ESI†), respectively, whereby HRESIMS data of 1 served as a reference (Fig. S4, ESI†). HPLC fractions containing 0.6 mg of 3 and 1.54 mg of 4, respectively, were subjected to NMR analysis (NMR data for 1, 3, and 4 are available in Table 2 and in ESI,† Fig. S5–S21). As expected, the NMR data of 3 were highly similar to those of 1 (Table 2). However, the presence of two broad doublets (δH 7.24 and 7.37) in the 1H NMR spectrum as part of the characteristic AA′BB′ system indicated the incorporation of the 4-chloro-Phg unit. In the 13C NMR spectrum of 4, the splitting of the signals C-4/C-8, C-5/C-7 and C-6 with coupling constants JC,F = 8.6 Hz, 21.5 Hz and 245 Hz, respectively, indicated the coupling to the fluorine atom. Furthermore, the single peak at δF = −113.4 ppm gave direct proof of fluorine incorporation (Table 2 and Fig. S22, ESI†). The production rates of the PI derivatives were difficult to determine due to the inconsistent production behavior of the ΔpglAΔsnaE1 mutant. PI derivative concentrations were only measured for the pooled samples of C-PI, 3, and 4, which were produced in amounts of 1.52 μg mL−1, 1.55 μg mL−1, and 1.51 μg mL−1, respectively. Thus, the data indicate an incorporation of the unnatural derivatives at almost the same efficiency as the natural L-Phg precursor. Furthermore, the production of C-PI reached roughly half the amount produced by the wild-type strain (Fig. 4), which yielded an average of about 5 μg mL−1 of 1. Altogether, the lower PI derivative production yields are more likely due to the inconsistent production performance of the ΔpglAΔsnaE1 mutant strain, rather than that the acceptance of the halogenated derivatives is limiting.| Mutasynthon | Calculated mass of PI derivative | New PI-derivative/mass detected | Bioactivity against |
|---|---|---|---|
| B. subtilis | |||
| D-Phg | 866.4 | + | + |
| 2,5-Dihydro-D-Phg | 868.4 | + | n.t. |
| 4-Fluoro-L-Phg | 884.4 | + | + |
| 2-Fluoro-DL-Phg | 884.4 | + | n.t. |
| 4-Chloro-DL-Phg | 900.4 | + | + |
| 2-Chloro-DL-Phg | 900.4 | — | n.t. |
| 4-Bromo-DL-Phg | 944.3 | — | n.t. |
| 4-Amino-DL-Phg | 881.4 | — | n.t. |
| 4-Hydroxy-DL-Phg | 882.4 | + | n.t. |
| 2,2-Di-Phg | 942.4 | — | n.t. |
| 3-(Trifluoromethyl)-DL-Phg | 934.4 | — | n.t. |
| 4-(Trifluoromethyl)-L-Phg | 934.4 | — | n.t. |
| 4-Hydroxy-DL-2-fluoro-Phg | 882.4 | — | n.t. |
| Unit | Position | Pristinamycin I (1) | 6-Fluoropristinamycin I (4) | 6-Chloropristinamycin I (3) | |||
|---|---|---|---|---|---|---|---|
| 13C, mult. | 1H, mult. | 13C, mult. | 1H, mult. | 13C, mult.d | 1H, mult. | ||
| n.o. not observed.a Doublet with JC,F = 8.6 Hz.b Doublet with JC,F = 21.5 Hz.c Doublet with JC,F = 245 Hz.d Signals assigned from HSQC and HMBC correlations. | |||||||
| Phg | 1 | 168.5, C | 168.6, C | 169.6, C | |||
| 2 | 56.8, CH | 5.63, d (8.9) | 55.3, CH | 5.63, m | 57.0, CH | 5.63, s | |
| 2-NH | 8.48, d (8.9) | 8.52, br s | |||||
| 3 | 136.2, C | 132.7, C | 135.9, C | ||||
| 4/8 | 127.7, CH | 6.56, m | 130.0, CHa | 7.24, m | 130.6, CH | 7.24, br d (8.4) | |
| 5/7 | 128.7, CH | 7.35, m | 115.6, CHb | 7.20, m | 129.9, CH | 7.37, br d (8.4) | |
| 6 | 128.3, C | 7.34, m | 161.8, Cc | 135.2, C | |||
| Opp | 9 | 168.4, C | 168.8, C | 170.1, C | |||
| 10 | 56.1, CH | 5.21, br d (5.4) | 58.6, CH | 5.45, m | 58.0, CH | 5.29, br s | |
| 11 | 40.7, CH2 | 2.04, m | 40.7, CH2 | 2.08, m | 41.9, CH2 | 2.25, m | |
| 0.52, dd (15.0, 5.7) | 0.57, dd (15.5, 4.0) | 0.61, m | |||||
| 12 | 203.1, C | 203.7, C | 206.6, C | ||||
| 13 | 38.5, CH2 | 2.23, ddd (16.3, 11.9, 7.4) | 38.6, CH2 | 2.21, m | 39.6, CH2 | 2.33, m | |
| 2.14, br d (16.3) | 2.12, br d (16.3) | 2.24, m | |||||
| 14 | 36.0, CH2 | 4.54, br dd (13.6, 7.4) | 36.1, CH2 | 4.53, m | 37.7, CH2 | 4.70, m | |
| 2.63, ddd (13.6, 11.9, 4.5) | 2.63, m | 2.76, m | |||||
| Mdp | 15 | 170.5, C | 170.7, C | 173.5, C | |||
| 16 | 53.7, CH | 4.99, dd (11.6, 4.5) | 53.7, CH | 5.03, br s | 56.0, CH | 5.17, m | |
| 17 | 35.1, CH2 | 3.28, t (12.1) | 35.1, CH2 | 3.23, m | 35.1, CH2 | 3.26, m | |
| 2.83, m | 2.84, m | 3.05, dd (12.5, 4.3) | |||||
| 18 | 123.0, C | 123.1, C | 124.5, C | ||||
| 19/23 | 130.2, CH | 6.99, br d (8.6) | 130.2, CH | 6.99, br d (8.4) | 131.5, CH | 7.07, br d (8.4) | |
| 20/22 | 112.9, CH | 6.78, br d (8.6) | 112.9, CH | 6.74, br d (8.4) | 114.3, CH | 6.75, br d (8.4) | |
| 21 | 150.4, C | 150.3, C | 152.0, C | ||||
| 24/25 | 40.3, CH3 | 2.83, s | 40.4, CH3 | 2.83, s | 40.7, CH3 | 2.90, s | |
| 26 | 30.8, CH3 | 3.18, s | 30.8, CH3 | 3.17, br s | 31.3, CH3 | 3.25, br s | |
| Pro | 27 | 173.2, C | 172.9, C | 173.8, C | |||
| 28 | 56.8, CH | 4.64, t (7.4) | 56.9, CH | 4.64, t (7.4) | 58.8, CH | 4.63, t (7.4) | |
| 29 | 27.3, CH2 | 2.04, m | 27.4, CH2 | 2.08, m | 28.6, CH2 | 2.17, m | |
| 0.80, m | 0.88, m | 1.10, m | |||||
| 30 | 24.4, CH2 | 1.52, m | 24.4, CH2 | 1.56, m | 25.5, CH2 | 1.43, m | |
| 1.23, m | 1.20, m | ||||||
| 31 | 47.3, CH2 | 3.68, m | 47.3, CH2 | 3.65, m | 49.1, CH2 | 3.68, m | |
| 3.31, m | 3.28, m | ||||||
| Abu | 32 | 168.8, C | 168.9, C | 171.4, C | |||
| 33 | 50.8, CH | 4.77, dt (9.3, 7.7) | 50.8, CH | 4.73, q (7.5) | 52.8, CH | 4.81, m | |
| 33-NH | 8.28, d (9.3) | n.o. | |||||
| 34 | 23.5, CH2 | 1.68, dtq (13.2, 7.7, 7.3) | 23.7, CH2 | 1.67, dtq (13.2, 7.5, 7.3) | 25.3, CH2 | 1.74, m | |
| 1.53, m | 1.52, m | 1.63, m | |||||
| 35 | 10.1, CH3 | 0.79, t (7.3) | 10.0, CH3 | 0.79, t (7.3) | 10.1, CH3 | 0.92, t (7.4) | |
| Thr | 36 | 166.8, C | n.o. | 169.4, C | |||
| 37 | 54.4, CH | 4.91, dd (10.5, 1.1) | 54.7, CH | 4.9, d (10.3) | 56.8, CH | 4.99, br s | |
| 37-NH | 8.07, d (10.5) | 8.52, br s | |||||
| 38 | 71.6, CH | 5.66, qd (6.5, 1.1) | 71.7, CH | 5.63, m | 73.2, CH | 5.80, qd (6.4, 1.7) | |
| 39 | 15.9, CH3 | 1.11, d (6.5) | 16.0, CH3 | 1.10, d (6.5) | 16.4, CH3 | 1.28, d (6.4) | |
| Hpc | 40 | 169.0, C | n.o. | 171.0, C | |||
| 41 | 131.6, C | n.o. | 132.1, C | ||||
| 42 | 139.3, CH | 7.80, m | n.o. | 141.2, CH | 7.98, br s | ||
| 43 | 129.5, CH | 7.78, m | 128.2, CH | 7.30, m | 130.3, CH | 7.63, dd (8.1, 4.4) | |
| 44 | 126.5, CH | 7.58, dd (8.0, 1.7) | 126.5, CH | 7.23, m | 127.5, CH | 7.48, d (8.4) | |
| 45 | 157.5, C | n.o. | n.o. | ||||
| 45-OH | 11.97, s | 11.97, br s | |||||
2.4 Pristinamycin I mutasynthesis products are antimicrobially active
To test, whether the generated pristinamycin I derivatives are antimicrobially active, bioassays were carried out with crude extracts mutasynthesis samples containing 3 and 4. The antibiotic activity of crude extract samples was tested and confirmed against B. subtilis (data not shown). Bioactive samples were additionally tested against a panel of clinically relevant microorganisms from the WHO priority list of pathogens, including different Gram-positive and Gram-negative bacteria, as well as fungi. Namely, these were Staphylococcus aureus DSM 18827, CIP 111304, and CIP 108540, Enterococcus faecium DSM 20477, Pseudomonas aeruginosa DSM 1117, Escherichia coli DSM 1103, Proteus vulgaris DSM 2140, Candida albicans DSM 1386, and Trichophyton rubrum DSM 16111 (Table S1, ESI†). Of the three tested S. aureus strains, two strains (CIP 111304 and CIP 108540) were specifically selected due to their genetically encoded resistance to SA and SB antibiotics. CIP 111304 harbors the genes vat(A) and vgb(A), each conferring resistance against SA and SB antibiotics, respectively, whereas CIP 108540 contained the SA resistance genes vga(A), vga(B), and vat(B), as well as the SB resistance genes erm(A) and erm(B).47 DSM18827 was selected as a multi-resistant test strain48 (Table S1, ESI†). Of the different test strains, Enterococcus faecium was inhibited by the crude extract samples containing 3 and 4, respectively, to a similar extent as observed with pure pristinamycin I substance and Synercid®, which were used as positive controls (Fig. S22A, ESI†). Bioassays with 1, 3 and 4 were also carried out in combination with pure pristinamycin II compound (10 μg per compound and filter disc). Here, E. faecium and the S. aureus test strains were inhibited by the respective pristinamycin I derivative plus pristinamycin II combination to a similar extent as with the respective positive control samples, which were pure pristinamycin I + II mixture and Synercid®, respectively (Fig. 5 and Fig. S22A–D, ESI†). Altogether, this showed that 3 and 4 have antimicrobial activities similar to that of the natural compound 1. Furthermore, MIC assays and cytotoxicity tests were performed with pure 4 as this compound was available in sufficient amounts (approx. 1 mg of the pure compound) to allow for additional bioactivity tests. MIC analysis was performed using a panel of standard test organisms, including yeast, fungi, Gram-negative bacteria, as well as B. subtilis, S. aureus, and Mycobacterium smegmatis as Gram-positive test bacteria. Here, the derivative 4 inhibited the growth of S. aureus at a concentration of 33.2 μg mL−1 and that of B. subtilis at a concentration of 4.2 μg mL−1, which were 16.7 μg mL−1 and 2.1 μg mL−1 for the pristinamycin I control, respectively (Table S2, ESI†). Since the MIC values of 4 and 1 resided in a similar range, the generated derivative 4 is obviously similarly bioactive as the natural compound 1. Cytotoxicity assays were carried out with 4 and 1 against two standard human cell lines (KB3.1 and L929), respectively. As with the pristinamycin I (1) control, no effect on proliferation was observed for 4 (Table S3, ESI†). Thus, altogether the 4 derivative shows an equal bioactivity pattern as the natural pristinamycin I (1). So far, this represents the only example of successful mutasynthesis of streptogramin antibiotics.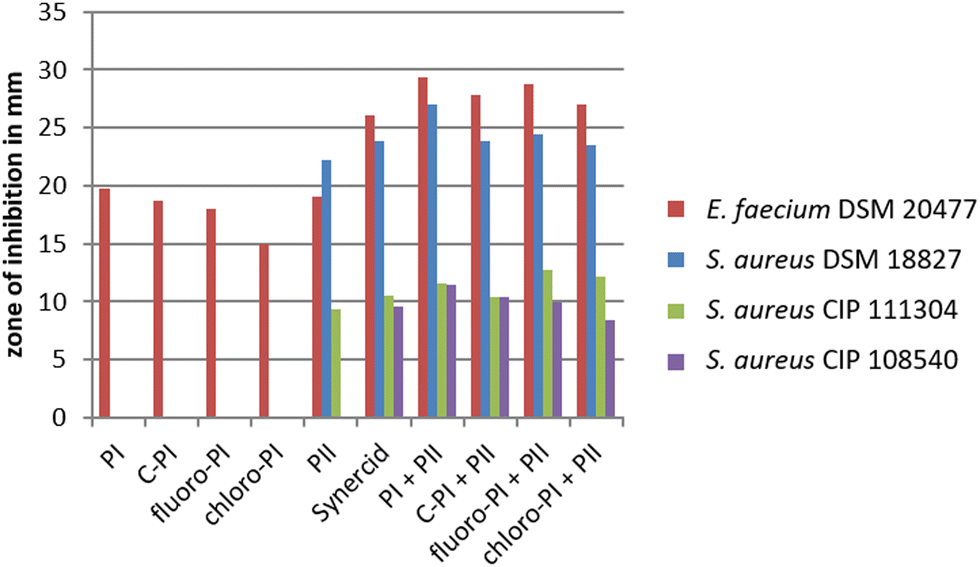 | ||
| Fig. 5 Diameter of inhibition zones caused by pristinamycin I derivatives. Crude extract samples of C-PI, 6-fluoropristinamycin I (6-fluoro-PI), 6-chloropristinamycin I (6-chloro-PI) were used as well as pure pristinamycin I (PI) and II (PII). All SB samples were applied with and without the addition of pristinamycin II. Synercid® filter discs were used as a positive control. | ||
In a recent study from Walker and Clardy, 202149 on a machine learning bioinformatics approach to predict bioactivities from BGCs, it has been found that BGCs containing Phg encoding genes are explicitly associated with antibacterial activities. Unlike proteinogenic amino acids, Phg does not have a β-carbon, so it has fewer rotatable bonds than proteinogenic aromatic amino acids like e.g. phenylalanine. Therefore, Phg-containing peptides should be more rigid, reducing the entropic cost for binding to a target.50 Thus overall, we predict to be Phgs a good target residue for derivatization to improve compound properties.
2.5 Target related studies with pristinamycin I derivatives
To investigate the inhibitory effect of the pristinamycin I derivatives on protein biosynthesis as an antibiotic target, in vitro transcription/translation assays were performed using semi-purified pristinamycin I derivatives 3 and 4 and respective control samples (pristinamycin I (1) and II). Assays were carried out and evaluated as reported before.51 Based on previous studies, ivTT fluorescence values of 0–20% indicate strong inhibition of the assay, 0–40% are considered to represent specific inhibition, whereas values above 40% do not indicate inhibition of the ivTT assay. In ivTT assays with pristinamycin derivatives, it was found that pristinamycin II resulted in strong inhibition of the ivTT assay, whereas neither pristinamycin I (1) nor the halogenated pristinamycin I derivatives 3 and 4 led to an inhibition (Fig. S23, ESI†). This might be explained by the insufficiency of the ivTT assay to sense streptogramin B-mediated protein synthesis inhibition as it has been reported previously that translational inhibition was also not detected in cell-free assays for streptogramin B antibiotics, such as virginiamycin S, quinupristin (2) or pristinamycin I (1).52–542.6 Extended mutasynthesis with E. coli-derived mutasynthons
To generally extent the possibility of accessing further, potentially not commercially available Phg derivatives, we established an E. coli BL21(DE3) whole cell biotransformation route from halogenated phenylalanines as precursors to produce Phg derivatives (Fig. S3, ESI†). We have demonstrated in a previous study that E. coli BL21(DE3) is a suitable microorganism to transform L-phenylalanine derivatives to the corresponding mandelic acid by using a heterologously expressed hydroxymandelate synthase gene hmaS from A. mediterranei.55 To expand the production platform to Phg, we further expressed the genes for mandelate oxidase (hmo) from S. coelicolor and leucine dehydrogenase (bcd) from B. thuringensis in a second E. coli BL21(DE3) strain. To overcome a NADH cofactor limitation, the glucose dehydrogenase (gdh) was additionally expressed in E. coli BL21(DE3). To test the substrate spectrum of the Hmo-LeuDH-GDH cascade, a variety of halogenated mandelic acid derivatives were tested. Fluorinated Phgs were detected in the supernatant with a high enantiomeric excess of more than 95%, while chlorinated Phgs were detected with a lower enantiomeric excess of 50% for 2-chloro-L-Phg (Table S4, ESI†).Mutasynthesis is usually carried out with chemically pure mutasynthons as building blocks that are supplied to the respective biosynthesis mutant. In order to establish a completely fermentative biosynthetic route from the production of the mutasynthon to the final mutasynthesis end product, we developed a mixed mutasynthesis set-up, involving samples from the E. coli biotransformation approach that were fed to S. pristinaespiralis ΔpglAΔsnaE1 as pristinamycin I mutasynthesis strain. This should allow for a direct incorporation of non-commercially available mutasynthons and in principle also provide a more sustainable production route for drug derivatization. To test for the feasibility of such an approach, 100 mL of S. pristinaespiralis ΔpglAΔsnaE1 main cultures were supplemented with 1 mL of sterile filtered culture supernatants of E. coli BL21(DE3) pET28-hmo/pACYC-bcd-gdh, which contained approx. 1 mg mL−1 of 4-fluoro-Phg as representative Phg mutasynthon for pristinamycin I derivatization. LC-MS analysis of crude extracts from ΔpglAΔsnaE1 fed with 4-fluoro-Phg biotransformation supernatant samples led to mass signals characteristic of 6-fluoro-PI (Fig. S24, ESI†). Thus, 4-fluoro-Phg from E. coli supernatant samples was successfully incorporated into pristinamycin I, which proved the feasibility of the adapted mutasynthesis procedure. Thereby, the pristinamycin derivative production rates were comparable to those when supplemented with commercially available mutasynthons (Fig. S24A–F, ESI†). This shows that the biotransformation-based mutasynthesis approach is comparably efficient to conventional mutasynthesis. The extended version of the mutasynthesis approach including biotransformation-derived mutasynthon samples was termed “biotransformation-coupled mutasynthesis”.
3. Conclusions
The streptogramin family of antibiotics represents a rare and underexplored family of antibiotics with pristinamycin I (1) as a representative substance in clinical application as an antibiotic of last resort against multi-drug resistant bacterial infections. Substance derivatization is difficult to implement chemically due to the structural complex nature structure of the streptogramin molecule(s). So far, the mutasynthesis of streptogramin antibiotics has been prevented by the lack of knowledge on precursor biosynthesis and the encoded genes. In this study, we present the first successful mutasynthesis approach for the derivatization of the streptogramin antibiotic pristinamycin I (1) based on a modification of the Phg residue. Two halogenated pristinamycin I derivatives, 6-chloropristinamycin I (3) and 6-fluoropristinamycin I (4), were generated by mutasynthesis, their chemical structures were confirmed by NMR and the bioactivity of both substances was demonstrated with antibacterial bioassays. The pristinamycin I mutasynthesis approach was extended by a biotransformation process using a genetically modified E. coli strain as a Phg mutasynthon producer, which provided Phg derivatives that were directly fed to the mutasynthesis strain. This mutasynthesis 2.0 process represents a novel extension of the original mutasynthesis method and the feasibility of the process has been demonstrated by the successful production of 46-fluoropristinamycin I (4).4. Material and methods
4.1 Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table S1 (ESI†). For routine cloning work, Escherichia coli Novablue (Novagen) (Sigma-Aldrich, Germany) was used. S. pristinaespiralis PR11 (Sanofi-Aventis) was used for pristinamycin production analysis and S. pristinaespiralis MpglA41 served as the host strain for the generation of the S. pristinaespiralis mutasynthesis strain. The biological samples used for the experiments were obtained from the Leibniz Institut DSMZ – Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany.4.2 Media and cultivation conditions
Strain cultivation procedures for S. pristinaespiralis were carried out as described before.56 For cultivation and harvesting of genomic DNA, S. pristinaespiralis strains were grown in 100 mL of S-medium57 in 500 mL Erlenmeyer flasks (with steel springs) on an orbital shaker (180 rpm) at 28 °C. Kanamycin (50 μg mL−1), apramycin (50 μg mL−1), and thiostrepton (50 μg mL−1) were used for selection when appropriate. For pristinamycin production analysis, S. pristinaespiralis strains were grown in 100 mL of the HT7T medium.584.3 Preparation and manipulation of DNA
Total DNA isolation from S. pristinaespiralis was performed as described by Kieser et al., 200057 and with the DNA Nucleospin Microbial DNA kit (Bioanalysis, Macherey-Nagel, Germany), respectively. Plasmid isolation was performed using the peqGold Plasmid MiniPrep kit (VWR, Life Science, USA), Pure Yield Plasmid MidiPrep System kit (Promega, USA), or according to Sambrook et al., 1989.59 PCR products were purified from 1% agarose gel using the Wizard® SV Gel and PCR Clean-Up System kit (Promega, USA). Enzymes, including restriction endonucleases, ligase, and Q5 DNA polymerase were used according to the manufacturer's recommendations (New England Biolabs, USA; Thermo Fischer Scientific, USA). The primers used for PCR were obtained from MWG Biotech AG (MWG; Ebersberg, Germany) and are listed in Table S1 (ESI†). The primers used for PCR were obtained from MWG Biotech AG (MWG; Ebersberg, Germany) and are listed in Table S1 (ESI†).4.4 Cloning procedure for obtaining mutasynthesis strain S. pristinaespiralis ΔpglAΔsnaE1
The mutasynthesis strain S. pristinaespiralis ΔpglAΔsnaE1 was generated using the following procedure: A 2.4 kb fragment (snaE1′), covering an internal part of the snaE1 gene, was amplified with PCR using genomic DNA from strain S. pristinaespiralis PR11 as template and primer pair MsnaE1fw/rv (Table S1, ESI†). The snaE1′ amplificate was subcloned into the EcoRV-restricted E. coli vector pDRIVE, resulting in the construct pDRIVE/snaE1′. The snaE1′ fragment was isolated from pDRIVE/snaE1′ as an EcoRI fragment and then ligated into the EcoRI-restricted E. coli vector pK18, resulting in the construct pK18/snaE1′. A 1.1 kb thiostrepton resistance cassette (tsrR) was isolated as a SnaBI/AleI fragment from pDRIVE/thio and cloned into the StuI restriction site of snaE1′, resulting in the mutational construct pK18/snaE1tsr (Fig. S25, ESI†). The plasmid was transferred to the pglA deletion mutant S. pristinaespiralis MpglA41 by protoplast transformation, followed by selection for apramycin/thiostrepton-resistant and kanamycin-sensitive transformants, which resulted in the double mutant S. pristinaespiralis ΔpglAΔsnaE1. The correctness of the mutant was verified by PCR using genomic DNA from ΔpglAΔsnaE1. For verification of the snaE1 gene inactivation, primer pairs were used, which annealed to the tsrR cassette (thio1/2) and to internal parts of the snaE1 gene (KsnaE1fw/rv), respectively. A 0.4 kb fragment was amplified with the primer pair thio1/2, a 2.1 kb fragment with primer pair KsnaE1fw/rv, a 0.8 kb fragment with the primer pair KsnaE1fw/thio2, and a 1.7 kb fragment with the primer pair thio1/KsnaE1rv, confirming the correctness of the S. pristinaespiralis ΔpglAΔsnaE1 mutant (Fig. S26, ESI†).4.5 Mutasynthesis with Phg derivatives
For mutasynthesis experiments with S. pristinaespiralis, strains were grown in 100 mL of HT7T medium as a preculture. After 72 hours, 10 mL of preculture was used to inoculate 100 mL of fresh HT7T medium as the main culture. For feeding experiments, the main cultures were supplemented with different mutasynthons (Table 1) (solubilized with 70 μL of 1 N NaOH and neutralized with 50 μL of 1 N HCl per mg compound) to reach a final concentration of 10 μg mL−1. The cells were grown for 168 hours at 28 °C under shaking at 180 rpm. To perform mutasynthesis with E. coli biotransformation samples, main cultures were prepared as described above and supplemented with 1 mL of E. coli BL21(DE3) pET28-hmo/pACYC-bcd-gdh culture supernatant containing approx. 1 mg mL−1 of the respective mutasynthon.4.6 Compound extraction and purification
For initial compound detection and bioassay testing with S. pristinaespiralis samples, 5 mL of culture was extracted with 5 mL of ethyl acetate (1:1) for 1 h at room temperature (RT) in an overhead shaker. After centrifugation at 5000 rpm for 20 min, ethyl acetate phases were concentrated in vacuo completely and then dissolved in 500 μL of methanol. Crude extracts were used for bioassays, high-performance liquid chromatography (HPLC) and high-performance liquid chromatography-mass spectrometry (HPLC-MS, HPLC-MS/MS) analysis. Batch cultures of S. pristinaespiralis ΔpglAΔsnaE1 fed with chlorinated or fluorinated mutasynthons were grown, extracted, and treated for compound purification following Protocol S1 and S2, respectively. In total, 12 l and 6 l of culture were extracted for 3 and 4 isolation, respectively. Pure fractions were used to elucidate the structure of halogenated derivatives using NMR.
4.7 HPLC and HPLC-ESI-MS(/MS) analyses of pristinamycin variants
HPLC analyses of pristinamycin derivatives were performed using an HP1090M system with ChemStation 3D software rev. A.08.03 (Agilent Technologies, Waldbronn, Germany) on a Waters Symmetry C18 (3.0 × 150 mm, 5 μm) with a flow rate of 850 μL min−1. Chromatography was done by linear step gradient elution from 80% solvent A (water with 0.1% phosphoric acid) and 20% solvent B (acetonitrile to 50% solvent B over 12 min, 50% solvent B to 100% solvent B over 3 min followed by a 3 min hold of 100% solvent B). The injection volume was 5 μL. Multiple wavelength monitoring was performed at 210, 230, 260, 280, 310, 360, 435 and 500 nm. UV-Vis spectra were measured from 200 to 600 nm. The evaluation of the chromatograms (210 nm only) was done by means of an in-house HPLC-UV-Vis database. HPLC-MS and HPLC-MS/MS analyses of pristinamycin derivatives were performed using an Agilent 1200 series chromatography system (binary pump, high performance autosampler, DAD-detector) coupled with an LC/MSD Ultra Trap System XCT 6330 (Agilent Technologies, Waldbronn, Germany). The sample (2.5 μL) was injected on a Nucleosil 100 C18 column (100 × 2 mm, 3 μm) fitted with a precolumn (10 × 2 mm, 3 μm) at a flow rate of 400 μL min−1 and a linear gradient from 90% solvent A (0.1% formic acid in water) to 100% solvent B (0.06% formic acid in acetonitrile) over 15 minutes at 40 °C. UV-Vis-detection was done at 220, 260, 280, 360 and 435 nm. Electrospray ionization was performed in positive and negative ultra-scan modes (alternating) with a capillary voltage of 3.5 kV and a drying gas temperature of 350 °C. Detection of m/z values was conducted with Agilent DataAnalysis for 6300 Series IonTrap LC/MS Version 3.4 (Bruker Daltonik). Upon HPLC-MS analysis, pristinamycin I derivatives were identified by comparison of their UV-Vis spectra, retention times and molecular masses with an authentic pristinamycin I (PI) standard (Sanofi-Aventis) and predicted masses for 1 = m/z 867.4034 [M + H]+, and derivatives 3 = m/z 901.3646 [M + H]+, and 4 = m/z 885.3941 [M + H]+.Pristinamycin I (1): white powder; 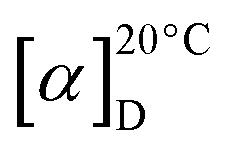 −50 (c 0.6 g/100 mL, MeOH); UV (MeOH) λmax (logε) 202 (4.8), 260 (4.2), 304 (3.9); ESI-MS: m/z 865.32 [M−H]− and 867.56 [M + H]+; HR-ESI-MS: m/z 867.4034 [M + H]+ (calculated for sum formula C45H55N8O10, 867.4036).
−50 (c 0.6 g/100 mL, MeOH); UV (MeOH) λmax (logε) 202 (4.8), 260 (4.2), 304 (3.9); ESI-MS: m/z 865.32 [M−H]− and 867.56 [M + H]+; HR-ESI-MS: m/z 867.4034 [M + H]+ (calculated for sum formula C45H55N8O10, 867.4036).
6-Chloro-PI (3): white to slightly brown powder; UV (MeOH) λmax (logε) 201 (4.9), 255 (4.3), 302 (4.0); 1H NMR data (700 MHz, CH3OH-d4): see Table 2; 13C NMR data (175 MHz, CH3OH-d4): see Table 2; ESI-MS: m/z 899.47 [M−H]− and 901.44 [M + H]+; HR-ESI-MS: m/z 901.3645 [M + H]+ (calculated for C45H54N8O10Cl, 901.3646).
6-Fluoro-PI (4): white powder; 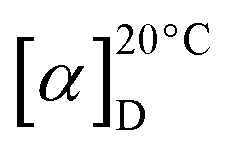 −45 (c 0.1 g mL−1, MeOH); UV (MeOH) λmax (logε) 203 (4.8), 260 (4.2), 303 (3.9); ESI-MS: m/z 883.55 [M−H]− and 885.54 [M + H]+; 1H NMR data (700 MHz, DMSO-d6): see Table 2; 13C NMR data (175 MHz, DMSO-d6): see Table 2; 19F NMR (470 MHz, DMSO-d6): δF 113.38 ppm; HR-ESI-MS: m/z 885.3945 [M + H]+ and 907.3762 [M + Na]+ (calculated C45H54N8O10F, 885.3941 and C45H53N8O10FNa, 907.3748).
−45 (c 0.1 g mL−1, MeOH); UV (MeOH) λmax (logε) 203 (4.8), 260 (4.2), 303 (3.9); ESI-MS: m/z 883.55 [M−H]− and 885.54 [M + H]+; 1H NMR data (700 MHz, DMSO-d6): see Table 2; 13C NMR data (175 MHz, DMSO-d6): see Table 2; 19F NMR (470 MHz, DMSO-d6): δF 113.38 ppm; HR-ESI-MS: m/z 885.3945 [M + H]+ and 907.3762 [M + Na]+ (calculated C45H54N8O10F, 885.3941 and C45H53N8O10FNa, 907.3748).
Atom numbering refers to Oku et al. 2004.60
4.8 Structure elucidation
NMR spectra were recorded using an Avance III 700 spectrometer with a 5 mm TCI cryoprobe (1H 700 MHz, 13C 175 MHz) and an Avance III 500 spectrometer (1H 500 MHz, 13C 125 MHz) (both Bruker BioSpin, Rheinstetten, Germany). Chemical shifts δ were referenced to DMSO-d6 (δH = 2.50 ppm; 13C, δC = 39.51 ppm), CHCl3-d (δH = 7.27 ppm; 13C, δC = 77.0 ppm) and trifluoroacetic acid (δF = −74.95 ppm in DMSO).ESI mass spectra were recorded with an UltiMate 3000 Series uHPLC (Thermo Fisher Scientific, Waltman, MA/USA) by utilizing a C18 Acquity UPLC BEH column (50 × 2.1 mm, 1.7 μm; Waters, Milford, USA) connected to an amaZon speed ESI-Iontrap-MS (Bruker Daltonics, Bremen, Germany). The HPLC parameters were set as follows: solvent A: H2O + 0.1% formic acid, solvent B: acetonitrile (MeCN) + 0.1% formic acid, gradient: 5% B for 0.5 min, increasing to 100% B over 19.5 min, keeping 100% B for a further 10 min, flow rate 0.6 mL min−1, and DAD detection 190–600 nm.
HRESIMS was performed using a maXis ESI-TOF (electrospray ionization-time of flight) mass spectrometer (Bruker GmbH, Bremen, Germany) coupled to an Agilent 1260 series HPLC-UV system equipped with a C18 Acquity UPLC BEH 2.1 × 50 mm, 1.7 μm (Waters) column; DAD-UV detection at 200–600 nm; solvent A (H2O) and solvent B (MeCN) supplemented with 0.1% FA as a modifier; flowrate of 0.6 mL min−1, 40 °C oven temperature, gradient elution system with stationary phase of 0.5 min at 5% B, followed by an increase from 5% to 100% B in 19.5 min and holding at 100% B for 5 min.
4.9 Computational docking studies of pristinamycins with the ribosome
For computational docking studies with pristinamycins, SeeSAR version 13.0.1 was used; BioSolveIT GmbH, Sankt Augustin, Germany, 2023, https://www.biosolveit.de/SeeSAR. PDB files of the co-crystal structures of quinupristin (2) with the ribosome of E. coli (PDB: 4U26), D. radiodurans (PDB: 1MS1), and H. marismortui (PBD: 1YJW)34,35,44 were downloaded from https://www.rcsb.org/. Fragment growing and truncation approaches were performed based on the structure of quinupristin (2) and poses of pristinamycin I (1) as well as new derivatives with modifications of the Phg moiety were generated and re-docked into the binding pockets of quinupristin (2).4.10 Disk diffusion bioassays
General antibiotic activity of pristinamycin variants was analyzed by disc diffusion assays using Bacillus subtilis ATCC 6633 as the test organism and was carried out as reported before.61 Additionally, a panel of pathogenic organisms including, Gram-positive and Gram-negative bacteria, as well as eukaryotic organisms was used (Table S1, ESI†). 30 μL of S. pristinaespiralis culture extracts were pipetted on a filter disc, which was placed on a Bacillus subtilis test plate. Synercid® filter discs (15 μg; Oxoid) were used as a positive control and 30 μL of methanol as a negative control. The plates were incubated overnight at 37 °C. The bioactivity of the samples was determined qualitatively by measuring the diameter of the inhibition zone. For more quantitative activity tests of halogenated pristinamycin I derivatives 3 and 4 using streptogramin susceptible pathogenic test organisms (Enterococcus faecium DSM 20477 and Staphylococcus aureus (DSM 18827, CIP 111304, CIP 108540)), the amount of used extract was calculated to contain approximately 10 μg of the respective derivative and was added to filter discs with or without 10 μg of pristinamycin II, respectively.4.11 Minimal inhibition concentration (MIC) tests
Compound 4 was tested in MIC assays in serial dilutions to determine the minimum inhibitory concentration against standard test organisms, which included fungi, Gram-positive and Gram-negative test bacteria (Table S2, ESI†). Appropriate antibiotics were used as positive controls. Assays were performed following the protocol described by Becker et al.624.12 Cytotoxicity tests
Compound 4 was tested in cytotoxicity assays against two different mammalian cell lines, including human endocervical adenocarcinoma KB 3.1 and mouse fibroblasts L929 cell lines, with epothilone B as a positive control (Table S3, ESI†). Assays were performed following the protocol described by Becker et al.624.13 In vitro transcription and translation (ivTT) assay with pristinamycin I derivatives
Semi-purified HPLC fractions of 1, 3, and 4 were tested in ivTT assays. Assays were carried out as reported before51 and were conducted in triplicates. Each assay had an end concentration of approximately 0.5 μg mL−1 of 1, 3, 4, or PI standard (Sanofi-Aventis) with or without the addition of 0.5 μg mL−1 pristinamycin II standard (Sanofi-Aventis), respectively. Fluorescence of generated eGFP was measured using a Varioskan LUX plate reader (Thermo Scientific) to quantify the effect of tested samples on transcription/translation.4.14 Construction of a whole cell biotransformation route for Phg mutasynthon production
In order to biotechnologically produce Phg mutasynthons, a synthetic enzymatic cascade was applied (Fig. S27, ESI†). A whole cell biotransformation route with resting cells was established by heterologous expression of the genes encoding hydroxymandelate synthase (hms) from Amycolatopsis mediterranei, mandelate oxidase (hmo) from S. coelicolor, leucine dehydrogenase (bcd) from Bacillus thuringensis, and glucose dehydrogenase (gdh) from B. megaterium in E. coli BL21(DE3). The construction of the plasmids used is described in Protocol S3. For the formation of cells used for whole cell biotransformation, production of Hms was done as described before.55 Coproduction of the Hmo, Bcd and Gdh was performed in 2 L shaking flasks containing 400 mL of LB medium (kanamycin (50 μg mL−1) and chloramphenicol (30 μg mL−1). The media were inoculated with an overnight culture to an OD600 of 0.02 and incubated at 37 °C and 180 rpm to an OD600 of 0.8. Expression was induced with 0.1 mM IPTG and further incubated at 30 °C and 100 rpm for 4 h. Cells were harvested by centrifugation (10 min, RT, 4000 × g) and washed twice with 200 mM Tris–HCl buffer (pH 7.5). The optical density at 600 nm was monitored photometrically using a Cary 50 Bio spectrophotometer (Varian, Mulgrave, Australia). The biotransformation was performed in two steps in 250 mL shaking flasks with a reaction volume of 20 mL. The transformation of the phenylalanine derivative to the corresponding mandelic acid was performed as described before.55 The supernatant was isolated after 24 h of incubation by centrifugation (10 min, RT, 4000 × g) and was used directly as a substrate for the transformation to the Phg synthesis with E. coli BL21(DE3) pET28-hmo/pACYC-bcd-gdh. 20 mM glucose and 50 mM ammonium acetate were added after 1 h and 16 h to the HMO-LeuDH-GDH reaction to ensure the cofactor supply. After 24 h of biotransformation, the supernatant was isolated by centrifugation (10 min, RT, 4000 × g), sterile filtrated (Filtropur S (0.45 μm), Sarstedt) and stored at −20 °C until use. The respective enantiomeric excess (ee) of the produced Phgs was determined using HPLC analysis with comparison to standards of available racemic and enantiomeric pure Phgs (Fig. S28, ESI†). The separation of Phg enantiomers was achieved by using a Chirex 3126 (D)-penicillamine column (150 × 4.6 mm, Phenomenex, Aschaffenburg, Germany) with 2 mM copper(II) sulfate and 15% methanol as mobile phase at 30 °C. The isocratic flow was set to 1 mL min−1 and 4 μL of sample was injected.Author contributions
OH carried out a mutasynthesis experiment with S. pristinaespiralis. LW and J-WY performed biotransformation experiments for Phg-derivative supply. KH, FS, and AK performed chemical analysis (HPLC, MS/MS, NMR). KH and FS carried out MHC and cytotoxicity tests. PK carried out SeeSAR modeling analysis. J-WY designed and supervised biotransformation studies. YM designed and supervised mutasynthesis studies and coordinated the study.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
We thank Sabine Gronow and the DZIF Pathogen Repository team for supplying us with pathogenic strains from the DSMZ culture collection. Pure pristinamycin I and II compounds were kindly provided by Sanofi-Aventis Pharma. We acknowledge technical assistance from Meike Döppner with ivTT assays, Wera Collisi for conducting the MIC and cytotoxicity assays, Esther Surges with NMR and Aileen Gollasch with HRMS measurements. This work was supported by the Baden-Württemberg-Stiftung (BWST_WSF-035). We also acknowledge funding received from the German Center for Infection Research (DZIF) (TTU 09.819) and from the Leibniz Association through the Collaborative Excellence funding program (K445/2022).References
- F. Haney and E. W. Hancock, Addressing Antibiotic Failure—Beyond Genetically Encoded Antimicrobial Resistance, Front. Drug Discov., 2022, 2, 892975 CrossRef.
- T. Maxson and D. A. Mitchell, Targeted Treatment for Bacterial Infections: Prospects for Pathogen-Specific Antibiotics Coupled with Rapid Diagnostics, Tetrahedron, 2016, 72, 3609–3624 CrossRef CAS PubMed.
- J. F. Prescott, The resistance tsunami, antimicrobial stewardship, and the golden age of microbiology, Vet. Microbiol., 2014, 171, 273–278 CrossRef PubMed.
- C. J. L. Murray, et al., Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- M. S. Butler, V. Gigante, H. Sati, S. Paulin, L. Al-Sulaiman, J. H. Rex, P. Fernandes, C. A. Arias, M. Paul, G. E. Thwaites, L. Czaplewski, R. A. Alm, C. Lienhardt, M. Spigelman, L. L. Silver, N. Ohmagari, R. Kozlov, S. Harbarth and P. Beyer, Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed, Antimicrob. Agents Chemother., 2022, 66, e0199121 CrossRef PubMed.
- F. Kloß and S. Gerbach, Obstacles and perspectives of new antimicrobial concepts within research and development, Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz, 2018, 61, 595–605 CrossRef PubMed.
- A. R. Coates, G. Halls and Y. Hu, Novel classes of antibiotics or more of the same?, Br. J. Pharmacol., 2011, 163, 184–194 CrossRef CAS PubMed.
- E. A. Barka, P. Vatsa, L. Sanchez, N. Gaveau-Vaillant, C. Jacquard, J. P. Meier-Kolthoff, H. P. Klenk, C. Clément, Y. Ouhdouch and G. P. van Wezel, Taxonomy, Physiology, and Natural Products of Actinobacteria, Microbiol. Mol. Biol. Rev., 2016, 80, 1–43 CrossRef PubMed.
- A. Gavriilidou, S. A. Kautsar, N. Zaburannyi, D. Krug, R. Müller, M. H. Medema and N. Ziemert, Compendium of specialized metabolite biosynthetic diversity encoded in bacterial genomes, Nat. Microbiol., 2022, 7, 726–735 CrossRef CAS PubMed.
- R. H. Baltz, Renaissance in antibacterial discovery from actinomycetes, Curr. Opin. Pharmacol., 2008, 8, 557–563 CrossRef CAS PubMed.
- M. Miethke, M. Pieroni, T. Weber, M. Brönstrup, P. Hammann., L. Halby, P. B. Arimondo, P. Glaser, B. Aigle, H. B. Bode, R. Moreira, Y. Li, A. Luzhetskyy, M. H. Medema, J. L. Pernodet, M. Stadler, J. R. Tormo, O. Genilloud, A. W. Truman, K. J. Weissman, E. Takano, S. Sabatini, E. Stegmann, H. Brötz-Oesterhelt, W. Wohlleben, M. Seemann, M. Empting, A. K. H. Hirsch, B. Loretz, C. M. Lehr, A. Titz, J. Herrmann, T. Jaeger, S. Alt, T. Hesterkamp, M. Winterhalter, A. Schiefer, K. Pfarr, A. Hoerauf, H. Graz, M. Graz, M. Lindvall, S. Ramurthy, A. Karlén, M. van Dongen, H. Petkovic, A. Keller, F. Peyrane, S. Donadio, L. Fraisse, L. J. V. Piddock, I. H. Gilbert, H. E. Moser and R. Müller, Towards the sustainable discovery and development of new antibiotics, Nat. Rev. Chem., 2021, 5, 726–749 CrossRef CAS PubMed.
- D. Pavlović, S. Mutak, D. Andreotti, S. Biondi, F. Cardullo, A. Paio, E. Piga, D. Donati and S. Lociuro, Synthesis and Structure-Activity Relationships of α-Amino-γ-lactone Ketolides: A Novel Class of Macrolide Antibiotics, ACS Med. Chem. Lett., 2014, 5, 1133–1137 CrossRef PubMed.
- H. Sun, Z. Liu, H. Zhao and E. L. Ang, Recent advances in combinatorial biosynthesis for drug discovery, Drug Des., Dev. Ther., 2015, 9, 823–833 CAS.
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. Süssmuth, Mutasynthesis of glycopeptide antibiotics: variations of vancomycin's AB-ring amino acid 3, 5-dihydroxyphenylglycine, J. Am. Chem. Soc., 2004, 126, 5942–5943 CrossRef CAS PubMed.
- K. C. Nicolaou, H. J. Mitchell, N. F. Jain, N. Winssinger, R. Hughes and T. Bando, Total synthesis of vancomycin, Angew. Chem., Int. Ed., 2004, 38, 240–244 CrossRef.
- M. J. Moore, S. Qu, C. Tan, Y. Cai, Y. Mogi, D. Jamin Keith and D. L. Boger, Next-generation total synthesis of vancomycin, J. Am. Chem. Soc., 2020, 142, 16039–16050 CrossRef CAS PubMed.
- S. Pelzer and A. Dziarnowski, Sulfation: a new biocombinatorial tool, Chem. Biol., 2006, 13, 113–114 CrossRef CAS PubMed.
- W. Wohlleben, Y. Mast, G. Muth, M. Röttgen, E. Stegmann and T. Weber, Synthetic biology of secondary metabolite biosynthesis in actinomycetes: Engineering precursor supply as a way to optimize antibiotic production, FEBS Lett., 2012, 586, 2171–2176 CrossRef CAS PubMed.
- K. L. Rinehart, Mutasynthesis of new antibiotics, Pure Appl. Chem., 1977, 49, 1361–1384 CAS.
- J. Kennedy, Mutasynthesis, chemobiosynthesis, and back to semi-synthesis: combining synthetic chemistry and biosynthetic engineering for diversifying natural products, Nat. Prod. Rep., 2008, 25, 25–34 RSC.
- H. B. Bode and R. Müller, The Impact of Bacterial Genomics on Natural Product Research, Angew. Chem., Int. Ed., 2005, 44, 6828–6846 CrossRef CAS PubMed.
- A. Kirschning and F. Hahn, Merging chemical synthesis and biosynthesis: a new chapter in the total synthesis of natural products and natural product libraries, Angew. Chem., 2012, 51, 4012–4022 CrossRef CAS PubMed.
- L. Gou, Q. Wu, S. Lin, X. Li, J. Liang, X. Zhou, D. An, Z. Deng and Z. Wang, Mutasynthesis of pyrrole spiroketal compound using calcimycin 3-hydroxy anthranilic acid biosynthetic mutant, Appl. Microbiol. Biotechnol., 2013, 97, 8183–8191 CrossRef CAS PubMed.
- M. Kaniusaite, T. Kittila, R. J. Goode, R. B. Schittenhelm and M. J. Cryle, Redesign of substrate selection in glycopeptide antibiotic biosynthesis enables effective formation of alternate peptide backbones, ACS Chem. Biol., 2020, 15, 2444–2455 CrossRef CAS PubMed.
- K. J. Weissman, Mutasynthesis – uniting chemistry and genetics for drug discovery, Trends Biotechnol., 2007, 25, 139–142 CrossRef CAS PubMed.
- S. Eichner, T. Knobloch, H. G. Floss, J. Fohrer, K. Harmrolfs, J. Hermane, A. Schulz, F. Sasse, P. Spiteller, F. Taft and A. Kirschning, The Interplay between Mutasynthesis and Semisynthesis: Generation and Evaluation of an Ansamitocin Library, Angew. Chem., Int. Ed., 2012, 51, 752–757 CrossRef CAS PubMed.
- L. Toscano, G. Fioriello, R. Spagnoli, L. Cappelletti and G. Zanuso, New fluorinated erythromycins obtained by mutasynthesis, J. Antibiot., 1983, 36, 1439–1450 CrossRef CAS PubMed.
- Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. M. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Structure, Biosynthetic Origin, and Engineered Biosynthesis of Calcium-Dependent Antibiotics from Streptomyces coelicolor, Chem. Biol., 2002, 9, 1175–1187 CrossRef CAS PubMed.
- J. Delzer, H. P. Fiedler, H. Müller, H. Zähner, R. Rathmann, K. Ernst and W. A. König, New nikkomycins by mutasynthesis and directed fermentation, J. Antibiot., 1984, 37, 80–82 CrossRef CAS PubMed.
- L. Winand, P. Schneider, S. Kruth, N. J. Greven, W. Hiller, M. Kaiser, J. Pietruszka and M. Nett, Mutasynthesis of Physostigmines in Myxococcus xanthus, Org. Lett., 2021, 23, 6563–6567 CrossRef CAS PubMed.
- U. Galm, Marco A. Dessoy, J. Schmidt, L. A. Wessjohann and L. Heide, In Vitro and In Vivo Production of New Aminocoumarins by a Combined Biochemical, Genetic, and Synthetic Approach, Chem. Biol., 2004, 11, 173–183 CrossRef CAS PubMed.
- D. Ulanova, J. Novotná, Y. Smutná, Z. Kameník, R. Gazák, M. Sulc, P. Sedmera, S. Kadlcík, K. Plhácková and J. Janata, Mutasynthesis of lincomycin derivatives with activity against drug-resistant staphylococci, Antimicrob. Agents Chemother., 2010, 54, 927–930 CrossRef CAS PubMed.
- S. Reissier and V. Cattoir, Streptogramins for the treatment of infections caused by Gram-positive pathogens, Expert Rev. Anti-Infect. Ther., 2021, 19, 587–599 CrossRef CAS PubMed.
- D. Tu, G. Blaha, P. B. Moore and T. A. Steitz, Structures of MLSBK Antibiotics Bound to Mutated Large Ribosomal Subunits Provide a Structural Explanation for Resistance, Cell, 2005, 121, 257–270 CrossRef CAS PubMed.
- J. M. Harms, F. Schlünzen, P. Fucini, H. Bartels and A. Yonath, Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin, BMC Biol., 2004, 2, 4 CrossRef PubMed.
- Y. Mast and W. Wohlleben, Streptogramins – Two are better than one!, Int. J. Med. Microbiol., 2014, 304, 44–50 CrossRef CAS PubMed.
- N. J. Johnston, T. A. Mukhtar and G. D. Wright, Streptogramin Antibiotics: Mode of Action and Resistance, Curr. Drug Targets, 2002, 3, 335–344 CrossRef CAS PubMed.
- M. S. Svetlov, E. A. Syroegin, E. V. Aleksandrova, G. C. Atkinson, S. T. Gregory, A. S. Mankin and Y. S. Polikanov, Structure of Erm-modified 70S ribosome reveals the mechanism of macrolide resistance, Nat. Chem. Biol., 2021, 17, 412–420 CrossRef CAS PubMed.
- S. Schwarz, J. Shen, K. Kadlec, Y. Wang, G. B. Michael, A. T. Feßler and B. Vester, Lincosamides, streptogramins, phenicols, and pleuromutilins: mode of action and mechanisms of resistance, Cold Spring Harbor Perspect. Med., 2016, 6, a027037 CrossRef PubMed.
- Y. Mast, T. Weber, M. Gölz, R. Ort-Winklbauer, A. Gondran, W. Wohlleben and E. Schinko, Characterization of the ‘pristinamycin supercluster’ of Streptomyces pristinaespiralis, Microb. Biotechnol., 2011, 4, 192–206 CrossRef CAS PubMed.
- Y. Mast, W. Wohlleben and E. Schinko, Identification and functional characterization of phenylglycine biosynthetic genes involved in pristinamycin biosynthesis in Streptomyces pristinaespiralis, J. Biotechnol., 2011, 155, 63–67 CrossRef CAS PubMed.
- D. Moosmann, V. Mokeev, A. Kulik, N. Osipenkov, S. Kocadinc, R. Ort-Winklbauer, F. Handel, O. Hennrich, J. W. Youn, G. A. Sprenger and Y. Mast, Genetic engineering approaches for the fermentative production of phenylglycines, Appl. Microbiol. Biotechnol., 2020, 104, 3433–3444 CrossRef CAS PubMed.
- N. Osipenkov, A. Kulik and Y. Mast, Characterization of the phenylglycine aminotransferase PglE from Streptomyces pristinaespiralis, J. Biotechnol., 2018, 278, 34–38 CrossRef CAS PubMed.
- J. Noeske, J. Huang, N. B. Olivier, R. A. Giacobbe, M. Zambrowski and J. H. Cate, Synergy of streptogramin antibiotics occurs independently of their effects on translation, Antimicrob. Agents Chemother., 2014, 58, 5269–5279 CrossRef PubMed.
- J. Meng, R. Feng, G. Zheng, M. Ge, Y. Mast, W. Wohlleben, J. Gao, W. Jiang and Y. Lu, Improvement of pristinamycin I (PI) production in Streptomyces pristinaespiralis by metabolic engineering approaches, Synth. Syst. Biotechnol., 2017, 2, 130–136 CrossRef PubMed.
- C. Mahlert, S. A. Sieber, J. Grünewald and M. A. Marahiel, Chemoenzymatic approach to enantiopure streptogramin B variants: characterization of stereoselective pristinamycin I cyclase from Streptomyces pristinaespiralis, J. Am. Chem. Soc., 2005, 127, 9571–9580 CrossRef CAS PubMed.
- S. K. Gupta, P. Sharma, J. B. Barrett, L. M. Hiott, T. A. Woodley, J. G. Frye and C. R. Jackson, Draft genome sequence of a human-associated streptogramin-resistant Staphylococcus aureus, J. Glob. Antimicrob. Resist., 2019, 16, 72–73 CrossRef PubMed.
- B. M. Grüner, S. R. Han, H. G. Meyer, U. Wulf, S. Bhakdi and E. K. Siegel, Characterization of a catalase-negative methicillin-resistant Staphylococcus aureus strain, J. Clin. Microbiol., 2007, 45, 2684–2685 CrossRef PubMed.
- A. S. Walker and J. Clardy, A Machine Learning Bioinformatics Method to Predict Biological Activity from Biosynthetic Gene Clusters, J. Chem. Inf. Model., 2021, 61, 2560–2571 CrossRef CAS PubMed.
- R. S. Al Toma, C. Brieke, M. J. Cryle and R. D. Süssmuth, Structural aspects of phenylglycines, their biosynthesis and occurrence in peptide natural products, Nat. Prod. Rep., 2015, 32, 1207–1235 RSC.
- F. Handel, A. Kulik, K. W. Wex, A. Berscheid, J. S. Saur, A. Winkler, D. Wibberg, J. Kalinowski, H. Brötz-Oesterhelt and Y. Mast, Ψ-Footprinting approach for the identification of protein synthesis inhibitor producers, NAR Genom. Bioinform., 2022, 4, lqac055 CrossRef PubMed.
- G. Chinali, E. Nyssen, M. Di Giambattista and C. Cocito, Inhibition of polypeptide synthesis in cell-free systems by virginiamycin S and erythromycin. Evidence for a common mode of action of type B synergimycins and 14-membered macrolides, Biochim. Biophys. Acta, Gene Struct. Expression, 1988, 949, 71–78 CrossRef CAS PubMed.
- T. A. Mukhtar, K. P. Koteva and G. D. Wright, Chimeric Streptogramin-Tyrocidine Antibiotics that Overcome Streptogramin Resistance, Chem. Biol., 2005, 12, 229–235 CrossRef CAS PubMed.
- C. Cocito and F. Vanlinden, Inhibitory action of virginiamycin components on cell-free systems for polypeptide formation from Bacillus subtilis, Arch. Mikrobiol., 1983, 135, 8–11 CrossRef CAS PubMed.
- J.-W. Youn, C. Albermann and G. A. Sprenger, In vivo cascade catalysis of aromatic amino acids to the respective mandelic acids using recombinant E. coli cells expressing hydroxymandelate synthase (HMS) from Amycolatopsis mediterranei, Mol. Catal., 2020, 483, 110713 CrossRef CAS.
- Y. Mast, J. Guezguez, F. Handel and E. Schinko, A Complex Signaling Cascade Governs Pristinamycin Biosynthesis in Streptomyces pristinaespiralis, Appl. Environ. Microbiol., 2015, 81, 6621–6636 CrossRef CAS PubMed.
- T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater and D. A. Hopwood, Practical Streptomyces genetics, John Innes Foundation, Norwich, 2000 Search PubMed.
- M. Folcher, H. Gaillard, L. T. Nguyen, K. T. Nguyen, P. Lacroix, N. Bamas-Jacques, M. Rinkel and C. J. Thompson, Pleiotropic functions of a Streptomyces pristinaespiralis autoregulator receptor in development, antibiotic biosynthesis, and expression of a superoxide dismutase, J. Biol. Chem., 2001, 276, 44297–44306 CrossRef CAS PubMed.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, New York, 2nd edn, 1989 Search PubMed.
- N. Oku, S. Takemura, H. Onaka and Y. Igarashi, NMR characterization of streptogramin B and L-156,587, a non-synergistic pair of the streptogramin family antibiotic complexes produced inductively by a combined culture of Streptomyces albogriseolus and Tsukamurella pulmonis, Magn. Reson. Chem., 2022, 60, 261–270 CrossRef CAS PubMed.
- I. Handayani, H. Saad, S. Ratnakomala, P. Lisdiyanti, W. Kusharyoto, J. Krause, A. Kulik, W. Wohlleben, S. Aziz, H. Gross, A. Gavriilidou, N. Ziemert and Y. Mast, Mining Indonesian microbial biodiversity for novel natural compounds by a combined genome mining and molecular networking approach, Mar. Drugs, 2021, 19, 316 CrossRef CAS PubMed.
- K. Becker, S. Pfütze, E. Kuhnert, R. J. Cox, M. Stadler and F. Surup, Hybridorubrins A–D: Azaphilone heterodimers from stromata of Hypoxylon fragiforme and insights into the biosynthetic machinery for azaphilone diversification, Chem. – Eur. J., 2020, 27, 1438–1450 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00143a |
| This journal is © The Royal Society of Chemistry 2023 |