
A bio-inspired, ultra-tough, high-sensitivity, and anti-swelling conductive hydrogel strain sensor for motion detection and information transmission†
Xiang
Di
,
Jiawen
Hou
,
Mingming
Yang
,
Guolin
Wu
 * and
Pingchuan
Sun
*
* and
Pingchuan
Sun
*
Key Laboratory of Functional Polymer Materials, Institute of Polymer Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: guolinwu@nankai.edu.cn; spclbh@nankai.edu.cn
First published on 3rd October 2022
Abstract
Conductive hydrogels are excellent candidates for the next-generation wearable materials and are being extensively investigated for their potential use in health monitoring devices, human–machine interfaces, and other fields. However, their relatively low mechanical strength and performance degradation due to swelling have presented challenges in their practical application. Inspired by the multiscale heterogeneous architecture of biological tissue, a dynamic cross-linked, ultra-tough, and high-sensitivity hydrogel with a swelling resistance characteristic was fabricated by the principle of multiple non-covalent interaction matching and a step-by-step construction strategy. A heterogeneous structure was constructed by the combination of a ‘soft’ hydrophobic-conjugated micro-region structural domain with inter/intra-molecular hydrogen bonding and π–π stacking along with ‘rigid’ cross-linking via strong ionic coordination interactions. Reversible cross-linking synergies and variations in the content of rigid and flexible components guaranteed the hydrogel to undergo flexible and efficient modulation of the structures and gain excellent mechanics, including elongation at break (>2000%), toughness (∼60 MJ m−3), and recovery (>88%). Notably, hydrogels displayed good anti-swelling properties even in solutions with different pH (pH 2–11) and solvents. Moreover, the hydrogel further exhibited fast response (47.4 ms) and high sensitivity due to the presence of dynamic ions (Fe3+, Na+, and Cl−); therefore, it was assembled into a sensor to detect various human motions and used as a signal transmitter for the encryption and decryption of information according to Morse code. This study provides basis for the development of a variety of robust and flexible conductive hydrogels with multifunctional sensing applications in next-generation wearable devices.
New conceptsInspired by the structure of biological tissues, we proposed a facile step-by-step construction strategy to fabricate a dynamic cross-linked, ultra-tough, and high-sensitivity hydrogel with swelling resistance through the principle of multiple non-covalent interaction matching, which was constructed by the combination of inter/intra-molecular hydrogen bonding and π–π stacking of hydrophobic–conjugated microregion structural domains as the ‘soft’ skeleton, and strong ionic coordination interaction as the ‘rigid’ reversible sacrificial bond. Reversible cross-linking synergies and variations in the content of rigid and flexible components enabled efficient modulation of the hydrogel structure and mechanics, including excellent elongation at break (>2000%), toughness (∼60 MJ m−3) and recovery properties (>88%). Notably, the prepared hydrogels exhibited good resistance to swelling in different solvents and pH solutions. The presence of dynamic ions not only rendered the hydrogel with anti-freezing performance but also led to high sensitivity, fast response (47.4 ms), and cyclic stability, which can be used for healthcare monitoring, encrypting, and decrypting information according to Morse code. Thus, the nature-inspired design and step-by-step building strategy will balance strength, detection accuracy, and swelling resistance, also provide a new insight into the fabrication of flexible smart hydrogel sensors. |
Introduction
Hydrogel sensors, also termed as ‘electronic skin’, can detect and respond to external state changes or stresses and convert them into recordable signals such as current, resistance, and capacitance.1–7 The next-generation wearable materials formed using ionic conductive hydrogels with excellent mechanical, electrical, and physiological characteristics similar to human tissue have generated extensive interest due to their advanced applications, including health monitoring, motion recognition, and information encryption transmission.8–13 However, these sensors generally exhibit low toughness, limited responsivity, and rapid attenuation of signal detection under repeated large deformation.14–16 Furthermore, anti-swelling properties are also necessary to cope up with complex solvent environments and to maintain their stability.13,17 Therefore, fabrication of conductive hydrogels with superior toughness, excellent sensitivity, and swelling resistance is required for future conductive wearable sensors.In recent years, bionic hydrogels with enhanced mechanical strength have been fabricated via effective energy dissipation or by tailoring uniform structures such as hydrophobically associated hydrogels,18 double-network hydrogels,19 nanocomposite hydrogels,20 polyampholyte hydrogels,21 and others.22 Hydrophobic interaction, a type of physical interaction with tunable bonding strength, is strong and stable when exposed to an aqueous environment due to the contraction of hydrophobic chains. It has aided the development of tough hydrogels; however, the tensile strength of most of these hydrogels could hardly exceed 1 MPa.23–25 Besides, non-covalent interactions or reversible metal ion ligands have also been utilized as physical cross-linkers often in the form of sacrificial bonds to regulate the mechanical performance of materials.26–28 It was speculated that hydrophobic association combined with ionic coordination bond could significantly improve the mechanical strength and toughness of hydrogels. For example, Li et al. proposed a simple strategy to design a new type of physical hydrogel crosslinked via both hydrophobic association (poly(stearyl methacrylate)) and ionic coordination bond between carboxyl groups and Fe3+, which could realize excellent mechanical properties and good self-recovery capabilities.29 Inspired by cartilage, Gao et al. fabricated a conductive hydrogel by generating hydrophobic association interactions (hexadecyltrimethylammonium) and metal coordination, achieving ultrahigh toughness and high tensile strength.30 The mechanical performance of gels, such as the tensile properties, self-recovery, and toughness, have been greatly enhanced in recent years; however, achievement of adjustable strength and toughness of hydrogels based on skillful selection of mutually matching non-covalent interactions is of great scientific importance, which has rarely been investigated to date.
In general biological tissues, such as tendons, cartilage, and ligaments, exhibit a perfect combination of strength and toughness as well as swelling resistance due to their fine hierarchical microstructure and dense connective tissue composed of abundant interwoven elastic fibers.31–33 For instance, tendon, as a conduit for effective energy transfer by muscle contraction, is mainly composed of a synergy between the ‘soft’ extracellular matrix and ‘hard’ collagen fibers, in which the collagen binds into progressively larger subunits of a hierarchical structure. Human tissue ligaments also possess a similar synergistic structure of soft and hard components. To mimic the hierarchical heterogeneous structure of bio-tissues with soft and hard domains, it is possible to build the hydrogels with incredibly enhanced mechanical properties.34 Notably, the conditions for the construction of different non-covalent interactions vary, thus providing the possibility of constructing soft and hard synergistic heterogeneous structures in hydrogels similar to the microstructure of biological tissue through a step-by-step construction approach. Moreover, most hydrogels with hydrophilic polymer chains inevitably ‘swell’ under changes in osmotic pressure or physiological conditions, forming a loose network structure with detrimental effects on their mechanical properties.35–38 According to the literature, the anti-swelling performance of biological tissues is also related to its special hierarchical heterogeneous microstructure.30,35,37
Herein, inspired by the heterogeneous network structure of biological tissues, a pure dynamic physically cross-linked conductive hydrogel with high elongation at break, toughness, and responsiveness based on the principle of multiple heterogeneous non-covalent interaction matching and a step-by-step construction strategy was successfully fabricated. The network was composed of interconnected ‘soft’ hydrophobic entangled polymer chain and a ‘rigid’ cross-linked ionic body. The excellent mechanical properties, including tensile strength, elasticity modulus, toughness, and great recoverability of the hydrogel, were ascribed to the efficient energy dissipation capacity and combination of a rigid and flexible reversible network. Furthermore, the strong coordination effect and the introduction of hydrophobic association interactions led to excellent anti-swelling properties even in solutions with different pH (pH 2–11) and solvents (water, saline, acetone, and ethanol). The dynamic mechanical responsive behavior and heterogeneous structural evolution of the material were also characterized by rheology and multiple-quantum (MQ) nuclear magnetic resonance (NMR) spectroscopy. Furthermore, the free-moving ions (Fe3+, Na+, and Cl−) in the system not only endowed the gel with freeze-resistant properties, but also provided excellent conductive behavior, together with high sensitivity, fast response (47.4 ms), and cyclic stability. The hydrogel sensor was used to monitor human activities at multiple scales such as joint movements (finger, wrist, and elbow bending) and small deformations (light coughing and swallowing). Combined with Morse code, the application of this sensor in the field of information encryption and decryption was further explored. Therefore, the prepared heterogeneous network-structured hydrogel provides insight into the fabrication process of flexible sensors with excellent application prospects in the fields of biosensors, health monitoring, and information encryption transmission.
Results and discussion
Synthesis of ultra-strong and tough dual-network hydrogels
Biological tissue (tendons, ligaments, and cartilages) usually exhibits a perfect combination of strength and toughness as well as excellent resistance to swelling due to synergistic interaction between the soft extracellular matrix and hard collagen fibers.39–41 Inspired by biological tissue, a double-network dynamic cross-linked hydrogel was designed and prepared via a three-step method, in this study. First, a novel hydrophobic nucleic acid–base monomer (BA-A, namely, 4-((3-(adenin-9-yl)propanoyl)oxy)butyl acrylate) was successfully synthesized. The synthesis routes of BA-A and PAM-BA-A-AA (namely, poly(acrylamide-co-4-((3-(adenin-9-yl)propanoyl)oxy)butyl acrylate-co-acrylic acid)), and their molecular structures were confirmed by NMR spectroscopy and Fourier transform infrared (FTIR) spectroscopy (Scheme 1a and Fig. S1, ESI†). Then, the hydrophilic monomers (namely, acrylamide (AM) and acrylic acid (AA)) and hydrophobic monomer (BA-A) in sodium dodecyl sulfate (SDS)-NaCl, were copolymerized with SDS micelles using potassium persulfate (KPS) as a thermal initiator. BA-A conjugated within the surfactant SDS micelles acted as a physical cross-linking point and formed the pristine hydrogel (P-hydrogel), which resembled the role of the soft extracellular matrix in biological tissue, maintaining the entire gel backbone and providing toughness (Scheme 1b). The AM and AA monomers were applied as hydrophilic and functional components, respectively, to form the second cross-linking point. The P-hydrogel was then immersed in Fe3+ solution for 24 h to generate the transition-state double cross-linked coordination hydrogel (C-hydrogel), which formed ionic coordination bonds between carboxyl groups and Fe3+ in the network structure. Owing to the high osmotic pressure inside the hydrogel network, a large amount of Fe3+ penetrated the gel system, resulting in the formation of single, double, or triple coordination complexes. The role of these strong metal coordination interactions is equivalent to that of collagen fibers, the ‘hard’ component of the biological tissue structure, endowing the hydrogel with sufficient strength. To obtain more triple coordination complexes and stronger hydrophobic associations for the full combination of strength and toughness of hydrogel, the C-hydrogel was immersed in excess distilled water to remove excess Fe3+, which resulted in the formation of strongly double cross-linked equilibrium swollen hydrogel (E-hydrogel) with a reconfigured structure in the equilibrium state. The formation of the hydrogel network structure was verified by FTIR spectra (Fig. S1, ESI†).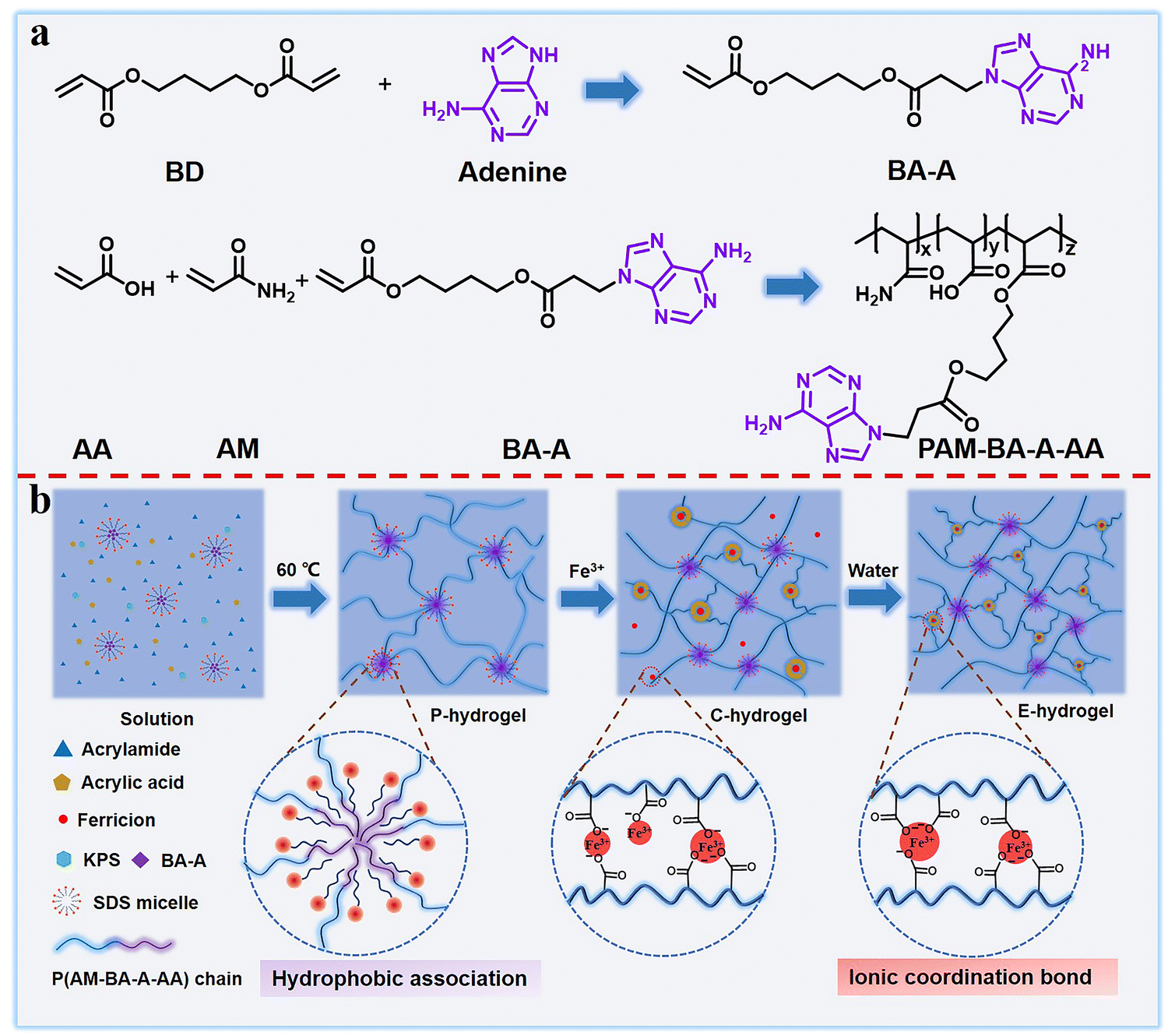 | ||
| Scheme 1 (a) Synthesis route of BA-A and PAM-BA-A-AA and (b) schematic showing the preparation process of double-network dynamic cross-linked hydrogels. | ||
Mechanical properties
The direct factor affecting the mechanical performance of hydrogels is the composition difference. For more accurate understanding of the strengthening mechanism of the hydrogel, a series of samples with different components was prepared (see Table S1, ESI†). The effect of hydrophobic association interactions on the mechanical performance of materials is shown in Fig. 1a. With the increase in the content of BA-A/AM from 0% to 2%, the mechanical properties (toughness, strength, and elongation) first increased and then decreased due to the synergistic interaction between metal coordination and the increase of physical hydrophobic linkage, which shortened the distance between polymer chains and further enhanced the performance of the hydrogel. The trend of this result is similar to the effect of hydrophobic association on P-hydrogels (Fig. S3, ESI†). Owing to the synergistic effect of metal coordination and hydrophobic association, the hydrogel performance was significantly improved after the introduction of Fe3+ (Fig. S4, ESI†). Moreover, the concentration of Fe3+ in the immersing process also affected the mechanical strength. That is, when concentration of the Fe3+ solution was increased from 0.01 M to 0.06 M, the corresponding strength and elastic modulus got enhanced; however, further increase in the concentration to 0.12 M resulted in the degeneration of its high elongation at break (Fig. 1b). This result is attributed to the formation of more ionic coordinates between Fe3+ and –COO– groups (bidentate coordinates and/or even monodentate coordinates) at a high concentration of ferric solution, leading to lower secondary cross-linking of hydrogels.42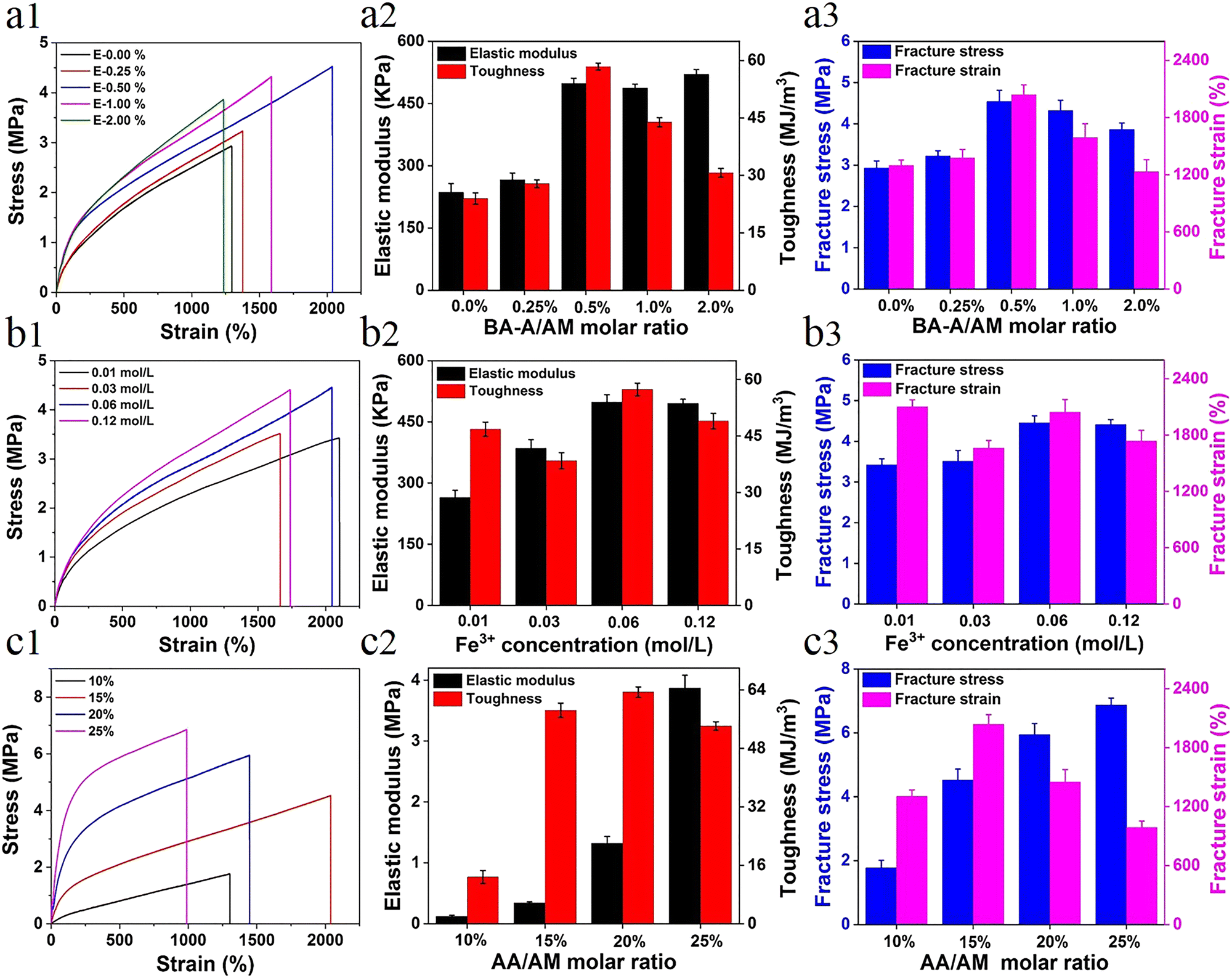 | ||
| Fig. 1 Tensile curves with different contents of (a1) BA-A/AM, (b1) Fe3+, and (c1) AA/AM; variation of the content of elastic modulus and toughness of (a2) BA-A/AM, (b2) Fe3+, and (c2) AA/AM; and variation of the content of fracture stress and strain of (a3) BA-A/AM, (b3) Fe3+, and (c3) AA/AM. | ||
Another factor affecting the performance of hydrogels was the content of ionic coordination bonds, mainly determined by the AA/AM molar ratio in this system. With the increase in the AA/AM molar ratio, the fracture stress and elastic modulus enhanced rapidly with an elastic modulus of ∼3.87 MPa and a tensile strength of ∼6.8 MPa (Fig. 1c). Toughness and strain first increased and then decreased due to the improvement in ionic coordination cross-linking density. With the increase in the molar ratio of AA/AM to 20% or 25%, more ionic coordination bonds were formed, leading to the rigidity of the hydrogel and reduction in its deformation ability.42 As the hydrogels exhibited optimal mechanical properties (tensile strength ∼4.52 MPa, elastic modulus 0.49 MPa, fracture strain ∼2044%, and toughness ∼58.4 MJ m−3) under the conditions of 0.5 mol% of BA-A/AM, 15 mol% of AA/AM, and 0.06 M of Fe3+ solution, these ratios of E-hydrogel were selected as optimized values for the subsequent studies unless otherwise stated.
Owing to the synergistic effect of hydrophobic conjugation (inter/intra-molecular hydrogen bonding and π–π stacking) and metal coordination, the prepared hydrogel showed remarkable mechanical properties (Fig. S2, ESI†). The hydrogel could tolerate diverse deformations such as stretching, twisting stretching, and knotting (Fig. 2a). Remarkably, the strips could be stretched to almost five times their original length and withstand loads of up to 3.5 kg without rupture, demonstrating ultra-mechanical strength (Fig. 2b). The hydrogel did not get punctured in the penetration test and, after 10 min, it showed excellent recovery, indicating that the hydrogel possessed puncture resistance and good recovery properties (Fig. 2c). The characterization of the mechanical performance of all hydrogels showed that the elastic modulus, toughness, strength, and elongation of P-hydrogels were only ∼10.2 kPa, 1.514 MJ m−3, 257 kPa, and 1045%, respectively (Fig. 2d–f). In contrast, the elastic modulus, toughness, strength, and elongation of E-hydrogels were ∼498.3 kPa, 58.415 MJ m−3, 4.52 MPa, and 2044%, respectively (Fig. 2b and c), i.e., 48.85, 38.58, 17.6, and 1.94 times higher than those of the P-hydrogel. These results show that the introduction of ionic coordination interaction could significantly improve the mechanical performance of hydrogels.
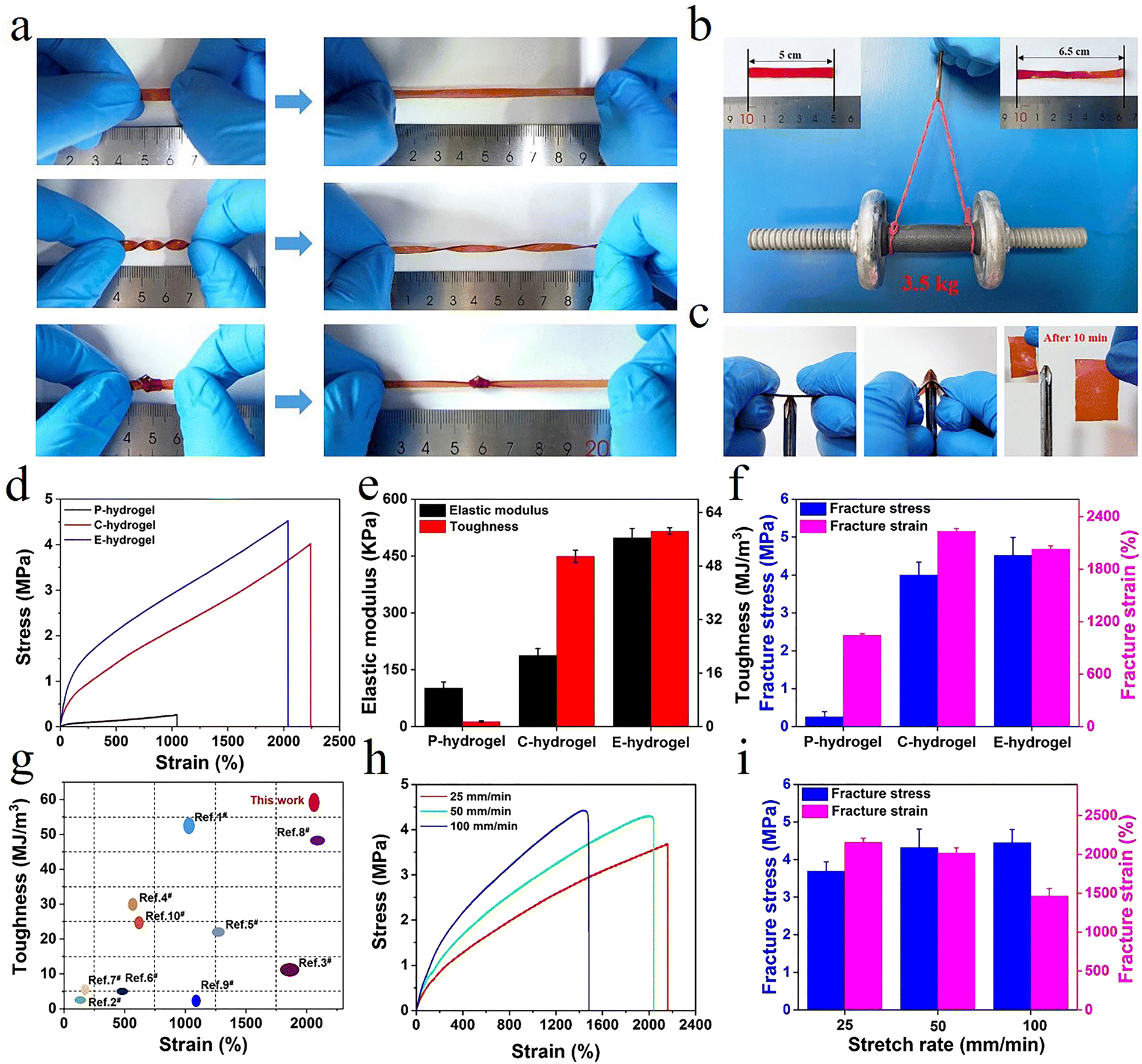 | ||
| Fig. 2 (a and b) The E-hydrogel was stretched, twisted, and knotted, and a 3.5 kg load was applied; (c) anti-puncture and self-healing capabilities; (d) tensile curves, (e) elastic modulus and toughness, (f) fracture stress and strain of hydrogels; (g) comparison of the mechanical behavior between E-hydrogel and other previously reported hydrogels (relevant data are summarized in Table S2, ESI†); (h) tensile curves of E-hydrogel at different stretching rates; and (i) corresponding fracture stress and strain. | ||
Moreover, the performance of the E-hydrogel was also distinctly better than that of the C-hydrogel due to excess Fe3+ diffusing out of the network, where the ionic coordination bonds were reorganized to form more tridentate coordination complexes in the system. These results were confirmed by a reduction in the E-hydrogel volume and a change in color from red to deep red, along with a stable elastic modulus and transmittance after immersion in water (Fig. S10, ESI†),42 manifesting the formation of more tridentate coordination complexes that reached a saturated state in the E-hydrogel network. Furthermore, the network structure of the E-hydrogel also became denser and more homogeneous than that of C-hydrogel (Fig. 3d–f). Low-field NMR spectroscopy and rheological experiments also demonstrated a reduction in structural defects and the uniformity and stability of the network. Compared with other high-strength hydrogels reported in the literature (Table S2, ESI†), hydrophobic association and interaction with metal coordination endowed the gel with an improved network structure and better mechanical properties than C-hydrogel (Fig. 2g and 3a–c). Owing to the dynamically reversible nature of metal coordination bonds, the mechanical characteristics of gels also depended on the variation of the stretching rate. The tensile curves of the E-hydrogel shifted upward with the increase in the stretching rate (Fig. 2h). Elastic modulus and strength increased, yet elongation decreased with the stretching rate (Fig. 2i), indicating that some moderately strong coordination interactions became coordination bonds with higher strength at high tensile rates. Therefore, they could be used as permanent crosslinks to resist deformation.27
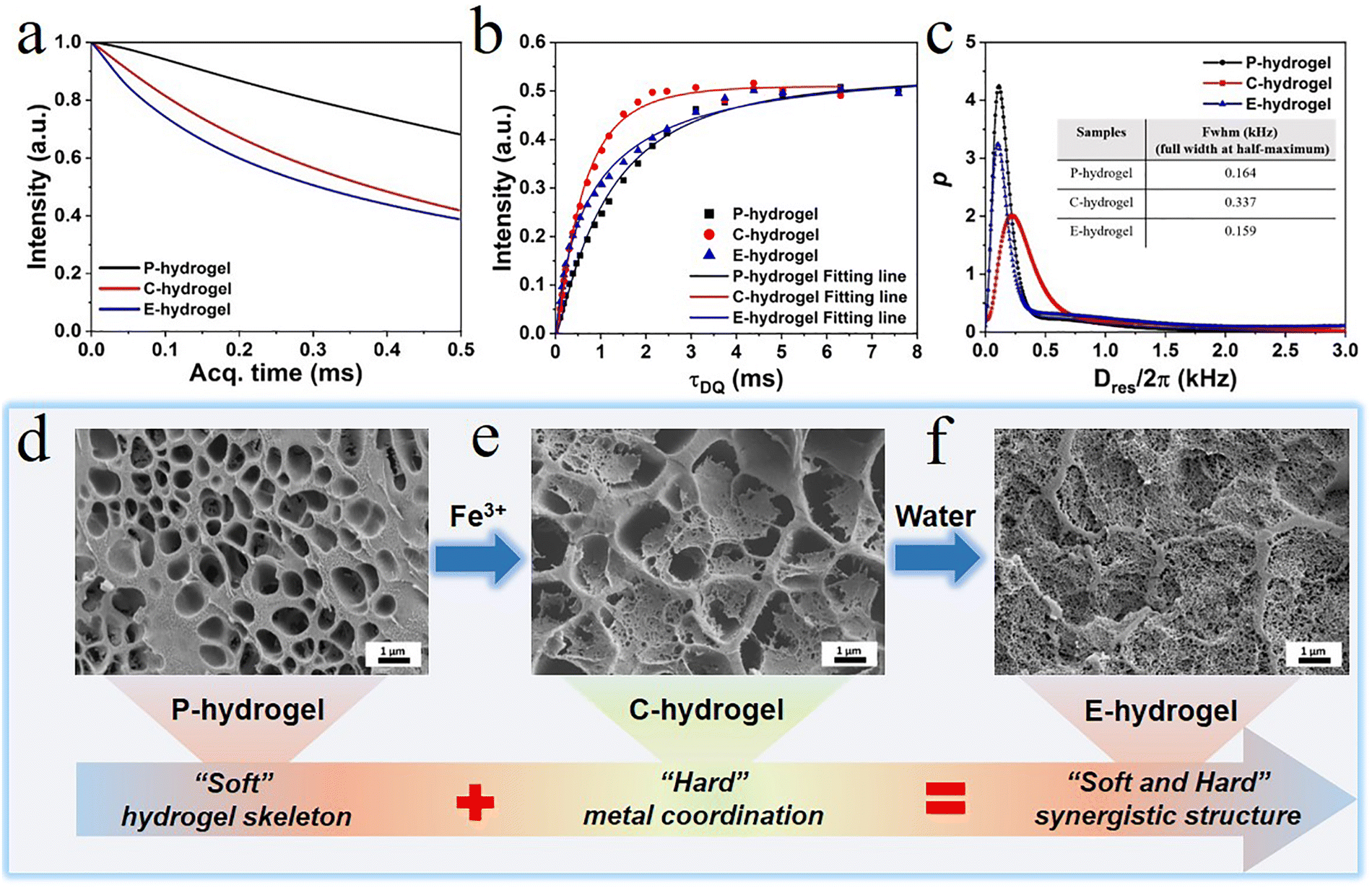 | ||
| Fig. 3 (a) Low-field MSE-FID NMR signal; (b) normalized DQ intensity as a function of DQ excitation time; (c) Dres distribution curves as obtained from the fitting on the normalized DQ curves; and SEM images of (d) P hydrogel, (e) C-hydrogel, and (f) E-hydrogel. | ||
Low-field NMR spectroscopy and the microstructural analysis
To elucidate the relationship between internal structure and performance, the as-prepared hydrogel, in which a physically cross-linked system with trivalent Fe3+-acrylic acid coordination was imposed on the hydrophobic association network, was characterized by MQ NMR spectroscopy and rheology (Fig. 3). MQ NMR spectroscopy was used to study the anisotropic structure and the associated chain segment mobility. Qualitative results of rigid and flexible phases were obtained (Fig. 3a). The free-induction decay (FID) of E-hydrogels was significantly faster than that of P-hydrogels due to the fact that the Fe3+-acrylic acid coordination (rigid components) in the network showed a rapidly decaying FID signal; however, water (soft components) in the hydrogel displayed a slow decay of proton signals due to the averaging effect of molecular motion. Relative to the C-hydrogel, the fast decay of E-hydrogel signal is ascribed to the reorganization of some unsaturated coordinates (monodentate and bidentate) to form tridentate complexes, leading to the increased content of rigid chain components.The normalized DQ intensity as a function of DQ excitation time for P.C.E-hydrogel was obtained (Fig. 3b). The rate of normalized DQ intensity strength growth provides information on cross-link density; the faster the growth rate, the higher the crosslink density.43–45 The growth rate of C-hydrogels and E-hydrogels was higher than that of P-hydrogels in the stage (0–3 ms), indicating that the introduction of Fe3+ coordination increased the cross-linking density and shortened the distance between polymer chains.46,47 The growth rate of C-hydrogel gradually exceeded that of E-hydrogel due to the presence of a large amount of Fe3+ in the system, forming more unstable coordination bonds than in the E-hydrogel. Therefore, the C-hydrogel in the transition state showed an inhomogeneous network and defects in its internal structure. The Dres distribution curves further illustrated the network structure parameters of the three gels (Fig. 3c), where Dres is the apparent residual dipolar coupling due to the anisotropic segmental dynamics imposed by the cross-linking/entanglement structures.48 The Dres distribution in the cross-linked hydrogel can well reveal the structural heterogeneity in terms of segmental lengths between cross-linking nodes in the sample. In most cases, heterogeneity, either structural/conformational difference among polymer chains or dynamics gradient/variation in different components, is present in the system. The full width at half-maximum (FWHM) of the Dres distribution can usually be applied to indicate the width of the distribution curve. A smaller value of its FWHM indicates a narrower distribution, indicating a more homogeneous hydrogel network.48 The FWHM values of the P.C.E-hydrogel were 0.164, 0.337, and 0.159 kHz, respectively (the inset picture of Fig. 3c). With the incorporation of Fe3+ ions in the hydrogel, the FWHM value of C-hydrogel became larger relative to the P-hydrogel (Fig. 3c) due to the partial formation of Fe3+ coordination complex (monodentate, bidentate, or tridentate), which imposed additional constraints on the polymer chain segments. Compared to the tridentate complex, the monodentate and bidentate complexes acted adversely to enhance the mechanical properties,42 and thus the C-hydrogel network structure exhibited inhomogeneity. The C-hydrogels immersed in deionized water formed E-hydrogels with narrower FWHM values and led to enhancement of structural heterogeneity due to the more stable cross-linking of trivalent complexes in the network. This resulted in enhanced constraints of polymer segments, which was expected to benefit the formation of a homogeneous coordination network with strong binding strength.48
The morphology of gel sections was observed by scanning electron microscopy (SEM) to explore changes in the physical network structure. The P-hydrogel formed by hydrophobic association, multiple hydrogen bonds, π–π stacking, and chain entanglement exhibited a homogenous network structure, and its function resembled the role of the soft extracellular matrix in biological tissue, maintaining the entire gel backbone and providing toughness (Fig. 3d and Fig. S2, ESI†). After immersing in Fe3+ solution, a dense structure, similar to the hard component collagen fibers in biological tissue, was formed in the network skeleton of the P-hydrogel, which was caused by strong metal coordination interactions, offering sufficient strength to the hydrogel. However, the uneven distribution of the tight network structure indicated the presence of defects in the gel network. These results could be attributed to excess Fe3+ in the system, leading to the formation of diverse types of coordination complexes (monodentate, bidentate, or tridentate). It is noteworthy that the monodentate and bidentate complexes are unfavorable for the mechanical performance compared to tridentate complexes.42 When immersed in water, the hydrogel shrank and the color became darker (Fig. S10, ESI†). Furthermore, the microstructure of the E-hydrogel became much denser and more uniformly distributed than that of the C-hydrogel, which is in agreement with the structure obtained by low-field NMR spectroscopy. A more tightly cross-linked microstructure could significantly enhance the stiffness of the gel, and the smaller pore structure could effectively disperse energy and toughen the hydrogel. Therefore, the step-by-step strategy can aid in successful construction of a soft and hard hierarchical microstructure, thus achieving the combination of strength and toughness of hydrogels.
For the systematic exploration of the energy dissipation mechanism of E-hydrogels, cyclic loading–unloading tests of E-hydrogel with different strains were conducted under the tensile rate of 50 mm min−1. The hysteresis loops increased with the increase in strain from 25% to 700% (Fig. S5a and b, ESI†), indicating that damage to the internal network structure preceded macroscopic fracture, and the degree of damage continued to increase with increasing elongation. Moreover, a clear overlap between the two adjacent hysteresis loops was observed, thus reflecting the self-recovery of E-hydrogels.35
The relationship between dissipated energy (U) and strain was assessed (Fig. S5c, ESI†). Dissipated energy gradually increased throughout the deformation process, indicating the absence of obvious defects in the internal structure, which is consistent with the results of low-field NMR spectroscopy and SEM. The relationship between U/W (fracture energy, W) and strain of E-hydrogel was also assessed (Fig. S5d, ESI†). The two phases in the curve indicated that the changes in the network structure might be caused by different physical interactions. The larger the value of U/W, the more serious the damage of the network structure. Under a small strain (0–150%), the entanglement and hydrogen bonding dissociation of polymer chains were responsible for the gradual increase of U. As the strain exceeded the yield point, the disentanglement of the polymer network began to increase, initiating the fracture of strong metal coordination interactions to dissipate energy.49 With the gradual increase in strain, unlike the immediate response of chemical cross-linking, there is a hysteresis in the re-coordination of Fe3+–COO– and the reconstruction of the chain entanglement structure, leading to structural failure by slow increase in the U/W stage. The yield points of P-hydrogels, C-hydrogels, and E-hydrogels were 175%, 125%, and 150%, respectively (Fig. S5d–f, ESI†). Compared with E-hydrogel, the yield point of P-hydrogel was 175%, and the value of U/W gradually decreases when the deformation is higher than the yield point. This is attributed to the fact that in the single network structure of P-hydrogel, the hydrogen bonding and hydrophobic conjoining interactions enable the material to effectively dissipate energy when subjected to gradually increasing deformation, and thus it exhibits better resilience. The presence of excess Fe3+ in the C-hydrogel resulted in the formation of a large number of single, double, or triple coordination complexes. Therefore, during the stretching process, there were unsaturated Fe3+–COO– coordination complexes dissipating energy at the same time, and thus the yield point was lower than that of E-hydrogel. Overall, multiple physical interactions endowed E-hydrogels with excellent fatigue resistance and self-healing ability.
The self-recovery ability of the E-hydrogel was systematically explored. Both E-hydrogel and C-hydrogel exhibited distinct hysteresis loops from those of P-hydrogel at the strain of 200% (Fig. S6, ESI†). The hysteresis energy values of the E-hydrogel (1.76 MJ m−3) and C-hydrogel (0.64 MJ m−3) were much higher than those of the P-hydrogel (0.03 MJ m−3), confirming that metal coordination interactions led to the more efficient dissipation of energy in the hydrogel. Owing to the formation of a more compact network and more efficient energy dissipation pathways during equilibrium, the E-hydrogel exhibited higher stress (1.77 MPa) and toughness (1.76 MJ m−3) compared with the C-hydrogel.50 The self-recoverability of the E-hydrogel without any external repair was observed (Fig. S6b, ESI†). After the first loading–unloading cycle, a clear hysteresis cycle was observed, with the second cycle loading curve showing a much smaller hysteresis compared to the first curve, indicating the time dependence of dissociation and recombination of dynamic bonds. After resting for 15 min, the gel quickly recovered its elastic modulus (69.6%) and dissipated energy (64.3%). Thus, the hydrogel showed rapid recovery, mainly attributed to the elastic contraction of the polymer network as well as the strong ionic coordination bonds acting as reversible sacrificial bonds.
Following 60 min of rest, the recovery rate of elastic modulus and dissipated energy further reached 88.2% and 85.4%, respectively, indicating the occurrence of network reconstruction at the molecular level (hydrophobic association, multiple hydrogen bonds, and metal coordination bonds) and recovery of mechanical performance (stress, strain, elastic modulus, and toughness) was not instantaneous and there was a certain hysteresis. Even when the deformation was 500%, recovery rates of 61.5% (elastic modulus) and 72.17% (dissipated energy) were achieved (Fig. S7, ESI†), further verifying that the hydrogel possessed excellent self-recovery ability. The E-hydrogel also exhibited good fatigue resistance (Fig. S6c, ESI†). The mechanical strength and hysteresis loop could be effectively restored to their original state after 24 h; the same result was observed at 500% cyclic loading–unloading test (Fig. S7c, ESI†), further illustrating that the physically cross-linked E-hydrogel showed remarkable resilience and fatigue resistance compared with chemically cross-linked hydrogels. The above-mentioned characteristics are of great significance for the preparation of hydrogel sensors with ultra-long service life.
Dynamic rheological measurements were performed to investigate the flow characteristics and structure behavior of prepared hydrogels (Fig. S9, ESI†). The shear strain dependence of moduli (G′ and G′′) for P.C.E-hydrogels was assessed (Fig. S9a, ESI†). The storage and loss moduli of C-hydrogel and E-hydrogel were both higher than those of P-hydrogel, manifesting that the coordination effect significantly enhanced the elastic behavior of the material. The frequency dependence of the hydrogel rheological behavior was assessed (Fig. S9b, ESI†). For all hydrogels, G′ values were higher than the G′′ values throughout the frequency test range, indicating a stable elastic behavior of the hydrogels. Furthermore, G′′ values of C-hydrogel and E-hydrogel were higher than G′ values of P-hydrogel, denoting that the coordination effect of Fe3+ significantly influenced the material properties. The mechanical response of the P.C.E-hydrogel could also be investigated by the frequency dependence of complex viscosity (η*, Pa s). For a fully covalent cross-linked network, a certain quantitative relationship was observed between complex viscosity and angular frequency, i.e., a linear relationship between log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) η* and logω, and the slope of the line was −1.48,49 In this system, with the incorporation of second strong metal coordination bond, the network structure of the hydrogel became almost similar to that of a covalently bonded cross-linked network. When immersed in water, excess Fe3+ diffused out of C-hydrogel and the ionic coordinates could reorganize to form more tridentates in E-hydrogel, which was necessary to obtain the near-perfect elastic network. As a result, the slope of the P.C.E-hydrogels gradually became closer to −1 over the probed frequency range (Fig. S9c, ESI†). The microscopic self-recovery behavior was also investigated (Fig. S9d, ESI†). The hydrogels were subjected to oscillatory forces with alternative amplitudes. When subjected to small amplitude oscillatory strains (0.5%), G′ was greater than G′′, indicating the formation of gel structures with complete networks under small oscillatory strains. At large amplitude (50%) oscillatory strains, G′ and G′′ dropped sharply and were accompanied by inversion, indicating a disruption of the gel network that led to the transformation of the gel into a sol–gel. The self-recovery mechanism was due to the reconstruction of reversible ionic coordination complexation in the gel network system, in agreement with the loading–unloading results.
η* and logω, and the slope of the line was −1.48,49 In this system, with the incorporation of second strong metal coordination bond, the network structure of the hydrogel became almost similar to that of a covalently bonded cross-linked network. When immersed in water, excess Fe3+ diffused out of C-hydrogel and the ionic coordinates could reorganize to form more tridentates in E-hydrogel, which was necessary to obtain the near-perfect elastic network. As a result, the slope of the P.C.E-hydrogels gradually became closer to −1 over the probed frequency range (Fig. S9c, ESI†). The microscopic self-recovery behavior was also investigated (Fig. S9d, ESI†). The hydrogels were subjected to oscillatory forces with alternative amplitudes. When subjected to small amplitude oscillatory strains (0.5%), G′ was greater than G′′, indicating the formation of gel structures with complete networks under small oscillatory strains. At large amplitude (50%) oscillatory strains, G′ and G′′ dropped sharply and were accompanied by inversion, indicating a disruption of the gel network that led to the transformation of the gel into a sol–gel. The self-recovery mechanism was due to the reconstruction of reversible ionic coordination complexation in the gel network system, in agreement with the loading–unloading results.
Anti-swelling properties
In high water content polymer materials, an increase in the swelling ratio leads to a significant decline in properties such as mechanical strength and self-healing behavior, severely limiting the application of hydrogels. Therefore, effective control of the swelling ratio not only ensures a stable performance, but also offers significant practical value (Fig. S8, ESI†). The weight swelling ratio of the P-hydrogel and the E-hydrogel in deionized water was assessed (Fig. 4a). The swelling rate of the P-hydrogel reached an equilibrium of ∼5500%, much higher than that of the E-hydrogel, and the latter displayed excellent swelling resistance performance. This result is attributed to the more tridentate structure with strong binding strength between carboxylic acid and Fe3+, leading to an increase in the cross-linking intensity and a more denser network structure. The swelling curves of E-hydrogel in different solvents were recorded (Fig. 4b). The swelling ratio of E-hydrogel in physiological saline environment decreased by ∼−9% after 10 days, which was mainly due to the high concentration in the physiological saline, causing the gel to appear in a slightly dehydrated state. The hydrogel underwent shrinking in ethanol (∼−32%) and acetone (∼−51%) because of solvent exchange. The weight swelling ratio of E-hydrogel in solutions with different pH was also observed (Fig. 4c). After soaking for 15 days, the hydrogel maintained good swelling resistance in a wide pH range (4–10). The equilibrium swelling ratio was less than 20% even in a solution with pH 3.0. When the pH was below 2.0 or above 10.0, the hydrogel exhibited a high swelling ratio due to the fact that, under strong acid–base conditions, the hydrophobic association in the network structure was disrupted and the metal–ligand bonding interaction was dissociated.30 The hydrogels were immersed in solutions with different pH for 24 h to investigate the effect of pH on the tensile properties. The hydrogels exhibited excellent mechanical properties over a wide pH range of 2–11, demonstrating the potential of application in more complex scenarios. Therefore, effective control of the swelling ratio not only ensures a stable performance, but also endows it with a great potential in motion detection in water, such as swimming or diving (Fig. 4d).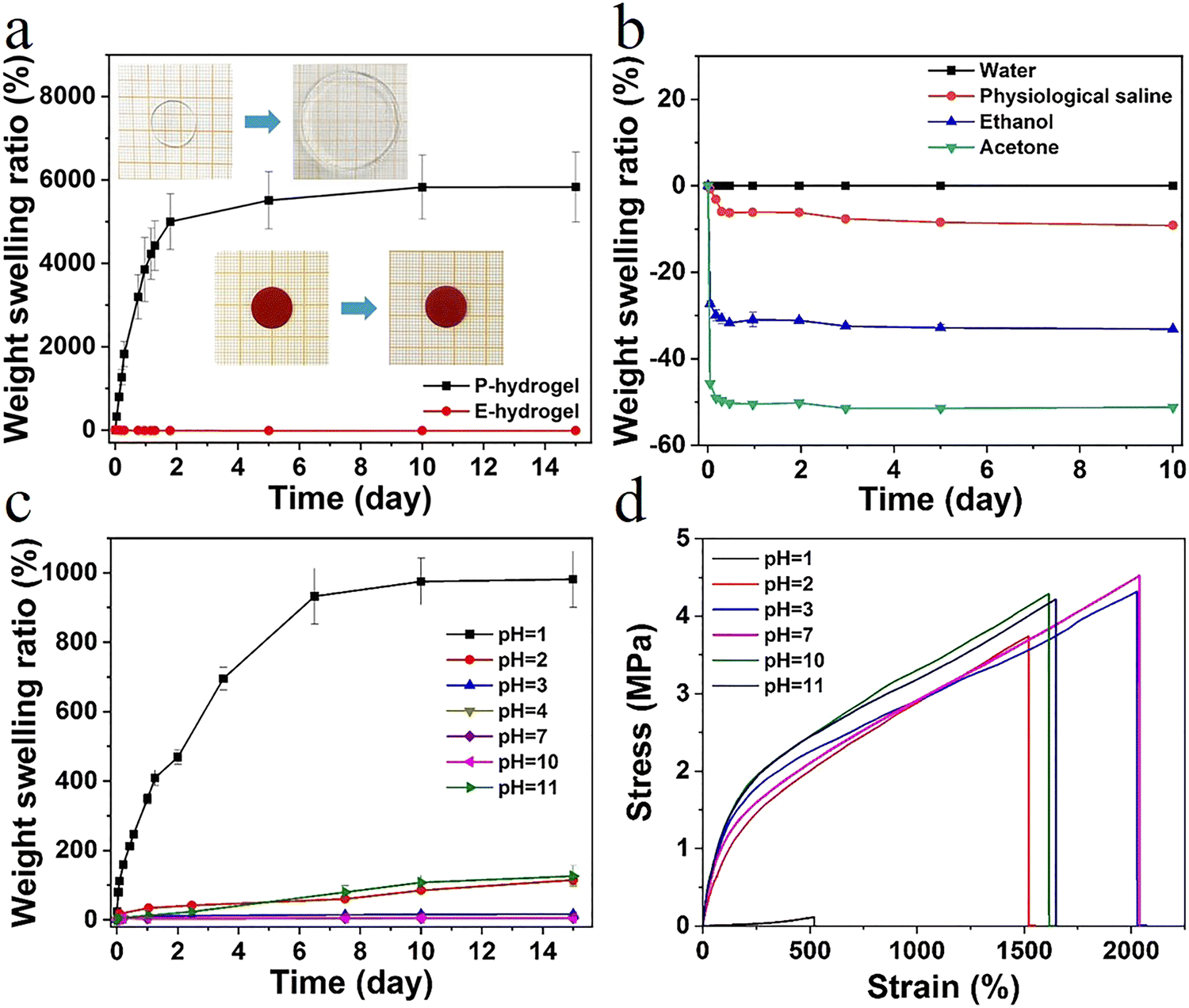 | ||
| Fig. 4 (a) Weight swelling ratio of P.E-hydrogel in deionized water; inset shows the extent of swelling after 15 days, (b) weight swelling ratio of E-hydrogel in different solutions (water, physiological saline, ethanol, and acetone), and (c) in different pH solutions, (d) mechanical properties of E-hydrogel at different pH after swelling for 24 h. | ||
Sensitive responsive properties of the hydrogel sensor
The hydrogel also exhibited excellent electrical conductivity due to the presence of Fe3+, Cl−, and Na+ in the network structure. When the hydrogel was connected to the channel of LED light, the brightness synchronously changed with the variation of deformation. After deformation, changes in channels and lengths of ion transport resulted in changes in conductivity (Fig. S12, ESI†). The ionic interaction in the gel system also conferred the hydrogel with outstanding antifreezing properties. The crystallization temperature of E-hydrogel was −14.3 °C, offering the possibility of operating well in low-temperature environments (Fig. S11, ESI†).In order to accurately explore the sensitive responsive properties of the sensor, the relative resistance changes of the hydrogel during repeated stretching under small strain (0.2–7%) and large strain (10–300%) were also measured, respectively. This is in line with the range of human body part movement monitoring deformation,51,52 providing a basis for motion monitoring and the application in the field of speech recognition (Fig. 5a–d). Furthermore, the relative resistance signal remained unchanged with the increase in the tensile speed from 25 to 200 mm min−1, further indicating that the sensor showed a fast response to the stretching speed (Fig. 5e). To assess the long-term stability and repeatability, the sensor was subjected to consecutive tensile cycles at 50% and 200% strain. No significant loss of electrical signal was observed with a levelled baseline throughout the stretching process, demonstrating excellent stability and cycling durability (Fig. 5f and Fig. S14, ESI†). In contrast, evident baseline drift was observed for many previously reported hydrogel strain sensors.53–55 The response and recovery times of the sensor were 48.8 ms and 47.7 ms, respectively, higher than those of previously reported hydrogels (Fig. 5g and i). Another important parameter, gauge factor (GF), was also calculated to characterize the sensitivity of the sensor. The GF values were 0.862 and 0.668 in the strain range of 0–5% and 5–50%, respectively, which are significantly improved compared to those for the other reported high-strength hydrogels (Fig. 5h). Furthermore, the sensor displayed excellent electric sensitivity, fast responsiveness, and good stretchability (Fig. 5i and Table S3, ESI†), making it an ideal candidate for the monitoring of human health conditions and different exercise states.
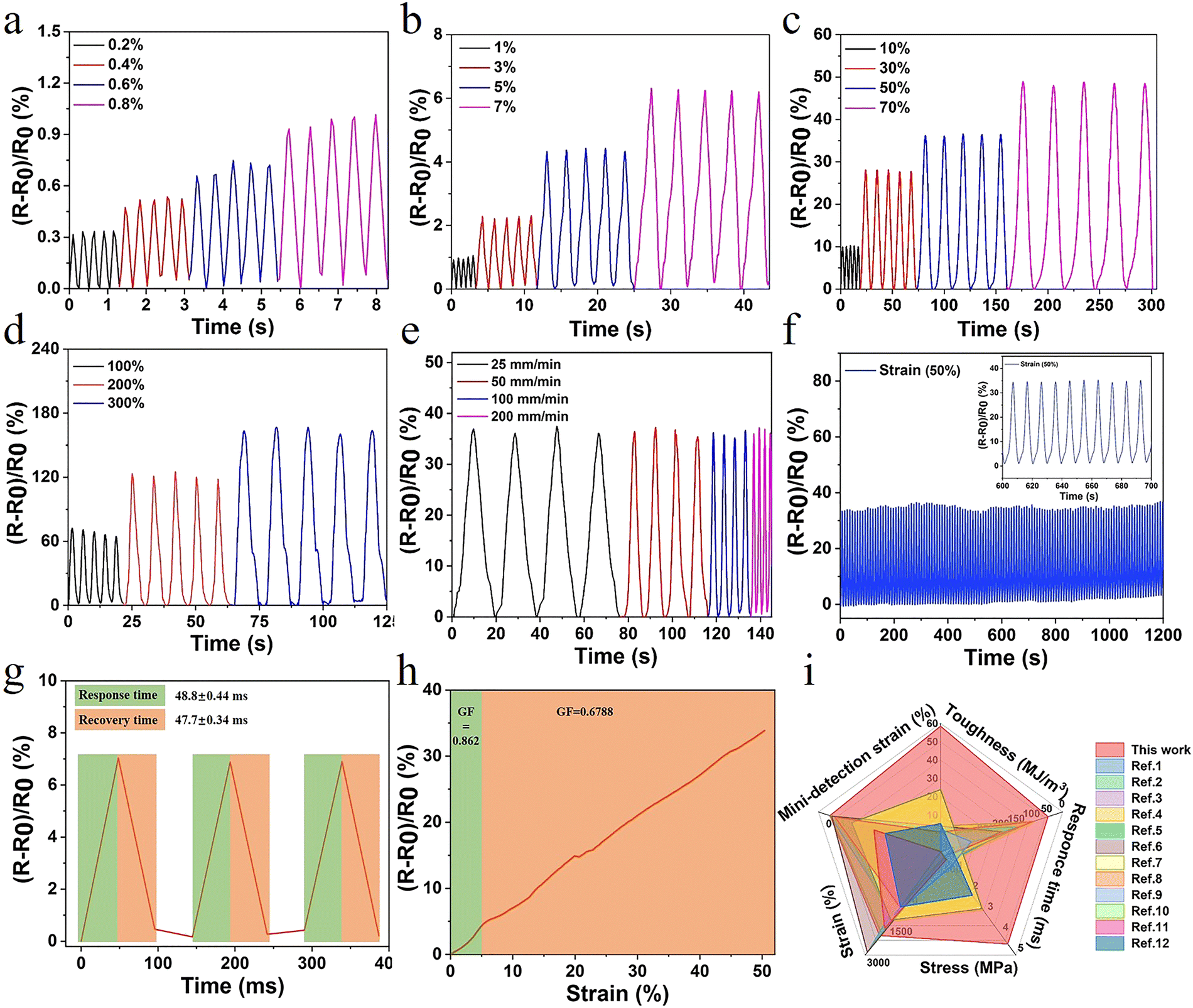 | ||
| Fig. 5 (a–d) Relative resistance when subjected to multiple scale strain; (e) relative resistance changes at varied tensile rates (strain at 50%); (f) stability of relative resistance signals of the sensor at the strain of 50% for 120 cycles; and (g) response and recovery times; (h) effects of applied strain on the gauge factor; and (i) comparison with currently reported study in terms of toughness, stress, strain, response time, and detection of strain (relevant data are summarized in Table S3, ESI†). | ||
Strength and toughness are essential for the long-term stable operation of sensors, while high sensitivity and fast response rates further ensure detection accuracy. Therefore, due to the high strength and sensitivity of the hydrogel prepared herein, it was assembled into a strain sensor to monitor different human movements. The hydrogel sensor was attached to different parts of the human body, including fingers, wrists, elbows, and knees (Fig. S13a–f, ESI†). The curve of relative resistance varied in real time with the state of motion, indicating good sensor stability. The sensor monitored the different respiratory states of the wearer, enabling a preliminary diagnosis of the health status of the human body (Fig. S13d, ESI†). Moreover, sensors fixed to the position of the human laryngeal node could be applied to monitor the vibration and slight deformation caused by the laryngeal node such as swallowing and light cough action (Fig. S13g and h, ESI†). These results reveal the excellent sensitivity and superior usability of sensor, with potential applications for human motion detection and personal medical diagnostics.
Hydrogel sensors with high sensitivity and long-lasting stability offer the possibility of achieving information encryption and decryption applications. International Morse code is an on–off signal code, which uses dots and dashes to express different letters, numbers, and punctuation marks in sequences (Fig. 6a). Therefore, in this system, the change in relative resistance caused by a small degree of finger bending and a large bending angle was set as a dot and a dash, respectively. The relative resistance of the hydrogel exhibited a real-time synchronous change with the deformation. When the finger was bent at different angles, the resistance changed accordingly (Fig. 6b). Combined with the international Morse code table, the encryption and decryption of information were achieved. Selected representative letters, such as A,B,C; R,S,T; and X,Y,Z, were represented by changes in the sensor resistance according to the Morse code table (Fig. 6c). The signal displayed good repeatability and stability. The message “I LOVE NKU” was output in this way (Fig. 6d). It was possible to record numbers and different symbols. Therefore, information could be recorded and transmitted using this hydrogel, opening a new area for the application of hydrogel sensors.
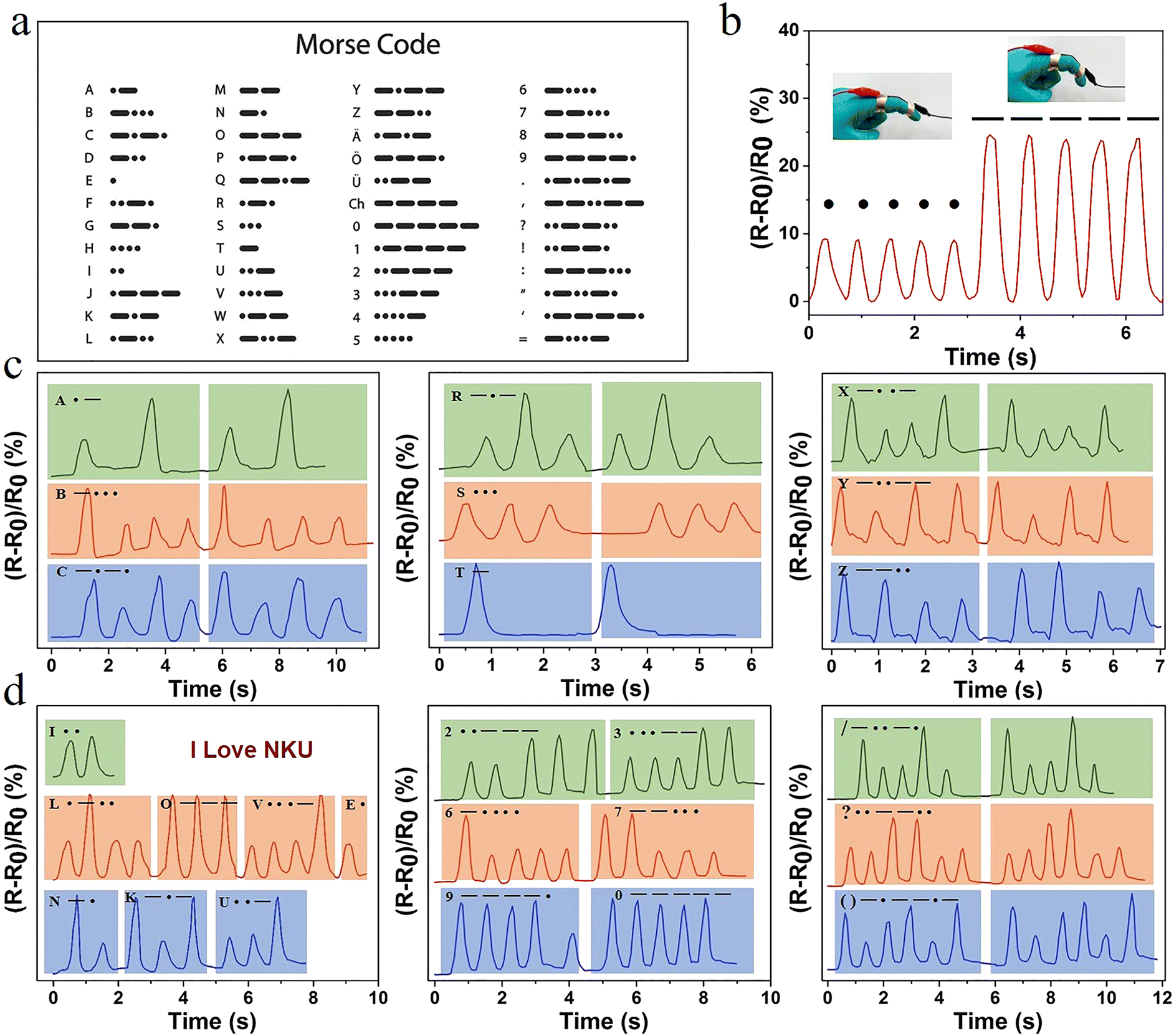 | ||
| Fig. 6 (a) The corresponding symbols of Morse code in the alphabet, numbers, and symbols table; (b) changes of relative resistance following the movement of fingers with different degrees of bending to record points and lines in Morse code; and (c) recording of different English letters, and (d) the words “I LOVE NKU”, different numbers, and symbols. | ||
Conclusions
Inspired by the structure of biological tissues, a high-performance ionic conductive hydrogel was successfully developed combining inter/intra-molecular hydrogen bonding and π–π stacking of hydrophobic-conjugated microregion structural domains as the ‘soft’ skeleton, and strong ionic coordination interaction as the ‘rigid’ reversible sacrificial bond through a step-by-step building. The excellent mechanical properties, including elongation at break (>2000%), toughness (∼60 MJ m−3), and recovery (>88%), of the hydrogel were attributed to the favorable synergistic interactions of soft and hard networks as well as to the efficient energy dissipation mechanism. Most importantly, the flexible adjustable structure and mechanics of hydrogels could be easily achieved by simply adjusting the content of both the rigid and flexible components. Notably, the prepared hydrogels exhibited good resistance to swelling in different solutions, maintaining mechanical strength and toughness even after 24 h of swelling in solutions with different pH (2–11). Dynamic response behaviors and heterogeneous structural evolution were systematically observed by rheology and MQ NMR spectroscopy. The presence of dynamic ions (Na+, Cl−, Fe3+) in the network not only rendered the hydrogel with anti-freezing performance, but also led to high sensitivity (GF, 0.862), fast response (47.4 ms), and cyclic stability. As a portable sensor, the hydrogel could monitor multiple ranges of human activity such as joint movements (finger, wrist, and elbow flexion) and small deformations (swallowing and light coughing). Owing to the stable and distinct signal display under different deformations, the application of this sensor in the field of information encryption and decryption was achieved through Morse code. The nature-inspired design was able to balance excellent strength, high detection accuracy, and anti-swelling properties. Therefore, this study provides insight into the fabrication of flexible sensors and expansion applications in health monitoring, motion recognition, and information encryption transmission.Author contributions
Conceptualization: X. D. and G. W. Methodology: X. D., J. H., M. Y. Investigation: X. D., J. H., M. Y. Writing: X. D., G. W. and P. S. Review and editing: G. W. and P. S. Supervision and funding acquisition: P. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21534005 and 22073051).References
- C. Qian, T. Higashigaki, T. A. Asoh and H. Uyama, ACS Appl. Mater. Interfaces, 2020, 12, 27518–27525 CrossRef CAS PubMed.
- N. Sun, F. Lu, Y. Yu, L. Su, X. Gao and L. Zheng, ACS Appl. Mater. Interfaces, 2020, 12, 11778–11788 CrossRef CAS PubMed.
- S. Wang, G. Guo, X. Lu, S. Ji, G. Tan and L. Gao, ACS Appl. Mater. Interfaces, 2018, 10, 19133–19142 CrossRef CAS PubMed.
- T. Yang, W. Wang, H. Zhang, X. Li, J. Shi, Y. He, Q. S. Zheng, Z. Li and H. Zhu, ACS Nano, 2015, 9, 10867–10875 CrossRef CAS PubMed.
- H. Huang, L. Han, X. Fu, Y. Wang, Z. Yang, L. Pan and M. Xu, Adv. Electron. Mater., 2020, 6, 2000239 CrossRef CAS.
- Y. Niu, H. Liu, R. He, M. Luo, M. Shu and F. Xu, Small, 2021, 17, e2101151 CrossRef PubMed.
- H. Liu, M. Li, C. Ouyang, T. J. Lu, F. Li and F. Xu, Small, 2018, 14, e1801711 CrossRef PubMed.
- L. Han, L. Yan, M. Wang, K. Wang, L. Fang, J. Zhou, J. Fang, F. Ren and X. Lu, Chem. Mater., 2018, 30, 5561–5572 CrossRef CAS.
- D. Zhang, Y. Tang, Y. Zhang, F. Yang, Y. Liu, X. Wang, J. Yang, X. Gong and J. Zheng, J. Mater. Chem. A, 2020, 8, 20474–20485 RSC.
- W. Zhang, P. Feng, J. Chen, Z. M. Sun and B. X. Zhao, Prog. Polym. Sci., 2019, 88, 220–240 CrossRef CAS.
- H. Liu, M. Li, S. Liu, P. Jia, X. Guo, S. Feng, T. J. Lu, H. Yang, F. Li and F. Xu, Mater. Horiz., 2020, 7, 203–213 RSC.
- Y. Zhou, C. Wan, Y. Yang, H. Yang, S. Wang, Z. Dai, K. Ji, H. Jiang, X. Chen and Y. Long, Adv. Funct. Mater., 2019, 29, 1806220 CrossRef.
- J. Liu, Z. Chen, Y. Chen, H. U. Rehman, Y. Guo, H. Li and H. Liu, Adv. Funct. Mater., 2021, 31, 2101464 CrossRef CAS.
- S. X. Pan, M. Xia, H. H. Li, X. L. Jiang, P. X. He, Z. G. Sun and Y. H. Zhang, J. Mater. Chem. C, 2020, 8, 2827–2837 RSC.
- B. Yang and W. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 16765–16775 CrossRef CAS PubMed.
- H. L. Huang, L. Han, J. F. Li, X. B. Fu, Y. L. Wang, Z. L. Yang, X. T. Xu, L. K. Pan and M. Xu, J. Mater. Chem. A, 2020, 8, 10291–10300 RSC.
- X. Liu, Q. Zhang and G. Gao, ACS Nano, 2020, 14, 13709–13717 CrossRef CAS PubMed.
- J. Hao and R. A. Weiss, Macromolecules, 2011, 44, 9390–9398 CrossRef CAS.
- T. Nonoyama, S. Wada, R. Kiyama, N. Kitamura, M. T. Mredha, X. Zhang, T. Kurokawa, T. Nakajima, Y. Takagi, K. Yasuda and J. P. Gong, Adv. Mater., 2016, 28, 6740–6745 CrossRef CAS PubMed.
- X. Di, Y. Kang, F. Li, R. Yao, Q. Chen, C. Hang, Y. Xu, Y. Wang, P. Sun and G. Wu, Colloids Surf., B, 2019, 177, 149–159 CrossRef CAS PubMed.
- C. K. Roy, H. L. Guo, T. L. Sun, A. B. Ihsan, T. Kurokawa, M. Takahata, T. Nonoyama, T. Nakajima and J. P. Gong, Adv. Mater., 2015, 27, 7344–7348 CrossRef CAS PubMed.
- T. Liu, M. Liu, S. Dou, J. Sun, Z. Cong, C. Jiang, C. Du, X. Pu, W. Hu and Z. L. Wang, ACS Nano, 2018, 12, 2818–2826 CrossRef CAS PubMed.
- H. Guo, T. Nakajima, D. Hourdet, A. Marcellan, C. Creton, W. Hong, T. Kurokawa and J. P. Gong, Adv. Mater., 2019, 31, e1900702 CrossRef PubMed.
- T. Liu, F. Wang, Q. Wu, T. Chen and P. Sun, Mater. Horiz., 2021, 8, 3096–3104 RSC.
- J. Xu, X. Ren and G. Gao, Polymer, 2018, 150, 194–203 CrossRef CAS.
- J. Cao, J. Li, Y. Chen, L. Zhang and J. Zhou, Adv. Funct. Mater., 2018, 28, 1800739 CrossRef.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- N. Yuan, L. Xu, H. Wang, Y. Fu, Z. Zhang, L. Liu, C. Wang, J. Zhao and J. Rong, ACS Appl. Mater. Interfaces, 2016, 8, 34034–34044 CrossRef CAS PubMed.
- Z. Qin, R. Niu, C. Tang, J. Xia, F. Ji, D. Dong, H. Zhang, S. Zhang, J. Li and F. Yao, Macromol. Mater. Eng., 2018, 303, 1700396 CrossRef.
- J. Xu, R. Jin, X. Ren and G. Gao, J. Mater. Chem. A, 2019, 7, 25411–25448 Search PubMed.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- M. T. I. Mredha, Y. Z. Guo, T. Nonoyama, T. Nakajima, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1704937 CrossRef PubMed.
- X. Di, J. Li, M. Yang, Q. Zhao, G. Wu and P. Sun, J. Mater. Chem. A, 2021, 9, 20703–20713 RSC.
- X. Li, H. Wang, D. Li, S. Long, G. Zhang and Z. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 31198–31207 CrossRef CAS PubMed.
- D. G. Barrett, G. G. Bushnell and P. B. Messersmith, Adv. Healthcare Mater., 2013, 2, 745–755 CrossRef CAS PubMed.
- H. Fan, J. Wang and Z. Jin, Macromolecules, 2018, 51, 1696–1705 CrossRef CAS.
- H. J. Zhang, Y. R. Cheng, X. J. Hou, B. Yang and F. Guo, New J. Chem., 2018, 42, 9151–9158 RSC.
- Y. J. Liu, W. T. Cao, M. G. Ma and P. Wan, ACS Appl. Mater. Interfaces, 2017, 9, 25559–25570 CrossRef CAS PubMed.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef CAS.
- M. A. Meyers, P.-Y. Chen, A. Y.-M. Lin and Y. Seki, Prog. Mater. Sci., 2008, 53, 1–206 CrossRef CAS.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- F. Wang, S. Chen, Q. Wu, R. Zhang and P. Sun, Polymer, 2019, 163, 154–161 CrossRef CAS.
- R. Zhang, S. Yu, S. Chen, Q. Wu, T. Chen, P. Sun, B. Li and D. Ding, J. Phys. Chem. B, 2014, 118, 1126–1137 CrossRef CAS PubMed.
- K. Saalwächter, P. Ziegler, O. Spyckerelle, B. Haidar, A. Vidal and J.-U. Sommer, J. Chem. Phys., 2003, 119, 3468–3482 CrossRef.
- W. Chasse, J. Lopez Valentin, G. D. Genesky, C. Cohen and K. Saalwachter, J. Chem. Phys., 2011, 134, 044907 CrossRef PubMed.
- K. Saalwächter and J.-U. Sommer, Macromol. Rapid Commun., 2007, 28, 1455–1465 CrossRef.
- X. Zou, X. Kui, R. Zhang, Y. Zhang, X. Wang, Q. Wu, T. Chen and P. Sun, Macromolecules, 2017, 50, 9340–9352 CrossRef CAS.
- T. Liu, S. Zou, C. Hang, J. Li, X. Di, X. Li, Q. Wu, F. Wang and P. Sun, Polym. Chem., 2020, 11, 1906–1918 RSC.
- Y. Hu, Z. Du, X. Deng, T. Wang, Z. Yang, W. Zhou and C. Wang, Macromolecules, 2016, 49, 5660–5668 CrossRef CAS.
- C. Hu, Y. Zhang, X. Wang, L. Xing, L. Shi and R. Ran, ACS Appl. Mater. Interfaces, 2018, 10, 44000–44010 CrossRef CAS PubMed.
- S. Xia, S. Song, Y. Li and G. Gao, J. Mater. Chem. C, 2019, 7, 11303–11314 RSC.
- Y. Yang, Y. Yang, Y. Cao, X. Wang, Y. Chen, H. Liu, Y. Gao, J. Wang, C. Liu, W. Wang, J.-K. Yu and D. Wu, Chem. Eng. J., 2021, 403, 126431 CrossRef CAS.
- S. Xia, Q. Zhang, S. Song, L. Duan and G. Gao, Chem. Mater., 2019, 31, 9522–9531 CrossRef CAS.
- Q. Zhang, X. Liu, L. Duan and G. Gao, J. Mater. Chem. A, 2020, 8, 4515–4523 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh00456a |
| This journal is © The Royal Society of Chemistry 2022 |