
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the CO2 capture and reduction on K-promoted Cu/Al2O3 by spatiotemporal operando methodologies†
Donato
Pinto
 ,
Victor
van der Bom Estadella
and
Atsushi
Urakawa
*
,
Victor
van der Bom Estadella
and
Atsushi
Urakawa
*
Catalysis Engineering, Department of Chemical Engineering, Van der Maasweg 9, 2629 HZ, Delft, The Netherlands. E-mail: A.Urakawa@tudelft.nl
First published on 18th July 2022
Abstract
Integrated CO2 capture and conversion processes bring the promise of drastic abatement of CO2 emission together with its valorisation to chemical building blocks such as CH4 and CO. Isothermal CO2 capture and reduction (CCR) on a K-promoted Cu/Al2O3 was recognised as an effective catalytic strategy for removing CO2 from diluted stream and converting it to syngas (H2 + CO) employing green H2 as reducing agent. The dual functionality of the catalyst is the key of this dynamic process, in which the alkaline metal introduces the capture functionality and copper ensures the selective conversion of the captured CO2 to CO. However, the highly dynamic state of the catalyst at reaction conditions represents a barrier for the identification of the catalytic mechanism of CCR, which is vital for rational process improvement and design. In this work, we conducted a mechanistic investigation of CCR by means of spatiotemporal operando methodologies, gaining insights into dynamic variation of temperature, gas concentration and reactive surface species in the CCR reactor. The results show the unique potassium state exothermically captures CO2 as surface carbonates which can be reduced to CO rapidly under H2 atmosphere. When the surface carbonates are transformed to formates the reaction path is altered and the reduction to CO becomes slower. By designing controlled catalytic experiments, we further demonstrate the active involvement of CO in the capture mechanism and the effectiveness of CO2 capture in presence of an oxidised surface, extending the perspectives and suitability of CCR to treat actual complex effluent streams.
Introduction
The urgency of reducing anthropogenic emissions of CO2 has given impulse to the elaboration of carbon capture and utilisation (CCU) strategies. Such strategies are designed to substantially reduce the CO2 emissions of industrial processes and power plants by separating CO2 from exhaust gases and, after purification and compression, making it available as feedstock for chemicals and fuels production. Compared to the alternative carbon capture and storage (CCS) that treat CO2 as a waste, CCU technologies extract value from the captured CO2 by employing it as a carbon source to produce carbon-neutral commodities.1–4Several technological options have been investigated and developed for CO2 capture, including amine-based liquid solutions, solid sorbents, separation membranes, ionic liquids, cryogenic separation and biological systems among others.5–8 Technologies employing amine-based solutions, in particular monoethanolamine, are available at commercial stage, although their employment put some constraints on the capture process.9 The flue gases containing CO2 need to be cooled down to an optimal temperature of operation, which is around 40–60 °C for amines. Moreover, O2, dust, NOx and SOx compounds often have to be separated from the flue gas, since they act as poisoning agent provoking the decomposition of the solvent. After saturation of the solution with CO2, an energy-intensive CO2 desorption and solvent regeneration has to be performed.
Solid sorbents, including metal oxides, zeolites, carbon, polymers and metal organic frameworks, partially overcome the constraints introduced by an absorbent in the liquid phase, with great promises to reduce capital costs and energy requirements for regeneration.10–12 Alkaline metal oxides, especially CaO, present high affinity towards CO2 capture and are tested in carbonation cycles.13 However, their thermal regeneration requires high temperatures, leading to particle sintering and capture efficiency losses.
Despite the promising readiness of the available technologies, the operational costs related to CO2 capture still represent the main limitation. Such costs become prohibitive to capture diluted CO2, as in the case of flue gases emitted by power plants, cement and steel industry, in which the capture cost can exceed 100 USD per ton of CO2.14 Furthermore, high purity CO2 needs to be separated and compressed to be sourced to the chemical industry. The current CO2 utilisation involves mainly urea synthesis and other small-scale applications (food industry, methanol, carboxylic acids synthesis, etc.), that accounts for transforming an amount of CO2 equivalent to 0.5% of its annual emissions.15,16 Consequently, it is more desirable to design processes which can directly and successfully handle diluted sources of CO2, as flue gases from power plants, or even make use of the hundreds of ppm concentration in air, in order to reduce the costs related to purification and compression steps.
Anticipating wider availability of economic green H2 in the near future, the combination of CO2 capture with its direct conversion to more appealing carbon-containing molecules, can bring additional value to the product.
In the last decade, several research groups have proposed and demonstrated integrated CO2 capture and conversion processes.17,18 By using a properly functionalized solid catalyst, CO2 from diluted streams is captured and stored in the material. Successively, employing H2 as a reducing agent, the captured CO2 can be selectively converted to more appealing carbon-containing molecules like CO and CH4 with a wider range of applications in chemical industry than CO2. With economic green H2 availability, such processes will become competitive options to close the carbon-cycle.
Farrauto and coworkers19 proved the feasibility of the concept by means of a bifunctional catalyst, in which a combination of alkaline metal oxide (CaO) and transition metal (Ru) supported on γ-Al2O3 provided efficient capture of CO2 from diluted streams and its subsequent conversion to CH4 when the atmosphere was switched to H2. A key characteristic of the process is that both capture and conversion steps can take place at the same pressure and in isothermal conditions. Thanks to the nanodispersion of the alkali component on the support, the regeneration of the sorbent in H2 was achieved at temperatures as low as ∼300 °C, permitting to reduce the energy requirements compared to the case of bulk CaO regeneration. A similar integrated process, referred to as CO2 capture and reduction (CCR), was demonstrated by our research group employing catalysts containing abundant chemical elements.20 Potassium was introduced as efficient promoter for CO2 sorption, while the combination of Fe, Cu and Cr exhibited high performance targeting the selective formation of CO, in view of obtaining a valuable syngas mixture (H2 + CO) in the product stream.
The interest in the catalytic process has grown recently with the exploration of alternative catalytic formulations. Several alkali (Li, Na, K, Cs) and alkali earth promoters (Ca, Ba) can be employed to introduce the CO2 capture functionality.21–24 The conversion of the captured CO2 is selectively driven towards the desired product by proper selection of the active metal phase. In particular, Cu-based catalysts exhibited selective reduction to CO,20,25 while Ru and Ni were employed for methanation.26–29
Recently, Kosaka et al.30 demonstrated the beneficial effect of increasing reaction pressures (up to 9 bar) to enhance the performances of direct CO2 capture at the level of the atmosphere and CH4 formation on a Na-promoted Ni/Al2O3 catalyst.
Despite a trending research directed towards optimisation of CCR processes, knowledge regarding the fundamental catalytic mechanism is still limited. Ex situ investigation of the catalytic materials inherently lacks information about the active catalytic state. Hyakutake et al.25 employed a K-promoted Cu/Al2O3 catalyst as model systems to investigate the peculiar characteristics of the active phase for CO2 capture and reduction. Operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) suggested the formation of surface intermediates assigned to formates on potassium, while operando XRD analysis revealed a highly dynamic system in reaction conditions, with a complex state composed of an highly amorphous potassium state and nanodispersed copper. At the operation temperatures, the dynamic nature of the catalytic system varies in time and space, requiring spatiotemporal analytical techniques to elucidate the CCR chemistry and to identify the catalytic roles played by the K and Cu phases.
In this contribution, we aimed to deepen the comprehension on the catalytic mechanism responsible for CO2 capture and reduction on a K-promoted Cu/Al2O3 system, by identifying active reaction paths of the CCR catalysis. Spatiotemporal sampling of temperature and gas composition along the catalytic bed were used, while operando DRIFTS was used to identify spatiotemporal variations of reactive surface species. Unprecedented mechanistic insights were gained by the substitution of CO2 with CO at the inlet stream and from the evaluation of how CCR operates when Cu is oxidised.
Results and discussion
A Cu–K/γ-Al2O3 catalyst (11 wt% Cu, 10 wt% K) was synthesised by a two-step incipient wetness impregnation method, following the procedure already reported for similar materials.25 The successful outcome of the synthesis was verified by X-ray diffraction (Fig. S1†).Fig. 1 shows a representative concentration profile during the CCR catalytic test performed at 350 °C on the Cu–K/γ-Al2O3 catalyst.
In a typical CCR catalytic test, 0.25 g of catalyst are first activated by exposure to a reducing stream at 450 °C (100% H2, 50 mL min−1, 1 h). During this pretreatment, CuO is fully reduced to metallic Cu, while the highly dispersed K2CO3 phase is partially decomposed. The presence of copper enhances the decomposition of K2CO3 at lower temperature compared to bulk K2CO3 or K/Al2O3 system, as confirmed by TGA analysis (Fig. S3†). As a result, active sites for the capture of CO2 are generated. Then, the catalyst is cyclically exposed to the alternating reactive gases. First, a CO2 feed (10% in He, 15 mL min−1) is passed to the catalyst bed, from which CO2 is captured until saturation of the active sites. For this catalytic test (Fig. 1), an excess of CO2, compared to the CO2 capture capacity of the catalyst, is passed to the reactor in order to study the reaction paths before and after catalyst saturation. It is possible to maximize the full CO2 capture period by adjusting gas flow rate, catalyst amount and CO2 concentration. An inert flush phase (He, 30 mL min−1) is then introduced to the reactor to remove weakly-adsorbed species and avoid mixing of reactants and related gas-phase reactions for more precise mechanistic studies. After that, H2 is introduced to the reactor (100%, 15 mL min−1) to remove the adsorbed CO2 in the form of CO and regenerate the catalytically active phase, followed by an additional inert flush phase (He, 30 mL min−1). Fig. 1 reports the average of the cycles with stable catalytic activity achieved after the first non-reproducible cycle. Compared to the CO2 profile obtained for a blank experiment performed on the inactive catalyst bed at room temperature, the delay in the appearance of CO2 at the outlet (20 s) suggests that CO2 is aggressively captured by the catalyst. Quantitative evaluation of the outlet stream composition clarifies that ca. 100% of CO2 was captured during this initial period. No CO signal is detected in this time interval within the detection limit (ca. 100 ppm), resulting in virtually a COx-free reactor effluent.
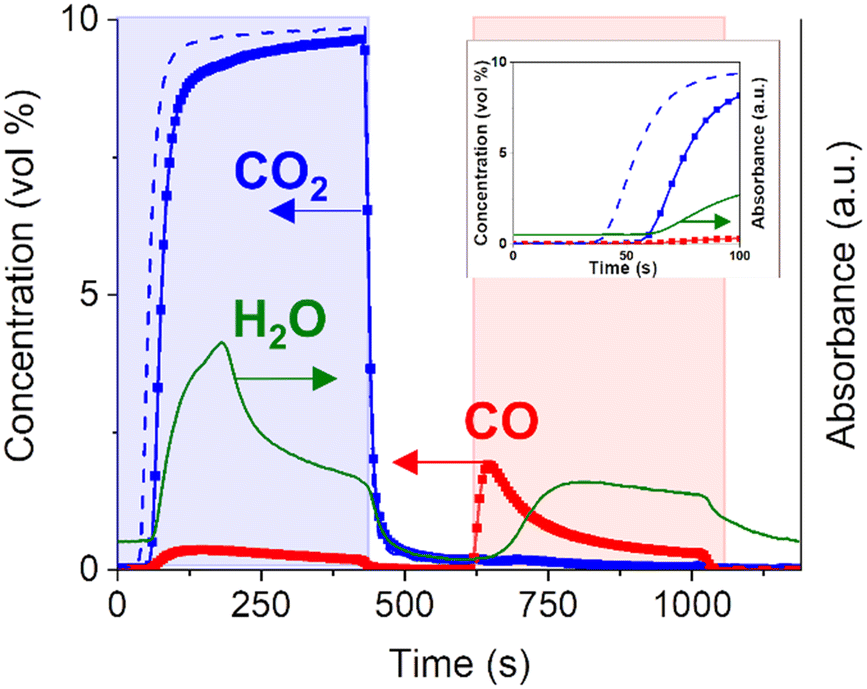 | ||
| Fig. 1 Outlet gas concentrations during CCR performed at 350 °C. 1 CCR cycle consists of CO2 feed (0–420 s, 15 mL min−1, 10% in He), inert flush (420–595 s, 30 mL min−1, He), H2 (595–1015 s, 15 mL min−1, 100%), inert flush (1015–1190 s, 30 mL min−1, He) and the cycle is repeated. Results are averaged over 18 CCR cycles. CO2 blank signal (dotted line) obtained as average of 2 CCR cycles during blank experiment at room temperature. The inset shows the concentration profiles during the initial phase of the CO2 capture phase (0–100 s). | ||
Once saturation of the capture sites is achieved, most of the CO2 entering the reactor is directly released to the reactor outlet apart from a small fraction reacting to produce CO. The detection of CO only after the end of the initial active capture period suggests that the active sites generated on this catalyst may be active for both CO2 and CO capture. Interestingly, the outlet gas analysis (Fig. 1) highlighted the formation of water during the capture phase. The formation of water in the CO2 atmosphere indicates the presence of active H or OH species, generated during catalyst exposure to gaseous H2 and stored on the catalyst surface. The identification of such species seems directly related to the catalytically active phase and will be targeted in the following sections.
Upon switching to the reduction (H2) feed, the captured CO2 is instantaneously and selectively reduced to CO and no other carbon-containing products such as CH4 are observed within the detection limit (Fig. 1). The sharp peak of CO detected at the outlet is not accompanied by water formation. The water signal starts to rise with a significant delay (ca. 60 s) compared to the start of the H2 phase (595 s). This excludes a direct correlation of water formation to the main reduction mechanism of captured CO2 towards CO formation.
The presence of unconverted H2 and CO in the effluent stream generates a syngas, whose quality, after separation of water in the product stream, is generally defined by the H2/CO ratio. The beauty of the CCR is the flexibility of the process, since this ratio can be tuned by the H2 flow rate, the reduction period, the reduction rate defined by the catalyst and also the reaction temperature.
Effect of reaction temperature
In general, reaction temperature is one of the process parameters greatly affecting CCR performance. Fig. 2 compares the CCR activity at three different temperatures, namely 350, 400 and 450 °C. At higher temperatures, the CO2 is detected in the reactor effluent during the capture phase increasingly delayed, revealing an higher capture capacity of the material. Increasing the temperature is beneficial for the CCR reaction. At higher temperature, a larger portion of potassium active sites for capture is likely regenerated in H2 atmosphere at each cycle. This is in line with the increased weight loss detected at 450 °C during H2-TGA, which is related to decomposition of the potassium phase (Fig. S2†). As a consequence, the amount of CO2 removed from the feed gas increases from 54 μmol gcat−1 (350 °C), 95 μmol gcat−1 (400 °C) to 130 μmol gcat−1 (450 °C).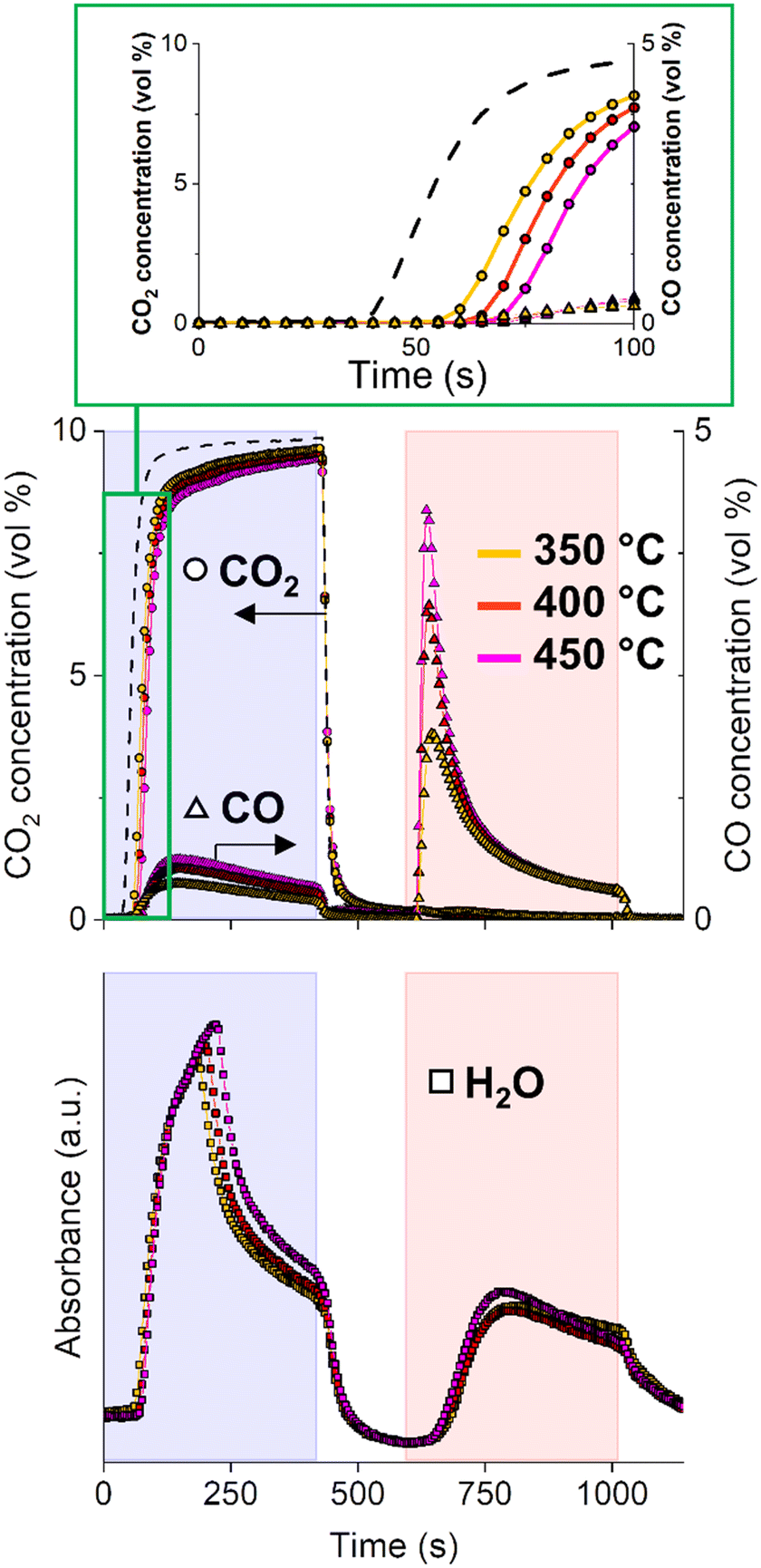 | ||
| Fig. 2 Outlet gas concentrations of CO2 (○) and CO (△) and signal of H2O (□) during CCR performed at 350 °C (yellow profiles), 400 °C (red profiles), 450 °C (magenta profiles). Results are averaged of 18 CCR cycles. CO2 blank signal (dotted line) obtained as average of 2 CCR cycles during blank experiment at room temperature. | ||
After the initial active capture, the amount of CO produced by direct interaction of CO2 with the catalyst slightly increases with temperature. Some considerations can be made from the qualitative analysis of the water signals evolved during the CO2 stream at different temperatures. On the one hand, the slight increase in water signal with temperature, associated to the increased amount of CO formation, indicates that a CO2 hydrogenation path to CO is active, which is enhanced at higher temperatures in accordance with the endothermic RWGS reaction.31 As mentioned earlier, CO is only detected after saturation of the capture sites, when the CO2 signal starts to rise. The initial absence of CO detection suggests that CO has affinity towards the capture sites and may directly participate in the capture mechanism.
The analysis of the gaseous species at the reactor outlet permits to identify three main reactions taking place during the CO2 capture phase:
| CO2 + □ = CO2(a) + H2O | (1) |
| CO2 + 2H(a) = CO + H2O | (2) |
| CO + □ = CO(a) | (3) |
When the feed gas is switched to H2, the comparison of the catalytic activity profiles at different temperatures clarifies the presence of two distinct mechanisms involved in the formation of CO.
The first mechanism is associated with the initial fast release of CO. At higher temperatures, the peak of CO reaches higher values, consistent with the higher amount of CO2 captured. Interestingly, this initial CO release is not accompanied by any water formation (vide supra). The absence of a marked release of water at this stage suggests the absence of CuO formation during the CO2 capture stage, as evidenced in a previous work.25 This also excludes the relevance of a redox reaction involving a Cu/CuxO cycle as mechanism for CO2 reduction for this catalytic system. Rather, CO2 is reduced by active H species formed by H2 dissociation on Cu (reaction (4)). However, this reaction does not follow a standard hydrogenation path (RWGS, reaction (5)) leading to CO and water release as a consequence of CO2 reduction. Rather, during the initial reduction phase of CCR, the excess of active H species generated in the reduction phase may induce the destabilisation of the surface intermediates formed during CO2 capture, provoking the fast selective release of CO (reactions (6) and (7)). Results from the investigation of surface species and their dynamics in capture and reduction phases are fundamental to elucidate the type of mechanism involved.
| H2 = 2H(a) | (4) |
| CO2(a) + H2 = CO + H2O | (5) |
| CO2(a) + 1/2H2 = OH(a) + CO | (6) |
| CO(a) + 1/2H2 = H(a) + CO | (7) |
Spatiotemporal operando studies of temperature, concentration and surface species in the reactor
The analysis of the catalytic activity profiles characterised by the outlet stream concentrations offers relevant indications on the type of reactions involved in CCR. However, the information retrieved at the reactor outlet lacks spatial resolution, potentially missing any temperature or concentration gradients developed in the reactor bed. Moreover, such results are detached from any consideration on the catalyst state and dynamic of surface species involved in the capture of CO2 and selective conversion to CO. In particular for the Cu–K/Al2O3 system, the unique catalytic state reached in operation conditions does not permit to make conclusive statements based on ex situ data.25 To obtain deeper understanding of the reaction mechanisms involved in CCR, we performed operando spatiotemporal sampling of temperature and gas concentration along the reactor bed. In addition, with the aim of identifying the surface reaction mechanisms involved in the CCR reaction, we performed spatiotemporal operando DRIFTS and compared the dynamics of surface species with the evolution of the catalytic activity.Fig. 3 contains the results obtained from spatiotemporal sampling of concentration and temperature by means of a moving capillary system under CCR with Cu–K/γ-Al2O3 at 350 °C. The reaction condition employed was identical to that of the experiment shown in Fig. 1. A schematic of the sampling positions is represented in Fig. 3A. The sampling positions inside the bed are indicated with the relative position, where positions 0 and 1 correspond to quartz wool at the front and after the catalyst bed, respectively. Fig. 3B highlights the evolution of CO2 sampled at different positions along the catalyst bed during the initial stages of the capture phase and analysed by mass spectrometry. Moving along the bed (relative position from 0 to 1), the CO2 is detected at progressively increasing times, confirming the existence of an active adsorption front proceeding until saturation of the active sites at each position.
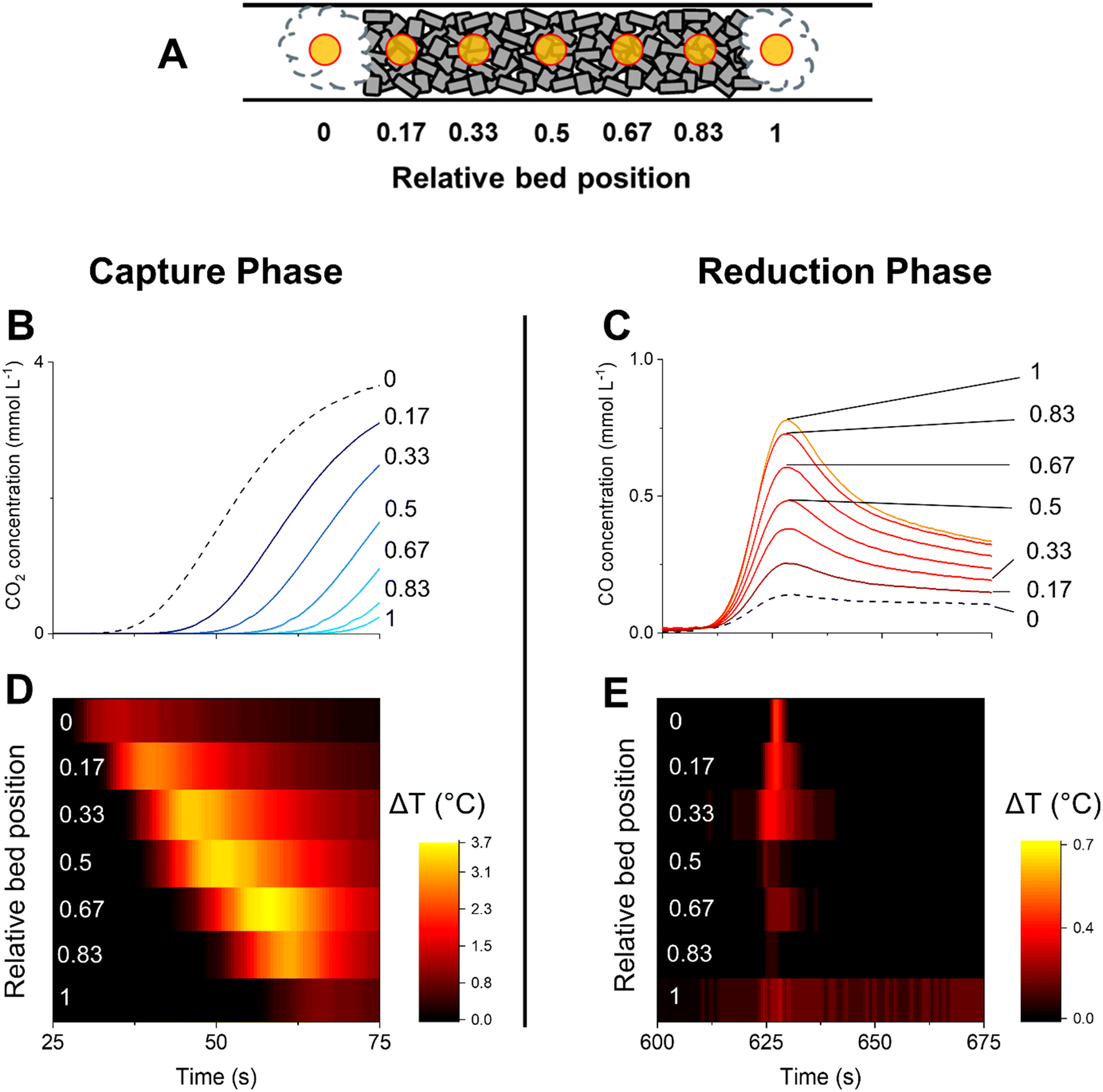 | ||
| Fig. 3 Spatiotemporal profiling of temperature and gas phase composition (MS) along the catalyst bed during CCR at 350 °C. Schematic of different sampling positions (A) shown in the relative position (0: beginning of the catalyst bed, 1: end of the catalyst bed). Position 0 and 1 correspond to quartz wool at the front and after the catalyst bed, respectively. CO2 (B) and CO (C) concentration profiles obtained for the initial stage of the capture phase and reduction phase, respectively. Dashed black line correspond to the signal collected at position 0 in the quartz wool in front of the bed. Temperature of the catalyst bed sampled at different positions from the front to the end of the catalyst bed during the initial stage of the capture phase (D) and reduction phase (E). | ||
At the same time, a temperature increase is detected before the detection of gaseous CO2 at each position (Fig. 3D vs. B). The capture is then associated with an exothermic process which can result from the adsorption of CO2 from the gas phase. This is consistent with the exothermic reaction of CO2 with K2O or KOH to form carbonate or bicarbonate species (reactions (8)–(11)).32 However, the generation of water during the capture reaction suggests that the mechanism of CO2 capture may involve a potassium hydroxide phase with formation of carbonates species (reaction (10)).
| K2O + CO2 = K2CO3 ΔH300K = −386 kJ mol−1 | (8) |
| K2O + 2CO2 + H2O = 2KHCO3 ΔH300K = −176 kJ mol−1 | (9) |
| 2KOH + CO2 = K2CO3 + H2O ΔH300K = −154 kJ mol−1 | (10) |
| KOH + CO2 = KHCO3 ΔH300K = −156 kJ mol−1 | (11) |
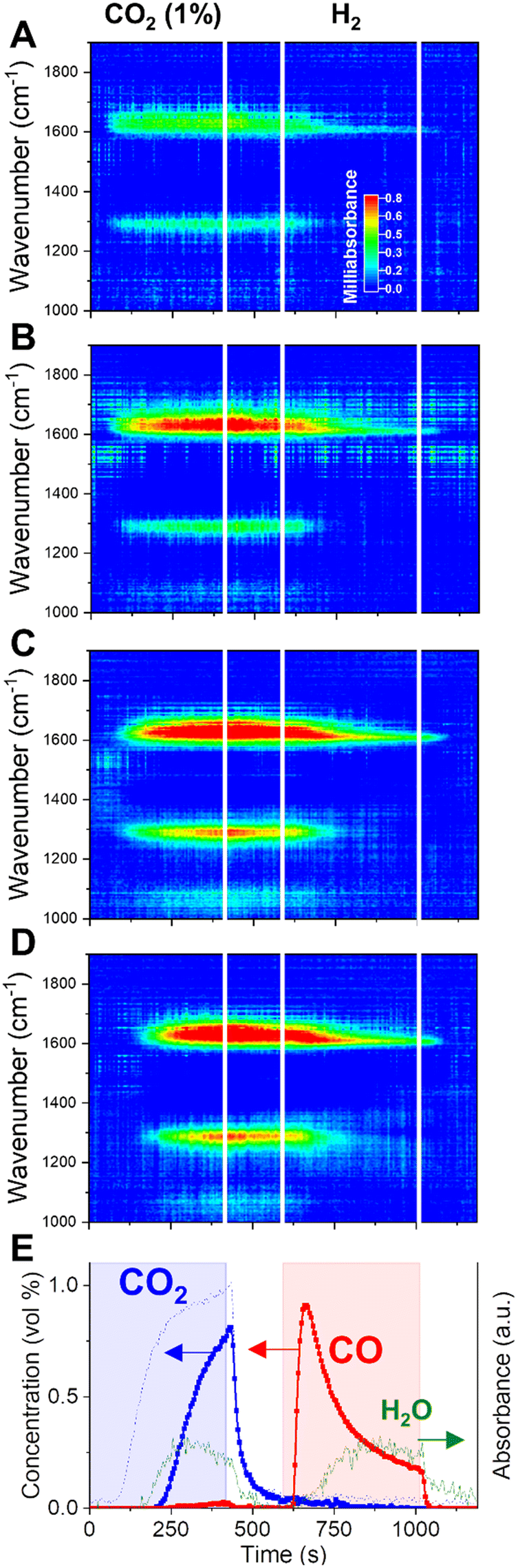 | ||
| Fig. 4 Dynamic evolution of surface species elucidated by operando DRIFTS during CCR at 350 °C at different positions (A = 3.3 mm, B = 6.4 mm, C = 9.5 mm, D = 12.6 mm) of the catalyst bed from front (A) to back (D). Outlet gas composition (E) obtained by averaging 5 cycles at 350 °C. CO2 blank signal (dotted line) obtained as average of 2 CCR cycles during a blank experiment at room temperature. | ||
When CO2 is fed to the catalyst bed (0–420 s), we observe the rise of specific vibrations in the CO stretching region (ν(CO) = 1640 and 1290 cm−1). The outlet stream concentrations (Fig. 4E) clarify that those species are appearing on the surface before the rise of CO2 signal at the outlet (ca. 200 s). When the saturation is reached at one position of the bed, the surface intermediates start forming at positions further in the bed. The delay observed at each position for the appearance of the ν(CO) stretching bands reproduces quite well the spatially-resolved profile of CO2 along the catalytic bed obtained with operando gas sampling experiments (Fig. 3B). This confirms that those signals are directly related to the surface intermediates formed during CO2 capture.
Such species were previously assigned to specific formates on potassium. Contrarily to what happens in H2 + CO2 cofeed operation,33 the rise of CH stretching bands (2700–2900 cm−1) during the CO2 capture is not observed here, indicating that formates may be not a stable intermediate for the capture process. This is in line with the indication of low thermal stability of potassium formate species on Al2O3-supported catalysts, which would decompose to carbonates at temperature lower than 350 °C.34,35 A similar consideration applies to eventual bicarbonate groups on potassium.36 Rather, the absorption bands centered at 1640 and 1290 cm−1, arising during CO2 capture, reflect the splitting of the doubly degenerated asymmetric CO stretching vibration of carbonates, as a result of the lowered symmetry when coordinated to a surface metal cation. The high value of the splitting Δν > 300 cm−1, reported for similar catalytic systems,37,38 identifies the observed surface species as bidentate carbonates.39,40 This represents a strong indication that the principal capture mechanism consists of the exothermic reaction of a KOH phase and CO2 to form carbonates and water (reaction (10)). When the atmosphere is switched to inert flushing (at 420 s), neither a temperature change nor massive COx release is detected by spatial sampling (Fig. S4†), confirming a strong chemical interaction stabilising the adsorbed COx species on the catalyst surface. Nevertheless, a slight decrease in intensity of the DRIFTS bands is observed, indicating that the inert flushing may provoke only a limited removal of the surface intermediates formed during capture.
H2 is essential to remove the surface species generated during CO2 capture. Switching to H2 flow (Fig. 3C), a sudden release of CO is detected instantaneously for each position along the catalytic bed. The removal of adsorbed CO2 in the form of CO is a fast process. Spatiotemporal sampling of temperature during the reduction phase (Fig. 3E) indicates that the CO release is associated with only limited exothermicity and temporally confined at the beginning of the reduction phase. Such results confirm the absence of a redox cycle involving extensive copper oxidation, which would have in turn resulted in a highly exothermic reaction. Rather, H2 is activated on the catalyst by dissociation on Cu and provokes the fast decomposition of the CO2-derived surface intermediates.
In terms of surface species, it can be observed from the DRIFTS data in Fig. 4 that, at the switching to H2 atmosphere (595 s), the bidentate carbonates formed during the capture phase are rapidly decomposed, as indicated by the fast decrease in the intensity of the corresponding ν(CO) stretching bands centered at 1640 and 1290 cm−1. The fast removal of the surface intermediates for capture matches temporarily the fast release of CO detected in the outlet stream (Fig. 4E). Thus, a fast decomposition of the surface carbonates, induced by the presence of H2, constitutes the principal route for the fast CO production in CCR and the regeneration of the active KOH phase (reaction (12)).
| K2CO3 + H2 = 2KOH + CO | (12) |
CO capture
To prove the participation of CO in the capture mechanism, we designed a CCR experiment in which the CO2 phase is substituted by a diluted CO stream (0.5% in He) and performed operando DRIFTS. A direct comparison of dynamic surface species in presence of CO2 or CO as reagents is aimed by employing the same catalyst bed of the experiment in Fig. 4 and analysing the position at 9.5 mm from the front end of the catalyst bed (the Fig. 4C position, the total bed length of 14 mm). The position was selected for providing the best combination in terms of higher absorbance intensity and temporal resolution to discriminate the dynamic evolution of surface species, compared to positions at the front of the bed.The results are reported in Fig. 5. When the inlet CO2 is substituted by a CO stream, to our surprise, very similar catalytic behaviour and surface species dynamics are observed. During the capture phase (0–420 s), CO is effectively captured from the inlet stream. Such result indicates that the active sites on the catalyst surface, formed during the activation of potassium phase in H2, have high affinity for both CO2 and CO.
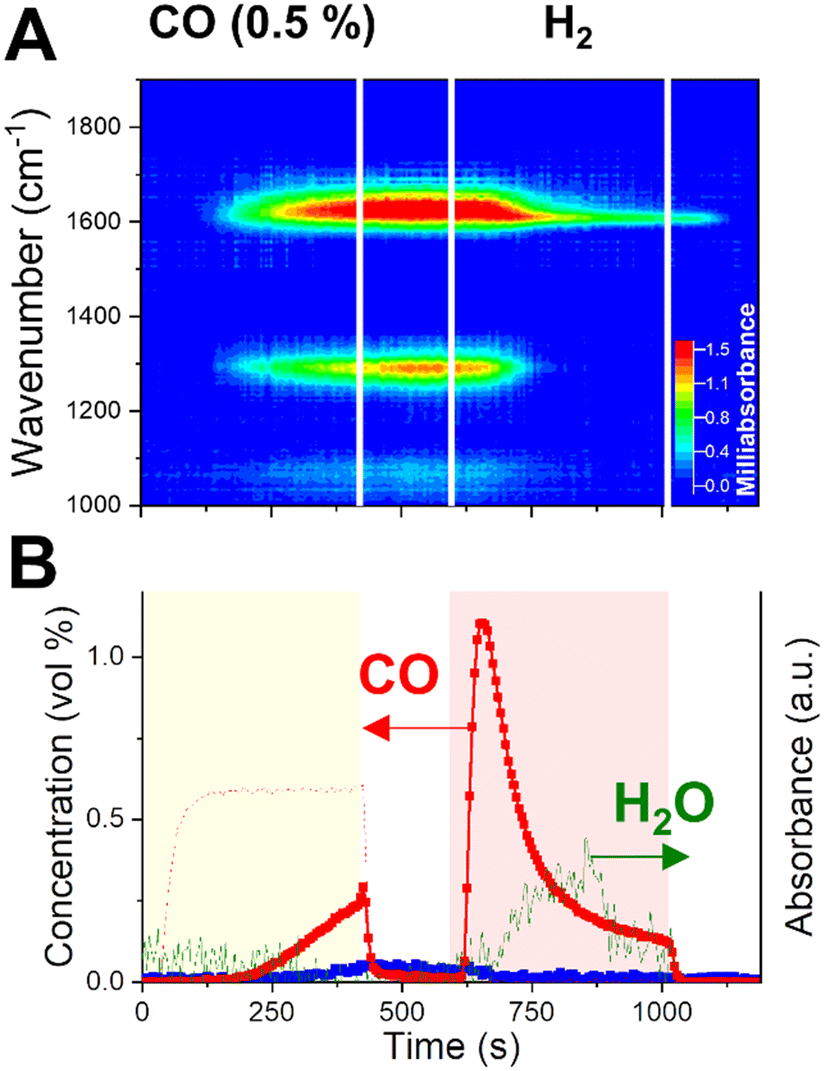 | ||
| Fig. 5 Dynamic evolution of surface species elucidated by operando DRIFTS (A) during CO-CCR performed at 350 °C. Outlet gas composition (B) was obtained from averaging 5 cycles. CO blank signal (dotted line) obtained as average of 2 CCR cycles during a blank experiment at room temperature. | ||
Fig. 5A reports the operando DRIFTS spectra obtained for one position towards the end of the catalyst bed. The CO capture is accompanied by the rise of the same absorption bands in the CO stretching region as observed during CO2 capture (Fig. 4) and assigned to bidentate carbonates. At this position, such bands rise with a significant delay (ca. 200 s) with respect to the start of the CO feed, confirming the progress of a capture front along the bed until saturation of the active sites.
In contrast to the CO2 capture (Fig. 4E), the absence of water release upon CO capture indicates a different type of interaction between CO and the catalytic surface. Alkali hydroxides such as NaOH and KOH are known to efficiently react with CO to form K2CO3 with release of H2 (reaction (13)).44,45
| 2KOH + CO = K2CO3 + H2 | (13) |
Simultaneously, the same bands appear in the ν(CH) stretching region (Fig. S6†) as observed in the CO2-CCR experiment (Fig. S5†) and they are assigned to potassium formates species. However, contrary to the CO2 experiment, the water signal rise is limited and drops before the end of the H2 phase, implying the importance of extra oxygen atom in CO2 for water formation.
CO2 capture on oxidised catalytic surface
Previous operando XRD investigation by Hyakutake et al.25 elucidated that, during CCR operation, a highly dispersed K phase is generated and strongly interacts with the metallic Cu, provoking its nanodispersion. The combination of dispersed potassium phase and metallic copper has been proven a critical property to induce the efficient CCR catalytic activity. Metallic Cu provides sites for H2 dissociation and ensures the regeneration of the catalytically active phase by enhancing the decomposition of the potassium carbonate phase. However, it is not clear whether Cu is playing a fundamental role in the CO2 capture mechanism. To answer this, we performed a modified CCR experiment by introducing a diluted O2 flow (5% in He) instead of the inert flushing phase of the end of each cycle. At the reaction temperature, it is expected that the introduction of oxygen in the reactor leads to the complete oxidation of the copper phase, while keeping intact the active potassium phase generated by decomposition of the potassium carbonate precursor in H2. As a consequence, CO2 capture takes place on a oxidised surface, which contains CuO.Operando DRIFTS results obtained at a single position (9.5 mm in the bed, similar to Fig. 5A) are presented in Fig. 6A. Importantly, the capture of CO2 is still observed in presence of an oxidised catalytic surface, as demonstrated by the delayed appearance of CO2 at the reactor outlet. In terms of surface species, the typical CO stretching bands associated to bidentate carbonates are observed in the capture phase, apart for the band centered at 1650 cm−1, whose peak presents a blue-shift compared to the experiments with CO2 or CO capture on a reduced surface. This result may indicate a specific influence of metallic Cu in the bonding of carbonates species to the surface.
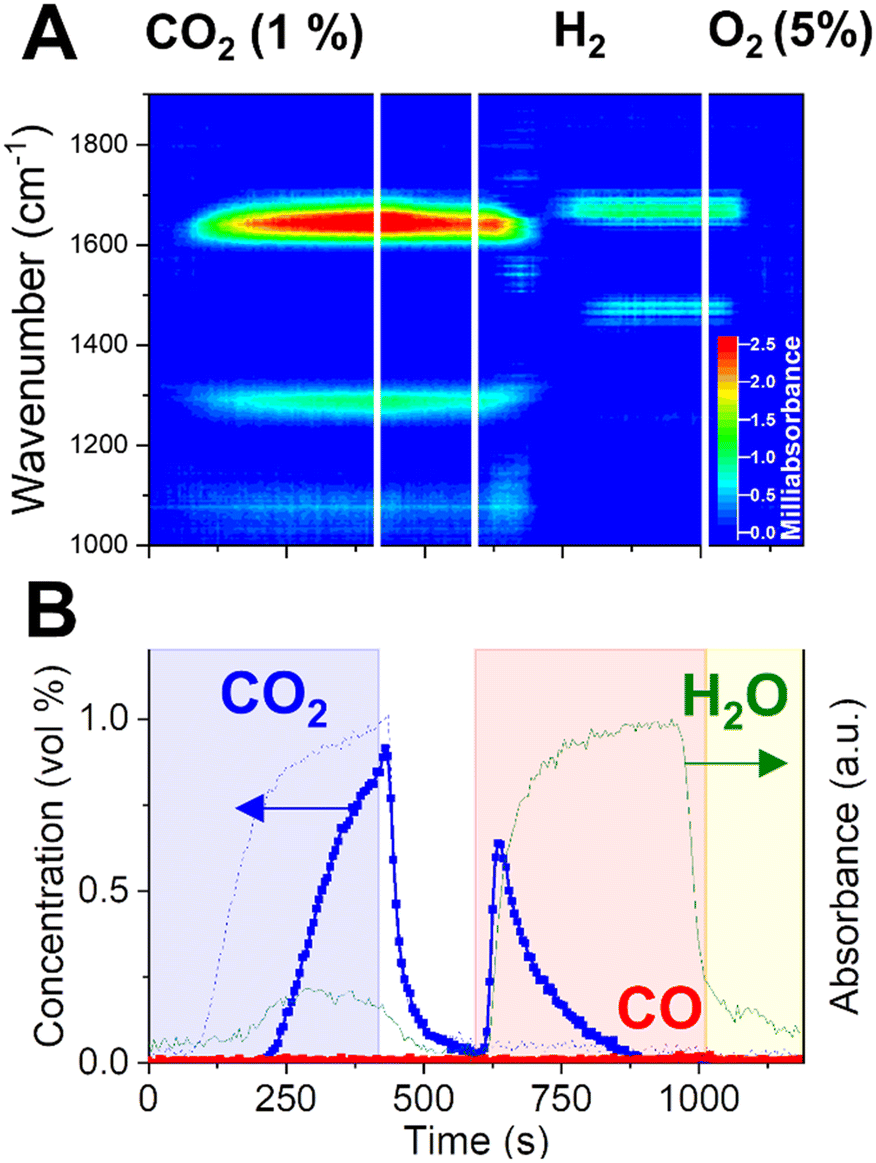 | ||
| Fig. 6 Dynamic evolution of surface species elucidated by operando DRIFTS (A) during CCR performed at 350 °C with substitution of last He gas phase with O2 (5% in He). Outlet gas composition (B) was obtained from averaging 5 cycles at 350 °C. CO2 blank signal (dotted line) obtained as average of 2 CCR cycles during a blank experiment at room temperature. | ||
The amount of CO2 captured and the evolution of the H2O signal at the outlet are comparable to the ones observed in the regular CCR experiment. In contrast, there is no CO detected when CO2 interacts with an oxidised Cu surface. No modifications of the potassium phase are expected as a consequence of the oxygen treatment. The temperature of the reaction and the cyclic exposure to the highly reducing H2 phase may not induce dehydration of KOH to K2O, for which only limited evidences were reported.46,47 The short O2 treatment (175 s) employed in this experiment is not expected to specifically promote such phase transition. In agreement with the regular CCR operation, H2O is detected during CO2 capture and appears as a specific product of the capture reaction. KOH species are then expected to react with CO2 to form potassium carbonates species observed on the surface and to release water (reaction (10)).
Furthermore, no detected CO in the capture phase indicates that the responsible reaction path is prohibited in presence of CuO. Since no evidences of extensive copper oxidation were found during regular CCR operation in this work and previous work, we are tempted to assign the CO formation in normal CCR to an hydrogenation reaction (reaction (2)), resulting from the interaction of CO2 with specific H species activated on Cu. Such species may be oxidised during the forced Cu oxidation in the experiment shown in Fig. 6. Proof of this oxidation process is the water release observed at the reactor outlet during the O2 flushing phase (1015–1190s).
Upon switching to H2 (at 595 s), the presence of CuO activates its reduction accompanying significant production of water. Notably, no CO is released, and only removal of the captured species in the form of CO2 is observed. The extensive release of water may destabilise the surface intermediate and promoting a water-gas shift type of reaction resulting in the removal of unconverted CO2.
The most important observation is that the selective conversion of captured CO2 to CO is lost in presence of oxidised Cu. Operando DRIFTS clarifies that, after the fast removal of surface carbonates and corresponding evolution of CO2, new surface bands appear on the catalyst surface, which can be possibly assigned to adsorbed water due to high amount of water released from CuO reduction. However, it is proved that the Cu–K-based catalyst is able to efficiently capture CO2 in presence of oxygen and water in the stream,20 which is a functionality of interest for realistic application as the removal of COx species from flue gases in combustion and chemical plants.
Conclusions
This work investigated a Cu–K/Al2O3 catalyst with the aim of clarifying the principal reaction routes active in the CO2 capture and its selective reduction to CO. The results indicate that the catalytically active phase for capture, first generated during activation in H2 at 450 °C, may consist of highly dispersed KOH species. Operando sampling of gaseous concentration and temperature inside the catalyst bed revealed that, when the catalyst is exposed to CO2 atmosphere, an exothermic reaction takes place progressively along the bed until saturation of the active site, accompanying a release of water. In addition, spatiotemporal operando DRIFTS confirmed that CO2 is fixated on the catalyst surface in the form of bidentate carbonates species. During the CO2 capture, the presence of metallic Cu and reactive hydrogen stored in the catalyst stimulates a CO2 hydrogenation path to CO as side reaction. This side reaction takes place after completion of the active CO2 capture period. By substituting CO2 with CO in the feed, we demonstrated that CO can be also effectively captured by the catalyst material. However, this capture step does not involve the formation of water.Switching the reactant feed to H2, the regeneration of the catalyst and the selective release of CO take place. Combination of the spatiotemporal operando methodologies revealed that surface carbonates are rapidly decomposed, selectively generating CO and restoring the active phase for CO2 capture. No water is released during the fast removal of surface carbonates, in agreement with a K2CO3 decomposition route and excluding the intervention of extensive copper oxidation in the reaction. After the initial fast release of CO, a tailing CO release is observed. This slow CO formation is caused by the decomposition of the surface formates formed on the potassium-containing species.
Intentionally oxidising the surface by O2 before CO2 capture, we proved that CO2 capture can still take place in presence of CuO with formation of surface carbonates and release of water, reinforcing the hypothesis that the active phase for capture consists of KOH species. However, in the H2 atmosphere, the excess water generated by CuO reduction leads to carbonates decomposition with release of unconverted CO2. These insights have practical implications in designing better CCR catalysts and defining feed gas composition and operation conditions.
Experimental
Catalyst synthesis
Aluminum oxide (γ-phase, Thermo Fisher, catalyst support), was employed as support material. Cu and K were added by incipient wetness impregnation using copper nitrate trihydrate (Merck, >99.5%) and potassium carbonate anhydrous (Sigma, >99.0%) as precursors. Copper nitrate was first impregnated on the support, the resulting mixture was dried overnight at 80 °C and then calcined at 500 °C for 5 h. The obtained Cu/Al2O3 material was further impregnated with a K2CO3 aqueous solution to obtain the final composition (Cu/K/Al2O3, 11/10/79 wt%).Catalyst characterization
Powder X-ray diffractograms were acquired on a Bruker D8 Advance Diffractometer with Bragg–Brentano geometry using monochromatic Co Kα radiation (λ = 1.7902 Å). BET surface area of the catalysts was determined from N2 adsorption isotherms at 77 K using a Micromeritics TriStar II 3020 instrument.Catalytic testing
The reaction setup configuration was similar to the one described in previous works.25 The gas controlling part consisted of mass flow controllers (MFCs, Bronkhorst) and two electric 4-way valves to switch among different gas flows at the inlets. After being pelletized, crushed and sieved in 200–300 μm range, 250 mg of catalyst material were placed in a tubular quartz tube reactor (4 mm ID, 6 mm OD). Blank experiments were conducted by passing the reaction gas mixture on the fresh, non-activated catalyst at room temperature. Prior to the catalytic experiment, the catalyst was activated by reduction under 50 mL min−1 of pure H2 at 450 °C. The temperature of the bed was controlled by a thermocouple inserted in the quartz reactor. Catalytic performance was evaluated under CCR conditions at 350, 400 and 450 °C and ambient pressure. 15 mL min−1 of 9.9 vol% CO2 in He, referred to as capture phase, was alternated to a reduction phase consisting of 15 mL min−1 of H2 (100 vol%). An inert phase of He (30 mL min−1, 100 vol%) was flushed between the capture and reducing phases (and vice versa). The effluent gas mixture composition was quantified by FT-IR spectroscopy (ALPHA Bruker) with a time resolution of 5 s. Valve switching and spectral acquisition were synchronized by LabView software. The data presented were obtained from the average of multiple cycles of operation, after a stable and reproducible composition of the effluent was achieved.Spatiotemporal sampling of temperature and gas composition
For the spatiotemporal analysis of temperature and gas composition along the catalyst bed, the reaction setup was modified introducing a stainless steel capillary in the reactor through the centre of the packed bed. The capillary (inner diameter = 500 μm, outer diameter = 700 μm, REACNOSTICS GmbH) had 4 holes for gas sampling, equally distanced at the same axial position, with diameter of 50 μm. For measuring the temperature at the sampling position, a thermocouple was inserted through the capillary, with the tip aligned with the sampling holes. The capillary end was sealed with epoxy glue, fixing the thermocouple in place. The other end of the capillary was connected to a mass spectrometer (Omnistar Pfeiffer Vacuum). The reactor effluent was analysed by the FT-IR as explained previously. The same experimental conditions employed for the catalytic testing were applied.Operando DRIFTS
The DRIFTS measurements were carried out in a custom-made reaction cell, similar to a previously reported design,48 mounted in a Harrick praying mantis optical system. The reaction cell consisted of a 2 × 2 mm rectangular channel, sealed with a ZnSe window on the top, which limited the operation temperature to 350 °C. A catalyst pellet mass of 160 mg was tested, which translated to a bed length in the reaction cell of ca. 14 mm. Acquiring DRIFT spectra at different positions on the catalytic bed was possible by moving the entire reactor cell, mounted on an actuator. Prior to the reaction, the catalyst was reduced at 350 °C for 1 h in H2 (30 mL min−1, 100 vol%). For the regular CCR experiments, CO2 (10 mL min−1, 1 vol% in He, 420 s), He (30 mL min−1, 100 vol%, 175 s) and H2 (10 mL min−1, 100 vol%, 420 s) and He (30 mL min−1, 100 vol%, 175 s) phases were alternatingly passed to the reactor. CO (10 mL min−1, 0.5 vol% in He, 420 s) and O2 (30 mL min−1, 5 vol% in He, 175 s) phases were also employed in dedicated experiments. Spectra were acquired using a Bruker INVENIO R FT-IR spectrometer, equipped with a liquid-nitrogen-cooled MCT detector. Before measurements, a background was taken over the completely reduced catalyst. DRIFT spectra were averaged over consecutive, reproducible cycles to improve signal to noise ratio (S/N). The reactor effluent was analysed by means of FT-IR spectroscopy as explained previously.Conflicts of interest
There are no conflicts to declare.References
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- C. J. Quarton and S. Samsatli, Appl. Energy, 2020, 257, 113936 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- S.-i. Nakao, K. Yogo, K. Goto, T. Kai and H. Yamada, Advanced CO2 capture technologies: absorption, adsorption, and membrane separation methods, Springer, 2019 Search PubMed.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- Y. Belmabkhout, V. Guillerm and M. Eddaoudi, Chem. Eng. J., 2016, 296, 386–397 CrossRef CAS.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. W. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- IEA, CCUS in Clean Energy Transitions, International Energy Agency, Paris, 2020 Search PubMed.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- H.-J. Ho, A. Iizuka and E. Shibata, Ind. Eng. Chem. Res., 2019, 58, 8941–8954 CrossRef CAS.
- P. Melo Bravo and D. P. Debecker, Waste Dispos. Sustain. Energy, 2019, 1, 53–65 CrossRef.
- I. S. Omodolor, H. O. Otor, J. A. Andonegui, B. J. Allen and A. C. Alba-Rubio, Ind. Eng. Chem. Res., 2020, 59, 17612–17631 CrossRef CAS.
- M. S. Duyar, M. A. A. Treviño and R. J. Farrauto, Appl. Catal., B, 2015, 168–169, 370–376 CrossRef CAS.
- L. F. Bobadilla, J. M. Riesco-García, G. Penelás-Pérez and A. Urakawa, J. CO2 Util., 2016, 14, 106–111 CrossRef CAS.
- S. Cimino, F. Boccia and L. Lisi, J. CO2 Util., 2020, 37, 195–203 CrossRef CAS.
- E. L. G. Oliveira, C. A. Grande and A. E. Rodrigues, Sep. Purif. Technol., 2008, 62, 137–147 CrossRef CAS.
- A. Porta, R. Matarrese, C. G. Visconti, L. Castoldi and L. Lietti, Ind. Eng. Chem. Res., 2021, 60, 6706–6718 CrossRef CAS.
- H. Sun, J. Wang, J. Zhao, B. Shen, J. Shi, J. Huang and C. Wu, Appl. Catal., B, 2019, 244, 63–75 CrossRef CAS.
- T. Hyakutake, W. van Beek and A. Urakawa, J. Mater. Chem. A, 2016, 4, 6878–6885 RSC.
- L. Proaño, E. Tello, M. A. Arellano-Trevino, S. Wang, R. J. Farrauto and M. Cobo, Appl. Surf. Sci., 2019, 479, 25–30 CrossRef.
- A. Porta, C. G. Visconti, L. Castoldi, R. Matarrese, C. Jeong-Potter, R. Farrauto and L. Lietti, Appl. Catal., B, 2021, 283, 119654 CrossRef CAS.
- A. Bermejo-López, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, J. CO2 Util., 2019, 34, 576–587 CrossRef.
- L. Hu and A. Urakawa, J. CO2 Util., 2018, 25, 323–329 CrossRef CAS.
- F. Kosaka, Y. Liu, S.-Y. Chen, T. Mochizuki, H. Takagi, A. Urakawa and K. Kuramoto, ACS Sustainable Chem. Eng., 2021, 9, 3452–3463 CrossRef CAS.
- Y. A. Daza and J. N. Kuhn, RSC Adv., 2016, 6, 49675–49691 RSC.
- Y. Duan, D. R. Luebke and H. H. Pennline, Int. J. Clean Coal Energy, 2012, 1, 1–11 CrossRef.
- A. Bansode, B. Tidona, P. R. von Rohr and A. Urakawa, Catal. Sci. Technol., 2013, 3, 767–778 RSC.
- K. Kobl, L. Angelo, Y. Zimmermann, S. Sall, K. Parkhomenko and A.-C. Roger, C. R. Chim., 2015, 18, 302–314 CrossRef CAS.
- P. Baraldi, Spectrochim. Acta, Part A, 1979, 35, 1003–1007 CrossRef.
- M. Hartman, K. Svoboda, B. Čech, M. Pohořelý and M. Šyc, Ind. Eng. Chem. Res., 2019, 58, 2868–2881 CrossRef CAS.
- K. Coenen, F. Gallucci, B. Mezari, E. Hensen and M. van Sint Annaland, J. CO2 Util., 2018, 24, 228–239 CrossRef CAS.
- N. D. Parkyns, J. Chem. Soc. A, 1969, 410–417 RSC.
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 CrossRef CAS.
- G. J. Millar, C. H. Rochester and K. C. Waugh, J. Chem. Soc., Faraday Trans., 1992, 88, 1477–1488 RSC.
- G. J. Millar, C. H. Rochester and K. C. Waugh, J. Catal., 1995, 155, 52–58 CrossRef CAS.
- I. L. C. Freriks, P. C. de Jong-Versloot, A. G. T. G. Kortbeek and J. P. van den Berg, J. Chem. Soc., Chem. Commun., 1986, 253–255 RSC.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Catal. Lett., 2000, 68, 45–48 CrossRef CAS.
- M. C. Boswell and J. V. Dickson, J. Am. Chem. Soc., 1918, 40, 1779–1786 CrossRef CAS.
- S. Kumar, V. Drozd and S. K. Saxena, Catalysts, 2012, 2, 532–543 CrossRef CAS.
- C. Di Blasi, A. Galgano and C. Branca, Energy Fuels, 2009, 23, 1045–1054 CrossRef CAS.
- T. Otowa, R. Tanibata and M. Itoh, Gas Sep. Purif., 1993, 7, 241–245 CrossRef CAS.
- A. Urakawa, N. Maeda and A. Baiker, Angew. Chem., Int. Ed., 2008, 47, 9256–9259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00228k |
| This journal is © The Royal Society of Chemistry 2022 |