
Stiffening of DU145 prostate cancer cells driven by actin filaments – microtubule crosstalk conferring resistance to microtubule-targeting drugs†
Andrzej
Kubiak
 a,
Matteo
Chighizola
b,
Carsten
Schulte
b,
Natalia
Bryniarska
c,
Julita
Wesołowska
d,
Maciej
Pudełek
e,
Małgorzata
Lasota
f,
Damian
Ryszawy
e,
Agnieszka
Basta-Kaim
c,
Piotr
Laidler
f,
Alessandro
Podestà
*b and
Małgorzata
Lekka
*a
a,
Matteo
Chighizola
b,
Carsten
Schulte
b,
Natalia
Bryniarska
c,
Julita
Wesołowska
d,
Maciej
Pudełek
e,
Małgorzata
Lasota
f,
Damian
Ryszawy
e,
Agnieszka
Basta-Kaim
c,
Piotr
Laidler
f,
Alessandro
Podestà
*b and
Małgorzata
Lekka
*a
aDepartment of Biophysical Microstructures, Institute of Nuclear Physics, Polish Academy of Sciences, PL-31342 Kraków, Poland. E-mail: Malgorzata.Lekka@ifj.edu.pl
bC.I.Ma.I.Na. and Dipartimento di Fisica “Aldo Pontremoli”, Università degli Studi di Milano, via Celoria 16, 20133 Milano, Italy. E-mail: alessandro.podesta@mi.infn.it
cLaboratory of Immunoendocrinology, Department of Experimental Neuroendocrinology, Maj Institute of Pharmacology, Polish Academy of Sciences, 12 Smętna St., 31-343 Kraków, Poland
dLaboratory of in vivo and in vitro imaging, Department of Experimental Neuroendocrinology, Maj Institute of Pharmacology, Polish Academy of Sciences, 12 Smętna St., 31-343 Kraków, Poland
eDepartment of Cell Biology, Faculty of Biochemistry, Biophysics, and Biotechnology, Jagiellonian University, Kraków, Poland
fChair of Medical Biochemistry Jagiellonian University Medical College, Kopernika 7, 31-034 Kraków, Poland
First published on 26th February 2021
Abstract
The crucial role of microtubules in the mitotic-related segregation of chromosomes makes them an excellent target for anticancer microtubule targeting drugs (MTDs) such as vinflunine (VFL), colchicine (COL), and docetaxel (DTX). MTDs affect mitosis by directly perturbing the structural organisation of microtubules. By a direct assessment of the biomechanical properties of prostate cancer DU145 cells exposed to different MTDs using atomic force microscopy, we show that cell stiffening is a response to the application of all the studied MTDs (VFL, COL, DTX). Changes in cellular rigidity are typically attributed to remodelling of the actin filaments in the cytoskeleton. Here, we demonstrate that cell stiffening can be driven by crosstalk between actin filaments and microtubules in MTD-treated cells. Our findings improve the interpretation of biomechanical data obtained for living cells in studies of various physiological and pathological processes.
Introduction
Microtubules (MTs) are filamentous structures constituting one of the cell cytoskeleton's major components. They are cylinders composed most often of 13 longitudinally arranged protofilaments. Individual protofilaments are composed of tubulin heterodimers built up from α- and β-tubulin and linked together by non-covalent bonds forming α/β-tubulin heterodimers.1 Inside the cell, microtubules start from a microtubule-organising centre (MTOC) and span the entire cytoplasm, ending at a cell membrane. MTs provide functional pathways for cellular cargo transport and are involved in intracellular signalling.1 The most important role of microtubules is the participation in mitosis. They form a mitotic spindle used by cells to separate replicated chromosomes into two newly created nuclei during this process.2,3 Thus, any agents affecting microtubule dynamics will introduce severe consequences for cell division by disabling mitosis completion, thereby inhibiting cell proliferation. Such effect has been in the centre of attention of clinical oncologists for many years, leading to the use of taxanes and vinca alkaloids in anticancer therapy of various cancers, including lung, breast, prostate, gastric, oesophagal, and bladder, as well as squamous cell carcinoma of the head and neck.4–6Since the invention of atomic force microscopy (AFM7) and its first use for the characterisation of mechanical properties of cells,8–10 the spectrum of possible AFM applications has widened enormously. A variety of AFM-based mechanical measurements indicate that cell rigidity changes are related to the cell cytoskeleton, mainly to actin filaments.11,12 The microtubular network alone has already been the object of AFM-based nanomechanical studies. In most cases, research was focused on the mechanical properties of cells treated by taxol-based compounds.13–20 Less frequently, the effect of compounds such as colchicine, nocodazole, or vinca alkaloids has been evaluated using AFM.14,21 Among these measurements, only a few papers show the biomechanical changes of cancerous cells treated with microtubule-targeting drugs.11,14,15,21 For example, no stiffness changes were reported for cells treated with colchicine within a time frame of 2 and 4 hours for applied concentrations of 500 μM and 10 μM, respectively.14,21 Vincristine (belonging to vinca alkaloids) induced stiffening of cells treated with 3 nM, 12 nM and 60 nM of drug concentrations for 48 hours.15 Paclitaxel (belonging to taxanes) affects cells’ mechanical properties in various manners – in some cells, no effect was observed14 while in others, an increasing cell rigidity was detected.15,21,22 In the results published so far, high MTD concentrations were used, and the applied action time was mostly smaller than 48 hours.18 Gathered evidence shows that AFM-based mechanical measurements can contribute to a better understanding of the physical mechanisms involved in the cellular response to various conditions, including chemical treatments.
Antimitotic agents belonging to microtubule-targeting drugs (MTDs), such as docetaxel, improve prostate cancer patients’ survival23 but, during treatment, cancer cells often develop drug resistance. It is still elusive why some cells are resistant to the MTDs, despite the known antitumor effect. Multidrug resistance linked with overexpression of ABC transporters is one of the possible explanations. Another possible explanation is a change in tubulin isotypes’ expression, which prevents interaction between the drug and its target.24 The mechanisms of MTDs binding to α/β tubulin heterodimers affect their dynamics by stabilising or destabilising the microtubular network.25 The molecular mechanisms involved in the binding of MTDs are, theoretically, independent of the cell types indicating that all cancers can be treated with such drugs. However, a group of cancers does not respond positively to such a treatment, resulting in cells resistant to the drug treatment, as in prostate cancers. Recent studies further indicate a potential involvement of cytoskeletal remodelling, and thus a modulation of biomechanical properties, in the development of drug resistance against various cancer drugs.26,27 Therefore, we were interested in scrutinizing whether the biomechanical cues underlie the cellular response of prostate cancer cells to drugs against microtubules.
Here, we combine AFM and cell biological methods to study changes in the mechanical properties of prostate DU145 cancer cells treated with three MTD drugs, i.e., vinflunine (VFL), docetaxel (DTX), and colchicine (COL) at concentrations below IC50, for which a high level of proliferating and non-apoptotic DU145 cells exists. We identified that the biomechanical changes in DU145 cells engage the reorganisation of actin filaments and microtubules in response to MT drugs.
Experimental section
Microtubule-targeting drugs (MTDs)
The MTDs chosen for our study were vinflunine, docetaxel, and colchicine. Vinflunine ditartrate (VFL, Toronto Research Chemicals) was dissolved in deionised water (Cobrabid purification system, 18 Ω cm−1) to prepare stock solutions of 1 mM vinflunine concentration. They were stored at −18 °C. Docetaxel and colchicine (DTX and COL, respectively, Sigma-Aldrich) were dissolved in ethanol. Drugs were added to the cell cultures 24 hours after cell seeding, followed by applying a set of methods (Fig. 1a) to characterise the properties of DU145 prostate cancer cells.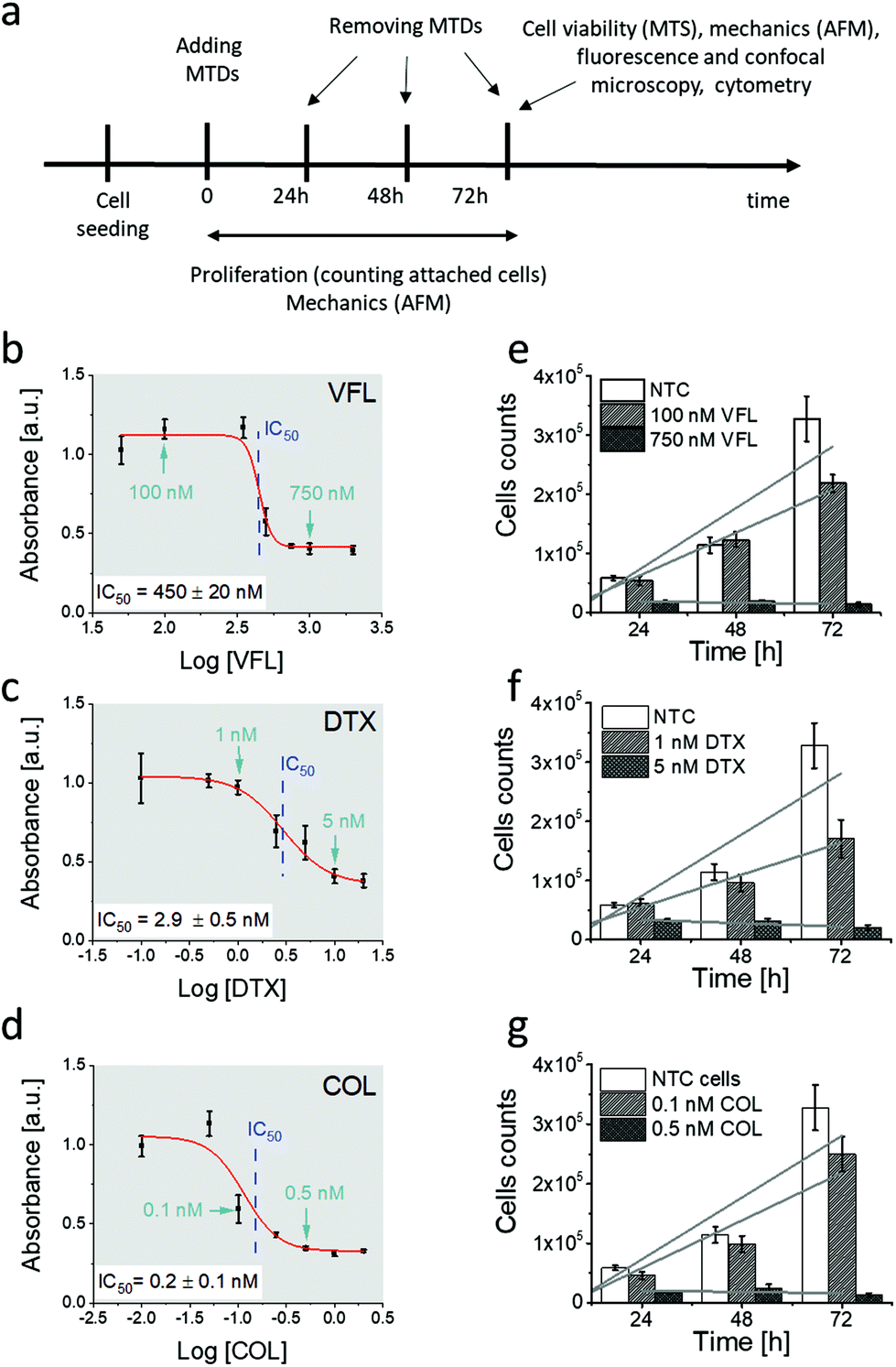 | ||
| Fig. 1 Prostate DU145 cells respond differently to distinct MTDs drug concentrations at different time-points. (a) Illustration of experimental steps and methods applied. (b–d) Dose–response curves for cells exposed to VFL (b), DTX (c), and COL (d). Calculated from the fit, IC50 values were 450 ± 20 nM (VFL), 2.9 ± 0.5 nM (DTX) and 0.2 ± 0.1 (COL). (e–g) Proliferation level of prostate DU145 cells determined for low and high concentrations for VFL (e), DTX (f), and COL (g). A slope of linear fits determines a rate of proliferation. NTC cells denote non-treated DU145 cancer cells. Data are shown as mean ± standard deviation (s.d.) from at least n = 3 measurements; IC50 is expressed as a fitted value ± standard error, (s.e.). Arrows indicate the position of low and high concentrations of drugs chosen for further experiments. | ||
Cell cultures
The DU145 cell line (human prostate cancer cells, ATCC, LGC Standards) was cultured in Dulbecco's Modified Eagle's Medium (DMEM, Sigma-Aldrich) supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS, LGC Standards) without antibiotics. Cells were grown in 25 cm2 culture flasks (Saarstedt), passaged every 3–4 days by trypsinisation with trypsin-EDTA solution (Sigma-Aldrich), and moved to the corresponding Petri dishes. To study the effect of anticancer drugs targeting MTs, VFL, DTX, and COL were added to cells cultured in Petri dishes. Final concentrations of anticancer drugs applied to cells were 100 nM and 750 nM for VFL, 1 nM and 5 nM for DTX, and 0.1 nM and 0.5 nM for COL. As a control, non-treated cells were used.Assessment of drug cytotoxicity by MTS assay
Cytotoxicity of drugs was assessed by using a Cell Growth Determination Kit, an MTT based colorimetric assay (Sigma-Aldrich). 3000 DU145 cells per well were seeded on a 96-well cell culture plate (TPP). After the overnight culture of cells, the culture medium was exchanged to that containing the corresponding drug (VFL, DTX, or COL). After 68 hours of culture with drugs, 10 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was added to each well. After 4 hours of incubation (at time point 72 h), formazan crystals were dissolved in DMSO, and absorbance was measured using a spectrophotometer (ELISA SPECTROstar Nano, BMG LABTECH) at 570 nm wavelength for dissolved formazan and 690 nm for a background.Proliferation of cells
To assess anticancer drugs’ effect on DU145 cell proliferation, cells were seeded at a density of 3000 cells per cm2 in cell culture dishes with Ø = 35 mm (Sarstedt). They were kept in a DMEM medium containing no phenol red (Sigma-Aldrich), 10% heat-inactivated FBS, and 2 mM L-glutamate (Sigma-Aldrich). After 24 hours, the culture medium was replaced by a medium containing a specific drug concentration. Then, cells were maintained in such a medium for 24 h, 48 h, and 72 h. At each time point cells were detached and counted using the Bürker chamber.Apoptosis analysis by flow cytometry
DU145 cells were seeded and cultured on a six-well plate with investigated drugs. After 72 h of incubation, a part of the cell plates was treated with 20 μM cytochalasin D for 30 min. According to the manufacturer's protocol, apoptosis was analysed with a PE Annexin V Apoptosis Detection Kit (BD Biosciences). Briefly, treated cancer cells in six-well plates were collected, washed twice with ice-cold phosphate-buffered saline (PBS, Sigma-Aldrich), and centrifuged at 1000g for 5 min, and then 1 × 105 cells were re-suspended in 100 μl of binding buffer containing 5 μl of Annexin V-FITC and 5 μl of propidium iodine (PI) in the dark. After 15 min, 400 μl of buffer solution was added before the cytometric analysis. The quantification of apoptotic cells was analysed by flow cytometry on a BD FACSCantoTM (Becton Dickinson, New York, USA).Atomic force microscopy – indentation measurements
To assess the relative changes in the cells’ mechanical properties at different time-points, AFM indentation experiments were carried out using pyramidal and custom spherical probes.28 Mechanical properties of DU145 cells were determined by analysing force curves (additional experimental details are included in ESI Notes S5 and S6†). According to consolidated procedures,29,30 the raw force curves (raw cantilever deflection versus z-piezo displacement) were converted into force versus indentation curves in the proper units (nN and nm, respectively).31 The spring constant of the cantilever was measured using the thermal noise calibration.32,33 The deflection sensitivity was calibrated by recording raw force curves on non-deformable substrates (coverslip without cells in this case) and evaluating the inverse of the uncalibrated slope of the contact region. Alternatively, the deflection sensitivity was also calibrated in situ and non-invasively by imposing that the result of the thermal noise calibrated should be compatible with the previously characterised spring constant, according to the SNAP procedure.34 Assuming that Hertz contact mechanics35 can describe the elastic response of the cells, Young's modulus was determined.In the case of a conical approximation of the probing tip, the relationship between load force F and the resulting indentation δ is:
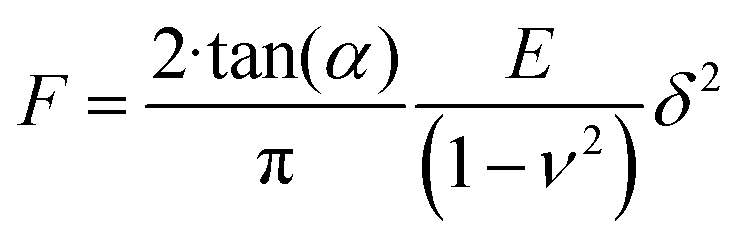 | (1) |
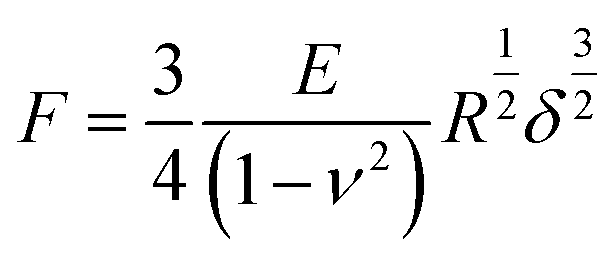 | (2) |
Characterising mechanical properties of different cellular regions
To evaluate Young's modulus changes occurring in different regions of the cell, information on both a local height of the cell body and the value of the apparent Young's modulus was determined from each single force curve and further used to provide the topographical and the Young's modulus maps30 (Fig. 6c–e). A finite-thickness correction was applied to extract accurate values of the Young's modulus from the force curves36 coming from experiments with colloidal probes because the rigid substrate underneath the cells can cause an apparent increase of the Young's modulus that depends on the local height of the sample, therefore masking fine mechanical modifications that take place inside the cell body (Fig. S5†). The correction was not applied to pyramidal sharp tips since the finite-thickness effect becomes relevant with a large contact area (ESI Note S6†).Force curves were linearised to identify multiple elastic regimes inside the cell (typically attributed to an upper layer made of the actin cortex and an inner layer, including the region around the cell nucleus). Two linear regions were typically identified in the linearised curves: a first region corresponding to the 5–15% relative indentation range (to the local values of cell height), the shallow indentation range, and a second inner region, corresponding to the 20–40% relative indentation range, the deep indentation range. The Hertz model with the applied finite thickness corrections was applied to the force curves in the two indentation ranges, obtaining two maps representing the surface's rigidity and inner part of the cell, respectively (Fig. 6c and d).
To characterise the elastic properties of different parts of the cell's body, we created masks to select force curves belonging only to specific regions of interest (nuclear and peripheral region with varying indentation (Fig. 6b). Nuclei are usually located in the tallest cell region. Since the nucleus elasticity could be hidden by that of the stiff actin cortex, we decided to build the mask by multiplying point by point, the topographic and the elasticity maps. We found this procedure more reliable than identifying the nuclei as merely belonging to the tallest regions in the topographic maps. The masks allowing to segregate the force curves belonging to the nuclear regions were built exploiting both the elastic and the topographical maps.
Statistical analysis for the determination of Young's modulus for given cellular regions
The Young's modulus of each cellular region typically follows a lognormal distribution, which appears as Gaussian distributions in semilog10 scale. A Gaussian fit in semilog10 scale provides the peak value E′ and the geometric standard deviation σg10, from which the median Emed and the standard deviation of the median σmed can be calculated:37| Emed = 10E′ | (3) |
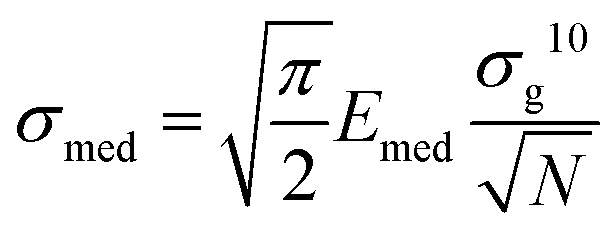 | (4) |
 | (5) |
Then, the mean Emean of the moduli median, measured for different clusters under a given condition for the different cellular regions, was evaluated as: 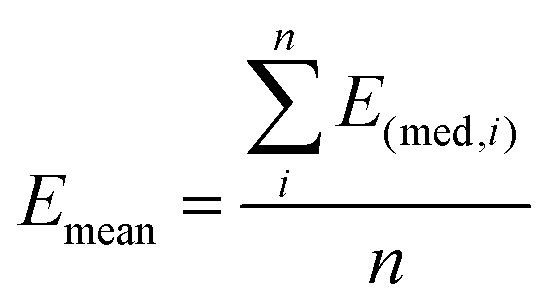 where i is the number of clusters investigated. The total error σE associated with Emean, representative of a specific condition and cellular region, was calculated by summing in quadrature the propagated error of the medians σS and the standard deviation of the mean σmean:
where i is the number of clusters investigated. The total error σE associated with Emean, representative of a specific condition and cellular region, was calculated by summing in quadrature the propagated error of the medians σS and the standard deviation of the mean σmean:
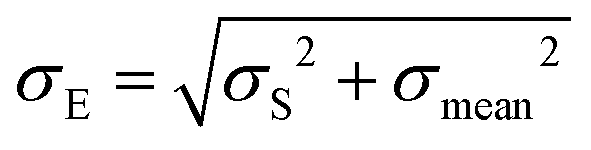 | (6) |
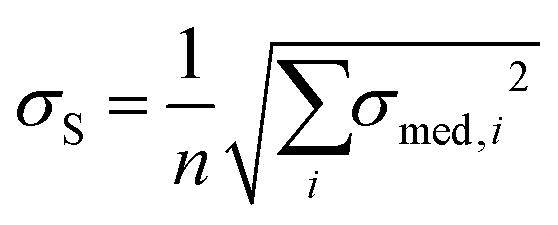 and
and 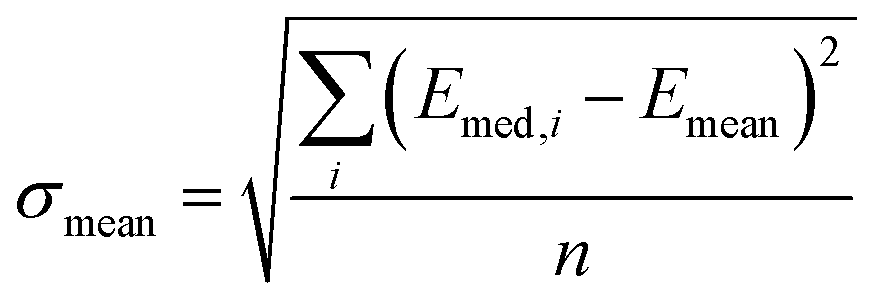 , and n is the number of clusters investigated.
, and n is the number of clusters investigated.
Immunofluorescence staining
To assess the impact of microtubule-targeting drugs on the microtubule network organisation in DU145 cells, fluorescence microscopy was employed. Cells were seeded in density 3000 cells per cm2 on glass coverslips and allowed to stabilise at the substrate overnight. Afterward, the culture medium was exchanged for a fresh medium containing final concentrations of microtubule-targeting drugs. After 72 hours, the culture medium was removed, and cells were rinsed 3 times with Dulbecco's Phosphate Buffered Saline (DPBS, Sigma-Aldrich). Next, cells were fixed with 3.7% formaldehyde solution and washed with DPBS to remove the remaining formaldehyde. Permeabilization of the cell membranes was realised by treating fixed cells with a 0.2% Triton X-100 solution, followed by washing with DPBS. Actin filaments were stained with phalloidin conjugated with Alexa Fluor 488 dye (0.33 μM) for 45 minutes. Afterward, cells were incubated overnight with anti-β-tubulin antibody conjugated with fluorescent dye Cy3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200) (Sigma-Aldrich), followed by nuclei counterstaining using Hoechst 33342 (0.2 μg mL−1).
:200) (Sigma-Aldrich), followed by nuclei counterstaining using Hoechst 33342 (0.2 μg mL−1).
Fluorescence and confocal microscopy
Epi-fluorescent images were acquired using a fluorescence microscope (Olympus IX53) equipped with a fluorescence camera XC10 (1.4 mln pixels). The focal plane was set close to the cell surface. 2D and 3D images were acquired by using a Leica TCS SP8 X confocal laser-scanning microscope (Leica Microsystems CMS GmbH, Germany) using 63× HC PL APO CS2 1.40 NA oil immersion objective. Image acquisition was performed bi-directionally along the X-axis at 200 Hz scan speed and image format 1024 × 1024 (pixel size 72 nm), zoom 2.5, and line average 2. Three-dimensional images were reconstructed using Leica Application Suite X software (3D Movie Editor, Leica Microsystems CMS GmbH, Germany) or ImageJ 1.51n (Wayne Rasband, National Institute of Health, USA) on default parameters (except non-linear intensity adjustment – gamma parameter was 0.6).Surface area of single cells
The surface area of individual DU145 cells was quantified manually using a module included in the CellSens Dimension software (Olympus) from 5 epi-fluorescent images of actin filaments. Data are expressed as a mean ± standard error from n = 29 cells.Calcein efflux monitoring
Cells were seeded into 6-well plates at a density of 60000 cells per cm2 and treated with appropriate concentrations of MTDs for 24 h. After incubation with 0.75 μM calcein-AM (acetoxymethyl ester derivative of calcein, Sigma-Aldrich) for 30 min, the cells were washed with a warm PBS buffer and incubated in a fresh medium for another 3 h. After harvesting with trypsin/EDTA solution, the cells were centrifuged and re-suspended in FluoroBrite® DMEM (supplemented with 10% FBS and 1% GlutaMAX; Gibco) and acquired with an ImageStreamX MkII system and ISX software (Luminex Corp.) to determine the efficiency of dye efflux. Data processing was performed with IDEAS 6.2 software. The analysis was performed on “in-focus” images of single cells. In-focus single cells were classified based on their bright-field area and high bright-field aspect ratio (width to height ratio). At least 10000 images were collected for data analysis.
Statistics
For cell biology methods (proliferation and MTS assay, cytometry, morphometric analysis) data are presented as a mean ± standard deviation (s.d.) (Fig. 1 and 3e,f) calculated from three independent experiments. Data describing the morphometric properties of cells (ESI Fig. S1–S3†) are expressed as a mean ± s.d. calculated for n denoting the number of cells measured (specified in the figure legends). Original Young's modulus values were calculated as a mean ± s.d. considering the number of cells or clusters measured (ESI Tables S1 and S2†). Relative Young's modulus changes are presented in Fig. 5 and 6, ESI Fig. S4† which were accompanied by an error calculated using the error propagation method. In all cases, statistical significance was estimated by an unpaired t-Student test at a level of 0.05. The following notation was applied: * p < 0.05; ** p < 0.01; *** p < 0.001.Results and discussion
Adherent DU145 cells are partially resistant to low MTD concentrations
We started our analysis with the evaluation of MTDs’ effect on the viability and proliferation of DU145 cells (Fig. 1a).To verify how low and high MTD concentrations deviate from the IC50, the MTS assay was applied to cells exposed to different drug concentrations, i.e., 50 nM to 2000 nM for VFL, 0.1 nM to 20 nM for DTX, and 0.01 nN to 2 nM for COL. MTS assay quantifies the fraction of metabolically active cells (through dehydrogenase activity). Recorded absorbance was normalised to NTC cells’ viability level and plotted against MTD drug concentration in the logarithmic scale (Fig. 1b–d). IC50, obtained from the fit of a dose–response function, was 450 ± 20 nM (VFL, Fig. 1b), 2.9 ± 0.5 nM (DTX, Fig. 1c), and 0.2 ± 0.1 nM (COL, Fig. 1d), respectively.
The proliferation rate was assessed by counting cells attached to the glass surface after their incubation with MTDs for 24 h, 48 h, and 72 h. Results show a strong decrease in the cell number for high drug concentrations, i.e., for 750 nM for VFL, 5 nM DTX and 0.5 nM COL (Fig. 1e–g), regardless of the culture time. The number of cells drops to the level of 10%–20% with respect to the number of alive, non-treated cancer (NTC) cells. For low MTD concentrations, i.e., 100 nM for VFL, 1 nM for DTX, and 0.1 nM COL, the number of cells after 24 and 48 hours of culture remained at the same level as for NTC cells regardless of the drug type. The decrease in the number of cells was observed only after 72 hours. A drop by 33%, 48%, and 23% for VFL, DTX, and COL, respectively, was accompanied by the smallest proliferation rate (ESI Note 1†).
In our study, low MTD concentrations are below IC50, while high concentrations are above IC50. 72 h exposure of DU145 cells to low concentrations of MTDs reveals a cell population that contains a strongly dominating fraction of proliferating cells, while at high concentrations, many of the cells died through both apoptosis and necrosis. Knowing that, in our next step, we quantified the number of apoptotic/necrotic cells under conditions of MTD treatment for 72 hours using flow cytometry assay via Annexin V-FITC/PI dual-labelling (Fig. 2).
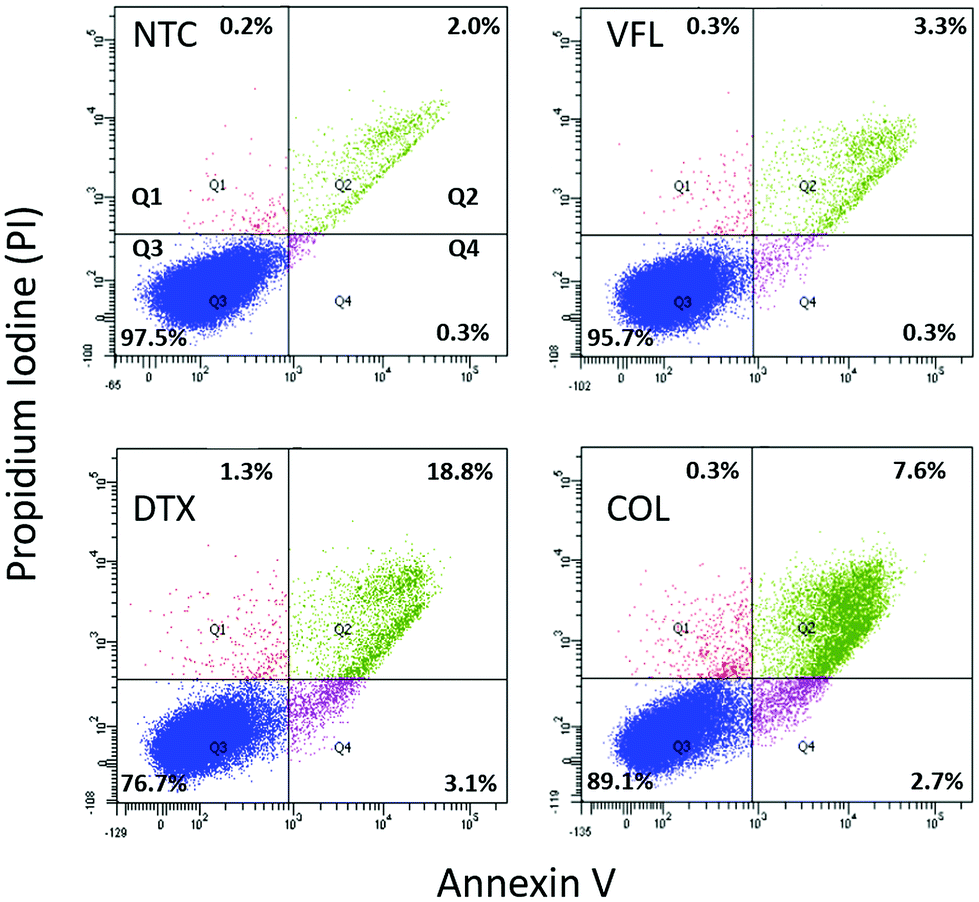 | ||
| Fig. 2 MTD-based induction of cell death in DU145 cells. The images from the flow cytometry analysis of Annexin V and PI fluorescence (a dot-plot graph) showing NTC and cells treated with VFL (100 nM), DTX (1 nM), and COL (0.1 nM) for 72 h (Q1 – necrotic, Q2 – late apoptotic, Q3 – alive, and Q4 – early apoptotic cells). | ||
Non-treated DU145 cell cultures grown for 72 h revealed 97.5% alive cells and 2.3% of apoptotic (0.3% and 2.0% in early and late apoptosis, respectively), and 0.2% of necrotic cells. The treatment of DU145 cells with low concentrations of MTD drugs shows drug-dependent changes, i.e., the amount of alive cells decreases to 95.7%, 89.1%, and 76.7% for VFL (100 mM), COL (0.1 nM), and DTX (1 nM), correspondingly. Consequently, the number of both apoptotic and necrotic cells increased. However, regardless of the treatment, a low level of necrotic cells was observed (0.2%–1.3%). As expected, fluorescence-activated cell sorting (FACS) analysis showed that MTDs induced apoptosis in DU145 cells after 72 hours. The number of apoptotic cells increased from 2.3% (NTC) to 3.6%, 10.3%, and 21.9% for VFL, COL, and DTX treated cells. These results suggest that only DTX treatment effectively promoted apoptosis of DU145 cells.
All measurements assessing the viability of cells show that DU145 prostate cells seem to be more resistant to low concentrations of VFL, COL, and DTX since a large number of cells remain alive (within the range of 76.7%–97.5%).
NTC and VFL-treated cells show a similar level of calcein efflux
To elaborate on how the viability of cells is linked with the capability of DU145 cells to release MTDs, in our next step, we decided to trace the acetoxymethyl ester derivative of calcein using flow cytometry in NTC and MTD treated cells (Fig. 3).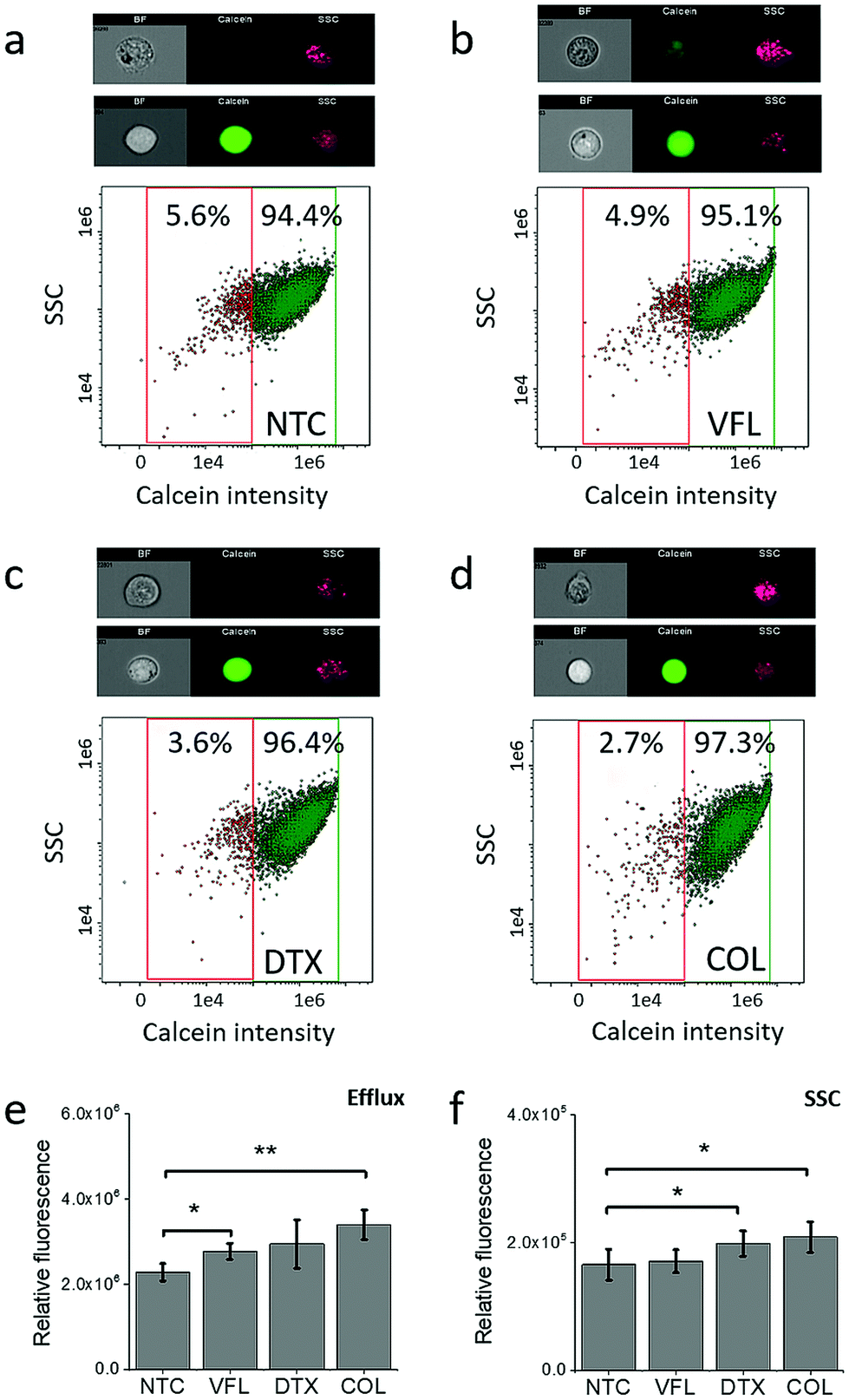 | ||
| Fig. 3 Calcein fluorescence intensity changes with the MTD type. (a–d) Correlative dot plots of side scatter light (SSC) and calcein intensity within the cell population 72 h after MTD treatment. The high SSC/high calcein intensity subpopulation was observed after cell treatments with VFL and COL. ImageStream obtained cell images are representative of the cells gated in NTC (green) and MTDs-treated (magenta) cell populations. (e and f) Average fluorescence intensity of calcein calculated as a mean ± s.d. (n = 3 independent measurements). Statistical significance was estimated by an unpaired t-Student test at the level of 0.05. | ||
Calcein-AM is a non-fluorescent, uncharged molecule that can freely permeate the plasma membrane of viable cells. Inside the cell, it is converted to a fluorescent form, calcein, upon cleavage of the lipophilic blocking groups by nonspecific esterase in the cytoplasm.37,38 Cytometric study shows that within the analysed population of DU145 cells (NTC, 5.6%, Fig. 3a), there are minoritarian groups of cells actively pumping calcein out, i.e., side population. The side population denotes the shift of fluorescence proportional to the loss of calcein pumped out or accumulated inside the cells.39 The size of this group decreases for cells treated with MTDs. The largest drop, about two-fold compared to non-treated DU145 cells, was observed for COL (2.7%, Fig. 3d) and DTX (3.6%, Fig. 3c) treated cells. In the case of VFL treatment, the population of these cells was only slightly smaller (4.9%, Fig. 3b). With the decreasing number of cells and calcein pumping to the extracellular microenvironment, a shift of a cytometric population towards higher calcein intensity appears. The observed MTD-related retention of calcein in treated DU145 cells could be indirectly attributed to drug accumulation (Fig. 3e and f).
Microtubule rearrangements depend on MTD binding sites
The MTDs such as vinflunine, docetaxel, or colchicine are known to bind at distinct locations in the α/β tubulin heterodimer (Fig. 4a) and affect the organisation of the microtubular network. Thus, we visualised MTs inside the DU145 cells after 72 hours of incubation with a specific drug at low and high concentrations (Fig. 4b–h).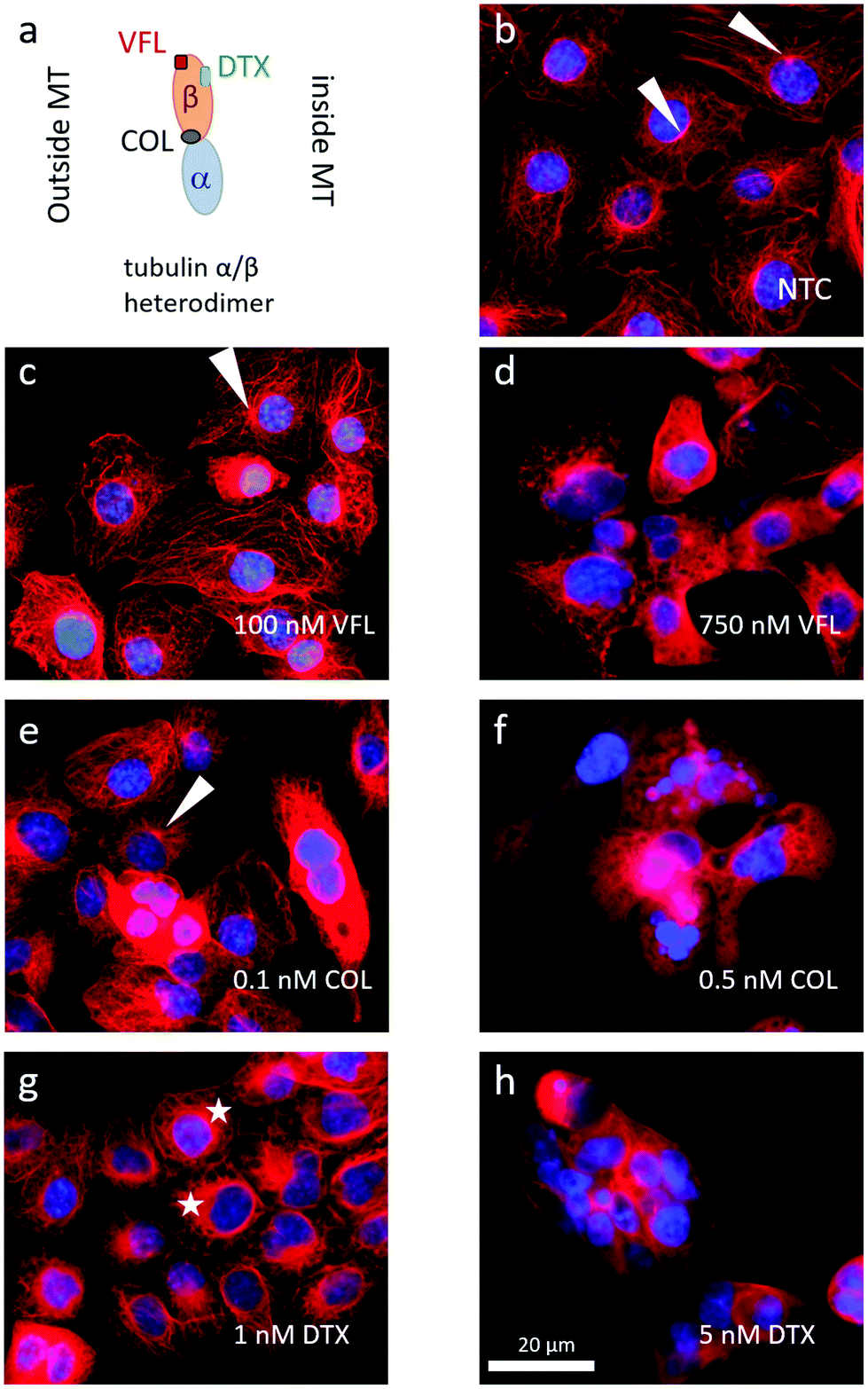 | ||
| Fig. 4 MTDs bind to distinct sites located at the α/β tubulin heterodimer affecting the microtubular network organisation differently. (a) A scheme illustrating the binding of VFL, DTX, and COL to the α/β tubulin heterodimer. (b–h) Epi-fluorescent images showing the organisation of microtubules in DU145 cells upon treatment for 72 hours with low and high MTDs concentrations (white arrows show MTOCs formed close to cell nuclei while stars indicate MT rings formed around cell nuclei). | ||
In NTC cells, microtubules exhibit multi-branched tubulin networks with clearly visible MTOCs located close to the cell nucleus (indicated by white arrows, Fig. 4b). Many MTs alone form long fibers that span from MTOC towards the cell membrane. For cells treated with low MTD concentrations, the degree of microtubule reorganisation is drug-dependent (Fig. 4c, e and g). Some cells treated with 100 nM VFL showed still visible MTOCs (Fig. 4c). Here, the proliferating and non-apoptotic DU145 cells constitute the majority of the cell population (Fig. 1 and 2). Overall, the impact of VFL on the microtubular network of DU145 cells was rather mild, and it was strongly preserved (Fig. 4c). The effect of 0.1 nM COL on DU145 cells induced a rearrangement of the microtubule network (Fig. 4e). Here, MTOCs were visible only in few cells. In the case of prostate cancer cells exposed to 1 nM DTX for 72 h, MTOCs became even less visible, and instead, the microtubular network seems to shrink (Fig. 4g). The MTs aggregation in the form of thicker rings surrounding the cell nucleus (marked with stars) was accompanied by a smaller surface area of DTX-treated cells: 6950 μm2 ± 355 μm2 (n = 29 cells), 6900 μm2 ± 200 μm2 (n = 29 cells), 6950 μm2 ± 305 μm2 (n = 29 cells), and 4085 μm2 ± 230 μm2 (n = 31 cells) for NTC and VFL, COL and DTX-treated DU145 cells.
Under the DTX condition, the lower proliferation rate and the larger number of apoptotic cells (roughly 22% of cells underwent apoptosis, Fig. 2) came along with an altered organisation of MTs. Changes in the MT network can be rated as follows (from the strongest to weakest): DTX > COL > VFL. This is in accordance with the results of the MTS and PE Annexin V assays, from which one can expect large changes in cells treated with DTX, compared to COL, or VFL (Fig. 1 and 2).
For high MTD concentrations, a clustering of the surviving cells was recorded (Fig. 4d, f and h). In the majority of cells, the microtubular network was fully disorganised (disassembled). This could be linked with apoptosis, particularly in the case of DTX and COL treated cells, for which a destruction of MTs was observed and expected.40
Changes in the microtubular network were accompanied by morphological alterations observed for cell nuclei (ESI Note 2, ESI Fig. S1†). High MTD doses cause a fragmentation of nuclei, especially for DTX and COL, while low MTD doses reveal a strong decrease of nuclei surface area and roundness for DTX treated cells (ESI Fig. S1c and d†). The results obtained for low doses confirm the hypothesis that drugs affecting microtubules at small concentrations are not sufficient to destroy them, but they probably tend to change MT organisation.6
Only VFL-treated DU145 cells remain stiffer after 72 h of culture
Changes in MT organisation roughly correlate with the results of cell viability. Simultaneously, according to already published results,16,17,22 MT organisation is also expected to affect the mechanical properties of cells. To investigate the role of biomechanics in drug resistance of cancer cells, we employed AFM to characterise the mechanical properties of treated cells.29,41–44 In particular, we used AFM to obtain an overview of the temporal evolution of cellular rigidity upon exposure to different concentrations of MTDs (Fig. 5).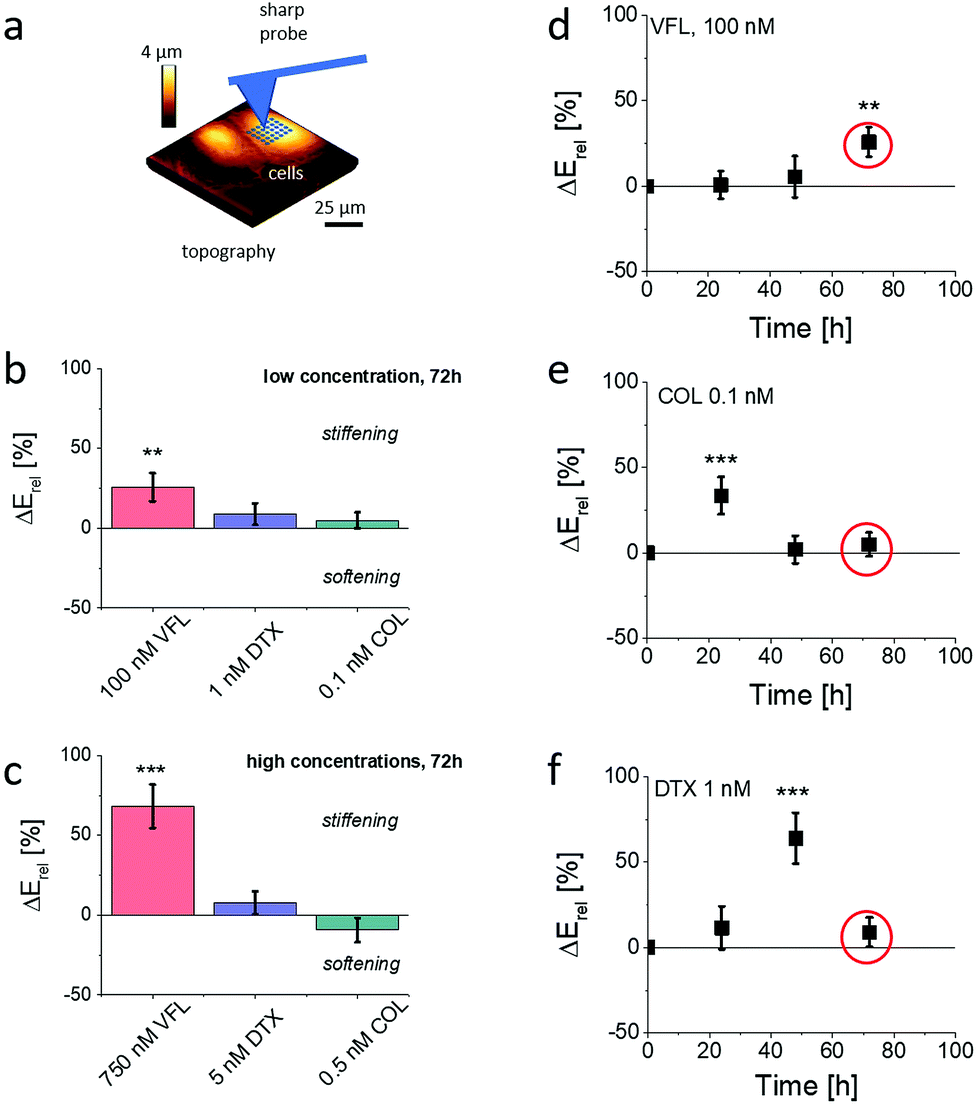 | ||
| Fig. 5 Mechanical properties of DU145 cells depend on the dose and MTD type. (a) Measurements were conducted using AFM in indentation mode using pyramidal probes. Cells were probed above the cell nucleus. (b and c) The 72 h treatment of DU145 cells exposed to MTDs shows a large change in Young's modulus for cells treated with VFL, regardless of the applied drug concentration, indicating cell stiffening. DTX action leads to weak stiffening regardless of the applied drug concentration, while COL treatment to stiffening (b, high drug doses) or softening (c, low drug dose) of DU145 cells. (c–e) Relative Young's modulus change, ΔE, was determined for cells treated with low MTDs doses in relation to NTC cells after 24 h, 48 h, and 72 h of the incubation with the corresponding MT drug. Depending on the MTDs, a maximal increase of cell deformability was observed. The circle indicates values for which a decrease in cell proliferation was noticed. All data were normalised by the Young's modulus of NTCs measured at the same time-point. Statistical significance was estimated by an unpaired t-Student test at the level of 0.05 (** p < 0.01, *** p < 0.001). | ||
The relative change ΔErel of Young's modulus E, defined as ΔErel = (Edrug − ENTC)/ENTC, is quantified using Hertz contact mechanics (Materials and methods, ESI Note 4†). We first carried out characterisation of the relative changes in the mechanical properties of cells, measured within the cell center, i.e., above the cell nucleus at different time-points, i.e., after 24 h, 48 h, and 72 h of culture in the presence of MTDs. The results show that the MTD type, dose, and time affect prostate DU145 cells’ biomechanical properties. Each MTD applied to cells at the low concentration reveals different time-related characteristics (Fig. 5d–f). Vinflunine added to cell culture at a final concentration of 100 nM induced stiffening of cells after 72 h of culture. Colchicine-related maximum in cell rigidity was observed relatively soon, i.e., after 24 h of growth in the presence of the drug. After 72 h of culture, the mechanical properties of DU145 cells were similar to those observed for non-treated cells. In the case of DTX-treated cells, the maximal cell rigidity was observed after 48 h of culture, followed by the decrease of the Young's modulus to the level of NTC cells. Interestingly, the maximum rigidity is not related to the proliferation rate (Fig. 1e–g), showing the largest decrease of the number of cells after 72 h of culture. Similarly, the mechanical properties were not related to the number of apoptotic cells, which was the lowest in VFL-treated cells and the largest in DTX-treated cells.
Already published data indicate that for high concentrations, microtubule-targeted drugs can be broadly grouped into microtubule-stabilising agents and destabilising agents, depending on whether they promote microtubule assembly or whether they trigger disassembly into tubulin dimers.3 The consequence in either case is the MTs rearrangement. Here, high MTD concentrations cause heavy damage to the microtubular network (Fig. 4d, f and h) for all tested MTDs (particularly for DTX and COL). The observed strong effects on MT damage were not related to relative changes in mechanical properties, neither for low nor high MTD concentrations.
The AFM indentation experiments showed that prostate DU145 cells stiffen (positive ΔE, p < 0.01) in response to 72 h VFL treatment. After the same time, for cells treated with DTX and COL the changes in mechanical properties were not statistically significant (p > 0.2). These results contradict the fluorescence images of microtubular networks, showing that, for proliferating cells, the morphology of 100 nM VFL-treated cells is very similar to that of NTC cells. In comparison, the largest morphological changes are observed for 1 nM DTX and 0.1 nM COL-treated cells, for which less obvious changes in cell mechanics are noticed.
Considering the occurrence of maximal cell rigidity for the VFL treatment for 72 h without an evident strong effect on the MT organisation, we focused our further experiments on cells exposed to low concentrations of MTDs after 72 h, also because the proliferation rate decreased at this time point.
Stiffening of VFL-treated cells occurs within the nuclear region
To probe more thoroughly the mechanical changes occurring after 72 h treatment with MTDs, we performed combined AFM topographical and mechanical measurements using a spherical probe. This enabled us to analyse the cells’ elastic response at different indentation depths according to an already documented methodology30 (Fig. 6a, Materials and methods). Each recorded force curve was linearised, and two linear regions, corresponding to different Hertzian regimes, were typically identified.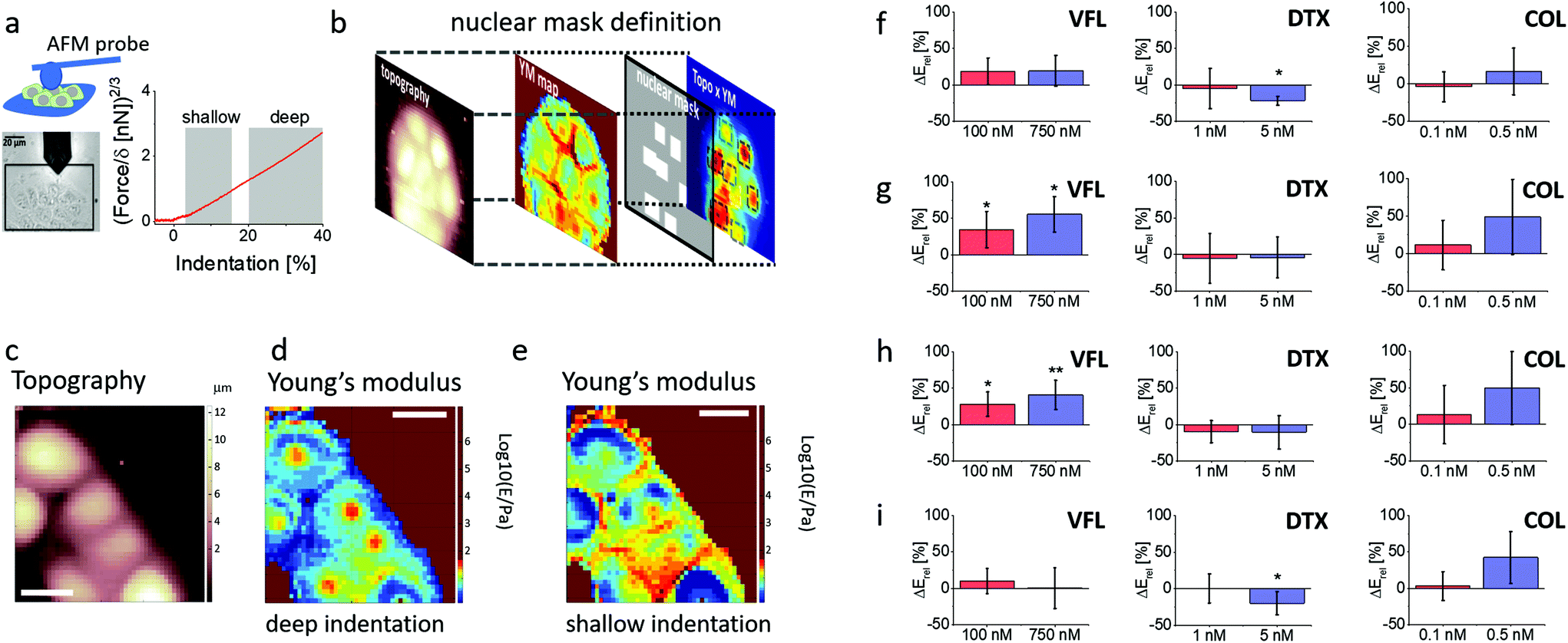 | ||
| Fig. 6 Multicomponent mechanical analysis of cell clusters after 72 h exposure to MTDs. (a) Exemplary optical image of prostate cancer cell clusters with highlighted regions selected for the indentation measurements (the colloidal probe at the end of the AFM cantilever is visible as a darker circular shadow). Exemplary linearised force curves collected for VFL treated cells with highlighted shallow and deep indentation regions. (b) Nuclear mask definition used further to sort the data recorded around cell nuclei. (c) Topography of a cell cluster created based on contact point positions. (d and e) Young's modulus maps obtained at (d) deep and (e) shallow indentations. (f–i) Relative Young's modulus change, ΔErel, determined for DU145 cells treated with specific MTD concentrations for the whole cell (f), the nuclear region at shallow (g), and deep (h) indentations, and at the cell periphery at shallow indentation (i). Error bars were calculated using the error propagation method. Statistical significance was estimated by an unpaired t-Student test at the level of 0.05 (* p < 0.05; ** p < 0.01). | ||
The first regime (shallow indentation corresponding to the 5–15% relative indentation range normalised to the local cell height) is expected to represent mainly the mechanical response of the cell membrane coupled with the actin cortical layer; the second regime (deep indentation) corresponding to the 20–40% relative indentation range is expected to be strongly affected by the response of the nucleus and the surrounding MT network.
Given the peculiar organisation of the MT network around the nucleus, we defined nuclear masks to focus our attention on the nuclear region of the cells (Fig. 6b). This methodology enabled us to analyse the deformability of different cellular regions and layers, according to the following classification: whole cell, from cell clusters (as DU145 cells tend to grow as clusters), nuclear region probed at the deep and shallow indentations, and cell periphery probed at shallow indentation (Fig. 6c–e). The applied finite thickness correction minimised the underlying glass substrate's effect and local cell height variations (ESI Note 6; ESI Fig. S5†). Elasticity maps revealed that in the case of shallow indentations, a large increase of Young's modulus was observed at the cell periphery (Fig. 6e), while the nuclear region appeared softer. At deep indentations, the cell periphery appeared to have a lower Young's modulus as compared to the nuclear regions (Fig. 6d). The obtained Young's moduli for DU145 cells show changes that are strongly dependent on the cellular regions and the used drug (Fig. 6f–i; ESI Table S2†). Mechanical properties of COL-treated cells show a very weak tendency to stiffen, especially for 0.5 nM drug concentration; however, experimental errors are large enough to exclude statistical significance (p varies between 0.0584 and 0.8905). This leads to the conclusion that colchicine does not affect the mechanical properties of prostate cells. Relative Young's modulus changes in DTX-treated prostate cancer cells do not reveal any alterations in mechanical properties at low (1 nM) concentration. Changes in cell mechanics were observed for high (5 nM) concentration, but instead of stiffening, a softening was noted within a peripheral region. No difference compared to NTC was observed within a nuclear region in both indentation regimes. DTX-induced softening at the cell periphery (Fig. 6i) dominates the overall cell softening, observed when the whole cells were measured (Fig. 6f). In the case of VFL, changes in cell mechanics were more pronounced for nuclear regions at both applied MTD drug concentrations and indentation regions. The lack of ΔE variations within the cell periphery and at the whole-cell level indicates that the mechanical properties measured for individual cells, independently of the indentation depth, are not affected in these zones by the changes induced by the MTD within the nuclear regions. In summary, DTX and VFL have two different effects on DU145 cells. DTX induces cell softening only at the cell periphery, while VFL generates the stiffening of cells within a nuclear region.
MT network around cell nuclei is not responsible for the stiffening of VFL-treated cells
In our first approach, we assumed that the changes in mechanical properties of DU145 cells induced by the presence of MTDs might be associated with alterations of the MT component of the cytoskeletal network. The epi-fluorescent images suggested the presence of MT rings around cell nuclei that could be associated with cell stiffening after DTX treatment (Fig. 4g). However, the results of the biomechanical measurements indicated the cell's stiffening within the nuclear region in VFL-treated DU145 cells (Fig. 5 and 6). A higher density of MTs in the nuclear region could suggest a link between MT crowding and stiffening induced by MTDs in DU145 cells. To test this hypothesis, we employed confocal microscopy to assess whether the microtubules’ and actin filaments’ spatial organisation around the cell nucleus is different in treated and non-treated prostate cancer DU145 cells (Fig. 7a and b). The results show no evidence of the MT-related rings observed in epi-fluorescence images, which could be attributed to a denser microtubular network localised mainly around the cell nucleus (see Fig. 7a, DTX-treated cells). Again, also in these more detailed confocal images, the state of the microtubule organisation under the different experimental conditions is in contradiction with the biomechanical results. One can assume that a denser MT network would lead to cell stiffening, in particular, within the nuclear region of DTX-treated cells. For these cells, stiffening was observed at the peripheral parts of the cells.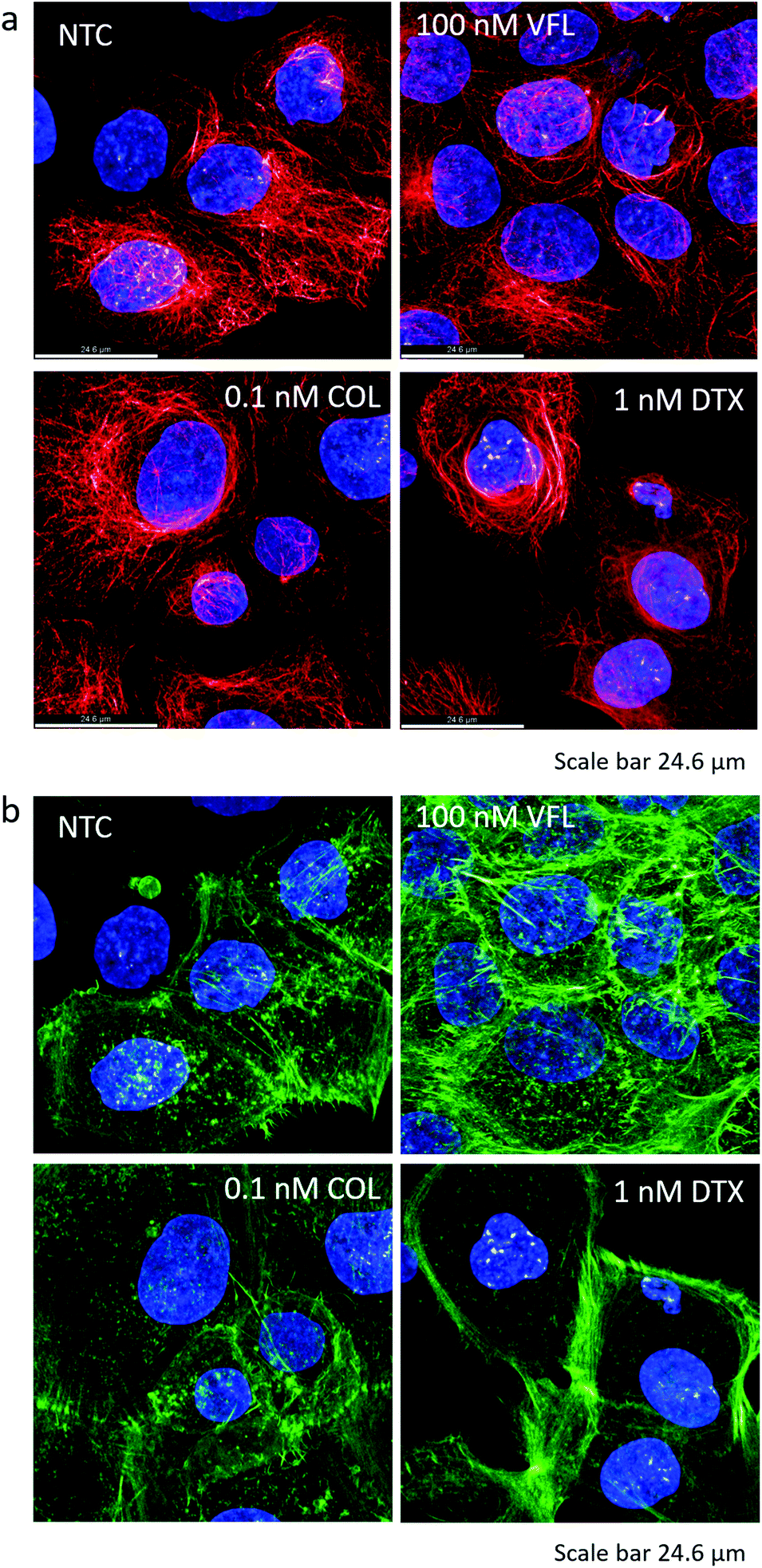 | ||
| Fig. 7 Confocal microscopy of microtubular networks (a) and the corresponding actin filaments (b) of MTD treated DU145 cells (staining: microtubules β-tubulin antibody – Cy3; F-actin – phalloidin conjugated with Alexa Fluor 488; cell nuclei – Hoechst 33342). | ||
However, this apparent lack of correlation between the MT reorganisation around the cell nucleus and the biomechanical properties guided us to investigate the actin filaments’ organisation. The actin cytoskeleton is known to have a major contribution to cell biomechanics. Thus, in our next step, we verified whether changes in the mechanical properties of cells might be related to actin filaments’ organisation (Fig. 7b).
For cancerous cells, the disorganisation of actin filaments and lack of stress fibres are frequently observed.45,46 Phalloidin practically binds only to the polymerised form of actin, not to actin monomers. In non-treated prostate DU145 cells, the actin cytoskeleton showed only a few filaments spanning over the cell body, accompanied by a small number of actin-rich filopodia. Mostly, short filaments (detectable in confocal images as a shadow) were formed. After 72 hours of incubation with MTDs, the amount of actin bundles increased in VFL-treated cells, predominantly in the perinuclear region, i.e., the formation of perinuclear actin caps over the cell nucleus,47,48 and in filopodia. The effect is similar to that observed for endothelial cells treated with 200 nM nocodazole.45 The actin cytoskeleton of COL-treated cells remained similar to non-treated cells. Instead, in DTX-treated cells, thick actin filaments were visible at the cell periphery, but the perinuclear region was almost void of them. Densification of actin filaments is compatible with the already mentioned shrinking of DU145 cells visible after DTX treatment. These results are in agreement with biomechanical data showing the stiffening of the nuclear region after VFL treatment. They underline that in AFM-based measurements of cell elasticity, actin filaments are the major contribution (confirmed by cross-sectional images of these cells showing the domination of actin in VFL-treated cells, ESI Fig. S6†).
As a final step of our study, we conducted experiments in which we interfered with actin filaments’ organisation by cytochalasin D (cyto D) treatment. Cyto D is a cell-permeable fungal toxin causing the actin filaments’ disassembling by inhibiting actin polymerisation.46 In our experiments, 20 μM cyto D was added for 30 minutes to the following group of cells: NTC, VFL-, DTX-, and COL-treated cells. Adding cyto D to the cell cultures decreased the number of alive cells under all conditions. The flow cytometry assay via Annexin V-FITC/PI dual-labelling showed a significant increase in the number of dead cells in an MTD-dependent manner, roughly from 2 to 10% (ESI Fig. S7†). As expected, confocal images display a strong reorganisation of actin filaments after cyto D treatment. They show a depolymerised actin cytoskeleton, also accompanied by disorganisation of MT networks, especially at the cell periphery and close to the cell surface (ESI Fig. S8 and S10†). Biomechanical measurements of cyto D-treated and non-treated DU145 cells (with or without additional VFL treatment) reveal a mechanical response of cells to cyto D for VFL-treated cells (Fig. 8).
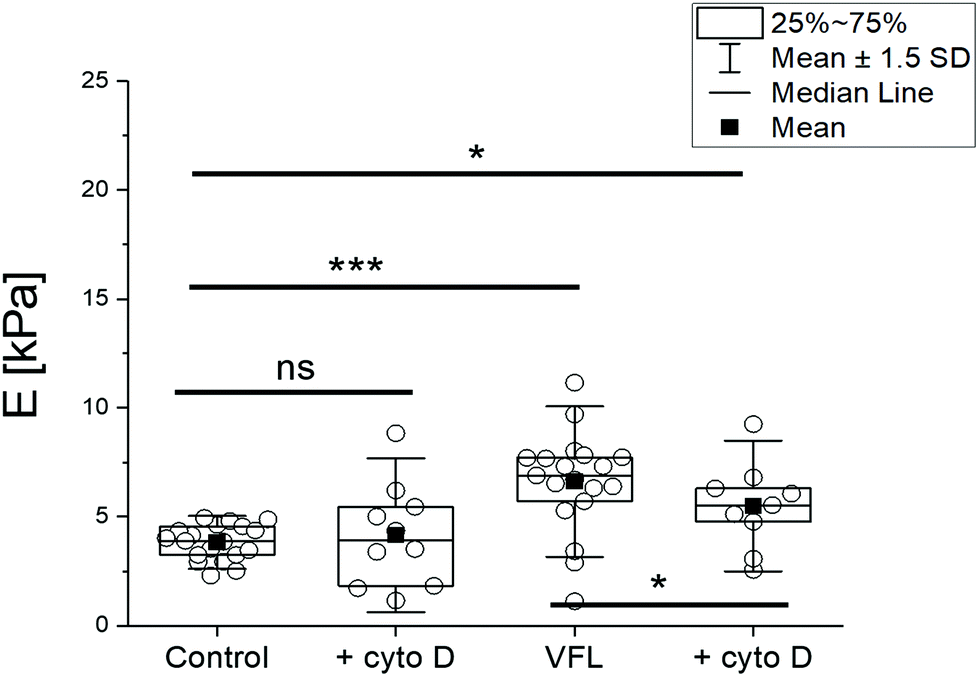 | ||
| Fig. 8 Biomechanical properties of cytochalasin D treated cells, obtained for the indentation depth of 400 nm. Cells were initially treated with 20 μM cyto D for 30 min and then with 380 μM cyto D for 15 min. Box plots representing basic statistical parameters overplayed with recorded data. Each point is a median, calculated for a single elasticity map (16 force curves). Statistical significance was estimated by an unpaired t-Student (mean values) and Mann–Whitney (median) tests at the level of 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001). | ||
The stress fibres of perinuclear actin caps are connected with a nuclear envelope and, in parallel, with focal adhesions;47,48 thus, the action of cyto D is supposed to lead to alterations in perinuclear actin cap-related cellular mechanical properties. A low amount of actin filaments can explain the lack of response to cyto D in the NTC DU145 cells within the probed region (over the cell nuclear area), as suggested from the analysis of all recorded Z-stack positions by confocal microscopy (ESI Fig. S10†). The reduction of the perinuclear stiffness in VFL-treated cells after cyto D (p < 0.05) is consistent with the presence, respectively absence, of perinuclear actin caps over the cell nucleus under the different experimental conditions (Fig. 7, ESI Fig. S10†).
Discussion
Microtubules, and proteins associated with them, are critical for successful cell division, intracellular transport, signaling, and the regulation of focal adhesion dynamics in both normal and cancer cells. Their deregulation observed in cancer makes both tubulin and microtubules a prominent target for various chemotherapies.49 Numerous studies have reported a cell-, dose-, and time-dependent cellular response to microtubule-targeting drugs.6,50–54 Although it is obvious that cells treated with high toxic concentrations are characterised by large damage causing their functional impairments, the effect of low drug concentrations on cells is less visible.All MTDs interact with microtubules by binding to specific locations at the α/β tubulin heterodimer, thus, affecting the MT dynamics by stabilising or destabilising the microtubular network.25,55–57 MT stabilising and destabilising drugs disrupt microtubule functioning, which is accomplished by various mechanisms.
Vinca alkaloids destabilise microtubules by binding to a site located at the inter-dimeric interface between α and β tubulin heterodimers.25 They induce the formation of alternate microtubules leading to the dissociation of existing microtubules.58,59 Taxanes reversibly bind to β-tubulin at the binding site located at the interior lumen of microtubules and enhance the polymerisation process.60 Colchicine binds to a site located at the interface between α and β-tubulin subunits, adjacent to the GTP-binding site of α-tubulin.61 It blocks the availability of α/β tubulin heterodimers for protofilaments or MTs by changing the dimer conformation.51 As a consequence, colchicine causes microtubule depolymerisation by inhibiting lateral contacts between protofilaments.62 Thus, we could expect that vinca alkaloids and colchicine should increase the softening of cells, while taxanes should enhance the stiffening of cells.
In this study, we have focused on the effect of three selected members of MTDs, i.e., DTX, VFL, and COL, on prostate DU145 cancer cells. This cell line is considered to be one of the cell lines used as the standard prostate cancer cells in therapeutic research.63–65 Among three MTDs applied to these cells, we focused particularly on vinflunine effects on DU145 cells as it is a new drug, not frequently used in cancer therapy (https://www.clinicaltrials.gov/ct2/results?cond=vinflunine&term=&cntry=&state=&city=&dist=). Thus, we compared the results to those obtained for drugs binding to non-vinca alkaloid binding sites present in the tubulin dimer.
Proliferation and MTS assays conducted for cells cultured on glass surfaces for low and high MTD concentrations show confirmed dose-dependent effects. Applying high MTD concentrations, regardless of their type, leads to cell death showing a significant decrease in the number of attached cells within a whole-time range (24 h–72 h). MTS assay carried out for cells substantially confirms the proliferation assay results. For low MTD concentrations, a large drop in the number of attached cells visible after 72 h of the exposure of cells to specific MTDs and weakly affected viability of cells indicated that the cell population is not uniform. It contains cells that detached from the surface and cells that still stay attached.
Calcein efflux demonstrated that VFL-treated cells are still able to pump out the drug, while this process is limited in the case of cells treated with DTX and COL (Fig. 3). These results indirectly show the accumulation of MTDs inside the cancer cells at drug concentrations below IC50. In contrast to DTX and COL, VFL impacts the cellular bioenergetics very weakly, which does not lead to drug accumulation. Disturbances within MTs and mitochondrial dynamics may be responsible for diminishing intracellular energy deposits necessary for effective drug efflux by transmembrane protein complexes.66,67 These results agree with the migratory capability of the DU145 cells showing lower migration speed for cells treated with MTDs (ESI Fig. S3†). Mean displacement remained at the same level of 24–30 μm.
The alterations in the cell cytoskeleton structure induced by MTDs, in both microtubular and actin filaments, generate the question: how these changes are correlated with the cell biomechanical properties. As we mentioned in the Introduction, the microtubular network alone has already been the object of AFM-based nanomechanical studies, even though in a majority of studies, biomechanical changes are correlated with the actin filaments. Taxol is the most common MTD drug used in AFM-based elasticity measurements. Less frequently, colchicine or nocodazole has been applied.13–17,21 The only drug belonging to vinca alkaloids applied in the AFM measurements was vincristine that induces cell stiffening.15 These results agree with vinflunine's effect on prostate DU145 cells observed in our study for comparable conditions (concentration of 60 nM, here 100 nM and incubation time 48 h, here 72 h). Differences between the literature data and our results observed for DTX and COL might reflect a distinct response of prostate DU145 cells. Other studies have already demonstrated cancer cells’ susceptibility to cell death induced by these drugs.53,54
According to our results, low concentrations of MTDs applied to prostate DU145 cancer cells induced changes in the organisation of the microtubular network and the mechanical properties of cells in a surprising manner. For the VFL, which weakly affected the MT organisation, the largest cell stiffening is observed. Cells became more rigid in the nuclear region. DTX-treated cells soften at high (above IC50) concentrations at which large damage of MTs is detected. For COL, which had the intermediate effect on the microtubular network, the mechanical properties of cells remained almost unchanged. From these data, we cannot support the hypothesis that the mechanical resistance of prostate cells is associated only with the changes in the MT organisation, nor from the impairments in efflux pumps that transport the drugs out of the cancer cells.66,67
By analysing the confocal images (including cross-sections), we found the prerequisites, albeit MTDs should affect only MTs in DU145 cells, that the actin cytoskeleton is more likely to be responsible for MTD-induced modulations of the mechanical properties measured by AFM. Our biomechanical data are showing a stiffening of cells in the nuclear regions of VFL-treated cells. In this region, confocal microscopy images revealed a larger density of actin filaments upon VFL treatment. Lack of evident MT reorganisation around the cell nucleus in VFL-treated DU145 cells guided us, therefore, towards the hypothesis of a crosstalk phenomenon between actin filaments and microtubules68–73 involved in the development of resistance to vinflunine. Actin filaments influence the microtubular network through filament growth57 or crosslinking and anchoring of microtubules within its network.74 Thus, any disruption of the microtubular network will affect the actin cytoskeleton organisation and vice versa, and potentially impact the chemoresistance of cancer cells.27,68,75 The action filaments – microtubule cross talk phenomenon is known to be involved in cell migration and invasiveness.69–73 For DU145 cells treated with VFL, the migration speed was slightly lower than that of non-treated cancer cells (p < 0.0396). Here, we observe the formation of predominantly perinuclear actin filaments at low VFL concentrations for which there are no obvious changes in the microtubular network. In DTX-treated cells, the actin filaments were accumulated in the cell periphery, while in the case of COL, no strong formation of actin filaments was observed. Cells treated with DTX and COL were less migratory (p < 0.0001 and p < 0.0015, respectively).
Various works reported a RhoA signaling-dependent impact of microtubule depolymerising or destabilising agents on actin filament formation and contraction, indicating further the close correlation between these cytoskeletal elements.76–79 However, compared to our results, these studies report larger MTD concentrations and shorter time of action. Taking into account the relatively mild (visible) effect of the low MTD concentrations on the microtubular network in our experiments, we may have a contribution from an actin cytoskeleton-dependent adaptive mechanism27,68,75 helping DU145 cells to resist MTDs’ effect.
An open question remains on the specific way the interaction between microtubules and actin filaments mediates the MTD effect (in particular vinflunine) in the DU145 cells. It has been reported that the microtubule-actin filament cross talk can be of mechanical80 and biochemical81 origins. Changes in cytoskeleton organisation observed in confocal microscopy and significant changes in DU145 cells’ mechanical properties treated with cyto D and vinflunine might suggest that both mechanical and biochemical couplings overlay and are linked to specific proteins. One interesting direct cross talk mediating protein in this context is, e.g., plectin, which can bind microtubules and actin.82 Its expression level in prostate cancer cells has been correlated with the metastatic potential.66,80 Furthermore, transient microtubule plus-end capturing by focal adhesions regulates focal adhesion turnover. This capture is mediated by a microtubule stabilisation complex close to the membrane realised by numerous proteins, such as EB-1, CLASPs, KIF21A, MICAL3, ELKS, and LL5β at the microtubule side, linked by liprins to Kank and talin in the focal adhesions. The sequestering of Rho guanine nucleotide exchange factor GEF-H1 in this region regulates RhoA signalling and myosin II-mediated actin filament contraction. In turn, these processes govern focal adhesion maturation, stress fibre formation, and cell migration.83 This leads to the importance of epithelial–mesenchymal transition (EMT)84,85 during cancer cells’ response to antitumor drugs.
Epithelial cells have to undergo EMT during tumour progression, i.e., they must lose many of their epithelial characteristics to acquire the malignant mesenchymal phenotype with front-rear polarity that enables them to disseminate from the primary tumour. EMT requires changes in the cell shape and a vast remodelling of the actin cytoskeleton with the formation of stress fibres and integrin adhesion complexes.84 EMT is involved in developing drug resistance of not only MTDs but also of cisplatin, doxorubicin, EGFR, PI3K inhibitors, or paclitaxel in several cancers.85 In a seminal study by Kajiyama et al., epithelial ovarian carcinoma cells resistant to paclitaxel have been found to exhibit morphological and molecular alterations typical of EMT.86Vice versa, a reversion of EMT by PKA activation rendered immortalised human mammary epithelial cells more sensitive to paclitaxel and doxorubicin.87,88 A recent study demonstrated that high ROCK-myosin II activity and cytoskeletal reorganisation in melanoma cells are essential for the acquisition of resistance towards MAPK inhibitors.75 Also, as recently reported, SMAD3 stimulates the transcription process of STYK1, thereby promoting both the EMT process and paclitaxel resistance of ovarian cancer cells.89 Although not studied here, a similar effect can be expected in DU145 cells treated with VFL, COL, and DTX. The tested MTDs, through their impact on the actin cytoskeleton, have the potential to influence the mentioned microtubule/actin crosstalk and EMT-related processes. Finding the responsible mechanistic link for the effects of the MTDs in prostate cancer cells that we analysed in our work will be a major focus for our future studies.
Conclusions
We have demonstrated that the nanomechanical characterisation of living cells is a powerful tool for monitoring the effect of antitumor drugs. We investigated the elasticity of prostate DU145 cancer cells in combination with optical (fluorescence) microscopy and biochemical tests, assessing the cytoskeletal organisation, as well as the cell viability and proliferation. These cells were incubated for 72 hours with low VFL, DTX, and COL concentrations, representing three classes of MT drugs. Each binds to specific target sites located at the α/β tubulin dimer inducing different cellular responses. The findings show that prostate DU145 cancer cells stiffen in response to the stress induced by the presence of MTDs, particularly to vinca alkaloids. The characteristics of the cell stiffening were dependent on the MTD type, and differed regarding the location within the cell (peripheral or nuclear regions). We hypothesise that the primary mechanism responsible for cell rigidity is associated with the crosstalk between microtubules and actin cytoskeleton. Deciphering the changes in nanomechanical properties as a function of the cellular response to antitumour drugs enables us to understand the role of nanomechanics in triggering mechanisms leading to the development of the cell resistance to the drugs.Author contributions
AK conducted the majority of the experiments, analysed the data, interpreted the data and wrote the manuscript; MC, CS and AP contributed multilayer-based analysis of mechanical properties of cells, conducted biomechanical experiments on cells treated with colchicine, and interpreted obtained results; JW, NB and AK-B recorded fluorescence images using confocal microscopy, created 3D movies based on z-stacks, and contributed to data interpretation; MP and DR performed, analysed and interpreted data from cytometry; ML and PL and AP designed the study, decided on the choice of AFM probes, interpreted the data, wrote the manuscript, and provided resources and funding for the study. All authors contributed to manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AK and NB acknowledge the support of InterDokMed project no. POWR.03.02.00-00-I013/16. AP and ML acknowledge the European Union's support under the Marie Skłodowska-Curie grant agreement No. 812772 (Phys2BioMed). AP and CS acknowledge the European Union's support under the FET Open grant agreement No. 801126 (EDIT). DR and MP acknowledge the Polish National Science Centre's support (grant no. UMO-2015/19/D/NZ3/00273). ML and AK are grateful to Mrs Klaudia Suchy for help in cell cultures and to Dr Justyna Bobrowska to record epi-fluorescence images of the cell nucleus.References
- M. Schoumacher, R. D. Goldman, D. Louvard and D. M. Vignjevic, J. Cell Biol., 2010, 189, 541–556 CrossRef CAS.
- P. Singh, K. Rathinasamy, R. Mohan and D. Panda, IUBMB Life, 2008, 60, 368–375 CrossRef CAS PubMed.
- M. A. Jordan, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2, 1–17 CrossRef CAS PubMed.
- J. A. Yared and K. H. R. Tkaczuk, Drug Des., Dev. Ther., 2012, 6, 371–384 CAS.
- V. M. Malikov and S. Y. Yunusov, Chem. Nat. Compd., 1977, 13, 497–512 CrossRef.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930–933 CrossRef PubMed.
- M. Lekka, P. Laidler, D. Gil, J. Lekki, Z. Stachura and A. Z. Hrynkiewicz, Eur. Biophys. J., 1999, 28, 312–316 CrossRef CAS PubMed.
- W. H. Goldmann and R. M. Ezzell, Exp. Cell Res., 1996, 226, 234–237 CrossRef CAS PubMed.
- M. Lekka, Nat. Nanotechnol., 2012, 7, 691–692 CrossRef CAS PubMed.
- M. E. Grady, R. J. Composto and D. M. Eckmann, J. Mech. Behav. Biomed. Mater., 2016, 61, 197–207 CrossRef CAS.
- J. C. Martens and M. Radmacher, Pflugers Arch.: Eur. J. Physiol., 2008, 456, 95–100 CrossRef CAS PubMed.
- X. P. Wang, T. S. Chen, L. Sun, J. Y. Cai, M. Q. Wu and M. Mok, Micron, 2008, 39, 1216–1221 CrossRef CAS PubMed.
- C. Rotsch and M. Radmacher, Biophys. J., 2000, 78, 520–535 CrossRef CAS PubMed.
- N. P. B. Au, Y. Fang, N. Xi, K. W. C. Lai and C. H. E. Ma, Nanomedicine, 2014, 10, 1323–1333 CrossRef CAS PubMed.
- X. Yun, M. Tang, Z. Yang, J. J. Wilksch, P. Xiu, H. Gao, F. Zhang and H. Wang, RSC Adv., 2017, 7, 43764–43771 RSC.
- K. S. Kim, C. H. Cho, E. K. Park, M. H. Jung, K. S. Yoon and H. K. Park, PLoS One, 2012, 7, e30066 CrossRef CAS PubMed.
- A. Kubiak, T. Zieliński, J. Pabijan and M. Lekka, Int. J. Mol. Sci., 2020, 21, 8786 CrossRef CAS PubMed.
- C. C. K. Lin, C. H. Yang and M. S. Ju, Cancer Chemother. Pharmacol., 2020, 86, 245–255 CrossRef CAS PubMed.
- G. T. Charras and M. A. Horton, Biophys. J., 2002, 82, 2970–2981 CrossRef CAS PubMed.
- N. Shamitko-Klingensmith, J. W. Boyd and J. Legleiter, AIMS Biophys., 2016, 3, 261–285 CAS.
- J. Ren, H. Huang, Y. Liu, X. Zheng and Q. Zou, PLoS One, 2015, 10(5), e0126107 CrossRef.
- D. P. Petrylak, C. M. Tangen, M. H. A. Hussain, P. N. Lara, J. A. Jones, M. E. Taplin, P. A. Burch, D. Berry, C. Moinpour, M. Kohli, M. C. Benson, E. J. Small, D. Raghavan and E. D. Crawford, N. Engl. J. Med., 2004, 351, 1513–1520 CrossRef CAS PubMed.
- S. Ranganathan, C. A. Benetatos, P. J. Colarusso, D. W. Dexter and G. R. Hudes, Br. J. Cancer, 1998, 77, 562–566 CrossRef CAS PubMed.
- P. Barbier, P. O. Tsvetkov, G. Breuzard and F. Devred, Phytochem. Rev., 2014, 13, 157–169 CrossRef CAS.
- J. L. Orgaz and V. Sanz-Moreno, Mol. Cell. Oncol., 2020, 3, e1735911 CrossRef PubMed.
- J. L. Young, X. Hua, H. Somsel, F. Reichart, H. Kessler and J. P. Spatz, Nano Lett., 2020, 20, 1183–1191 CrossRef CAS PubMed.
- M. Indrieri, A. Podesta, C. Bongiorno, D. Marchesi and P. Milani, Rev. Sci. Instrum., 2011, 82, 023708 CrossRef CAS PubMed.
- J. Zemła, J. Bobrowska, A. Kubiak, T. Zieliński, J. Pabijan, K. Pogoda, P. Bobrowski and M. Lekka, Eur. Biophys. J., 2020, 49, 485–495 CrossRef PubMed.
- L. Puricelli, M. Galluzzi, C. Schulte, A. Podestà and P. Milani, Rev. Sci. Instrum., 2015, 86, 033705 CrossRef PubMed.
- H. J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- M. Chighizola, L. Puricelli, L. Bellon and A. Podesta, J. Mol. Recognit., 2020, 34, 2879 Search PubMed.
- H. J. Butt and M. Jaschke, Nanotechnology, 1995, 6, 1–7 CrossRef.
- H. Schillers, C. Rianna, J. Schäpe, T. Luque, H. Doschke, M. Wälte, J. J. Uriarte, N. Campillo, G. P. A. Michanetzis, J. Bobrowska, A. Dumitru, E. T. Herruzo, S. Bovio, P. Parot, M. Galluzzi, A. Podestà, L. Puricelli, S. Scheuring, Y. Missirlis, R. Garcia, M. Odorico, J. M. Teulon, F. Lafont, M. Lekka, F. Rico, A. Rigato, J. L. Pellequer, H. Oberleithner, D. Navajas and M. Radmacher, Sci. Rep., 2017, 7, 5117 CrossRef PubMed.
- I. N. Sneddon, Int. J. Eng. Sci., 1965, 3, 47–57 CrossRef.
- E. K. Dimitriadis, F. Horkay, J. Maresca, B. Kachar and R. S. Chadwick, Biophys. J., 2002, 82, 2798–2810 CrossRef CAS.
- H. Cramér, Mathematical Methods of Statistics (PMS-9), 2016 Search PubMed.
- Z. Holló, L. Homolya, C. W. Davis and B. Sarkadi, Biochim. Biophys. Acta, Biomembr., 1994, 1191, 384–388 CrossRef.
- M. A. Goodell, K. Brose, G. Paradis, A. S. Conner and R. C. Mulligan, J. Exp. Med., 1996, 183, 1797–1806 CrossRef CAS PubMed.
- M. Oropesa Ávila, A. Fernández Vega, J. Garrido Maraver, M. Villanueva Paz, I. De Lavera, M. De La Mata, M. D. Cordero, E. Alcocer Gómez, A. Delgado Pavón, M. Álvarez Córdoba, D. Cotán and J. A. Sánchez-Alcázar, Cytoskeleton, 2015, 72, 435–446 CrossRef.
- J. Zemła, J. Danilkiewicz, B. Orzechowska, J. Pabijan, S. Seweryn and M. Lekka, Semin. Cell Dev. Biol., 2018, 73, 115–124 CrossRef.
- Q. S. Li, G. Y. H. Lee, C. N. Ong and C. T. Lim, Biochem. Biophys. Res. Commun., 2008, 374, 609–613 CrossRef CAS PubMed.
- M. Lekka, J. Pabijan and B. Orzechowska, Biochim. Biophys. Acta, Gen. Subj., 2019, 1863, 1006–1014 CrossRef CAS PubMed.
- Y. M. Efremov, A. A. Dokrunova, A. V. Efremenko, M. P. Kirpichnikov, K. V. Shaitan and O. S. Sokolova, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 3117–3125 CrossRef CAS PubMed.
- K. M. Smurova, A. A. Birukova, A. D. Verin and I. B. Alieva, Biochem. Suppl. Ser. A: Membr. Cell Biol., 2008, 2, 119–127 CrossRef.
- J. A. Cooper, J. Cell Biol., 1987, 105, 1473–1478 CrossRef CAS PubMed.
- S. B. Khatau, D. H. Kim, C. M. Hale, R. J. Bloom and D. Wirtz, Nucleus, 2010, 1, 337–342 CrossRef PubMed.
- D. H. Kim, A. B. Chambliss and D. Wirtz, Soft Matter, 2013, 9, 5516–5523 RSC.
- P. Giannakakou, D. Sackett and T. Fojo, J. Natl. Cancer Inst., 2000, 92, 182–183 CrossRef CAS PubMed.
- M. A. Jordan, R. J. Toso, D. Thrower and L. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9552–9556 CrossRef CAS PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CrossRef CAS PubMed.
- K. E. Gascoigne and S. S. Taylor, Cancer Cell, 2008, 14, 111–122 CrossRef CAS PubMed.
- D. A. Brito and C. L. Rieder, Cell Motil. Cytoskeleton, 2009, 66, 437–447 CrossRef CAS PubMed.
- L. Li, S. Jiang, X. Li, Y. Liu, J. Su and J. Chen, Eur. J. Med. Chem., 2018, 151, 482–494 CrossRef CAS PubMed.
- C. Kanthou and G. M. Tozer, Int. J. Exp. Pathol., 2009, 90, 284–294 CrossRef CAS PubMed.
- K. Jiang and A. Akhmanova, Curr. Opin. Cell Biol., 2011, 23, 94–101 CrossRef CAS PubMed.
- M. P. López, F. Huber, I. Grigoriev, M. O. Steinmetz, A. Akhmanova, G. H. Koenderink and M. Dogterom, Nat. Commun., 2014, 5, 4778 CrossRef PubMed.
- J. Correia and S. Lobert, Curr. Pharm. Des., 2005, 7, 1213–1228 CrossRef PubMed.
- R. J. Toso, M. A. Jordan, K. W. Farrell, B. Matsumoto and L. Wilson, Biochemistry, 1993, 32, 1285–1293 CrossRef CAS PubMed.
- J. J. Manfredi, J. Parness and S. B. Horwitz, J. Cell Biol., 1982, 94, 688–696 CrossRef CAS PubMed.
- R. B. G. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS PubMed.
- B. Bhattacharyya, D. Panda, S. Gupta and M. Banerjee, Med. Res. Rev., 2008, 28, 155–183 CrossRef CAS PubMed.
- M. Luty, K. Piwowarczyk, A. Łabędź-Masłowska, T. Wróbel, M. Szczygieł, J. Catapano, G. Drabik, D. Ryszawy, S. Kędracka-Krok, Z. Madeja, M. Siedlar, M. Elas and J. Czyż, Cancers, 2019, 11, 77 CrossRef CAS PubMed.
- E. Tsakalozou, A. M. Eckman and Y. Bae, Biochem. Res. Int., 2012, 2012, 832059 Search PubMed.
- J. Liu, X. Tan, W. Zhao, J. Liu, X. Xing, G. Fan, P. Zhang, Z. Zhang, Y. Zhong and D. Kong, Curr. Cancer Drug Targets, 2018, 19, 321–329 CrossRef PubMed.
- J. I. Fletcher, M. Haber, M. J. Henderson and M. D. Norris, Nat. Rev. Cancer, 2010, 10, 147–156 CrossRef CAS PubMed.
- T. Hu, Z. Li, C. Y. Gao and C. H. Cho, World J. Gastroenterol., 2016, 22, 6876–6889 CrossRef CAS PubMed.
- M. Dogterom and G. H. Koenderink, Nat. Rev. Mol. Cell Biol., 2019, 20, 38–54 CrossRef CAS PubMed.
- S. Z. Wu and M. Bezanilla, J. Cell Biol., 2018, 217, 3531–3544 CrossRef CAS PubMed.
- T. K. Akhshi, D. Wernike and A. Piekny, Cytoskeleton, 2014, 71, 1–23 CrossRef CAS PubMed.
- E. E. Joo and K. M. Yamada, Bioarchitecture, 2016, 6, 53–59 CrossRef PubMed.
- O. C. Rodriguez, A. W. Schaefer, C. A. Mandato, P. Forscher, W. M. Bement and C. M. Waterman-Storer, Nat. Cell Biol., 2003, 5, 599–609 CrossRef CAS PubMed.
- S. Etienne-Manneville, Traffic, 2004, 5, 470–477 CrossRef CAS PubMed.
- J. C. M. Meiring, B. I. Shneyer and A. Akhmanova, Curr. Opin. Cell Biol., 2020, 62, 86–95 CrossRef CAS PubMed.
- J. L. Orgaz, E. Crosas-Molist, A. Sadok, A. Perdrix-Rosell, O. Maiques, I. Rodriguez-Hernandez, J. Monger, S. Mele, M. Georgouli, V. Bridgeman, P. Karagiannis, R. Lee, P. Pandya, L. Boehme, F. Wallberg, C. Tape, S. N. Karagiannis, I. Malanchi and V. Sanz-Moreno, Cancer Cell, 2020, 37, 85–103 CrossRef CAS PubMed.
- T. Enomoto, Cell Struct. Funct., 1996, 21, 317–326 CrossRef CAS PubMed.
- A. Graness, K. Giehl and M. Goppelt-Struebe, Cell. Signalling, 2006, 18, 433–440 CrossRef CAS PubMed.
- J. N. Heck, S. M. Ponik, M. G. Garcia-Mendoza, C. A. Pehlke, D. R. Inman, K. W. Eliceiri and P. J. Keely, Mol. Biol. Cell, 2012, 23, 2583–2592 CrossRef CAS PubMed.
- M. Krendel, F. T. Zenke and G. M. Bokoch, Nat. Cell Biol., 2002, 4, 294–301 CrossRef CAS PubMed.
- D. E. Ingber, J. Cell Sci., 2003, 116, 1397 CrossRef CAS PubMed.
- Q. Zhang, M. K. Magnusson and D. F. Mosher, Mol. Biol. Cell, 1997, 8, 1415–1425 CrossRef CAS PubMed.
- T. C. Burch, M. T. Watson and J. O. Nyalwidhe, PLoS One, 2013, 8(5), e65005 CrossRef PubMed.
- C. Garcin and A. Straube, Essays Biochem., 2019, 63(5), 509–520 CrossRef CAS PubMed.
- J. P. Thiery, Nat. Rev. Cancer, 2002, 2, 442–454 CrossRef CAS PubMed.
- R. Kalluri and R. A. Weinberg, J. Clin. Invest., 2009, 119, 1420–1428 CrossRef CAS PubMed.
- H. Kajiyama, K. Shibata, M. Terauchi, M. Yamashita, K. Ino, A. Nawa and F. Kikkawa, Int. J. Oncol., 2007, 31, 227–283 Search PubMed.
- T. Shibue and R. A. Weinberg, Nat. Rev. Clin. Oncol., 2017, 14, 611–629 CrossRef PubMed.
- D. R. Pattabiraman, B. Bierie, K. I. Kober, P. Thiru, J. A. Krall, C. Zill, F. Reinhardt, W. L. Tam and R. A. Weinberg, Science, 2016, 351, aad3680 CrossRef PubMed.
- Y. Shi, J. Zhang, M. Liu, Y. Huang and L. Yin, J. Cell Biochem., 2019, 120, 10796–10811 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr06464e |
| This journal is © The Royal Society of Chemistry 2021 |