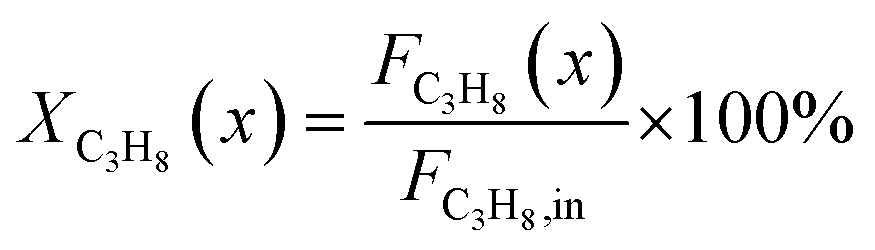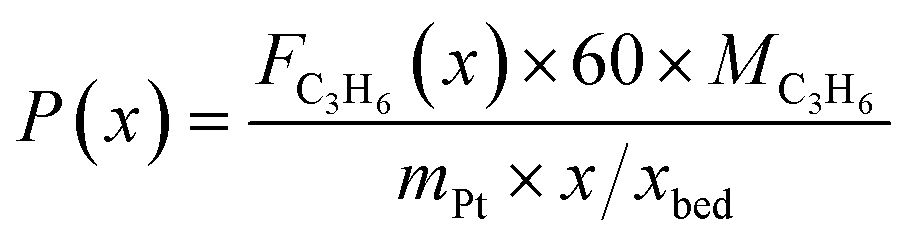DOI:
10.1039/D0FD00010H
(Paper)
Faraday Discuss., 2021,
229, 359-377
Capturing spatially resolved kinetic data and coking of Ga–Pt supported catalytically active liquid metal solutions during propane dehydrogenation in situ†
Received
14th January 2020
, Accepted 12th February 2020
First published on 16th March 2020
Abstract
Supported liquid phase catalysis has great potential to unify the advantages from both homogeneous and heterogeneous catalysis. Recently, we reported supported catalytically active liquid metal solutions (SCALMS) as a new class of liquid phase catalysts. SCALMS enable high temperature application due to the high thermal stability of liquid metals when compared to supported molten salts or ionic liquids. The highly dynamic liquid metal/gas interface of SCALMS allows for catalysis over single atoms of an active metal atom within a matrix of liquid gallium. In the present study, kinetic data is acquired along the catalyst bed in a compact profile reactor during propane dehydrogenation (PDH) over gallium–platinum SCALMS. The reactor design allows for the analysis of the temperature and gas phase composition along the catalyst bed with a high spatial resolution using a sampling capillary inside the reactor. The concentration profiles suggest enhanced deactivation of the catalyst at the end of the bed with a deactivation front moving from the end to the beginning of the catalyst bed over time on stream. Only minor amounts of side products, formed via cracking of propane, were identified, supporting previously reported high selectivity of SCALMS during alkane dehydrogenation. The acquired data is supported by in situ high-resolution thermogravimetry coupled with mass spectrometry to monitor the activity and coking behaviour of SCALMS during PDH. The results strongly suggest an enhanced formation of coke over Al2O3-supported SCALMS when compared to using SiO2 as the support material.
Introduction
Industrial application of Pt-based catalysts for technical propane dehydrogenation (PDH; eqn (1)) requires frequent regeneration of the catalysts due to strong deactivation. The deposition of carbonaceous species is generally accepted to be the culprit for the rapid decay of the catalyst activity.1–4 Coke formation is mostly caused by overcracking of hydrocarbons.5–8 Even though regeneration of the catalyst via oxidative treatments may be realised within short time frames,3 the associated downtime of the reactor drastically reduces the process efficiency due to increased capital and operational expenditures. Hence, research on catalyst design for effective PDH focuses on high resistance against carbon deposition.1,2,9–11
We have recently proposed supported catalytically active liquid metal solutions (SCALMS) as a novel supported liquid phase catalysis concept.12 Application of classical SLP catalysts with organic liquids, ionic liquids, or molten salts as the liquid phase on porous supports, is typically restricted to relatively low temperature applications (≤300 °C) due to the limited thermal stability of the applied liquid phases.13–16 Conversely, SCALMS employ liquid metals allowing for high temperature applications, because virtually no decomposition occurs for a solution of elementary metals.10–12 While other concepts for catalysis over liquid metals require large volumes of liquid metal in a reactor,17,18 SCALMS materials are composed of dispersed supported droplets of a liquid alloy consisting of a catalytically active metal and an excess of a low melting point metal, e.g. Ga.10–12,19 The catalytic reaction in SCALMS occurs exclusively at the liquid metal/gas interface.10,12,19–25 Contrary to conventional SLP catalysis, the reactants and products are insoluble in the liquid phase. In addition, the liquid metal/gas interface is highly dynamic on an atomic scale. The topmost layer of the Ga-rich alloy droplets is depleted of active metal atoms in the absence of substrates.10,12,20,21 Nevertheless, the active metal atoms may diffuse to the surface of the droplets and interact with substrates via adsorption, which is a prerequisite for the catalytic activity of SCALMS. In the case of PDH over Ga–Rh SCALMS, ab initio molecular dynamics simulations suggest that the presence of a propane molecule at the liquid metal/gas interface may trigger the diffusion of Rh atoms to the surface of the liquid alloy droplet. Adsorption of the substrate then retains the Rh atom at the surface and is followed by C–H bond breakage resulting in two hydrogen atoms and propylene bound to a single Rh atom. Subsequently, diffusion of propylene to adjacent Ga atoms is suggested by the simulations. Lastly, propylene desorbs and the two hydrogen atoms bound to Rh combine and desorb as H2, while the Rh immediately moves away from the surface of the droplet into the Ga matrix.10 Hence, dehydrogenation of propane over SCALMS is suggested to require only a single active metal atom in a Ga matrix, which is in line with Biloen et al.26 Consequently, side reactions, which require a second vicinal active site, may be suppressed during PDH over SCALMS.
Herein, we employ Ga–Pt SCALMS during PDH using Al2O3 and SiO2 as carrier materials. We recently hypothesised that the coke formation during PDH over a related catalyst, namely GaRh/Al2O3 SCALMS, at increased reaction temperatures of 550 °C is governed by the acidic sites of the support material.11 Hence, the coking behaviour of both Ga–Pt SCALMS catalysts was compared by means of in situ high-resolution thermogravimetric analysis coupled with mass spectrometry (HRTGA-MS). Further, spatially resolved kinetic data was acquired along the catalyst bed in a compact profile reactor (CPR), which may provide the foundation for a comprehensive kinetic analysis of the activity and deactivation mechanisms of SCALMS during PDH.
Experimental section
Preparation of supported catalytically active liquid metal solutions (SCALMS)
At first, (Et3N)GaH3 was synthesised according to the published procedure,27 while triethylammonium chloride (Sigma-Aldrich) was used instead of trimethylammonium chloride. The compound was not isolated and used as ethereal solution. The Ga content of this solution was determined as follows: 1 mL of ethereal solution was quenched in 10 mL of HCl (∼2 M) and the resulting mixture shortly boiled to evaporate the diethyl ether. The homogeneous solution was diluted to 500 mL and the Ga content determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES). Given the low stability of gallane complexes, the (Et3N)GaH3 solution was analysed and used immediately after its preparation. The second part of the synthesis procedure includes the decoration of the support material with Ga according to a previously published procedure.10,12 Either 20 g of SiO2 (Sigma-Aldrich; particle size: 200–500 μm; BET surface area: 500 m2 g−1) or 100 g of Al2O3 (Sigma-Aldrich; grade: Brockmann I, activated, standard; particle size: 50–150 μm; BET surface area: 155 m2 g−1; pH: 7.0 ± 0.5) were employed as carrier materials and dried by heating under vacuum (1 mbar) at 350 °C overnight in a Schlenk flask. After cooling-down, the support was suspended in dry diethyl ether (50 mL for SiO2 and 200 mL for alumina) under an inert argon atmosphere. The amount of an ethereal solution of (Et3N)GaH3 added to this suspension was that required in order to obtain the targeted loading of Ga (ca. 5–6 wt% of Ga) with respect to the support. After complete removal of diethyl ether under vacuum at ca. −30 °C, the flask was heated up to 300 °C (ca. 10 °C min−1) until no gaseous products were observed indicating the termination of the gallane decomposition. The resulting grey solid was further heated to 300 °C under vacuum (1 mbar) overnight and stored under Ar after cooling-down to room temperature. In the last step of the synthesis of Ga–Pt SCALMS, 10 g of Ga decorated material were suspended in 50 mL of isopropanol in a 100 mL round flask under stirring. The required volume of a stock solution of H2PtCl6 (nominal Pt concentration of 3.6 mg mL−1) in distilled water was added to this solution to obtain the targeted atomic Ga/Pt ratio. The flask was connected to a rotary evaporator and the solvent evaporated at 50 °C and 25 mbar. The resulting solid was calcined at 500 °C for 120 min.
Metal content analysis
The Ga and Pt loading and the corresponding Ga/Pt ratios of the SCALMS were determined by ICP-AES using a Ciros CCD (Spectro Analytical Instruments GmbH). The solid samples were digested in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 volumetric ratio of concentrated HCl:HNO3:HF using microwave heating up to 220 °C for 40 min. The instrument was calibrated for Pt (214.423 nm) and Ga (417.206 nm) with standard solutions of the particular elements prior to the analyses.
:1:1 volumetric ratio of concentrated HCl:HNO3:HF using microwave heating up to 220 °C for 40 min. The instrument was calibrated for Pt (214.423 nm) and Ga (417.206 nm) with standard solutions of the particular elements prior to the analyses.
High-resolution thermogravimetric analysis coupled with mass spectrometry (HRTGA-MS)
PDH over SCALMS was also characterised by means of in situ HRTGA-MS using a XEMIS sorption analyser (Hiden Isochema).28 The XEMIS sorption analyser has a superior resolution of ±0.1 μg when compared to classical thermogravimetric analysers and can be operated under a vacuum or pressures up to 200 bar and temperatures up to 500 °C. A total of 200 mg of as prepared GaPt/Al2O3 or GaPt/SiO2 SCALMS was loaded and dried under a flow of He (200 mLN min−1; 500 °C for 6 h with a heating ramp of 5 °C min−1). Desorption of physisorbed and chemisorbed water during in situ measurements would deter the measurement of the net weight change making this first step inevitable. Subsequently, PDH was conducted at 450 °C or 500 °C for 6 h using a feed gas composition of 10% C3H8/He (overall flow rate: 200 mLN min−1). The last experimental step during analysis by HRTGA-MS was temperature programmed oxidation (TPO) in 21% O2/He (100 mLN min−1), which was introduced immediately after catalytic application and cooling-down to 100 °C under He. TPO was conducted after an initial isotherm at 100 °C (0.5 h) during a ramp to 500 °C at 1 °C min−1 with a subsequent isotherm at 500 °C (12 h). A mass spectrometer (Hiden Analytical) continuously analysed the off-gas in the mass-to-charge (m/z) range of 1–50.
Propane dehydrogenation (PDH) in a compact profile reactor (CPR)
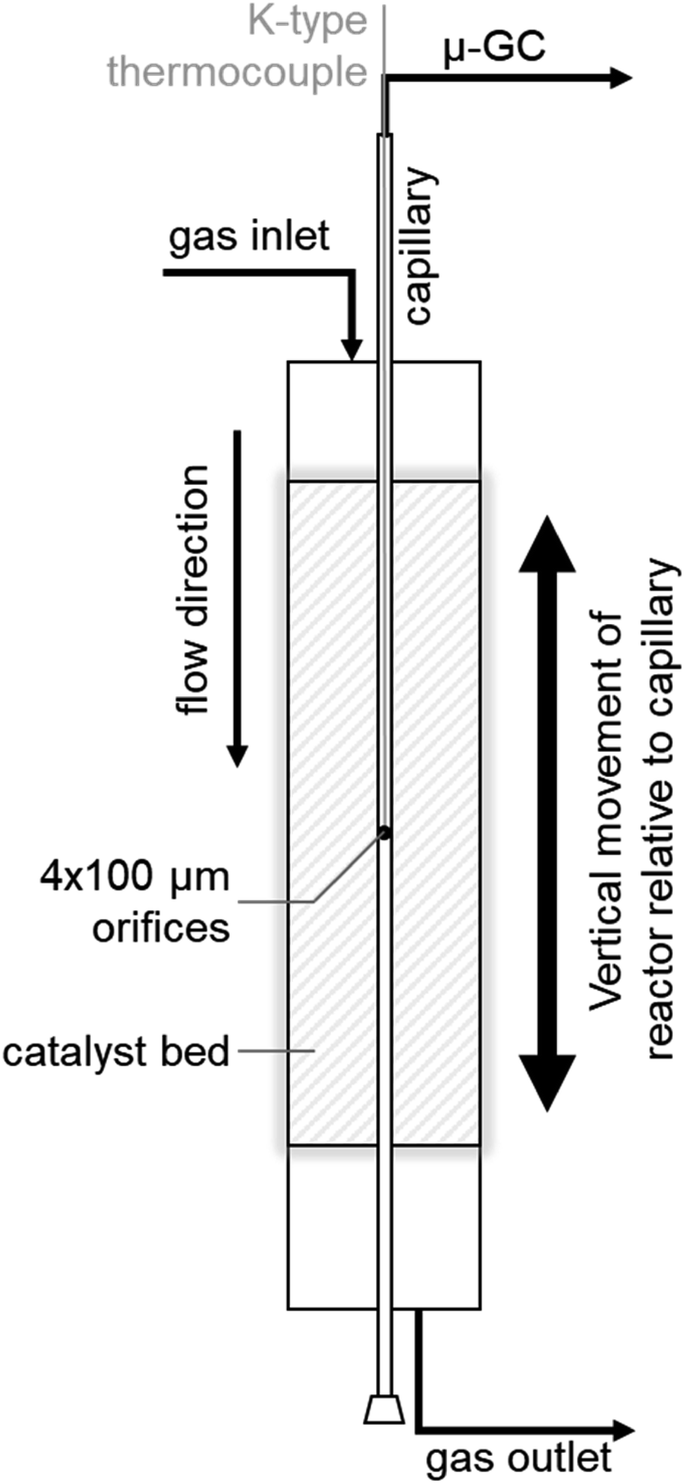
SCALMS were applied in PDH using a CPR (Reacnostics, Germany). The catalyst was placed in-between two quartz wool plugs inside a quartz tube reactor (length: 180 mm; outer diameter: 6 mm; inner diameter: 4 mm). A stainless steel capillary with four orifices (diameter: 100 μm) at a defined position is placed through the catalyst bed inside the quartz tube (Fig. 1). The reactor can be heated up to 550 °C and is mounted on a sledge allowing for movement of the reactor in the axial direction along the capillary. This relative movement enables spatially resolved sampling of the gas phase via the orifices, probing of the temperature inside the catalyst bed using a thermocouple inside the capillary (Fig. 1), as well as spatially resolved Raman spectroscopy via an external optical window. The latter was realised using a 532 nm solid state laser Raman set-up (Avantes: AvaRaman-532HERO-EVO, AvaSpec-HERO, AvaRaman-PRB-532) with a laser power of 15 mW, an exposure time of 30 s and averaging of 5 repetitions. A total of 333.3 mg of catalyst was loaded into the fixed-bed reactor. The resulting catalyst bed of 55 mm lies within the isothermal zone of the heating chamber (≥60 mm). The catalyst was heated to the desired temperature (450, 500, or 550 °C) at 10 °C min−1 under a flow of He (Air Liquide, purity 4.6). After 35 min at the final temperature, the feed stream was changed to 12% C3H8 (Air Liquide, purity 3.5) in He for PDH. The total gas hourly space velocity (WHSV) of 4500 mLN mLcat−1 h−1 was constant throughout the experiments. The reaction gas mixture was actively sampled through the orifices of the capillary by the pump of an I-GRAPHX PR micro gas chromatograph (I-GraphX GmbH, Germany).
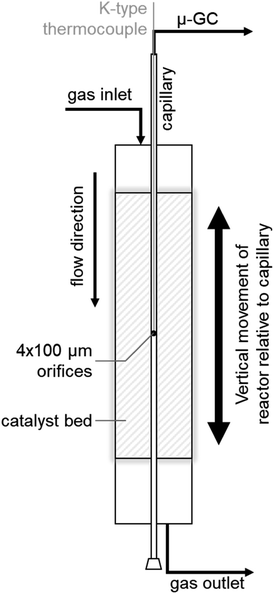 |
| | Fig. 1 Graphical depiction of the compact profile reactor (CPR) allowing for spatially resolved concentration profiles along the catalyst bed inside a quartz tube reactor. | |
Results and discussion
Elemental analyses of the as prepared SCALMS samples by means of ICP-AES resulted in a Ga loading of 5.80 and 4.14 wt% on SiO2 and Al2O3, respectively. Together with a Pt loading of 0.29 and 0.14 wt%, molar ratios of Ga/Pt of 55 and 86 are obtained for SiO2 and Al2O3, respectively. Both Ga-rich alloy compositions are expected to be fully liquid at temperatures exceeding 200 °C.29
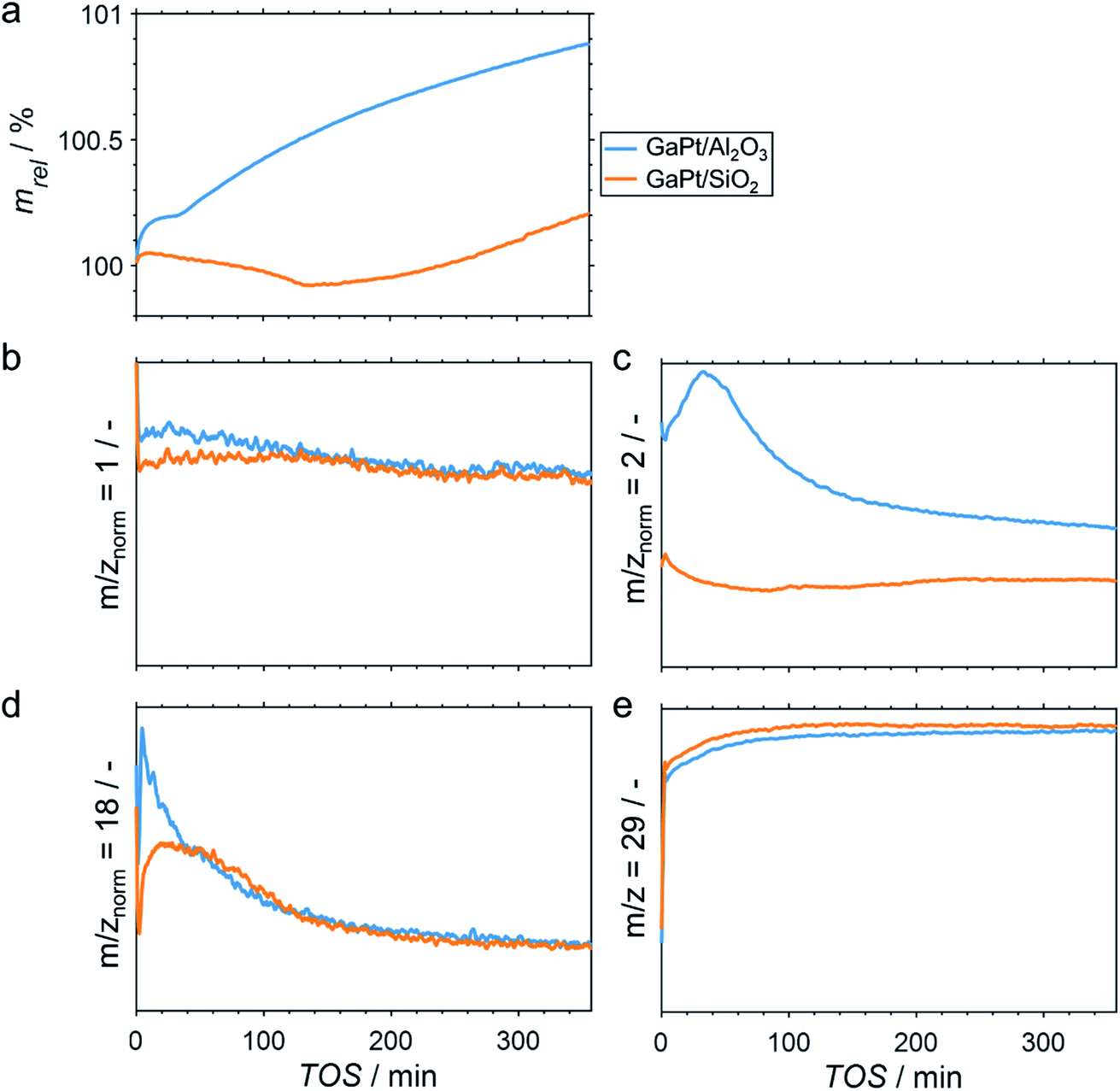
The performance of GaPt/SiO2 and GaPt/Al2O3 SCALMS during PDH at 450 and 500 °C was qualitatively evaluated by means of in situ HRTGA-MS using a XEMIS sorption analyser. We recently hypothesised that the coking behaviour of the catalysts may be dominated by the applied carrier materials.11 Coking can be monitored using in situ HRTGA-MS as well, but at least three processes may affect the sample weight during PDH over SCALMS: gas–metal interaction such as adsorption, formation of carbonaceous deposit, and reduction of oxidic gallium species (GaOx) by the dehydrogenation product H2.11 The latter species are present in the as-prepared SCALMS due to the passivation of metallic Ga during the synthesis of SCALMS or the subsequent exposure to air. Abstraction of oxygen from these GaxO species is enhanced by the presence of Pt atoms10,30,31 and consequently decreases the sample weight. The activity of the catalysts during PDH was qualitatively monitored via mass spectrometry (MS). While the fragmentation patterns of propane and propylene show high similarities, the mass-to-charge ratios (m/z) of 1 and 2 can be assigned to propylene (Fig. S1†). During PDH, H2 is formed at an equimolar ratio together with propylene (eqn (1)) and has a parent ion peak at m/z = 2, which will dominate this mass-to-charge ratio during PDH. Equal signal strengths are expected for the fragments of H2 and propylene for m/z = 1 (Fig. S1†). Hence, m/z = 2 resembles the formation of H2 from propane, while m/z = 1 describes the formation of propylene and H2. Normalisation of the ion signal strengths to the parent ion peak of propane (m/z = 29) isolates the signal from flow effects due to the large dead volume of the HRTGA-MS.
During PDH at 500 °C, the sample weight of the SCALMS increases upon first exposure of the pre-dried sample to 10% propane in He (Fig. 2a). The weight increase is due to the first interaction of propane with the catalytically active metal atoms of the alloy at the liquid–gas interface of SCALMS.10–12 Adsorbed propane may not only be dehydrogenated to propylene and H2, but also result in initial coke formation contributing to this first increase of the sample weight. The weight increase is more pronounced for the GaPt/Al2O3 SCALMS than for the GaPt/SiO2 SCALMS (0.19 vs. 0.04%). As the former catalyst contains lower quantities of Pt when compared to the SiO2-supported SCALMS (0.14 vs. 0.29 wt%), this may be a first indication of enhanced coking over GaPt/Al2O3. Within the first 30 min time on stream (TOS), the sample weight stabilises (GaPt/Al2O3) or even decreases (GaPt/SiO2) due to the in situ reduction of GaOx species by the dehydrogenation product H2.11 This reduction is also evidenced when analysing the off-gas by means of MS. The simultaneous formation of H2O (m/z = 18) and a consumption of in situ formed H2 (m/z = 2) is identified for both SCALMS (Fig. 2c and d). A continuous formation of H2O with an exponential decay over TOS is evidenced for the GaPt/Al2O3 sample. The formation of H2 reaches a maximum after 30 min TOS, which may indicate initial consumption of H2 during an almost spontaneous reduction of GaOx species. Contrarily, the formation of H2O peaks after 20 min TOS and the formation of H2 decreases for the SiO2-supported catalyst suggesting an initially hindered reduction of GaOx species.
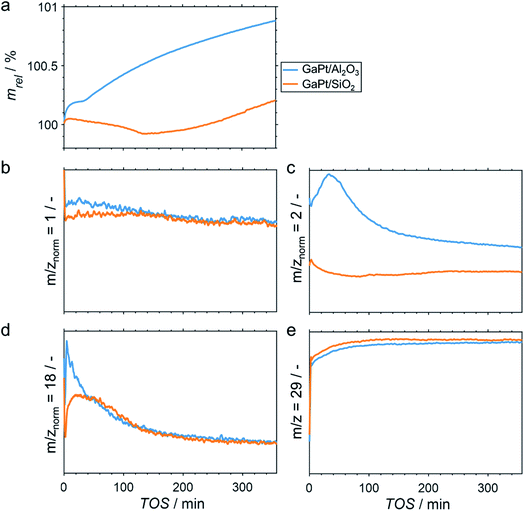 |
| | Fig. 2 (a) Sample weight relative to the weight of the dried SCALMS during PDH over GaPt/SiO2 and GaPt/Al2O3 SCALMS with atomic Ga/Pt ratios of 55 and 86, respectively, at 500 °C; (b–d) mass-to-charge ratios of 1 (propylene and H2), 2 (almost exclusively H2), and 18 (exclusively H2O) relative to the mass-to-charge ratio of 29 (exclusively propane); and (e) mass-to-charge ratio of 29 (exclusively propane) as monitored via in situ high-resolution thermogravimetry coupled with mass spectrometry. Conditions of the experiment: 180 mLN min−1 He; 20 mLN min−1 C3H8; WHSV 60000 mLN g−1 h−1. | |
The net weight change of the GaPt/Al2O3 SCALMS is dominated by the formation of coke after 30 min TOS (Fig. 2a) even though the reduction of the GaOx species is not completed (Fig. 2d). The formation of coke seemingly decelerates with extended TOS, which is in contrast to previously observed constant coke formation over a GaRh/Al2O3 SCALMS.11 When using SiO2 as a support material, the weight only increases after 140 min TOS indicating a higher resistance of this SCALMS against coking. A linear weight increase after 100 min TOS may be exclusively due to the continuous deposition of coke. Similar results were obtained when analysing PDH over the same SCALMS materials in situ at 450 °C (Fig. S2†). However, the duration of the reduction of GaOx species was extended due to the reduced reaction temperature. In general, 450 °C is within a rather moderate temperature range for PDH. Coke formation dominates the weight increase after 180 min TOS for the GaPt/Al2O3 sample, while the weight decreases throughout PDH for the GaPt/SiO2 catalyst suggesting low or zero coke formation.
The performance of SCALMS was analysed by MS. The conversion of propane to the desired dehydrogenation product propylene (m/z = 1 normalised to the parent ion peak of propane m/z = 29) decreases continuously upon first exposure to propane (Fig. 2b). Initial deactivation of catalysts during PDH is well established in the literature1–3 and has recently been reported for GaRh/Al2O3 SCALMS.10,11 Blockage of active sites by carbonaceous deposits may be at play, which may (in part) lead to the initial weight increase of the SCALMS (Fig. 2a). However, the herein observed deactivation is less pronounced, which can be assigned to the low conversion during HRTGA-MS. Both SCALMS materials display a relatively stable performance even though a continuous build-up of coke is indicated by the weight increase for the GaPt/Al2O3 catalyst. Comparison of m/z = 1 and 2 allows for analysis of the ratio of formed propylene and H2, which theoretically forms in an equimolar ratio during PDH (eqn (1)). The profile of H2 formation (m/z = 2) differs from the one of propylene and H2 (m/z = 1; Fig. 2c), which is in part due to the aforementioned reduction of GaOx species consuming H2.11 Further, H2 is a side product of coking. Significantly enhanced formation of H2 over GaPt/Al2O3 strongly suggests enhanced coking when compared to the GaPt/SiO2 sample (Fig. 2c), which is in line with the observed weight increase (Fig. 2a).
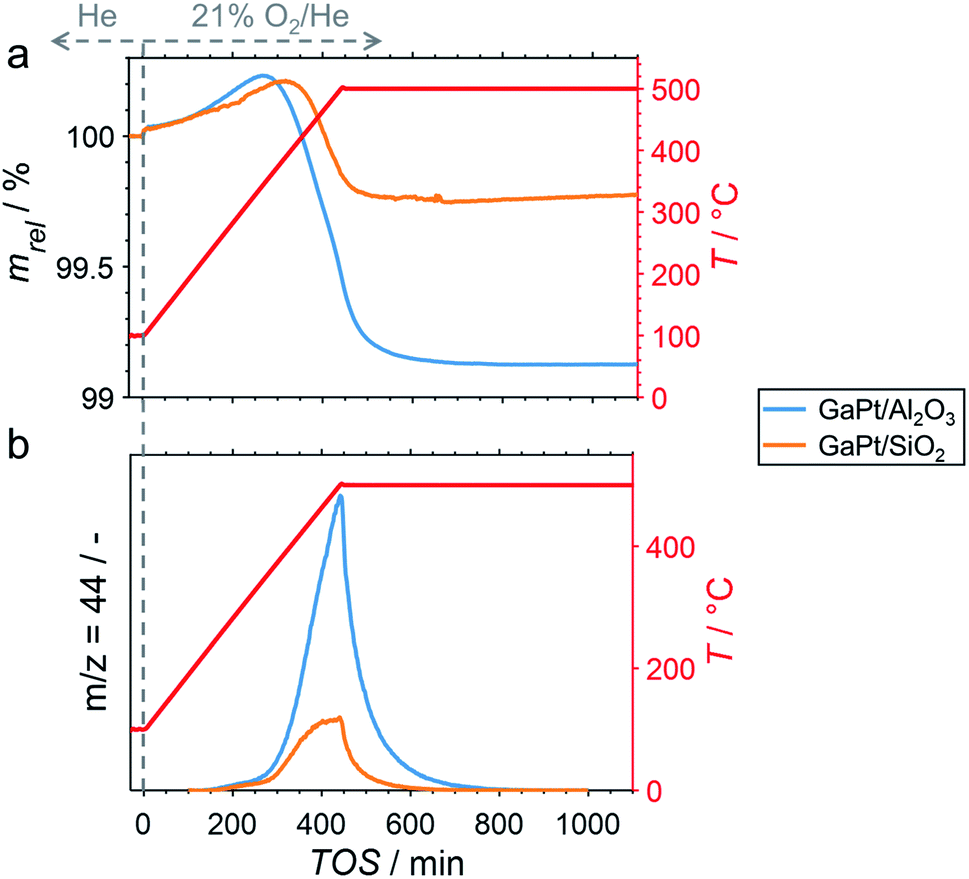
After cooling-down the reactor to 100 °C under a continuous flow of He, the potential formation of coke during PDH was analysed by TPO in 21% O2/He. The weight increase upon first exposure to the oxidative atmosphere is mostly due to adsorption of O2 to form GaOx species (Fig. 3a).11 In addition, the increase may in part originate from the formation of oxygen-containing functionalities (C(Ox)) on the surface of coke.11,32–36 The temperature increase to 500 °C at 1 °C min−1 results in an enhanced weight increase of the spent catalysts (Fig. 3a), which is followed by decomposition of C(Ox) functionalities. This volatilisation of coke results in a net decrease of weight and can be monitored via the formation of CO2 by means of MS (Fig. 3b). As expected, the GaPt/SiO2 sample displayed a less pronounced weight loss of 0.25%, while the sample weight of the GaPt/Al2O3 catalyst after PDH at 500 °C decreased by 0.89%. The same trend can be observed for the formation of CO2. Hence, the formation of coke over Al2O3-supported SCALMS is clearly enhanced when compared to SiO2 as the carrier material. Once again, similar results were obtained during TPO of SCALMS after PDH at 450 °C (Fig. S3†). The major reason for this pronounced affinity towards coking may be the increased concentration of acidic sites in the Al2O3 material. It is well established in the literature, that such acidic sites promote cracking of hydrocarbons and the formation coke.4–8
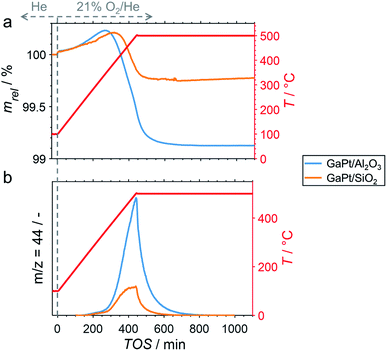 |
| | Fig. 3 (a) Sample weight relative to the weight prior to exposure to 21% O2/He at 100 °C, and (b) formation of CO2 during TPO (1 °C min−1) of spent GaPt/SiO2 and GaPt/Al2O3 SCALMS with atomic Ga/Rh ratios of 55 and 86, respectively, after PDH at 500 °C for 24 h as monitored via in situ high-resolution thermogravimetry coupled with mass spectrometry. Conditions of the experiment: 100 mLN min−1 He (TOS < 0); 79 mLN min−1 He and 21 mLN min−1 O2 (TOS > 0); WHSV 30000 mLN g−1 h−1. | |
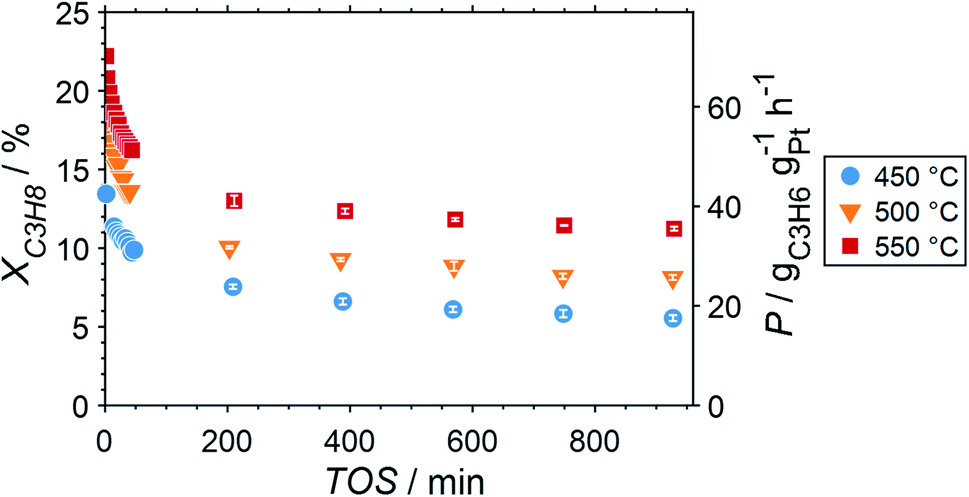
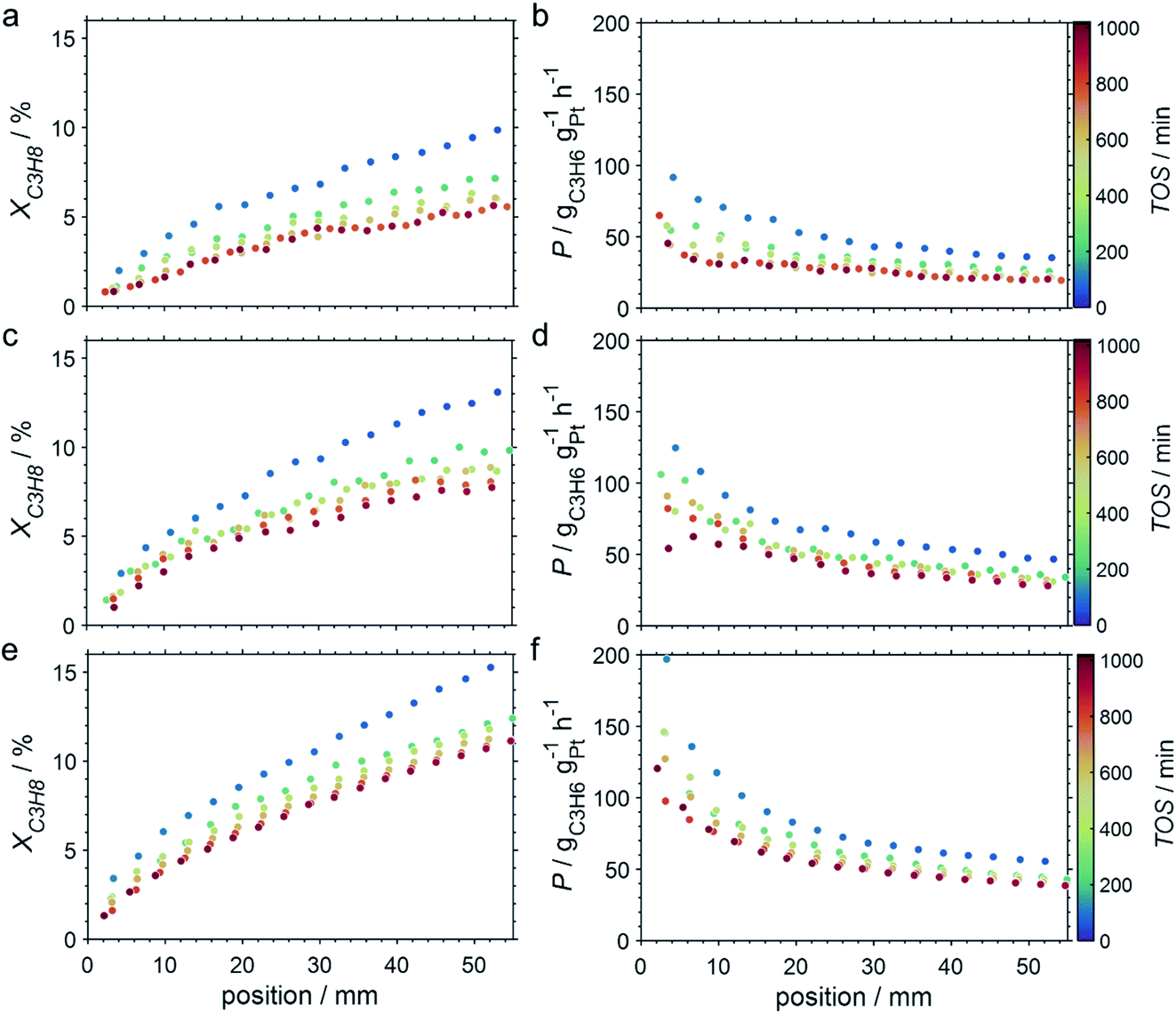
Due to the lower affinity towards the formation of coke during PDH, the performance of the GaPt/SiO2 catalyst with an atomic Ga/Pt ratio of 55 was evaluated using the CPR (Fig. 1) at reaction temperatures of 450, 500, and 550 °C. The initial conversion of propane at the end of the catalyst bed (length: 55 mm) increases with temperature from ∼13 to ∼22% (Fig. 4). The initial conversion levels at given conditions in the CPR are in the range of the thermodynamic equilibrium conversion for pure propane,37,38 but the heavy dilution of the feed stream with inert gas shifts the thermodynamic equilibrium in the present study to higher conversion levels. A rapid decay of the initial high activity is observed for all reaction temperatures. The initial activity of Ga–Pt SCALMS reduces between 41–45% within a start-up time of 200 min. The final activity after >900 min TOS is reduced by 59, 55, and 49% of the initial activity for a reaction temperature of 450, 500, and 550 °C, respectively. Hence, the SCALMS may be operated at a relatively stable operation point after the initial start-up period. Such deactivation behaviour of catalysts during PDH has often been described in the literature.1–3 The herein conducted in situ HRTGA-MS measurements suggest a moderate deposition of coke during PDH over GaPt/SiO2 SCALMS at 450–500 °C (Fig. 2). However, even small amounts of monoatomic carbon may result in strong deactivation. Al2O3-supported SCALMS was shown to deactivate with a similar profile10,11 and has been demonstrated to be prone to coking due to the increased acidity of the support material (see above). On the other hand, structural reorganisation with consequent morphology change of the liquid metal droplets may be another, potentially major, culprit for the observed deactivation of the SCALMS samples.10 However, SEM imaging of such materials has not yet produced conclusive results,10,12i.e., the exact mechanisms at play are still under investigation. Finally, a contribution of Pt–GaOx species, a known catalyst for PDH,39,40 may also explain the higher initial activity. The reduction of said species by in situ formed H2 to the metallic state upon exposure to propane has been observed by means of in situ HRTGA-MS measurements (Fig. 2).11 Apparently, this process transforms the initial SCALMS material into less active species and consequentially exacerbates the activity of the catalysts. Nevertheless, the rather stable operation after the initial start-up period results in productivity (P) values that are comparable with the literature for Pt catalysed PDH (Fig. 4).41–45
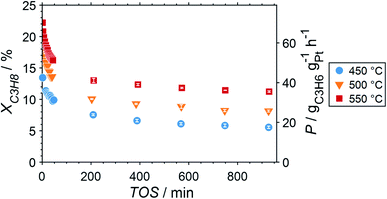 |
| | Fig. 4 Conversion of propane and productivity at the end of the catalyst bed during PDH over GaPt/SiO2 SCALMS with an atomic ratio of Ga/Pt of 55 at 450–550 °C. The error bars represent the standard deviation of 6 consecutive measurements in-between profile acquisitions. Conditions of the experiment: 22 mLN min−1 He; 3 mLN min−1 C3H8; WHSV 4500 mLN g−1 h−1. | |
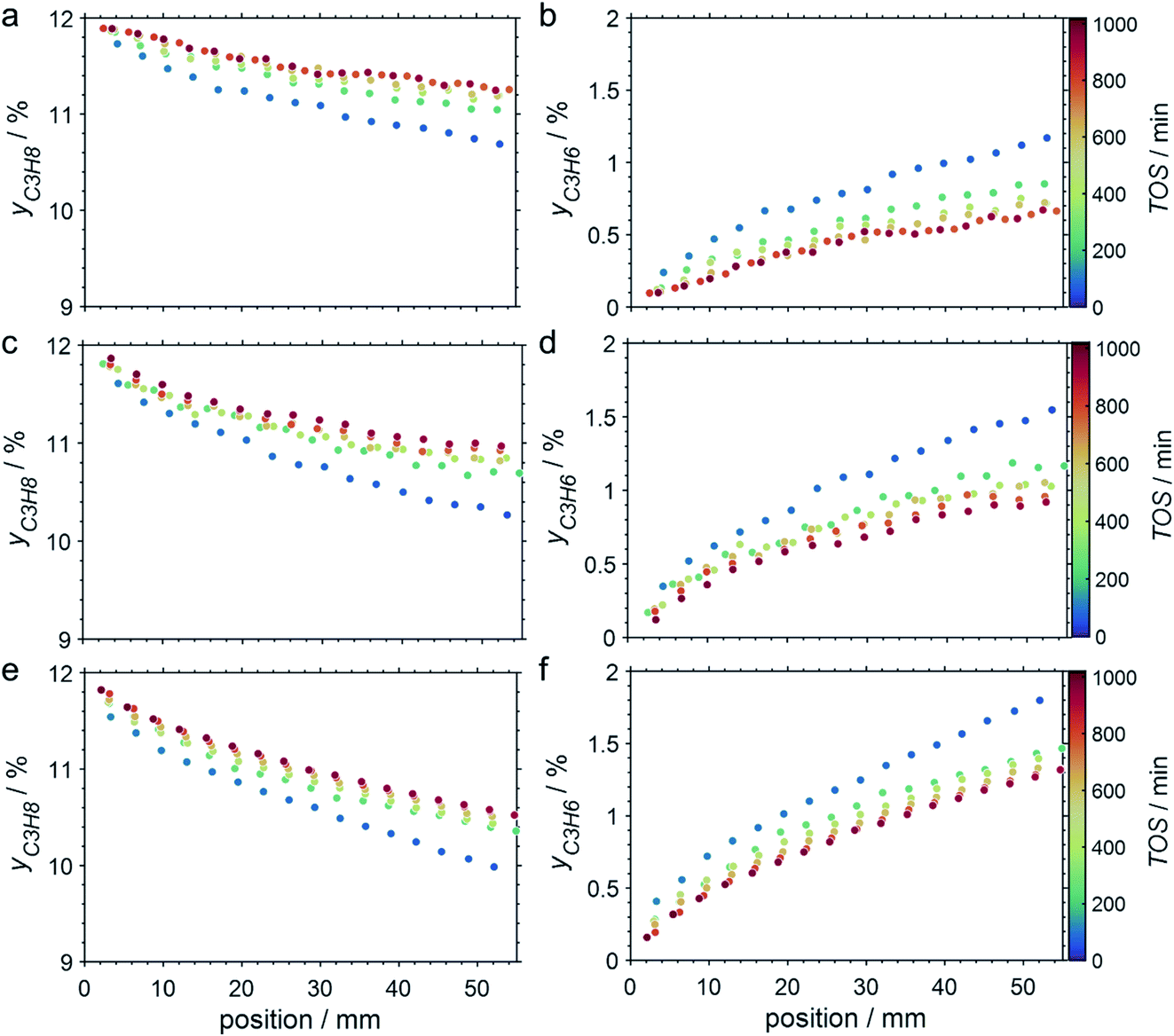
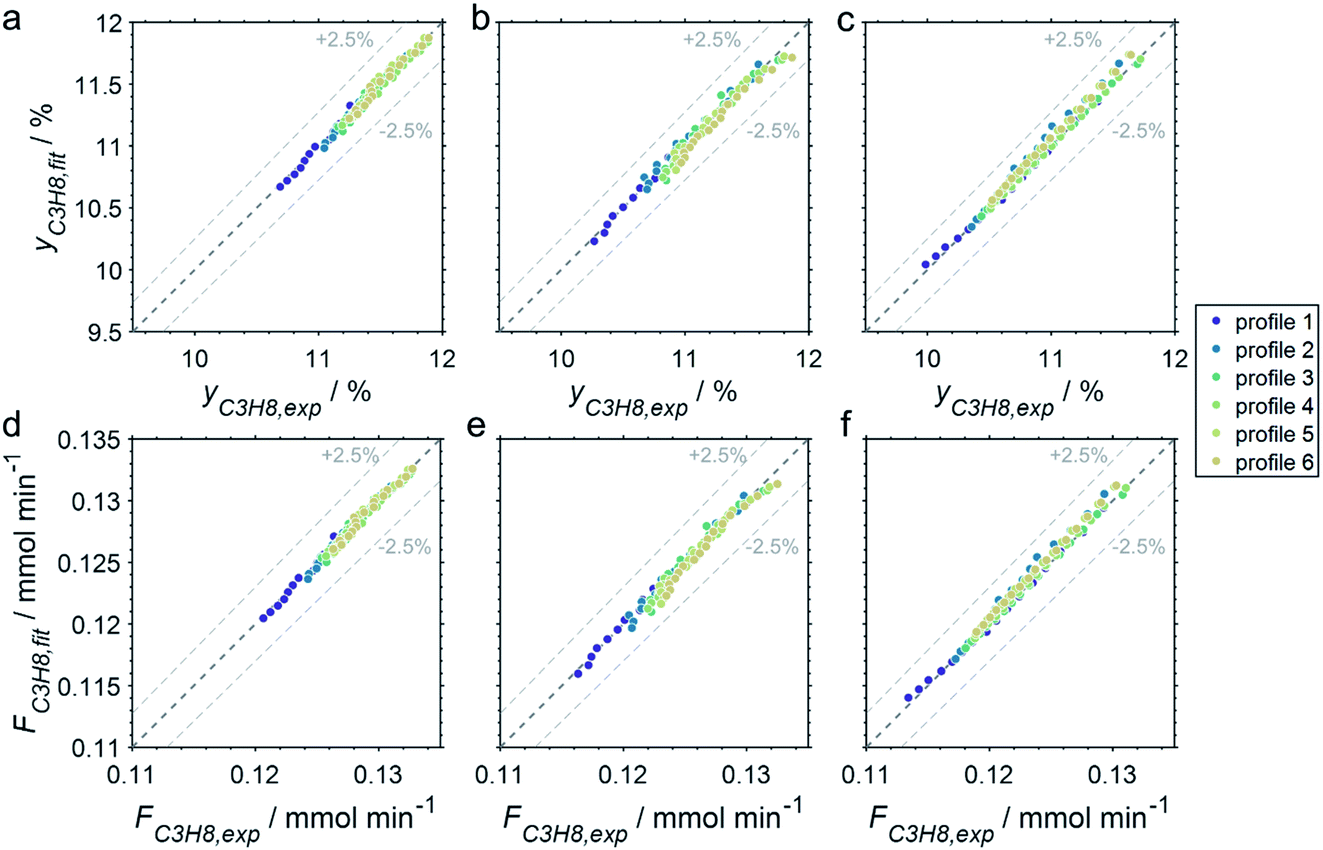
The CPR (Fig. 1) allows for analysis of spatially resolved concentration profiles during PDH over the applied novel GaPt/SiO2 SCALMS. Only small amounts of catalyst are required to obtain valuable and meaningful kinetic information on activity and deactivation alike. Herein, only 333.3 mg of catalyst were loaded for each reaction temperature (450, 500, and 550 °C), which remains the only parameter to be varied to generate valuable kinetic data. As expected, a decreasing gas phase concentration of propane (Fig. 5a, c and e) and an increasing concentration of propylene (Fig. 5b, d and f) are monitored along the catalyst bed at all reaction temperatures and throughout the experiments. The profiles of the gas fraction of propane (yC3H8) and the molar flow of propane (FC3H8) can be mathematically described by a simple power function (eqn (2) and (3)) for all profiles at all three reaction temperatures (Fig. 6). This behaviour exemplarily demonstrates the high consistency and significance of the data acquired in the CPR.
| | | yC3H8(x) = 12% − a × xb%1−b | (2) |
| | | FC3H8(x) = (0.134 − c × xd mm−d) mmol min−1 | (3) |
with
x being the position in the catalyst bed in mm, 4.59 × 10
−2 <
a < 2.33 × 10
−1, 0.54 <
b < 0.72, 4.68 × 10
−4 <
c < 2.32 × 10
−3, and 0.54 <
d < 0.72.
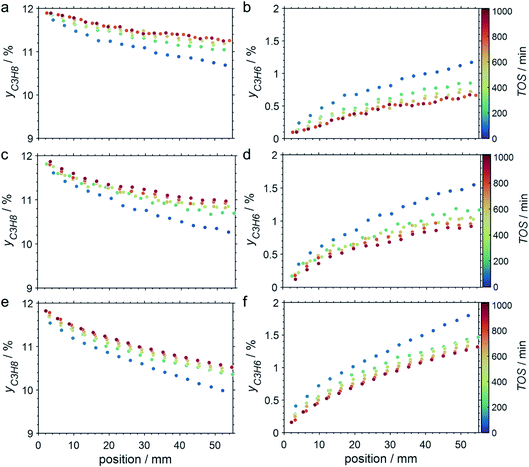 |
| | Fig. 5 Concentration profiles of propane (a, c, and e) and propylene (b, d and f) as a function of the catalyst bed length at various points in time during PDH over GaPt/SiO2 SCALMS with an atomic ratio of Ga/Pt of 55 at (a and b) 450 °C, (c and d) 500 °C, and (e and f) 550 °C. Conditions of the experiment: 22 mLN min−1 He; 3 mLN min−1 C3H8; WHSV 4500 mLN g−1 h−1. | |
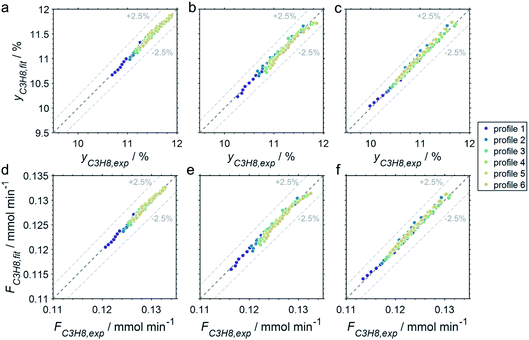 |
| | Fig. 6 Parity plots of the experimental and fitted (a–c) gas fractions and (d–f) molar flows of propane of the six profiles acquired at approx. 82, 259, 437, 614, 791, and 969 min time on stream during PDH over GaPt/SiO2 SCALMS with an atomic ratio of Ga/Pt of 55 at (a and d) 450, (b and e) 500, and (c and f) 550 °C. | |
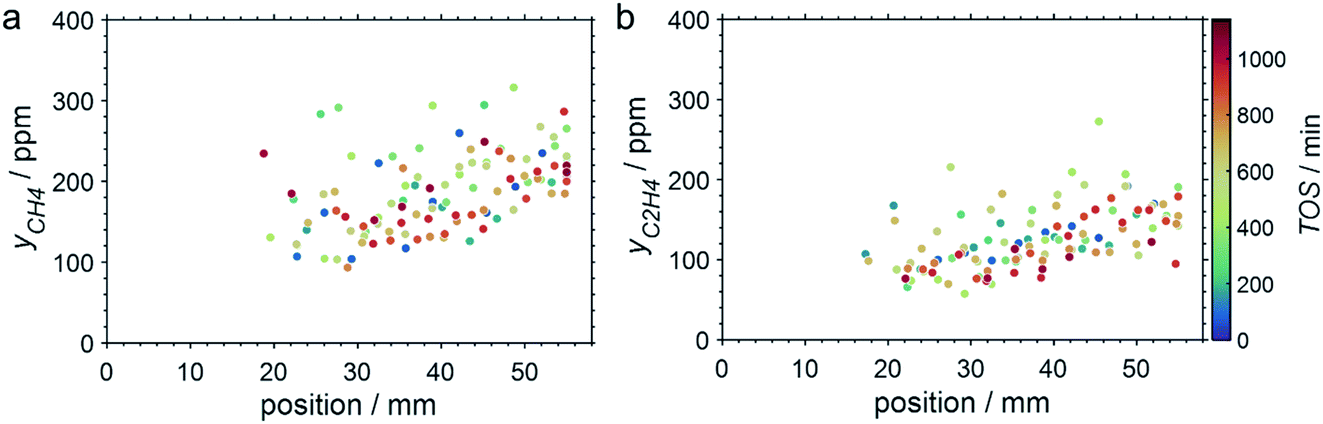
SCALMS have been reported to display a superior alkene selectivity during alkane dehydrogenation when compared to conventional solid-phase heterogeneous catalysts.10,12 Herein, only negligible amounts of side products were identified in the μ-GC. In fact, reasonable signal-to-noise ratios were obtained for methane and ethylene during PDH at 550 °C only and exclusively in the second half of the catalyst bed. Both compounds are expected side products of PDH and may form via the cracking of propane (eqn (4)).5 However, concentrations as low as 100–250 ppm were identified (Fig. 7). Hence, the (gas phase) selectivity towards propylene from propane can be expected to be close to 100% for 450–500 °C. At the highest reaction temperature of 550 °C, the propylene selectivity still exceeds 97% after 900 min TOS.
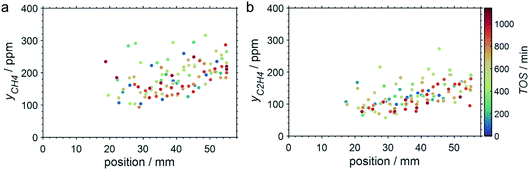 |
| | Fig. 7 Concentration profiles of (a) methane and (b) ethylene as a function of the catalyst bed length at various points in time during PDH over GaPt/SiO2 SCALMS with an atomic ratio of Ga/Pt of 55 at 550 °C. Conditions of the experiment: 22 mLN min−1 He; 3 mLN min−1 C3H8; WHSV 4500 mLN g−1 h−1. | |
The captured, spatially resolved concentration profiles of the reactant propane and product propylene, indicate a continuous increase of the conversion of propane along the catalyst bed length with increased residence time (Fig. 5). However, deactivation over TOS is suggested by the steady increase and decrease of the particular profiles of gas fractions. In particular the first profile deviates from the second. This change in catalytic performance can be easily described with the corresponding conversion of propane (eqn (5)) during PDH (Fig. 8a, c and e). Strong deactivation of SCALMS over TOS is observed throughout the catalyst bed in the first three profile acquisitions. Only the first profiles of the conversion of propane have a linear dependency of conversion and bed length from 15–40 mm of the catalyst bed. It is noted, that the profiles were acquired from the end of the catalyst bed to the beginning, which enhances potential effects of rapid deactivation during acquisition of the profile. At 450 and 500 °C, the obtained profiles are comparable from the third acquisition onwards (Fig. 8a and c). During PDH at 550 °C, already the second profile resembles the others, while the initial slope decreases with TOS resulting in deviation between the profiles at 15–25 mm bed length (Fig. 8e). This observation points towards a deactivation front moving from the end to the beginning of the catalyst bed during the experiments, i.e., enhanced deactivation of the catalyst along the catalyst bed over TOS. This spatial dependency of the deactivation of SCALMS can be demonstrated with the integral productivity along the catalyst bed length (eqn (6)), which describes the effective utilisation of Pt atoms for the conversion of propane to propylene (Fig. 8b, d and f). Firstly, a continuous decrease along the catalyst bed at all reaction temperatures, once again, indicates lower efficiency of the catalyst at the end of the catalyst bed. On the one hand, this behaviour is anchored in the kinetics of PDH if the reaction rate is dependent on the partial pressure of the reactant or products. However, the point of divergence in consecutive profiles of the productivity shifts from the end to the beginning of the catalyst bed over TOS indicating the dependency of deactivation on TOS and the position in the catalyst bed. For example, the point of divergence between the second and third profile at 550 °C is identified at approx. 40 mm bed length, while it shifts to approx. 22 mm for the fourth and fifth profile (Fig. 8f). Hence, the catalytic performance of the remaining catalyst bed is comparable and the increase in conversion is only due to the different activity levels of the SCALMS in the beginning of the catalyst bed.
| | 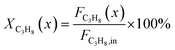 | (5) |
| | 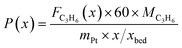 | (6) |
with
x being the position in the catalyst bed in mm,
Fi being the molar flow of compound i in mol min
−1,
FC3H8,in being the molar flow of propane in the feed stream in mol min
−1,
MC3H6 being the molar weight of propylene in g mol
−1,
mPt being the mass of Pt in the loaded SCALMS (0.3333 g × 0.29/100), and
xbed being the total length of the catalyst bed (55 mm).
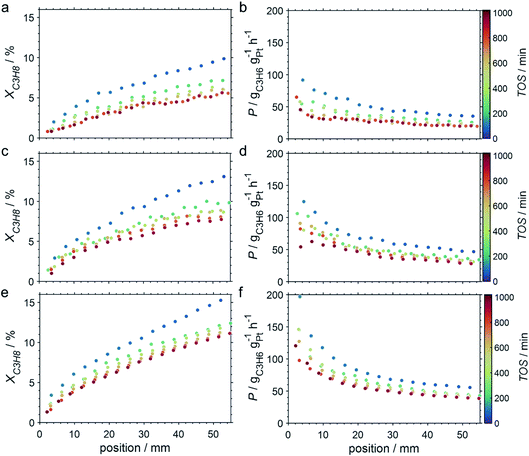 |
| | Fig. 8 Conversion profiles of propane, and productivity as a function of the catalyst bed length at various points in time during PDH over GaPt/SiO2 SCALMS with an atomic ratio of Ga/Pt of 55 at (a and b) 450 °C, (c and d) 500 °C, and (e and f) 550 °C. Conditions of the experiment: 22 mLN min−1 He; 3 mLN min−1 C3H8; WHSV 4500 mLN g−1 h−1. | |
As demonstrated using HRTGA-MS (Fig. 2, 3, S2 and S3†), coking of the SCALMS catalyst is feasible,10,11 even for SiO2 supported GaPt droplets. Carbonaceous deposits are generally known to have the potential to exacerbate the activity of catalysts due to chemical or physical blockage of active sites.46,47 Enhanced coke formation during PDH at 550 °C is supported by the formation of considerable amounts of the cracking products methane and ethylene (Fig. 7), but coking may also be at play at lower reaction temperatures. Coking is generally expected to be pronounced under the (relatively) high conversion environment at the end of the catalyst bed,48 which is also indicated by the concentration profiles acquired using the CPR suggesting a coking front moving from the end to the beginning of the catalyst bed over TOS. Such a spatial dependency of coke formation has been reported for oxidative dehydrogenation of ethane to ethylene over a MoO3/Al2O3 catalyst.49 Geske et al. employed a similar reactor set-up and analysed the formation of coke via in situ Raman spectroscopy. Catalytic partial oxidation of methane using Pt coated, cylindrical α-Al2O3 foam monoliths has also been reported to result in an enhanced deposition of carbonaceous species in the axial direction of the reactor.50 Once again, the formation of coke was spatially analysed by means of Raman spectroscopy after catalytic testing. In the present study, the strong fluorescent character of SiO2 prohibited a spatially resolved analysis of coke deposits in the GaPt/SiO2 samples. However, coke deposition is clearly identified by the colour change of the SCALMS catalysts in the quartz capillary reactors after catalytic application (Fig. 9). After PDH at 550 °C, the catalyst has the darkest shade of brown/grey and even some carbonaceous residuals are identified in the quartz wool plug at the end of the catalyst bed demonstrating the high coking affinity of the reaction gas mixture during PDH at 550 °C.
 |
| | Fig. 9 Picture of the three quartz capillary reactors after PDH over GaPt/SiO2 SCALMS with an atomic ratio of Ga/Pt of 55 at 450 (left), 500 (middle), and 550 °C (right) visualising the temperature dependent deposition of carbonaceous species on the catalyst. It is noted, that small amounts of coke also formed on the second part of the quartz wool plug at the end of the catalyst bed after operation at 550 °C. The first part of this quartz wool plug is seemingly not affected, most likely as it is still within the isothermal zone of the fixed-bed reactor. | |
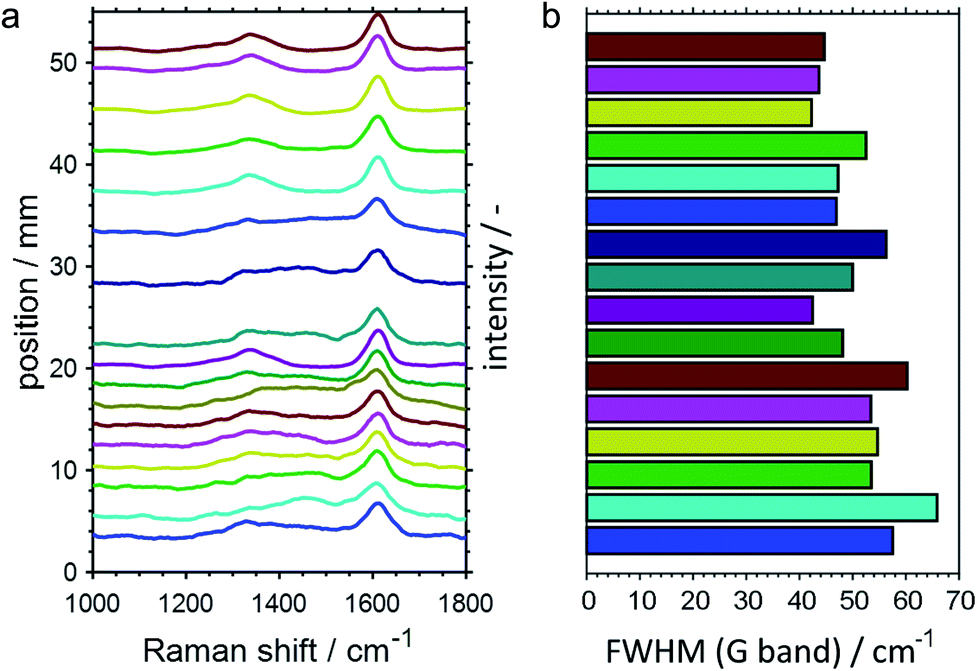
In addition to the SiO2-supported catalysts, the CPR was also employed for studying PDH over GaPt/Al2O3 SCALMS for the medium operation temperature of 500 °C. However, the increased coking affinity of this catalyst (Fig. 2 and 3) prevented sampling of reaction gas mixture via the orifices in the capillary over prolonged TOS, which was most likely caused by clogging of the 100 μm openings by carbon deposits from the catalyst bed. As aforementioned, the employment of the Al2O3 carrier material is hypothesised to cause this enhanced formation of carbon when compared to the SiO2 carrier, which enabled the spatially resolved analysis of the coking behaviour of the GaPt/Al2O3 SCALMS by means of Raman spectroscopy. In contrast to the SiO2-supported catalyst, a sufficient amount of carbon was deposited on this catalyst to quench the moderate fluorescence of the Al2O3 support material. The acquired spatially resolved Raman spectra strongly support the herein hypothesised enhanced coking at the end of the catalyst bed during PDH (Fig. 10a). Firstly, a peak evolves along the catalyst bed at the typical Raman shift for the D band of carbon (∼1360 cm−1),51 which is accompanied by a generally increased signal-to-noise ratio of the spectra. Secondly, the full width at half maximum (FWHM) of the G band (∼1605 cm−1)51 decreases along the catalyst bed (Fig. 10b) indicating a spatially dependent increase in the quantity or a change in the nature of the carbon deposits, e.g. an increasing degree of graphitisation. Even though the low signal strength avoids a more detailed analysis of the Raman spectra, the results demonstrate the spatial dependency of the coking behaviour in a GaPt/Al2O3 SCALMS catalyst bed during PDH, which is most likely also the case for the SiO2-supported catalyst as indicated by the catalytic data (Fig. 7 and 8).
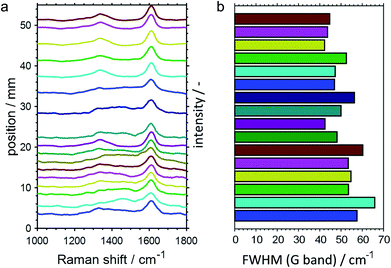 |
| | Fig. 10 (a) Spatially resolved Raman spectra of the catalyst bed after propane dehydrogenation over GaPt/Al2O3 SCALMS with an atomic ratio of Ga/Pt of 86 with (b) the corresponding full width at half maximum (FWHM) of the G band of carbon at a Raman shift of 1605 cm−1. Conditions of the experiment: 500 °C; 22 mLN min−1 He; 3 mLN min−1 C3H8; WHSV 4500 mLN g−1 h−1. | |
The acquired data demonstrates the great potential of the CPR for acquisition of kinetic data describing spatially resolved activity, selectivity, and deactivation alike. However, the spatial dependency of deactivation of the catalyst on the TOS, results in superimposition of intrinsic kinetic data with deactivation. Hence, the acquired profiles are strongly affected by the enhanced deactivation over the catalyst bed length resulting in a perpetual change of the apparent kinetics. Nevertheless, the data may represent the foundation for a comprehensive study of intrinsic kinetics together with deactivation kinetics, which exceeds the scope of the present study.
Summary and conclusion
The in situ characterisation of novel SCALMS during PDH by means of HRTGA-MS provides valuable insights on the effect of the carrier material on coke formation. Only small amounts of coke (∼0.25 wt%) were formed during PDH over Ga–Pt SCALMS at 500 °C employing SiO2 as the carrier material. In contrast, more than three times the amount of coke were formed during PDH over Al2O3-supported SCALMS due to the enhanced acidity of the carrier material. The coke is seemingly formed continuously during PDH over both SCALMS. In addition, spatially resolved kinetic data was acquired during PDH over GaPt/SiO2 SCALMS using an innovative CPR. The performance of the catalyst was relatively stable after a start-up period of ∼200 min with rapid deactivation of the catalyst. Structural rearrangement of the liquid Ga-rich GaPt droplets and/or rapid initial carbon deposition may cause this decay in activity. After this initial period, the concentration profiles are highly consistent, which may enable the future development of kinetic models including deactivation terms in more comprehensive studies. The results indicate an enhanced deactivation of the SCALMS at the end of the catalyst bed. In fact, a deactivation front moving from the end to the beginning of the catalyst bed over time on stream is highly likely, which may be related to accelerated coking of the catalyst under high conversion environments. Such a spatially dependent deposition of carbon deposits was successfully demonstrated by coupling the CPR with Raman spectroscopy for the GaPt/Al2O3 SCALMS due to the increased coking affinity of this catalyst. Only minor amounts of side products from propane cracking were identified supporting the previously reported superior selectivity of SCALMS during alkane dehydrogenation.
Conflicts of interest
There are no conflicts to declare.
Note after first publication
The data in Fig. 10, and the related discussion, was presented at the Faraday Discussions meeting and added to the manuscript post-meeting.
Acknowledgements
Financial support by the European Research Council is gratefully acknowledged (Project 786475: Engineering of Supported Catalytically Active Liquid Metal Solutions).
References
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, J. Catal., 2010, 276, 268–279 CrossRef CAS.
- H. N. Pham, J. J. H. B. Sattler, B. M. Weckhuysen and A. K. Datye, ACS Catal., 2016, 6, 2257–2264 CrossRef CAS PubMed.
- T. Otroshchenko, S. Sokolov, M. Stoyanova, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, Angew. Chem., Int. Ed., 2015, 54, 15880–15883 CrossRef CAS PubMed.
- Q. Li, Z. Sui, X. Zhou, Y. Zhu, J. Zhou and D. Chen, Top. Catal., 2011, 54, 888–896 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- P. Borges, R. Ramos Pinto, M. A. N. D. A. Lemos, F. Lemos, J. C. Védrine, E. G. Derouane and F. Ramôa Ribeiro, J. Mol. Catal. A: Chem., 2005, 229, 127–135 CrossRef CAS.
- T. Noda, K. Suzuki, N. Katada and M. Niwa, J. Catal., 2008, 259, 203–210 CrossRef CAS.
- J. Im and M. Choi, ACS Catal., 2016, 6, 2819–2826 CrossRef CAS.
- N. Raman, S. Maisel, M. Grabau, N. Taccardi, J. Debuschewitz, M. Wolf, H. Wittkämper, T. Bauer, M. Wu, M. Haumann, C. Papp, G. Görling, E. Spiecker, J. Libuda, H.-P. Steinrück and P. Wasserscheid, ACS Catal., 2019, 9, 9499–9507 CrossRef CAS PubMed.
- M. Wolf, N. Raman, N. Taccardi, M. Haumann and P. Wasserscheid, ChemCatChem, 2020, 12, 1085–1094 CrossRef CAS PubMed.
- N. Taccardi, M. Grabau, J. Debuschewitz, M. Distaso, M. Brandl, R. Hock, F. Maier, C. Papp, J. Erhard, C. Neiss, W. Peukert, A. Görling, H. P. Steinrück and P. Wasserscheid, Nat. Chem., 2017, 9, 862–867 CrossRef CAS PubMed.
- J. M. Herman, A. P. A. F. Rocourt, P. J. Van den Berg, P. J. Van Krugten and J. J. F. Scholten, Chem. Eng. J., 1987, 35, 83–103 CrossRef CAS.
- A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Eur. J. Inorg. Chem., 2006, 2006, 695–706 CrossRef.
- M. Lijewski, J. M. Hogg, M. Swadźba-Kwaśny, P. Wasserscheid and M. Haumann, RSC Adv., 2017, 7, 27558–27563 RSC.
- J. M. Marinkovic, A. Riisager, R. Franke, P. Wasserscheid and M. Haumann, Ind. Eng. Chem. Res., 2018, 58, 2409–2420 CrossRef.
- M. Plevan, T. Geißler, A. Abánades, K. Mehravaran, R. K. Rathnam, C. Rubbia, D. Salmieri, L. Stoppel, S. Stückrad and T. Wetzel, Int. J. Hydrogen Energy, 2015, 40, 8020–8033 CrossRef CAS.
- D. C. Upham, V. Agarwal, A. Khechfe, Z.
R. Snodgrass, M. J. Gordon, H. Metiu and E. W. McFarland, Science, 2017, 358, 917–921 CrossRef CAS.
- G. Rupprechter, Nat. Chem., 2017, 9, 833–834 CrossRef CAS.
- M. Grabau, S. Krick Calderón, F. Rietzler, I. Niedermaier, N. Taccardi, P. Wasserscheid, F. Maier, H.-P. Steinrück and C. Papp, Surf. Sci., 2016, 651, 16–21 CrossRef CAS.
- M. Grabau, J. Erhard, N. Taccardi, S. K. Calderon, P. Wasserscheid, A. Gorling, H. P. Steinruck and C. Papp, Chem.–Eur. J., 2017, 23, 17701–17706 CrossRef CAS PubMed.
- T. Bauer, S. Maisel, D. Blaumeiser, J. Vecchietti, N. Taccardi, P. Wasserscheid, A. Bonivardi, A. Görling and J. Libuda, ACS Catal., 2019, 9, 2842–2853 CrossRef CAS PubMed.
- C. Hohner, M. Kettner, C. Stumm, C. Schuschke, M. Schwarz and J. Libuda, Top. Catal., 2019, 62, 849–858 CrossRef CAS.
- M. Kettner, S. Maisel, C. Stumm, M. Schwarz, C. Schuschke, A. Görling and J. Libuda, J. Catal., 2019, 369, 33–46 CrossRef CAS.
- D. Esposito, Nat. Catal., 2019, 2, 179 CrossRef.
- P. Biloen, F. M. Dautzenberg and W. M. H. Sachtler, J. Catal., 1977, 50, 77–86 CrossRef CAS.
- D. F. Shriver, A. E. Shirk and J. A. Dilts, Inorg. Synth., 1977, 17, 42–45 CAS.
- D. L. Minnick, T. Turnaoglu, M. A. Rocha and M. B. Shiflett, J. Vac. Sci. Technol., A, 2018, 36, 050801 CrossRef.
-
T. B. Massalski, H. Okamoto, P. R. Subramanian and L. Kacprzak, in Binary Alloy Phase Diagrams, ed. T. B. Massalski, H. Okamoto, P. R. Subramanian and L. Kacprzak, ASM International, Russell Township, USA, 2nd edn, 1990, pp. 1840–1842 Search PubMed.
- E. A. Redekop, V. V. Galvita, H. Poelman, V. Bliznuk, C. Detavernier and G. B. Marin, ACS Catal., 2014, 4, 1812–1824 CrossRef CAS.
- K. Föttinger, Catal. Today, 2013, 208, 106–112 CrossRef.
- A. E. Lear, T. C. Brown and B. S. Haynes, Symposium (International) on Combustion, 1991, 23, 1191–1197 CrossRef.
- C. Le Minh, R. A. Jones, I. E. Craven and T. C. Brown, Energy Fuels, 1997, 11, 463–469 CrossRef.
-
C. Le Minh, C. Li and T. C. Brown, in Catalyst Deactivation 1997, ed. C. H. Bartholomew and G. A. Fuentes, 1997, vol. 111, pp. 383–390 Search PubMed.
- C. Li, C. Le Minh and T. C. Brown, J. Catal., 1998, 178, 275–283 CrossRef CAS.
- C. Li and T. C. Brown, Carbon, 2001, 39, 725–732 CrossRef CAS.
-
D. E. Resasco and G. L. Haller, in Catalysis, ed. J. J. Spivey and S. K. Agarwal, The Royal Society of Chemistry, Cambridge, 1994, vol. 11 Search PubMed.
- R. K. Grasselli, D. L. Stern and J. G. Tsikoyiannis, Appl. Catal., A, 1999, 189, 9–14 CrossRef CAS.
- K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem.
Soc., 2018, 140, 11674–11679 CrossRef CAS PubMed.
- M. W. Schreiber, C. P. Plaisance, M. Baumgartl, K. Reuter, A. Jentys, R. Bermejo-Deval and J. A. Lercher, J. Am. Chem. Soc., 2018, 140, 4849–4859 CrossRef CAS PubMed.
- B. K. Vu, M. B. Song, I. Y. Ahn, Y.-W. Suh, D. J. Suh, W.-I. Kim, H.-L. Koh, Y. G. Choi and E. W. Shin, Appl. Catal., A, 2011, 400, 25–33 CrossRef CAS.
- Z. Han, S. Li, F. Jiang, T. Wang, X. Ma and J. Gong, Nanoscale, 2014, 6, 10000–10008 RSC.
- D. Akporiaye, S. F. Jensen, U. Olsbye, F. Rohr, E. Rytter, M. Ronnekleiv and A. I. Spjelkavik, Ind. Eng. Chem. Res., 2001, 40, 4741–4748 CrossRef CAS.
- D. Hullmann, G. Wendt, U. Šingliar and G. Ziegenbalg, Appl. Catal., A, 2002, 225, 261–270 CrossRef CAS.
- Y. Zhang, Y. Zhou, J. Shi, S. Zhou, X. Sheng, Z. Zhang and S. Xiang, J. Mol. Catal. A: Chem., 2014, 381, 138–147 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- J. van Doorn and J. A. Moulijn, Catal. Today, 1990, 7, 257–266 CrossRef CAS.
- J. Towfighi, M. Sadrameli and A. Niaei, J. Chem. Eng. Jpn., 2002, 35, 923–937 CrossRef.
- M. Geske, O. Korup and R. Horn, Catal. Sci. Technol., 2013, 3, 169–175 RSC.
- O. Korup, C. F. Goldsmith, G. Weinberg, M. Geske, T. Kandemir, R. Schlögl and R. Horn, J. Catal., 2013, 297, 1–16 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fd00010h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Narayanan
Raman
a,
Nicola
Taccardi
a,
Raimund
Horn
b,
Marco
Haumann
a,
Narayanan
Raman
a,
Nicola
Taccardi
a,
Raimund
Horn
b,
Marco
Haumann