
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReagent controlled stereoselective synthesis of teichoic acid α-(1,2)-glucans†
Liming
Wang
,
Francesca
Berni
,
Jacopo
Enotarpi
,
Hermen S.
Overkleeft
,
Gijs
van der Marel
and
Jeroen D. C.
Codée
 *
*
Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: jcodee@chem.leidenuniv.nl
First published on 27th February 2020
Abstract
The stereoselective construction of 1,2-cis-glycosidic linkages is key in the assembly of biologically relevant glycans, but remains a synthetic challenge. Reagent-controlled glycosylation methodologies, in which external nucleophiles are employed to modulate the reactivity of the glycosylation system, have become powerful means for the construction of 1,2-cis-glycosidic linkages. Here we establish that nucleophilic additives can support the construction of α-1,2-glucans, and apply our findings in the construction of a D-alanine kojibiose functionalized glycerol phosphate teichoic acid fragment. This latter molecule can be found in the cell wall of the opportunistic Gram-positive bacterium, Enterococcus faecalis and represents a structural element that can possibly be used in the development of therapeutic vaccines and diagnostic tools.
Introduction
α-Glucans, polysaccharides composed of glucose monosaccharides connected through α-glycosidic linkages, are widely found in nature.1 The most abundant α-glucans are the α-1,4-glucans, that occur as starch and glycogen, and can be found as extracellular polysaccharides surrounding bacteria, such as Mycobacterium tuberculosis.2 α-1,3-Glucans are prominent components of fungal cell walls and play a role in the interaction between the pathogenic fungus Aspergillus fumigatus and the host immune system.3 α-1,2-Glucans, also referred to as kojioligosaccharides, have been found in koji extract, sake, beer and honey and they have also been encountered in bacterial cell walls.4 Kojibiose, kojitriose and kojitetraose have been found as part of the lipoteichoic acid (LTA) of Enterococcus faecalis, which is a commensal Gram-positive bacterium.5 Although this bacterium is generally of low virulence, it is the causative agent of life-threatening infections in immunocompromised patients and one of the most prominent hospital bacteria.6 LTA is an anionic polymer featuring a glycolipid tail, through which it is anchored in the cell wall of the bacteria. The polyanionic part is composed of glycerol phosphate repeating units, which can be functionalised with glycans and D-alanine esters at C2 of the glycerol moieties (Fig. 1).7 Kojibiose is a prominent appendage of E. faecalis LTA, and it can be decorated with D-alanyl esters at both C6-hydroxyl groups of the disaccharide.7D-Alanylation plays a critical role in the virulence of the bacteria and it renders the bacteria less susceptible to (cationic) antimicrobial peptides. E. faecalis LTA fragments, containing a single α-glucosyl substituent have previously been explored as components in synthetic vaccine conjugates,8 and antibodies raised by these conjugates have been shown to be opsonophagocytic and capable of inducing killing of the bacteria in both active and passive immunization strategies.9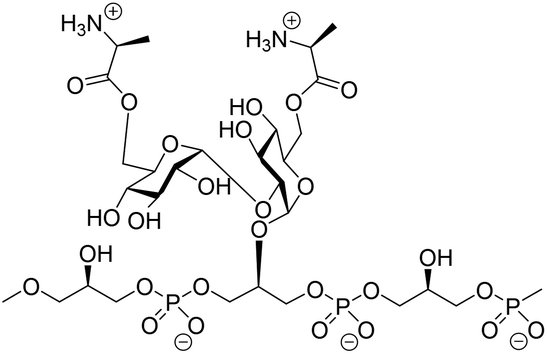 | ||
| Fig. 1 The structure of E. faecalisD-alanine kojibiose functionalized LTA. | ||
The microheterogeneity of naturally occurring LTA makes it challenging if not impossible- to establish clear structure–activity relationships for these molecules, and therefore organic synthesis is the method of choice to generate well-defined single LTA molecules.9,10 Only a few chemical syntheses of α-(1,2)-glucans have been reported. Takeo and Suzuki reported a procedure to assemble kojitriose, kojitetraose and kojipentaose based on Koenings-Knorr-type glycosylations, using anomeric chlorides under the activation of silver perchlorate.11 Hogendorf et al. relied on Crich's α-glucosylation protocol, which involves the use of benzylidene glucose donors for the synthesis of a kojibiose glycerol building block.9 We have previously shown that additive-controlled glycosylations12 of per-benzylated glucosyl imidate donors can be used effectively for the introduction of α-(1,4)- and α-(1,3)-glucosyl linkages, which we used to assemble a set of M. tuberculosis α-(1,4)-glucans and A. fumigatus α-(1,3)-glucans.13 In this strategy we exploited a suite of orthogonal benzyl ethers to discriminate between different hydroxyl functionalities that had to be protected permanently (using a benzyl (Bn) group), and hydroxyl groups that were used for chain elongation (protected with 2-methylnaphthyl (Nap) ethers) and hydroxyl groups from which branching occurred (using para-methoxybenzyl (PMB) ethers). The use of solely benzylic ethers to protect the hydroxyls ensured that the nature of the protecting groups does not induce variation in the reactivity of the building blocks and that we could rely on reactivity tuning by the used external nucleophile to control the stereoselective construction of the α-glucan linkages. This strategy could provide an effective entry to construct α-(1,2)-linkages and generate (substituted) kojioligosaccharides such as encountered in E. faecalis LTA. We here describe the use of additive-controlled glycosylation methodology for the construction of α-(1,2)-glucans and we show the application of the methodology in the preparation of a D-alanine-kojibiose functionalized LTA fragment.
Results and discussion
We first explored the assembly of spacer-functionalized kojioligosaccharides. To achieve this aim, donor 6 (Scheme 1) was designed and assembled. The building block bears three permanent Bn-ethers to mask the C3, C4 and C6 hydroxyls and a Nap-ether as a temporary protecting group at the C2-OH. The synthesis of this building block used benzylidene protected glucose building block 1 as starting compound. Selective protection of the C3-OH as the benzyl ether was achieved through the formation of an intermediate tin ketal which was treated with benzyl bromide and cesium fluoride to give compound 2. Protection of the remaining C2-OH with a Nap ether then provided compound 3. Selective opening of the benzylidene and protection of the liberated primary alcohol provided the fully benzylated thiodonor 5. The anomeric thioacetal in this building block was hydrolyzed using N-iodosuccinimide (NIS) in acetone/water to liberate the anomeric hydroxyl group, which could then be turned into the required N-phenyltrifluoroacetimidate 6. | ||
Scheme 1 Synthesis of donor 6. (a) (i) Bu2SnO, toluene, 120 °C; (ii) CsF, BnBr, 76%. (b) NapBr, NaH, DMF, 91%. (c) BF3·Et2O, TES, DCM. (d) BnBr, NaH, DMF, 88%. (e) (i) NIS, acetone/H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v); (ii) 2,2,2-trifluoro-N-phenylacetimidoyl chloride, Cs2CO3, acetone, 81% (over two steps). :1, v:v); (ii) 2,2,2-trifluoro-N-phenylacetimidoyl chloride, Cs2CO3, acetone, 81% (over two steps). | ||
With donor 6 in hand, the assembly of kojioligosaccharides was explored as depicted in Scheme 2. Firstly, donor 6 was glycosylated with azidopropanol, a primary and relatively reactive alcohol, using the TMSI-Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O activation couple. TMSI transforms the anomeric imidate into the α-glucosyl iodide, which under the agency of Ph3PO is isomerized into its less stable but more reactive β-counterpart. These conditions formed azidopropyl glucoside 7 in good yield and stereoselectivity (α:β = 10:1), in line with results previously described.13a,14 Deprotection of the C2-O-Nap ether under oxidative conditions (dichlorodicyanobenzoquinone (DDQ) in DCM/H2O, 10:1 v/v) provided alcohol 8 which was purified by silica gel column chromatography to deliver the pure α-anomer. Next the secondary and less reactive alcohol acceptor 8 was glycosylated with donor 6 under the agency of TfOH and DMF. This latter additive generates a mixture α/β-glucosyl imidinium ions, which are in rapid equilibrium. Substitution of the more reactive β-ion then leads to the stereoselective formation of α-glucosyl linkages. This additive has found wide application in the construction of various 1,2-cis-linked oligosaccharides.15 The condensation of 8 and 6 provided disaccharide 9 in 95% yield and a 15:1 α:β-ratio. Deprotection of the C2-O-Nap ether by DDQ provided disaccharide acceptor 10 and the third glycosylation, again performed under DMF mediated glycosylation conditions, then provided trisaccharide 11. The stereoselectivity of this glycosylation diminished slightly and 11 was formed as a 10:1 α:β-mixture. The erosion of stereoselectivity developed further upon elongation of the trisaccharide, as the DMF-mediated glycosylation of acceptor 12, obtained after DDQ treatment of 11, with per-benzyl donor 13 delivered tetrasaccharide 14 as a 2:1 α:β-mixture. Possibly the overall structure of the twisted α-(1,2)-oligosaccharide is at the root of the erosion of stereoselectivity, with increasing steric demands leading to lower yields and poorer selectivity.
O activation couple. TMSI transforms the anomeric imidate into the α-glucosyl iodide, which under the agency of Ph3PO is isomerized into its less stable but more reactive β-counterpart. These conditions formed azidopropyl glucoside 7 in good yield and stereoselectivity (α:β = 10:1), in line with results previously described.13a,14 Deprotection of the C2-O-Nap ether under oxidative conditions (dichlorodicyanobenzoquinone (DDQ) in DCM/H2O, 10:1 v/v) provided alcohol 8 which was purified by silica gel column chromatography to deliver the pure α-anomer. Next the secondary and less reactive alcohol acceptor 8 was glycosylated with donor 6 under the agency of TfOH and DMF. This latter additive generates a mixture α/β-glucosyl imidinium ions, which are in rapid equilibrium. Substitution of the more reactive β-ion then leads to the stereoselective formation of α-glucosyl linkages. This additive has found wide application in the construction of various 1,2-cis-linked oligosaccharides.15 The condensation of 8 and 6 provided disaccharide 9 in 95% yield and a 15:1 α:β-ratio. Deprotection of the C2-O-Nap ether by DDQ provided disaccharide acceptor 10 and the third glycosylation, again performed under DMF mediated glycosylation conditions, then provided trisaccharide 11. The stereoselectivity of this glycosylation diminished slightly and 11 was formed as a 10:1 α:β-mixture. The erosion of stereoselectivity developed further upon elongation of the trisaccharide, as the DMF-mediated glycosylation of acceptor 12, obtained after DDQ treatment of 11, with per-benzyl donor 13 delivered tetrasaccharide 14 as a 2:1 α:β-mixture. Possibly the overall structure of the twisted α-(1,2)-oligosaccharide is at the root of the erosion of stereoselectivity, with increasing steric demands leading to lower yields and poorer selectivity.
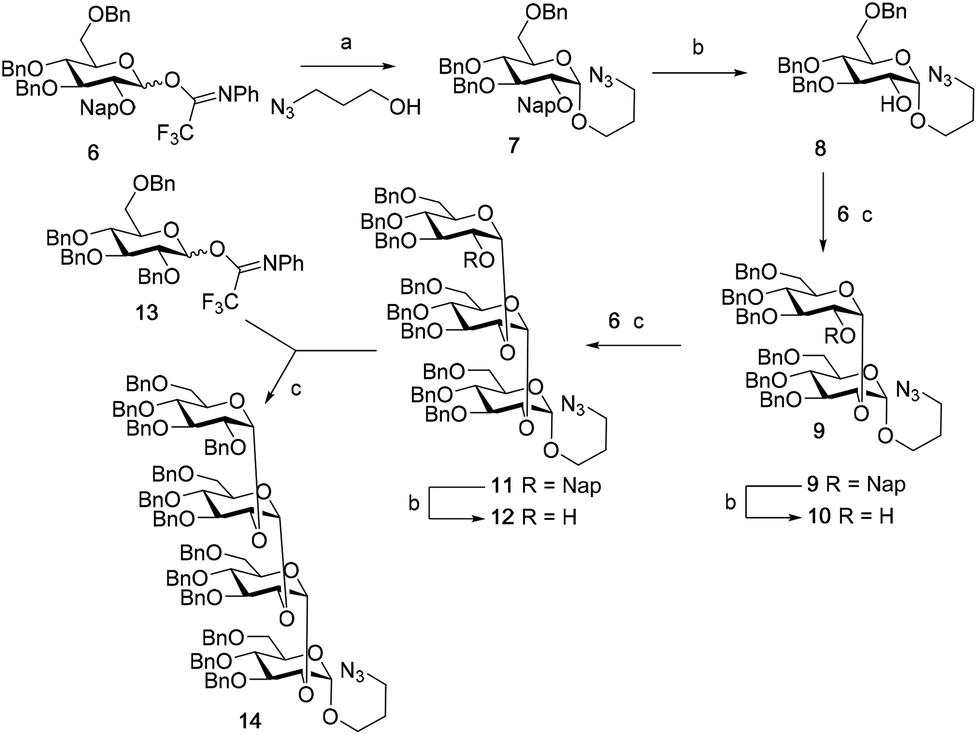 | ||
| Scheme 2 The assembly of kojioligosaccharides. (a) TMSI, Ph3PO, DCM, 36 h, 75%, α:β = 10:1. (b) DDQ, DCM:H2O = 10:1 (v:v), 8: 75%; 10: 49%; 12: 47%. (c) DMF, TfOH, DCM, 9: 95%, α:β = 15:1; 11: 67%, α:β = 10:1; 14: 67%, α:β = 2:1. | ||
Although a limitation of the DMF-mediated glycosylation protocol come forward in the above described kojitetraose assembly, the synthesis also showed that the additive controlled glycosylation approach can provide a rapid entry into the assembly of smaller kojibiose derivatives. Therefore, the assembly of an E. faecalisD-Ala-kojibiose LTA fragment was next explored. The synthesis of D-Ala-kojibiose LTA requires the availability of compound 19 featuring the kojibiose, decorated with two D-alanine esters at both primary alcohols, attached to the glycerol through an α-bond (Scheme 3). Compound 19 can be assembled from four building blocks: two benzyl type protected glucosyl donors, 15 and 16, glycerol 17 protected with a benzoyl ester (Bz) and allyl ether and N-benzyloxycarbonyl protected D-alanine 18 (Scheme 3). The glucosyl donors are protected with the triad of Bn, PMB and Nap-ethers to discriminate the alcohol groups that required permanent protection or needed to be removed to introduce the next glucose residue or introduce the D-alanine esters.
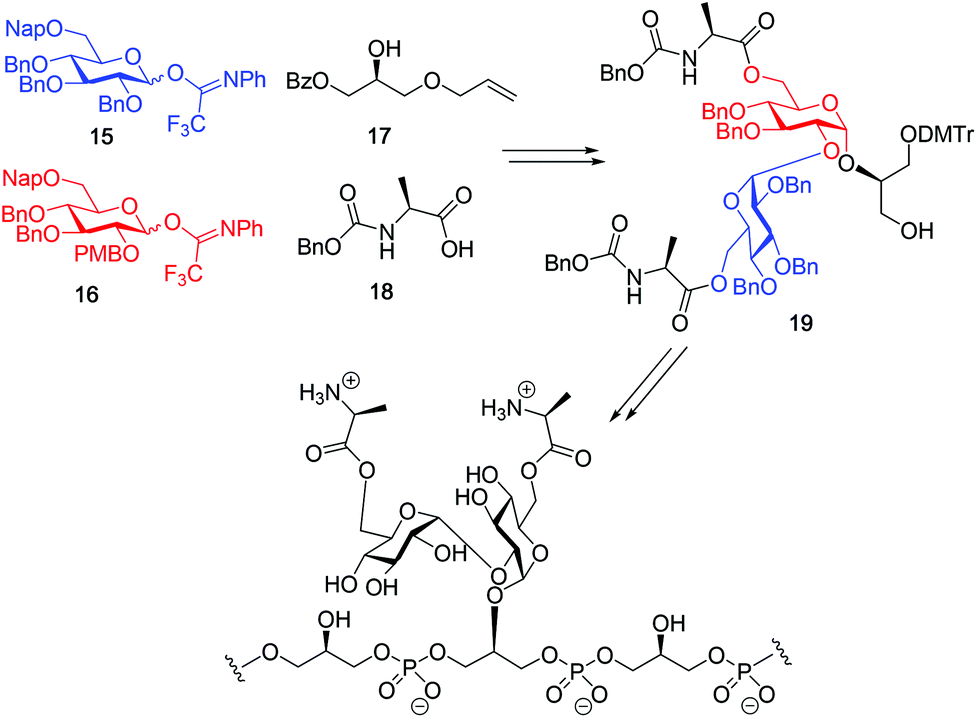 | ||
| Scheme 3 Building blocks applied in the here-presented studies in the synthesis of D-alanine kojibiose functionalized LTA-fragments of Enterococcus faecalis. | ||
The synthesis of the used building blocks is depicted in Scheme 4. Firstly, donor 15 was synthesized from compound 1 by benzylation of the C2 and C3-hydroxyls, regioselective opening of the benzylidene acetal and protection of the liberated primary alcohol with a Nap-ether, to give thio donor 22. Hydrolysis of the thioacetal and introduction of the 2,2,2-trifluoro-N-phenylacetimidate formed donor 15. Donor 16 was obtained from compound 2 following a similar sequence of reactions: protection of the C2-OH with a PMB-ether, regioselective opening of the benzylidene acetal and installation of the C6-O-Nap ether provided thioacetal 25, which was transformed into the corresponding imidate donor 16.
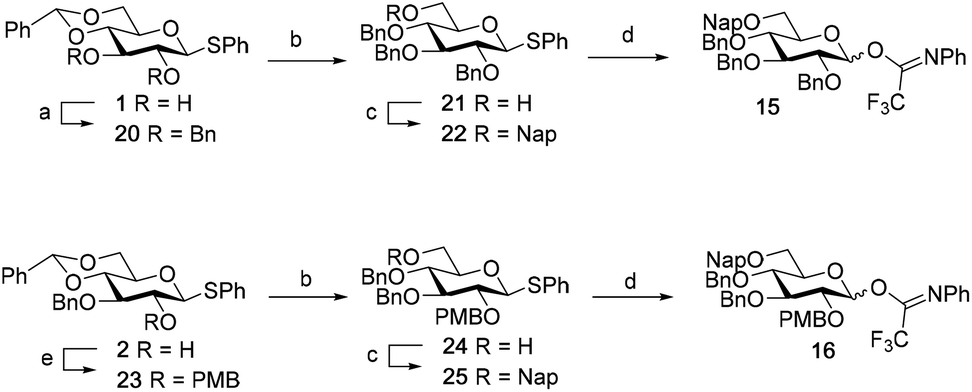 | ||
| Scheme 4 Synthesis of donor 15 and 16. (a) BnBr, NaH, DMF, 20: 96%. (b) BH3.THF, Cu(OTf)2, 21: 80%; 24: 76%. (c) NapBr, NaH, DMF, 22: 91%; 25: 93%. (d) 1) NIS, acetone:H2O = 10:1 (v:v); (2) 2,2,2-trifluoro-N-phenylacetimidoyl chloride, Cs2CO3, acetone, yield 15: 86%; 16: 84%. (e) PMBCl, NaH, DMF, 94%. | ||
With all the required building blocks available, we explored the assembly of a kojibiose LTA fragment as depicted in Scheme 5. First, glycerol building block 19 was assembled. To this end glycerol 17 was glycosylated with donor 16, of which the primary alcohol groups were protected by a benzoate ester and an allyl ether, using the TMSI-Ph3PO activation conditions to give the compound 26 in good yield and excellent selectivity. Next, the C2-O-PMB was selectively removed using a catalytic amount of HCl in a mixture of hexafluoro-iso-propanol (HFIP) and DCM to give C2-OH acceptor 27 in 95% yield.16 All other benzyl ethers remained unaffected under these conditions. Acceptor 27 was then glycosylated with donor 15 using the DMF-mediated glycosylation conditions to form the disaccharide 28 with good selectivity and excellent yield. To set the stage for the introduction of the D-alanyl esters, first the glycerol benzoate was replaced by a TBDPS ether, after which the two C-6-O-Nap ethers were removed to give diol 31. The introduction of the two D-alanine esters was accomplished using PyBOP as a condensing agent to form 6,6-di-alanyl-α-kojibiose 32 in 90% yield. To transform this building block into a synthon suitable for the assembly of TA-fragments, the allyl ether was replaced by a 4,4′-dimethoxytrityl (DMTr) group. Therefore, the allyl group was isomerized to the corresponding enol ether using an iridium catalyst, which was subsequently hydrolyzed using I2 in combination with sodium bicarbonate. The liberated primary alcohol was treated with DMTrCl in DCM to give compound 34, which was treated with HF-pyridine to give the 6,6-di-alanyl-α-kojibiose 19 in 86% yield. For the construction of the kojibiobiosyl glycerol trimer 40, we chose to use benzyl phosphoramidites, as opposed to the more commonly employed cyanoethyl reagents.10 The latter group however requires basic conditions for its removal, that may jeopardize the labile D-alanine esters in the target compound.17 Thus, compound 19 and phosporamidite 35 were coupled under the agency of dicyanoimidazole (DCI), to provide the intermediate phosphite triester, which was immediately oxidized to the phosphate using (1S)-(+)-(10-camphorsulfonyl)-oxaziridine (CSO), and the resulting dimer 36 was treated with a catalytic amount of p-toluenesulfonic acid (TsOH) in DCM/MeOH to provide compound 37 in 93% over three steps. A similar coupling of phophoramidite 38 and alcohol 37 then provided trimer 39 in 88% yield. Removal of all benzyl ethers, benzyl carbamates and benzyl phosphotriesters in 39 in a single hydrogenolysis event delivered 40.
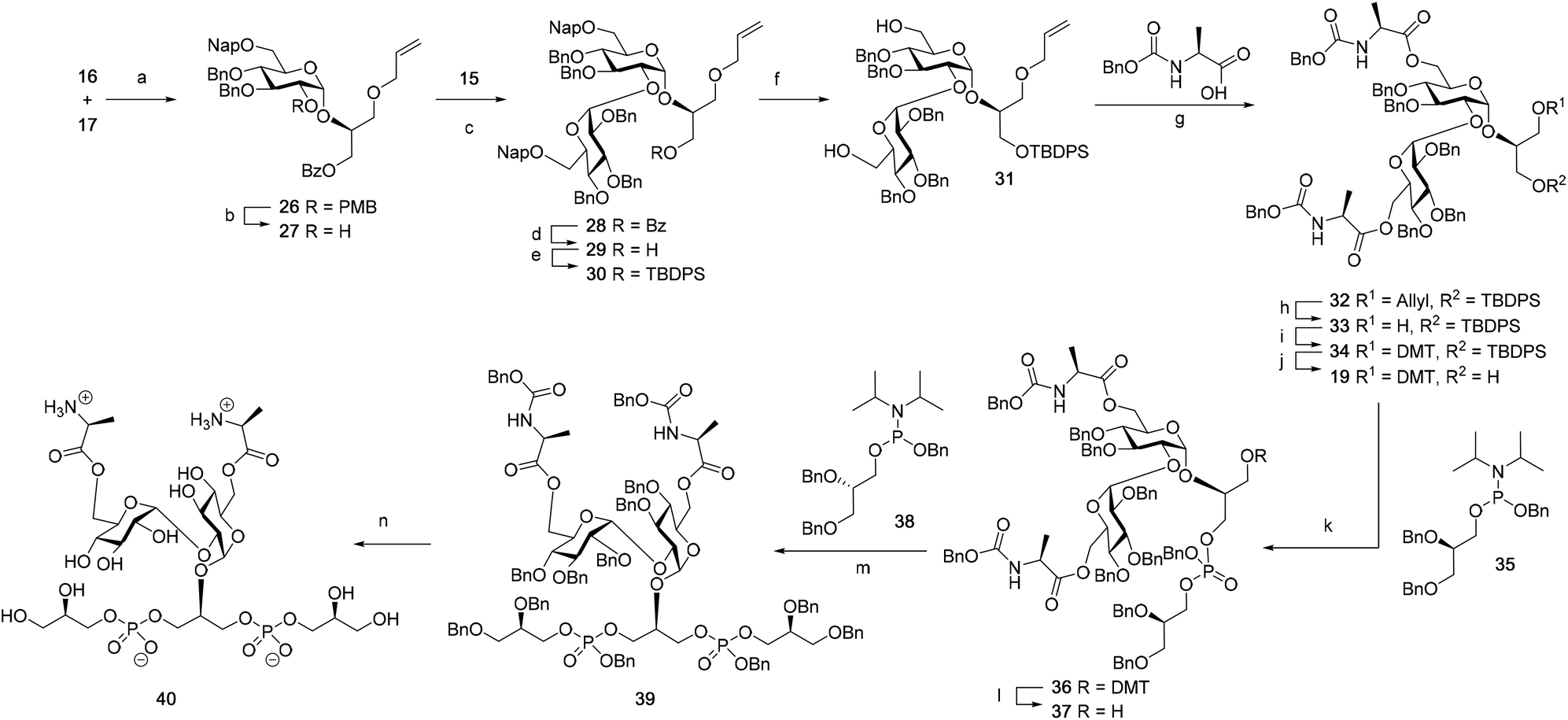 | ||
| Scheme 5 Synthesis of a di-alanyl-α-kojibiose containing LTA fragment. (a) TMSI, Ph3PO, DCM, 36 h, 76%, α:β > 20:1. (b) TES, HCl/HFIP, DCM:HFIP = 1:1 (v:v), 93%. (c) DMF, TfOH, 24 h, 95%, α:β > 10:1. (d) CH3ONa, DCM:CH3OH = 1:1 (v:v), 92%. (e) TBDPSCl, imidazole, DMF, 99%. (f) DDQ, DCM:H2O = 10:1 (v:v), 77%. (g) PyBOP, NMI, DCM, 90%. (h) (1) Ir(COD)(Ph2MeP)2PF6; (2) I2, NaHCO3, H2, THF, 85%. (i) DMTrCl, DIPEA, DCM, 95%. (j) HF-pyridine, THF/pyridine, 86%. (k) 1) DCI, CH3CN; (2) CSO. (l) TsOH, DCM/CH3OH = 1:1 (v:v), 93%. (m) DCI, CH3CN; (2) CSO, 88%. (n) H2, Pd/C, dioxane/H2O = 2:1 (v:v), 3 day, 95%. | ||
Conclusion
In conclusion, we have here explored additive controlled glycosylations for the construction of α-1,2-glucosyl linkages. After the successful construction of α-1,3-glucosyl and α-1,4-glucosyl linkages, described previously, we have shown that conditions, involving the activation of per-benzylated glucosyl imidate donors with stoichiometric TfOH in the presence of an excess DMF, can be profitably used for the construction of α-1,2-glucosyl linkages. The assembly of a linear kojitetraose however has brought to light that the stereoselectivity of the glycosylation reactions decrease with growing chain length. A kojibiose was assembled with excellent stereoselectivity (α:β = 15:1) and elongation of this disaccharide proceeded with good selectivity (α:β = 10:1), but extension of the trisaccharide with a fourth residue led to an anomeric mixture (α:β = 2:1). Using the methodology and building on a triad of benzyl-type protecting groups, a D-alanine-kojibiose functionalized LTA from the E. faecalis cell wall was synthesized. Through the use of mild global deprotection conditions the labile D-alanine esters were maintained in the final product. The generation of this structure will enable binding studies with antibodies, which may pave the way to the generation of a synthetic vaccine, based on this structure, directed at this important nosocomial pathogen.
Experimental
General experimental procedures
All reagents were of commercial grade and used as received. All moisture sensitive reactions were performed under an argon atmosphere. DCM used in the glycosylation reactions was dried with flamed 4 Å molecular sieves before being used. Reactions were monitored by TLC analysis with detection by UV (254 nm) and where applicable by spraying with 20% sulfuric acid in EtOH or with a solution of (NH4)6Mo7O24·4H2O (25 g L−1) and (NH4)4Ce(SO4)4·2H2O (10 g L−1) in 10% sulfuric acid (aq.) followed by charring at ∼150 °C. Column chromatography was carried out using silica gel (0.040–0.063 mm). Size-exclusion chromatography was carried out using Sephadex LH-20.1H and 13C spectra were recorded on a Bruker AV 400 and Bruker AV 500 in CDCl3 or D2O. Chemical shifts (δ) are given in ppm relative to tetramethylsilane as internal standard (1H NMR in CDCl3) or the residual signal of the deuterated solvent. Coupling constants (J) are given in Hz. All 13C spectra are proton decoupled. NMR peak assignments were made using COSY and HSQC experiments, where applicable Clean TOCSY, HMBC and GATED experiments were used to further elucidate the structure. The anomeric product ratios were analysed through integration of proton NMR signals.Experimental procedures and characterization data of products
For the synthesis procedure and data of known compounds 13,1317,1822,192320 see references. We used “a”, “b”, “c” and “d” to specify the H-1 and C-13 NMR signals of sugar rings from the “reducing” to the “non-reducing” end and “°” to specify the H-1 and C-13 NMR signals of the spacer.
Standard procedures
O (6 eq.) in dry DCM were stirred over fresh flame-dried molecular sieves 3A under nitrogen. Then TMSI (1.0 eq.) was added slowly in the mixture. The reaction was stirred at room temperature until TLC-analysis indicated the reaction to be complete. The solution was diluted and the reaction quenched with saturated Na2S2O3. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The products were purified by size exclusion and silica gel column chromatography.
:HFIP (1:1, 0.1 M). TES (1.0 eq.) and 0.1 M HCl/HFIP (0.1 eq.) were added to the mixture. The reaction stirred until TLC-analysis indicated full consumption of the starting material (15 min–2 h). Then the mixture was diluted with DCM and the reaction quenched with saturated NaHCO3. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography.
:H2O (10:1, 0.1 M). DDQ (1.1 eq.) was added to the mixture. The reaction stirred until TLC-analysis indicated full consumption of the starting material (±2 h). Then the mixture was diluted with DCM and the reaction quenched with saturated Na2S2O3. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography.
:EA = 40:1–20:1). Compound 6 (9.6 g, 92% yield, α:β = 1:1) was obtained as yellow syrup. 1H-NMR (CDCl3, 500 MHz, 60 °C) δ 7.79–7.74 (m, aromatic H), 7.49–7.01 (m, aromatic H), 6.83–6.73 (m, aromatic H), 6.40 (bs, 1 H, H-1α), 5.58 (bs, 1 H, H-1β), 4.96–4.53 (m), 4.02–3.95 (m), 3.79–3.59 (m), 3.40 (bs, 1 H).13C-APT (CDCl3, 125 MHz, 60 °C) δ 159.77, 159.73, 143.96, 143.73, 143.42, 138.99, 138.77, 138.38, 138.31, 135.81, 135.70, 133.57, 133.32, 130.33, 130.26 (aromatic C), 129.86, 129.43, 128.80, 128.46, 128.43, 128.33, 128.28, 128.06, 127.98, 127.94, 127.91, 127.84, 127.78, 127.69, 127.65, 127.60, 126.78, 126.72, 126.22, 126.20, 126.01, 125.97, 124.41, 124.28, 119.69, 119.58, 114.20, 114.17 (aromatic CH), 117.62 (q, CF3), 97.71, 94.00, 84.78, 81.75, 80.86, 79.41, 77.61, 77.36, 76.06, 75.31, 75.63, 75.55, 75.27, 75.02, 74.72, 73.82, 73.73, 73.48, 73.24, 68.73, 68.66, 55.83, 55.36. HR-MS: calculated for C46H42F3NO6 [M − [O(CNPh)CF3] + OH + NH4]+: 608.30066, found: 608.29853.
O (4.4 g, 15.8 mmol) and TMSI (371 μL, 2.6 mmol). The product was purified by silica gel column chromatography (pentane:EA = 10:1). Compound 7 (1300 mg, 75% yield, α:β = 10:1) was obtained as a colourless syrup. IR (neat, cm−1) ν 697, 735, 818, 1027, 1155, 1358, 1454, 2095, 2866, 2922. 1H-NMR (CDCl3, 400 MHz) δ 7.83–7.74 (m, 4 H, aromatic H), 7.49–7.12 (m, 18 H, aromatic H), 5.02 (d, J = 11.2 Hz, 1 H, 1 CHH), 4.92 (d, J = 12.4 Hz, 1 H, 1 CHH), 4.86–4.77 (m, 3 H, 3 CHH), 4.74 (d, J = 3.6 Hz, 1 H, H-1), 4.59 (d, J = 11.6 Hz, 1 H, 1 CHH), 4.48–4.41 (m, 2 H, 2 CHH), 3.99 (t, J = 9.2 Hz, 1 H, H-2), 3.75–3.59 (m, 6 H, H-6, H-5, H-4, H-2, H-1a°), 3.48–3.35 (m, 3 H, H-3°, H-1b°), 1.97–1.81 (m, 2 H, H-2°).13C-APT (CDCl3, 100 MHz) δ 138.93, 138.25, 138.14, 127.98, 135.72, 133.29, 133.17 (aromatic C), 128.52, 128.49, 128.46, 128.40, 128.06, 128.00, 127.84, 127.79, 127.70, 127.03, 126.29, 126.13, 126.06 (aromatic CH), 97.31 (C-1), 82.10 (C-3), 80.10 (C-2), 77.75 (C-4), 75.81, 75.24, 73.57, 73.53 (CH2), 70.43 (C-5), 68.50 (C-6), 64.83 (C-1°), 48.41 (C-3°), 28.96 (C-2°). HR-MS: calculated for C41H43N3O6 [M + NH4]+: 691.34901, found: 691.34807.
:H2O) and DDQ (482 mg, 2.1 mmol). The product was purified by silica gel column chromatography. Compound 8 (770 mg, 75% yield) was obtained as a colourless syrup. [α]20D +90.5 (c = 1, CHCl3). IR (neat, cm−1) ν 697, 735, 1027, 1066, 1038, 1359, 1454, 1497, 2095, 2870, 2923. 1H-NMR (CDCl3, 400 MHz) δ 7.39–7.13 (m, 15 H, aromatic H), 4.94–4.80 (m, 4 H, H-1, 3 CHH), 4.63 (d, J = 12.4 Hz, 1 H, 1 CHH), 4.52–4.48 (m, 2 H, 2 CHH), 3.85–3.61 (m, 7 H, H-6, H-5, H-4, H-3, H-2, H-3a°), 3.56–3.51 (m, 1 H, H-3b°), 3.43–3.32 (m, 2 H, H-1°), 2.18 (d, J = 7.6 Hz, 1 H, OH), 1.94–1.82 (m, 2 H, H-2°).13C-APT (CDCl3, 100 MHz) δ 138.69, 138.11, 137.93 (aromatic C), 128.50, 128.47, 128.02, 127.97, 127.96, 127.84, 127.80, 127.77 (aromatic CH), 98.66 (C-1), 83.31 (C-3), 77.46 (C-4), 75.43, 75.11, 73.59 (CH2), 72.94 (C-2), 70.82 (C-5), 68.47 (C-6), 65.15 (C-1°), 48.53 (C-3°), 28.86 (C-2°). HR-MS: calculated for C30H35N3O6 [M + NH4]+: 551.28641, found: 551.28604.
:MeOH = 1:1). Compound 9 (1160 mg, 95% yield, α:β = 15:1) was obtained as a colourless syrup. [α]20D +75.4 (c = 1, CHCl3). IR (neat, cm−1) ν 697, 736, 1029, 1069, 1359, 1454, 1497, 2098, 2866, 2922. 1H-NMR (CDCl3, 400 MHz) δ 7.83–7.71 (m, 4 H, aromatic H), 7.49–7.45 (m, 3 H, aromatic H), 7.34–7.21 (m, 23 H, aromatic H), 7.16–7.03 (m, 7 H, aromatic H), 5.07 (d, J = 3.2 Hz, 1 H, H-1b), 5.05 (d, J = 3.2 Hz, 1 H, H-1a), 5.01–4.79 (m, 8 H, 8 CHH), 4.63–4.39 (m, 5 H, 5 CHH), 4.31 (d, J = 12.4 Hz, 1 H, 1 CHH), 4.11 (t, J = 91.6 Hz, 1 H, H-3b), 4.06–3.99 (m, 2 H, H-3a, H-5a), 3.83 (dd, J1 = 3.2 Hz, J2 = 9.6 Hz, 1 H, H-2a), 3.78–3.61 (m, 7 H, H-6, H-5b, H-4b, H-4a, H-2b, H-3a°), 3.55–3.39 (m, 3 H, H-6, H-3b°), 3.29–3.19 (m, 2 H, H-1°), 1.84–1.70 (m, 2 H, H-2°). 13C-APT (CDCl3, 100 MHz) δ 138.81, 138.72, 138.32, 138.25, 138.00, 137.98, 135.69, 133.29, 133.11 (aromatic C), 128.49, 128.43, 128.42, 128.39, 128.29, 128.09, 128.03, 128.01, 127.97, 127.90, 127.81, 127.80, 127.71, 127.66, 127.62, 127.52, 126.87, 126.33, 126.12, 125.89 (aromatic CH), 96.06 (C-1a), 94.72 (C-1b), 82.17 (C-3b), 80.79 (C-3a), 79.14 (C-2b), 78.07 (C-4), 77.68 (C-4), 76.18, 75.71 (CH2), 75.45 (C-2a), 75.20, 74.95, 73.60, 73.45, 72.99 (CH2), 70.61 (C-5b), 70.44 (C-5a), 68.52 (C-6), 68.04 (C-6), 65.15 (C-1°), 48.36 (C-3°), 28.02 (C-2°). HR-MS: calculated for C68H71N3O6 [M + NH4]+: 1123.54269, found: 1123.54104.
:H2O) and DDQ (217 mg, 0.96 mmol). The product was purified by silica gel column chromatography. Compound 10 (416 mg, 49% yield) was obtained as a colourless syrup. [α]20D +90.8 (c = 1, CHCl3). IR (neat, cm−1) ν 697, 735, 1027, 1050, 1132, 1361, 1454, 2098, 2866, 2929.1H-NMR (CDCl3, 400 MHz) δ 7.38–7.06 (m, 30 H, aromatic H), 5.02–4.97 (m, 3 H, H-1a, H-1b, 1 CHH), 4.87–4.75 (m, 5 H, 5 CHH), 4.63–4.44 (m, 5 H, 5 CHH), 4.32 (d, J = 12.0 Hz, 1 H, 1 CHH), 3.94–3.65 (m, 11 H), 3.53–3.33 (m, 5 H, H-6), 1.91–1.84 (m, 2 H, H-2°).13C-APT (CDCl3, 100 MHz) δ 138.69, 138.61, 137.97, 137.86, 737.83 (aromatic C), 128.41, 128.39, 128.34, 128.30, 128.28, 128.16, 128.08, 127.97, 127.95, 127.87, 127.82, 127.78, 127.72, 127.65, 127.60, 127.55, 127.41 (aromatic CH), 96.21 (C-1b), 85.58 (C-1a), 83.28 (C-3b), 80.50 (C-3a), 77.79 (C-4), 76.94 (C-4), 76.00 (CH2), 75.37 (C-2a), 75.12, 74.75, 73.48, 73.29 (CH2), 72.79 (C-2b), 70.62 (C-5), 70.58 (C-5), 68.28 (C-6), 67.86 (C-6), 64.83 (C-1°), 48.33 (C-3°), 28.62 (C-2°). HR-MS: calculated for C68H71N3O11 [M + NH4]+: 983.48003, found: 983.47815.
:MeOH = 1:1). Compound 11 (372 mg, 67% yield, α:β = 10:1) was obtained as a colourless syrup. [α]20D +96.5 (c = 1, CHCl3). IR (neat, cm−1) ν 697, 735, 1029, 1069, 1359, 1454, 1497, 2098, 2845, 2921. 1H-NMR (CDCl3, 400 MHz) δ 7.82 (s, 1 H, aromatic H), 7.71–7.62 (m, 3 H, aromatic H), 7.52–7.50 (m, 1 H, aromatic H), 7.41–7.37 (m, 1H, aromatic H), 7.32–6.96 (m, 46 H, aromatic H), 5.49 (d, J = 3.6 Hz, 1 H, H-1c), 5.41 (d, J = 3.6 Hz, 1 H, H-1b), 5.23 (d, J = 2.0 Hz, 1 H, H-1a), 5.16 (d, J = 12.0 Hz, 1 H, 1 CHH), 4.99–4.34 (m, 18 H, 18 CHH), 44.18–3.52 (m, 21 H), 3.31–3.22 (m, 2 H, H-3°), 1.93–1.82 (m, 2 H, H-2°). 13C-APT (CDCl3, 100 MHz) δ 138.78, 138.76, 138.74, 138.45, 138.26, 138.16, 138.09, 137.98, 137.90, 136.09, 133.35, 132.94 (aromatic C), 128.72, 128.45, 128.44, 128.40, 128.38, 128.29, 128.26, 128.13, 128.06, 128.04, 127.93, 127.79, 127.72, 127.65, 127.60, 127.56, 127.51, 127.46, 126.11, 125.84, 125.77, 125.55 (aromatic CH), 95.84 (C-1c), 92.77 (C-1b), 92.09 (C-1a), 81.96 (C-3c), 81.45 (C-3a), 80.99 (C-3b), 79.97 (C-2c), 77.37 (2 C-4), 77.05 (C-4), 76.08, 75.99, 75.65, 74.94, 74.84 (CH2), 74.71 (C-2), 74.60 (C-2), 73.58, 73.50, 71.60 (CH2), 70.94 (C-5), 70.80 (C-5), 70.61 (C-5), 68.61 (C-6), 68.34 (C-6), 68.07 (C-6), 65.16 (C-1°), 48.31 (C-3°), 29.20 (C-2°). HR-MS: calculated for C95H99N3O16 [M + NH4]+: 1555.73636, found: 1555.73599.
:H2O) and DDQ (140 mg, 0.6 mmol). The product was purified by silica gel column chromatography. Compound 12 (300 mg, 47% yield) was obtained as a colourless syrup. [α]20D +122.1 (c = 1, CHCl3). IR (neat, cm−1) ν 697, 735, 1027, 1050, 1132, 1361, 1454, 2098, 2866, 2929.1H-NMR (CDCl3, 400 MHz) δ 7.37–6.98 (m, 45 H, aromatic H), 5.18 (d, J = 3.2 Hz, 1 H, H-1b), 5.06 (d, J = 3.2 Hz, 1 H, H-1a), 5.00–4.90 (m, 3 H, H-1a, 2 CHH), 4.84–4.30 (m, 16 H, 16 CHH), 4.01–3.34 (m, 22 H), 2.41 (s, 1 H, OH), 2.06–1.85 (m, 2 H, H-2°).13C-APT (CDCl3, 100 MHz) δ 138.89, 138.85, 138.56, 138.24, 138.22, 138.02, 137.96, 137.95, 137.89 (aromatic C), 128.56, 128.53, 128.51, 128.50, 128.42, 128.38, 128.36, 128.28, 128.08, 128.07, 127.86, 127.82, 127.79, 127.74, 127.67, 127.63, 127.60, 127.50 (aromatic CH), 96.03 (C-1c), 95.04 (C-1a), 91.66 (C-1b), 83.42 (C-3c), 80.90 (C-3a), 80.62 (C-3b), 77.89 (C-4), 77.73 (C-4), 76.84 (C-4), 76.30, 76.25, 75.20, 75.15, 74.94, 74.92 (CH2), 74.67 (C-2b), 74.00 (C-2a), 73.65, 73.59, 73.45 (CH2), 72.67 (C-2c), 70.69 (C-5), 70.64 (C-5), 70.57 (C-5), 68.38 (C-6), 68.00 (C-6), 67.97 (C-6), 64.50 (C-1°), 48.51 (C-3°), 28.77 (C-2°). HR-MS: calculated for C84H91N3O16 [M + NH4]+: 1415.67376, found: 1415.67378.
:MeOH = 1:1). Compound 14 (64 mg, 67% yield, α:β = 2:1) was obtained as a colourless syrup. IR (neat, cm−1) ν 695, 734, 1027, 1069, 1209, 1359, 1454, 1497, 1729, 2096, 2859, 2923. HR-MS: calculated for C118H125N3O21 [M + NH4]+: 1937.91438, found: 1937.91440.
:H2O (10:1, 15 mL). N-Iodosuccinimide (NIS) (660 mg, 2.93 mmol) was added in one portion and the reaction was stirred at room temperature for 2 hours. The solution was diluted with DCM and the reaction was quenched with saturated aqueous Na2S2O3. Then the organic layer was washed with water and brine. The organic layer was dried with anhydrous MgSO4, filtered and concentrated in vacuo, and the product purified by column chromatography. The lactol was obtained as a white solid. Next, the lactol was dissolved in acetone (15 mL). Cs2CO3 (713 mg, 2.19 mmol) and 2,2,2-trifluoro-N-phenylacetimidoyl chloride (355 μL, 2.19 mmol) were added to the solution respectively. The reaction was stirred overnight, then quenched with Et3N, filtered and concentrated in vacuo. The product was purified by column chromatography. Compound 15 (959 mg, 86% yield over two steps, α:β = 1:1) was obtained as yellow syrup. IR (neat, cm−1) ν 695, 734, 1027, 1073, 1153, 1208, 1314, 1454, 1597, 1716, 2869, 2915. 1H-NMR (CDCl3, 400 MHz) δ 7.82–6.50 (m, aromatic H), 6.53 (bs, 1 H, H-1), 5.67 (bs, 1 H, H-1), 5.00–4.61 (m), 4.52–4.46 (m), 4.06–3.97 (m), 3.81–3.66 (m). 13C-APT (CDCl3, 125 MHz) δ 143.78, 143.55, 138.67, 138.64, 137.92, 137.83, 135.45, 135.28, 133.35, 133.19, 133.16 (aromatic C), 129.51, 128.82, 128.64, 128.61, 128.57, 128.55, 128.49, 128.43, 128.36, 128.33, 128.13, 128.05, 127.99, 127.92, 127.88, 127.84, 127.81, 127.78, 127.00, 126.88, 126.49, 126.30, 126.26, 126.10, 126.08, 126.05, 124.39, 120.57, 119.44 (aromatic CH), 84.64, 81.62, 81.04, 79.37, 77.27, 76.94, 75.91, 75.82, 75.74, 75.44, 75.31, 75.17, 73.76, 73.65, 73.44, 73.19, 68.12.
:H2O (10:1, 77 mL). N-Iodosuccinimide (NIS) (3.0 g, 13.3 mmol) was added in one portion and the reaction was stirred at room temperature for 2 hours. The solution was diluted with DCM and the reaction was quenched with saturated aqueous Na2S2O3. Then the organic layer was washed with water and brine. The organic layer was dried with anhydrous MgSO4, filtered and concentrated in vacuo, and the product purified by column chromatography. The lactol was obtained as a white solid. Next, the lactol was dissolved in acetone (70 mL). Cs2CO3 (6.3 g, 19.5 mmol) and 2,2,2-trifluoro-N-phenylacetimidoyl chloride (3.2 mL, 19.5 mmol) were added to the solution respectively. The reaction was stirred overnight, then quenched with Et3N, filtered and concentrated in vacuo. The product was purified by column chromatography. Compound 16 (4.48 g, 84% yield over two steps, α:β = 1:1) was obtained as yellow syrup. 1H-NMR (CDCl3, 500 MHz, 60 °C) δ 7.79–7.74 (m, aromatic H), 7.49–7.01 (m, aromatic H), 6.83–6.73 (m, aromatic H), 6.40 (bs, 1 H, H-1α), 5.58 (bs, 1 H, H-1β), 4.96–4.53 (m), 4.02–3.95 (m), 3.79–3.59 (m), 3.40 (bs, 1 H). 13C-APT (CDCl3, 125 MHz, 60 °C) δ 159.77, 159.73, 143.96, 143.73, 143.42, 138.99, 138.77, 138.38, 138.31, 135.81, 135.70, 133.57, 133.32, 130.33, 130.26 (aromatic C), 129.86, 129.43, 128.80, 128.46, 128.43, 128.33, 128.28, 128.06, 127.98, 127.94, 127.91, 127.84, 127.78, 127.69, 127.65, 127.60, 126.78, 126.72, 126.22, 126.20, 126.01, 125.97, 124.41, 124.28, 119.69, 119.58, 114.20, 114.17 (aromatic CH), 117.62 (q, CF3), 97.71, 94.00, 84.78, 81.75, 80.86, 79.41, 77.61, 77.36, 76.06, 75.31, 75.63, 75.55, 75.27, 75.02, 74.72, 73.82, 73.73, 73.48, 73.24, 68.73, 68.66, 55.83, 55.36. HR-MS: calculated for C47H44F3NO7 [M − [O(CNPh)CF3] + OH + Na]+: 643.26662, found: 643.26659.
O), 134.30, 133.18 (aromatic CH), 129.87 (aromatic C), 129.80 (aromatic CH), 128.43 (C-5), 117.54 (C-6), 772.43, 71.02, 68.93 (C-2), 66.07. HR-MS: calculated for C13H16O4 [M + Na]+: 259.0941, found: 259.0948.
O (3.16 g, 11.4 mmol) and TMSI (270 μL, 1.9 mmol). The product was purified by silica gel column chromatography. Compound 26 (1100 mg, 76% yield, α:β > 20:1) was obtained as a colourless syrup. [α]20D +38.9 (c = 1, CHCl3). IR (neat, cm−1) ν 698, 713, 820, 1037, 1072, 1095, 1249, 1272, 1452, 1513, 1720, 2865, 2912. 1H-NMR (CDCl3, 400 MHz) δ 7.99–7.97 (m, 2 H, aromatic H), 7.76–7.63 (m, 4 H, aromatic H), 7.46–7.08 (m, 16 H, aromatic H), 6.92 (bd, 2 H, aromatic H), 6.85–6.82 (m, 2 H, aromatic H), 5.93–5.84 (m, 1 H, H-5°), 5.31–5.25 (m, 1 H, H-6a°), 5.19–5.16 (m, 2 H, H-1a, H-6b°), 4.99 (d, J = 10.8 Hz, 1 H, CHH), 4.81–4.77 (m, 2 H, 2 CHH), 4.70–4.63 (m, 3 H, 3 CHH), 4.57 (dd, J1 = 3.6 Hz, J2 = 11.6 Hz, 1 H, H-1a°), 4.45–4.35 (m, 3 H, 2 CHH, H-1b°), 4.28–4.24 (m, 1 H, H-2°), 4.04–3.99 (m, 4 H, H-3a, H-5a, H-4°), 3.74 (s, 3 H, OCH3), 3.73–3.58 (m, 4 H, H-2a, H-4a, H-3°), 3.50 (dd, 1 H, J1 = 2.8 Hz, J2 = 11.8 Hz, H-6aa), 3.37 (dd, 1 H, J1 = 2.0 Hz, J2 = 10.4 Hz, H-6ab).13C-APT (CDCl3, 100 MHz) δ 166.19 (CO), 159.30, 138.91, 138.22, 135.16 (aromatic C), 134.38 (C-5°), 133.11, 132.96 (aromatic C), 132.88 (aromatic CH), 130.27, 129.84 (aromatic C), 129.62, 129.60, 128.31, 128.28, 128.11, 127.85, 127.82, 127.67, 127.48, 127.44, 126.80, 126.03, 125.97, 125.83 (aromatic CH), 117.19 (C-6°), 113.78 (aromatic CH), 96.13 (C-1a), 81.83 (C-5a), 79.26 (C-2a), 77.47 (C-4a), 75.59, 74.83 (CH2), 73.59 (C-2°), 73.52, 72.43 (CH2), 72.35 (C-4°), 70.23 (C-3a), 69.59 (C-3°), 67.97 (C-6a), 65.01 (C-1°), 55.16 (OCH3). HR-MS: calculated for C52H54O10 [M + Na]+: 861.3609, found: 861.3647.
:HFIP), triethylsilane (190 μL, 1.19 mmol) and 0.1 M HCl/HFIP (1.2 ml, 0.12 mmol). The product was purified by silica gel column chromatography (PE:EA = 6:1). Compound 27 (800 mg, 93% yield) was obtained as a colourless syrup. [α]20D +73.0 (c = 1, CHCl3). IR (neat, cm−1) ν 710, 713, 738, 1027, 1070, 1095, 1272, 1452, 1721, 2866, 2915. 1H-NMR (CDCl3, 400 MHz) δ 8.02–8.00 (m, 2 H, aromatic H), 7.78–7.66 (m, 4 H, aromatic H), 7.49–7.09 (m, 14 H, aromatic H), 6.96–6.94 (m, 4 H, aromatic H), 5.93–5.84 (m, 1 H, H-5°), 5.32–5.26 (m, 1 H, H-6°a), 5.23–5.19 (m, 1 H, H-6°b), 5.12 (d, J = 3.2 Hz, 1 H, H-1a), 4.99 (d, J = 11.2 Hz, 1 H, 1 CHH), 4.81 (d, J = 11.2 Hz, 1 H, 1 CHH), 4.78 (d, J = 11.2 Hz, 1 H, 1 CHH), 4.69 (d, J = 12.4 Hz, 1 H, 1 CHH), 4.49–4.38 (m, 4 H, 3 CHH, H-1°),4.24–4.18 (m, 1 H, H-2°), 4.07–3.98 (m, 2 H, H-4°), 3.97–3.93 (m, 1 H, H-3a), 3.80–3.73 (m, 2 H, H-2a, H-5a), 3.68–3.57 (m, 4 H, H-3°, H-4a, H-6aa), 3.42 (dd, 1 H, J1 = 2.0 Hz, J2 = 10.4 Hz, H-6ab), 2.77 (d, 1 H, J = 8.0 Hz, OH). 13C-APT (CDCl3, 100 MHz) δ 166.29 (CO), 138.93, 138.25, 135.30 (aromatic C), 134.03 (C-5°), 133.24 (aromatic C), 133.18 (aromatic CH), 133.07, 129.83 (aromatic C), 129.75, 128.47, 128.41, 128.26, 127.99, 127.94, 127.80, 127.77, 127.61, 127.59, 126.86, 126.16, 126.05, 125.95 (aromatic CH), 117.92 (C-6°), 99.40 (C-1a), 83.53 (C-5a), 77.02 (C-4a), 76.14 (C-2°), 75.33, 74.95, 73.65 (CH2), 73.44 (C-2a), 72.46 (C-4°), 71.08 (C-3a), 69.23 (C-3°), 68.11 (C-6a), 64.80 (C-1°). HR-MS: calculated for C44H46O9 [M + Na]+: 741.3034, found: 741.3062.
:MeOH = 1:1). Compound 28 (2.06 g, 95% yield, α:β > 10:1) was obtained as a colourless syrup. Then the compound was purified by silica gel column chromatography to get pure α product. [α]20D +75.1 (c = 1, CHCl3). IR (neat, cm−1) ν 698, 713, 820, 1046, 1070, 1095, 1272, 1359, 1454, 1721, 2863, 2919.1H-NMR (CDCl3, 400 MHz) δ 8.00–7.81 (m, 2 H, aromatic H), 7.78–7.63 (m, 8 H, aromatic H), 7.44–6.91 (m, 34 H, aromatic H), 5.81–5.76 (m, 1 H, H-5°), 5.46 (d, J = 3.6 Hz, 1 H, H-1a), 5.29 (d, J = 3.6 Hz, 1 H, H-1b), 5.22–5.10 (m, 2 H, H-6°), 4.98–4.67 (m, 10 H, 10 CHH), 4.56–4.33 (m, 6 H, 4 CHH, H-1°), 4.29–4.23 (m, 1 H, H-2°), 4.14–4.04 (m, 4 H, H-5a, H-5b, H-3a, H-3b), 3.91–3.88 (m, 3 H, H-3a, H-4°), 3.80–3.43 (m, 9 H, H-3°, H-2b, H-4a, H-4b, H-6a, H-6b). 13C-APT (CDCl3, 100 MHz) δ 166.23 (CO), 138.69, 138.46, 138.34, 138.31, 138.27, 135.33, 135.22 (aromatic C), 134.27 (C-5°), 133.18, 133.15, 133.01, 132.99 (aromatic C), 132.91 (aromatic CH), 129.92 (aromatic C), 129.62, 128.41, 128.38, 128.35, 128.31, 128.19, 128.17, 128.15, 128.09, 127.92, 127.87, 127.74, 127.69, 127.63, 127.54, 127.51, 127.46, 127.43, 127.34, 126.88, 126.80, 126.09, 126.06, 125.98, 125.85, 125.84 (aromatic CH), 117.41 (C-6°), 99.70 (C-1b), 94.53 (C-1a), 82.10 (C-5b), 80.73 (C-5a), 79.35 (C-2b), 77.80 (C-4), 77.56 (C-4), 76.03, 75.54 (CH2), 75.39 (C-2a), 74.85, 74.82, 73.54, 73.53 (CH2), 73.14 (C-2°), 72.36, 72.22 (CH2), 70.46 (C-3a and 3b), 69.23 (C-3°), 68.98 (C-6a and 6b), 64.96 (C-1°). HR-MS: calculated for C82H82O14 [M + Na]+: 1313.5597, found: 1313.5624.
:CH3OH (1:1/v:v, 8.5 mL) stirring at room temperature. Then 5 drops of solution of CH3ONa in CH3OH (5.4 M) was added in the mixture. The reaction was stirred at rt until TLC-analysis showed complete conversion of the starting martial (3 h). Then the mixture was diluted with DCM. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography. Compound 29 (930 mg, 92% yield) was obtained as a colourless syrup. [α]20D +69.4 (c = 1, CHCl3). IR (neat, cm−1) ν 698, 737, 818, 1069, 1209, 1359, 1454, 2865, 2921.1H-NMR (CDCl3, 400 MHz) δ 7.83–7.69 (m, 8 H, aromatic H), 7.49–7.09 (m, 25 H, aromatic H), 7.03–6.92 (m, 6 H, aromatic H), 5.86–5.76 (m, 1 H, H-5°), 5.31 (d, J = 3.6 Hz, 1 H, H-1b), 5.22–5.10 (m, 3 H, H-6°, H-1a), 4.94–4.35 (m, 14 H, 14 CHH), 4.09–3.98 (m, 4 H, H-5a, H-5b, H-3a, H-3b), 3.89–3.45 (m, 14 H). 13C-APT (CDCl3, 100 MHz) δ 138.73, 138.52, 138.31, 138.21, 137.98135.41, 135.20 (aromatic C), 134.56 (C-5°), 133.33, 133.29, 133.18, 133.12 (aromatic C), 128.60, 128.45, 128.42, 128.31, 128.22, 128.07, 128.04, 128.01, 127.95, 127.84, 127.81, 127.78, 127.74, 127.67, 127.64, 127.49, 126.98, 126.26, 126.19, 126.06, 126.04, 125.97 (aromatic CH), 117.25 (C-6°), 95.62 (C-1b), 95.08 (C-1a), 82.22 (C-5a), 80.73 (C-5b), 79.24 (C-2b), 78.89 (C-2b), 78.28 (C-4), 77.71 (C-4), 76.15 (CH2), 75.73 (C-2°), 75.69, 75.16, 74.99, 73.70, 73.61, 72.86 (CH2), 72.34 (C-4°), 70.86 (C-3b), 70.59 (C-3a), 69.67 (C-3°), 68.64, 68.08 (C-6a and 6b), 63.61 (C-1°). HR-MS: calculated for C75H78O13 [M + Na]+: 1209.5335, found: 1209.5356.
:H2O) and DDQ (170 mg, 0.75 mmol). The product was purified by silica gel column chromatography. Compound 31 (280 mg, 77% yield, pentane:EA = 4:1, Rf = 0.33) was obtained as a colourless syrup. [α]20D +72.3 (c = 1, CHCl3). IR (neat, cm−1) ν 698, 737, 1027, 1072, 1209, 1361, 1454, 2858, 2929. 1H-NMR (CDCl3, 400 MHz) δ 7.66–7.63 (m, 4 H, aromatic H), 7.41–7.07 (m, 31 H, aromatic H), 5.88–5.78 (m, 1 H, H-5°), 5.44 (d, J = 3.2 Hz, 1 H, H-1a), 5.34 (d, J = 3.6 Hz, 1 H, H-1b), 5.23–5.13 (m, 2 H, H-6°), 4.99–4.58 (m, 10 H, 10 CHH), 4.11–4.00 (m, 3 H, H-5a, H-5b, H-2°), 3.92–3.88 (m, 3 H, H-3b, H-4°), 3.76–3.51 (m, 14 H), 1.76 (s, 1 H, OH), 1.64 (s, 1 H, OH), 1.04 (s, 9 H, 3 CH3). 13C-APT (CDCl3, 100 MHz) δ 138.69, 138.58, 138.44, 138.38, 138.35 (aromatic C), 135.62, 135.61 (aromatic CH), 134.52 (C-5°), 133.27, 133.24 (aromatic C), 129.81, 128.46, 128.44, 128.41, 128.38, 128.36, 128.05, 127.87, 127.84, 127.77, 127.70, 127.67, 127.62, 127.58 (aromatic CH), 117.28 (C-6°), 94.19 (C-1a), 93.40 (C-1b), 81.80 (C-5b), 80.69 (C-5a), 79.65 (C-2b), 77.31 (C-4), 77.25 (C-4), 76.09, 75.57 (CH2), 75.04 (C-2°), 74.93 (CH2), 74.90 (C-2a), 72.24, 71.93 (CH2), 71.25 (C-3b), 70.94 (C-3a), 69.84, 63.67, 61.68, 61.50 (CH2), 26.93 (3 CH3), 19.22. HR-MS: calculated for C69H80O13Si [M + Na]+: 1167.5260, found: 1167.5280.
O), 138.54, 138.32, 138.26, 138.19, 136.33 (aromatic C), 135.62, 135.60 (aromatic CH), 134.46 (C-5°), 133.20 (aromatic C), 129.86, 128.57, 128.46, 128.40, 128.35, 128.19, 128.10, 128.03, 127.88, 127.76, 127.70, 127.66, 127.60 (aromatic CH), 117.39 (C-6°), 96.64 (C-1a), 93.00 (C-1b), 81.86 (C-5b), 80.79 (C-5a), 79.60, 77.36, 76.12, 75.63, 74.96, 74.68, 72.22, 71.84, 69.30, 69.12, 68.71, 66.95, 63.73, 63.66, 63.50, 49.69 (2 CHNH), 29.34 (CH3), 26.95 (3 CH3), 19.23, 18.73 (CH3). HR-MS: calculated for C91H102N2O19Si [M + Na]+: 1577.67383, found: 1577.67285.
O), 138.38, 138.15, 137.92, 137.83, 137.55, 136.29 (aromatic C), 135.65, 135.62 (aromatic CH), 133.17, 133.09 (aromatic C), 128.71, 128.58, 128.52, 128.48, 128.43, 128.39, 128.22, 128.20, 128.18, 128.16, 127.98, 127.95, 127.93, 127.90, 127.77, 127.73 (aromatic CH), 96.63 (C-1a), 95.90 (C-1b), 82.27 (C-5b), 80.14 (C-5a), 79.57 (C-2°), 77.97 (C-2b), 77.67 (C-4), 77.59 (C-4), 76.41 (C-2a), 75.95, 75.61, 75.08, 75.03, 73.81 (CH2), 69.21 (C-3b), 69.06 (C-3a), 69.97, 63.84, 63.09, 63.03, 62.12 (CH2), 49.68 (CHNH), 49.60 (CHNH), 26.94 (3 CH3), 19.26, 18.70 (CH3), 18.57 (CH3). HR-MS: calculated for C88H98N2O19Si [M + Na]+: 1537.64253, found: 1537.64262.
O), 159.55 (aromatic C), 156.90, 156.87 (2 CO), 146.16, 139.99, 139.74, 139.68, 138.48, 139.32, 138.03, 137.98, 136.85, 136.69 (aromatic C), 136.40, 136.38 (aromatic CH), 134.06, 134.03 (aromatic C), 131.04, 131.00, 130.67, 130.65, 129.10, 129.07, 129.01, 128.95, 128.83, 128.67, 128.55, 128.40, 128.33, 128.26, 128.24, 128.17, 127.58, 114.08, 114.06 (aromatic CH), 94.97 (C-1a), 93.94 (C-1b), 87.37 (quaternary C), 82.55 (C-5a), 81.79 (C-5b), 80.87 (C-2b), 78.59 (C-4), 78.26 (C-4), 76.90 (C-2°), 76.58 (CH2), 76.03 (C-2a), 75.86, 75.64, 75.53, 72.41 (CH2), 70.29 (C-3b), 70.11 (C-3a), 66.93, 66.90, 64.59, 64.54, 63.86 (CH2), 55.53 (2 OCH3), 50.72 (2 CHNH), 27.39 (3 CH3), 19.73 (quaternary C), 17.97 (CH3), 17.93 (CH3). HR-MS: calculated for C109H116N2O21Si [M + Na]+: 1839.77321, found: 1839.77281.
:1/v:v, 6 mL) stirring at room temperature. Then HF-pyridine (0.4 mL) was added in the mixture. The reaction was stirred at rt until TLC-analysis showed complete conversion of the starting martial (10 h). Then the mixture was diluted with EtOAc. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography. Compound 19 (548 mg, 86% yield) was obtained as a colourless syrup. [α]20D +64.0 (c = 1, CHCl3). IR (neat, cm−1) ν 698, 751, 830, 1029, 1070, 1176, 1213, 1251, 1302, 1454, 1508, 1608, 1724, 2928.1H-NMR (d-acetone, 400 MHz) δ 7.49–7.10 (m, 44 H, aromatic H), 6.87–6.73 (m, 5 H, 4 aromatic H, 1 NH), 6.74 (d, J = 7.6 Hz, 1 H, NH), 5.52 (d, J = 2.8 Hz, 1 H, H-1a), 5.26 (d, J = 2.8 Hz, 1 H, H-1b), 5.10–5.02 (m, 5 H, 5 CHH), 4.93–4.80 (m, 4 H, 2 CHH), 4.70–4.62 (m, 3 H), 4.56–4.48 (m, 2 H), 4.42–4.21 (m, 7 H), 4.17–4.01 (m, 4 H), 3.88–3.86 (m, 2 H), 3.75–3.47 (m, 12 H), 3.19 (dd, J1 = 5.6 Hz, J2 = 10.0 Hz, 1 H), 1.47–1.38 (m, 6 H, 2 CH3). 13C-APT (d-acetone, 100 MHz) δ 173.50, 173.43 (2 CO), 159.60, 159.58 (aromatic C), 156.99, 156.89 (2 CO), 146.16, 140.03, 139.77, 139.58, 139.50, 138.07, 138.04, 136.93, 136.71 (aromatic C), 131.07, 130.99, 129.25, 129.14, 129.13, 129.09, 129.07, 129.05, 129.04, 128.99, 128.83, 128.76, 128.67, 128.64, 128.43, 128.34, 128.25, 128.19, 128.62, 114.06, 114.04 (aromatic CH), 95.52 (C-1a), 93.96 (C-1b), 87.35 (quaternary C), 82.63, 81.72, 80.85, 78.83, 78.59, 78.29, 76.48, 76.06, 75.86, 75.63, 72.42, 70.34, 70.20, 66.92, 65.28, 64.32, 63.92, 63.43, 55.54 (2 OCH3), 50.78 (CHNH), 50.74 (CHNH), 17.98 (2 CH3). HR-MS: calculated for C93H98N2O21 [M + NH4]+: 1596.70003, found: 1596.69828.
:1 (v:v, 1 mL) stirring at room temperature. Then TsOH (0.1 eq.) was added in the solution. The reaction was stirred at rt until TLC-analysis showed complete conversion of the starting martial (2–3 h). Then the reaction was quenched with Et3N, diluted with DCM. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The product was purified by size exclusion chromatography (DCM:MeOH = 1:1). Compound 37 (40 mg, 93%) was obtained as a colourless syrup. 1H-NMR (d-acetone, 400 MHz) δ 7.47–7.05 (m, 50 H, aromatic H), 6.95 (d, J = 7.6 Hz, 1 H, 1 NH), 6.74 (d, J = 7.6 Hz, 1 H, NH), 5.42–5.35 (m, 2 H, 2 H-1), 5.12–3.58 (m, 44 H), 2.89 (s, 1 H), 1.42–1.37 (m, 6 H, 2 CH3). 13C-APT (d-acetone, 100 MHz) δ 173.52, 173.33, 156.93, 156.82 (4 CO), 138.91, 139.64, 139.49, 139.39, 139.33, 138.03 (aromatic C), 129.36, 129.17, 129.09, 129.04, 129.02, 128.87, 128.85, 128.77, 128.71, 128.68, 128.60, 128.53, 128.52, 128.46, 128.35, 128.24, 128.22, 128.17, 128.14 (aromatic CH), 96.03 (C-1), 85.08 (C-1), 82.74, 81.47, 79.96, 78.89, 78.53, 77.80, 77.71, 77.67, 77.60, 76.43, 76.09, 75.93, 75.56, 73.72, 73.07, 72.50, 70.22, 70.15, 69.87, 69.84, 67.89, 67.73, 66.81, 64.14, 63.64, 61.32, 61.25, 63.43, 55.54, 50.76, 50.65 (CHNH), 50.74 (CHNH), 17.85, 17.79 (2 CH3).31P (d-acetone, 400 MHz) δ 0.77, 0.66.
:MeOH = 1:1). Compound 39 (44 mg, 88%) was obtained as a colourless syrup. 1H-NMR (d-acetone, 400 MHz) δ 7.45–7.06 (m, 65 H, aromatic H), 6.94–6.92 (m, 1 H, 1 NH), 6.73 (d, J = 7.6 Hz, 1 H, NH), 5.38–5.34 (m, 2 H, 2 H-1), 5.12–3.54 (m, 57 H), 1.42–1.37 (m, 6 H, 2 CH3). 13C-APT (d-acetone, 100 MHz) δ 173.39, 173.22, 156.79, 156.68 (4 CO), 139.87, 139.54, 139.51, 139.49, 139.33, 139.23, 137.88, 137.19, 137.14, 137.07, 137.02 (aromatic C), 129.26, 129.23, 129.11, 129.06, 129.05, 129.04, 129.00, 129.96, 129.93, 128.90, 128.87, 128.85, 128.83, 128.72, 128.68, 128.62, 128.55, 128.53, 128.45, 128.38, 128.35, 128.26, 128.20, 128.11, 128.07, 127.99, 127.97 (aromatic CH),96.37, 96.23 (C-1), 94.95, 94.80 (C-1), 82.60, 81.25, 80.24, 78.55, 78.24, 77.53, 77.50, 77.47, 77.45, 77.38, 76.27, 76.24, 76.07, 75.95, 75.79, 75.49, 75.43, 73.57, 72.97, 72.93, 72.87, 72.37, 72.35, 72.33, 72.31, 70.38, 70.07, 69.83, 69.79, 69.76, 69.73, 69.71, 69.67, 67.85, 67.80, 67.76, 67.69, 67.65, 66.68, 65.92, 65.87, 63.91, 63.55, 50.61 (CHNH), 50.51 (CHNH), 17.71, 17.64 (2 CH3). 31P (d-acetone, 400 MHz) δ 0.53, 0.57, 0.62, 0.65, 0.68, 0.78, 0.82. HR-MS: calculated for C120H130N2O29P2 [M + Na]+: 2147.81267, found: 2147.81610.
:1 (v:v, 1 mL) stirring at room temperature. Then Pd/C was added in the solution. The reaction was stirred at rt for 3 days under a H2 atmosphere, filtered and concentrated in vacuo. Compound 40 (∼5 mg, 93%) was obtained as a white solid. 1H-NMR (d-acetone, 400 MHz) δ 5.42 (d, 1 H, H-1), 5.11 (d, 1 H, H-1), 4.63–4.57 (m, 2 H), 4.42–4.38 (m, 2 H), 4.21–4.15 (m, 5 H), 3.98–3.56 (m, 20 H), 3.50–3.38 (m, 2 H), 1.87 (s, 1 H), 1.57–1.53 (m, 6 H, 2 CH3). 13C-APT (d-acetone, 100 MHz) δ 170.46, 170.38 (2 CO), 96.00, 94.42, 75.04, 74.70, 72.37, 72.17, 71.71, 71.54, 71.04, 70.85, 70.72, 70.68, 70.64, 70.61, 69.82, 69.43, 69.20, 66.42, 66.37, 66.32, 65.22, 64.90, 64.50, 62.47, 62.04, 62.01, 60.38, 50.45, 48.72, 16.06, 15.20. 31P (d-acetone, 400 MHz) δ 1.22, 1.13. HR-MS: calculated for C27H52N2O25P2 [M + H]+: 867.24071, found: 867.24106.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Chinese Scholarship Council (CSC grant to L. W.), the European Research Council (ERC-CoG-726072-‘GLYCONTROL’, to J. D. C. C.) and the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement Glycovax No. 675671.Notes and references
- S. Shimomura and T. Fukui, Biochemistry, 1980, 19, 2287–2294 CrossRef CAS PubMed.
- (a) Y. Ai, B. Nelson, D. F. Birt and J.-l. Jane, Carbohydr. Polym., 2013, 98, 1266–1271 CrossRef CAS PubMed; (b) J. Geurtsen, S. Chedammi, J. Mesters, M. Cot, N. N. Driessen, T. Sambou, R. Kakutani, R. Ummels, J. Maaskant, H. Takata, O. Baba, T. Terashima, N. Bovin, C. M. Vandenbroucke-Grauls, J. Nigou, G. Puzo, A. Lemassu, M. Daffe and B. J. Appelmelk, J. Immunol., 2009, 183, 5221–5231 CrossRef CAS PubMed; (c) S. Chen, Q. He and M. L. Greenberg, Mol. Microbiol., 2008, 68, 1061–1072 CrossRef CAS PubMed; (d) T. Sambou, P. Dinadayala, G. Stadthagen, N. Barilone, Y. Bordat, P. Constant, F. Levillain, O. Neyrolles, B. Gicquel, A. Lemassu, M. Daffé and M. Jackson, Mol. Microbiol., 2008, 70, 762–774 CrossRef CAS PubMed.
- (a) P. Zhang, L. Zhang and S. Cheng, Biosci. Biotechnol. Biochem., 1999, 63, 1197–1202 CrossRef CAS PubMed; (b) M. Nishimura, Physiol. Mol. Plant Pathol., 2016, 95, 14–19 CrossRef CAS; (c) A. Yoshimi, K. Miyazawa and K. Abe, J. Fungi, 2017, 3, 63 CrossRef PubMed; (d) S. Puanglek, S. Kimura and T. Iwata, Carbohydr. Polym., 2017, 169, 245–254 CrossRef CAS PubMed.
- (a) T. Watanabe and K. Aso, Nature, 1959, 183, 1740–1740 CrossRef CAS PubMed; (b) A. Sato and K. Aso, Nature, 1957, 180, 984–985 CrossRef CAS PubMed; (c) K. Matsuda, H. Watanabe, K. Fujimoto and K. Aso, Nature, 1961, 191, 278–278 CrossRef CAS PubMed; (d) T. Watanabe, Y. Kamo, K. Matsuda and W. F. Dudman, Carbohydr. Res., 1982, 110, 170–175 CrossRef CAS.
- (a) C. Weidenmaier and A. Peschel, Nat. Rev. Microbiol., 2008, 6, 276–287 CrossRef CAS PubMed; (b) I. B. Naumova, A. S. Shashkov, E. M. Tul'skaya, G. M. Streshinskaya, Y. I. Kozlova, N. V. Potekhina, L. I. Evtushenko and E. Stackebrandt, FEMS Microbiol. Rev., 2001, 25, 269–283 CrossRef CAS PubMed; (c) S. Wang, K. Liu, C. J. Seneviratne, X. Li, G. S. P. Cheung, L. Jin, C. H. Chu and C. Zhang, Biomed. Rep., 2015, 3, 697–702 CrossRef CAS PubMed.
- (a) J. E. Baik, Y. H. Ryu, J. Y. Han, J. Im, K.-Y. Kum, C.-H. Yun, K. Lee and S. H. Han, J. Endod., 2008, 34, 975–982 CrossRef PubMed; (b) F. Fabretti, C. Theilacker, L. Baldassarri, Z. Kaczynski, A. Kropec, O. Holst and J. Huebner, Infect. Immun., 2006, 74, 4164 CrossRef CAS PubMed.
- C. Theilacker, Z. Kaczynski, A. Kropec, F. Fabretti, T. Sange, O. Holst and J. Huebner, Infect. Immun., 2006, 74, 5703 CrossRef CAS PubMed.
- D. Laverde, D. Wobser, F. Romero-Saavedra, W. Hogendorf, G. van der Marel, M. Berthold, A. Kropec, J. Codee and J. Huebner, PLoS One, 2014, 9, e110953 CrossRef PubMed.
- W. F. Hogendorf, L. J. Bos, H. S. Overkleeft, J. D. Codee and G. A. Marel, Bioorg. Med. Chem., 2010, 18, 3668–3678 CrossRef CAS PubMed.
- (a) D. van der Es, W. F. J. Hogendorf, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Chem. Soc. Rev., 2017, 46, 1464–1482 RSC; (b) C. A. A. van Boeckel, G. M. Visser, J. P. G. Hermans and J. H. van Boom, Tetrahedron Lett., 1981, 22, 4743–4746 CrossRef CAS; (c) C. A. A. van Boeckel, G. M. Visser, J. P. G. Hermans and J. H. van Boom, Recl. Trav. Chim. Pays-Bas, 1983, 102, 526–537 CrossRef CAS; (d) P. Westerduin, G. H. Veeneman, Y. Pennings, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 1987, 28, 1557–1560 CrossRef CAS; (e) A. Stadelmaier, S. Morath, T. Hartung and R. R. Schmidt, Angew. Chem., Int. Ed., 2003, 42, 916–920 CrossRef CAS PubMed; (f) I. Figueroa-Perez, A. Stadelmaier, S. Morath, T. Hartung and R. R. Schmidt, Tetrahedron: Asymmetry, 2005, 16, 493–506 CrossRef CAS; (g) Y. Qiao, B. Lindner, U. Zähringer, P. Truog and R. R. Schmidt, Bioorg. Med. Chem., 2010, 18, 3696–3702 CrossRef CAS PubMed; (h) C. M. Pedersen, I. Figueroa-Perez, B. Lindner, A. J. Ulmer, U. Zähringer and R. R. Schmidt, Angew. Chem., Int. Ed., 2010, 49, 2585–2590 CrossRef CAS PubMed; (i) W. F. J. Hogendorf, N. Gisch, D. Schwudke, H. Heine, M. Bols and C. M. Pedersen, Chem. – Eur. J., 2014, 20, 13511–13516 CrossRef CAS PubMed; (j) A. Ali, R. J. B. H. N. van den Berg, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel and J. D. C. Codée, Tetrahedron Lett., 2009, 50, 2185–2188 CrossRef CAS; (k) W. F. J. Hogendorf, L. N. Lameijer, T. J. M. Beenakker, H. S. Overkleeft, D. V. Filippov, J. D. C. Codée and G. A. Van der Marel, Org. Lett., 2012, 14, 848–851 CrossRef CAS PubMed.
- (a) K. i. Takeo, Carbohydr. Res., 1981, 88, 158–161 CrossRef CAS; (b) K. i. Takeo and Y. Suzuki, Carbohydr. Res., 1987, 162, 95–109 CrossRef CAS.
- (a) J. C. Hu, A. W. Feng, B. Y. Chang, C. H. Lin and K. T. Mong, Org. Biomol. Chem., 2017, 15, 5345–5356 RSC; (b) H. Yao, M. D. Vu and X.-W. Liu, Carbohydr. Res., 2019, 473, 72–81 CrossRef CAS PubMed; (c) L. Yang, Q. Qin and X.-S. Ye, Asian J. Org. Chem., 2013, 2, 30–49 CrossRef CAS.
- (a) L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, Eur. J. Org. Chem., 2019, 1994–2003 CrossRef CAS PubMed; (b) L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed.
- S. E. Soliman and C. S. Bennett, Org. Lett., 2018, 20, 3413–3417 CrossRef CAS PubMed.
- (a) S. R. Lu, Y. H. Lai, J. H. Chen, C. Y. Liu and K. K. Mong, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS PubMed; (b) V. Dourtoglou and B. Gross, J. Carbohydr. Chem., 1983, 2, 57–73 CrossRef CAS; (c) Y. H. Lin, B. Ghosh and K. K. Mong, Chem. Commun., 2012, 48, 10910–10912 RSC; (d) C.-Y. I. Liu, S. Mulani and K.-K. T. Mong, Adv. Synth. Catal., 2012, 354, 3299–3310 CrossRef CAS; (e) B. Ghosh, Y. H. Lai, Y. Y. Shih, T. K. Pradhan, C. H. Lin and K. K. Mong, Chem. – Asian J., 2013, 8, 3191–3199 CrossRef CAS PubMed; (f) J.-H. Chen, J.-H. Ruei and K.-K. T. Mong, Eur. J. Org. Chem., 2014, 1827–1831 CrossRef CAS; (g) R. Wei, H. Liu, A. H. Tang, R. J. Payne and X. Li, Org. Lett., 2019, 21, 3584–3588 CrossRef CAS PubMed; (h) Y. Shingu, A. Miyachi, Y. Miura, K. Kobayashi and Y. Nishida, Carbohydr. Res., 2005, 340, 2236–2244 CrossRef CAS PubMed; (i) S. Koto, N. Morishima, M. Owa and S. Zen, Carbohydr. Res., 1984, 130, 73–83 CrossRef CAS; (j) Q. Lou, Q. Hua, L. Zhang and Y. Yang, Org. Lett., 2020, 22, 981–985 CrossRef CAS PubMed.
- A. G. Volbeda, H. A. V. Kistemaker, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov and J. D. C. Codée, J. Org. Chem., 2015, 80, 8796–8806 CrossRef CAS PubMed.
- (a) K. Fukase, T. Matsumoto, N. Ito, T. Yoshimura, S. Kotani and S. Kusumoto, Bull. Chem. Soc. Jpn., 1992, 65, 2643–2654 CrossRef CAS; (b) K. Fukase, T. Yoshimura, S. Kotani and S. Kusumoto, Bull. Chem. Soc. Jpn., 1994, 67, 473–482 CrossRef CAS; (c) A. Stadelmaier, S. Morath, T. Hartung and R. R. Schmidt, Angew. Chem., Int. Ed., 2003, 42, 916–920 CrossRef CAS PubMed.
- S. Casati, E. Santaniello and P. Ciuffreda, Tetrahedron: Asymmetry, 2012, 23, 395–400 CrossRef CAS.
- P. O. Adero, D. R. Jarois and D. Crich, Carbohydr. Res., 2017, 449, 11–16 CrossRef CAS PubMed.
- W. F. J. Hogendorf, L. J. V. d. Bos, H. S. Overkleeft, J. D. C. Codée and G. A. V. d. Marel, Bioorg. Med. Chem., 2010, 18, 3668–3678 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00240b |
| This journal is © The Royal Society of Chemistry 2020 |