
The mechanism of universal green antisolvents for intermediate phase controlled high-efficiency formamidinium-based perovskite solar cells†
Luyao
Wang
a,
Xin
Wang
a,
Lin-Long
Deng
 b,
Shibing
Leng
a,
Xiaojun
Guo
c,
Ching-Hong
Tan
d,
Wallace C. H.
Choy
*e and
Chun-Chao
Chen
*a
b,
Shibing
Leng
a,
Xiaojun
Guo
c,
Ching-Hong
Tan
d,
Wallace C. H.
Choy
*e and
Chun-Chao
Chen
*a
aSchool of Material Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: c3chen@sjtu.edu.cn
bPen-Tung Sah Institute of Micro-Nano Science and Technology, Xiamen University, Xiamen, 361005, China
cDepartment of Electronic Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China
dSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China
eDepartment of Electrical and Electronic Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong 852, SAR, China. E-mail: chchoy@eee.hku.hk
First published on 5th December 2019
Abstract
Antisolvent-assisted crystallization is adopted widely in the preparation of double-cation [e.g., cesium/formamidinium (Cs/FA), formamidinium/methylammonium (FA/MA)] and triple-cation (e.g., Cs/FA/MA) FA-based perovskite solar cells (PSCs) to enhance their power conversion efficiencies (PCEs) and device stability. Although many antisolvents have been applied to treat various perovskites with different compositions, their crystallization mechanisms have remained unclear. In this study, we investigated the effects of a series of green antisolvents—namely, the ethers diethyl ether, anisole, diisopropyl ether (DIE), and dibutyl ether—on the crystallization of perovskites. We found that the formation of an intermediate phase was heavily determined by the antisolvent's polarity. Indeed, through judicious control of the antisolvent's polarity, it was possible to form a pure intermediate phase, without a PbI2 or perovskite phase. Upon thermal annealing, the crystallization of perovskites was improved. Understanding the mechanism of formation of the intermediate phase led us to identify DIE as a green antisolvent with universal perovskite compatibility, achieving champion PCEs of 20.05%, 20.15%, and 21.26% for Cs/FA, FA/MA, and Cs/FA/MA, respectively. The PCE of large-area (1 cm2) PSCs reached 18.51%. Furthermore, the repeatability of these champion PCEs was greatly improved. This work contributes to the understanding of antisolvent's polarity as an important factor affecting the formation of a pure intermediate phase of FA-based perovskites, which also offers a greener approach for the production of PSCs.
New conceptsThe rapid development of formamidinium-based (FA) perovskite solar cells has dramatically improved the efficiency limit of perovskite solar cells. However, the utilization of antisolvent processing toward record-high efficiency solar cells still remains challenging. While we may know that the polarity of antisolvents is a key factor in determining the outcome of solar cell performance, a detailed mechanism of how an antisolvent participates in the precipitation crystallization process is still lacking. In our work, we propose a new concept in antisolvent studies, in which the polarity of the antisolvent can determine the phase purity of the intermediate phase of FA-perovskites. By utilizing diisopropyl ether (DIE) as a green antisolvent, double- and triple-cation FA-based perovskite solar cells are all capable of achieving excellent performance with the highest efficiency of 21.26% in triple-cation perovskite solar cells. Such universal compatibility of a green antisolvent in different FA-based perovskite solar cells hasn’t been demonstrated before. We therefore believe that this new concept can be very influential in the research field of perovskite solar cells. |
1. Introduction
Organic/inorganic hybrid perovskite solar cells (PSCs) have attracted an immense amount of attention in recent years because of the rapid advancement in their power conversion efficiencies (PCEs) from 3.8%1 to 25.2%.2 The high performance of PSCs has arisen from the excellent optoelectronic properties of their organic/inorganic hybrid perovskite materials, including their high degrees of optical absorption, large carrier diffusion lengths (>1 μm),3 long diffusion lifetimes (up to 30 μs),4 and tunable compositions for matching with various electron transport layers (ETLs) and hole transport layers (HTLs).5 A lot of recent studies on PSCs have focused on compositional engineering of perovskite materials. Single-cation perovskite absorbers—including methyl ammonium halides (MAPbX3),6 formamidinium lead halides (FAPbX3),7 and cesium lead halides (CsPbX3; X = I, Br, or Cl)8—often cannot satisfy all the demands of efficiency, stability, and simple processing at the same time.9 On the other hand, the use of mixed-cation perovskite absorbers, which have a greater flexibility in their compositional fine-tuning, can improve both the efficiency and durability of PSCs.10–12 Several approaches have been developed to fabricate PSCs, including one-step antisolvent-assisted precipitation crystallization,13,14 two-step sequential deposition,15,16 vapor-assisted solution processing,17,18 and thermal gas-assisted evaporation.19,20 Indeed, many state-of-the-art PSCs displaying efficiencies in the range 20–24% have been realized using antisolvent-assisted precipitation crystallization.10,21–26 Precipitation crystallization is generally a simple means of obtaining high-quality uniform perovskite layers suitable for devices providing high-efficiency outputs.Nevertheless, the use of antisolvent-assisted precipitation crystallization can raise problematic issues, including chemical hazards and poor compatibility. In terms of hazardous waste, the amount of antisolvent released to the ambient environment is typically four to five times as high as that of the solvent used to prepare the perovskite.23 If the mass production of PSCs reaches the stage where gigawatts of energy are produced, the tons of antisolvents released into the air during their fabrication will raise serious safety concerns. At present, chlorobenzene (CB) and toluene (TL) are commonly used as antisolvents to quench perovskite solutions prepared in γ-butyrolactone (GBL), N,N-dimethylformamide (DMF), and dimethylsulfoxide (DMSO). Notably, CB is a halogenated solvent that can cause dizziness, neurotoxicity, and other severe heath issues after long-term exposure;27,28 TL is a non-halogenated version of CB, but can cause asphyxiation, brain lesions, and congenital malformations upon inhalation.29 Recently, several green antisolvents—including diethyl ether (DE),30 diisopropyl ether (DIE),31 ethyl acetate (EA),14,32sec-butyl alcohol (SBA),33 and anisole (ANS)34,35—have been determined as potential replacements for the hazardous antisolvents. While all of them can be used toward achieving comparable efficiencies from a pure single-cation perovskite (MAPbI3),9 the poor compatibility of green antisolvents with triple-cation PSCs makes it difficult to precisely control the crystallization rates of FA-based perovskites. Although the Grätzel group achieved 20.4% efficiency by using ANS as the antisolvent for triple-mixed PSCs34 and the Park group obtained 19.5% efficiency from DE-treated PSCs,36 these efficiencies are lower than the state-of-the-art performance (21.17% efficiency) obtained when using CB as the antisolvent in the production of triple-mixed PSCs.37 So far, we only know how to select antisolvents by their polarity to help in precipitating the perovskite crystals. The effect of antisolvent's polarity on the formation of an intermediate phase has not been thoroughly explored. The intermediate phase staged in between a liquid precursor phase and a solid perovskite phase plays a very crucial role in determining the final quality of perovskite absorbers. Accordingly, finding a universal green antisolvent with controlled intermediate phase for different compositions of FA-based perovskites still remains a great challenge.
In this study, we employed a series of ethers—ANS, DE, DIE, and dibutyl ether (DBE)—as green antisolvents in the one-step precipitation crystallization fabrication of FA-based PSCs. By varying the polarity of the antisolvent, we could identify and control the intermediate phase formed during the precipitation crystallization growth of an FA-based perovskite film and, thus, provide a guideline for the future selection of suitable green antisolvents. Among the tested green antisolvents, DIE exhibited excellent compatibility in the preparation of various FA-based PSCs, due to its polarity (0.110) being appropriate to ensure a pure intermediate phase. When using DIE as a quenching solvent in the fabrication of triple-mixed (FA/MA/Cs) PSCs inside and outside a glove box, we obtain PCEs of up to 21.26 and 18.85%, respectively—these values are among the highest ever reported for triple-mixed PSCs based on mesoporous TiO2 substrates. In addition, we have also prepared pure FA/MA and FA/Cs PSCs displaying efficiencies consistently greater than 20%, demonstrating the excellent universal compatibility and repeatability of applying this green antisolvent. Overall, understanding the mechanism behind the intermediate phase growth has helped us to find such a suitable green antisolvent that will presumably be beneficial in the commercial production of PSC technologies.
2. Results and discussion
Fig. 1a illustrates the procedure of antisolvent-assisted precipitation crystallization. Here, the antisolvent plays important roles as the quenching solvent for the perovskite layer and in the formation of a pure or impure intermediate phase. Table S1 (ESI†) lists the boiling points and polarities of the antisolvents tested in this study. We employed X-ray diffraction (XRD) and Fourier transform infrared (FTIR) spectroscopy to examine how the antisolvents affected the growth dynamics of the perovskite films. Considering that the perovskite intermediate phase films are readily eroded by adventitious water from the ambient environment, we characterized the properties of the obtained intermediate phase powders by adding the antisolvent into the precursor solution and storing at 60 °C in a vacuum oven. Fig. 1b presents the XRD patterns of the intermediate phases of the triple-cation perovskite powders formed using the various antisolvents. To confirm that the powders that had been treated with CB, ANS, and DIE as antisolvents could produce intermediate phases, we recorded their FTIR spectra. Fig. 1c reveals a signal for NH stretching (ca. 3400–3250 cm−1) corresponding to FA+/MA+ in the intermediate phase. Another obvious peak at 1017 cm−1 represents S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, indicative of DMSO interacting with the Lewis-acidic MA+, FA+, and Pb2+ ions.38,39 The XRD patterns in Fig. 1b allowed us to distinguish the differences in the structures of the intermediate phases (CsI–MABr–FAI–PbI2–PbBr2–DMSO)22 formed using the various antisolvents. The intermediate phase powders formed after quenching with CB and ANS featured a perovskite(110) phase, while the intermediate phase powder formed using ANS featured an unwanted signal for PbI2. Because of their relatively slightly higher polarities, CB and ANS are miscible with both DMF and DMSO (Fig. S3, ESI†); such unwanted miscibility might accelerate the saturation of the precursor solution and avoid formation of the intermediate phase through the more complete removal of the DMF and DMSO. Upon thermal annealing, perovskite crystals induced by a more impure intermediate phase are more likely to have smaller grains and form more inhomogeneous films (Fig. 2). Fig. S3 (ESI†) indicates that DE, DIE, and DBE are not miscible with DMSO, making them more suitable antisolvents to ensure the formation of intermediate phases. Nevertheless, the intermediate phase treated with DE exhibited an obvious XRD signal for PbI2(DMSO)2; this signal was negligible for the DIE- and DBE-treated samples. We ascribe this difference to DE's better miscibility with DMF as compared to that of DIE and DBE, thereby, DE can extract DMF more quickly from the precursor solution and leave more redundant DMSO to coordinate with PbI2 forming PbI2(DMSO)2. Unfortunately, the existence of PbI2(DMSO)2 is harmful and likely to impede the crystallization of the pure perovskite phase, because it changes the stoichiometric ratio of the intermediate phase. Finally, we investigated the crystallization of annealed perovskite films. The XRD patterns in Fig. 1d reveal that the peaks of the perovskite film that had been treated with DIE—at 14.16, 24.4, and 28.1°, corresponding to the (100), (111), and (200) planes, respectively39,40—were more intense and sharper than those that had been quenched with the other antisolvents. The improved crystallinity of the thermally annealed perovskite film prepared using DIE was presumably the result of its pure intermediate phase. In other words, if no other contaminants exist in the intermediate phase, the crystallization of the perovskite phase can proceed more efficiently, resulting in a highly crystalline perovskite morphology. The XRD patterns of all of the samples featured an obvious peak for PbI2 at 12.72°, due to the excessive amount of PbI2 in the precursor recipe. As the polarity of the antisolvent decreased, the intensity of this signal for PbI2 decreased (cf. the patterns for the DIE- and DBE-treated films). The role of residual PbI2 remains debatable. For instance, the Jiang group found that the residual PbI2 could ensure defect passivation and suppress the recombination of the perovskite film,41 whereas the Jacobsson group found that the presence of the PbI2 phase had a negative influence on the formation of the perovskite phase.42 Furthermore, we observed that the perovskite film that had been treated with DBE featured an XRD peak for δ-perovskite (11.6°), the presence of which will lead to poor device performance. In fact, we find that the polarity of DBE is too low to be miscible with DMF, resulting in DMF not being completely removed during the antisolvent treatment. Upon high temperature annealing, the rapid evaporation of residual DMF from the intermediate phase can disturb the phase and morphology formation of the final perovskite film.43,44 We conclude that the antisolvent's polarity is an important factor affecting the formation of a pure intermediate phase, which will determine the crystallinity and residual PbI2 content of the final perovskite film.
O stretching, indicative of DMSO interacting with the Lewis-acidic MA+, FA+, and Pb2+ ions.38,39 The XRD patterns in Fig. 1b allowed us to distinguish the differences in the structures of the intermediate phases (CsI–MABr–FAI–PbI2–PbBr2–DMSO)22 formed using the various antisolvents. The intermediate phase powders formed after quenching with CB and ANS featured a perovskite(110) phase, while the intermediate phase powder formed using ANS featured an unwanted signal for PbI2. Because of their relatively slightly higher polarities, CB and ANS are miscible with both DMF and DMSO (Fig. S3, ESI†); such unwanted miscibility might accelerate the saturation of the precursor solution and avoid formation of the intermediate phase through the more complete removal of the DMF and DMSO. Upon thermal annealing, perovskite crystals induced by a more impure intermediate phase are more likely to have smaller grains and form more inhomogeneous films (Fig. 2). Fig. S3 (ESI†) indicates that DE, DIE, and DBE are not miscible with DMSO, making them more suitable antisolvents to ensure the formation of intermediate phases. Nevertheless, the intermediate phase treated with DE exhibited an obvious XRD signal for PbI2(DMSO)2; this signal was negligible for the DIE- and DBE-treated samples. We ascribe this difference to DE's better miscibility with DMF as compared to that of DIE and DBE, thereby, DE can extract DMF more quickly from the precursor solution and leave more redundant DMSO to coordinate with PbI2 forming PbI2(DMSO)2. Unfortunately, the existence of PbI2(DMSO)2 is harmful and likely to impede the crystallization of the pure perovskite phase, because it changes the stoichiometric ratio of the intermediate phase. Finally, we investigated the crystallization of annealed perovskite films. The XRD patterns in Fig. 1d reveal that the peaks of the perovskite film that had been treated with DIE—at 14.16, 24.4, and 28.1°, corresponding to the (100), (111), and (200) planes, respectively39,40—were more intense and sharper than those that had been quenched with the other antisolvents. The improved crystallinity of the thermally annealed perovskite film prepared using DIE was presumably the result of its pure intermediate phase. In other words, if no other contaminants exist in the intermediate phase, the crystallization of the perovskite phase can proceed more efficiently, resulting in a highly crystalline perovskite morphology. The XRD patterns of all of the samples featured an obvious peak for PbI2 at 12.72°, due to the excessive amount of PbI2 in the precursor recipe. As the polarity of the antisolvent decreased, the intensity of this signal for PbI2 decreased (cf. the patterns for the DIE- and DBE-treated films). The role of residual PbI2 remains debatable. For instance, the Jiang group found that the residual PbI2 could ensure defect passivation and suppress the recombination of the perovskite film,41 whereas the Jacobsson group found that the presence of the PbI2 phase had a negative influence on the formation of the perovskite phase.42 Furthermore, we observed that the perovskite film that had been treated with DBE featured an XRD peak for δ-perovskite (11.6°), the presence of which will lead to poor device performance. In fact, we find that the polarity of DBE is too low to be miscible with DMF, resulting in DMF not being completely removed during the antisolvent treatment. Upon high temperature annealing, the rapid evaporation of residual DMF from the intermediate phase can disturb the phase and morphology formation of the final perovskite film.43,44 We conclude that the antisolvent's polarity is an important factor affecting the formation of a pure intermediate phase, which will determine the crystallinity and residual PbI2 content of the final perovskite film.
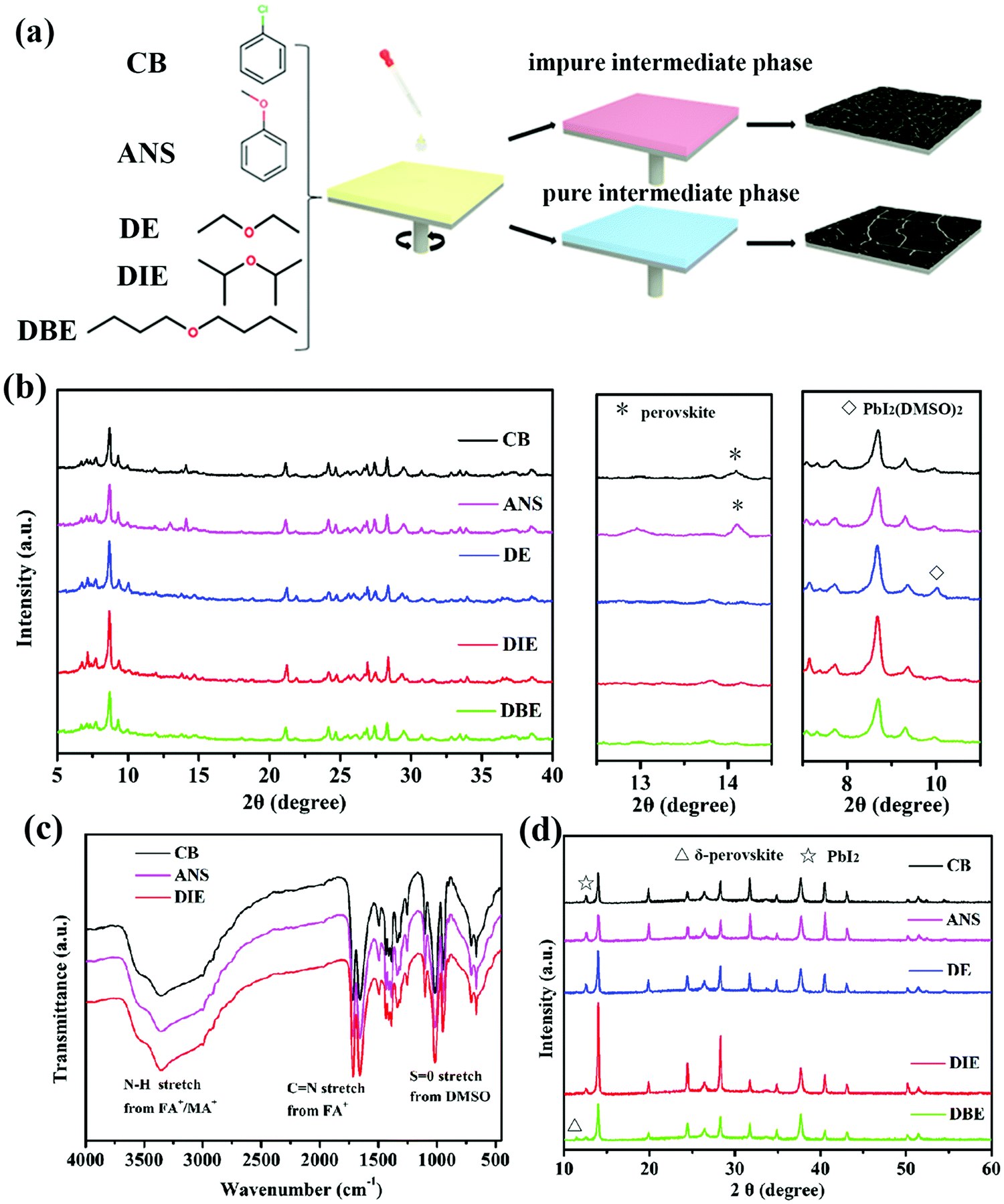 | ||
| Fig. 1 (a) Schematic representation of the antisolvent-assisted perovskite deposition process, with chemical structures of the antisolvents used in this study. (b) XRD patterns and (c) FTIR spectra of the intermediate phases obtained from the Cs0.05(MA0.17FA0.83)Pb(I0.83Br0.17)3 powder treated with the various antisolvents. (d) XRD patterns of the Cs0.05(MA0.17FA0.83)Pb(I0.83Br0.17)3 films prepared using CB, ANS, DE, DIE, and DBE as antisolvents. | ||
 | ||
| Fig. 2 (a–e) Top-view SEM images of the Cs0.05(MA0.17FA0.83)Pb(I0.83Br0.17)3 films prepared using (a) CB, (b) ANS, (c) DE, (d) DE, and (e) DBE as antisolvents. (f) Average grain sizes of the Cs0.05(MA0.17FA0.83)Pb(I0.83Br0.17)3 films prepared using the various antisolvents. | ||
To further explore the effects of the various antisolvents on the morphologies of the perovskite films, we observed the surface morphologies of Cs0.05(MA0.17FA0.83)Pb(I0.83Br0.17)3 triple-cation perovskite films that had been treated with CB, ANS, DE, DIE, and DBE as antisolvents. Fig. 2a–e reveal that these perovskite films all possessed dense and uniform surface morphologies. Interestingly, the grain sizes were different when using each of these five antisolvents; Fig. 2h and Fig. S1 (ESI†) present the grain size statistics of the five perovskite films. The average grain sizes of the perovskite films that had been treated with CB, ANS, DE, and DBE were 238, 228, 313, and 263 nm, respectively. In contrast, the average grain size of the perovskite layer that had been treated with DIE was much larger (353 nm). We used atomic force microscopy (AFM) to investigate the topographies of the perovskite films that had been quenched using the five different antisolvents (Fig. S2, ESI†). The root-mean-square (RMS) surface roughness of the perovskite film that had been treated with DIE (29.9 nm) was less than those of the films prepared using CB (36.1 nm), ANS (36.7 nm), DE (35.3 nm), and DBE (41.7 nm). Thus, DIE was capable of producing larger grains and a smoother surface when compared with the other antisolvents tested in this study. We conclude that a purer intermediate phase induced the crystallization of large grains and a smooth surface—two essential features for the generation and extraction of charge carriers.
We prepared PSCs having the architecture FTO/bl-TiO2/mp-TiO2/PVSK/Spiro-OMeTAD/Ag (Fig. 3a). Fig. 3b displays the current density–voltage (J–V) curves of the best devices prepared using the various antisolvents. The device performance of the DIE-treated PSCs was remarkably higher than that of the DBE-treated devices. The PCE of the best DIE-treated device was 21.26%; in contrast, the PCEs of the PSCs incorporating films quenched by CB, ANS, DE, and DBE were 20.02, 19.41, 20.20, and 18.64%, respectively. Using DIE as the antisolvent also resulted in a relatively high open-circuit voltage (Voc) of 1.16 V and fill factor (FF) of 0.79, presumably because of the higher crystallinity and smoother surface of the DIE-treated perovskite film. Fig. 3c and Table 1 present statistical data obtained from 15 devices fabricated using the various antisolvents. The application of DE provided PSCs displaying poor reproducibility, presumably because of its low boiling point, which made its use difficult to control. Devices fabricated with DIE had higher average PCEs, of up to 20.21%, when compared with those of the PSCs prepared with the other antisolvents. Fig. 3d displays the correlation between the PCE and the polarity of the antisolvent. We conclude that a polarity of 0.11 was the most suitable for obtaining high-efficiency PSCs. The measured short-circuit current densities (Jsc) were consistent with the integrated photocurrent densities obtained from the external quantum efficiency (EQE) spectra in Fig. 3e; the percentage difference between two values of Jsc was less than 5%. To examine the stability of their J–V performances, we tested the photocurrents of the devices under a bias of 0.96 V (Fig. 3f). The DIE-treated device displayed low hysteresis (1.6%, Fig. S4, ESI†) and a stable PCE output of 20.42%.
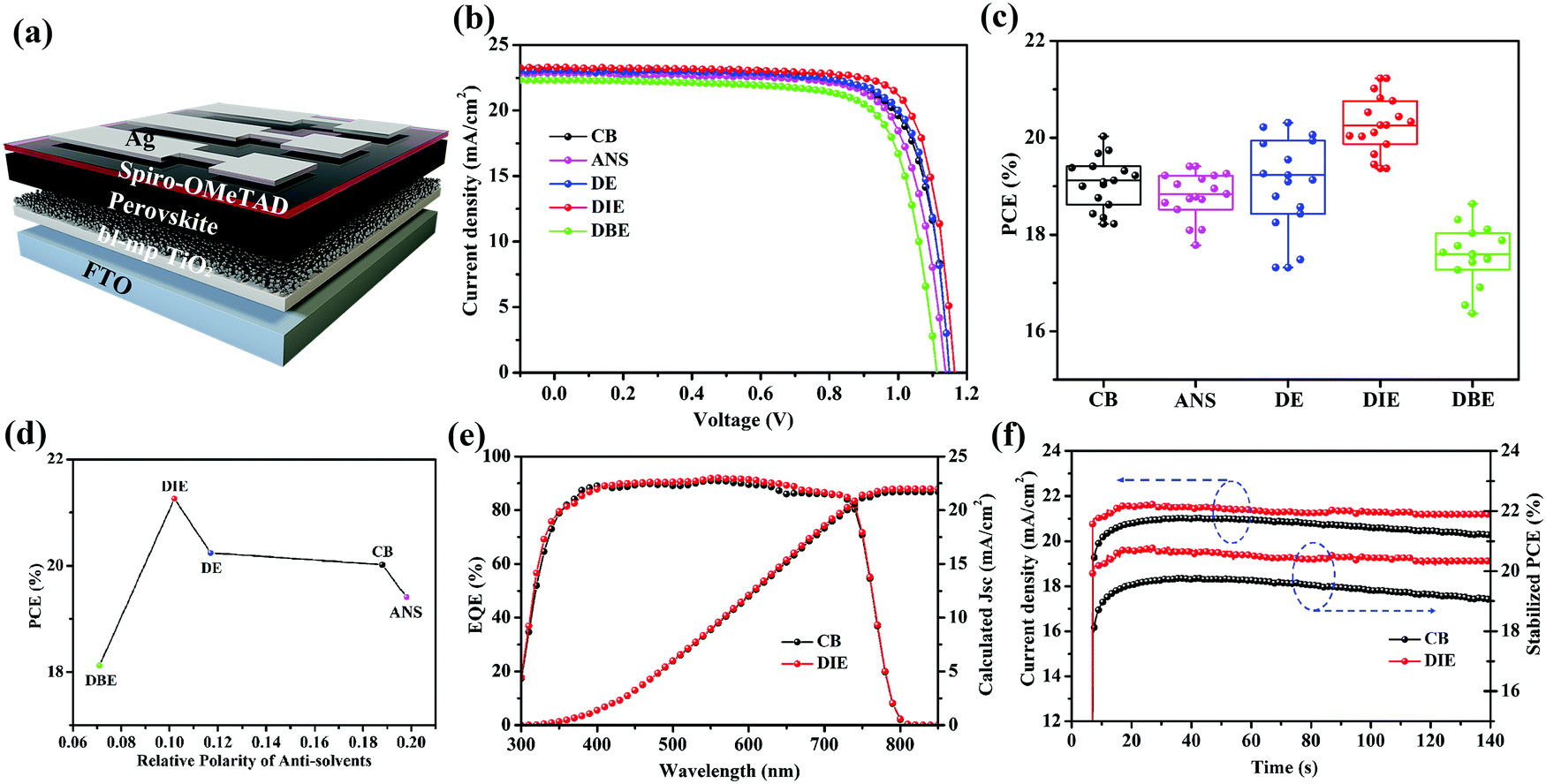 | ||
| Fig. 3 (a) Schematic representation of the PSC device structure. (b) J–V characteristics of the best Cs0.05(MA0.17FA0.83)Pb(I0.83Br0.17)3 perovskite devices and (c) PCE statistics from 15 devices fabricated using CB, ANS, DE, DIE, and DBE as antisolvents, recorded at a scan rate of 20 mV s−1. (d) Relationship between the polarity of the antisolvent and the PCE of the PSCs. (e) EQE spectra and integrated current densities of Cs0.05(MA0.17FA0.83)Pb(I0.83Br0.17)3 perovskites that had been treated with CB and DIE. (f) Stabilized PCEs of the corresponding triple-mixed PSCs. | ||
| Solvent | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| CB | 1.11 ± 0.02 | 22.76 ± 0.34 | 76.11 ± 0.95 | 19.08 ± 0.53 |
| ANS | 1.10 ± 0.02 | 22.23 ± 0.41 | 74.67 ± 1.14 | 18.43 ± 0.62 |
| DE | 1.12 ± 0.02 | 22.69 ± 0.33 | 75.55 ± 0.73 | 19.09 ± 0.95 |
| DIE | 1.14 ± 0.02 | 22.92 ± 0.27 | 78.12 ± 0.76 | 20.21 ± 0.41 |
| DBE | 1.07 ± 0.02 | 21.77 ± 0.45 | 74.16 ± 1.38 | 17.56 ± 0.59 |
To compare the charge generation and recombination dynamics in the triple-cation perovskite films treated with various solvents, we turned to time-resolved measurements using femto-second transient absorption (TA) and nano-second photoluminescence (TRPL) spectroscopies, accompanied by steady state UV-vis absorption and PL spectroscopies (Fig. 4). Fig. 4a shows the UV-vis absorption spectrum of the perovskite film treated with the CB antisolvent with an absorption band tail extending beyond 900 nm, consistent with the reported mixed cation perovskite layer.45 In comparison, the spectrum of the same perovskite treated with the DIE antisolvent, although shows a slightly higher intensity in the 400–500 nm region, reveals no significant difference across measured wavelengths. Steady state PL, employing an excitation wavelength of 460 nm, shows an intense peak at 765 nm for the perovskite film prepared using DIE and is stronger than that of the film prepared using CB (Fig. 4a). Note that the higher PL intensity of the perovskite film treated with DIE (∼29% higher) compared to the CB-treated film cannot be ascribed to its stronger absorbance which is only ∼7% higher, suggesting a larger emissive state population.
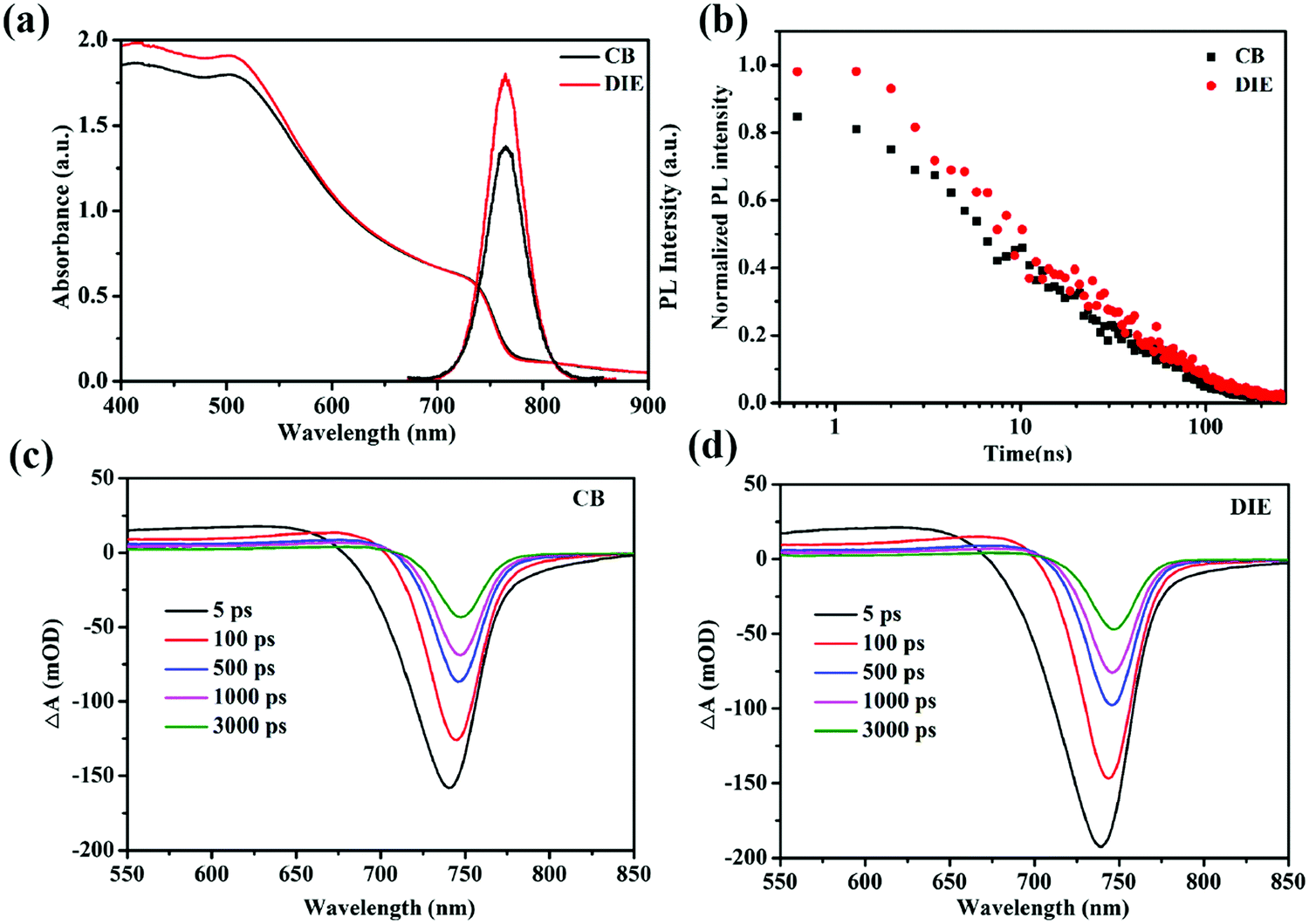 | ||
| Fig. 4 (a) UV-vis absorbance and PL spectra and (b) TRPL kinetics of triple-cation perovskite absorber films quenched using CB and DIE as antisolvents. TA spectra of triple-cation perovskite films treated with (c) CB and (d) DIE, employing an excitation wavelength of 515 nm at a power of 5.88 mW. | ||
TA spectra for the perovskite film treated with CB (Fig. 4c), employing a selective excitation wavelength of 515 nm, show a broad photo-induced band absorption signal at 550–720 nm which is assigned to the inter-band transition. The negative signal at 700–760 nm, corresponding to photobleaching (PB) and stimulated emission, can be assigned to the direct band to band filling transition. Both these signals are consistent with the reported TA spectra of the same mixed cation perovskite45,46 and are observed also in the TA spectra of the DIE-treated film. The red shifting of the PB signal measured at the timescale herein for the CB-treated film (∼7 nm), similar to that for the DIE-treated film, is consistent with the red shift reported previously for the same triple cation perovskite structure.47,48 This could be due to a band to band edge relaxation. The initial PB signal of the DIE-treated perovskite film is ∼19% higher than that of the CB-treated film.49–51 This difference could be the main reason for the observed difference in the PL intensity between these films, indicating an increase in the radiative transition. The excited states (Fig. S5, ESI†) for the DIE-treated film, probed at 750 nm, have a higher lifetime than those for the CB-treated film at the later timescale (a few nano-seconds), indicating a lower recombination rate for such states in the DIE-treated film.
TRPL, employing the same excitation as steady state PL at 460 nm, shows the decays of the excited states of the perovskite layer treated with CB and DIE antisolvents (Fig. 4b). The decays for DIE- and CB-treated films, fitted with a bi-exponent, have average lifetimes (τave) of 47.2 and 41.4 ns, respectively. The slightly higher lifetimes at both early and later timescales for the DIE-treated perovskite film (refer to Table S3, ESI†), consistent with the lifetime of our TA measurement at the later timescale, suggest a lower recombination rate for the states aforementioned. These states which have been studied previously could be photogenerated carriers.52–54 Presumably because fewer deep state defects exist in this film, defect-assisted charge-transfer loss is minimized in the DIE-treated film. These could be due to the fewer grain boundaries and smoother morphology of the DIE-treated film.
To explore the compatibility of DIE as an antisolvent for other perovskite systems, we also fabricated (FAPbI3)0.9(MAPbBr3)0.1 (FA/MA) and FA0.83Cs0.17PbI2.7Br0.3 (FA/Cs) PSCs. Fig. 5b exhibits the J–V curves and EQE spectra of the FA/MA and FA/Cs PSCs that had been prepared using DIE. Fig. 5a reveals the best PCE of 20.15% for the FA/MA PSC, with Jsc, Voc, and FF values of 23.90 mA cm−2, 1.08 V, and 0.78 in the reverse scan, respectively. When we also introduced DIE as the antisolvent for the FA/Cs perovskite system, the champion device provided the highest PCE of 20.05%, with Jsc, Voc, and FF values of 23.50 mA cm−2, 1.10 V, and 0.77 in the reverse scan, respectively. Meanwhile, the FA/MA and FA/Cs PSCs prepared using DIE also exhibited high repeatability (Fig. S6, ESI†). Fig. 5b presents the EQE spectra of three different perovskite devices; they provided values of Jsc consistent with those measured using the solar simulator. All three of the perovskite films exhibited uniform and enhanced crystallization, with an average perovskite layer thickness of approximately 500 nm (Fig. S6, ESI†). Based on the results in Fig. 5c, we conclude that DIE is a green and universally compatible antisolvent that can be used to obtain high PCEs in FA/MA/Cs, FA/MA, and FA/Cs PSCs.
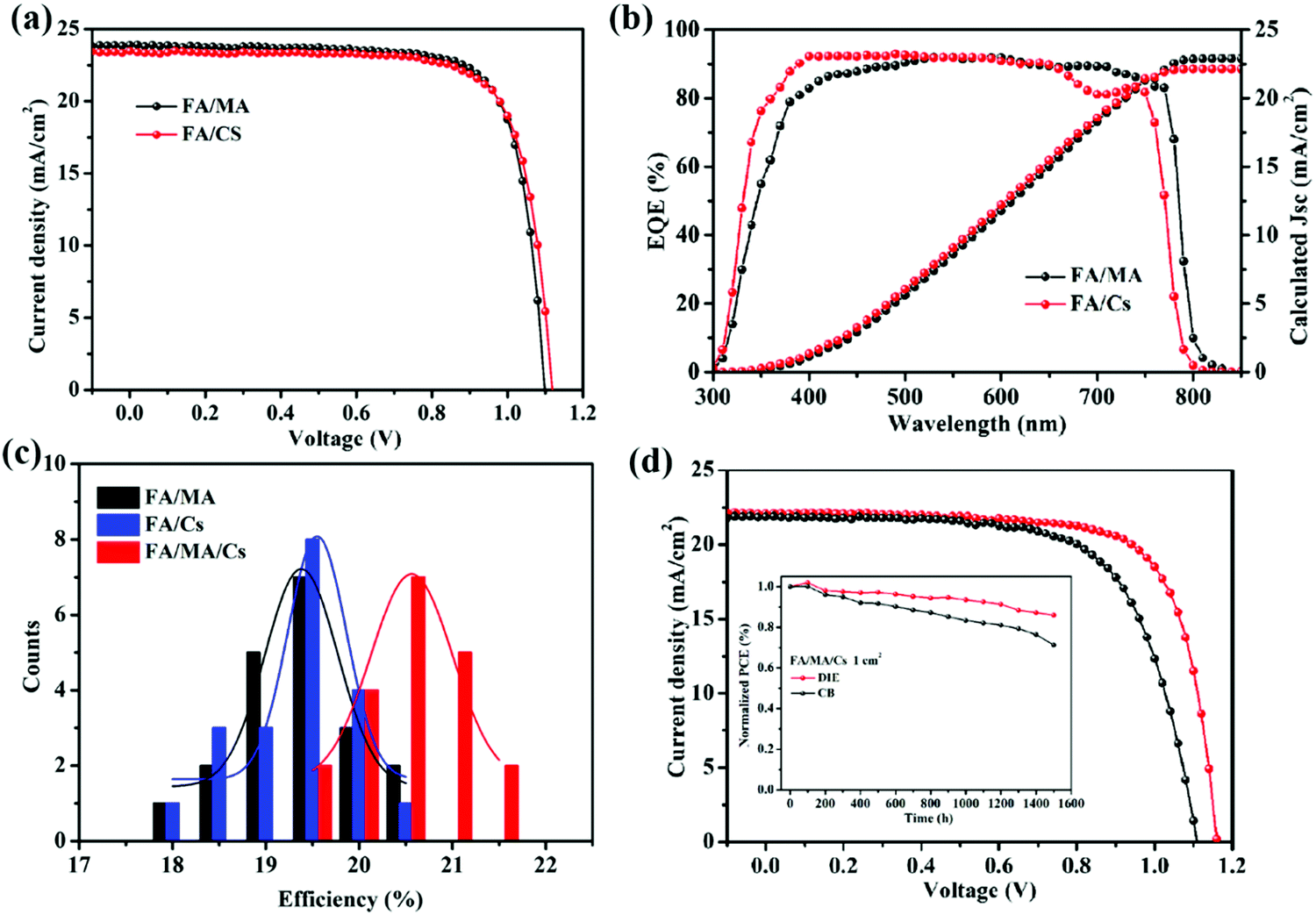 | ||
| Fig. 5 (a) Best J–V characteristics and (b) EQE spectra of FA/MA and FA/Cs perovskite devices that had been prepared using DIE as the antisolvent. (c) PCE distributions, based on 20 devices prepared from FA/MA/Cs, FA/MA, and FA/Cs that had been treated with DIE as the antisolvent. (d) Photovoltaic J–V curves and (inset) stability data for large-area (1 cm × 1 cm) FA/MA/Cs PSC that had been prepared using DIE as the antisolvent, recorded after storage in dry air (temp.: 20–25 °C; RH: 15–20%). | ||
To demonstrate the compatibility of DIE as a green antisolvent for practical applications, we fabricated PSCs having large active areas. Fig. 5d and Fig. S7 present the J–V curves of triple-mixed (FA/MA/Cs) PSCs having an active area of 1.00 cm2. The best-performing device provided a PCE of 18.51%, with a Voc of 1.16 V, a Jsc of 22.16 mA cm−2, and an FF of 0.72. In addition, the device displayed good stability, retaining 89% of its original PCE after 1500 h of storage in air [relative humidity (RH) ≤ 20%]. For comparison, the CB-treated large-area PSC also provided a lower PCE of 16.21%, but its average stability was only 71% of its original PCE after 1500 h of storage. Because DIE is a green solvent, we could fabricate its devices under ambient air conditions. Here, we adjusted the spin-coating conditions slightly to adapt to the air environment (RH: 30–35%), because solvent evaporation was slower. After optimizing the processing conditions, the best-performing cell provided a PCE of 18.85% with a Voc of 1.14 V, a Jsc of 22.56 mA cm−2, and an FF of 0.73 (Fig. S8, ESI†). This PCE is the highest ever reported for a PSC fabricated under ambient conditions using a green antisolvent. Accordingly, DIE is a green antisolvent that can be used to mass-produce high-performance, large-area PSC devices under conditions open to the air, potentially providing a new avenue for the commercialization of PSC technologies.
3. Conclusion
We have conducted a detailed study of antisolvent-assisted precipitation crystallization of perovskite films using CB (a hazardous antisolvent) and various ethers (ANS, DE, DIE, and DBE) as green antisolvents. Depending on the polarity of the antisolvents, we could control the formation of the intermediate phase. DIE has the most suitable polarity for inducing a pure intermediate phase and, therefore, its use as an antisolvent was beneficial for fabrication of efficient devices. Triple-cation PSCs (FA/MA/Cs) fabricated with DIE displayed a higher PCE (21.26%) than those obtained using CB, ANS, DE, and DBE as antisolvents. In addition, when we applied DIE as the antisolvent in the production of FA/MA and FA/Cs PSCs, we obtained PCEs of 20.15 and 20.05%, respectively—among the highest PCEs ever recorded for these systems. To explore the prospective application of DIE processing for industrial production, we fabricated triple-cation PSCs having an active area of 1 cm × 1 cm and obtained a PCE of 18.51%. Furthermore, PSC processing with DIE as the antisolvent under ambient conditions (RH: 30–35%) resulted in a PCE of 18.85%—the new record efficiency for a PSC treated with a green antisolvent under ambient conditions. The results of this study suggest an alternative green approach for the fabrication of high-performance PSCs under ambient conditions with low health risks.4. Experimental section
Materials
PbI2 (99.8%) was purchased from TCI Chemicals. DMF (99.8%), DMSO (99.8%), and CB (99.8%) were obtained from Sigma–Aldrich. DE (99.5%), DIE (99.8%), DBE (99%), lithium bis(trifluoromethylsulfonyl)imide (Li-TFSI, 95%), and 4-tert-butylpyridine (TBP, 96%) were received from J&K Chemical. 2,2′,7,7′-tetrakis-(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD, 99%) was purchased from Lumtec. Formamidinium iodide (FAI), methylammonium iodide (CH3NH3I, 99.5%), and methylammonium bromide (MABr) were purchased from Xi’an Polymer Light Technology (China).Device fabrication—substrate preparation
A fluorine-doped tin oxide (FTO)-coated glass substrate was cleaned thoroughly with detergent, deionized water, EtOH, acetone, and isopropanol. The substrate was treated with UV-ozone for 10 min prior to use. A thin compact TiO2 blocking layer (bl-TiO2) was deposited (5000 rpm, 30 s) on the cleaned FTO substrate from a solution of 0.2 M titanium diisopropoxide bis(acetylacetonate) [Ti(acac)2, 75 wt% in isopropanol; Sigma-Aldrich] in n-butyl alcohol, followed by annealing (120 °C, 5 min) and cooling to room temperature. This process was repeated three times and then the sample was annealed at 500 °C for 60 min. The resulting film was immersed in 20 mM TiCl4 (>98%, Sigma-Aldrich) at 70 °C for 20 min, followed by washing with DI water. A mesoporous TiO2 (mp-TiO2) layer was deposited on the bl-TiO2 by spin-coating a solution of TiO2 paste (Dyesol 18NR-T) in anhydrous EtOH (5 mL); the sample was subsequently annealed at 500 °C for 30 min. Li-Doping of the mp-TiO2 was performed by spin-coating with 0.1 M Li-TFSI in MeCN, followed by sintering at 450 °C for 30 min.Device fabrication—perovskite film preparation
The [CsPbI3]0.05[(FAPbI3)0.85(MAPbBr3)0.15]0.95 triple-mixed precursor solution was made by mixing 1.18 M PbI2, 1.08 M FAI, 0.22 M MABr, and 0.22 M PbBr2 in DMF and DMSO (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Then 48 ul of CsI (1.5 M in DMSO) solution was added into the mixed FA/MA precursor solution to obtain the perovskite precursor solution of desired compositions. The dissolved hybrid solution was spin-coated on the mp-TiO2 layer at 800 rpm for 10 s and then at 6000 rpm for 25 s. In the second step, DIE (0.5 mL) was added dropwise on the substrate 5 s before finishing the spinning procedure. [This procedure was also performed for the other antisolvents: namely, CB/ANS (0.2 mL), DE (0.5 mL), and DBE (0.7 mL) were quickly added dropwise on the rotating substrate 5 s before completing the spinning procedure.] The films were then annealed at 150 °C for 10 min. FA0.83Cs0.17PbI2.7Br0.3 solution: A 1.45 M perovskite precursor solution was prepared using DMF and DMSO (4:1, v/v) as a mixed solvent. The precursor solutions were spin-coated on the mp-TiO2 substrates through a two-step procedure (10 s at 1000 rpm, 30 s at 6000 rpm), with dropwise addition of DIE as the antisolvent 10 s before finishing the procedure. The films were then annealed at 100 °C for 60 min.
:1, v/v). Then 48 ul of CsI (1.5 M in DMSO) solution was added into the mixed FA/MA precursor solution to obtain the perovskite precursor solution of desired compositions. The dissolved hybrid solution was spin-coated on the mp-TiO2 layer at 800 rpm for 10 s and then at 6000 rpm for 25 s. In the second step, DIE (0.5 mL) was added dropwise on the substrate 5 s before finishing the spinning procedure. [This procedure was also performed for the other antisolvents: namely, CB/ANS (0.2 mL), DE (0.5 mL), and DBE (0.7 mL) were quickly added dropwise on the rotating substrate 5 s before completing the spinning procedure.] The films were then annealed at 150 °C for 10 min. FA0.83Cs0.17PbI2.7Br0.3 solution: A 1.45 M perovskite precursor solution was prepared using DMF and DMSO (4:1, v/v) as a mixed solvent. The precursor solutions were spin-coated on the mp-TiO2 substrates through a two-step procedure (10 s at 1000 rpm, 30 s at 6000 rpm), with dropwise addition of DIE as the antisolvent 10 s before finishing the procedure. The films were then annealed at 100 °C for 60 min.
Device fabrication—HTL and top electrode
The HTL solution was prepared from a solution of Spiro-OMeTAD (72.3 mg) in 1 mL of CB mixed with 4-tert-butyl pyridine (28.8 μL) and Li-TFSI solution (17.5 μL, 520 mg mL−1 in acetonitrile). This solution was spin-coated on the perovskite films and then a gold film (ca. 100 nm) was deposited through thermal evaporation onto the devices.Measurements and characterization
XRD was performed using a Rigaku Ultima IV diffractometer and Kα radiation. UV-vis absorption spectra were recorded using a lambda 35 UV-vis spectrometer. SEM images were obtained using a JEOL JSM-7800 Prime scanning electron microscope. TRPL and steady state PL spectra were recorded using a FLS1000 photoluminescence spectrometer and an excitation wavelength of 460 nm. Femtosecond transient absorption (fs-TA) spectroscopy measurements were performed by using a commercial pump–probe spectrometer (HARPIA, Ultrafast Systems); a pump pulse (515 nm, 5.88 mW, the size of the laser spot was about 150 μm) was utilized to excite the perovskite films, the range of probe beam (0.27 mW) was 550 nm to 950 nm, and the time resolution was about 175 fs. Photovoltaic performance was measured under AM 1.5G illumination at 100 mW cm−2 using an Abet Technologies Sun 2000 solar simulator, calibrated with a standard VLSI Si reference solar cell (SRC-1000-TC-K-QZ). The J–V curves were measured in ambient air without encapsulation. The test conditions involved a reverse [forward bias (1.2 V) → short circuit (−0.1 V)] or forward scan with a step of 20 mV s−1, obtained using a Keithley 2400 source meter. During the measurement process, the cell was covered with a mask having an aperture of 0.1 cm2. Topographic AFM images were recorded using a Bruker Dimension Icon system operated in tapping mode under ambient conditions. EQE spectra were recorded using an Enlitech LQE-50-FLIPCE measurement system. FTIR spectra were recorded using a Thermo-Nicolet IR200 instrument.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was funded by the National Natural Science Foundation of China (Grant No. 51950410581), the Shanghai Pujiang Program, and the Open Fund of Zhejiang Tsinghua Institute of Flexible Electronics Technology.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- National Renewable Energy Laboratory, Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- N.-G. Park, Mater. Today, 2015, 18, 65–72 CrossRef CAS.
- Y. Chen, H. Yi, X. Wu, R. Haroldson, Y. Gartstein, Y. Rodionov, K. Tikhonov, A. Zakhidov, X.-Y. Zhu and V. Podzorov, Nat. Commun., 2016, 7, 12253 CrossRef CAS PubMed.
- R. K. Misra, S. Aharon, B. Li, D. Mogilyansky, I. Visoly-Fisher, L. Etgar and E. A. Katz, J. Phys. Chem. Lett., 2015, 6, 326–330 CrossRef CAS PubMed.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC.
- R. J. Sutton, G. E. Eperon, L. Miranda, E. S. Parrott, B. A. Kamino, J. B. Patel, M. T. Hörantner, M. B. Johnston, A. A. Haghighirad and D. T. Moore, Adv. Energy Mater., 2016, 6, 1502458 CrossRef.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449–453 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- T. M. Brenner, D. A. Egger, L. Kronik, G. Hodes and D. Cahen, Nat. Rev. Mater., 2016, 1, 15007 CrossRef CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y. B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 9898–9903 CrossRef CAS PubMed.
- M. Yin, F. Xie, H. Chen, X. Yang, F. Ye, E. Bi, Y. Wu, M. Cai and L. Han, J. Mater. Chem. A, 2016, 4, 8548–8553 RSC.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H.-S. Duan, H.-H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2013, 136, 622–625 CrossRef PubMed.
- R. Sedighi, F. Tajabadi, S. Shahbazi, S. Gholipour and N. Taghavinia, ChemPhysChem, 2016, 17, 2382–2388 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395 CrossRef CAS PubMed.
- Z. Xiao, C. Bi, Y. Shao, Q. Dong, Q. Wang, Y. Yuan, C. Wang, Y. Gao and J. Huang, Energy Environ. Sci., 2014, 7, 2619–2623 RSC.
- M. M. Tavakoli, P. Yadav, D. Prochowicz, M. Sponseller, A. Osherov, V. Bulović and J. Kong, Adv. Energy Mater., 2019, 9, 1803587 CrossRef.
- S. Paek, P. Schouwink, E. N. Athanasopoulou, K. T. Cho, G. Grancini, Y. Lee, Y. Zhang, F. Stellacci, M. K. Nazeeruddin and P. Gao, Chem. Mater., 2017, 29, 3490–3498 CrossRef CAS.
- T. Singh, S. Öz, A. Sasinska, R. Frohnhoven, S. Mathur and T. Miyasaka, Adv. Funct. Mater., 2018, 28, 1706287 CrossRef.
- J. J. Yoo, S. Wieghold, M. C. Sponseller, M. R. Chua, S. N. Bertram, N. T. P. Hartono, J. S. Tresback, E. C. Hansen, J.-P. Correa-Baena, V. Bulović, T. Buonassisi, S. S. Shin and M. G. Bawendi, Energy Environ. Sci., 2019, 12, 2192–2199 RSC.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- W. Hu, W. Zhou, X. Lei, P. Zhou, M. Zhang, T. Chen, H. Zeng, J. Zhu, S. Dai, S. Yang and S. Yang, Adv. Mater., 2019, 8, 1806095 CrossRef PubMed.
- C.-C. Chueh, K. Yao, H.-L. Yip, C.-Y. Chang, Y.-X. Xu, K.-S. Chen, C.-Z. Li, P. Liu, F. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 3241–3248 RSC.
- H. W. Choi, T. Zhou, M. Singh and G. E. Jabbour, Nanoscale, 2015, 7, 3338–3355 RSC.
- J. M. Donald, K. Hooper and C. Hopenhayn-Rich, Environ. Health Perspect., 1991, 94, 237–244 CAS.
- N. Ahn, D.-Y. Son, I.-H. Jang, S. M. Kang, M. Choi and N.-G. Park, J. Am. Chem. Soc., 2015, 137, 8696–8699 CrossRef CAS PubMed.
- L.-Y. Wang, L.-L. Deng, X. Wang, T. Wang, H.-R. Liu, S.-M. Dai, Z. Xing, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Nanoscale, 2017, 9, 17893–17901 RSC.
- T. Bu, L. Wu, X. Liu, X. Yang, P. Zhou, X. Yu, T. Qin, J. Shi, S. Wang, S. Li, Z. Ku, Y. Peng, F. Huang, Q. Meng, Y.-B. Cheng and J. Zhong, Adv. Energy Mater., 2017, 7, 1700576 CrossRef.
- F. Zhang, J. Lian, J. Song, Y. Hao, P. Zeng and H. Niu, J. Mater. Chem. A, 2016, 4, 3438–3445 RSC.
- M. Yavari, M. Mazloum-Ardakani, S. Gholipour, M. M. Tavakoli, S. H. Turren-Cruz, N. Taghavinia, M. Grätzel, A. Hagfeldt and M. Saliba, Adv. Energy Mater., 2018, 8, 1800177 CrossRef.
- K. Schutt, P. K. Nayak, A. J. Ramadan, B. Wenger, Y.-H. Lin and H. J. Snaith, Adv. Funct. Mater., 2019, 29, 1900466 CrossRef CAS.
- M. M. Byranvand, S. Song, L. Pyeon, G. Kang, G.-Y. Lee and T. Park, Nano Energy, 2017, 34, 181–187 CrossRef CAS.
- Q. Jiang, Z. Chu, P. Wang, X. Yang, H. Liu, Y. Wang, Z. Yin, J. Wu, X. Zhang and J. You, Adv. Mater., 2017, 29, 1703852 CrossRef PubMed.
- S. Paek, P. Schouwink, E. N. Athanasopoulou, K. Cho, G. Grancini, Y. Lee, Y. Zhang, F. Stellacci, M. K. Nazeeruddin and P. Gao, Chem. Mater., 2017, 29, 3490–3498 CrossRef CAS.
- H. B. Lee, M.-K. Jeon, N. Kumar, B. Tyagi and J.-W. Kang, Adv. Funct. Mater., 2019, 29, 1903213 CrossRef.
- J. Lu, X. Lin, X. Jiao, T. Gengenbach, A. D. Scully, L. Jiang, B. Tan, J. Sun, B. Li, N. Pai, U. Bach, A. N. Simonov and Y.-B. Cheng, Energy Environ. Sci., 2018, 11, 1880–1889 RSC.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2017, 2, 16177 CrossRef CAS.
- T. J. Jacobsson, J.-P. Correa-Baena, E. Halvani Anaraki, B. Philippe, S. D. Stranks, M. E. Bouduban, W. Tress, K. Schenk, J. L. Teuscher and J.-E. Moser, J. Am. Chem. Soc., 2016, 138, 10331–10343 CrossRef CAS PubMed.
- Y. Yu, S. Yang, L. Lei, Q. Cao, J. Shao, S. Zhang and Y. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 3667–3676 CrossRef CAS PubMed.
- J.-W. Lee, Z. Dai, T.-H. Han, C. Choi, S.-Y. Chang, S.-J. Lee, N. De Marco, H. Zhao, P. Sun, Y. Huang and Y. Yang, Nat. Commun., 2018, 9, 3021 CrossRef PubMed.
- S. Liu, W. Huang, P. Liao, N. Pootrakulchote, H. Li, J. Lu, J. Li, F. Huang, X. Shai, X. Zhao, Y. Shen, Y.-B. Cheng and M. Wang, J. Mater. Chem. A, 2017, 5, 22952–22958 RSC.
- C. Liang, D. Zhao, P. Li, B. Wu, H. Gu, J. Zhang, T. W. Goh, S. Chen, Y. Chen, Z. Sha, G. Shao, T. C. Sum and G. Xing, Nano Energy, 2019, 59, 721–729 CrossRef CAS.
- J. C. Brauer, Y. H. Lee, M. K. Nazeeruddin and N. Banerji, J. Phys. Chem. Lett., 2015, 6, 3675–3681 CrossRef CAS PubMed.
- C. Fei, M. Zhou, J. Ogle, D.-M. Smilgies, L. Whittaker-Brooks and H. Wang, J. Mater. Chem. A, 2019, 7, 23739–23746 RSC.
- L. Wang, C. McCleese, A. Kovalsky, Y. Zhao and C. Burda, J. Am. Chem. Soc., 2014, 136, 12205–12208 CrossRef CAS PubMed.
- Y. Wang, J. Wan, J. Ding, J.-S. Hu and D. Wang, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef CAS.
- E. M. Talbert, H. F. Zarick, A. Boulesbaa, N. Soetan, A. A. Puretzky, D. B. Geohegan and R. Bardhan, Nanoscale, 2017, 9, 12005–12013 RSC.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed.
- N. D. Pham, C. Zhang, V. T. Tiong, S. Zhang, G. Will, A. Bou, J. Bisquert, P. E. Shaw, A. Du, G. J. Wilson and H. Wang, Adv. Funct. Mater., 2019, 29, 1806479 CrossRef.
- Z. Yang, C.-C. Chueh, P.-W. Liang, M. Crump, F. Lin, Z. Zhu and A. K. Y. Jen, Nano Energy, 2016, 22, 328–337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh01679a |
| This journal is © The Royal Society of Chemistry 2020 |