
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew keys for old locks: carborane-containing drugs as platforms for mechanism-based therapies
Philipp
Stockmann
 ,
Marta
Gozzi
,
Robert
Kuhnert
,
Menyhárt B.
Sárosi
* and
Evamarie
Hey-Hawkins
*
,
Marta
Gozzi
,
Robert
Kuhnert
,
Menyhárt B.
Sárosi
* and
Evamarie
Hey-Hawkins
*
Universität Leipzig, Fakultät für Chemie und Mineralogie, Institut für Anorganische Chemie, Johannisallee 29, 04103 Leipzig, Germany. E-mail: menyhart.sarosi@uni-leipzig.de; hey@uni-leipzig.de; Fax: +49-341-9739319; Tel: +49-341-9736151
First published on 19th June 2019
Abstract
Icosahedral carboranes in medicine are still an emerging class of compounds with potential beneficial applications in drug design. These highly hydrophobic clusters are potential “new keys for old locks” which open up an exciting field of research for well-known, but challenging important therapeutic substrates, as demonstrated by the numerous examples discussed in this review.
 Philipp Stockmann | Philipp Stockmann received his MSc from Leipzig University (Germany) in 2017. He worked as a Research Assistant in computational chemistry investigating in silico design of carborane-containing drug molecules. Currently, he holds a position as a doctoral student at Leipzig University under the supervision of Prof. Evamarie Hey-Hawkins. His research focuses on carborane chemistry with medicinal applications. |
 Marta Gozzi | Marta Gozzi received her BSc degree from the University of Bologna (Italy). She joined the Erasmus Mundus program Advanced Spectroscopy in Chemistry and received a double MSc degree (University of Lille1 and Leipzig University). She carried out her doctoral studies at Leipzig University (with Prof. E. Hey-Hawkins), and received her doctoral degree in early 2019. Her research interests are in the field of bioorganometallic chemistry, particularly on metallacarborane complexes for application in medicine. She contributed book chapters to Boron-based Compounds: Potential and Emerging Applications in Medicine, and the Handbook of Boron Chemistry in Organometallics, Catalysis, Material Science and Medicine. |
 Robert Kuhnert | Robert Kuhnert studied chemistry at TU Bergakademie Freiberg (Germany) and Leipzig University (Germany). He received his masters degree 2013 and started his doctoral degree in the group of Prof. Evamarie Hey-Hawkins at Leipzig University investigating carboranes as phenyl mimetics for lipoxygenase inhibitors and boron carriers for boron neutron capture therapy (BNCT). After he received his doctoral degree in 2017, he was scientific manager of the European network on Smart Inorganic Polymers (SIPs). Since October 2018, he is laboratory head of a medium-sized chemical company in Bitterfeld (Germany). |
 Menyhárt B. Sárosi | Menyhárt B. Sárosi received his doctoral degree in chemistry from Babeş-Bolyai University (2011) funded by the Sectoral Operational Programme Human Resources Development, Contract POSDRU 6/1.5/S/3 – ‘‘Doctoral studies: through science towards society” (Romania). Afterwards, he was a postdoctoral researcher financed by the European Union and the Free State of Saxony (ESF-NFG 100148835) at the Faculty of Chemistry and Mineralogy, Leipzig University. Since 2015, he is a junior research group leader supported by the German Research Foundation (DFG, SA 2902/2-1) mainly interested in the molecular modelling of carborane-containing drugs and their interactions with biomolecules. |
 Evamarie Hey-Hawkins | Evamarie Hey-Hawkins has been Professor of Inorganic Chemistry at Leipzig University, Germany, since 1993. She received her diploma (1982) and doctoral degree (1983) at Philipps University Marburg, Germany. After stays in the UK (1984/85) and Australia (1985/87), she completed her habilitation in Marburg (1988). Her scientific interests are organophosphorus chemistry, biologically active boron compounds, and heterometallic transition metal complexes with applications in catalysis and materials science. She has published over 480 papers, edited a book on Boron-Based Compounds: Potential and Emerging Applications in Medicine (with Clara Viñas Teixidor), is a member of the European Academy of Sciences, and has received several prestigious awards. |
Key learning points1. Highlight important receptors (“old locks”) as targets for efficient therapeutic treatments and propose carboranes as new class of drugs (“new keys”).2. Present the benefits of carboranes as building block in drug design and outline novel carborane-based receptor ligands. 3. Guideline for readers through the versatility of potential medical applications of carborane-containing agents. 4. Current challenges of this novel strategy and the possibilities for structural modifications for enhancing drug properties and effects. 5. Molecular docking strategies available for carborane-based receptor ligands. |
1. Introduction
The first time the term “chemotherapy” appeared in the scientific community was at the beginning of the 20th century, when Paul Ehrlich introduced the concept of therapeutic treatments based on chemical substances (“magic bullets”, in German Zauberkugel) with selective affinity, or toxicity, for specific biological targets, such as pathogens. For his discoveries, he received the Nobel Prize in Physiology and Medicine in 1908, and is nowadays considered to be the father of chemotherapy. Over the last 100 years, chemicals-based therapies have considerably evolved, and are world-wide approved for treatment of tumours, viral, parasitic, or bacterial infections, inflammation, pain, and combinations thereof, although the underlying principle is, for all of them, Ehrlich's heritage of mechanism-based therapies.1 Structurally, most of the drugs on the market are small molecules (MW < 1000 Da), usually organic compounds, like doxorubicin, a topoisomerase II inhibitor used for the treatment of leukaemia, Hodgkin's lymphoma, breast, bladder and other cancers, the anti-malarial drug chloroquine, or the non-steroidal anti-inflammatory (NSAID) drug ibuprofen.2 Among metal-based drugs, a few platinum-containing complexes are approved for treatment of ovarian, testicular, cervical, breast and other cancers: examples are cisplatin, oxaliplatin and carboplatin.3 Another big class of drugs are therapeutic peptides (MW up to 2000 Da), such as insulin and adrenocorticotropic hormone (ACTH), which are mostly used as “replacement therapies” to supply hormone deficiency.4In the last 20 years, great interest has arisen for hybrid organic–inorganic compounds as novel small molecule drugs, which contain boron–carbon–hydrogen clusters, known as dicarba-closo-dodecaboranes (also known as carbaboranes or carboranes, Fig. 1). Although carborane-based drugs are yet to be approved for therapeutic treatments, many compounds were shown to be very promising drug candidates for applications in several fields: for boron neutron capture therapy (BNCT), e.g. carboranyl nucleosides and sugars,5 or enzyme inhibitors, e.g. cobalt bis(dicarbollide) complexes ([commo-3,3′-Co(1,2-C2B9H11)2]−, known as COSAN species) for potent and selective inhibition of HIV-1 protease,6 or combinations thereof, as in the case of carborane-based epidermal growth factor receptor (EGFR) inhibitors, recently reported by Couto et al.7
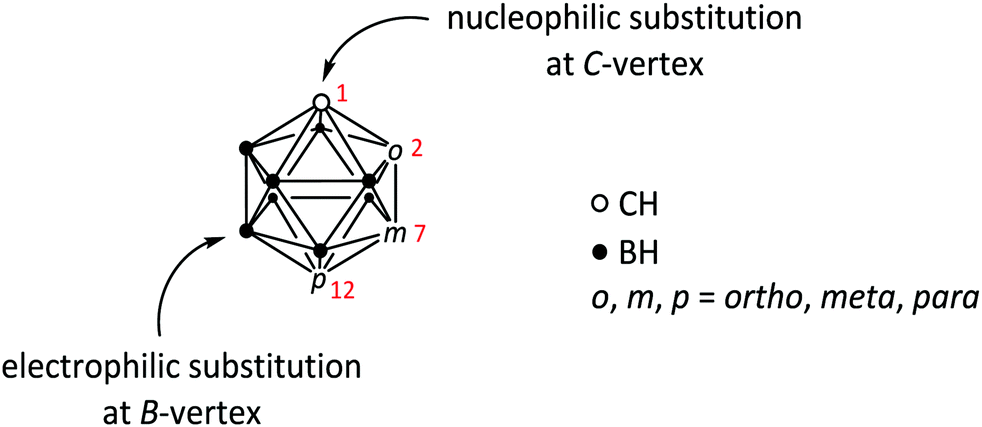 | ||
| Fig. 1 General structure of icosahedral carboranes of type closo-C2B10H12. Numbers in red indicate the numbering of the C-vertices of the cluster in the respective isomer: 1,2 (ortho, o), 1,7 (meta, m) and 1,12 (para, p). For detailed explanation of the numbering system in carborane clusters see ref. 1. The two most common types of substitution reactions (nucleophilic at C- and electrophilic at B-vertices) are indicated. | ||
Similar to purely organic and organometallic drugs, the medicinal chemistry of carboranes typically follows target-vector recognition approaches.8 There is in fact a rich literature on carborane-containing compounds, which were designed for interacting with a specific target, such as an enzyme receptor, thereby deserving the classification as “new keys”, in the sense of novel lead structures, for “old locks”, i.e. long- and well-known useful therapeutic targets, such as the EGF receptor.7
To understand the great potential of using these inorganic clusters for drug design, one must consider two main perspectives: on the one side, the physico-chemical and biophysical properties of carboranes, and on the other side, the type and strength of non-covalent binding interactions of a potential drug structure with the chosen biological target.
Carboranes are an extensive class of compounds, ranging from 4-vertex (sub-icosahedral) to 12-vertex (icosahedral) and 14-vertex (supra-icosahedral) structures.9 In the present review, we focus our attention solely on the 12-vertex clusters, dicarba-closo-dodecaboranes, of the general formula closo-C2B10H12.† Icosahedral carboranes are clusters of ten BH units and two CH vertices (Fig. 1).9 The carbon atoms in the cluster can be organised in a 1,2-, 1,7- and 1,12-fashion, giving rise to three isomers, namely ortho-, meta-, or para-carborane. All three isomers are commercially available; the ortho isomer is synthesised from decaborane (B10H14) and acetylene, in the presence of a Lewis base; thermal isomerisation to the meta and para isomers occurs at 450–500 °C and 700 °C, respectively.9 Of the three isomers, the ortho one is the most reactive, due to higher inductive electron attraction (–I) than in the meta and para isomers.9 This means that ortho-carboranes are generally easier to functionalise than the other two isomers, which require harsher reaction conditions. Also, ortho-carborane is more prone to undergo deboronation (i.e. loss of a BH unit) upon nucleophilic attack, to give the corresponding 11-vertex nido-[7,8-C2B9H12]− species.8,9 Due to the different nature of the cluster-bound hydrogen atoms, i.e. acidic (δ+) C–H, vs. hydridic (δ−) B–H, substitution reactions at either C- or B-vertices follow different mechanisms, which means that substituents can be attached to selected cluster positions (regioselectivity).8 The carborane cluster occupies a volume similar to that of adamantane, and is a little larger than a rotating benzene ring. Therefore, carboranes are often described as three-dimensional benzene analogues, in which the 26 skeletal electrons occupy 13 bonding molecular orbitals (MOs). The analogy with the benzene ring is indeed a strong motivation for the use of carborane clusters in drug design, following the well-known concept of bioisosteric replacement, inherited from the medicinal organic and organometallic chemistry.10 However, in contrast to aromatic hydrocarbons, which feature delocalised carbon–carbon π bonding, the aromaticity in polyhedral boron clusters utilises σ bonding interactions, involving a set of orbitals tangential to the polyhedral surface, and another set directed towards the centre.11 This means that the physico-chemical properties of the carborane cluster are substantially different from those of their organic counterparts, which largely substantiates the “benzene-inspired” medicinal chemistry of carboranes. There is in fact a rich literature on hybrid organic–inorganic compounds, which have been rationally designed to interact with a specific enzyme, based on well-known purely organic inhibitors, where the carborane cluster replaces either a phenyl ring or an adamantyl group.7,12–14 Following this approach, one might address common problems associated with the biological stability of organic drugs, e.g. fast metabolism and rapid clearance from body fluids, while preserving, or modulating, the target-vector recognition capabilities of the drug.15 Properties of carborane clusters, which are particularly relevant for application in medicine, are (i) their high hydrophobicity, due to the low polarity of the hydridic B–H groups, which proved to be beneficial for enhancing transport across cellular membranes and the blood–brain barrier (BBB),16 (ii) their inorganic nature, which prevents them from undergoing conventional enzymatic degradation, thereby strongly promoting drug stability under biological conditions, (iii) the possibility of three-dimensional orthogonal functionalisation of the cluster, superior to benzene systems, which are limited to two dimensions, leading to higher structural flexibility and, therefore, possibilities of fine-tuning the structure of the final compound, and (iv) the multitude of possible non-covalent interactions with biological targets: B–H⋯H–X (X = N, C, and S) dihydrogen bonds, B–H⋯Na+ bridging interactions, and C–H⋯X (X = O, N, S, F, π system) hydrogen bonds.8 Another favourable property of carborane clusters for drug design is their general tendency to self-assemble in aqueous solution, spontaneously forming nano- to micrometre sized particles.17 Nanometre sized particles potentially represent a very efficient way for the selective delivery of chemotherapeutics to cancer cells, via exploitation of the so-called “enhanced permeability and retention” (EPR) effect.18 The self-assembly behaviour is relatively broadly investigated and well characterised for anionic boron cluster compounds (ABCCs), e.g. COSAN species,19 and only recently are studies appearing on neutral carborane-containing compounds, such as the carboranyl-cysteine published by He et al.20 However, studies that specifically relate this spontaneous behaviour with the biological activity of carborane-containing drug candidates are still very rare in the literature.
Nowadays it is fairly routine to predict the binding mode and binding affinity of promising drug candidates to their target receptors with the help of computational tools. Molecular docking is one of the most often used methods in structure-based drug design for ranking potential ligands and for selecting the most promising ones. First, the possible conformations of a potential ligand within the binding site of a receptor are sampled, then the conformations are ranked using a scoring function. An ideal molecular docking protocol should be able to reproduce the experimental binding mode and select it as the preferred conformation. Molecular docking of carboranes or even boron-containing compounds is not straightforward, since the corresponding atom types, bonding and non-covalent interactions are not commonly available. Nevertheless, the number of publications containing molecular docking of carborane-derivatives is increasing.7,12,14,15,21–25
In recent years, several reviews have appeared in the literature on the topic of carboranes in medicine (see for example Scholz et al.,8 Issa et al.,16 Leśnikowski,6 and Schwarze et al.26). Therefore, in this review we focus our attention on the most recent developments on hybrid organic–inorganic compounds containing a closo-carborane cluster, which were designed as “new keys” for target-vector recognition mechanisms. Specifically, we review those compounds which have appeared in the literature since the extensive review work of Scholz et al. and Issa et al. (2011),8,16 and which were designed to trigger inhibition or activation of a specific enzyme. Moreover, the related molecular docking studies are also mentioned.7,12,14,15,21–25
The cannabinoid receptor type 1 (CB1) agonists designed by Vázquez et al. in 2012 are not discussed here, since after the rationally designed synthesis of the potential drug candidates no evaluation of their biological activity has been published so far.27
2. Carborane-based receptor ligands
2.1 Estrogen receptor ligands
Endo and co-workers pioneered in the early 2000s the structure-based drug design of carborane derivatives, supported by molecular docking studies.28 Their idea was to make use of the high hydrophobicity of the closo-carborane cluster, to modulate the binding affinity of receptor–ligand complexes. They focused their attention on potential novel ligands for the nuclear (intracellular) estrogen receptors (ERs). Particularly, they designed carborane-containing analogues of 17β-estradiol (Fig. 2), the major female sex hormone, which plays a central role in growth, differentiation and physiology of both male and female reproductive systems, as well as in bone marrow maintenance.29 More recently, the same group worked on the synthesis and biological evaluation of carboranyl-based compounds with selective affinity for specific ER subtypes.30 Nuclear ERs exist in fact as two major subtypes, α and β, which are composed of several domains: DNA binding, hormone binding and activation of transcription domains. A variety of nuclear responses are mediated through the binding of estrogens (17β-estradiol, estrone, estriol, estetrol), or estrogen analogues, to the ligand binding domain (LBD), which transactivates gene expression upon ligand binding. LBDs of ERα and ERβ are rather similar; therefore, many ligands bind unselectively to both receptor subtypes. However, ERβ selectivity for novel ER modulators has emerged as attractive therapeutic approach for the regulation of immune responses associated with chronic inflammation. Following this trend, Endo and co-workers synthesised a small series of tetrahydrofluorenone analogues, varying the length and the position of alkyl substituents at the cluster vertices, with n-propyl derivative 2 exhibiting up to 7 times higher affinity toward ERβ (Fig. 2). The rational design idea was that the aliphatic group should disfavour binding to the ERα, due to steric hindrance with the Met421 residue in the LBD, which is replaced in the ERβ subtype with Ile373. Besides this, the ERα Leu384 is replaced by Met336 in ERβ. Therefore, a wide range of ERβ-selective ligands obtain their ER subtype selectivity through steric, electronic or hydrophobic interactions with these essential amino acid residues. Based on the theory to specifically decrease the affinity for ERα by introducing ligand substituents that interfere sterically with Met421 of ERα, various ERβ-selective derivatives have been reported. With emulating, for example, structural properties of non-carborane receptor subtype-selective agents, such as androstane (a C19 steroid with a gonane core) skeleton analogues 3 or the modification of the carborane cage by halogenation 4 (Fig. 3), binding affinities to ERα were reduced with up to nearly 60 times higher affinity towards ERβ.29 Although the selectivity of the cyclohexanol derivative 3 is significantly higher than 4, the still low binding affinity toward ERβ does not allow for an adequate application as a potential drug. Thus, recent studies held the idea of modification through iodinated carborane cages, resulting in further possible ERβ-selective ligands, such as the 9,10-diiodo-m-carborane 5 (Fig. 3) with a 14 times higher affinity towards ERβ than ERα.31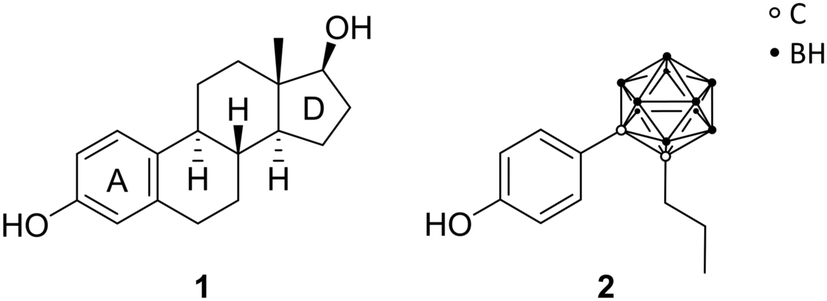 | ||
| Fig. 2 Structures of 17β-estradiol 1 and the n-propyl ortho-carborane derivative 2. | ||
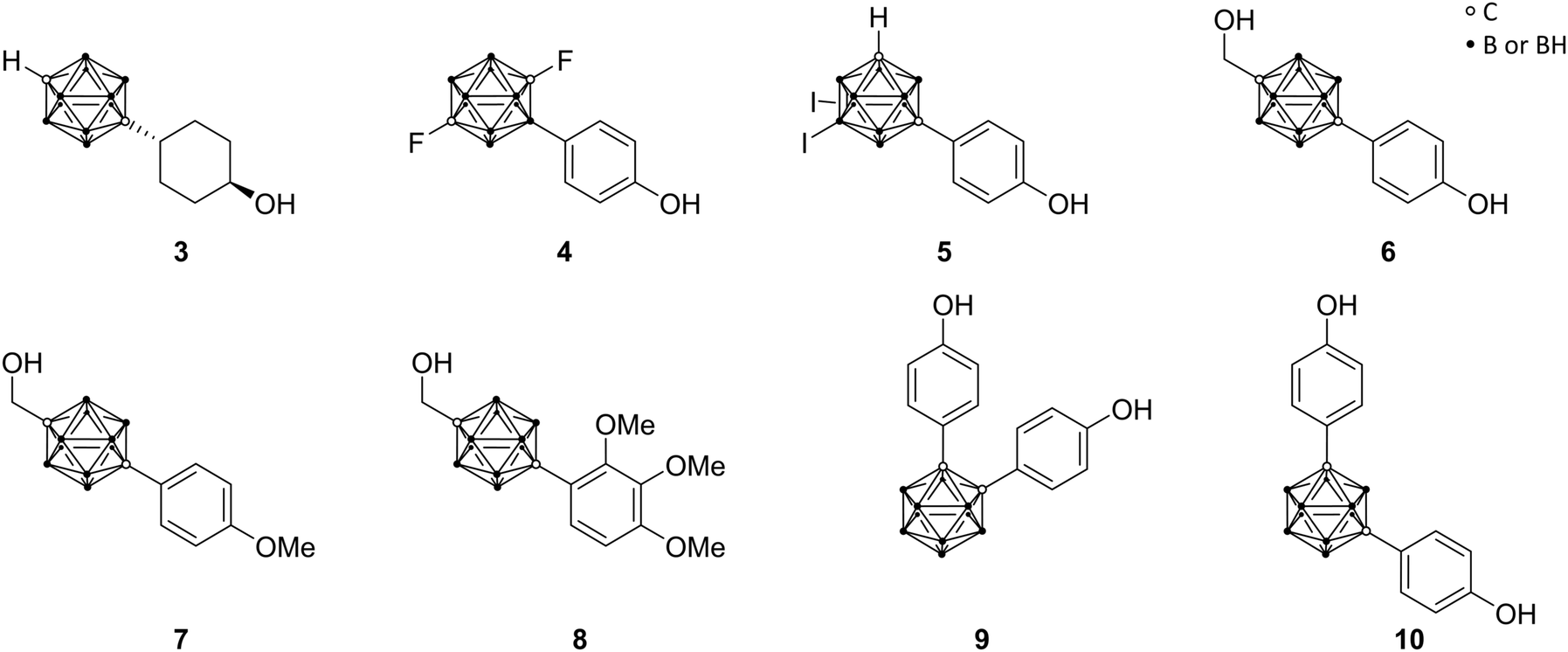 | ||
| Fig. 3 Molecular structures of estrogen receptor ligands; androstane analogue 3, halogenated analogues 4 and 5, BE120 6 and BE120-precursor 7, trimethoxy derivative of BE120 8, and bisphenolic carborane derivatives 9 (BE360) and 10. | ||
The first successful agent BE120 (1-hydroxymethyl-12-(4-hydroxy-phenyl)-1,12-dicarba-closo-dodecaborane) 6 (Fig. 3), an estradiol analogue bearing a para-carborane for mimicking the C and D ring of the steroid skeleton, exhibited a higher binding affinity (Ki = 0.1 nM) than estradiol (KD = 0.4 nM) and acted as an ERα super agonist.28 Revisiting BE120, Endo and co-workers recently presented different derivatives of 1-(4-methoxyphenyl)-12-hydroxy-methyl-p-carborane 7 (Fig. 3), a precursor for BE120, and their anti-proliferative activities. Studies revealed that with an increasing number of phenolic methoxy groups the cell growth inhibitory activity is enhanced. Moreover, the trimethoxy derivative 8 (Fig. 3) provided the most potent activity for cell growth inhibition within an assay of 39 human cancer cell lines (the concentration for 50% of maximal inhibition of cell proliferation – Gl50 value: 5.8 μM) and additionally inducing apoptosis for a certain breast cancer cell line, making this concept interesting for further research.32
On the basis of the known selective ER modulators (SERMs) raloxifene ([6-hydroxy-2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]-[4-(2-piperidin-1-ylethoxy)phenyl]methanone) and tamoxifen ((Z)-2-[4-(1,2-diphenylbut-1-enyl)phenoxy]-N,N-dimethylethylamine) and their structural features, various bisphenolic and related carborane derivatives were designed. Ligands based on meta- and ortho-carborane proved to be favoured as modulators. SERMs act as antagonists or agonists for targeting tissue-specific expressed ER subtypes. Tissue-specific expression of co-regulatory proteins and the variation of ligand-induced ER conformational changes are reasons for SERM specificity. Simple bis(4-hydroxyphenyl)-carborane derivatives were synthesised, such as the ortho-carborane-based BE360 9 (Fig. 3), exhibiting strong ER binding affinity and modulating properties in vivo with the significant prevention of bone loss without any estrogenic activity in the uterus, as revealed by studies on ovariectomised mice. Being 1000 times less potent than estradiol, BE360 exhibited an almost equal agonistic potency in bone tissue as the osteoporosis drug raloxifene. Then again, the m-carborane-based analogue 10 (Fig. 3) showed potent ERα agonistic activity and represented therefore a versatile scaffold for further research.29
2.2 Androgen receptor ligands
Prompted by the encouraging results of ER agents, similar targets were investigated by the same authors, particularly the androgen receptor, as member of the nuclear steroid hormone receptor family. Specific for the androgens testosterone and dihydrotestosterone (DHT), the androgen receptor (AR) regulates, similar to ER, various physiological functions in male reproductive, bone and muscle tissues. Prostate cancer, one of the major causes of death in adult males, is promoted through agonists of the androgen receptor, including the natural hormones, based on irregular behaviour of AR-mediated functions. This leads to a demand for AR ligands as antagonistic agents for prostate cancer treatments.Clinically used non-steroidal AR antagonists, as compounds without a steroid scaffold, such as flutamide (11) and bicalutamide (12) (Fig. 4), are used as models for further research. Adapted from this, Endo and co-workers concentrated on para-carborane derivatives bearing an electron-withdrawing group at the carborane-bound benzene ring (BA 321 13, BA341 14, Fig. 4). Studies revealed up to ten times higher binding activity to AR than hydroxyflutamide, the major active metabolite of flutamide, and both carborane derivatives 13 and 14 exhibited a dose-dependent inhibition of DHT activity and potent antagonistic activities.29 However, recent in vivo studies revealed that the carborane 13 acts as an androgen in bone tissues, but not in the reproductive system. Therefore, 13 occurs to be a potential AR modulator for bone homeostasis in elderly men with age-related bone weakness.28
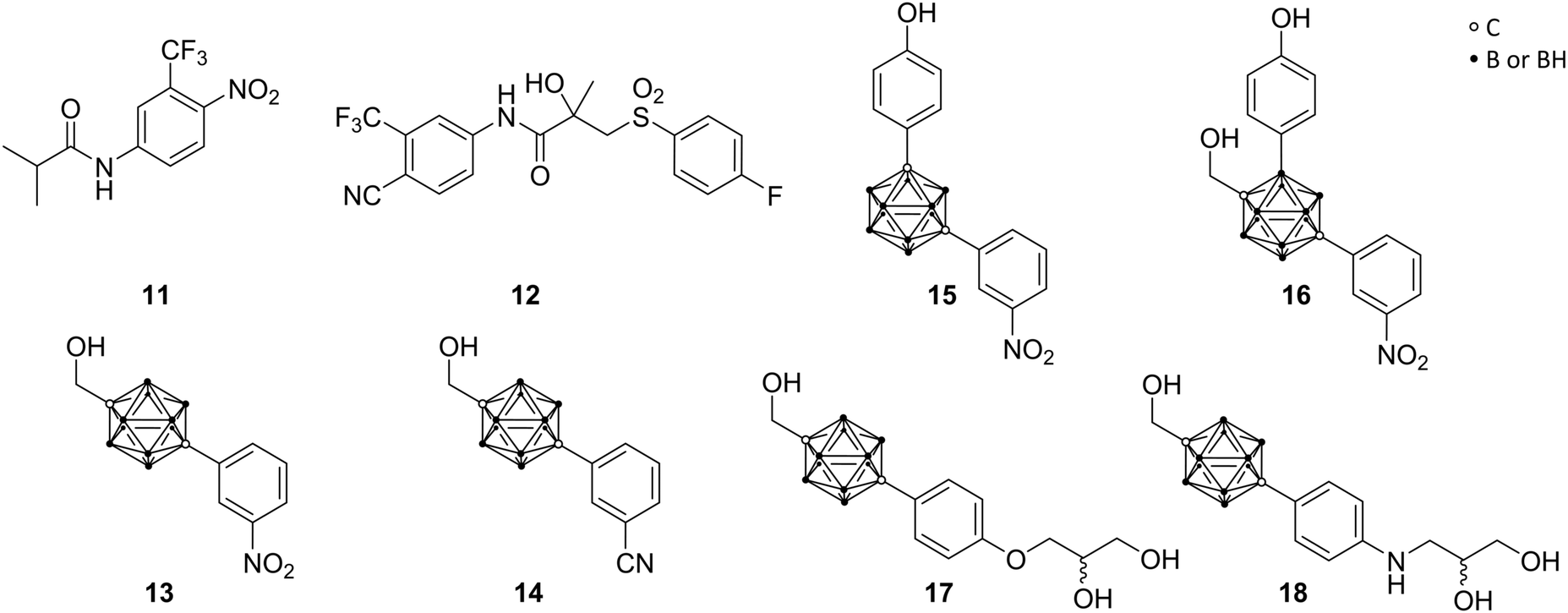 | ||
| Fig. 4 Structures of androgen receptor (AR) ligands flutamide 11, bicalutamide 12, BA321 13, BA341 14, the phenolic derivatives 15 and 16, and the glycerol and aminoglycerol carborane derivatives 17 and 18. | ||
Relying on the idea of opposing effects of estrogen agonists toward agonistic actions of androgens, Endo and co-workers attempted to design AR-ER dual ligands. Based on meta- and para-carborane structures, compounds 15 and 16 were evaluated (Fig. 4). 15 exhibited higher ER-agonistic activity at lower concentration than AR-antagonistic activity, and was therefore classified as ER agonist. On the contrary, 16 showed opposite behaviour, being closer to an AR antagonist.28
In turn, prostate cancer frequently progresses to advanced stages through the occurrence of hormone-independent cancer cells or endogenous androgen metabolites, promoting tumour growth. Since the mechanism of operation for androgen receptor is yet not entirely understood, studies report that mutations can occur. A point mutation of Thr877 to alanine (T877A) within the AR-LBD was found to be strictly related to aggressive advance of the disease. 13 and 14 exhibited high inhibitory activity toward wild-type prostate cancer cells, whereas they show agonistic activity towards human T877A-mutated prostate cancer cell lines (LNCap cells). The aforementioned organic drug bicalutamide 12 acts as AR antagonist in both types of prostate cancer cells, wild and T877A, whereby resistances also have been reported. Endo and co-workers followed the idea of the design of unaffected antagonistic activity of full antagonists for wild-type and mutant cells.22
Based on crystallographic data of the complex structure of bicalutamide bound to the mutant AR-LBD, they were able to show that the cyano group of the ligand binds to Gln711 and Arg752 through a water molecule. Therefore, following the approach of mimicking the binding mode of bicalutamide, glycerol and amino glycerol groups were introduced at the phenolic para position of the para-carborane derivative. The novel compounds 17 and 18 (Fig. 4) exhibited anti-androgenic activity by inhibiting the mutant androgen receptor in LNCaP cell lines. 17 and 18 exhibited antagonistic activities in LNCaP-proliferation assay (IC50 = 0.39 μM and 0.42 μM, respectively), similar to that of the (R)-bicalutamide, the active enantiomer (IC50 = 0.43 μM). Furthermore, none of the carborane derivatives showed inhibition of cell proliferation in PC3 cell lines (AR-independent prostate cancer cell line), confirming that inhibition of cell proliferation in AR-dependent cell lines is caused by inhibition of AR activation.22 Docking simulations of the two enantiomers of 17 helped to elucidate the observed isomer-dependent AR antagonistic activity. The chirality of the secondary hydroxy group of 17 resulted in different orientation of the para-carborane cages of the two enantiomers and the steric repulsion between (R)-17 and helix-12 of the AR-LBD was proposed as the cause of the potent anti-androgenic activity.22
2.3 Vitamin D receptor ligands
As a ligand-inducible nuclear receptor specific for vitamin D (formally known as calcitriol 19 or 1,25(OH)2D3, the metabolically activated form of vitamin D3), the vitamin D receptor (VDR) is expressed in most tissues of the human body. The receptor is involved in many essential physiological processes, by regulating the expression of specific target genes involved in, for example, cell proliferation and differentiation, immune regulation, bone metabolism, and homeostasis of calcium and phosphate. Playing, thus, a pivotal role in the pathogenesis and therapy of diseases, VDR ligands have been evaluated as potential therapeutic agents for the treatment of arthritis, osteoporosis, psoriasis, and cancers.29Reported by Fujii et al., the first VDR ligands bearing a carborane cage are non-secosteroidal ligands imitating calcitriol.29 Following the pattern of increasing the hydrophobicity of nuclear receptor ligands for more effective hydrophobic interactions, the C and D rings of calcitriol, similar to ER receptor ligands, were replaced with the carborane moiety. Furthermore, the three necessary hydroxyl groups should be preserved (Fig. 5). The carborane analogues exhibited, compared to the natural hormone, a moderate activity.28 Crystallographic data of the complexed LBD with the carborane-based 1,25(OH)2D3 analogue 20 confirmed hydrophobic interactions with the hydrophobic region of the VD-LBD (protein data bank code: 3VJS).29 Otero et al. investigated the first secosteroidal vitamin D analogue with a carborane unit (21), which was found to efficiently bind to the VDR-LBD (protein data bank code: 5E7V).25 The binding mechanism could be observed by co-crystallisation of the carborane analogue and VDR with 21 mimicking the 1,25(OH)2D3 hydroxyl interactions through boron-mediated dihydrogen bonds. Further, biological properties were analysed on different cell lines, such as MCF-7, HaCaT keratinocytes and HEK 293, showing decreased cell proliferation and differentiation-inducing activity compared to 1,25(OH)2D3. 21 has been designed with the help of molecular docking simulations.25 The docked pose of the carborane analogue corresponded well with the subsequently obtained crystal structure. Unfortunately, the source of the boron parameters used for docking has not been specified.
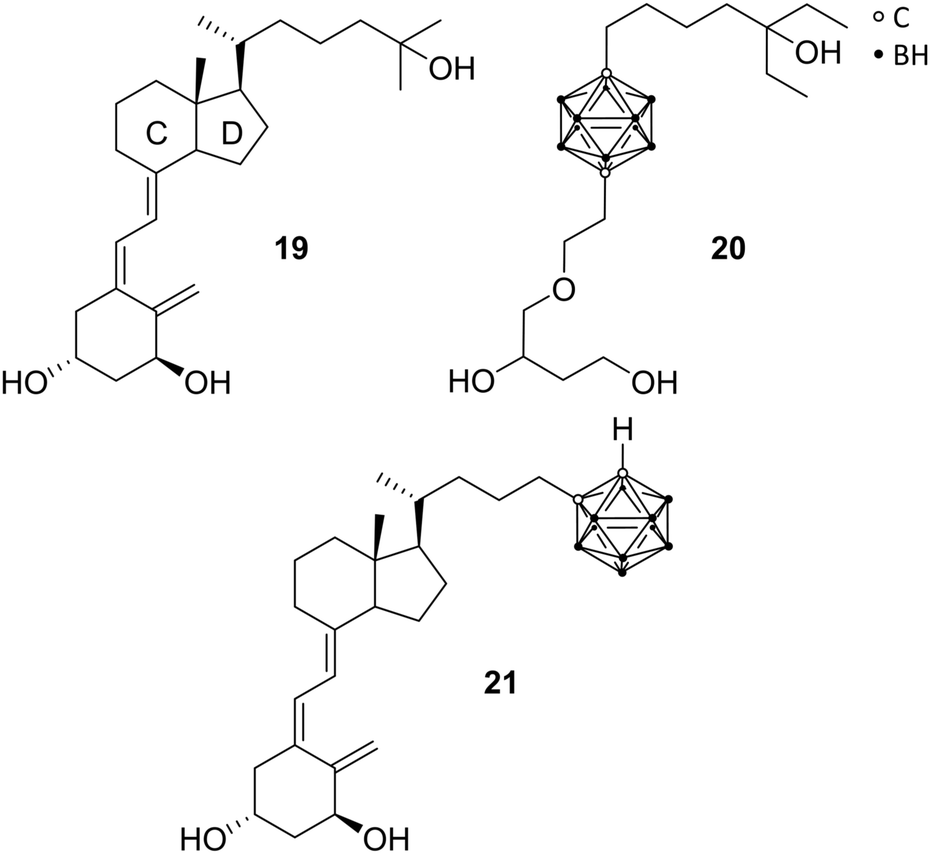 | ||
| Fig. 5 Structures of calcitriol 19 and vitamin D receptor ligands 20 and 21. | ||
2.4 Purinergic receptor ligands
The P1 receptors, also called adenosine receptors (AdRs), are purinergic G-protein coupled receptors. AdRs are preferentially activated by adenosine and are implicated in various pathological states and physiological processes, making them therapeutic targets. Over the last decades, selective and potent exogenous AdR agonists and antagonists as well as inhibitors for extracellular adenosine metabolism were designed.33 Keeping in mind that AdR ligands bear a promising approach for several clinical applications, the problem of the omnipresence of purinergic receptors in the human body is difficult to overcome. The approach of modulating the cell function by purine-based derivatives yielded numerous compounds but gave rise to only a few agents as approved drug or still in clinical trials. Therefore, there is an urgent need for new agents, which target tissue-specific AdRs.34Bednarska et al. focused on modified adenosine analogues, carrying a para-carborane moiety, such as 22 and 23 (Fig. 6). These compounds were able to induce inhibitory activity for production of reactive oxygen species (ROS) in human neutrophils (specialised immune cells) stimulated with fMLP (N-formyl-met-leu-phe; termed as chemotactic peptide) in the low micromolar range, while adenosine as well as the appropriate nido derivatives are inefficient inhibitors at these low concentrations. Interestingly, the inhibitory effect of 22, a para-carborane bearing adenosine derivative, was considerably higher than that of adenosine, with IC50 values of 0.72 and 1.81 μM, respectively. Furthermore, 22 still showed inhibition of ROS production in low nanomolar ranges (up to 10 nM), with adenosine exhibiting no effect at these concentrations.33 Indeed, these compounds opened up new possibilities for the design of novel AdR modulators and agents affecting inflammatory mechanisms, as recent in silico studies on AdRs A2A and A3 have demonstrated.35
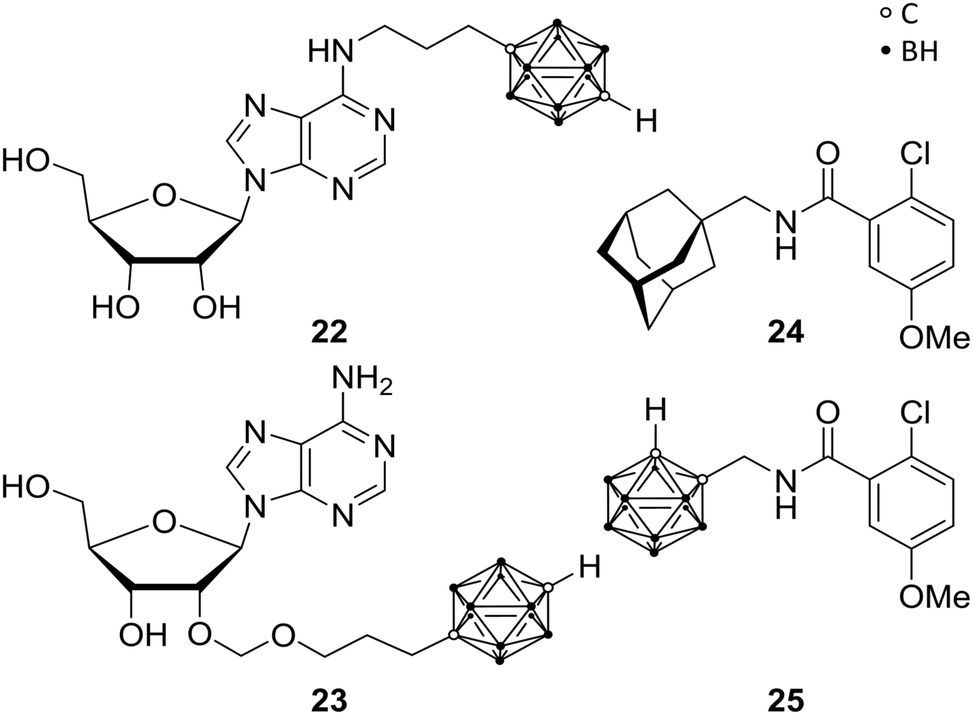 | ||
| Fig. 6 Adenosine receptor ligands 22 and 23 and P2X7R ligands 24 and 25. | ||
In the wide field of human purinergic receptors, different individual subtype receptors are gaining interest as a potential biological target, through their ubiquity and variety of functions. Wilkinson et al. focused on the P2X purinergic receptor 7, the P2X7R. P2X7R is a protein being composed of ion channels, which are cation selective and ligand gated, opening in response to ATP binding.34 Upon iterative and persistent exposure of P2X7 to agonistic agents, non-selective pore formation is induced, leading to permeability for cations (Mw (cation) < 900 Da).36 The latter induce apoptosis and the secretion of cytokines (interleukin 1β), which are correlated to neuronal disorders, such as hyperalgesia, depression and neurodegeneration. P2X7R antagonistic ligands are, therefore, feasible anti-analgesic, anti-depressant or neuroprotective agents. Furthermore, as a potent stimulant of immunity and inflammation, and a promoter of cancer cell growth, P2X7R might be the target for, e.g., anti-inflammatory and anti-cancer therapies. Based on previous studies,37 Wilkinson et al. replaced the adamantyl moieties of 24 in order to increase metabolic stability.38 They synthesised a mono-functionalised ortho-carboranyl benzamide (25, Fig. 6) as potential anti-depressant targeting the central nervous system (CNS). 25 showed slight enhancement of the inhibitory activity towards human P2Y7R (hP2Y7R) in comparison with its adamantyl analogue 24 (Fig. 6), with IC50 values of 8.7 and 10.5 nM, respectively. Subsequently, animal behaviour tests were performed on mice, assessing and approving CNS activity and the ability of crossing the blood–brain barrier (BBB). Despite their inhibitory potency in vitro against hP2X7R, 24 and 25 showed no anti-depressant efficacy in vivo. Not crossing the BBB and/or interspecific distinctions and species-specific differences between organisms may be possible reasons for failure. Interestingly, when 25 is converted to its nido-carborane derivative, significant anti-depressant activity is observed with increased efficacy by two orders of magnitude, despite decreased lipophilicity by its anionic charge. Thus, the nido analogue of 25 is representing the first carborane-containing agent showing the ability to modify CNS activity. Nevertheless, 25 exhibited significantly higher values of lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (partition coefficient), i.e. higher lipophilicity than the reported CNS ligands which are effective in vivo, suggesting a potential inhibition of the BBB penetration despite the fact that normally higher lipophilicity enhances crossing the BBB.39 Thus, presented closo-carborane derivatives can be considered as starting point for prospective CNS agents or, furthermore, as potential precursor for their corresponding nido analogues.34
P (partition coefficient), i.e. higher lipophilicity than the reported CNS ligands which are effective in vivo, suggesting a potential inhibition of the BBB penetration despite the fact that normally higher lipophilicity enhances crossing the BBB.39 Thus, presented closo-carborane derivatives can be considered as starting point for prospective CNS agents or, furthermore, as potential precursor for their corresponding nido analogues.34
2.5 Hypoxia-inducible factor ligands
Angiogenesis, the formation of new blood vessels from pre-existing vessels, is a vital process in the human body and, therefore, similarly essential for tumour cells in growth and metastasis. Erythropoietin (EPO) and the vascular endothelial growth factor (VEGF), as major angiogenesis factors in tumours, are activated gene hormones and are physiologically expressed in response to the stimulation by the hypoxia-inducible factor 1 (HIF-1). The heterodimeric transcription factor HIF-1, consisting of two subunits (α and β), is formed under hypoxic conditions. Under normal, aerobic conditions (normoxia) no dimer is formed, due to hydroxylation of the proline residues in the HIF-1α protein, which is recognised by tumour suppressor proteins for further degradation through the ubiquitin-proteasome system. It was found that HIF-1α is overexpressed in various cancers and, thus, represents a promising target for a new type of approach in cancer therapy.40Nakamura and co-workers used LW6, a phenoxyacetanilide derivative (LW6, 26, Fig. 7), as structural model for boron- and carborane-based analogues. Particularly, the adamantyl moiety of LW6 was replaced by an ortho-carborane cage to obtain derivatives such as 27 and 28 (Fig. 7), which showed a more potent HIF-1 inhibitory activity (IC50 = 0.7 and 1.7 μM, respectively) than LW6 (IC50 = 3.1 μM), using a reporter assay in HRE(×5)-Luc transfected HeLa cells (HRE, hypoxia response element). 27 showed the ability to suppress the accumulation of HIF-1α under hypoxic conditions. Further studies revealed a similar inhibitory activity on heat shock protein 60 (HSP60) chaperone activity through the higher binding affinity evoked by the carborane cage, resulting in a potential mechanistic connection between HIF-1 and HSP60.40 Further modifications of the LW6 model led to structures such as 29 and 30 (Fig. 7), containing either an ortho- or a meta-carborane cluster, which feature different phenylamino moieties and substituted phenyl or alkyl groups at the two C-vertices.40,42 Generally, it was found that increasing the steric demand of the alkyl substituent attached to the meta-carborane moiety enhanced the inhibitory activity. Particularly, 29, with an isobutyl group, showed substantial inhibitory activity for HIF-1 transcription (IC50 = 0.55 μM). Phenoxyacetanilides with meta-carborane backbone showed strong inhibitory potential with IC50 values in the low-micromolar range. The meta-carborane derivatives are generally more potent than their ortho-carboranyl counterparts, except for one ortho analogue, 30. The derivative 30, containing an ethylene linker for a rigid conformation, exhibited the highest inhibitory activity with an IC50 of 0.53 μM (compound 27 was used as reference: IC50 = 2.2 μM). The authors stated that sterically bulky groups are necessary for an efficient inhibitory potential. Furthermore, these compounds inhibited cell growth and showed cytotoxic effects for HeLa cells in the low micromolar range.40
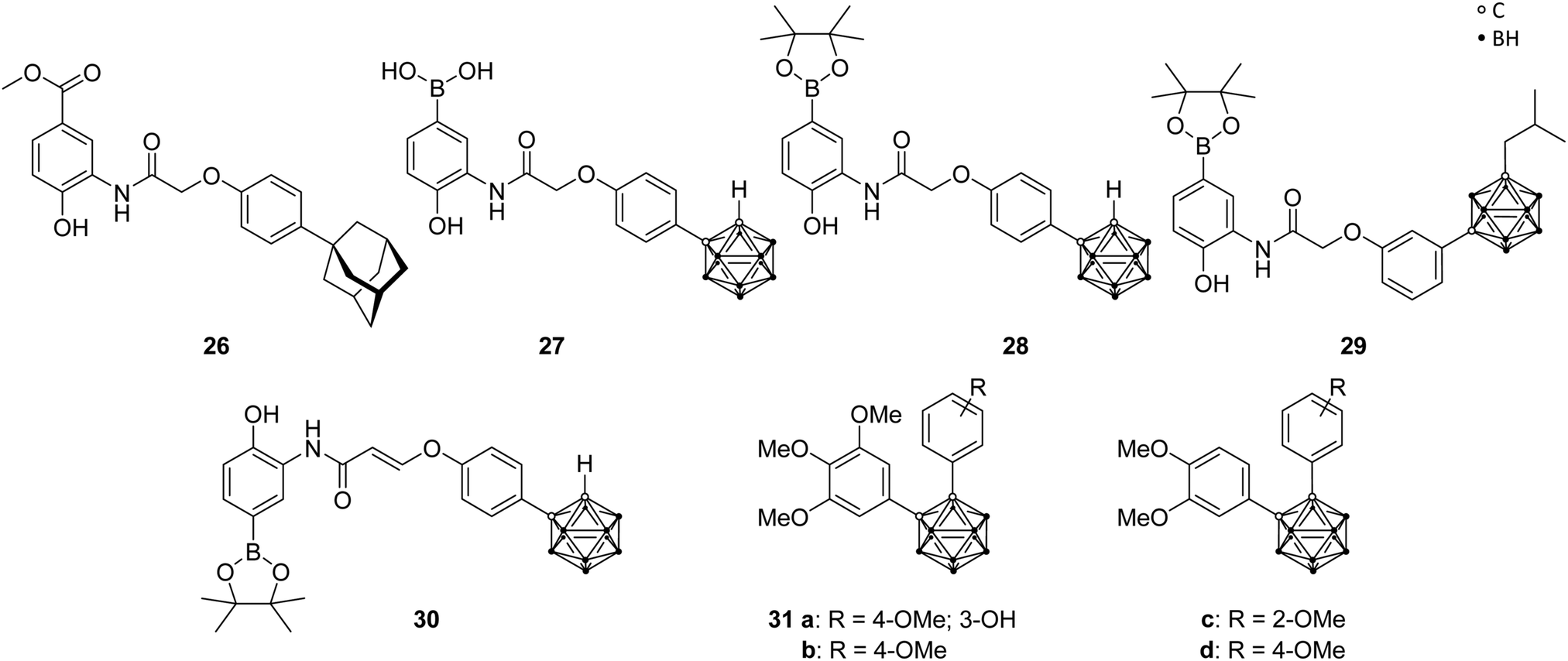 | ||
| Fig. 7 Hypoxia-inducible factor ligand structures of LW6 26, the carborane analogues 27–30 and the carboranyl combretastatin analogues 31. | ||
Additionally, recent studies examined analogues of combretastatin A-4, a tubulin-binding cis-stilbene which induces apoptosis of cancer cells by disrupting microtubule dynamics. By replacing the cis-stilbene scaffold with an ortho-carborane, various compounds were synthesised, among which 31a–d (Fig. 7) exhibited a significant inhibition of HIF-1 transcriptional activity (IC50 of 0.6, 1.2, 0.8 and 0.4 μM, respectively), in hypoxic cancer cell line assay, with slightly higher potency than the known HIF inhibitor YC-1, used as positive control (IC50 = 1.5 μM). Additional investigations were able to indicate tubulin, a cell protein, as primary target for carboranyl combretastatin A-4 analogues, and the inhibitory activity toward HIF-1 is, therefore, mediated via the tubulin pathway, setting a novel concept for prospective anti-cancer therapy.13
2.6 Epidermal growth factor receptor ligands
Couto et al. recently reported on carborane-based epidermal growth factor receptor (EGFR) inhibitors. The EGFR is a transmembrane glycoprotein, which acts as receptor for epidermal growth factors (EGFs) of extracellular protein ligands, and is composed of a hydrophobic transmembrane section, an intracellular tyrosine kinase domain and an extracellular binding domain. Upon binding of EGFs, downstream signals are activated, which stimulate physiological processes, such as cell proliferation, differentiation and cell growth, and are, thus, part of the regulatory functions in cells. Overexpression of EGFR was found in various solid tumours, e.g. breast, colon, lung and glioma. Thus, perturbing the EGFR signalling through inhibition of the tyrosine kinase activity or blocking of the EGFR binding site is a promising therapeutic approach for EGFR-related tumour growth.7Previous investigations had shown that carboranyl anilinoquinazoline derivatives of the known EGFR inhibitor erlotinib (32, Fig. 8) exhibited selective cytotoxic effects against EGFR-overexpressing C6 rat glioma cells.41 Couto et al. evaluated, with further help of molecular docking studies, the mechanism of action by investigating the inhibitory activity on wild-type EGFR and T790M, an erlotinib-resistant mutational form of EGFR. T790M-mutation involves the replacement of threonine 790 with methionine. As a result, the larger methionine residue prevents binding of a tyrosine kinase inhibitor, such as erlotinib 32. EGFR resistance to tyrosine kinase inhibitors has been described for numerous cancers. The activities for the wild-type EGFR (wt-EGFR) are similar for C6 glioma cell lines. Among the tested ligands, the carborane derivative 33 (Fig. 8) exhibited the strongest inhibitory effect on wt-EGFR (IC50 = 2.3 nM), almost ten times more potent than erlotinib (IC50 = 22.9 nM). Docking of 33 into wt-EGFR, using non-covalent interaction parameters derived from quantum chemical calculations for the carborane cluster, revealed the perfect fit of 33 into the catalytic cleft.7 The inhibitory activity for EGFR was measured using a cell-free kinase inhibition assay. Ligand 33 showed the ability to cross the brain–blood barrier (ΔlgP = 1.49; ΔlgP = lgP(octanol/PBS) − lgP(cyclohexane/PBS)) and sufficient inhibitory activity for the T790M-mutated EGFR, with erlotinib showing no effect on the mutated cells. Thus, 33 is a promising lead structure for the design of dual agents, which target the EGFR and for potential BNCT applications.7
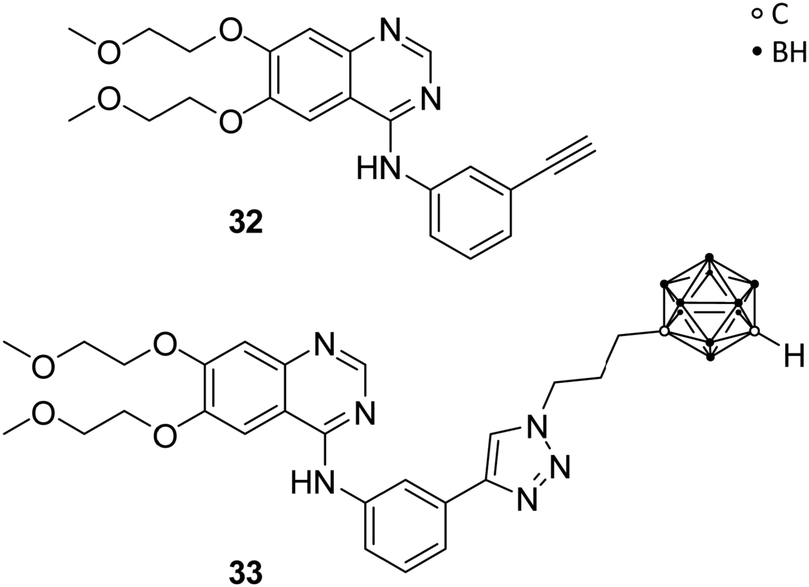 | ||
| Fig. 8 Structures of EGFR inhibitors erlotinib 32 and the carborane derivative 33. | ||
2.7 Thymidine kinase ligands
Recently, the impact of the human thymidine kinase 1 (hTK1) in cancer research intensified, as increased levels of hTK1 in blood serum were found when malignant tumours were present and, interestingly, even before the occurrence of physical symptoms. The enzyme hTK1 is embedded as vital member of the synthetic recycling pathway in the nucleotide synthesis and, despite its normal activity during the S-phase of the cell cycle, high hTK1 activity can occur in cancer cells during other cell cycle phases.42Since thymidine is a major endogenous hTK1 substrate, Agarwal et al. prepared numerous derivatives, bearing a carborane cage attached to N-3 of the thymidine skeleton (Fig. 9) via various linkers, such as amidinyl, guanidyl, tetrazolylmethyl, and tetrazolyl groups, to improve hTK1 substrate activities and water solubility compared with previously presented carboranyl thymidine derivatives, such as N5-2OH 34. Unfortunately, the introduction of carboranes led to problematic solubility and the compounds showed no stability in aqueous solution, limiting biological investigations. The guanidyl- (35) and amidinyl-substituted carboranyl thymidine analogues (36) exhibited low phosphorylation rates (<30% relative to thymidine), in phosphoryl transfer assays. 36a and 36b showed >1900 and >1500 times better solubility in phosphate-buffered saline solutions than 34.43 However, further improvement of the reported ligands on phosphorylation rates, solubility and metabolic stability were needed.43
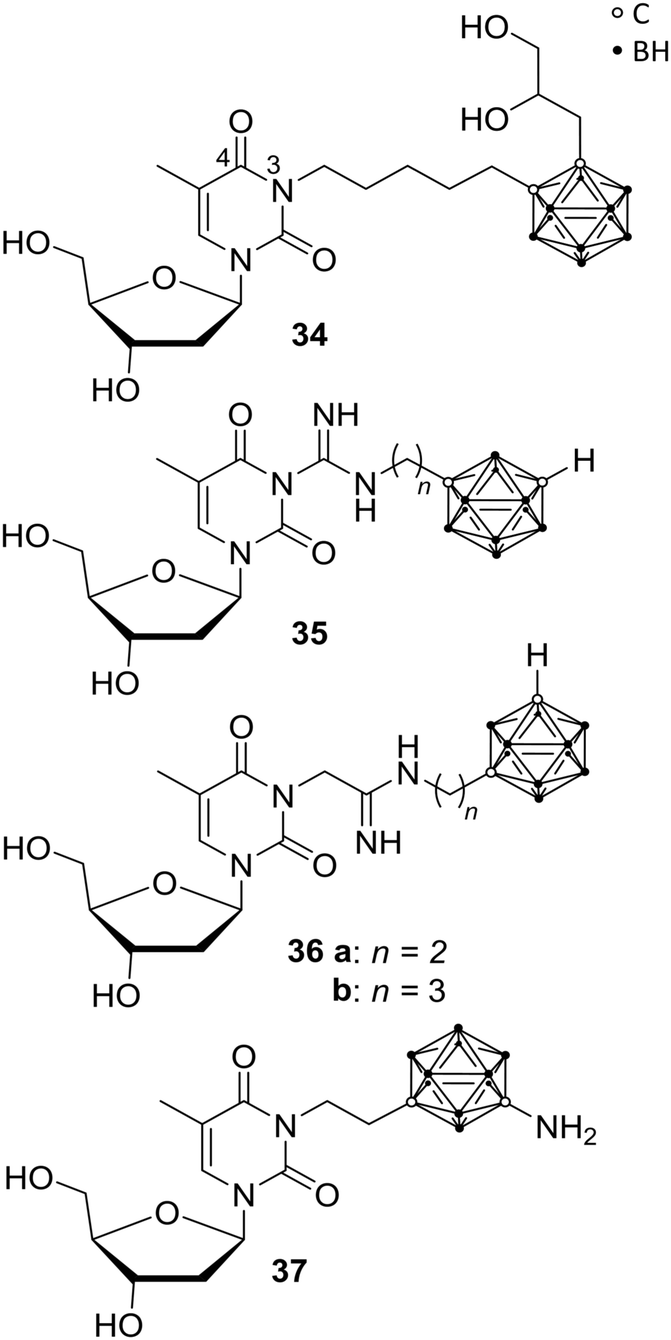 | ||
| Fig. 9 Thymidine kinase inhibitors N5-2OH 34, the guanidyl 35 and amidinyl derivatives 36 and YB18A 37. | ||
In recent studies, Agarwal et al. presented further carborane-based thymidine kinase 1 inhibitors, including derivative YB18A 37 reported earlier.44,45 Phosphorylation rates (rPr) relative to that of thymidine were determined, exhibiting low values for most of the new ligands. The rPr values of amino meta-carborane derivative YB18A 37 were twice as high as for N5-2OH 34 (78.7 and 40.0%, respectively; with thymidine set as 100%). Biodistribution studies of these compounds in mice bearing intracerebral RG2 gliomas showed very good tumour-to-blood ratios of up to 62.5 and tumour-to-brain ratios of up to 4.9. The boron concentration of 37 in the tumour tissue was determined by ICP-OES (inductively coupled plasma – optical emission spectrometry) to be 32.5 μg g−1 tumour.45 Thus, these compounds might also be suitable candidates for BNCT.
In addition, Agarwal et al. were able to show that a substitution at the 4-position of the pyrimidine ring eliminated any activity. It was presumed that, based on the examinations, the linker length between pyrimidine scaffold and carborane is defining the location of the cluster inside or outside of the binding pocket and, further, the carborane-attached amino group of 37 interacts with amino acid residues of the receptor, resulting in improved hTK1 binding affinity and, subsequently, enhanced substrate features and antagonistic effects toward thymidine.45
2.8 Nicotinamide phosphoribosyltransferase ligands
The nicotinamide phosphoribosyltransferase enzyme (NAMPT) has a pivotal role in cellular proliferation and maintenance, metabolism and inflammatory response and, thus, holds a promising opportunity as target in treatments for several diseases, such as diabetes, arthritis and cancer. Overexpression of this enzyme leads to severe symptoms, such as sepsis and pulmonary inflammation. NAMPT is a rate-limiting enzyme, which catalyses the synthesis of nicotinamide mononucleotide (NMN), within the nicotinamide adenine dinucleotide (NAD+) biosynthesis and salvage pathway. NAMPT is able to regulate immune response and, furthermore, has been reported to be a cytokine that promotes B-cell maturation and inhibits neutrophil apoptosis.Lee et al. reported novel NAMPT ligands based on FK866 (38, Fig. 10), comparing all three carborane isomers.14 The organic NAMPT inhibitor 38 had been previously identified through ligand library screening, showing induction of apoptosis by lowering of NAD+ levels, the oxidised form of nicotinamide adenine dinucleotide, without affecting the cell energy metabolism.46 Through co-crystallisation of the enzyme-drug complex, narrow tunnels at each of the two active sites of the protein dimeric surface were identified as fundamental for binding affinity. Hence, molecular docking studies were carried out, showing hydrophobic interactions with several hydrophobic residues, including tyrosine, valine, and isoleucine. However, the introduction of carborane led to 100-fold increased inhibitory potency towards NAMPT and a 10-fold increase in anti-proliferative activity against cancer cells over their purely organic counterparts. Despite the fact that para-carboranes are the most hydrophobic isomers, therefore presumably interacting more strongly with hydrophobic sites of NAMPT, the meta-derivative MC4 m-39 (Fig. 10) demonstrated by far the most potent effect compared to FK866 against human cell lines for lung (A549), colon (DLD1), and breast tumours (T47D) in the low nanomolar range (<0.3 nM).14
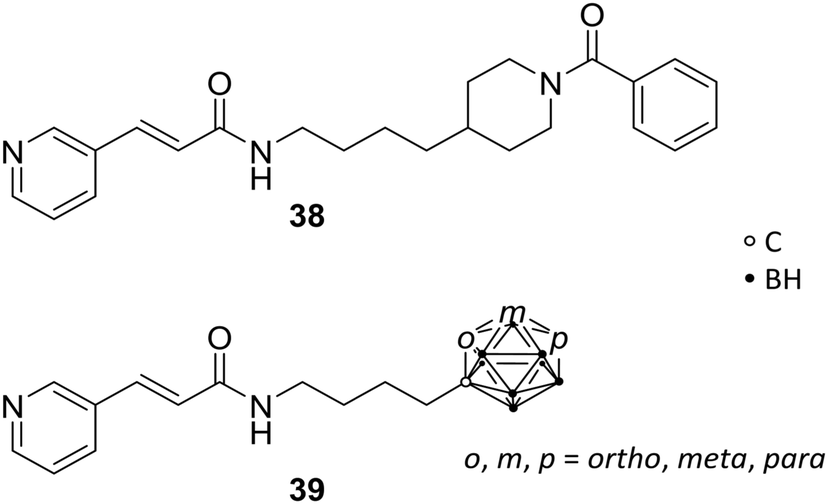 | ||
| Fig. 10 Molecular structures of nicotinamide phosphoribosyltransferase inhibitor FK866 38 and the carborane analogues 39. | ||
Further in vitro and in vivo studies of MC4 revealed stronger inhibitory activity than FK866 on CLP-induced (cecal ligation and puncture) mortality, TNFα levels, pulmonary myeloperoxidase activity, alveolar injury, and interleukin 6 and interleukin 1 beta messenger RNA levels. 39 proved to be a potential novel agent for sepsis and inflammation treatments in prospective clinical applications.47
2.9 Carbonic anhydrase ligands
Human carbonic anhydrases (CA) are metalloenzymes, containing a Zn2+ ion at the catalytic centre, which exhibits an important role in numerous physiological and pathological functions (e.g. in epilepsy and tumourigenicity). Therefore, CA inhibitors represent attractive scaffolds for drug design. CA exists as several isozymes, which differ in some amino acid residues at the entrance of the binding site and, therefore, allow for selective targeting of CA subtypes. Brynda et al. reported rational drug design, based on manual molecular docking studies of CA subtype II (CAII). The introduction of carboranes led to highly efficient inhibitors, with IC50 values in the low micro- to higher nanomolar range, and selectivity indices (SI) of up to 18 for specific CA subtypes. Through manual molecular docking into the binding site of CAII, lead structure 40 was designed (Fig. 11), bearing a methylene-bridged sulfamide moiety attached to the ortho-carborane cluster. 40 exhibited inhibitory activities towards CAII (Ki = 0.70 μM) and CAIX (Ki = 0.38 μM), with SI of 2 for CAIX.12 CAIX is a transmembrane protein, with an extracellular catalytic site, and is overexpressed in hypoxic solid tumours.48 Further, co-crystallisation of the ligand-protein complex confirmed the predicted interactions and binding at the active site (PDB code 4MDG), leading to the evaluation of further carborane-based derivatives of 40. A second sulfamide group (41, Fig. 11) increased inhibitory activity towards both tested isozymes, with the same SI as for 40. Higher SIs were only achieved with nido derivatives.12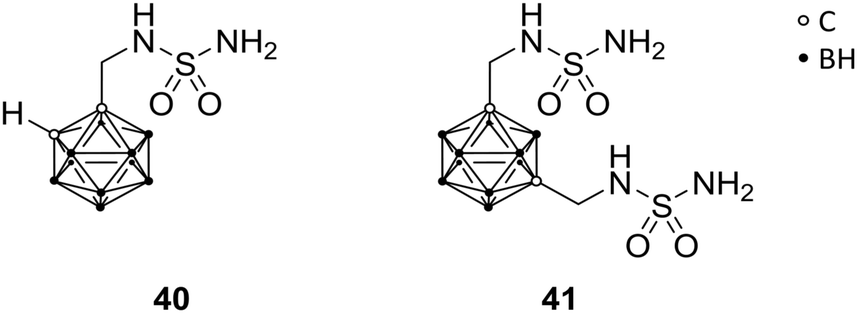 | ||
| Fig. 11 Carbonic anhydrase inhibitors 40 and 41. | ||
2.10 Glutamate carboxypeptidase II ligands
The prostate-specific membrane antigen (PSMA), also referred to as the glutamate carboxypeptidase II (GCPII), is highly expressed in nervous and prostatic tissues. It is a membrane-residing zinc metallopeptidase responsible for neuronal signalling through hydrolysis of N-acetylaspartylglutamate (NAAG), a major peptide neurotransmitter. Also, GCPII is highly overexpressed in androgen-independent and metastatic tumours, like prostate carcinoma, and therefore a biomarker for prostate cancers.49Ligand–enzyme interactions in the two major binding pockets of GPCII were determined by X-ray structures of ligand-GCPII complexes. The S1 pocket, situated at the entrance funnel of the active site of GPCII, shows a size-limited hydrophobic cavity, bearing a positively charged arginine residue. The S1′ pocket features a glutamate-recognition site. Among the numerous reported inhibitors, Youn et al. designed carboranyl analogues of the urea-based (S)-2-{3-[(S)-1-carboxy-5-(4-iodobenzamido)pentyl]ureido}pentanedioic acid (DCIBzL) 42 (Fig. 12), replacing the iodophenyl group with ortho- or meta-carborane. DClBzL shows a singular and very specific binding mode in GPCII's active site, whereas the iodophenyl moiety fits into the S1 binding pocket, thus promoting high binding affinity. The carborane analogues 43 (ortho) and 44 (meta) were found to be potent inhibitors with IC50 values in the low nanomolar range (15.6 nM and 20.5 nM, respectively, Fig. 12) in fluorescence polarisation assays, although less potent than DClBzL, used as control (IC50 < 1 nM). The compounds were co-crystallised and crystal structure analysis revealed the carborane being situated at the entrance funnel and outside the S1 binding pocket, due to its sterically demanding bulky structure. Consequently, the location of the carborane moiety leads to the assumption that the cluster can be further functionalised in order to provide even more potent inhibitors or, also, introducing cluster-halogen substituents for imaging and anti-tumour therapy.50
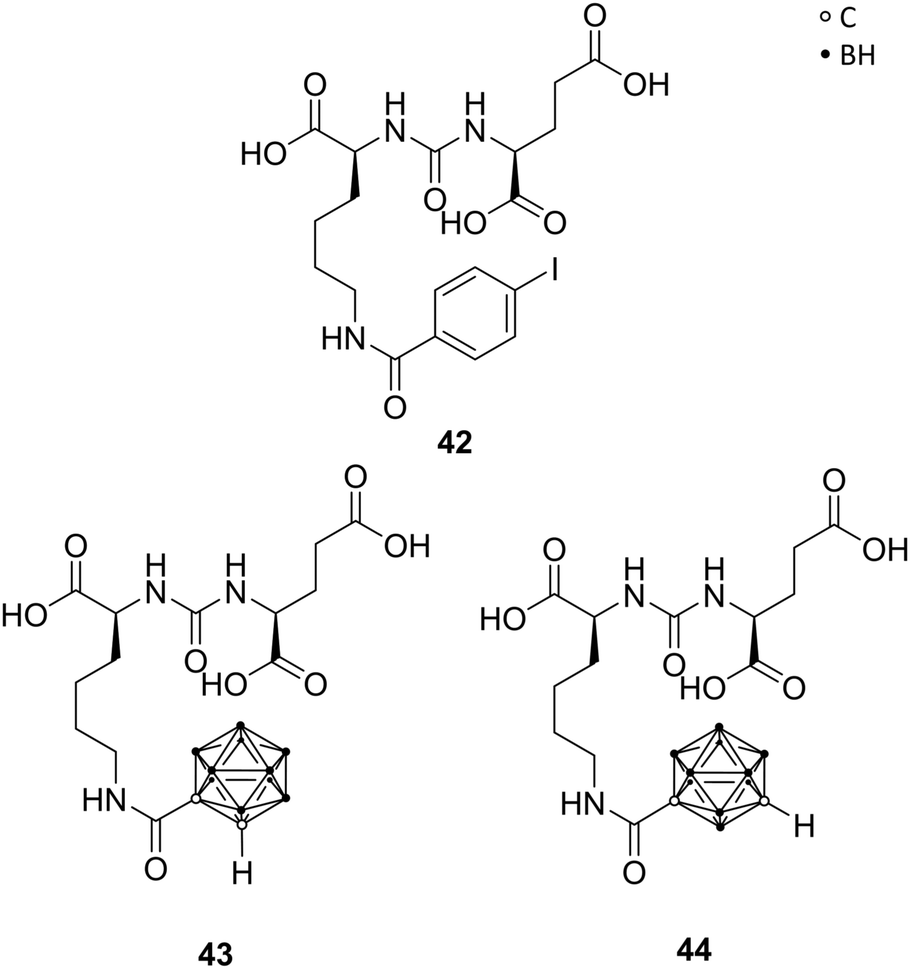 | ||
| Fig. 12 Structures of the GCPII inhibitors DCIBzL 42 and the carborane derivatives 43 and 44. | ||
2.11 Cyclooxygenase ligands
Cyclooxygenase (COX) is an enzyme that catalyses the key steps in the biosynthesis of prostaglandins, and exists as two different isoforms, COX-1 and COX-2. COX-1, the so-called ‘housekeeping’ enzyme, is constitutively expressed, whereas COX-2 is highly inducible at sites of inflammation. COX inhibitors are among the most used medications, primarily for the treatment of inflammation. The well-known non-steroidal anti-inflammatory drugs (NSAIDs; e.g., aspirin, ibuprofen, and indomethacin) are generally non-selective inhibitors of both COX isozymes. COX-2 is upregulated in various tumours and tumour microenvironment, leading to tumour-promoted inflammation. Therefore, the COX-2 variant emerged as promising drug target. Since the active site of COX-2 is approximately 25% larger than that of COX-1, a size-extension of classical NSAIDs successfully yielded inhibitors with COX-2 selectivity. Indomethacin (45, Fig. 13) offers two possible positions, the chlorophenyl ring or the carboxylic acid moiety, in the periphery of the indole core for the introduction of bulky substituents, in order to induce the desired selectivity.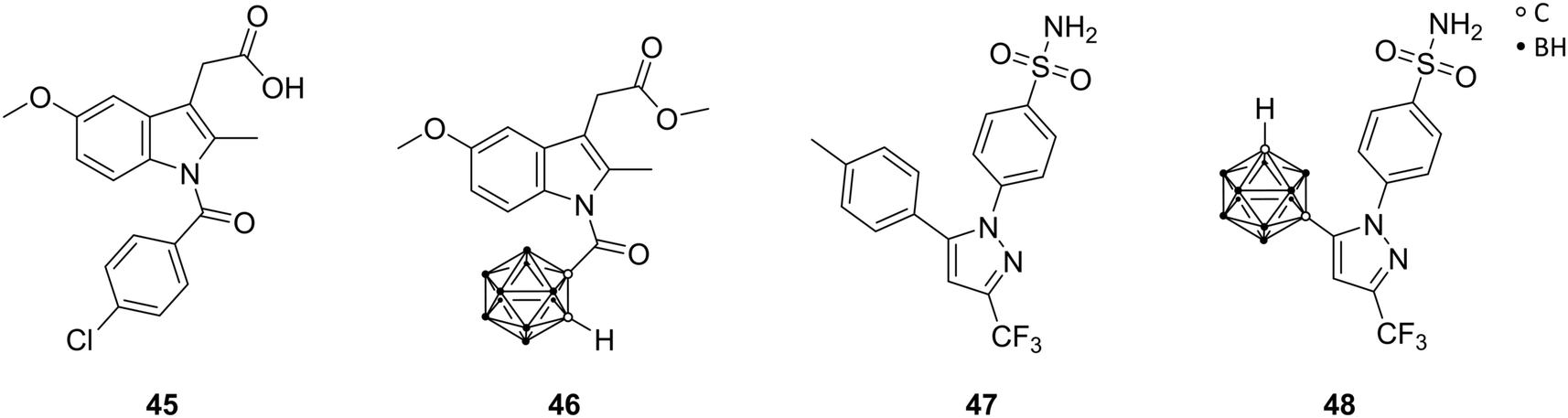 | ||
| Fig. 13 Structures of indomethacin 45, the methyl ester derivative 46 and celecoxib 47 and its carborane analogue 48. | ||
Scholz et al. described carborane-based cyclooxygenase inhibitors derived from indomethacin 45, via replacement of the chlorophenyl moiety by a carborane cage. Previous studies had shown that modifications of the chlorophenyl ring are synthetically more challenging but revealed also very potent inhibitors. The presence of two additional chlorine substituents in meta position to the chlorine atom of 45 in para position created a highly selective COX-2 inhibitor. The chlorine residues increased both size and electron deficiency of the benzoyl group. These two effects are probably responsible for the introduction of COX-2 selectivity and, thus, suitable for a replacement by a carborane moiety. The ortho-carborane indomethacin methyl ester derivative 46 (Fig. 13) showed high selectivity towards the cancer-specific isoform COX-2 in radioactive assays (conversion of [14C]-arachidonic acid) with IC50 values in the mid nanomolar range (84 nM). The meta and para isomers of 46 exhibited practically no inhibitory activity.51 Following studies by Neumann et al. revealed that the electron-withdrawing effect of the carbonyl group directly attached to the carborane promotes rapid deboronation of 46 in aqueous solution. Tailored synthesis and evaluation of the deboronated nido species of 46 revealed increased COX-2 inhibition in vitro (IC50[closo-46] = 72 nM; IC50[nido-46] = 51 nM) and higher water solubility.24 Docking into the COX-2 structure (PDB code 3LN1) using boron parameters predicted a binding pose fairly similar to the subsequently determined crystal structure (PDB code 4Z0L), placing the carborane cluster in the vicinity of Leu531. Leu531 rotates during crystallisation and opens up a hydrophobic sub-pocket to accommodate the nido-carborate. Furthermore, the COX-2 membrane-binding domain shifted considerably. This large shift in the protein structure could not be accounted for in the utilised docking protocol, giving similar predictions for both enantiomers of the deboronated nido species of 46.24 However, docking into the 4Z0L structure predicted a higher binding probability for the S enantiomer of the nido species of 46, probably due to the planar chirality of the cluster.
Recent studies by Buzharevski et al. presented celecoxib analogues, bearing carboranes as pharmacophores. Celecoxib (47, Fig. 13) belongs to the family of COX-2-selective inhibitors (COXIBs), which were developed to address the side effects (bleeding, ulcers in the gastrointestinal tract) of the non-selective NSAIDs, due to inhibition of constitutive COX-1. The carborane derivatives reported by Buzharevski et al. showed promising cytostatic activities against various melanoma and colorectal adenocarcinoma cell lines. Inhibited cell proliferation accompanied by caspase-independent apoptotic cell death was found to be the main cause of decreased cell viability upon treatment with the most efficient celecoxib analogue (48, Fig. 13).52
2.12 Lipoxygenase ligands
5-Lipoxygenase (5-LO) catalyses the conversion of arachidonic acid into leukotrienes. These fatty acid metabolites are involved in normal host defence, inflammatory responses, and cellular signalling, including apoptosis, but they are also associated with the development of numerous pathologies, e.g. asthma, several types of cancer, atherosclerosis, and cardiovascular disorders. The activity of 5-LO is regulated by the 5-lipoxygenase activating protein (FLAP). An early inhibitor of FLAP-mediated activation of 5-LO is Rev-5901 49 (Fig. 14), a rapidly metabolised drug. To overcome metabolic instability, Kuhnert et al. used carboranes as sterically demanding bioisosteres for replacement of phenylene moieties, resulting in analogues CarbORev-5901 50 and CarbSRev-5901 51 (Fig. 14). The binding of 50 and 51 to FLAP was investigated by means of docking simulations.15 The inhibitory activity of 5-LO was decreased relative to Rev-5901, but cytotoxicity toward the melanoma A375 cell line was strongly increased. A375 cells are nitric oxide (NO)-producing cells: 50 and 51 inhibited the generation of NO and reactive oxygen and nitrogen species (ROS/RNS); Rev-5901 acted oppositely, indicating that the structural modification of Rev-5901 with meta-carborane leads to a different mechanism of action. Thus, presented 5-LO ligands are considered as promising candidates for the treatment of NO-dependent tumours, known to be highly metastatic.15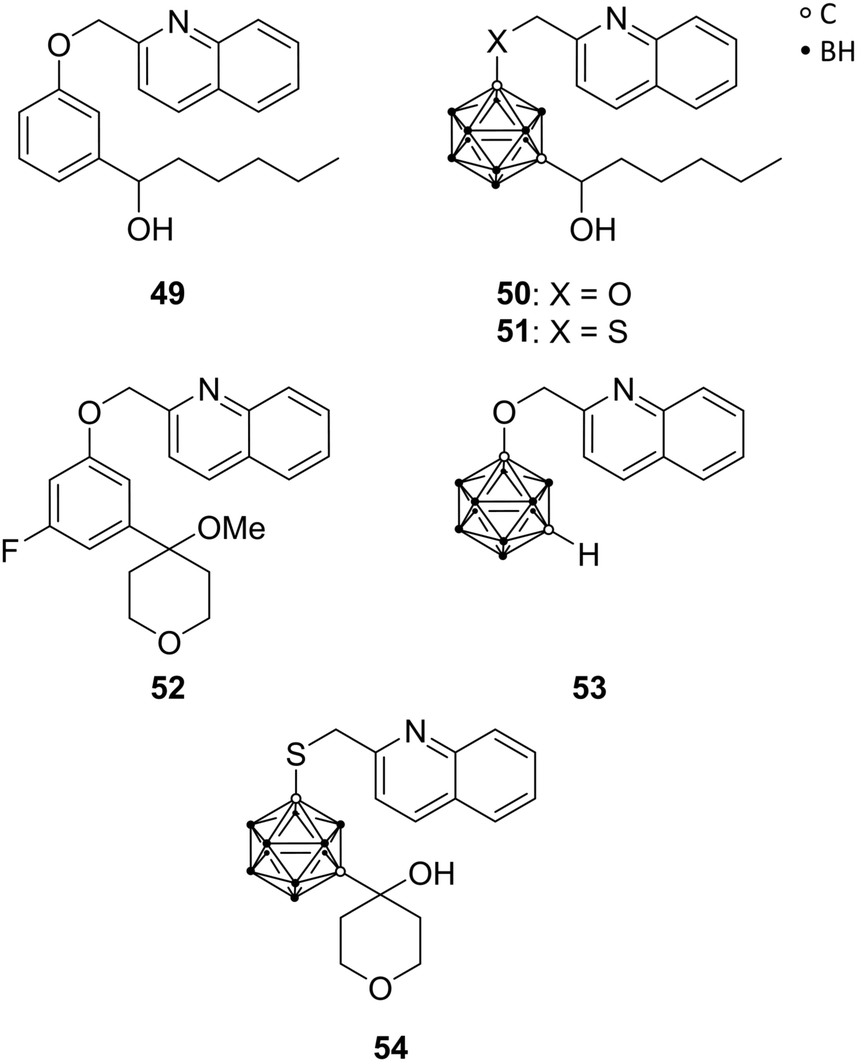 | ||
| Fig. 14 Structures of 5-lipoxygenase inhibitors Rev-5901 49, CarbORev-5901 50, CarbSRev-5901 51, ZD-2138 52, meta-carborane analogue of 2-(phenoxymethyl)-quinoline 53 and the 4-hydroxytetrahydropyranyl carborane derivative 54. | ||
Kuhnert et al. were inspired by another inhibitor of the 5-LO pathway, ZD-2138 52 (Fig. 14).23 The hydroxyalkyl substituent of 52 was introduced into further carborane derivatives (Fig. 14) in order to improve aqueous solubility as well as its inhibitory and cytotoxic potential through increased receptor binding affinity. Docking studies of the envisioned derivatives into the FLAP–MK591 crystal structure, co-crystallised complex of FLAP and the 5-LO specific inhibitor MK591, identified the isosteric replacement of the phenylene moiety in ZD-2138 by meta-carborane functionalised with 2-methylquinoline as the most promising approach, similar to CarbORev-5901. IC50 values for 5-LO product formation in intact polymorphonuclear leukocytes (granulocytes, PMNL) were determined, and the monofunctionalised meta-carborane analogue of 2-(phenoxymethyl)quinoline 53 exhibited the highest inhibitory activity of all tested compounds toward the 5-LO pathway with a value of 2.30 μM (Rev-5901: IC50 = 0.58 μM). Screening for anti-tumour activity on various tumour cell lines, selected based on 5-LO expression and the well-known inflammatory background in melanoma (A375, B16 and its metastatic clone B16F10) and colon cancer pathogenesis (CT26CL25 and HCT116), surprisingly revealed that the most potent 5-LO inhibitor (53) was completely inactive (all tumour activity values >50 μM), indicating that 5-LO inhibitory activity could not be correlated with the anti-tumour activity. Compound 54 (Fig. 14) functionalised with a 4-hydroxytetrahydropyranyl moiety, revealed remarkably higher cytotoxic activity against melanoma and colon cancer cells, being more potent than 50 and 51, despite exhibiting lower 5-LO inhibitory activity. This effect could be partly explained by the co-inhibition of another cellular target of compound 54, namely, heat shock protein 90 (HSP90). Docking simulations were useful for identifying the off-target receptors of 54. The multichaperone protein HSP90 facilitates cell growth, metastasis and angiogenesis and has anti-apoptotic effects.23
2.13 ATP-Binding cassette transporter G2 ligands
Neoplastic cells often develop resistance against chemotherapeutic treatments, leading to insufficiency of anti-cancer therapies. Specific transmembrane transporter proteins are involved in the efflux of chemotherapeutic agents from the intracellular space and can cause multidrug resistances (MDRs). Several ATP-binding cassette transporters (ABCs) are known ATP-dependent efflux-pump proteins for xenobiotics, and were found to be overexpressed in various cancer cell lines. By upregulation of those ABCs, chemotherapeutics can be excreted from the cell, decreasing the concentration of the drug to a non-therapeutic level. Multidrug resistance is associated with three transport proteins, namely the permeability glycoprotein (Pgp, also referred to as multidrug resistance protein 1, MDR1, and ABCB1), the ABCC1, and the ABC transporter G2 (ABCG2, also known as the breast cancer resistance protein BCRP), which gained attention within the last years as targets for efficient and promising strategies of cancer chemotherapy. With the focus on selective inhibition of ABCG2-mediated excretion of xenobiotics, the efficacy of anti-cancer treatments can be enhanced.53Köhler et al. prepared ABCG2-selective inhibitors, with further regards to their selectivity against the MDR-involved MDR1. Based on a known heteroaryl-phenyl lead structure 55 (Fig. 15), a series of analogues was designed, including two meta-carborane derivatives, 56a and b (Fig. 15), as phenyl or adamantyl mimetics. Both carborane derivatives 56a and 56b exhibited enhanced activity compared to 55 and their adamantyl model towards ABCG2-overexpressed MDCK II BCRP cells (BCRP transfected canine kidney cell lines) using the Hoechst 33342 assay. The functionalisation with carboranes led to efficient inhibition of ABCG2 at low micromolar concentrations. Both derivatives, 56a/b, showed lower inhibitory potency (IC50 = 0.508 and 0.807 μM, respectively) than 55 (IC50 = 0.067 μM) for inhibition of ABCG2 activity and, interestingly, targeted MDR1 less significantly than their adamantyl analogue.54 However, for selective ABCG2 inhibition the phenyl-heteroaryl-phenylamide framework of 55 and 56 is essential, as SAR studies revealed.
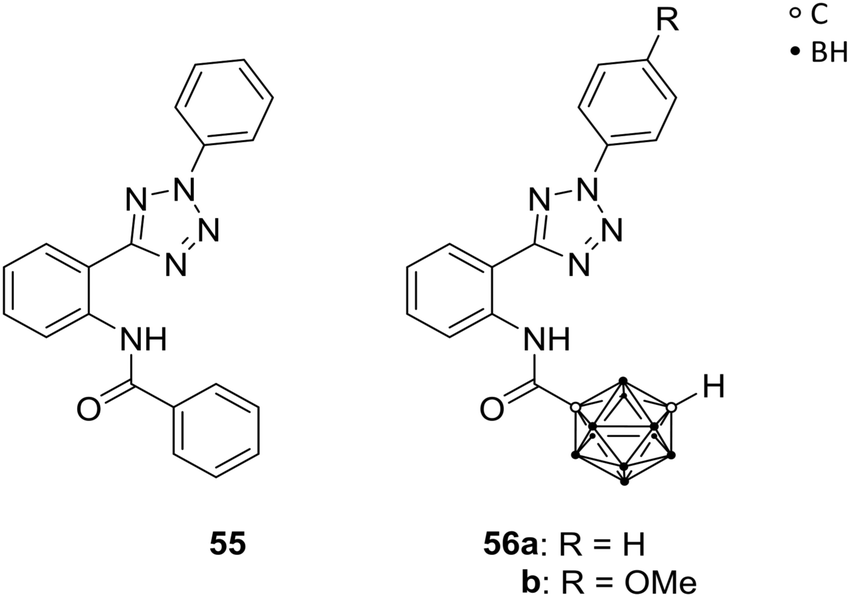 | ||
| Fig. 15 ABCG2 inhibitor 55 and the carborane analogues 56. | ||
2.14 Indoleamine-2,3-dioxygenase-1 ligands
Austin et al. selected the indoleamine-2,3-dioxygenase-1 (IDO1) for ligand–enzyme interaction studies, which contains two hydrophobic binding pockets and, thus, the authors designed highly hydrophobic carborane-based pharmacophores.55 IDO1 is a haem-containing enzyme that catalyses the first and rate-limiting step in the major tryptophan metabolic pathway during inflammation and disease, the kynurenine pathway. Involved in immune response modulation, IDO1 shows the ability to limit the functions of T cells and can engage immune tolerance mechanisms. Thus, it is believed that IDO1 is activated in tumour cell progression for downregulating the immune system-response against malignant cells.56 Thus, IDO1 inhibitors possess the potential as immunoregulatory chemotherapeutics, restoring regular physiological immune reactions. Moreover, IDO1 can be downregulated by COX-2 inhibitors, as IDO1 expression in tumour cells is driven by COX-2 expression.57Determination of the crystal structure of IDO1 resulted in better insight into the enzyme structure and, thus, of the active site, which features a small hydrophobic cavity near the active site's entrance and a larger hydrophobic, haem catalytic site-containing pocket. The pyranonaphthoquinone derivatives dehydro-α-lapachone (57, Fig. 16) and the diastereomeric cis-hydroxy-benzylamino analogues 58 (Fig. 16) were evaluated recently as potent inhibitors for IDO1. Austin et al. replaced the phenylene moiety of 58 with ortho-carborane. The carborane derivatives 59 and 60 (Fig. 16) displayed inhibitory activity towards human recombinant enzyme in the micromolar range, with IC50 values of 0.78 and 1.77 μM, respectively, and are thus half as potent as the racemic parental analogues 58 (IC50 = 0.35–0.50 μM).55
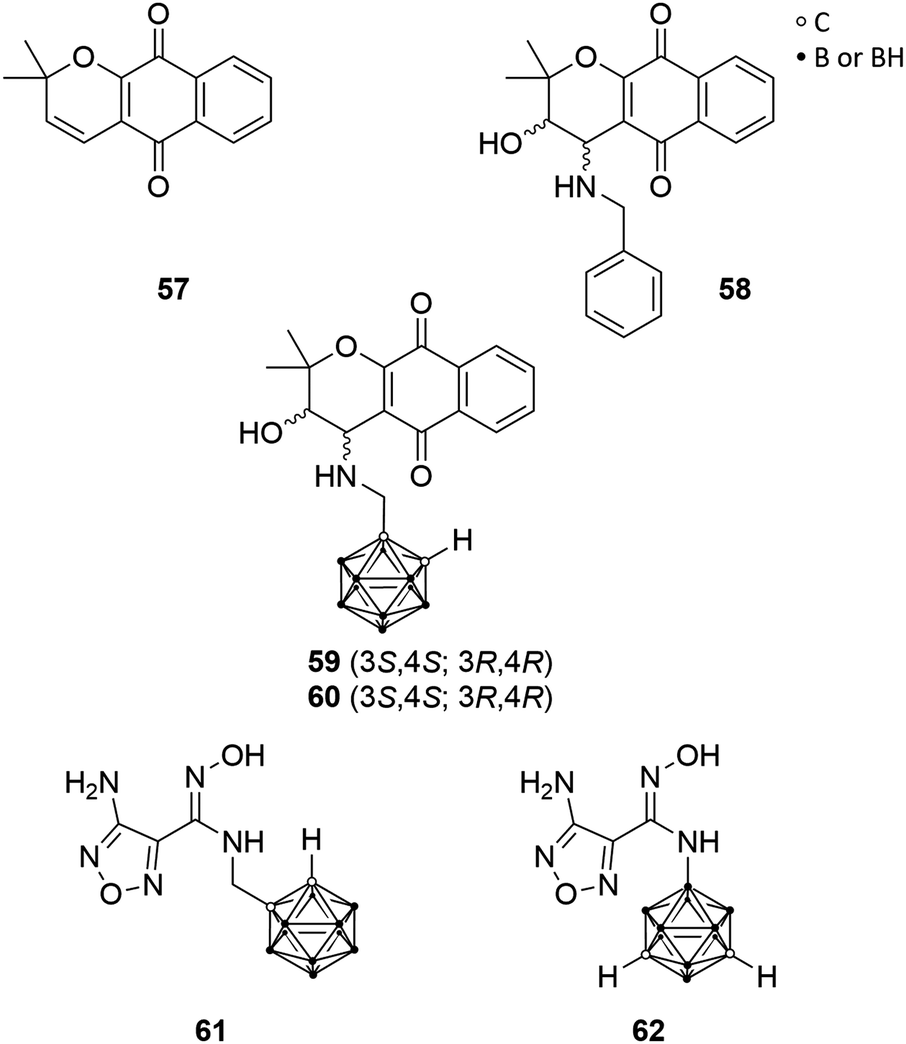 | ||
| Fig. 16 Indoleamine-2,3-dioxygenase-1 ligands; structures of dehydro-α-lapachone 57, the benzylamino derivative 58, the carborane derivatives 59/60 and carborane-based hydroxyamidine derivatives 61 and 62. | ||
Furthermore, Austin et al. designed two novel carborane-based ligands on the basis of smaller sized hydroxyamidines, which presented high inhibitory potency toward IDO1. Hydroxyamidine analogue 61 (Fig. 16), bearing a methylene spacer between the cluster and the hydroxyamidine group, showed inhibition activity in the low micromolar level (IC50 = 1.90 μM), whereas 62 (Fig. 16), the boron-substituted meta-carborane derivative, was inactive. This may be attributed to a different binding mode of 62 due to minor conformational freedom and enhanced steric hindrance, due to the missing methylene linker, as revealed by docking simulations. Nevertheless, these novel carborane-based IDO1 ligands may be feasible as prospective IDO1 inhibitors and lead structure.21
2.15 N-Alkyl-deoxynojirimycins as glycoprocessing enzyme ligands
Deoxynojirimycin analogues (DNMs), in particular N-alkylated deoxynojirimycin derivatives, represent a major class of glycoprocessing enzyme inhibitors. A variety of glycosidase and glycosyltransferase inhibitory agents are approved drugs for different applications, e.g. for the treatment of lysosomal storage disorder through glucosylceramide synthase inhibition (N-butyl-DNM) or anti-diabetics (hydroxyethyl-DNM) as intestinal glycosidase inhibitor.58Hoogendoorn et al. prepared ortho-carboranyl N-alkyl-deoxynojirimycins that are suitable for the simultaneous inhibition of several glycoprocessing enzymes: glucosylceramide synthase (GCS), lysosomal glucosyl-ceramidase (GBA) and neutral glucosylceramidase (GBA2). Based on previous results, the authors evaluated N-substituted DNMs with different hydrophobic moieties, including an adamantyl group. The adamantane-based N-alkylated DNM (AMP-DNM 63, Fig. 17) showed higher potency as GCS inhibitor than the clinically used N-butyl-DNM, with also good activities toward GBA, GBA2 and intestinal glycosidase. The C5-epimer of 63, L-AMP-DNM 64 (Fig. 17), differs in its action compared to AMP-DNM, enhancing the inhibition of GCS, but lowering the GBA inhibitory activity. Thus, a hydrophobic and sterically demanding carborane cluster was introduced in both epimeric forms to improve the inhibitory potency. A series of carboranyl derivatives of D-DNM (65a–c) and L-DNM (66a–c) were designed (Fig. 17), varying the length of the alkyl spacer. Despite the fact that no improvement in the inhibitory activity on GCS, GBA and GBA2 was accomplished, the most efficient compound 66c inhibits all three enzymes in low micromolar ranges, below 0.5 μM, similar to 63 and 64.59
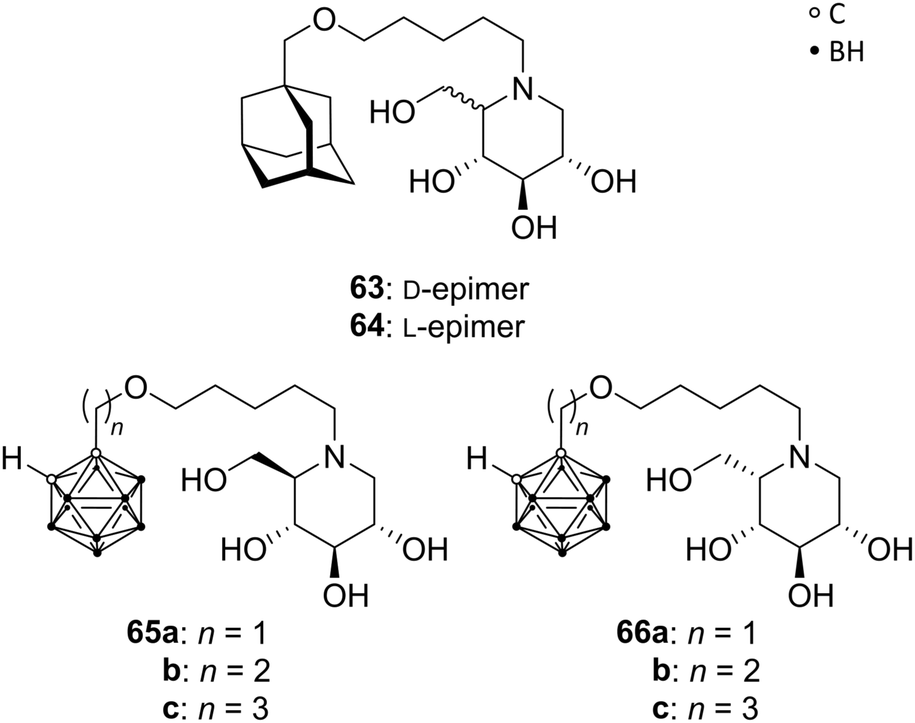 | ||
| Fig. 17 Structures of AMP-DNMs 63 and 64 and carborane-based analogues 65 and 66. | ||
3. Conclusions
With respect to medicinal organic chemistry, carboranes in medicine are still an emerging class of compounds. Researchers have just begun to scratch the surface of the potential beneficial applications of these highly hydrophobic clusters in drug design. Mechanisms of action in the cells, as well as the cause–effect relationship of the biological activities with respect to the specific biological targets, is still largely underexplored. Often, introduction of a closo-carborane cluster into the skeleton of a purely organic agonist, antagonist or inhibitor, completely changes the activity profile of the drug, and also modulates its effects on cell viability in several types of tumours. Therefore, icosahedral carboranes deserve the classification as “new keys for old locks” and open up an exciting field of research for well-known, but challenging, important therapeutic substrates, as demonstrated by the various examples discussed in this review. For the efficient treatment of several pathologies (neuronal or immune disorders, cancer, pain, inflammation, etc.), there is in fact urgent need for novel chemotherapeutics to overcome the problems associated with the traditional organic-based drugs. In this scenario, carboranes are definitely very promising, robust and chemically stable keys.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M.-B. S. and E. H.-H. are grateful for the financial support provided by the German Research Foundation (DFG, SA 2902/2-1 and HE 1376/38-1).Notes and references
- P. Imming, The Practice of Medicinal Chemistry, Elsevier, 2015, pp. 3–13 Search PubMed.
- C. O. Wilson, J. M. Beale and J. H. Block, Wilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical Chemistry, Lippincott Williams & Wilkins, Baltimore MD, 12th edn, 2011 Search PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- R. F. Barth, P. Mi and W. Yang, Cancer Commun., 2018, 38, 35 CrossRef PubMed.
- Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758 CrossRef PubMed.
- M. Couto, M. F. García, C. Alamón, M. Cabrera, P. Cabral, A. Merlino, F. Teixidor, H. Cerecetto and C. Viñas, Chem. – Eur. J., 2018, 24, 3122–3126 CrossRef CAS PubMed , and references therein.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed , and references therein.
- R. N. Grimes, Carboranes, Academic Press, Amsterdam, 2016, and references therein Search PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- W. H. Eberhardt, B. Crawford and W. N. Lipscomb, J. Chem. Phys., 1954, 22, 989–1001 CrossRef CAS.
- J. Brynda, P. Mader, V. Šícha, M. Fábry, K. Poncová, M. Bakardiev, B. Grüner, P. Cígler and P. Řezáčová, Angew. Chem., Int. Ed., 2013, 52, 13760–13763 CrossRef CAS PubMed.
- H. Nakamura, L. Tazaki, D. Kanoh and S. Sato, Pure Appl. Chem., 2015, 87, 143–154 CAS.
- M. W. Lee, Y. V. Sevryugina, A. Khan and S. Q. Ye, J. Med. Chem., 2012, 55, 7290–7294 CrossRef CAS PubMed.
- R. Kuhnert, M.-B. Sárosi, S. George, P. Lönnecke, B. Hofmann, D. Steinhilber, B. Murganić, S. Mijatović, D. Maksimović-Ivanić and E. Hey-Hawkins, ChemMedChem, 2017, 12, 1081–1086 CrossRef CAS PubMed.
- F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed , and references therein.
- R. Fernandez-Alvarez, V. Ďorďovič, M. Uchman and P. Matějíček, Langmuir, 2018, 34, 3541–3554 CrossRef CAS PubMed , and references therein.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225–2238 CrossRef CAS PubMed.
- M. Tarrés, E. Canetta, E. Paul, J. Forbes, K. Azzouni, C. Viñas, F. Teixidor and A. J. Harwood, Sci. Rep., 2015, 5, 7804–7811 CrossRef PubMed.
- T. He, J. C. Misuraca and R. A. Musah, Sci. Rep., 2017, 7, 16995–17005 CrossRef PubMed.
- C. J. D. Austin, M. Moir, J. Kahlert, J. R. Smith, J. F. Jamie, M. Kassiou and L. M. Rendina, Aust. J. Chem., 2015, 68, 1866–1870 CrossRef CAS.
- A. Kaise, K. Ohta, S. Fujii, A. Oda, T. Goto and Y. Endo, Bioorg. Med. Chem., 2018, 26, 3805–3811 CrossRef CAS PubMed.
- R. Kuhnert, M.-B. Sárosi, S. George, P. Lönnecke, B. Hofmann, D. Steinhilber, S. Steinmann, R. Schneider-Stock, B. Murganic, S. Mijatovic, D. Maksimovic-Ivanic and E. Hey-Hawkins, ChemMedChem, 2018, 14, 255–261 CrossRef PubMed.
- W. Neumann, S. Xu, M. B. Sárosi, M. S. Scholz, B. C. Crews, K. Ghebreselasie, S. Banerjee, L. J. Marnett and E. Hey-Hawkins, ChemMedChem, 2016, 11, 175–178 CrossRef CAS PubMed , and references therein.
- R. Otero, S. Seoane, R. Sigüeiro, A. Y. Belorusova, M. A. Maestro, R. Pérez-Fernández, N. Rochel and A. Mouriño, Chem. Sci., 2016, 7, 1033–1037 RSC.
- B. Schwarze, M. Gozzi and E. Hey-Hawkins, in Boron-Based Compounds, ed. E. Hey-Hawkins and C. Viñas Teixidor, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 60–108 and references therein Search PubMed.
- N. Vázquez, V. Gómez-Vallejo and J. Llop, Tetrahedron Lett., 2012, 53, 4743–4746 CrossRef.
- Y. Endo, in Boron-Based Compounds, ed. E. Hey-Hawkins and C. Viñas Teixidor, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 1–19 and references therein Search PubMed.
- S. Fujii, Med. Chem. Commun., 2016, 7, 1082–1092 RSC , and references therein.
- K. Ohta, T. Ogawa, A. Kaise, A. Oda and Y. Endo, Chem. Pharm. Bull., 2014, 62, 386–391 CrossRef CAS PubMed.
- K. Ohta, T. Ogawa and Y. Endo, Bioorg. Med. Chem. Lett., 2017, 27, 4030–4033 CrossRef CAS PubMed.
- A. Kaise, K. Ohta and Y. Endo, Eur. J. Med. Chem., 2016, 122, 257–263 CrossRef CAS PubMed.
- K. Bednarska, A. B. Olejniczak, A. Piskala, M. Klink, Z. Sulowska and Z. J. Lesnikowski, Bioorg. Med. Chem., 2012, 20, 6621–6629 CrossRef CAS PubMed.
- A. Adamska-Bartłomiejczyk, K. Bednarska, M. Białek-Pietras, Z. M. Kiliańska, A. Mieczkowski, A. B. Olejniczak, E. Paradowska, M. Studzińska, Z. Sułowska, J. D. Żołnierczyk and Z. J. Lesnikowski, in Boron-Based Compounds, ed. E. Hey-Hawkins and C. Viñas Teixidor, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 20–34 Search PubMed.
- M. Vincenzi, K. Bednarska and Z. J. Leśnikowski, Molecules, 2018, 23, 1846–1866 CrossRef PubMed.
- C. Volonté, S. Apolloni, S. D. Skaper and G. Burnstock, CNS Neurol. Disord.: Drug Targets, 2012, 11, 705–721 CrossRef.
- A. Baxter, J. Bent, K. Bowers, M. Braddock, S. Brough, M. Fagura, M. Lawson, T. McInally, M. Mortimore, M. Robertson, R. Weaver and P. Webborn, Bioorg. Med. Chem. Lett., 2003, 13, 4047–4050 CrossRef CAS PubMed.
- S. M. Wilkinson, H. Gunosewoyo, M. L. Barron, A. Boucher, M. McDonnell, P. Turner, D. E. Morrison, M. R. Bennett, I. S. McGregor, L. M. Rendina and M. Kassiou, ACS Chem. Neurosci., 2014, 5, 335–339 CrossRef CAS PubMed , and references therein.
- F. Di Virgilio, D. Dal Ben, A. C. Sarti, A. L. Giuliani and S. Falzoni, Immunity, 2017, 47, 15–31 CrossRef CAS PubMed.
- G. Li, H. S. Ban and H. Nakamura, in Boron-Based Compounds, ed. E. Hey-Hawkins and C. Viñas Teixidor, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 35–59 and references therein Search PubMed.
- M. Couto, I. Mastandrea, M. Cabrera, P. Cabral, F. Teixidor, H. Cerecetto and C. Viñas, Chemistry, 2017, 23, 9233–9238 CrossRef CAS PubMed.
- M. M. Alegre, R. A. Robison and K. L. O'Neill, J. Oncol., 2012, 2012, 1–5 CrossRef PubMed.
- H. K. Agarwal, C. A. McElroy, E. Sjuvarsson, S. Eriksson, M. V. Darby and W. Tjarks, Eur. J. Med. Chem., 2013, 60, 456–468 CrossRef CAS PubMed.
- Y. Byun, J. Yan, A. S. Al-Madhoun, J. Johnsamuel, W. Yang, R. F. Barth, S. Eriksson and W. Tjarks, J. Med. Chem., 2005, 48, 1188–1198 CrossRef CAS PubMed.
- H. K. Agarwal, A. Khalil, K. Ishita, W. Yang, R. J. Nakkula, L.-C. Wu, T. Ali, R. Tiwari, Y. Byun, R. F. Barth and W. Tjarks, Eur. J. Med. Chem., 2015, 100, 197–209 CrossRef CAS PubMed.
- M. Hasmann and I. Schemainda, Cancer Res., 2003, 63, 7436–7442 CAS.
- P. Huang, M. W. Lee, K. Sadrerafi, D. P. Heruth, L. Q. Zhang, D. Maulik and S. Q. Ye, Drug Des., Dev. Ther., 2017, 11, 629–641 CrossRef CAS PubMed.
- P. C. McDonald and S. Dedhar, Subcell. Biochem., 2014, 75, 255–269 CAS.
- P. Sácha, J. Zámecník, C. Barinka, K. Hlouchová, A. Vícha, P. Mlcochová, I. Hilgert, T. Eckschlager and J. Konvalinka, Neuroscience, 2007, 144, 1361–1372 CrossRef PubMed.
- S. Youn, K. Im Kim, J. Ptacek, K. Ok, Z. Novakova, Y. Kim, J. Koo, C. Barinka and Y. Byun, Bioorg. Med. Chem. Lett., 2015, 25, 5232–5236 CrossRef CAS PubMed.
- M. Scholz, A. L. Blobaum, L. J. Marnett and E. Hey-Hawkins, Bioorg. Med. Chem., 2012, 20, 4830–4837 CrossRef CAS PubMed.
- A. Buzharevski, S. Paskas, M.-B. Sárosi, M. Laube, P. Lönnecke, W. Neumann, S. Mijatovic, D. Maksimovic-Ivanic, J. Pietzsch and E. Hey-Hawkins, ChemMedChem, 2019, 14, 315–321 CrossRef CAS PubMed.
- S. C. Köhler, K. Silbermann and M. Wiese, Eur. J. Med. Chem., 2016, 124, 881–895 CrossRef PubMed.
- S. C. Köhler, S. Vahdati, M. S. Scholz and M. Wiese, Eur. J. Med. Chem., 2018, 146, 483–500 CrossRef PubMed.
- C. J. D. Austin, J. Kahlert, F. Issa, J. H. Reed, J. R. Smith, J. A. Ioppolo, J. A. Ong, J. F. Jamie, D. Hibbs and L. M. Rendina, Dalton Trans., 2014, 43, 10719–10724 RSC.
- D. H. Munn and A. L. Mellor, Trends Immunol., 2016, 37, 193–207 CrossRef CAS PubMed.
- M. Hennequart, L. Pilotte, S. Cane, D. Hoffmann, V. Stroobant, E. D. Plaen and B. van den Eynde, Cancer Immunol. Res., 2017, 5, 695–709 CrossRef CAS PubMed.
- T. Wennekes, R. J. B. H. N. van den Berg, W. Donker, G. A. van der Marel, A. Strijland, J. M. F. G. Aerts and H. S. Overkleeft, J. Org. Chem., 2007, 72, 1088–1097 CrossRef CAS PubMed.
- S. Hoogendoorn, E. D. Mock, A. Strijland, W. E. Donker-Koopman, H. van den Elst, R. J. B. H. N. van den Berg, J. M. F. G. Aerts, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2015, 4437–4446 CrossRef CAS.
Footnote |
| † For nomenclature and numbering of the carborane clusters refer to the IUPAC project 2012-045-1-800 by Beckett et al., nomenclature for boranes and related species, 2018. |
| This journal is © The Royal Society of Chemistry 2019 |