
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoronic acids as building blocks for the construction of therapeutically useful bioconjugates
João P. M.
António
 a,
Roberto
Russo
ab,
Cátia Parente
Carvalho
a,
Pedro M. S. D.
Cal
b and
Pedro M. P.
Gois
*a
a,
Roberto
Russo
ab,
Cátia Parente
Carvalho
a,
Pedro M. S. D.
Cal
b and
Pedro M. P.
Gois
*a
aResearch Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal. E-mail: pedrogois@ff.ulisboa.pt
bInstituto de Medicina Molecular, Faculty of Medicine, Universidade de Lisboa, Lisbon, Portugal
First published on 3rd June 2019
Abstract
Bioconjugates are multifunctional constructs in which biomolecules like peptides, proteins, vitamins and nucleic acids are endowed with the properties of specific payloads. These constructs recently emerged as a new generation of high-precision therapeutics, with several representatives reaching the market. This success stimulated an intense search for new biocompatible synthetic methodologies to connect both components and to control the bioconjugate's function. Despite the remarkable advances made in this field, most of the technologies developed for the construction of bioconjugates were engineered to yield stable constructs that can endure complex physiological conditions. Because of this, the use of reversible covalent bonds in the synthesis of bioconjugates has been rather overlooked, notwithstanding the potential of this strategy to generate stimuli responsive constructs that may operate in areas like the selective delivery of drugs, live-cell imaging and new theranostic approaches. Boronic acids are a well-known class of reagents that have been widely used in modern synthesis for the formation of C–C and C–heteroatom bonds. Apart from this, boronic acids exhibit an exquisite reversible coordination profile that can be explored as a molecular construction tool featuring specific mechanisms to control the structure and biological properties of bioconjugates. In this review, the use of boronic acids in the construction of therapeutically useful bioconjugates will be discussed, focusing on the molecular mechanisms that allow the use of these reagents as bioconjugation warheads, as central pieces of linker structures and as functional payloads.
 João P. M. António | João P. M. António is currently a PhD student at iMed.ULisboa in the Faculty of Pharmacy of the University of Lisbon under the supervision of Prof. Pedro Gois and co-supervision of Dr Gonçalo Bernardes (University of Cambridge, United Kingdom). He received his MSc degree in Pharmaceutical Sciences from the Faculty of Pharmacy of the University of Lisbon in 2014 under the supervision of Prof. Pedro Gois and co-supervision of Dr Philippe Hubert (University of Liège, Belgium). His research interests focus on the development of new strategies for protein modification, construction of stimuli-responsive linkers for bioconjugation and discovery of new scaffolds with potential application in medicinal chemistry. |
 Roberto Russo | Roberto Russo is a Marie Skłodowska-Curie fellow currently working on his PhD in the group of Prof. Pedro Gois at the Faculty of Pharmacy of the University of Lisboa. He received his MSc degree from the University of Milano in 2015 and his BSc degree from the University of Bologna in 2013, both in Chemistry. His research, in the framework of the ITN ProteinConjugates, focuses on the development of new stimuli-responsive linker strategies for protein functionalization based on boronic acids, encompassing organic synthesis, biological evaluation and mechanistic studies. |
 Cátia Parente Carvalho | Cátia P. Carvalho graduated from the University of Algarve, Portugal, with a BS degree in Biochemistry. She then moved to Spain to complete a second MSc and PhD in the field of photo-responsive supramolecular chemistry at the University of Huelva. In 2016, after finishing her PhD, she moved to Prof. Pedro Gois group to develop innovative modular boronate-based fluorescent probes for biological applications, at iMed.ULisboa in the University of Lisbon. She is currently working as a Postdoctoral Specialist at the National Centre for Biomolecular Research of Masaryk University in the Czech Republic. |
 Pedro M. S. D. Cal | Pedro M. S. D. Cal received his integrated MSc degree in Pharmaceutical Sciences (2010) and his PhD degree in Pharmacy (2014) from the University of Lisbon, PT, under the supervision of Prof. Pedro Gois. During his PhD he also worked in Professor Stephen Caddick's lab at the University College of London, UK. In 2015, he joined the group of Dr Gonçalo J. L. Bernades as a postdoctoral researcher, working from either the Instituto de Medicina Molecular, PT, or the University of Cambridge, UK. In 2018 he was awarded a Junior Research Position at the Instituto de Medicina Molecular, PT. His main research interests are chemical biology, antibody–drug conjugates, microenvironment-driven protein modification, design of antibiotics and machine learning tools. He is also part of a team of researchers that is on the path to spin-off some of the research developed in Dr Bernardes’ lab. |
 Pedro M. P. Gois | Pedro M. P. Gois studied chemistry at the New University of Lisbon from where he also received in 2005 his PhD degree in Organic Chemistry under the supervision of Prof. Carlos Afonso. From May 2005 to May 2008 he worked as a postdoctoral research fellow at the University of Sussex with Prof. F. Geoffrey N. Cloke FRS, at the University College of London with Prof. Stephen Caddick and at the Instituto Superior Técnico (Technical University of Lisbon) with Prof. Carlos Afonso. In May 2008, he joined the Faculty of Pharmacy of the University of Lisbon as an assistant research fellow of the medicinal chemistry group (iMed.ULisboa—Research Institute for Medicines), and in July 2013, Gois was appointed principal investigator in the same institution and head of the Bioorganic Group. In 2017 he received his habilitation in Pharmacy and was appointed assistant professor of the Faculty of Pharmacy. |
1. Introduction
Rational design of bioconjugates emerged in recent years as a leading strategy to construct innovative hybrid materials that may be used to devise new therapeutic options for unmet medical needs like cancer, inflammation and neurodegenerative diseases. The clinical success of bioconjugates is intimately related to the chemistry used to connect their functional components. For instance, the biomolecule's activity and pharmacokinetic properties should not be significantly altered by the attachment of the payload and the conjugate's stability should be maintained in circulation until it reaches the diseased site, where the payload must be activated preferentially as a response to a different chemical environment. With the objective of addressing the different aspects of bioconjugates’ construction, many efforts have been devoted towards discovering new conjugation strategies and innovative linker technologies to generate more homogeneous and stimuli-responsive conjugates. In recent years, boronic acids’ (BAs’) exquisite coordination profile, very favourable physicochemical properties under physiological conditions and reduced toxicity have brought BAs into the limelight of chemical biology. BAs' ability to form reversible covalent bonds enables the synthesis of bioconjugates with suitable properties for use in areas like bioconjugation, live-cell-imaging, theranostics and selective drug delivery.1–41.1. Boronic acids’ structure and coordination profile
Since their discovery, BAs have become one of the most prevalent classes of reagents in modern synthesis, enabling the effective construction of different types of C–C and C–heteroatom bonds. Apart from their use as reagents, BAs also exhibit unique properties that have recently been well appreciated in the areas of medicinal chemistry, chemical biology and materials science.BAs feature a vacant p-orbital centred on the boron atom which readily establishes reversible covalent bonds with oxygen and nitrogen nucleophiles. This Lewis acid–base reaction results in the central boron atom interconversion between an uncharged trigonal planar structure and an anionic tetravalent borate species, with a geometry and an isoelectronic structure that are equivalent to those of a neutral sp3-hybridized carbon. BAs also bind vicinal nucleophiles to generate cyclic boronate esters. In recent years, this reaction has become a powerful synthetic strategy to construct self-organizing systems, sensors and many different types of functional materials (Fig. 1).5–8
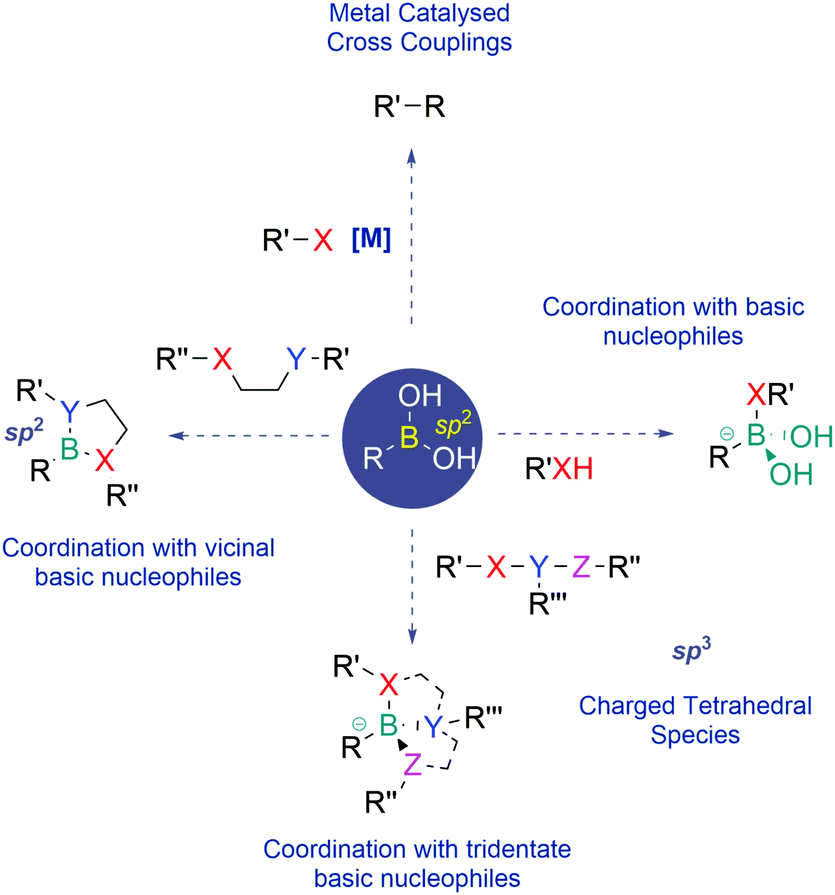 | ||
| Fig. 1 General reactivity profile of boronic acids. X, Y and Z = oxygen or nitrogen. | ||
Due to their mild Lewis acidity, BAs are typically uncharged under physiological conditions and retain their ability to reversibly coordinate oxygen-based nucleophiles, namely at the active site of proteases. The discovery that BAs may be used as transition-state analogue protein inhibitors triggered burgeoning interest among the medicinal chemistry community that has been using boronated compounds mostly due to their ability to emit alpha particles under neutron bombardment (boron neutron capture therapy).9–18 Currently, BAs are a preeminent class of functions in medicinal chemistry, with FDA-approved representatives like Bortezomib, a first-in-class proteasome inhibitor used in the clinic as an anticancer agent in patients with multiple myeloma, and Tavaborole, a leucyl-tRNA synthetase inhibitor commercialized for the treatment of onychomycosis.
1.2. Bioconjugate engineering
Bioconjugates are multifunctional constructs in which biomolecules like peptides, proteins, small vitamins and nucleic acids are endowed with the properties of specific payloads (e.g., biological activity and fluorescence).19–21The synthesis of bioconjugates by direct chemical installation of molecular payloads onto biomolecules is often impossible due to the lack of suitable chemical functions that may be used in the bioconjugation process.3,22–27 Therefore the construction of these hybrid materials depends on the use of chemical spacers to connect the payload and the biomolecule.28–31 In this review the use of BAs in the construction of bioconjugates will be summarized, with a particular focus on the molecular mechanisms underlying the role of the BA moiety in the construction and function of the bioconjugate, namely as a bioconjugation warhead, as a payload and as part of a bioconjugate linker (Fig. 2).
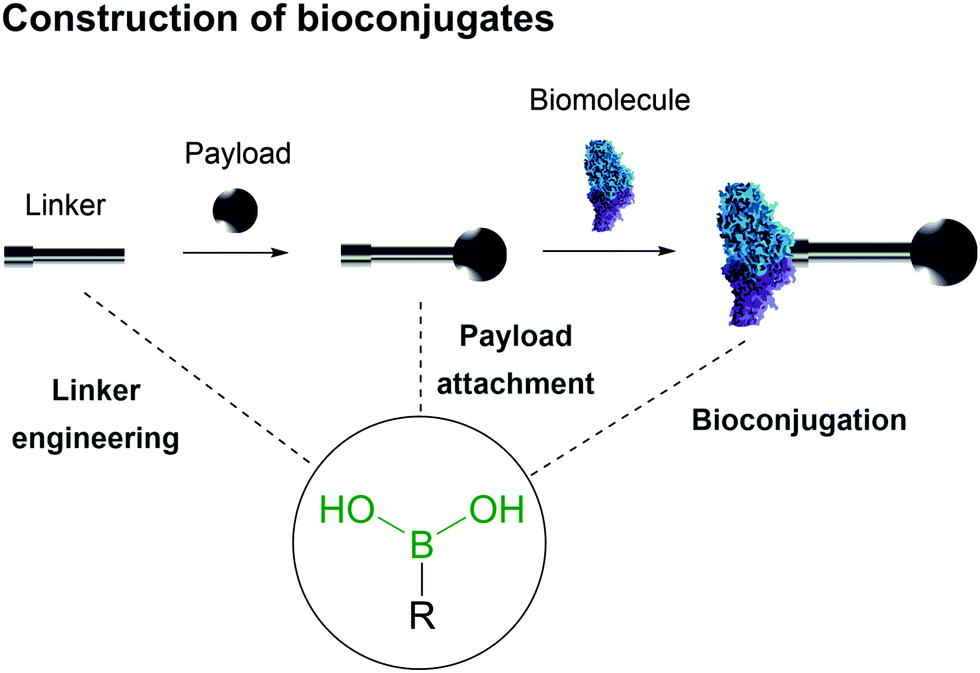 | ||
| Fig. 2 Boronic acid's intrinsic characteristics are useful for design of all three components of a bioconjugate. | ||
2. Boronic acids as payloads
A central component of bioconjugates is the payload that elicits the therapeutic properties desired for the construct. In this context, BAs are a very promising class of therapeutic agents, with several representatives reaching the market in areas such as oncology and antimicrobials. In addition to this, because the BA function can be synthetically introduced into the structures of organic molecules using methodologies like Miyaura borylation,32,33 the range of BA based payloads that can be used to generate bioconjugates is very diverse and includes not only drugs but also pro-drugs and fluorescent molecules.2.1. Boronic acids as drugs
Several boron-containing bioactive molecules have been developed over the past decades, with a particular focus on the design of protein inhibitors.34–39 As of the end of 2018, almost 300 protein structures have been crystallized with BA inhibitors and are available in the Protein Data Bank.40 From the analysis of these structures it emerges that the inhibitory activity of BAs is, among their various targets, mainly directed towards serine proteases. The most common inhibition mechanism is the formation of a tetracoordinated boronate complex by coordination with the hydroxyl side chain of an active serine residue, although other mechanisms of action have also been identified. Despite the promising results shown in vitro,34 only a few drugs containing the BA moiety have made it to the clinic, namely the antifungal agent Tavaborole (KERYDIN®),41 Crisaborole (Eucrisa®)42 for the topical treatment of eczema, the β-lactamase inhibitor Vaborbactam (Vabomere®) which is used in combination with meropenem to treat urinary tract infections and pyelonephritis,43 and the proteasome inhibitors Bortezomib (Velcade®)44 and Ixazomib (Ninlaro®)45 for the treatment of multiple myeloma (Fig. 3). One of the reasons behind the poor translation of positive in vitro results to the clinic lies in the reactivity of BAs in their open shell form, as they are able to interact with different naturally-occurring nucleophiles,46 resulting in poor pharmacokinetic profiles. The two FDA-approved BA-based proteasome inhibitors Bortezomib and Ixazomib offer an interesting case study for understanding the BA ligand's role in the pharmacokinetics of a drug. Both drugs target the β5-subunit of the 20S proteasome, exhibiting similar low-nanomolar IC50 values. Bortezomib, approved in 2003, is used in the clinic as a BA and requires subcutaneous administration.47 On the other hand, Ixazomib, approved in 2015, became the first orally-available proteasome inhibitor due to the derivatization of its BA moiety with citric acid, thus restraining its activity and improving the pharmacokinetic properties of the drug.48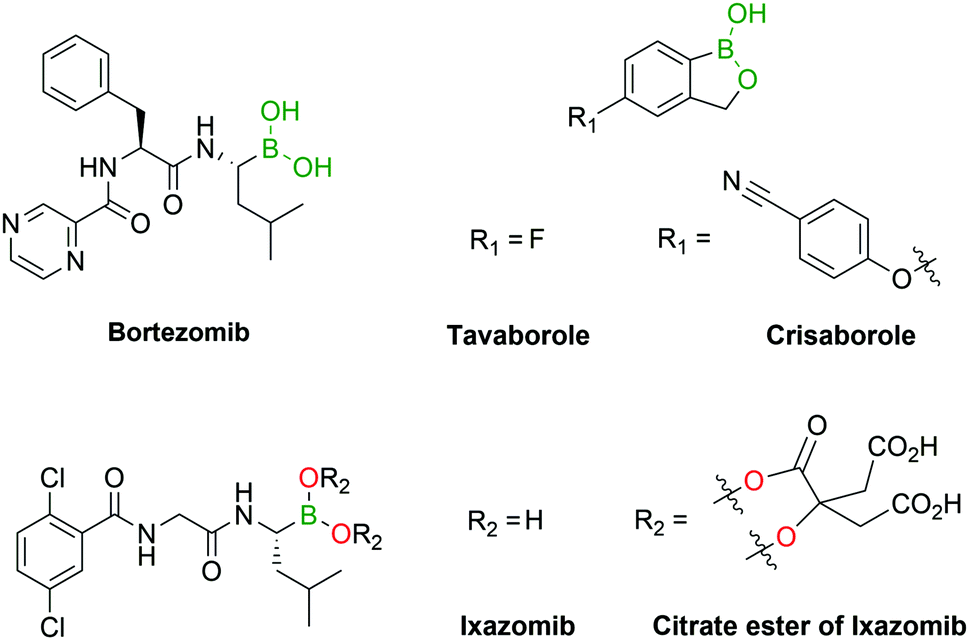 | ||
| Fig. 3 Examples of boronic acids in pharmacologically relevant compounds. | ||
Another alternative to solve BAs’ pharmacokinetic issues is their incorporation into heterocycles. Recent reports have demonstrated that certain B–N heterocycles are able to improve BAs’ stability and selectivity while maintaining their comparable inhibitory activity towards their target.49 On the other hand, despite some reports of BAs’ ability to coordinate threonine50 and histidine,51–58 the development of BA-based inhibitors has recurrently focused on serine residues. Other nucleophilic residues such as lysine, histidine, arginine, tyrosine, threonine and cysteine have been fairly overlooked thus far and could represent interesting targets for the discovery of new inhibitors.
2.2. Boronic acids as prodrugs
Pharmaceutical agents are not the only valuable BA-based payloads, as the BA moiety has also been widely explored for the development of prodrugs and self-immolative groups. In these applications, the use of BAs is attractive because aromatic BAs such as phenylboronic acid (PBA) are readily converted to the corresponding phenols by reaction with H2O2 and other radical oxygen species (ROS). The concentration of ROS in tumour tissues is abnormally high (10 to 40 fold increase vs. healthy cells), thus representing a valuable biological trigger for payload release and activation.59 Exploiting this behaviour, various research groups have reported the construction of ROS-triggered prodrugs by replacing a phenol moiety in the original structure of a drug with a BA. For instance, Lu and co-workers60 reported the synthesis of a prodrug of SN-38, the active metabolite of the FDA-approved drug Irinotecan, through borylation of its phenol moiety (Fig. 4A). This molecule can be converted to the active drug with H2O2 concentrations as low as 0.5 mM and shows better in vivo results than Irinotecan itself in glioblastoma xenograft mouse models. High concentrations of ROS can also be found in inflamed tissues, which were addressed in 2015 by Kang and co-workers, through the preparation of a borylated precursor of p-hydroxybenzyl alcohol (HBA), an anti-inflammatory agent. The derivatization of HBA as a hydroxymethyl boronic ester led to a prodrug with a better circulation half-life than HBA itself and higher efficacy in the treatment of ischemia–reperfusion injury.61 The reactivity between BAs and ROS can also be used for imaging purposes, as shown by Wang and co-workers,62 who developed a BA derivative of coumarin. The borylation of coumarin turned off its fluorescent properties, which were restored in a concentration-dependent fashion upon incubation with H2O2 (Fig. 4B). Similar methodologies63–68 were developed by other groups using different fluorescent dyes.69–78 Following the same rationale, various self-immolative linkers triggered by H2O2 were reported. Cohen and co-workers published various examples of BA-based linkers in which the conversion of the BA into phenol triggers a self-immolative cascade (Fig. 4C).79 These mechanisms can be applied both as a prodrug strategy and as a delivery system in the construction of targeted drug conjugates. By combining some of the previous concepts, Kim and co-workers reported the synthesis of a hybrid construct between a coumarin and SN-38 for theranostic applications.80 When this compound is incubated with cancer cells, the H2O2-mediated oxidation of the borylated coumarin triggers an electron cascade that leads to the release of SN-38 and coumarin, providing both therapeutic effects and localized fluorescence. However, due to the large array of ROS present in living systems and the specific tasks each molecule is entrusted with, it is imperative to devise methods capable of discriminating between them. Until now, few BA-based methods have shown selectivity for a single species though in the future this approach could be a valuable tool to understand and visualize the molecular mechanisms behind ROS signalling and exploit them for targeted drug delivery.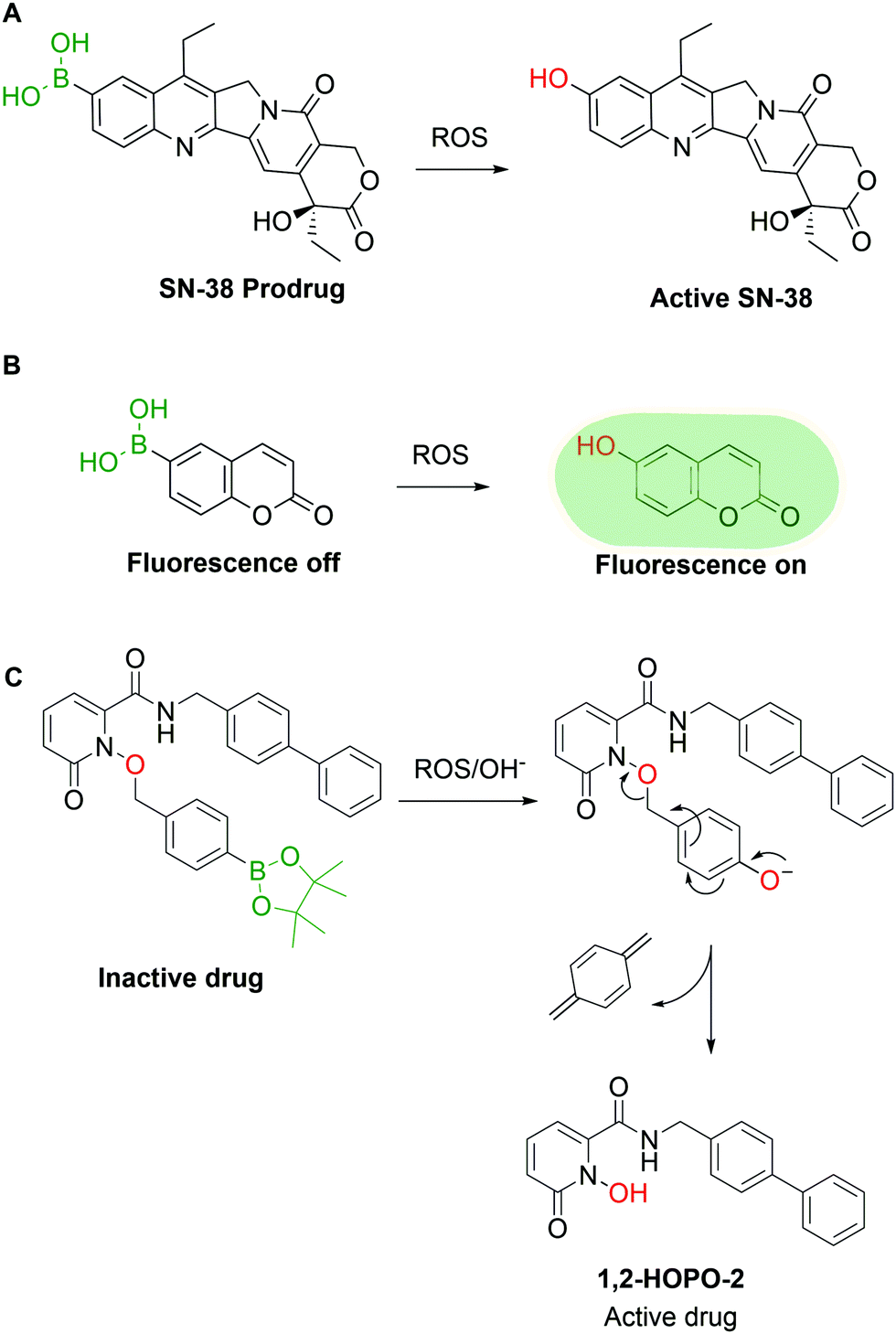 | ||
| Fig. 4 ROS-mediated activation of prodrugs/quenched probes; (A) SN-38 prodrug is transformed into SN-38 in the presence of ROS; (B) a quenched coumarin derivative is activated in the presence of ROS; and (C) ROS triggers a self-immolative cascade to release the active drug. | ||
3. Boronic acids as bioconjugation warheads
Bioconjugation emerged in recent years as an invaluable tool for performing chemical modification of complex molecules and for generating bioconjugates in which the biomolecules are endowed with the properties of the attached entities. Most of the chemical functions used in bioconjugation were designed to yield stable constructs capable of tolerating the complex conditions of the biological milieu. In contrast, the use of reversible ligation methods has been rather overlooked, despite their potential to prepare conjugates that may respond to a predetermined stimulus. In recent years, BAs have become a well appreciated functionality to design responsive bioconjugates, primarily because BAs can selectively establish reversible covalent bonds with a diverse set of biological entities under bioconjugation conditions.813.1. Boronate esters
BAs are able to react with various naturally-occurring nucleophiles including carbohydrates and catechols, because the boron centre can reversibly coordinate with vicinal hydroxyl groups to yield boronate esters (BEs) with reaction rates of 102–103 M−1 s−1.46,82–84The reversible nature of BEs in aqueous media was initially reported by Lorand and co-workers,85 and since then, extensive studies have been performed to understand the mechanisms underlying the formation and stability of BEs.46,86,87 Regarding the formation of BEs, the tetrahedral boronate anion has been postulated to favour their complexation at higher pH values.88 In what concerns the complex's stability, BEs tend to hydrolyse under more acidic conditions.89 Moreover, the stability greatly depends on the structure of the diol ligand as, typically, cyclic and hindered cis-1,2 diols yield more stable complexes.89 Despite the general agreement that the structural integrity of BEs is pH- and solvent-dependent, the factors that govern these processes are still the subject of considerable debate (Fig. 5).46,90
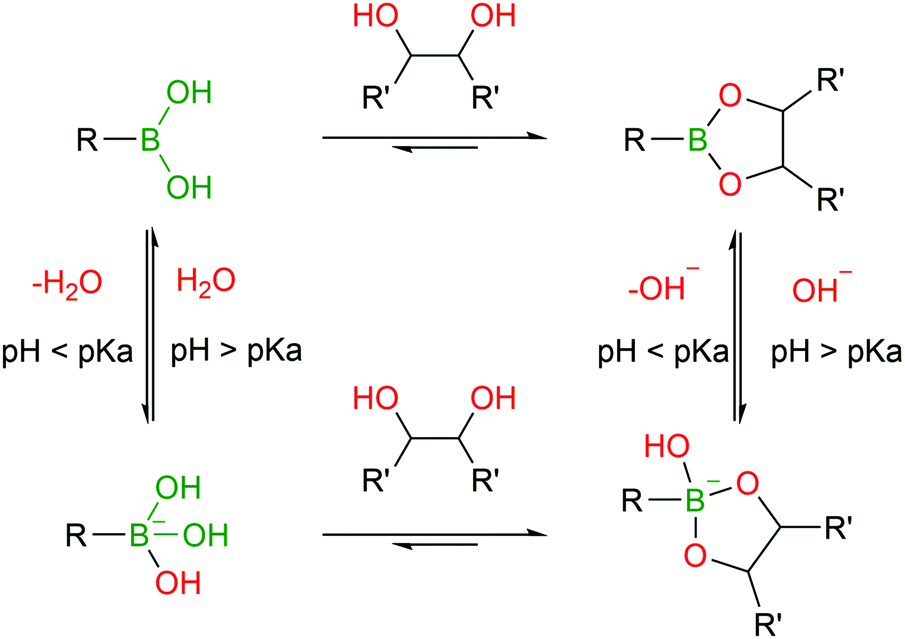 | ||
| Fig. 5 pH-Dependent formation equilibrium of a boronic ester in aqueous solution. | ||
Given their exquisite reactivity with various diols, BAs have been extensively used in the detection and quantification of carbohydrates. Despite being very important, these uses will not be covered here and the reader is directed to excellent existing reviews on this topic.86,91,92
Over the years, BEs have assumed a significant role in bioconjugation by enabling the construction of hybrid materials that exhibit a dynamic nature. The formation and trans-esterification of BEs under aqueous conditions are highly dependent on the diol–BA affinity. Therefore, BEs with good stability in aqueous solutions can still be displaced in the presence of diols with higher affinity for the same BA.
Taking advantage of the fast BE displacement in the presence of high-affinity carbohydrates, Markussen and co-workers developed a delivery system for insulin that responds to the increased concentration of D-sorbitol. This system was prepared by performing insulin functionalization at LysB29 with a modified glutamic acid displaying both a BA and a polyol that, upon interaction, generate an inactive multiprotein complex. These macromolecular structures exhibit an improved circulating half-life, though, in the presence of high concentrations of D-sorbitol (EC50 of 50 mM), they readily disassemble to release the active insulin monomers (Fig. 6).93 This technology could be the starting point for an interesting new concept for treatment of type II diabetes patients where a release of monomeric insulin (active form) occurs in response to an increase in serum carbohydrate concentration.94,95
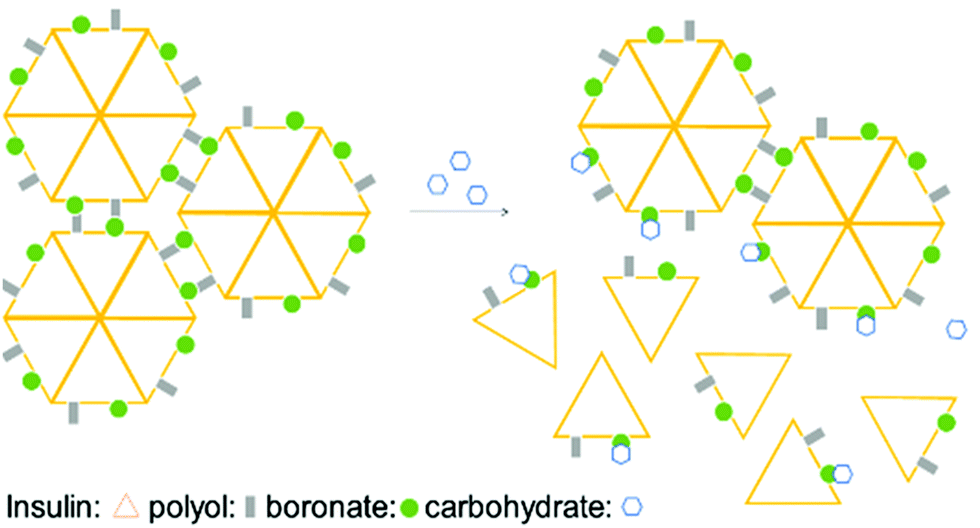 | ||
| Fig. 6 Illustration of insulin self-assembly under carbohydrate control. The inactive insulin multicomplex is disassembled in the presence of D-sorbitol, releasing the active insulin monomers. | ||
Similar strategies have been used to block the adhesion of cells to biological or biomedical surfaces by introducing PBA warheads onto a PEGylated polymer. In this approach, the PBA acts as a sugar-binding motif for carbohydrates on the cell surface (or hydroxyl groups on the surfaces of biomedical materials), while the PEG creates a hindered coating around the surface that blocks cell adhesion.96 This technology might have useful applications in decreasing the rejection rates of transfusions and transplants (natural or prosthetic) by blocking antibody adhesion to red blood cells or the transplanted organ/limb.
Owing to their ability to bind carbohydrates, BAs have become a very important tool for designing bioconjugates with therapeutic potential, because glycosylation patterns are important biomarkers in many different diseases.97–100 For instance, due to aberrant glycosylation, carbohydrate antigens are overexpressed in all tumour cells with higher frequency than other oncogene markers, thus making them effective targets for drug delivery.101 In humans, these glycophenotypes mainly present an abundance of sialic acid derivatives, which are associated with tumour growth in different types of cancer.102 Taking advantage of the high affinity of sialic acid for PBA (Kb 12.3 M−1vs. 1.71 M−1 of glucose at pH 7.4), Kataoka and co-workers developed PBA end-functionalized poly(ethyleneglycol)-b-poly(L-glutamic acid) copolymer micelles that target selectively sialic acid groups overexpressed on the surfaces of tumour cells (Fig. 7).103 In this example, the formation of the boronate complex on the cancer cell's surface enabled the effective delivery of micelles loaded with a (1,2-diaminocyclohexane)platinum(II) complex, which is a potent cytotoxic drug.
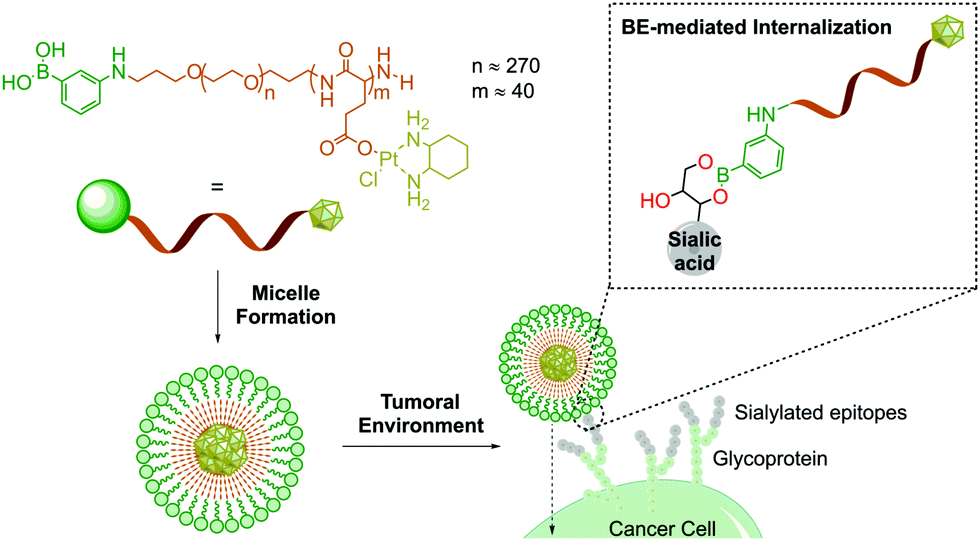 | ||
| Fig. 7 PBA-installed DACHPt-loaded micelles by self-assembly through polymer–metal complex formation between DACHPt and PBA–poly(ethylene glycol)-b-poly(L-glutamic acid) in distilled water. The PBA moieties on the surfaces of the micelles can bind to sialic acid. DACHPt stands for (1,2-diaminocyclohexane)platinum(II). | ||
In 2018, Best and co-workers reported the incorporation of BAs onto the surfaces of liposomes to achieve carbohydrate-mediated delivery along with nanoparticle disassembly.104 The insertion of BA-modified lipids into the liposome structure highly enhanced their cellular delivery, thanks to carbohydrate binding effects. Remarkably, the presence of carbohydrates also triggered the disassembly of the liposomes in a dose-dependent fashion, validating BA-liposomes as a useful tool for drug delivery and release.
Raines and co-workers disclosed a methodology to improve cellular delivery based on the formation of transient BEs with polysaccharides present on the cell surface. In particular, the authors explored the higher affinity of benzoxaborole for sialic acid (3.5 times higher than that of PBA) to deliver macromolecules into the cytoplasm of targeted cells. This approach was applied to the intracellular delivery of proteins, which is a challenging endeavour and typically requires the attachment of cationic domains to interact with the anionic cell membrane.105,106 In this work, benzoxaborole-modified RNAse A was efficiently delivered into the cytosol with a 4- to 5-fold increase in uptake versus the non-borylated protein.107
Continuing their work on benzoxaborole-based systems, Raines and co-workers reported an improved system for the traceless intracellular delivery of proteins based on a self-immolative benzoxaborole delivery vehicle. The self-immolative linker, an o-hydroxydihydrocinnamic acid derivative known as a trimethyl lock,108 is activated by intracellular esterases, followed by intramolecular lactonization and consequent release of the uncompromised protein (Fig. 8). With this technology, a 10-fold increase in cytosolic delivery was achieved for green fluorescent protein and cytotoxic ribonuclease in their native forms.109
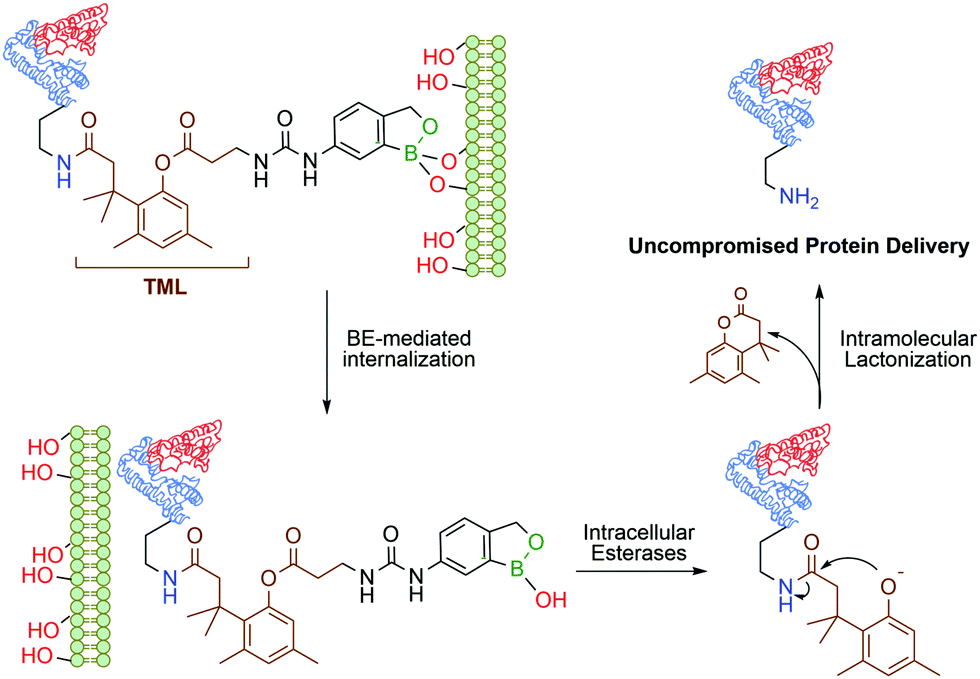 | ||
| Fig. 8 Boronate ester-based system for the traceless delivery of proteins into cells. The trimethyl lock linker promotes the intracellular release of the protein. | ||
In the context of protein modification, BAs were used in the traceless labelling of glycoproteins by taking advantage of the reversible formation of BEs. Affinity probes built through the attachment of a clickable handle to a PBA moiety via a reactive tosyl linker undergo a proximity-driven intramolecular SN2 reaction with a nucleophilic residue on the protein upon BE formation. Treatment with glycerol or sorbitol cleaved the BE and released the protein functionalized with the clickable handle, which was then used for pull-down assays (Fig. 9). Using this strategy, BA-tosyl-functionalized glass slides were used to develop microarrays for high-throughput screening of glycoproteins. This system was employed in the discovery of new glycoproteins in mixed protein pools and for the study of glycoprotein–protein interactions.110
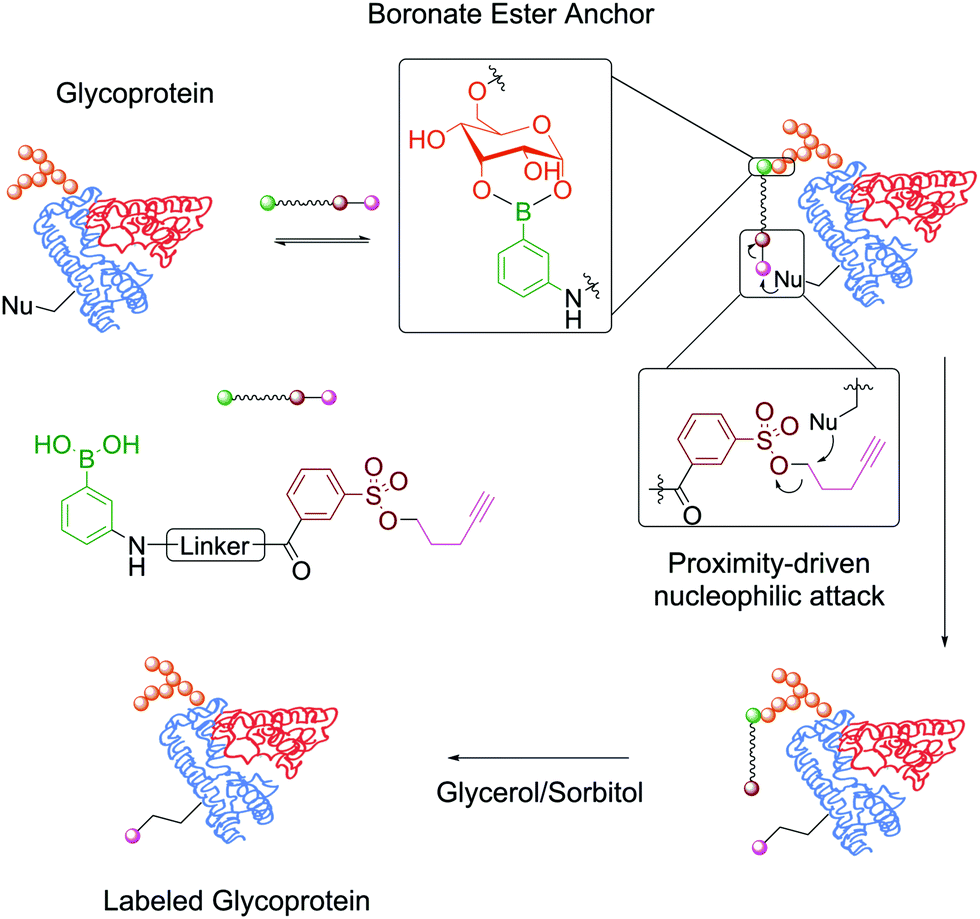 | ||
| Fig. 9 Attachment of a clickable handle through a proximity-driven SN2 reaction. This reaction was promoted by the formation of a BE with the glycoprotein's carbohydrates. | ||
The functionalization of protein amino acids featuring hydroxylated side chains with BAs would be a very practical bioconjugation strategy as it would target naturally-occurring residues like serine, threonine and tyrosine. However, the formation of a single B–O bond is often non-selective and typically lacks the stability required to endure the complex biological medium. To address this problem, Schepartz and co-workers developed a method capable of selectively recognizing protein tetraserine motifs based on the use of a bis-BA pro-fluorescent rhodamine compound. This bis-boronic acid (RhoBo) was employed as a selective small-molecule tag for proteins in living cells (Fig. 10). This non-toxic sensor exhibited cell-permeability and turn-on fluorescence upon tetraserine motif recognition, while displaying minimal affinity for monosaccharides and other endogenous molecules. The BE formation promoted a 3-fold increase in quantum yield, with a desirable bioimaging fluorescence emission wavelength (λmax = 520 nm). Notably, over 100 proteins within the human proteome contain the SSPGSS sequence that could potentially be recognized in the nanomolar concentration range by RhoBo.111
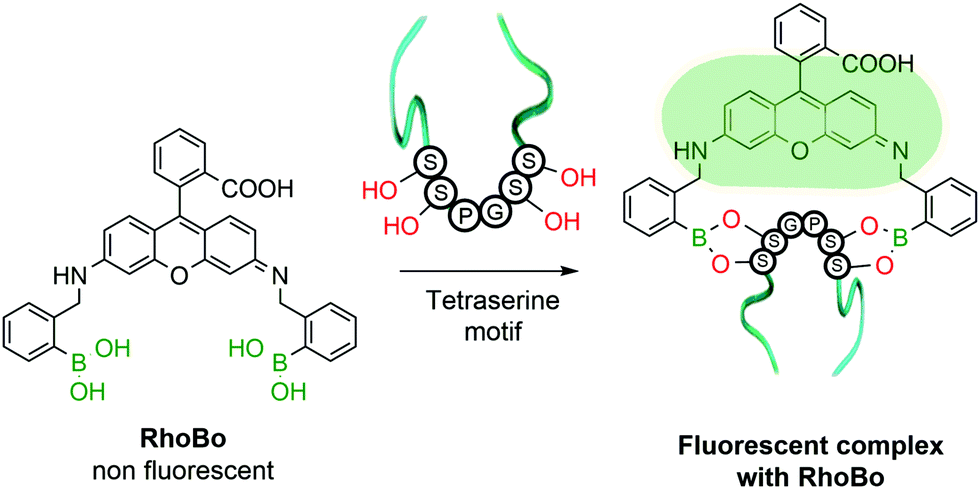 | ||
| Fig. 10 Representation of a RhoBo fluorescent complex upon tetraserine sequence recognition. | ||
In a similar trend and despite its increased acidity, tyrosine is equally unable to form stable conjugates directly with BAs. However, as reported by Tirelli and co-workers, this residue can be converted to its corresponding catechol derivative by the action of tyrosinase. The newly formed Tyr-derived catechol is a good BA ligand, with the ability to form BEs under bioconjugation conditions (Fig. 11). This approach can be applied to different proteins using the hemagglutinin-derived HA-tag that can introduce exposed Tyr groups onto proteins, thus making them suitable for this modification.112
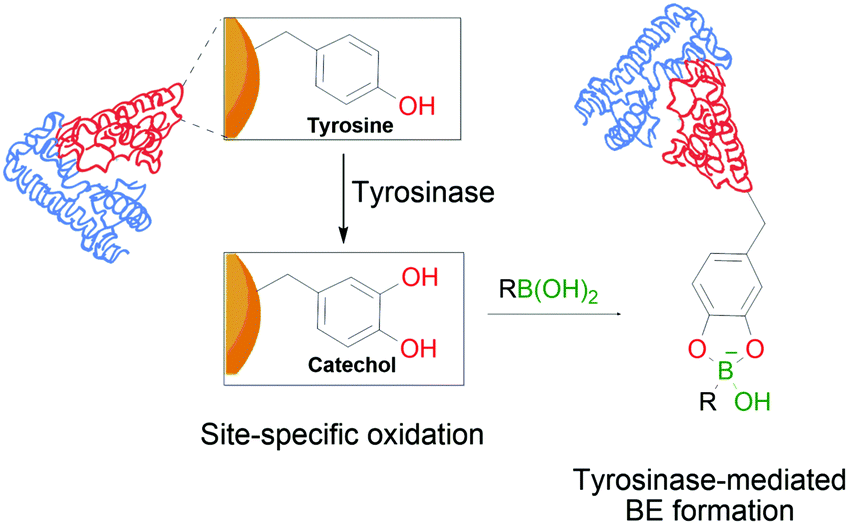 | ||
| Fig. 11 Oxidation of a tyrosine residue by tyrosinase to the corresponding catechol enables the formation of transient BEs on the protein's surface. | ||
The presence of a diol-containing ribose unit in the structure of ribonucleic acid (RNA) offers the possibility of generating BEs at this position. In fact, BAs have been used for RNA separation and purification and for design of different bioconjugates with potential therapeutic use.113–115 One particular area of interest which can be facilitated by the BE formation is the transport of ribonucleosides and ribonucleotides across cell membranes.116 Kataoka and co-workers reported a PBA-modified poly-lysine co-polymer with a C-terminus PEG chain that is able to bind reversibly to siRNAs. This copolymer forms micelles that enable the BE formation and electrostatic interactions with the phosphate anions of RNA, contributing to the stabilization of the siRNA–vector complex. Despite these strong interactions, the intracellular high concentration of ribonucleotides like ATP compromised the structural integrity of the complex and promoted the intended release of the siRNA.117
Zhuo and co-workers reported a similar bi-functional linker for gene transfection through the covalent incorporation of multiple PBA moieties into polyethylenimine polymers (Fig. 12). The cationic groups of the polymer interacted electrostatically with the phosphate anions of deoxyribonucleic acid (DNA), while the PBA was anchored to the cell surface through BE formation with glycoproteins, leading to an improved gene cellular uptake.118 Despite the polyethylenimine polymer being already one of the most successful non-viral gene vectors,119 the introduction of PBA into the polymer improved gene transfection by 2 to 3 orders of magnitude.
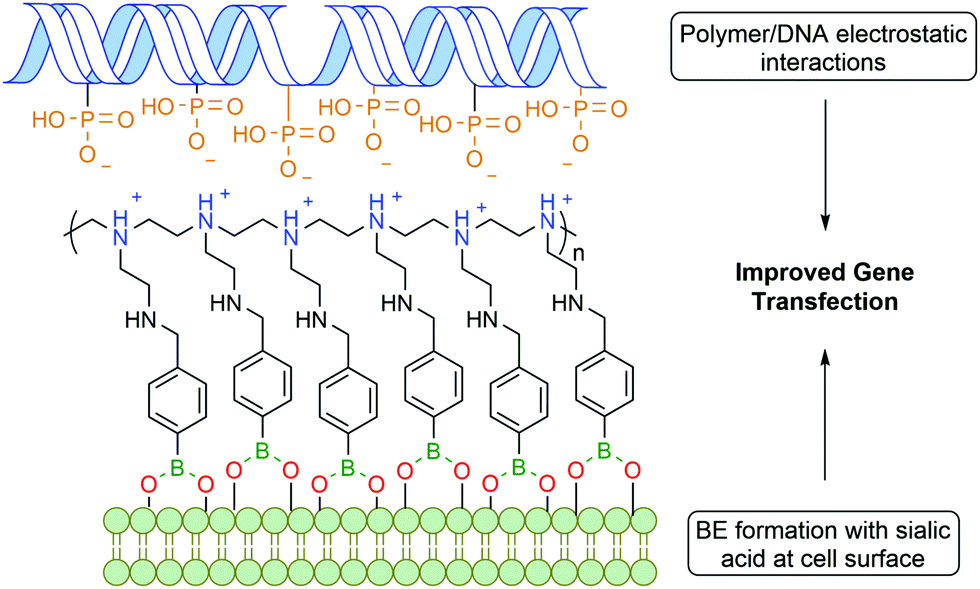 | ||
| Fig. 12 PBA improves gene transfection through interaction with surface glycans. | ||
In 2008 Schultz and co-workers developed a method to perform the site-specific incorporation of a non-natural boron-based amino acid (p-boronophenylalanine) into proteins using the amber stop codon (TAG) strategy.120 The obtained modified protein was not only able to react in classical boronate-mediated reactions, such as oxidation, reduction and Suzuki coupling, but also gained the ability to form BEs with poly-hydroxylated compounds that allowed a one-step traceless protein purification (Fig. 13). Furthermore, the authors used this technology to generate antibodies in an E. coli strain that encoded p-boronophenylalanine with the ability to form reversible boronate esters with acyclic glycans.121 These studies contributed to current and future applications in the design of therapeutic proteins122 and to the development of new fluorescent proteins as sensors for H2O2123 or peroxynitrite124 detection in vivo.
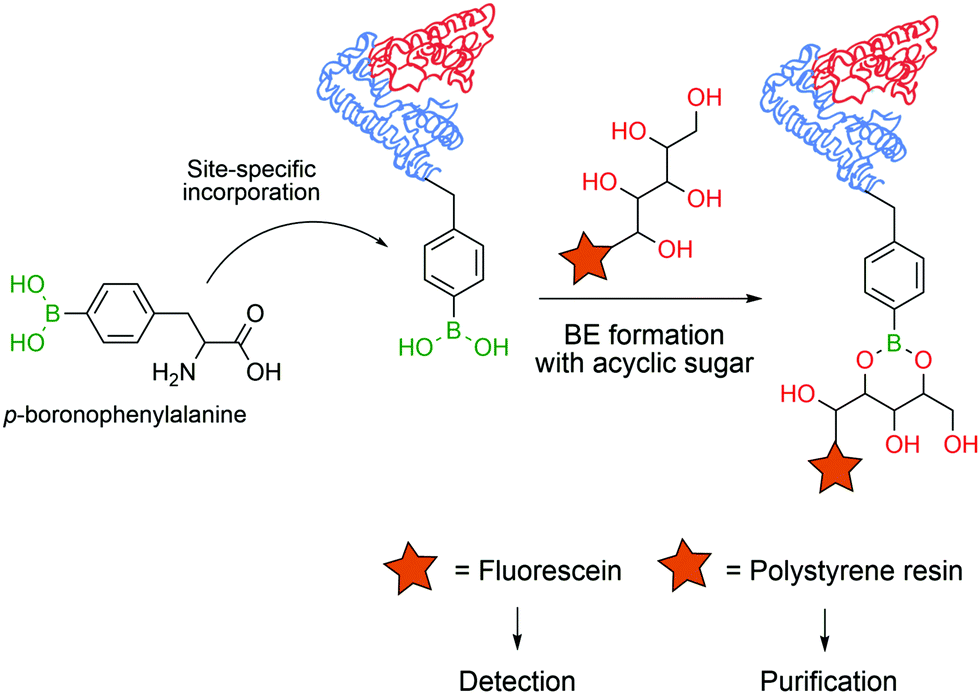 | ||
| Fig. 13 Incorporation of a p-boronophenylalanine non-canonical amino acid into proteins allows their detection and purification through BE formation. | ||
Despite their utility, BEs based on carbohydrates often lack the necessary stability to design bioconjugates that may operate in more complex biological settings. Aware of these limitations, Hall and co-workers reported a new click semi-bioorthogonal reaction system using a highly strained diol to improve the affinity of the BA–diol pair.125 Nopoldiol and 2-methyl-5-carboxyphenylboronic acid showed complete complexation within minutes at a low micromolar concentration in aqueous medium (ca. 6.9 M−1 s−1 and Keq ≈ 1.2 × 105 M−1 measured by 1H-NMR). Fluorescence quenching assays demonstrated that the kON and Keq values for the same pair can go up to 340 M−1 s−1 and 1.5 × 107 M−1. However, the authors suggested that these results might be significantly influenced by hydrophobic and π–π interactions between the fluorophores. Therefore, the actual kinetic rates should be somewhere in the middle of the underestimating, highly concentrated 1H-NMR experiments and the overestimating fluorescence quenching assays. The authors also noted that the introduction of a fluorine in the ortho position to the BA increased significantly the kON and Keq values but decreased the hydrolytic stability, making it unuseful for protein labelling. With this methodology, model proteins like thioredoxin and albumin were first modified with a BA linker, which then reacted with a nopoldiol fluorescent reporter under physiological conditions (Fig. 14A). Later, the same authors also described a synergistic “click” boronate/thiosemicarbazone system that enables a fast and irreversible bioorthogonal conjugation in live cells (Fig. 14B).126 This new system is similar to the previous one but, through the introduction of an additional hydrazone ligation, the reaction became irreversible. This click type reaction was successfully applied to site selective protein labelling on mammalian HEK293T cells.
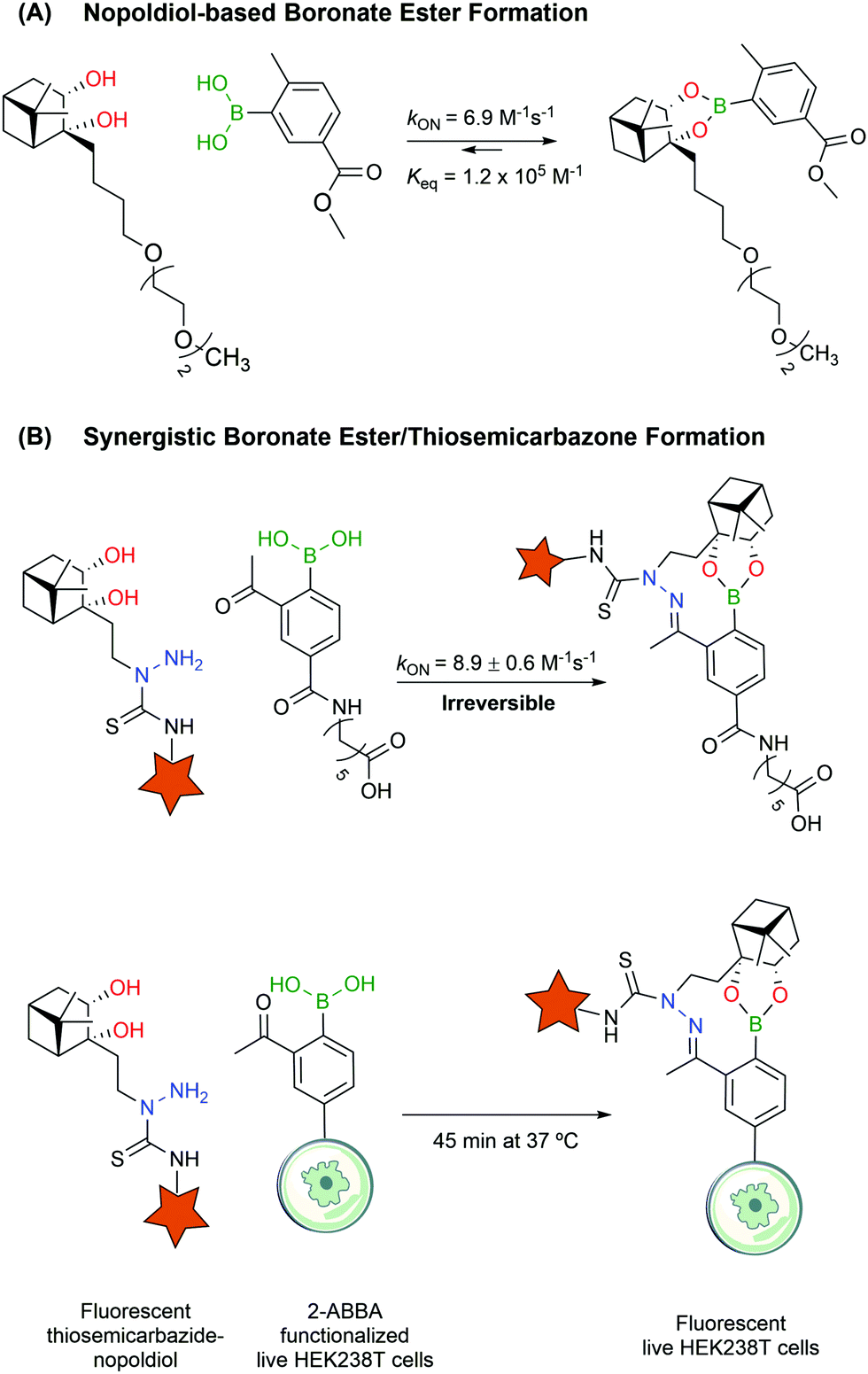 | ||
| Fig. 14 Formation of stable BEs. (A) The highly strained nopoldiol binds tightly but reversibly to PBA. (B) The introduction of a synergistic thiosemicarbazone renders the BE with nopoldiol irreversible. | ||
3.2. Iminoboronates
Bioconjugates with primary amines, either the α-amines of N-terminal functions or the ε-amines of lysine residues, are common targets of protein functionalization due to amines’ high nucleophilicity and solvent accessibility. These chemical functions can react with electrophiles like activated esters, sulfonyl chlorides, isocyanates and isothiocyanates.127 Alternatively, primary amines can also be selectively modified based on the generation of imines, though a second reductive step is commonly necessary to achieve an efficient conjugation, due to the inherent reversibility of this functionality (Fig. 15A).128,129 Therefore, the formation of stable imines in aqueous media would allow a direct, selective and potentially reversible strategy to functionalize the lysine residues and N-terminus.
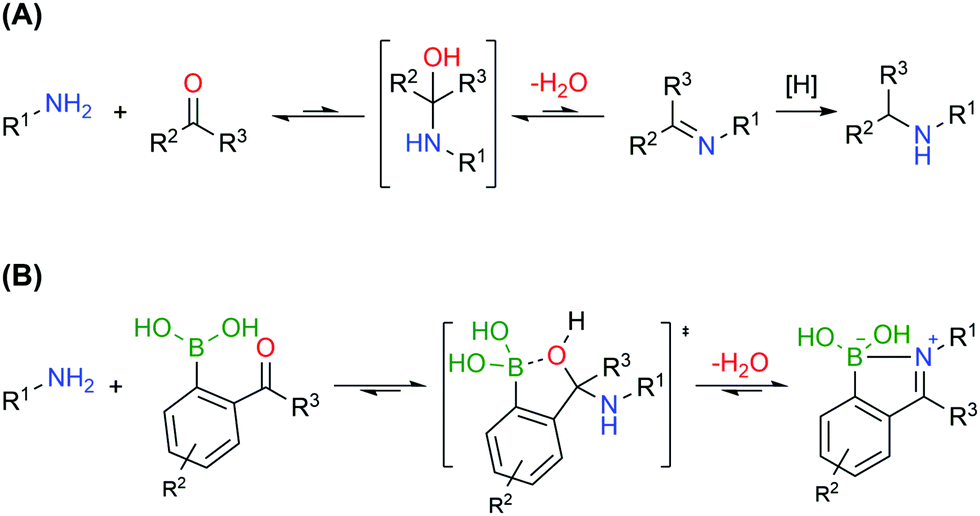 | ||
| Fig. 15 Strategies for amine condensation with carbonyl compounds. (A) Imine formation, reversibility in aqueous media and reductive amination. (B) Iminoboronate formation and reversibility in aqueous media. | ||
Similarly to oxygen-based nucleophiles, BAs can also coordinate with nitrogen Lewis bases to generate reversible B–N bonds (Fig. 15B) that have been used in the construction of self-assembled nanostructures, polymers, and carbohydrate sensors or to design molecules that explore B–N/C–C isosterism.130–132 Based on these studies, Gois and co-workers envisioned that the reaction of BAs substituted at the ortho position with a carbonyl group with alkylic primary amines could result in imines with improved stability in aqueous media due to the formation of an intramolecular B–N dative bond.133 In their initial studies, the authors showed that 2-formylbenzeneboronic acid (2-FBBA) and 2-acetylbenzeneboronic acid (2-ABBA) very effectively reacted with lysine in aqueous media to generate iminoboronates over a wide pH range (6–9), which, if not disturbed, could be maintained over 7 days. Density functional theory calculations performed on this system support a mechanism where the BA acts as an intramolecular Lewis acid that catalyses the nucleophilic attack of the amine to the carbonyl group, favouring the elimination of water to generate the imine and, more importantly, enables B–N bond formation (dB–N = 1.71 Å) that enhances the stability of the product (ΔE = −7.4 kcal mol−1) and contributes to an overall favourable energy balance of −10.0 kcal mol−1. In 2016, Anslyn and co-workers reported that iminoboronate esters predominantly give a solvent-inserted zwitterionic species in organic solvents, which must also be considered as a possible mechanism for the stabilization of iminoboronates.134
A direct comparison between 2-FBBA and 2-ABBA revealed that the ketone renders more stable iminoboronates, and for that reason, 2-ABBA was used in the successful functionalization of the primary amino groups exposed in the hormonal neuropeptide somatostatin and proteins like lysozyme, cytochrome c, ribonuclease A, and myoglobin. A very important aspect of this study was the discovery that, despite being more stable than imines, iminoboronates are still reversible in the presence of endogenous molecules such as glutathione (GSH), dopamine or fructose, which is a promising property to design therapeutic delivery systems and responsive materials (Fig. 16). More recently, iminoboronates have been shown to hydrolyse in more complex physiological media135 or in the presence of exogenous molecules such as tris(2-carboxyethyl)phosphine (TCEP).136
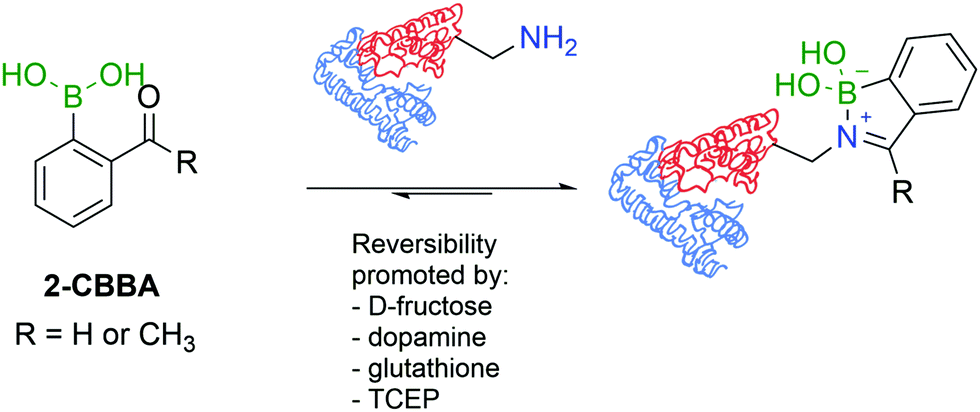 | ||
| Fig. 16 Iminoboronate protein modification and reversibility in aqueous media. | ||
The iminoboronate technology was then applied to tag folate derivatives with fluorescent probes which could be used to selectively label human non-small lung cancer cells (NCI-H460) that are known to overexpress folate receptors.137 Next, the same authors explored iminoboronate formation to prepare a folate–paclitaxel small molecule drug conjugate, which exhibited an IC50 of 20.7 nM against NCI-H460 cancer cells (Fig. 17). Additional studies performed on this system allowed the ligation of clickable handles to a model protein (lysozyme), which were then subjected to post-functionalization through a Cu-assisted or strain-promoted azide–alkyne cycloaddition. Finally, the 2-ABBA core was also modified with a polyethylene glycol chain and this reagent was used to functionalize insulin. Interestingly, in the presence of fructose, the construct was cleaved and the free insulin released.138
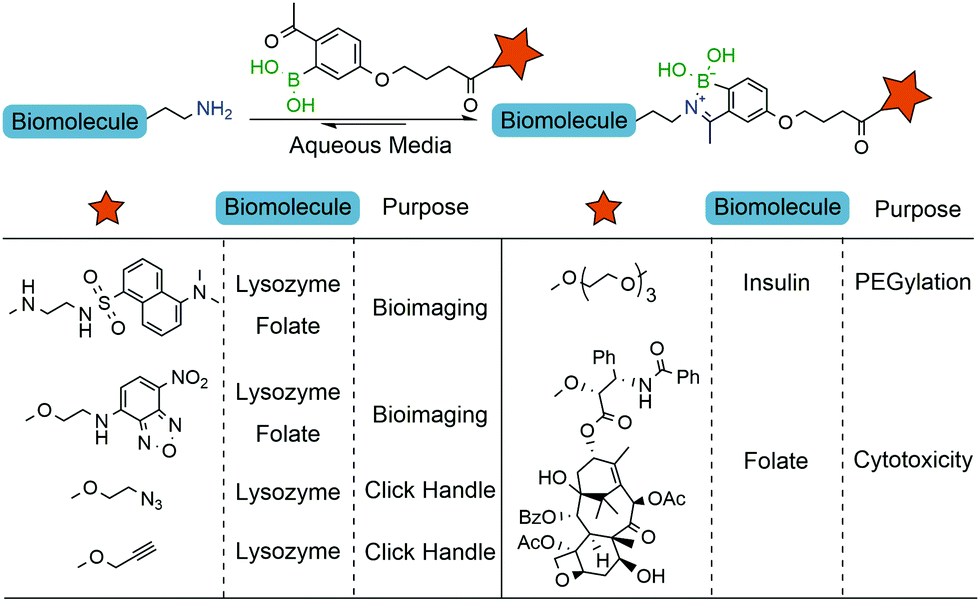 | ||
| Fig. 17 Iminoboronate-based bioconjugation to anchor fluorophores, clickable handles, PEG derivatives and paclitaxel. | ||
Motivated by the reversible nature of this lysine-targeting bioconjugation methodology, scientists at AstraZeneca reported the use of iminoboronates to enhance the on-target residence time of a protein–protein inhibitor. In this study, a known indole acid-based myeloid cell leukaemia 1 (Mcl-1) inhibitor was functionalized with a 2-FBBA warhead, which enabled iminoboronate formation with the non-catalytic Lys234 side chain of Mcl-1 and lined up the ligand's active motif to the respective binding site (Fig. 18).139
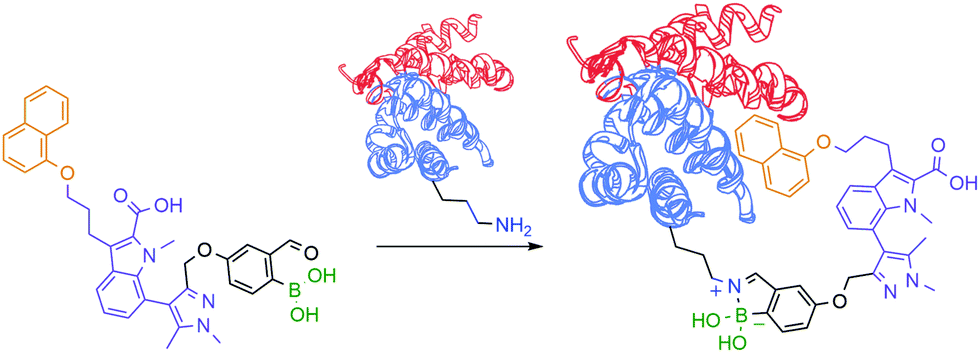 | ||
| Fig. 18 Improved “guided” inhibition of Mcl-1 based on a reversible iminoboronate modification of Lys234. | ||
Another example that exploits the reversible dynamic covalent features of iminoboronates was the use of BAs to block the ion current of individual nanopores through the modification of lysine residues. This study, developed by Cockroft and co-workers, describes the 2-ABBA-mediated modifications of three rings of lysine residues within the α-hemolysin channel: one at the cis-side, one at the trans-side and the other at the constriction site (Fig. 19). The modifications of the latter proved to be the most relevant to block ion current, as was anticipated. Based on the pH-responsiveness of the reaction, the authors were able to link ion current blockages and specific lysine modifications unambiguously. Assessments of which lysines were modified by applying different voltages to the electrodes surrounding the nanopore allowed concluding that the reactions at the trans-lysine ring were not observed upon addition of 2-ABBA from the cis-side, and vice versa. Furthermore, given that at a low concentration of the reagent a single lysine was dominantly modified, the authors were also able to perform interesting kinetic and ion current influence studies by individually modifying one of the seven constriction site-lysine residues.140 In their work, the authors showed a valuable and accessible alternative of genetic engineering of nanopores to study the ion current of transmembrane ion-channels.
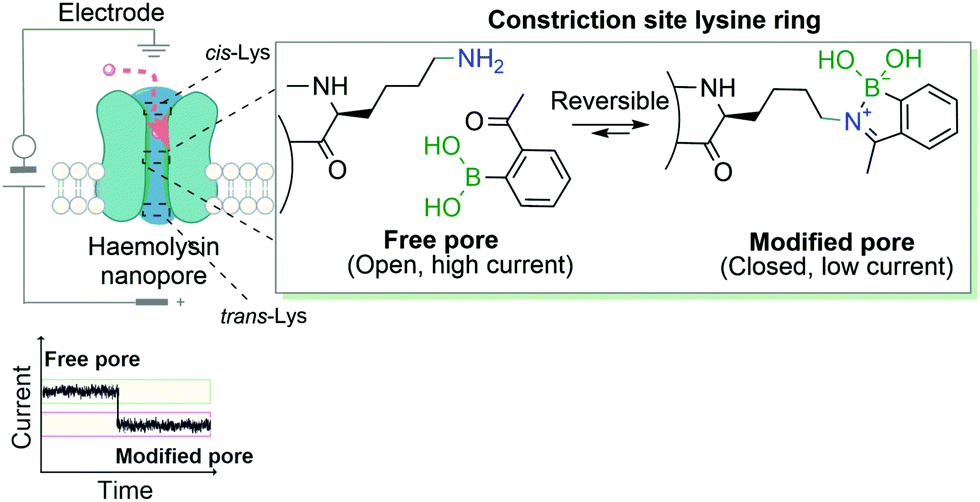 | ||
| Fig. 19 Transient iminoboronate formation enables the study of ion current in transmembrane ion channels. | ||
Most reports on bioconjugation using iminoboronates target lysine and N-terminal residues, though this technology has been successfully applied to other biological molecules besides proteins. Gao and co-workers successfully described the use of fluorescent 2-ABBA in the functionalization of membrane lipids phosphatidylethanolamine and lysine-modified phosphatidyl glycerol that are exclusively present on Gram positive bacteria (Fig. 20). Thus, by using this selective amine-oriented modification, it was possible to develop a novel imaging method to detect a specific bacterial infection by discriminating Gram-positive from Gram-negative bacteria and mammalian cells.141
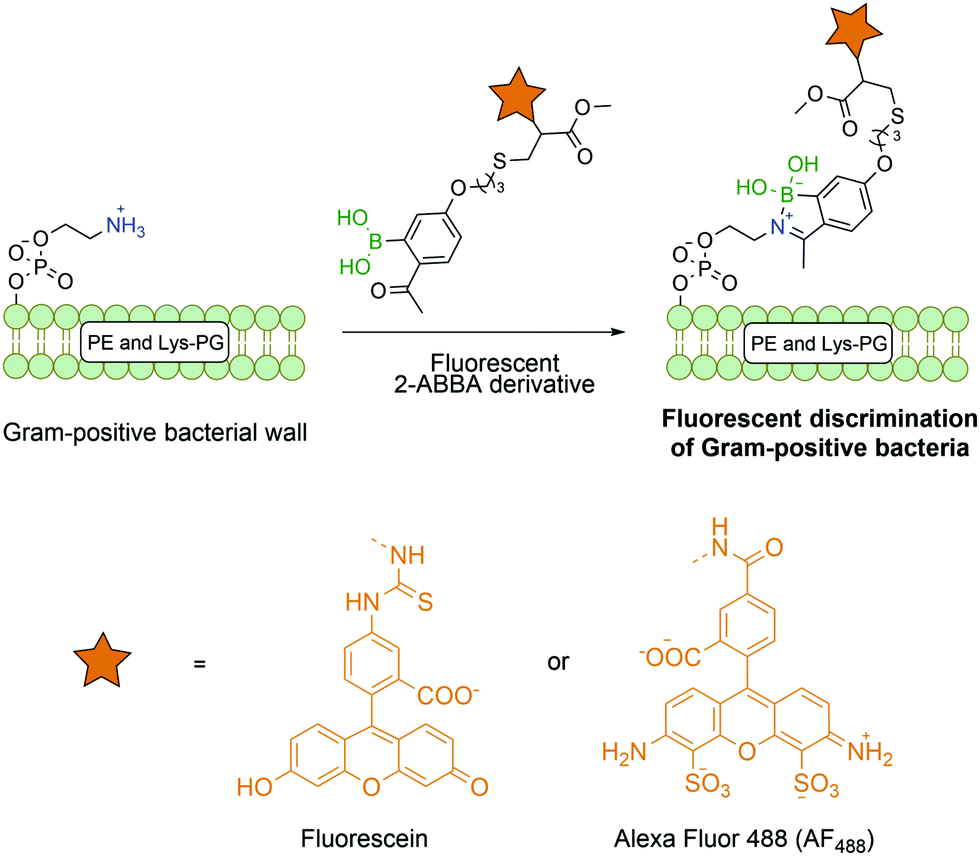 | ||
| Fig. 20 Fluorescent Gram-positive bacterial discrimination based on selective lipid-iminoboronate formation that is absent in Gram-negative bacteria and mammalian cells. | ||
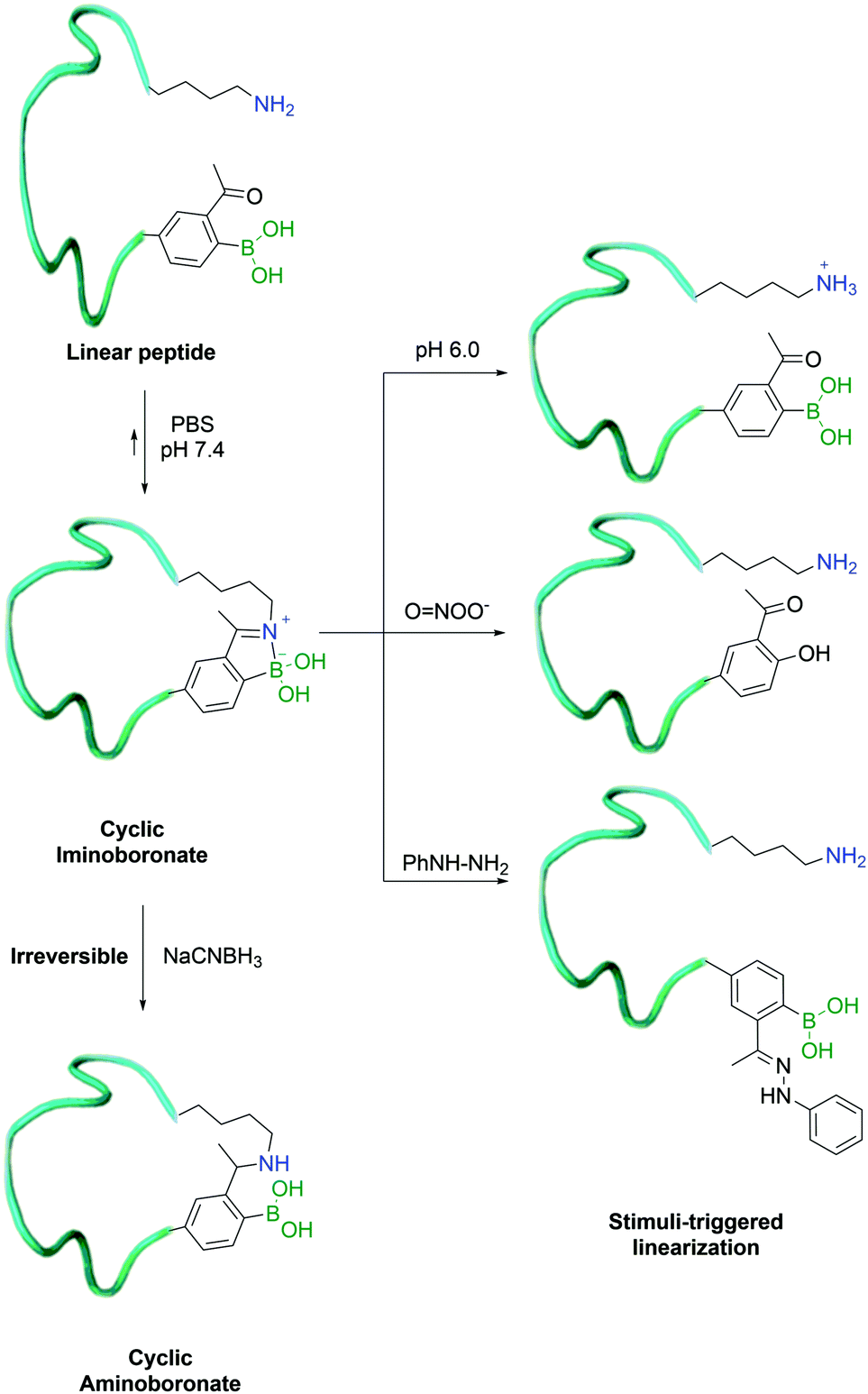 | ||
| Fig. 21 Iminoboronate formation enables the macrocyclization of a peptide which can be reverted in the presence of phenyl hydrazine, ROS and acidic pH. | ||
In 2017, Gao and co-workers developed an alternative method for their previously reported peptide cyclization strategy, using a shorter side-chain lysine homologue – diaminopropionic acid (Dap) – as an unnatural amino acid to enhance iminoboronate formation and consequently increase the efficiency of the process. In order to turn it into a hydrolytically stable product (aminoboronate), the authors used sodium cyanoborohydride to selectively reduce the imine bond formed between the β-amine of Dap and 2-ABBA. This was achieved even in the presence of in-chain lysine residues, which could then be exploited for further modification.142
3.3. Boron-stabilized heterocycles
An important addition to the iminoboronate technology resulted from the recognition that this function can react with adjacent nucleophiles.First reported by Wang and co-workers, the known procedure of cyanobenzothiazole–cysteine condensation was applied to iminoboronate chemistry, demonstrating that the adjacent BA could catalyse the reaction, improving its kinetics (Fig. 22A).143 Surprisingly, this work did not address any biological application as the reactions were conducted only in organic solvents. The ability of BAs to catalyse the nucleophilic addition of thiols to imines remained, however, an enticing concept for the modification of proteins given the fast kinetics and bioorthogonality of the reaction.
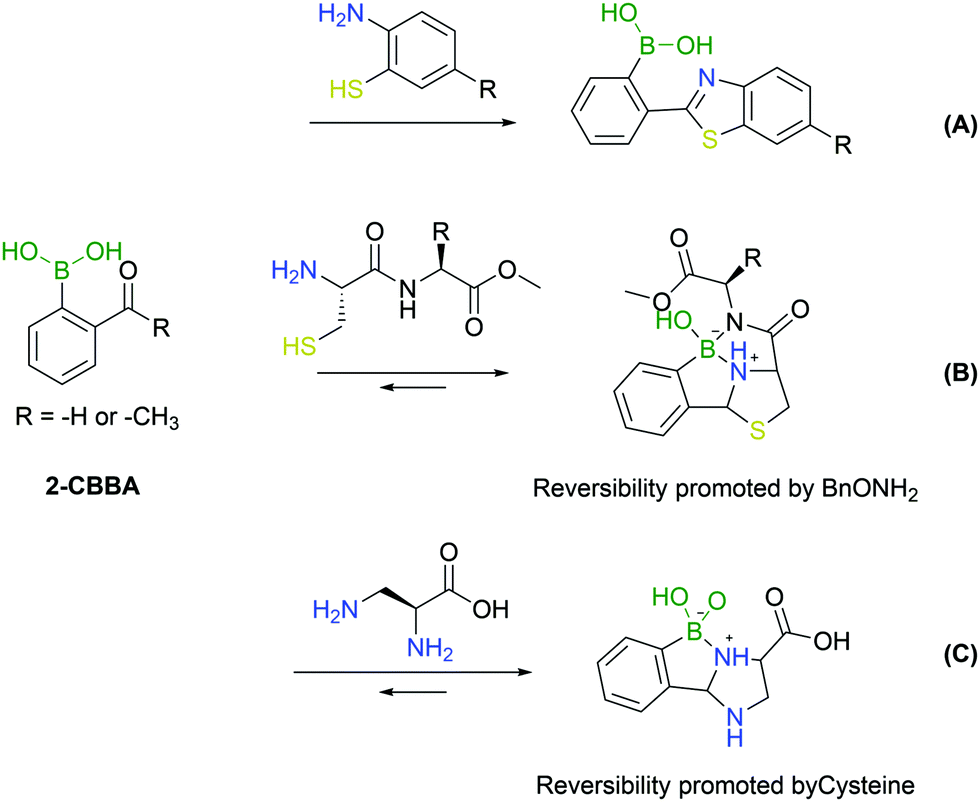 | ||
| Fig. 22 Iminoboronates as intermediaries for cyclic products. (A) Benzothiazole click reaction; (B) N-terminal cysteine modification to form boronated thiazolidines; and (C) N-terminal DAP modification to form boronated imidazolidines. | ||
A year later, Gao's and Gois’ groups independently reported the functionalization of the N-terminal cysteine using the 2-FBBA reagent.144,145 In this reaction, the B–N interaction affords a reactive imine that rapidly undergoes an intramolecular thiol addition. The boronated thiazolidine formation is one of the fastest bioorthogonal reactions for protein labelling (k2 = 5.5 × 103 M−1 s−1) and was applied successfully to several biomolecules. In particular, this method enabled a controlled dual functionalization of peptides featuring two different cysteines (N-terminal and in-chain residues). The boronated thiazolidine was also shown to be reversible upon incubation with a hydroxylamine derivative, which enabled the implementation of a sequential orthogonal functionalization of the peptide with two maleimide derivatives (Fig. 22B).145
The aforementioned N-terminal Dap residue was also used by Gao and co-workers to generate imidazolidino boronates. This is a similar strategy to what has been reported for N-terminal cysteine modification. Though, unlike the thiazolidine generation, this group can complex with boron with either the α- or β-amine from Dap, thus leading to different isomers. However, given the novel feature of the reversibility of imidazolidino boronates upon incubation with a specific endogenous molecule (cysteine), there can be specific biological applications where this strategy can present an added value (Fig. 22C).146
4. Boronic acids as linker components
Extensive research in the engineering of bioconjugates demonstrated that chemical linkers are not just molecular spacers used to connect both functional moieties but, perhaps as importantly, are important structures to control the conjugate's properties. Therefore, linkers often exhibit structures with a high functional density, and their construction comprises a sequence of complex and costly synthetic steps that are typically unsuited for a straightforward structural diversification. In this context, the coordination profile of BAs offers a unique platform to design chemical linkers with tuneable stability and reversibility.4.1. N,O-Iminoboronates
Continuing the development of iminoboronates and aiming to improve their stability, Gois and co-workers reported the synthesis of N,O-iminoboronates, a class of iminoboronates that showed improved resistance to hydrolysis (<12% hydrolysis in 7 days).147This new class of iminoboronates was prepared by the reaction of 2-FBBA or 2-ABBA with aminomethyl phenols, which provide an additional point of coordination to the boron centre, leading to a remarkably stable bicyclic structure. The constructs built with this core proved to be stable in human plasma (<20% degradation in 48 h) while still being hydrolysed at acidic pH, making them suitable linkers for payload delivery in cancer tissues. The value of this platform has been demonstrated by the synthesis of a folic acid conjugate for the delivery of a fluorescent probe to human breast cancer cells. The fluorescent probe was prepared using an aminophenol derivative of coumarin, whose fluorescence was turned off by its incorporation into the N,O-iminoboronate core (Fig. 23). As predicted, the folic acid-mediated internalization of the conjugate exposed it to the intracellular acidic pH that triggered the hydrolysis of the N,O-iminoboronate and the release of the fluorescent coumarin.
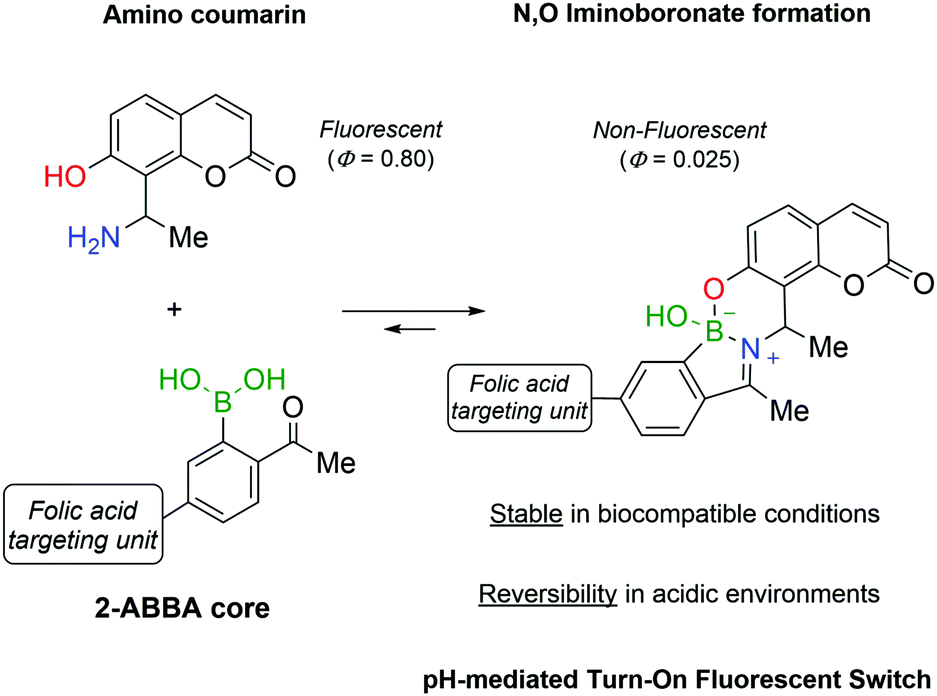 | ||
| Fig. 23 Aminomethyl phenols and 2-ABBA derivatives react to form a new class of N,O-iminoboronates which are stable at neutral and basic pHs but readily hydrolyse in acidic medium. This technology was used to generate a turn-on fluorescent switch using an amino coumarin and a folic acid–ABBA construct. | ||
4.2. Salicylhydroxamic acid–boronate esters
Stolowitz and co-workers discovered that salicylhydroxamic acid (SHA) formed a complex which was stable over a broad range of physiological pHs but readily hydrolysed under acidic conditions. To gain better insight into the newfound complexes, the authors used 11B-NMR to track the boron hybridization over time and at different pH values. The authors found that at neutral and alkaline pHs a single species is observed with an sp3 hybridization, indicating a tetrahedral coordination geometry, while at acidic pH (<5) a mixture of sp2, sp3 and PBA signals is visible, indicating possible hydrolysis of the complex. The preferential formation of a 6-membered ring in opposition to the respective 5-membered ring was also demonstrated (Fig. 24).148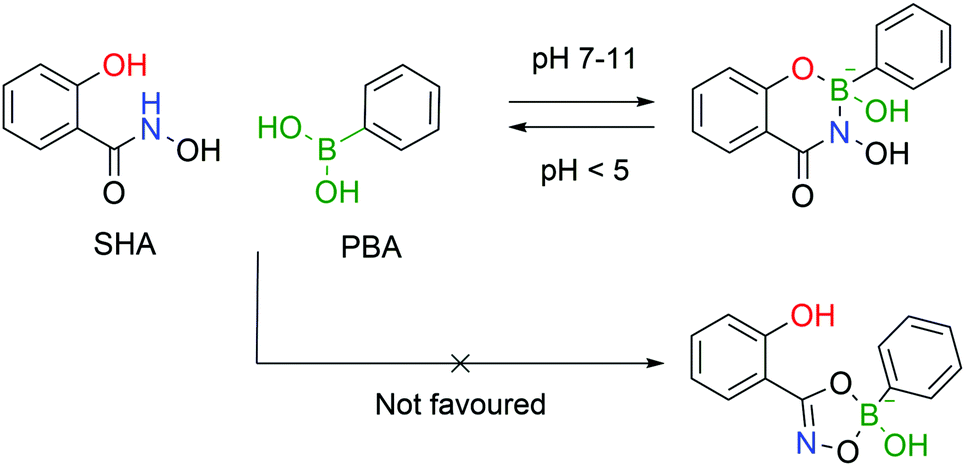 | ||
| Fig. 24 Complexation of SHA with PBA occurs readily at neutral and alkaline pHs but the formed complex is quickly hydrolysed under acidic conditions. The 6-membered ring is the preferential structure. | ||
The first application of this technology was developed by Wiley and co-workers for protein immobilization and purification on a chromatographic support. The authors envisioned that SHA-functionalized Sepharose beads could interact strongly (and selectively) with PBA-modified proteins, immobilizing them onto a chromatographic support, followed by a controlled release upon washing with an acidic eluent (Fig. 25). For such application, horseradish peroxidase (HRP) and alkaline phosphatase were randomly modified with BAs using NHS ester chemistry and evaluated for their affinity to SHA-beads. Both modified proteins were successfully immobilized onto the SHA–Sepharose column and were retained throughout successive washes with eluents at basic and neutral pHs. When the eluent's pH was decreased (pH < 3), PBA–HRP was successfully released from the Sepharose, while no release was observed for PBA–alkaline phosphatase. A possible explanation relies on the additional interactions of PBA with carbohydrates present on alkaline phosphatase's surface that blocked the release from Sepharose.148 The most important disadvantage of this technology is the strong dependence on the number of modifications in each protein. Despite the higher number of incorporated PBA moieties translating into an improved retention in the column, in the case of PBA–alkaline phosphatase it also affected its enzymatic activity (50% vs. unmodified enzyme). Therefore, to achieve optimal results, it is necessary to tune carefully the number of modifications in each protein. To solve this problem, the authors later reported a similar technology using proteins modified with a phenyl-diboronic acid moiety. This way, the number of modifications in each protein is maintained but the amount of BA–SHA interactions is doubled, thus creating stronger protein–Sepharose binding.149
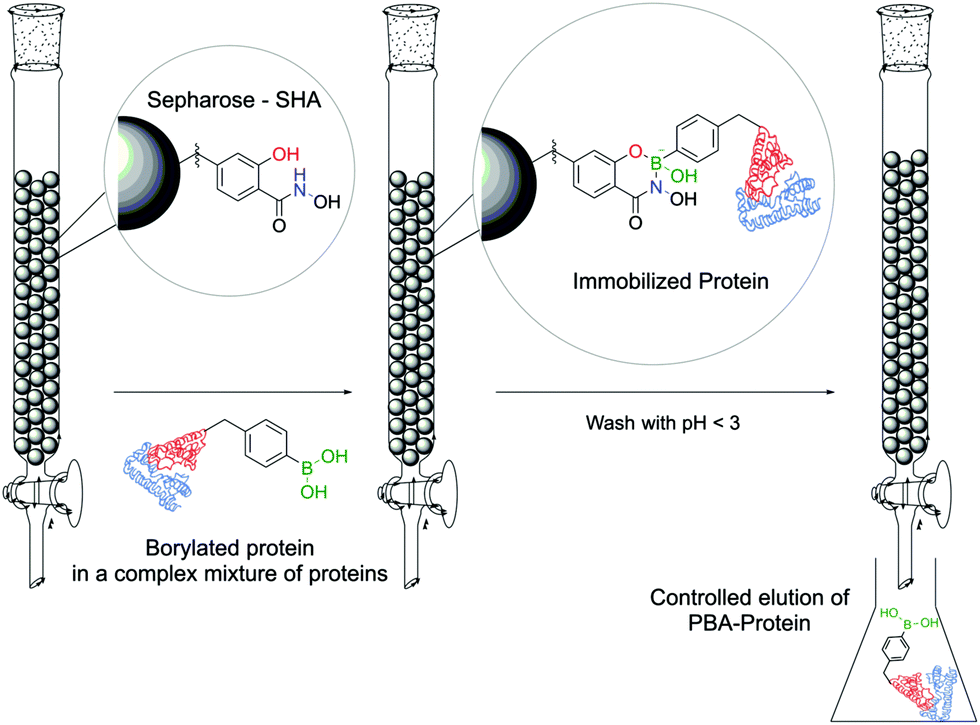 | ||
| Fig. 25 PBA-modified proteins can be selectively immobilized and purified on a SHA–Sepharose resin. | ||
In 2010, Jaffrey and co-workers adapted the SHA–BE formation technology to the development of a strategy for PBA-modified peptide dimerization in cells.150 To this end, a dimerizer molecule was synthesized by attaching two SHA groups to the ends of a spacer unit; the titration of this molecule with PBA confirmed that it is able to bind 2 molecules of PBA at the same time. This technology was applied to the formation of peptide dimers as agonists of the thrombopoietin receptor c-Mpl, as it is known that monomeric peptidic sequences only have marginal inhibitory activity. With this intent, one of the known peptidic agonists of c-Mpl, the EGPTLRQWLAARA peptide, was decorated with a PBA moiety at the C-terminus position and was incubated with cells overexpressing the target receptor. In the absence of the dimerizer, the agonist activity remained low, as that of the native peptide, while, in the presence of the dimerizer, a significant increase in activity was observed, confirming the validity of this approach for the formation of peptide dimers in a cellular environment. During this work, the authors determined the kinetic rate for the complexation to be approximately 7.01 ± 2.04 × 106 M−2 s−1.151
Adopting the same strategy for stochastic insertion of BAs onto protein surfaces used by Wiley and co-workers in their pioneering work on SHA–BE formation, Weil and co-workers used the PBA–SHA ligation's properties to develop hybrid zymogens through reversible attachment of protective dendrimers. Inspired by nature's processes for zymogen production, the authors envisioned that introduction of a poly(amide)amino (PAMAM) dendrimer on the surfaces of proteases would create a protective shell, which would hinder their activity.152 Moreover, taking advantage of the PBA–SHA complex's reversibility, upon zymogen's entrance into the cell through the lysosome, the low pH quickly hydrolyses the complex, releasing the protease in its active form. Three different proteases (i.e., trypsin, papain and DNAse I) were functionalized with PBA moieties followed by complexation at pH 7.4 with SHA-modified PAMAM dendrimers. After the modification, all three proteases were inactive as the dendrimers blocked the enzymes' active site. As expected, upon decreasing the pH to 5, the dendrimers dissociate from the proteases and they regain their enzymatic activity (Fig. 26). The authors also discovered that, besides acting as blocking agents, dendrimers facilitate their cellular uptake through electrostatic interaction between the cationic dendrimer and the negatively charged phospholipids.
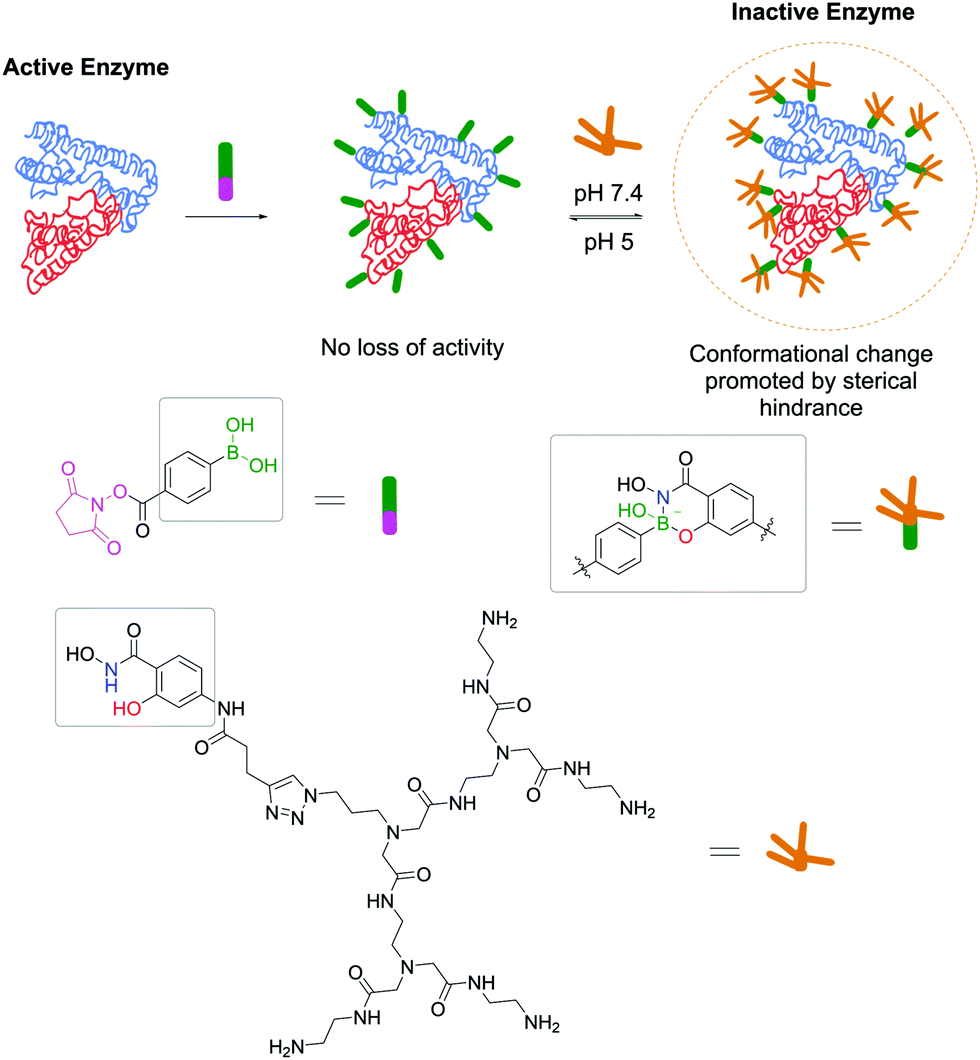 | ||
| Fig. 26 Reversible attachment of sterically-hindered dendrimers through a SHA–PBA linker promotes the controlled activation/deactivation of enzymes. | ||
In another work, the same group described the pH-responsive PEGylation of proteins to modulate their pharmacokinetic properties using a PBA–SHA complex as a dynamic switch.153 Furthermore, Weil and co-workers reported the development of a smart hydrogel based on the PBA–SHA complex's dynamic formation, which incorporates a cytotoxic enzyme as a structural feature.154 Cytochrome c, a pro-apoptotic enzyme when present in the cytoplasm, was modified with a PBA moiety and reacted with a bis-SHA–PEG. At neutral pH, the gel self-assembles and enfolds the cytotoxic enzyme in its interior, shielding it from uncontrolled activity. When the gel enters into tumour cells, the pH drop promotes the release of the active enzyme, triggering the apoptotic cascade and eventual cell death. Despite the presence of a sensitive protein in its constitution, the resulting gel presents interesting rheological properties (e.g., high mechanical strength and self-healing) and pH responsiveness due to the dynamic properties of the PBA–SHA link.
The latest development from the same authors comes in the form of a protein tag that allows for easy purification and, exploiting the PBA–SHA interaction, subsequent modification of proteins. In this work, the model protein lysozyme was site-selectively modified on one of its disulfide bridges with a vinyl sulfone rebridging agent bearing a PBA moiety, which allowed its straightforward purification on a carbohydrate-based column. Subsequently, the PBA-modified protein readily reacted with a SHA linker bearing a BODIPY dye, which allowed the evaluation of the stability and reversibility of the complex by microscale thermophoresis (Fig. 27).155
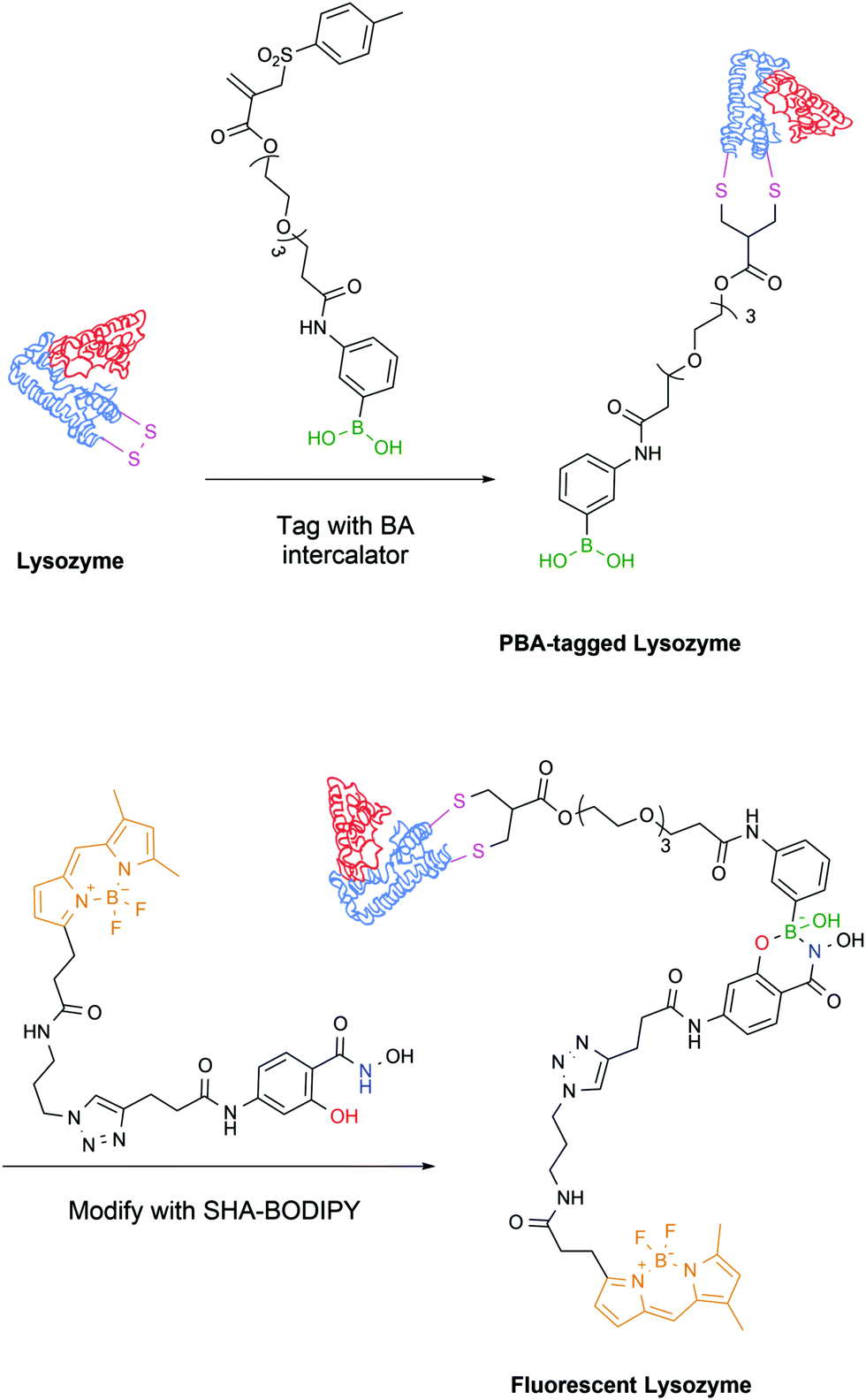 | ||
| Fig. 27 Lysozyme is modified with a rebridging agent, followed by complexation with SHA–BODIPY to yield a fluorescent conjugate. | ||
BA–SHA linkage has also found valid applications with smaller protein fragments, functioning as a linker unit for the attachment of a nucleic acid payload to tumour-targeting peptides.
Cristiano and co-workers reported the employment a PBA–SHA complex in the development of a non-viral approach for the targeted delivery of a DNA cationic polyplex.156 Gene therapy is based on the selective and efficient delivery of therapeutic genes into target populations of cells. Despite the advances in transfection and transduction technologies that improve the DNA expression in the target cells, most available techniques still use viral vectors to confer selectivity to the conjugate and to avoid the development of toxicity. To circumvent this limitation, the authors proposed the use of a CNGRC peptide, which is selective for CD13, a surface receptor over-expressed in tumour cells, as a targeting vector coupled via PBA–SHA bridging to a PEI–DNA polyplex. The mild conditions for the PBA–SHA complex formation allowed an easy functionalization of the peptide and the cationic polyplex, while maintaining the structural integrity of the vector (Fig. 28). By introducing a β-Gal plasmid into the vector, the transfection efficiency and biodistribution could be evaluated both in vitro and in vivo, through quantification of β-galactosidase expression. Additionally, by replacing the β-Gal plasmid with a Yellow Fluorescent Protein gene, it was possible to confirm by immunohistochemistry that the cellular target of CNGRC is indeed CD13 and that it is over-expressed only in tumour cells and endothelial cells of the tumour vasculature.
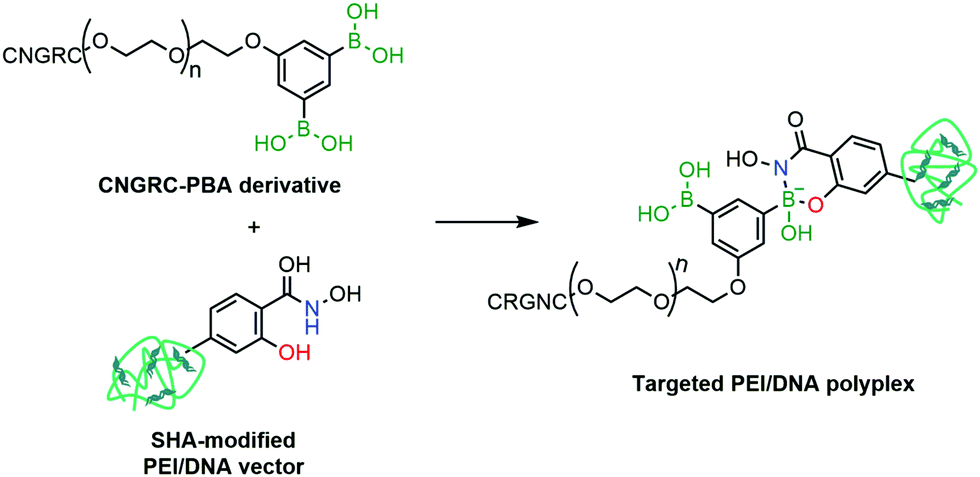 | ||
| Fig. 28 A CNGRC targeting peptide is attached to a PEI/DNA polyplex through a SHA–PBA linkage for targeted gene delivery. | ||
Subsequently, the same group reported similar methodologies based on the PBA–SHA link to deliver p53-expressing genes to tumour cells’ nuclei157 and to evaluate prostate-specific membrane antigen as an effective target for treatment of prostate cancer.158
4.3. Boronated oximes and diazaborines
Many of the aforementioned technologies attained substantial success, in part, due to their ability to form reversible covalent bonds. However, for many applications, the formation of more stable interactions is desirable, and to achieve such stability, one possible alternative is the replacement of conventional amines with α-effect nucleophiles such as hydroxylamines and hydrazines to generate oxime and hydrazone analogues of iminoboronates. Owing to their stability and tuneable reversibility, hydrazones and oximes have been employed in the development of bioconjugates, especially for controlled release and targeted drug delivery including the FDA-approved antibody–drug conjugate Mylotarg.159–161 Nonetheless, they have found limited success beyond controlled release of drugs, being mostly hampered by slow reactions rates under physiological conditions. To expand the range of potential applications in chemical biology, new compounds and catalysts were explored to improve the reaction rates.In early studies, Jencks and co-workers reported that aniline was capable of improving the formation of oximes at acidic pH by 3.5-fold.162 These findings were only much later applied to neutral conditions, and it was confirmed that aniline reacts quickly with the carbonyl, forming a very reactive imine intermediate that is readily attacked by the hydroxylamine to yield the corresponding oxime with very fast reaction rates (280 min vs. 7.8 days uncatalyzed).163 However, extremely high catalyst loadings are necessary, rendering this method unusable for in vivo applications. Other recent advances in oxime/hydrazone formation catalysis are depicted in another review and will not be further detailed herein.159
Despite preliminary works by Crisalli164,165 and Gillingham166 on the impact of adjacent groups on the formation rates of hydrazines and oximes, the major breakthrough came with the discovery that adjacent BAs accelerate by several orders of magnitude the condensation of oximes. In accordance with the mechanism elucidated by density functional theory for iminoboronates,133 Gillingham and co-workers proposed that the Lewis acidity of a BA in the ortho position to an aromatic aldehyde could facilitate the key dehydration step, thus improving the overall reaction rate, even under neutral conditions (Fig. 29A).167 The authors reported that 2-FBBA and benzyl hydroxylamine reacted in a 1 to 1 ratio, at low micromolar concentrations and in neutral pH buffer. Surprisingly, the rate constant determined through a fluorescence quenching assay was 1.1 × 104 M−1 s−1, which is several orders of magnitude higher than the reported values for oxime formation at neutral pH and in line with the fastest reactions in bioconjugation. This extraordinary increase was confirmed to be specifically due to the BA being in the ortho position as the same experiment yielded poor results when the BA was positioned in the meta and para positions. Also, the vacant p orbital in boron is crucial for the acceleration since 2-formylbenzene trifluoroborate reacted more slowly with benzyl hydroxylamine than with the corresponding BA. The reaction rates were found to be consistent with a two-step mechanism where the hydrolysis of the trifluoroborate to the BA is the rate limiting step.
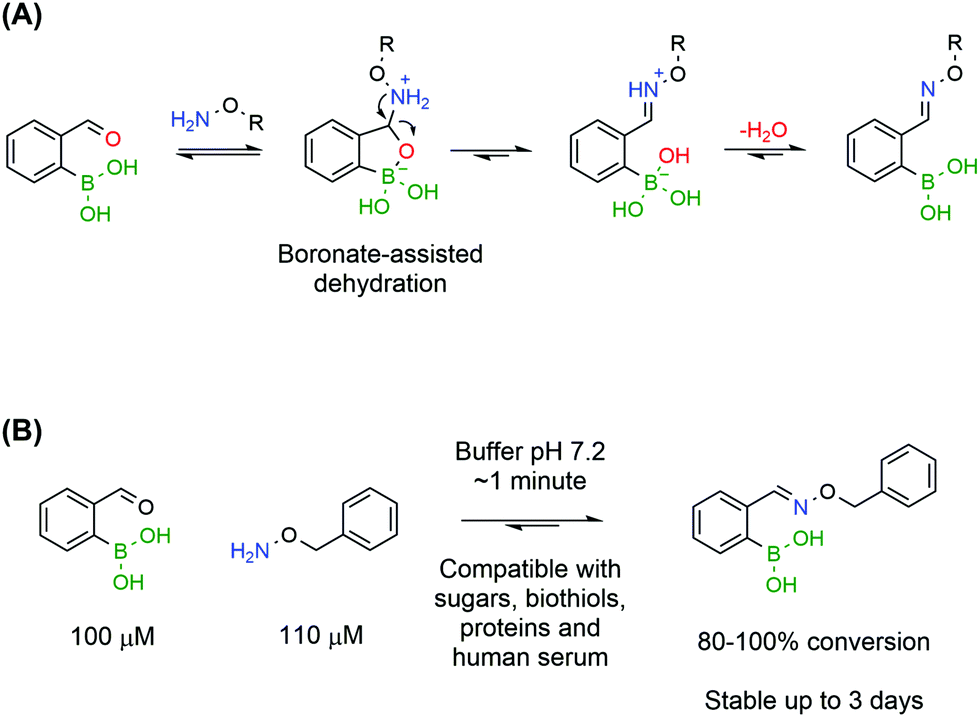 | ||
| Fig. 29 Boronated oximes: (A) oxime condensation is accelerated by the proximal BA, through a boronate-assisted dehydration; and (B) 2-FBBA and benzyl hydroxylamine react in buffer, pH 7.2, and in the presence of interfering biomolecules to yield the corresponding oxime in under 1 minute and with excellent conversion. | ||
The BA-assisted oxime condensation is a very robust reaction as it can be achieved with both aldehydes and ketones, although at slower rates with the latter, as well as with different and more complex hydroxylamines, including a hydroxylamine-bearing pentapeptide.167 This new class of compounds also inherits the hydrolytic stability of classical oximes and showed no relevant degradation after 3 days at neutral pH. Additionally, the reaction demonstrated an extraordinary tolerance to biological functional groups, maintaining the reaction efficiency in the presence of glutathione, sucrose, lysozyme and human serum, which is a good indicator of its bioorthogonality (Fig. 29). In the presence of human serum, oxidation of the boronic acid to the phenol was observed but the condensation was not compromised and the salicyloxime formed was stable.
Later work corroborated the fast oxime formation rates but produced different results regarding their stability.168 The oxime formed with 2-ABBA appears to be less stable than the one formed with 2-FBBA, which was confirmed by HPLC and UV absorption studies. This result was partially justified by 11B-NMR studies where the formation of an iminoboronate is visible only for 2-ABBA's oxime, which internally activates the imine for hydrolysis. The σ-donation effect of the methyl is also important as it increases the basicity of the imine nitrogen, rendering it more susceptible to nucleophilic attacks. Further works reported that ketoximes can rapidly equilibrate to the corresponding aldoximes in the presence of excess aldehyde.169
Subsequent studies demonstrated that 2-CBBA could also accelerate condensation of other α-nucleophiles such as hydrazines.168 2-ABBA was shown to react with acetylhydrazine and phenylhydrazine in neutral buffer with rate constants around 103 M−1 s−1, which is in the same range as oximes and other successful bioconjugation methodologies. BA-assisted hydrazone formation was tolerant to the presence of lysines, fructose, glutathione and bovine serum albumin and could be performed in small peptides and proteins while maintaining the fast reaction rates. Nevertheless, once again, the conjugate's stability was not satisfactory and hydrolysis appeared to be accelerated by the presence of the proximal BA.168 According to the authors, this characteristic can be explored in dynamic combinatorial chemistry where reversible covalent bonds are desirable to create virtual combinatorial libraries.170
An important advance was reported by Bane and co-workers, which demonstrated that hydrazones formed from 2-FBBA and 4-hydrazinylbenzoic acid could undergo a second internal dehydration step to form a stable boron–nitrogen heterocycle called 1,2-dihydro-1-hydroxy-2,3,1-benzodiazaborine.171
Diazaborines (DABs) are a class of boron–nitrogen heterocycles, which were initially reported in the early '60s172,173 and have since been employed essentially as antibiotics.174–178 Besides their pharmacological activity, DABs exhibit a skeleton with very interesting electronic properties due to their similarity to naphthalene.
The B–N covalent bond is a natural isostere of a C–C double bond as the nitrogen can donate its available electron pair to occupy the empty p-orbital of boron, forming a stable zwitterion. Despite its similarity to a C–C bond, the B–N bond offers interesting potentialities due to its intrinsic polarization, which has been extensively explored in materials chemistry.179 Additionally, analogues of aromatic compounds where a C–C bond is replaced with a B–N bond, such as DABs, also exhibit classical aromatic characteristics such as high stability. These aromatic properties, along with the very fast formation rates, make DABs very interesting candidates for in vivo chemistry (Fig. 30).
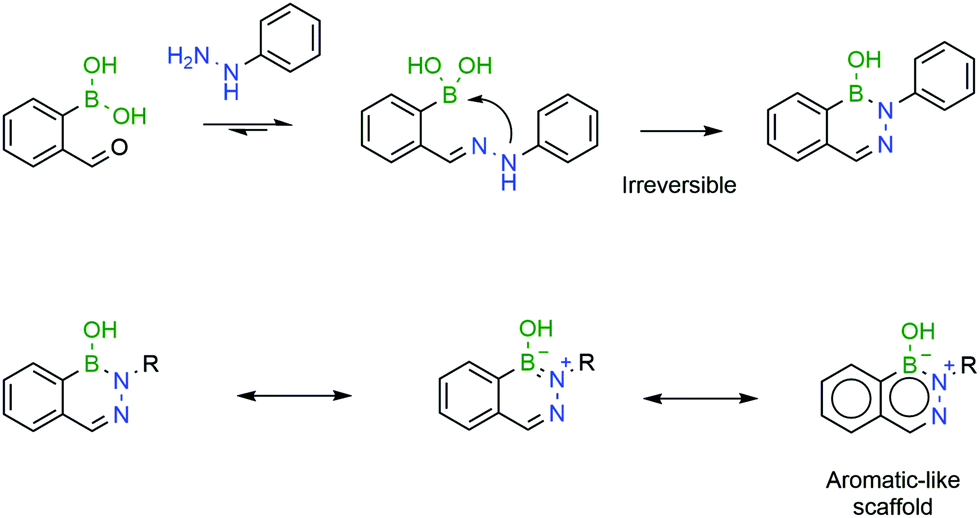 | ||
| Fig. 30 A second intramolecular attack renders the resulting DAB irreversible, due to its aromatic-like properties. | ||
Bane and co-workers reported that the reaction of 2-FBBA with 4-hydrazinylbenzoic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) at pH 7 and a low concentration proceeds extremely rapidly (>103 M−1 s−1) and yields a single product. However, the product was not the expected hydrazone, but the corresponding DAB. The rate-limiting step of the transformation was shown to be the internal dehydration step but, nevertheless, the reaction proceeded at very favourable rates. As expected, due to its aromatic-like character, the obtained DAB proved to be stable in solution over one month.171 Similar results were obtained by Gillingham's group, who reported that the reaction of 2-FBBA with phenylhydrazine proceeds well in human serum and in the presence of millimolar concentrations of glutathione and that DAB's formation is irreversible.180
:1 ratio) at pH 7 and a low concentration proceeds extremely rapidly (>103 M−1 s−1) and yields a single product. However, the product was not the expected hydrazone, but the corresponding DAB. The rate-limiting step of the transformation was shown to be the internal dehydration step but, nevertheless, the reaction proceeded at very favourable rates. As expected, due to its aromatic-like character, the obtained DAB proved to be stable in solution over one month.171 Similar results were obtained by Gillingham's group, who reported that the reaction of 2-FBBA with phenylhydrazine proceeds well in human serum and in the presence of millimolar concentrations of glutathione and that DAB's formation is irreversible.180
Later, Gao and co-workers demonstrated that semicarbazide can also react with 2-CBBA to give the corresponding DABs.181 The authors proposed that the inherent oxidative instability and toxicity of phenylhydrazines could be a limitation for their use in bioconjugation and presented semicarbazide as a stable, safer and easier-to-modify alternative. The semicarbazide–DAB formation rate is in line with the previous reports and the corresponding products were stable under biological conditions.
However, a detailed study uncovered that, depending on pH and concentration, the reaction of 2-CBBA with carbohydrazides can yield different products.182 If the reaction is performed at high millimolar concentrations in aqueous or organic solvents, the major product is the DAB anhydrous dimer. Under dilute conditions, at higher pHs (8–9) DAB is the obtained product, preferentially in the tetravalent boronate form. However, at lower pHs, mixtures of DAB and hydrazone can be observed. To avoid such problems, Bane and co-workers proposed the use of α-amino carbohydrazides. The amine group in the α position forms an extra B–N bond, which stabilizes the DAB, via a tricyclic structure, which was confirmed by X-ray crystallography. The resulting molecule was shown to be stable for several weeks under acidic and basic conditions (Fig. 31).
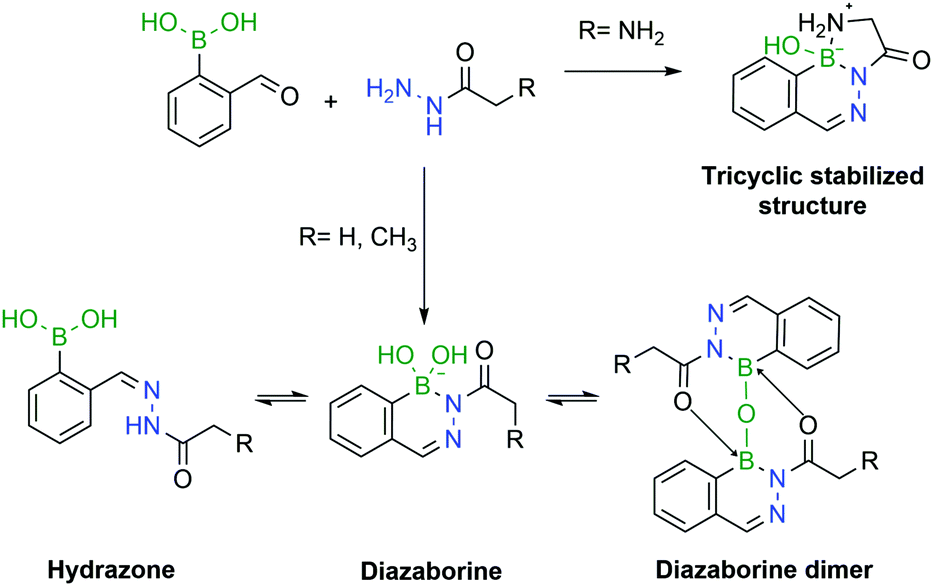 | ||
| Fig. 31 The pH is a crucial factor in the determination of the predominant species in solution. In the presence of an α-amino group, a tricyclic structure is formed, which locks the DAB conformation. | ||
With high formation rates, good tolerability to complex media and interesting stability, DAB displays all the necessary traits to be a useful technology for the construction of bioconjugates.
The first example of protein labelling using this technology was reported by Bane and co-workers in which bovine serum albumin (BSA) was modified with a fluorescent probe using DAB as a linker. BSA was covalently modified with a phenylhydrazine derivative through an N-hydroxysuccinimide (NHS) ester, and reacted with 2-FBBA modified with a coumarin at room temperature in phosphate buffer (pH 7). The resulting conjugate was analysed by fluorescence spectroscopy and SDS-PAGE, revealing that fluorescence could undoubtedly be observed on the modified protein (Fig. 32). This result demonstrated that not only are DABs a good option for the development of bioconjugates but their significant stability also allows downstream analysis even under harsh conditions (low pH of the SDS-PAGE resolving gel).171 A similar experiment was performed with the α-amino carbohydrazide in which BSA was functionalized with a fluorescent coumarin through DAB formation.182
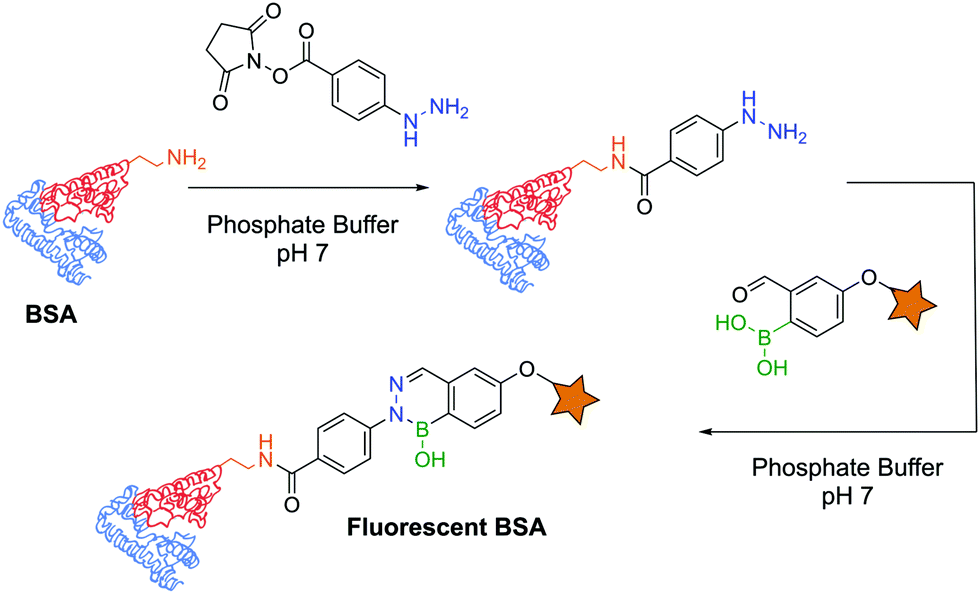 | ||
| Fig. 32 BSA functionalization with a fluorescent probe using a diazaborine linker under biocompatible conditions. | ||
Another example of DAB chemistry was reported by Gao and co-workers. Taking advantage of the dynamic rearrangement of bacterial peptidoglycan, the authors envisioned the possibility of incorporating a 2-ABBA-bearing amino acid, which could be subsequently labelled with a fluorescent semicarbazide.181 Starting from D-tyrosine, acetylation and borylation yielded a modified 2-ABBA-amino acid, which was exposed to different strains of pathogenic bacteria to be incorporated into their peptidoglycan (Fig. 33). Upon treatment with fluorescein–semicarbazide, these bacteria were analysed by fluorescence microscopy and flow cytometry. Different results were observed for each strain of bacteria, but in general the fluorescence was concentrated in the cell envelope, confirming the incorporation of the synthetic amino acid into the peptidoglycan's structure. Not only did this work demonstrate that DAB technology can operate under biocompatible conditions but it also achieved interesting conclusions concerning the dynamic processes occurring during peptidoglycan rearrangement. By analysing the fluorescence intensity observed for each strain of bacteria, it is possible to infer that E. coli's (Gram-negative) uptake of the synthetic amino acid is higher than those of S. aureus (Gram-positive) and P. aeruginosa (Gram-negative). This suggests that an underlying complex transport mechanism should be behind the uptake of the amino acids, which cannot be simply explained by the presence or absence of the cell wall.
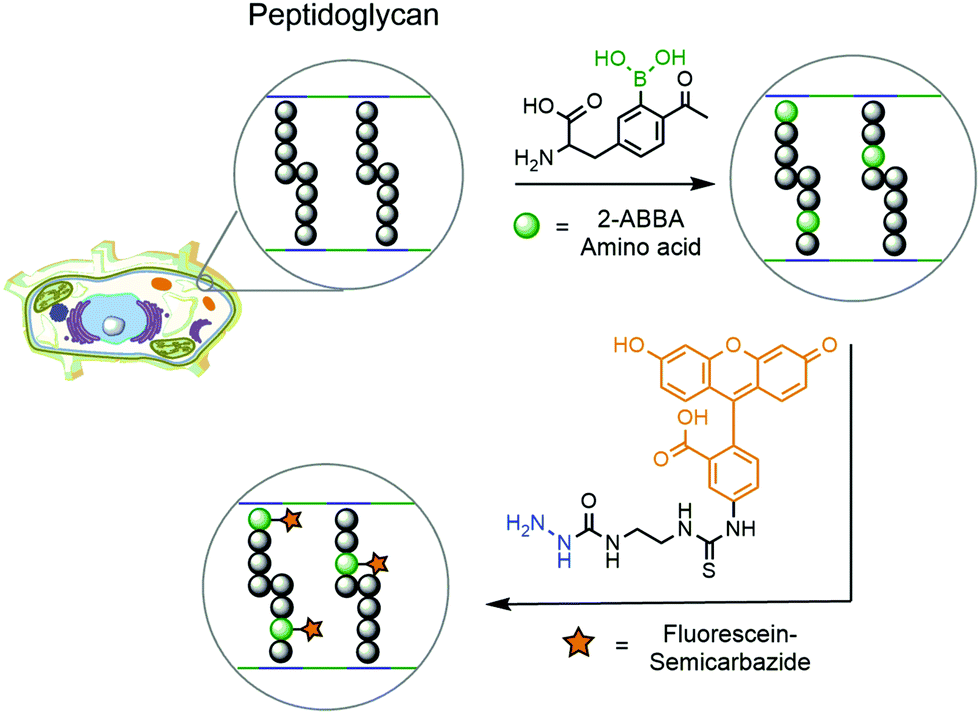 | ||
| Fig. 33 An unnatural 2-ABBA-amino acid is introduced into the bacterial cell wall taking advantage of the dynamic formation of peptidoglycan. This orthogonal handle is further modified with fluorescein–semicarbazide to generate fluorescent bacteria. | ||
Another appealing characteristic of DABs is their planar aromatic scaffold, leading Gillingham and co-workers to hypothesize that with the right tuning, DAB formation could produce fluorescent molecules. After designing a small library of compounds, the authors discovered that ortho-hydroxy phenylhydrazine would react with 4-dimethylamine 2-FBBA to yield a tricyclic planar compound with considerable fluorescence. The resulting DAB presented a 5-fold increase in fluorescence intensity and a higher quantum yield when compared with 4-dimethylamino 2-FBBA (Fig. 34).180
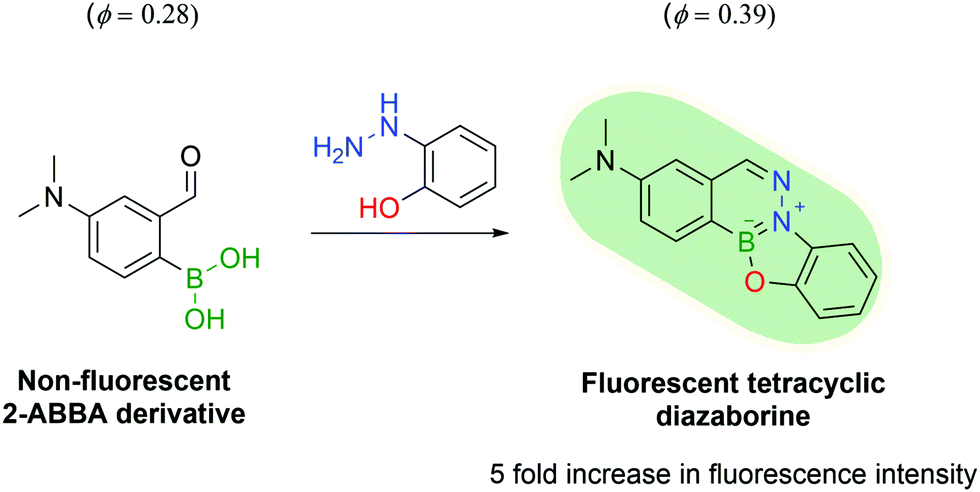 | ||
| Fig. 34 The reaction of a 2-ABBA derivative with 2-hydrazinyl phenol yields a fluorescent tetracyclic DAB. | ||
This first attempt at a turn-on fluorescent DAB paved the way for future research in this area and, one year later, Gao and co-workers reported a new fluorogenic DAB.183 By synthesizing a modified coumarin bearing a 2-ABBA side-chain, the fluorescence was quenched, and the authors envisioned that, upon reaction with semicarbazide, the molecule would regain its fluorescence. The resulting diazaborine showed a 7-fold increase in fluorescence intensity when compared to the starting reactants. This concept was used to tag bacteria using the aforementioned peptidoglycan reorganization model: an unnatural amino acid containing a semicarbazide side-chain was introduced into the bacterial wall, which could be further modified with the pro-fluorescent coumarin–ABBA to generate fluorescence in living bacteria (Fig. 35).
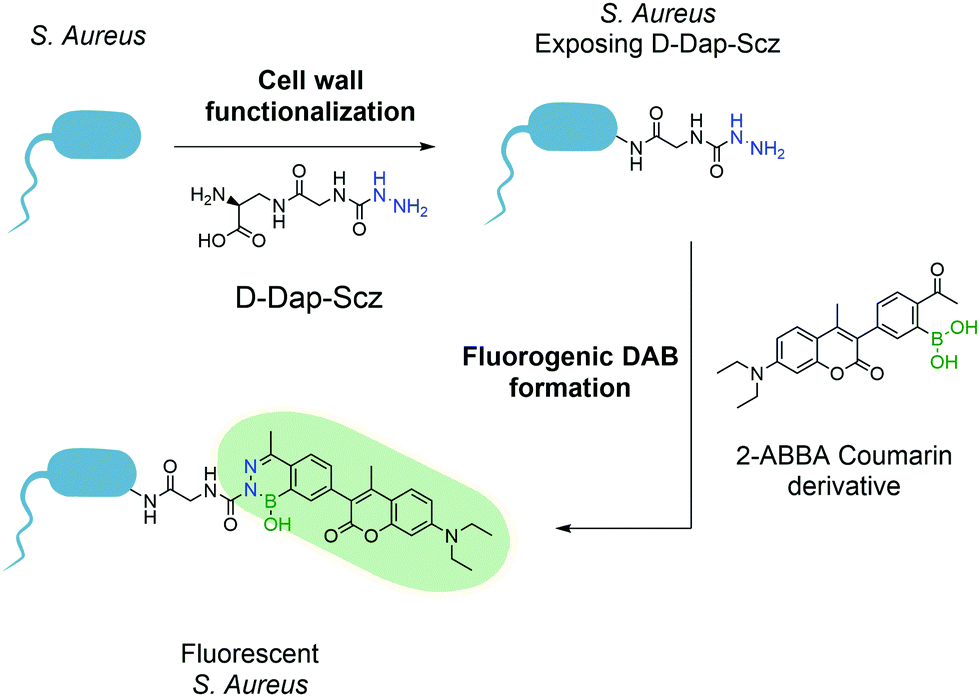 | ||
| Fig. 35 Incorporation of unnatural amino acid D-Dap-Scz enables the posterior functionalization with a coumarin derivative resulting in a fluorescent S. aureus. | ||
4.4. Multivalent boron complexes
Boron's exquisite coordination profile enables the construction of a multitude of scaffolds mostly based on oxygen and nitrogen ligands. Nonetheless, the previous examples have focused on direct coordination of solely one single or bidentated ligand to the boron centre. However, the possibility of coordinating multiple ligands at the same time opens up a new set of constructs with interesting novel characteristics and increased versatility due to the possible modular assembly.Initial advances were reported by James and co-workers with a three-component assembly of 2-FBBA, BINOL and a primary amine for NMR detection of enantiomeric purity.184–187 In the same line, Anslyn and co-workers reported an irreversible three-component reaction for peptide labelling in aqueous media. This one-pot assembly of 2-FBBA with a catechol and an N-hydroxylamine was shown to be fast, irreversible and orthogonal to other click reactions, yielding a BE that proved stable over 24 h over a wide range of pHs (1 to 13) and temperatures up to 50 °C.134 Using this system, the authors could label a small catechol-containing peptide with the fluorescent dye CF488A under aqueous conditions (Fig. 36).188
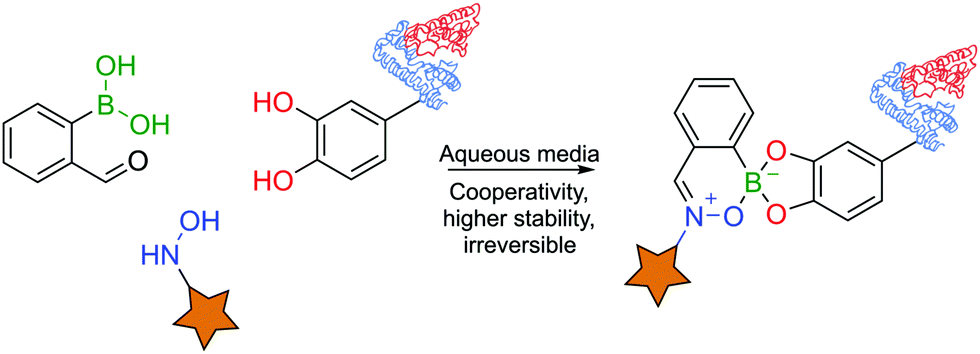 | ||
| Fig. 36 Irreversible three-component assembly of 2-FBBA, a hydroxylamine and a catechol. | ||
Inspired by the previous work, Shi and co-workers reported a three-component assembly for micelle functionalization and subsequent drug delivery. In this platform, 2-FBBA forms an iminoboronate with the amine group of poly(ethyleneglycol)-block-poly(L-lysine) (PEG-b-PLys), an important building unit for amphiphilic block-copolymer micelles. The third component is provided by the drug capecitabine (CAPE), which binds the boron centre with its vicinal OH groups, forming a stable 3-component assembly that is used for the functionalization of micelles. The resulting micelles proved to be stable under physiological conditions, while releasing their CAPE cargo when exposed to low pH or glutathione, attesting to this technology as a valid strategy for micelle-based drug delivery that circumvents the difficulties in loading these structures with hydrophilic drugs.189
Using a similar strategy, the same group reported a stimuli-sensitive hydrogel as a self-assembled multi-component supramolecular structure composed of an endogenous guanosine, 2-FBBA, tris(2-aminoethyl)amine and a cationic potassium core. Iminoboronates were introduced into the structure to facilitate the formation of the hydrogel and to allow a controlled hydrogel dissociation due to the dynamic interactions upon pH acidification and exposure to saccharide structures (Fig. 37).190 According to the authors, these structures can be promising candidates for future drug delivery applications.
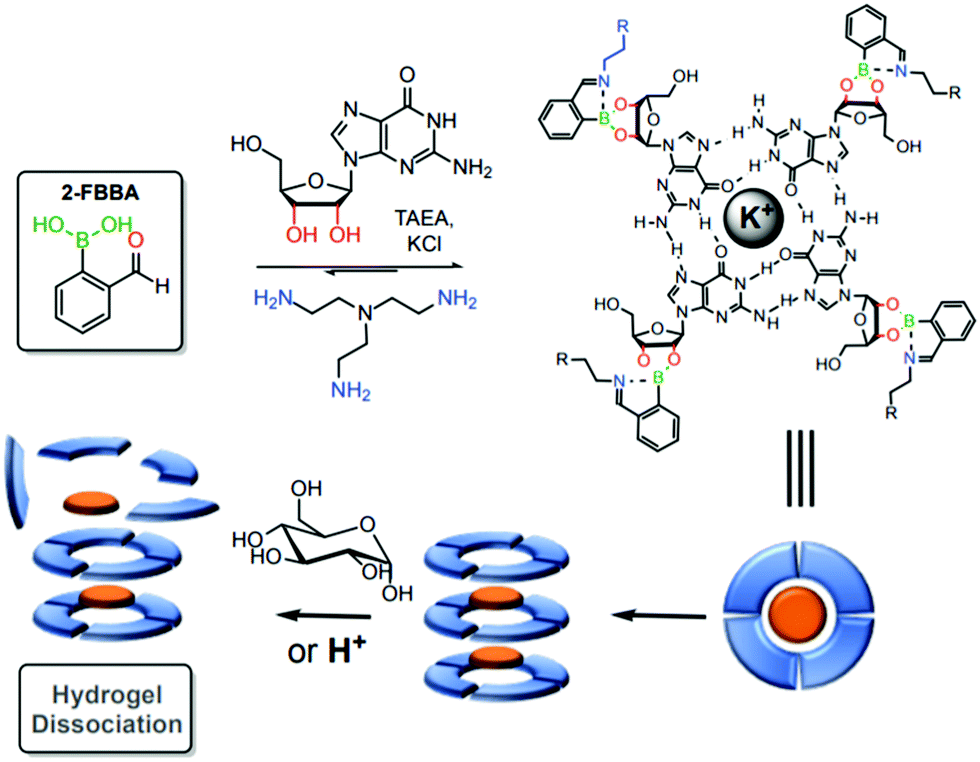 | ||
| Fig. 37 Construction of a responsive hydrogel through the reaction between 2-FBBA, guanosine and tris(2-aminoethyl)amine. This hydrogel is dissociated under acidic conditions and in the presence of high carbohydrate concentrations. | ||
A novel targeting drug conjugate reported by Gois and co-workers consists of a modular core obtained through a one-pot reaction of BAs with aminophenols and salicyl aldehydes.191 These constructs showed good stability in physiological medium, while maintaining GSH-mediated reversibility. The modular nature of these platforms allowed the straightforward assembly of a conjugate bearing a PEG unit, a folic acid targeting unit and the cytotoxic drug Bortezomib as the active payload. This construct showed high selectivity in vitro against folate-positive MDA-MB-231 cells with nanomolar IC50 values. The intracellular delivery of the cargo was confirmed by the synthesis of an analogous construct bearing, instead of Bortezomib, a self-immolative BA derivative of coumarin as the payload, whose release was followed by confocal microscopy (Fig. 38).
 | ||
| Fig. 38 Three-component assembly of modular cytotoxic platforms based on BAs. (A) Building blocks used in the assembly of the multicomponent construct. (B) Representative example of a targeting cytotoxic platform based on multicomponent assembly. | ||
Moreover, Gois and co-workers envisioned the possibility of generating a similar fluorescent construct through a one-pot reaction. The conjugation of BAs with Schiff-base ligands enabled the construction of innovative boronic acid salicylidenehydrazone (BASHY) dyes with suitable structural and photophysical properties for live cell bioimaging applications that can be fine-tuned with simple structural modifications.192–196 These dyes were employed by Gois, Bernardes and co-workers to site-selectively label Annexin V, an early marker of apoptosis, through maleimide technology for the detection of apoptotic cells. The BASHY dye maintained its photochemical and photophysical properties while attached to the biomolecules and the conjugate successfully targeted and detected the apoptotic cells, highlighting BASHY's utility as a promising fluorescent marker for biological imaging (Fig. 39).197
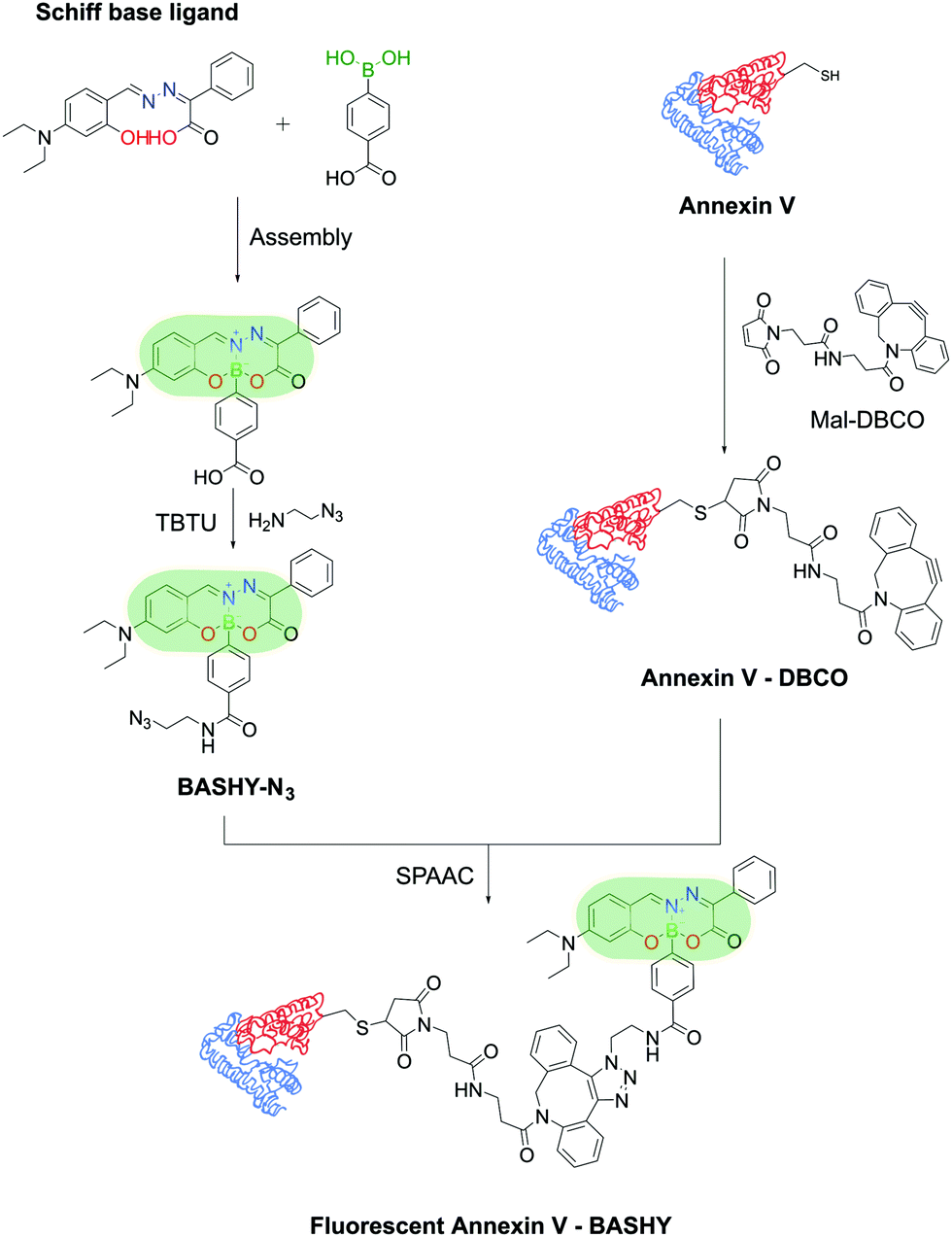 | ||
| Fig. 39 Reaction of a Schiff-base ligand and a functionalized PBA results in a fluorescent BASHY dye that was used to functionalize Annexin V and detect apoptotic cells. | ||
5. Conclusions and outlook
In this review, we summarized the recent applications of BAs in the synthesis of therapeutically useful bioconjugates, focusing on the molecular mechanisms enabled by the BA function when incorporated into the bioconjugate structure as the payload and as the bioconjugation warhead and when used as part of the linker unit.The Lewis acidity of BA, as a payload, allows a direct interaction with nucleophilic residues at the active site of therapeutically important enzymes. This approach has been extensively explored to prepare inhibitors for different proteases, which culminated in the discovery of Bortezomib and Ixazomib, two FDA-approved proteasome inhibitors used in the clinic for treatment of multiple myeloma. The chemical derivatization of the BA structure often enables the modulation of the drug affinity for a specific target. However, BA's promiscuous reactivity with many endogenous molecules has hampered its wider use, mostly due to its pharmacokinetic and toxicity concerns. Because of this, the discovery of new boron based therapeutics will certainly require the search for new strategies to control the reactivity of this function. In this context, BEs and boron heterocycles offer an excellent platform to discover new active molecules as they enable specific mechanisms to modulate the stereo-electronic properties of the boron centre.
Due to their ability to form reversible covalent bonds with diols, BAs have been used to target overexpressed glycans on cell surfaces and to promote the targeted delivery of cytotoxic drugs, proteins and gene vectors to cancer cells. Despite these promising results, the transient formation of BEs often lacks the affinity and selectivity required for a precise targeting, raising concerns whether these methods may develop off-target toxicity. Therefore, future developments in this area will probably focus on improving the affinity and stability of the BE, which may require the functionalization of the biological target with an unnatural handle that reacts selectively with a BA or the discovery of new BA reagents that exhibit high and specific affinities for naturally occurring carbohydrates.
The discovery that 2-formyl (or acetyl)-benzene BAs can efficiently promote the functionalization of biomolecules via the formation of B–N stabilized imines (iminoboronates) triggered much interest in the use of this method to modify the lysine and N-terminal residues of proteins and phospholipid derivatives. This bioconjugation method is particularly interesting because it is selective towards primary amines, exhibits fast reaction kinetics and is reversible in the presence of endogenous molecules like glutathione. Despite this success, the impossibility of controlling the site of modification and the fragile construct's stability have limited the use of this technology in the construction of reversible homogeneous conjugates. Recent advances in this area explored the use of iminoboronates as intermediates for the functionalization of N-terminal cysteines. This method showcased for the first time the potential use of BAs for the site-selective functionalization of peptides. Nevertheless, concerns regarding the stability of the constructs still persist, and clearly much work is still necessary to unravel bioconjugation methods that use BAs in the assembly of homogeneous bioconjugates for in vivo applications.
The linker technology is instrumental in controlling the fundamental properties of bioconjugates such as the required stability in circulation and the stimuli-responsiveness at the site of disease. In this context, the coordinative profile of BAs can be explored as an exceptional platform to develop functional linkers for the construction of bioconjugates. In this regard N,O-iminoboronates and the formation of BEs using salicyl hydroxamic acids have been explored to design linkers that are sensitive to acidic environments. Moreover, the condensation of 2-formyl (or acetyl)-benzene BAs with α-nucleophiles like hydrazines generates diazaborines, which have been shown to display very good stability under physiological conditions. BAs also engage in multicomponent reactions which allowed the assembly of reversible multivalent heterocycles that were used in the selective delivery of Bortezomib to cancer cells. These examples clearly demonstrate the potential of exploring BAs in this context, though the use of BAs as linkers in therapeutically useful conjugates still needs to be demonstrated in in vivo scenarios.
Advances in the molecular biology of complex diseases offer first-hand insights into new therapeutic targets and their molecular mechanisms which can be explored to develop innovative therapies such as bioconjugates. Extensive research on the design of these hybrid materials revealed that their therapeutic efficacy is highly dependent on the biological properties of the functional components as well as the chemistries used to connect them. Therefore, as shown throughout this review, the unique biological properties of BAs and the dynamic coordinative profile of this function will most certainly have an important impact on the discovery of future therapeutic bioconjugates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of Fundação para a Ciência e Tecnologia (FCT) Portugal (grants: PTDC/QEQ-QOR/1434/2014, PTDC/QUI-QOR/29967/2017, UID/DTP/04138/2013, SAICTPAC/0019/2015 (iMed.ULisboa), UID/QUI/00100/2013, SFRH/BPD/103172/2014, SFRH/BPD/115442/2016, PD/BD/128239/2016 – a MedChemTrain PhD grant to JPMA) and Marie-Sklodowska Curie ITN ProteinConjugates (MSCA-ITN-2015-ETN-675007). We thank Ricardo Lopes for the design of the graphical abstract and some of the figures present in the review.References
- D. G. Hall and B. Akgun, Angew. Chem., Int. Ed., 2018, 57, 13028–13044 CrossRef PubMed.
- S. Cambray and J. Gao, Acc. Chem. Res., 2018, 51, 2198–2206 CrossRef CAS PubMed.
- A. Bandyopadhyay and J. Gao, Curr. Opin. Chem. Biol., 2016, 34, 110–116 CrossRef CAS PubMed.
- Z. T. Ball, Acc. Chem. Res., 2019, 52(3), 566–575 CrossRef CAS PubMed.
- M. W. Tibbitt and B. Marco-Dufort, Mater. Today Chem., 2019, 12, 16–33 CrossRef.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrinyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- S. L. Diemer, M. Kristensen, B. Rasmussen, S. R. Beeren and M. Pittelkow, Int. J. Mol. Sci., 2015, 16, 21858–21872 CrossRef CAS.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- R. F. Barth, P. Mi and W. Yang, Cancer Commun., 2018, 35, 1–15 Search PubMed.
- R. F. Barth, Z. Zhang and T. Liu, Cancer Commun., 2018, 36, 1–7 Search PubMed.
- K. Nedunchezhian, N. Aswath, M. Thiruppathy and S. Thirugnanamurthy, J. Clin. Diagn. Res., 2016, 10, 1–4 CrossRef CAS.
- J. Hiratsuka, N. Kamitani, R. Tanaka, E. Yoden, R. Tokiya and M. Suzuki, Cancer Commun., 2018, 38, 1–10 CrossRef PubMed.
- R. F. Barth, J. A. Coderre, M. G. H. Vicente and T. E. Blue, Clin. Cancer Res., 2005, 11, 3987–4003 CrossRef CAS PubMed.
- M. Suzuki, I. Kato, T. Aihara, J. Hiratsuka, K. Yoshimura, M. Niimi, Y. Kimura, Y. Ariyoshi, S. Haginomori, Y. Sakurai, Y. Kinashi and S. Masunaga, J. Radiat. Res., 2014, 55, 146–153 CrossRef CAS PubMed.
- L. W. Wang, Y. Wan, H. Liu, F. I. Chou and S. H. Jiang, Cancer Commun., 2018, 37, 1–7 Search PubMed.
- R. L. Moss, Appl. Radiat. Isot., 2014, 88, 2–11 CrossRef CAS PubMed.
- K. Ono, Ther. Radiol. Oncol., 2018, 2, 1–7 CrossRef.
- H. R. Mirzaei, A. Sahebkar, R. Salehi, J. S. Nahand, E. Karimi, M. R. Jaafari and H. Mirzaei, J. Cancer Res. Ther., 2016, 12, 520–525 CrossRef CAS PubMed.
- C. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784–792 CrossRef CAS PubMed.
- T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS PubMed.
- J. Lotze, U. Reinhardt, O. Seitz and A. G. Beck-Sickinger, Mol. BioSyst., 2016, 12, 1731–1745 RSC.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- D. A. Shannon and E. Weerapana, Curr. Opin. Chem. Biol., 2015, 24, 18–26 CrossRef CAS PubMed.
- J. M. J. M. Ravasco, H. M. F. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- K. Renault, J. W. Fredy, P. Renard and C. Sabot, Bioconjugate Chem., 2018, 29, 2497–2513 CrossRef CAS.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 651–653 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CrossRef CAS.
- S. Davies, B. J. Stenton and G. J. L. Bernardes, Chimia, 2018, 72, 771–776 CrossRef CAS PubMed.
- W. K. Chow, O. Y. Yuen, P. Y. Choy, C. M. So, C. P. Lau, W. T. Wong and F. Y. Kwong, RSC Adv., 2013, 3, 12518–12539 RSC.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- W. Yang, X. Gao and B. Wang, Med. Res. Rev., 2003, 23, 346–368 CrossRef CAS PubMed.
- P. C. Trippier and C. McGuigan, MedChemComm, 2010, 1, 183–198 RSC.
- S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC.
- D. B. Diaz and A. K. Yudin, Nat. Chem., 2017, 9, 731–742 CrossRef CAS PubMed.
- F. Yang, M. Zhu, J. Zhang and H. Zhou, MedChemComm, 2018, 9, 201–211 RSC.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS.
- Protein Data Bank, https://www.rcsb.org/.
- B. E. Elewski, R. Aly, S. L. Baldwin, R. F. González Soto, P. Rich, M. Weisfeld, H. Wiltz, L. T. Zane and R. Pollak, J. Am. Acad. Dermatol., 2015, 73, 62–69 CrossRef CAS PubMed.
- T. Akama, S. J. Baker, Y.-K. Zhang, V. Hernandez, H. Zhou, V. Sanders, Y. Freund, R. Kimura, K. R. Maples and J. J. Plattner, Bioorg. Med. Chem. Lett., 2009, 19, 2129–2132 CrossRef CAS PubMed.
- S. J. Hecker, K. R. Reddy, M. Totrov, G. C. Hirst, O. Lomovskaya, D. C. Griffith, P. King, R. Tsivkovski, D. Sun, M. Sabet, Z. Tarazi, M. C. Clifton, K. Atkins, A. Raymond, K. T. Potts, J. Abendroth, S. H. Boyer, J. S. Loutit, E. E. Morgan, S. Durso and M. N. Dudley, J. Med. Chem., 2015, 58, 3682–3692 CrossRef CAS.
- J. Adams and M. Kauffman, Cancer Invest., 2004, 22, 304–311 CrossRef CAS.
- H. Avet-Loiseau, N. J. Bahlis, W. J. Chng, T. Masszi, L. Viterbo, L. Pour, P. Ganly, A. Palumbo, M. Cavo, C. Langer, A. Pluta, A. Nagler, S. Kumar, D. Ben-Yehuda, S. V. Rajkumar, J. San-Miguel, D. Berg, J. Lin, H. Van De Velde, D. L. Esseltine, A. di Bacco, P. Moreau and P. G. Richardson, Blood, 2017, 130, 2610–2618 CrossRef CAS PubMed.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- M. P. Curran and K. McKeage, Drugs, 2009, 69, 859–888 CrossRef CAS PubMed.
- M. Shirley, Drugs, 2016, 76, 405–411 CrossRef CAS PubMed.
- J. P. M. António, L. M. Gonçalves, R. C. Guedes, R. Moreira and P. M. P. Gois, ACS Omega, 2018, 3, 7418–7423 CrossRef PubMed.
- E. Kupperman, E. C. Lee, Y. Cao, B. Bannerman, M. Fitzgerald, A. Berger, J. Yu, Y. Yang, P. Hales, F. Bruzzese, J. Liu, J. Blank, K. Garcia, C. Tsu, L. Dick, P. Fleming, L. Yu, M. Manfredi, M. Rolfe and J. Bolen, Cancer Res., 2010, 70, 1970–1980 CrossRef CAS PubMed.
- E. F. Pai, R. C. Hynes, J. B. Jones, V. Martichonok, V. S. Stoll and B. T. Eger, Biochemistry, 2002, 37, 451–462 Search PubMed.
- E. Tsilikounas, C. Kettner and W. W. Bachovchin, Biochemistry, 1992, 31, 12839–12846 CrossRef CAS PubMed.
- E. Tsilikounas, C. A. Kettner and W. W. Bachovchin, Biochemistry, 1993, 32, 12651–12655 CrossRef CAS PubMed.
- L. H. Takahashi, R. Radhakrishnan, R. E. Rosenfield and E. F. Meyer, Biochemistry, 1989, 28, 7610–7617 CrossRef CAS PubMed.
- R. Bone, D. Frank, C. A. Kettner and D. A. Agard, Biochemistry, 1989, 28, 7600–7609 CrossRef CAS PubMed.
- R. Bone, A. B. Shenvi, C. A. Kettner and D. A. Agard, Biochemistry, 2005, 26, 7609–7614 CrossRef.
- W. W. Bachovchin, W. Y. L. Wong, S. Farr-jones, A. B. Shenvi and C. A. Kettner, Biochemistry, 1988, 27, 7689–7697 CrossRef CAS.
- Z. Chen, Z. Tian, K. Kallio, A. L. Oleson, A. Ji, D. Borchardt, D. Jiang, S. J. Remington and H. Ai, J. Am. Chem. Soc., 2016, 138, 4900–4907 CrossRef CAS PubMed.
- N. Aykin-Burns, I. M. Ahmad, Y. Zhu, L. W. Oberley and D. R. Spitz, Biochem. J., 2009, 418, 29–37 CrossRef CAS PubMed.
- L. Wang, S. Xie, L. Ma, Y. Chen and W. Lu, Eur. J. Med. Chem., 2016, 116, 84–89 CrossRef CAS PubMed.
- D. Lee, S. Park, S. Bae, D. Jeong, M. Park, C. Kang, W. Yoo, M. A. Samad, Q. Ke, G. Khang and P. M. Kang, Sci. Rep., 2015, 5, 1–13 CAS.
- L. Du, M. Li, S. Zheng and B. Wang, Tetrahedron Lett., 2008, 49, 3045–3048 CrossRef CAS.
- G. Cao, W. Mao, J. Han, Z. Zhao, M. Gao, X. Xu, S. Wang, H. Ye and C. Chu, Bioorg. Chem., 2018, 81, 362–366 CrossRef PubMed.
- K. Xu, L. He, X. Yang, Y. Yang and W. Lin, Analyst, 2018, 143, 3555–3559 RSC.
- D. Andina, J. C. Leroux and P. Luciani, Chem. – Eur. J., 2017, 23, 13549–13573 CrossRef CAS.
- Y. Liu, L. Bai, Y. Li, Y. Ni, C. Xin, C. Zhang, J. Liu, Z. Liu, L. Li and W. Huang, Sens. Actuators, B, 2019, 279, 38–43 CrossRef CAS.
- L. Zhang, M. Qian, J. Xia, L. Chen, H. Cui, Y. Xia, L. Xu, J. Wang and X. Peng, J. Photochem. Photobiol., A, 2019, 370, 12–17 CrossRef CAS.
- A. E. Hargrove, R. N. Reyes, I. Riddington, E. V. Anslyn and J. L. Sessler, Org. Lett., 2010, 12, 4804–4807 CrossRef CAS.
- L. Du Ý, N. Ni Ý, M. Li and B. Wang, Tetrahedron Lett., 2010, 51, 1152–1154 CrossRef PubMed.
- X. Q. Zhan, B. Y. Su, H. Zheng and J. H. Yan, Anal. Chim. Acta, 2010, 658, 175–179 CrossRef CAS PubMed.
- E. V. Lampard, A. C. Sedgwick, X. Sun, K. L. Filer, S. C. Hewins, G. Kim, J. Yoon, S. D. Bull and T. D. James, ChemistryOpen, 2018, 7, 262–265 CrossRef CAS.
- Y. Liu, J. Niu, J. Nie, F. Meng and W. Lin, New J. Chem., 2017, 41, 3320–3325 RSC.
- D. Srikun, E. W. Miller, D. W. Domaille and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 4596–4597 CrossRef CAS PubMed.
- A. R. Lippert, T. Gschneidtner and C. J. Chang, Chem. Commun., 2010, 46, 7510–7512 RSC.
- S. Lu, C. Jia, H. Huang, J. Tang and Y. Han, J. Fluoresc., 2015, 26, 121–127 Search PubMed.
- E. Lindberg and N. Winssinger, ChemBioChem, 2016, 1, 1612–1615 CrossRef PubMed.
- J. Xu, J. Zhai, Y. Xu, J. Zhu, Y. Qin and D. Jiang, Analyst, 2016, 141, 2380–2383 RSC.
- X. Shi, Z. Wang, X. Wang, Y. Chen and Z. Lu, Anal. Chem., 2017, 89, 5278–5284 CrossRef PubMed.
- J. L. M. Jourden and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 6795–6797 CrossRef PubMed.
- E. J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- S. D. Bull, M. G. Davidson, J. M. H. van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS PubMed.
- H. G. Kuivila, A. H. Keough and E. J. Soboczenski, J. Org. Chem., 1954, 19, 780–783 CrossRef CAS.
- A. Finch, P. J. Gardner, P. M. McNamara and G. R. Wellum, J. Chem. Soc. A, 1970, 3339–3345 RSC.
- W. L. A. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 CrossRef CAS.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- W. Zhai, X. Sun, T. D. James and J. S. Fossey, Chem. – Asian J., 2015, 10, 1836–1848 CrossRef CAS PubMed.
- T. D. James, M. D. Phillips and S. Shinkai, Boronic Acids in Saccharide Recognition, The Royal Society of Chemistry, 2006 Search PubMed.
- D. G. Hall, Structure, Properties, and Preparation of Boronic Acid Derivatives. Overview of Their Reactions and Applications, John Wiley & Sons, Ltd, 2006 Search PubMed.
- B. Pappin, M. J. Kiefel and T. A. Houston, Comprehensive Studies on Glycobiology and Glycotechnology, IntechOpen, 2012, pp. 37–54 Search PubMed.
- X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC.
- X. Wu, X.-X. Chen and Y.-B. Jiang, Analyst, 2017, 142, 1403–1414 RSC.
- W. J. Ramsay and H. Bayley, Angew. Chem., Int. Ed., 2018, 57, 2841–2845 CrossRef CAS PubMed.
- T. Hoeg-Jensen, S. Havelund, P. K. Nielsen and J. Markussen, J. Am. Chem. Soc., 2005, 127, 6158–6159 CrossRef CAS PubMed.
- L. Zhao, Q. Huang, Y. Liu, Q. Wang, L. Wang, S. Xiao, F. Bi and J. Ding, Materials, 2017, 10, 1–14 Search PubMed.
- M. Zuo, W. Qian, Z. Xu, W. Shao, X. Y. Hu, D. Zhang, J. Jiang, X. Sun and L. Wang, Small, 2018, 14, 1–10 Search PubMed.
- N. D. Winblade, I. D. Nikolic, A. S. Hoffman and J. A. Hubbell, Biomacromolecules, 2000, 1, 523–533 CrossRef CAS PubMed.
- Y. Tian and H. Zhang, Proteomics, 2013, 13, 504–511 CrossRef CAS PubMed.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- A. V. Everest-dass, E. S. X. Moh, C. Ashwood, A. M. M. Shathili and N. H. Packer, Expert Rev. Proteomics, 2018, 15, 341–352 CrossRef CAS.
- J. Munkley, Oncol. Lett., 2019, 17, 2569–2575 Search PubMed.
- S. Hakomori, Cancer Res., 1996, 56, 5309–5318 CAS.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS PubMed.
- S. Deshayes, H. Cabral, T. Ishii, Y. Miura, S. Kobayashi, T. Yamashita, A. Matsumoto, Y. Miyahara, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2013, 135, 15501–15507 CrossRef CAS PubMed.
- X. Zhang, D. S. Alves, J. Lou, S. D. Hill, F. N. Barrera and M. D. Best, Chem. Commun., 2018, 54, 6169–6172 RSC.
- Y. Zhang, J. J. Røise, K. Lee, J. Li and N. Murthy, Curr. Opin. Biotechnol., 2018, 52, 25–31 CrossRef CAS PubMed.
- A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25, 1602–1608 CrossRef CAS.
- G. A. Ellis, M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 3631–3634 CrossRef CAS PubMed.
- M. N. Levine and R. T. Raines, Chem. Sci., 2012, 3, 2412–2420 RSC.
- K. A. Andersen, T. P. Smith, J. E. Lomax and R. T. Raines, ACS Chem. Biol., 2016, 11, 319–323 CrossRef CAS PubMed.
- Y.-L. Yang, Y.-P. Lee, Y.-L. Yang and P.-C. Lin, ACS Chem. Biol., 2014, 9, 390–397 CrossRef CAS PubMed.
- T. L. Halo, J. Appelbaum, E. M. Hobert, D. M. Balkin and A. Schepartz, J. Am. Chem. Soc., 2009, 131, 438–439 CrossRef CAS PubMed.
- E. Montanari, A. Gennari, M. Pelliccia, L. Manzi, R. Donno, N. J. Oldham, A. Macdonald and N. Tirelli, Bioconjugate Chem., 2018, 29, 2550–2560 CrossRef CAS PubMed.
- M. Rosenberg, J. L. Wiebers and P. T. Gilham, Biochemistry, 1972, 11, 3623–3628 CrossRef CAS.
- P. R. Westmark and B. D. Smith, J. Pharm. Sci., 1996, 85, 266–269 CrossRef CAS PubMed.
- R. Tuytten, F. Lemière, W. Van Dongen, E. Witters, E. L. Esmans, R. P. Newton and E. Dudley, Anal. Chem., 2008, 80, 1263–1271 CrossRef CAS PubMed.
- A. R. Martin, J.-J. Vasseur and M. Smietana, Chem. Soc. Rev., 2013, 42, 5684–5713 RSC.
- M. Naito, T. Ishii, A. Matsumoto, K. Miyata, Y. Miyahara and K. Kataoka, Angew. Chem., Int. Ed., 2012, 51, 10751–10755 CrossRef CAS PubMed.
- Q. Peng, F. Chen, Z. Zhong and R. Zhuo, Chem. Commun., 2010, 46, 5888–5890 RSC.
- M. Neu, D. Fischer and T. Kissel, J. Gene Med., 2005, 7, 992–1009 CrossRef CAS PubMed.
- E. Brustad, M. L. Bushey, J. W. Lee, D. Groff, W. Liu and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 8220–8223 CrossRef CAS PubMed.
- C. C. Liu, A. V. Mack, E. M. Brustad, J. H. Mills, D. Groff, V. V. Smider and P. G. Schultz, J. Am. Chem. Soc., 2009, 131, 9616–9617 CrossRef CAS PubMed.
- K. Wals and H. Ovaa, Front. Chem., 2014, 2, 15 Search PubMed.
- F. Wang, W. Niu, J. Guo and P. G. Schultz, Angew. Chem., Int. Ed., 2012, 51, 10132–10135 CrossRef CAS PubMed.
- Z. J. Chen, W. Ren, Q. E. Wright and H. W. Ai, J. Am. Chem. Soc., 2013, 135, 14940–14943 CrossRef CAS PubMed.
- B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2016, 55, 3909–3913 CrossRef CAS PubMed.
- B. Akgun, C. Li, Y. Hao, G. Lambkin, R. Derda and D. G. Hall, J. Am. Chem. Soc., 2017, 139, 14285–14291 CrossRef CAS.
- P. Akkapeddi, S.-A. Azizi, A. M. Freedy, P. M. S. D. Cal, P. M. P. Gois and G. J. L. Bernardes, Chem. Sci., 2016, 7, 2954–2963 RSC.
- J. M. McFarland and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 13490–13491 CrossRef CAS PubMed.
- V. Raindlová, R. Pohl and M. Hocek, Chem. – Eur. J., 2012, 18, 4080–4087 CrossRef PubMed.
- M. Sánchez, H. Höpfl, M.-E. Ochoa, N. Farfán, R. Santillan and S. Rojas-Lima, Chem. – Eur. J., 2002, 8, 612–621 CrossRef.
- V. Barba, H. Höpfl, N. Farfán, R. Santillan, H. I. Beltran and L. S. Zamudio-Rivera, Chem. Commun., 2004, 2834–2835 RSC.
- V. Barba, R. Villamil, R. Luna, C. Godoy-Alcántar, H. Höpfl, H. I. Beltran, L. S. Zamudio-Rivera, R. Santillan and N. Farfán, Inorg. Chem., 2006, 45, 2553–2561 CrossRef CAS PubMed.
- P. M. S. D. Cal, J. B. Vicente, E. Pires, A. V. Coelho, L. F. Veiros, C. Cordeiro and P. M. P. Gois, J. Am. Chem. Soc., 2012, 134, 10299–10305 CrossRef CAS PubMed.
- B. M. Chapin, P. Metola, V. M. Lynch, J. F. Stanton, T. D. James and E. V. Anslyn, J. Org. Chem., 2016, 81, 8319–8330 CrossRef CAS PubMed.
- A. Bandyopadhyay and J. Gao, J. Am. Chem. Soc., 2016, 138, 2098–2101 CrossRef CAS PubMed.
- X. Liu, Z. Li, H. Xu, Y. Zhan, P. Ma, H. Chen and B. Jiang, Tetrahedron Lett., 2017, 58, 3101–3106 CrossRef CAS.
- P. M. S. D. Cal, R. F. M. Frade, V. Chudasama, C. Cordeiro, S. Caddick and P. M. P. Gois, Chem. Commun., 2014, 50, 5261–5263 RSC.
- P. M. S. D. Cal, R. F. M. Frade, C. Cordeiro and P. M. P. Gois, Chem. – Eur. J., 2015, 21, 8182–8187 CrossRef CAS PubMed.
- G. Akçay, M. A. Belmonte, B. Aquila, C. Chuaqui, A. W. Hird, M. L. Lamb, P. B. Rawlins, N. Su, S. Tentarelli, N. P. Grimster and Q. Su, Nat. Chem. Biol., 2016, 12, 931–936 CrossRef PubMed.
- S. Borsley and S. L. Cockroft, ACS Nano, 2018, 12, 786–794 CrossRef CAS PubMed.
- A. Bandyopadhyay, K. A. McCarthy, M. A. Kelly and J. Gao, Nat. Commun., 2015, 6, 6561 CrossRef CAS PubMed.
- K. Li and J. Gao, Synlett, 2017, 1913–1916 CAS.
- A. B. Draganov, K. Wang, J. Holmes, K. Damera, D. Wang, C. Dai and B. Wang, Chem. Commun., 2015, 51, 15180–15183 RSC.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589–4593 RSC.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 RSC.
- K. Li, C. Weidman and J. Gao, Org. Lett., 2018, 20, 20–23 CrossRef CAS PubMed.
- R. M. R. M. Lopes, A. E. Ventura, L. C. Silva, H. Faustino and P. M. P. Gois, Chem. – Eur. J., 2018, 24, 12495–12499 CrossRef CAS PubMed.
- M. L. Stolowitz, C. Ahlem, K. A. Hughes, R. J. Kaiser, E. A. Kesicki, G. Li, K. P. Lund, S. M. Torkelson and J. P. Wiley, Bioconjugate Chem., 2001, 12, 229–239 CrossRef CAS.
- J. P. Wiley, K. A. Hughes, R. J. Kaiser, E. A. Kesicki, K. P. Lund and M. L. Stolowitz, Bioconjugate Chem., 2001, 12, 240–250 CrossRef CAS.
- S. B. Y. Shin, R. D. Almeida, G. Gerona-Navarro, C. Bracken and S. R. Jaffrey, Chem. Biol., 2010, 17, 1171–1176 CrossRef CAS PubMed.
- M. Arzt, C. Seidler, D. Y. W. Ng and T. Weil, Chem. – Asian J., 2014, 9, 1994–2003 CrossRef CAS PubMed.
- D. Y. W. Ng, M. Arzt, Y. Wu, S. L. Kuan, M. Lamla and T. Weil, Angew. Chem., Int. Ed., 2014, 53, 324–328 CrossRef CAS PubMed.
- C. Seidler, D. Y. W. Ng, Y. Wu and T. Weil, Supramol. Chem., 2016, 28, 742–746 CrossRef CAS.
- C. Seidler, D. Y. W. Ng and T. Weil, Tetrahedron, 2017, 73, 4979–4987 CrossRef CAS.
- M. M. Zegota, T. Wang, C. Seidler, D. Y. Wah Ng, S. L. Kuan and T. Weil, Bioconjugate Chem., 2018, 29, 2665–2670 CrossRef CAS PubMed.
- S. Moffatt, S. Wiehle and R. J. Cristiano, Hum. Gene Ther., 2005, 16, 57–67 CrossRef CAS PubMed.
- S. Moffatt, S. Wiehle and R. J. Cristiano, Gene Ther., 2006, 13, 1512–1523 CrossRef CAS PubMed.
- S. Moffatt, C. Papasakelariou, S. Wiehle and R. Cristiano, Gene Ther., 2006, 13, 761–772 CrossRef CAS PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Chem. Soc. Rev., 2017, 46, 6638–6663 RSC.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- E. T. Kool, D. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed.
- P. Crisalli and E. T. Kool, Org. Lett., 2013, 15, 1646–1649 CrossRef CAS PubMed.
- P. Schmidt, L. Zhou, K. Tishinov, K. Zimmermann and D. Gillingham, Angew. Chem., Int. Ed., 2014, 53, 10928–10931 CrossRef CAS PubMed.
- P. Schmidt, C. Stress and D. Gillingham, Chem. Sci., 2015, 6, 3329–3333 RSC.
- A. Bandyopadhyay and J. Gao, Chem. – Eur. J., 2015, 21, 14748–14752 CrossRef CAS.
- D. Gillingham, Org. Biomol. Chem., 2016, 14, 7606–7609 RSC.
- M. Mondal and A. K. H. Hirsch, Chem. Soc. Rev., 2015, 44, 2455–2488 RSC.
- O. Dilek, Z. Lei, K. Mukherjee and S. Bane, Chem. Commun., 2015, 51, 16992–16995 RSC.
- M. J. S. Dewar and R. C. Dougherty, J. Am. Chem. Soc., 1964, 86, 433–436 CrossRef CAS.
- P. Tschampel and H. R. Snyder, J. Org. Chem., 1964, 29, 2168–2172 CrossRef CAS.
- G. Högenauer and M. Woisetschläger, Nature, 1981, 293, 662–664 CrossRef.
- C. Baldock, J. B. Rafferty, S. E. Sedelnikova, P. J. Baker, A. R. Stuitje, A. R. Slabas, T. R. Hawkes and D. W. Rice, Science, 1996, 274, 2107–2110 CrossRef CAS.
- D. Kanichar, L. Roppiyakuda, E. Kosmowska, M. A. Faust, K. P. Tran, F. Chow, E. Buglo, M. P. Groziak, E. A. Sarina, M. M. Olmstead, I. Silva and H. H. Xu, Chem. Biodiversity, 2014, 11, 1381–1397 CrossRef CAS PubMed.
- C. A. Jordan, B. A. Sandoval, M. V. Serobyan, D. H. Gilling, M. P. Groziak, H. H. Xu and J. L. Vey, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 1521–1530 CrossRef CAS PubMed.
- M. S. Ward, I. Silva, W. Martinez, J. Jefferson, S. Rahman, J. M. Garcia, D. Kanichar, L. Roppiyakuda, E. Kosmowska, M. A. Faust, K. P. Tran, F. Chow, E. Buglo, F. Zhou, M. P. Groziak and H. H. Xu, Bioorg. Med. Chem., 2016, 24, 3267–3275 CrossRef CAS PubMed.
- J. P. M. Antonio, G. D. V. Farias, F. M. F. Santos, R. Oliveira, P. M. S. D. Cal and P. M. P. Gois, Non-covalent Interactions in the Synthesis and Design of New Compounds, John Wiley & Sons, Ltd, 2016, pp. 23–48 Search PubMed.
- C. J. Stress, P. J. Schmidt and D. G. Gillingham, Org. Biomol. Chem., 2016, 14, 5529–5533 RSC.
- A. Bandyopadhyay, S. Cambray and J. Gao, J. Am. Chem. Soc., 2017, 139, 871–878 CrossRef CAS PubMed.
- H. Gu, T. I. Chio, Z. Lei, R. J. Staples, J. S. Hirschi and S. Bane, Org. Biomol. Chem., 2017, 15, 7543–7548 RSC.
- S. Cambray, A. Bandyopadhyay and J. Gao, Chem. Commun., 2017, 53, 12532–12535 RSC.
- A. M. Kelly, S. D. Bull and T. D. James, Tetrahedron: Asymmetry, 2008, 19, 489–494 CrossRef CAS.
- Y. Pérez-Fuertes, A. M. Kelly, A. L. Johnson, S. Arimori, S. D. Bull and T. D. James, Org. Lett., 2006, 8, 609–612 CrossRef PubMed.
- S. L. Yeste, M. E. Powell, S. D. Bull and T. D. James, J. Org. Chem., 2009, 74, 427–430 CrossRef CAS PubMed.
- A. M. Kelly, Y. Pérez-Fuertes, S. Arimori, S. D. Bull and T. D. James, Org. Lett., 2006, 8, 1971–1974 CrossRef CAS PubMed.
- M. K. Meadows, E. K. Roesner, V. M. Lynch, T. D. James and E. V. Anslyn, Org. Lett., 2017, 19, 3179–3182 CrossRef CAS.
- R. Ma, C. Zhang, Y. Liu, C. Li, Y. Xu, B. Li, Y. Zhang, Y. An and L. Shi, RSC Adv., 2017, 7, 21328–21335 RSC.
- Y. Li, Y. Liu, R. Ma, Y. Xu, Y. Zhang, B. Li, Y. An and L. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 13056–13067 CrossRef CAS PubMed.
- F. M. F. Santos, A. I. Matos, A. E. Ventura, J. Gonçalves, L. F. Veiros, H. F. Florindo and P. M. P. Gois, Angew. Chem., Int. Ed., 2017, 56, 9346–9350 CrossRef CAS.
- F. M. F. Santos, J. N. Rosa, N. R. Candeias, C. P. Carvalho, A. I. Matos, A. E. Ventura, H. F. Florindo, L. C. Silva, U. Pischel and P. M. P. Gois, Chem. – Eur. J., 2016, 22, 1631–1637 CrossRef CAS.
- V. G. Jiménez, F. M. F. Santos, S. Castro-Fernández, J. M. Cuerva, P. M. P. Gois, U. Pischel and A. G. Campaña, J. Org. Chem., 2018, 83, 14057–14062 CrossRef.
- M. M. Alcaide, F. M. F. Santos, V. F. Pais, J. I. Carvalho, D. Collado, E. Pérez-Inestrosa, J. F. Arteaga, F. Boscá, P. M. P. Gois and U. Pischel, J. Org. Chem., 2017, 82, 7151–7158 CrossRef CAS.
- F. M. F. Santos, Z. Domínguez, M. M. Alcaide, A. I. Matos, H. F. Florindo, N. R. Candeias, P. M. P. Gois and U. Pischel, ChemPhotoChem, 2018, 2, 1038–1045 CrossRef CAS.
- B. Zhang, S. Wang, J. Tan and X. Zhang, Dyes Pigm., 2018, 155, 186–193 CrossRef CAS.
- P. M. S. D. Cal, F. Sieglitz, F. M. F. Santos, C. Parente Carvalho, A. Guerreiro, J. B. Bertoldo, U. Pischel, P. M. P. Gois and G. J. L. Bernardes, Chem. Commun., 2017, 53, 368–371 RSC.
| This journal is © The Royal Society of Chemistry 2019 |