
Formation of a tetranickel octacarbonyl cluster from the CO2 reaction of a zero-valent nickel monocarbonyl species†
Changho
Yoo
ab and
Yunho
Lee
*ab
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea
bCenter for Catalytic Hydrocarbon Functionalization, Institute of Basic Science (IBS), Daejeon 305-701, Republic of Korea. E-mail: yunholee@kaist.ac.kr; Fax: +82 42 350 2810; Tel: +82 42 350 2814
First published on 21st March 2016
Abstract
The reaction of a zerovalent nickel complex {Na}{(PNP)Ni(CO)} (1) with CO2(g) results in the formation of a tetrameric cluster complex {(PNCOONaP)Ni(CO)2}4 (2) as the major product along with {(PNP)Ni}2-μ-CO3-κ2O,O (3), {(PNP)Ni}2-μ-CO2-κ2C,O (4) and (PNP)Ni(CO) (5). A labelling experiment with 13CO2(g) reveals that not only the carbamate moeity but also the CO ligand in 2 originate from CO2(g). The formation of both a carbonyl species 2 and a carbonate species 3 indicates that nickel-mediated reductive disproportionation of CO2 occurs. Alternatively, a direct CO2 oxidative reaction of 1 occurs to produce {(PNP)Ni}2-μ-CO2-κ2C,O (4) in a minor pathway. The synthesis and characterization of 2 and other products are described herein.
 Yunho Lee | Yunho Lee is an Associate Professor in the Department of Chemistry at the Korea Advanced Institute of Science and Technology (KAIST), Daejeon, Korea. He attended Chonbuk National University where he received a B.S. degree in Chemistry in 2000. Yunho left Korea to begin his doctoral studies under the guidance of Prof. Kenneth D. Karlin at Johns Hopkins University, Baltimore. After receiving his Ph.D. in 2007, Yunho was a postdoctoral fellow in the laboratory of Prof. Jonas C. Peters at the Massachusetts Institute of Technology and the California Institute of Technology. In the winter of 2010, Yunho returned to Korea and started his independent career as an Assistant Professor at KAIST. He received the Distinguished Lectureship Award from the Chemical Society of Japan in 2011 and the Young Inorganic Chemist Award from the Korea Chemical Society in 2015. He was promoted to associate professor in September 2015. His primary research interests focus on the fundamental transition metal coordination chemistry and its application in various small molecule conversions and catalysis inspired by metalloenzymes. |
Introduction
Carbon dioxide functionalization mediated by transition metal complexes has been widely explored in recent years to utilize CO2 as a synthetic C1 source.1 In particular, the carbon dioxide reactivity of a low-valent nickel species is receiving much attention due to its relevance to the biological CO2 conversion to CO occurring at the active site of the CODH (carbon monoxide dehydrogenase).2 At the C cluster of CODH, zerovalent nickel is hypothesized to be responsible for the initial binding of CO2. Further activation of carbon dioxide occurs revealing the bond formation between nickel and carbon to produce a Ni–COO–Fe species.2In organonickel chemistry, CO2 activation mediated by divalent nickel commonly reveals the formation of a nickel(II)–formate species via an insertion reaction to a Ni–H bond.3 In the case of a monovalent nickel species, the Caulton group reported the reaction of ((tBu2PCH2SiMe2)2N)NiI with CO2 revealing unusual transposition between the amide nitrogen and one CO2 oxygen to produce a nickel(I)–cyanate species.4 Recently, the Limberg group reported the reductive coupling of CO2 at the monovalent nickel center supported by a β-diketiminato ligand to give an oxalate-bridged dinickel(II) complex.5 The corresponding anionic analogue K2[LtBuNiI(N22−)NiILtBu] reveals the disproportionation of CO2 to CO and CO32− mediated by nickel(I) with an additional 1 electron donation from a bridging anionic N2 ligand.5 Binding of CO2 to a single transition metal ion was firstly demonstrated by Aresta and coworkers in the preparation of (PCy3)2Ni(η2-CO2).6 A similar zero-valent nickel–CO2 adduct was also reported by Hillhouse and coworkers using a bidentate phosphine ligand.7 Further reaction of these nickel–CO2 adducts resulted in CO production with associated phosphine oxidation.7,8 The Lee group reported a 5-coordinate nickel–η2-CO2 complex (PPMeP)Ni(CO2) (PPMeP = PMe[2-PiPr2-C6H4]2) and its reactivity toward triarylborane.9 Structural and theoretical analyses suggest that such a 5-coordinate nickel center supported by a tridentate ligand might significantly enhance the reactivity of a bound CO2 ligand compared to an analogous 4-coordinate species.9 Recently, few nickel(0) species supported by N-heterocyclic carbene ligands were also reported. The reaction of [(IPr)Ni]2 (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) with CO2 resulted in the generation of [(IPr)Ni]2(μ-CO)(μ-η2,η2-CO2) and (IPr)Ni(CO3) by reductive disproportionation.10 The CO2 reaction of a dinickel(0) complex supported by a binucleating bis(N-heterocyclic carbene) ligand revealed the formation of a dicarbonyl bridged dinickel(0) complex presumably with the formation of carbonate.11
The reaction of a nickel monocarbonyl species with CO2 is currently under investigation in our laboratory due to its relevance to CODH active site chemistry. Recently, we reported a series of mononuclear nickel complexes supported by an anionic PNP ligand (PNP− = N[2-PiPr2-4-Me-C6H3]2−). In the (PNP)Ni system, a formal nickel(0) state cannot be achieved without a π-accepting ligand, such as CO.12,13 Therefore, we have employed a zerovalent nickel monocarbonyl adduct, {Na}{(PNP)Ni(CO)} (1)13 to study CO2 conversion reactions. According to the solid-state structure, 1 has two sodium interactions with both a zero-valent nickel ion (dNi–Na2: 2.8523(5) Å) and an amido nitrogen (dN1–Na2: 2.463(1) Å) in its dimeric assembly (Fig. 1). This structural information suggests that the competition might occur between a nickel ion and an amido nitrogen during the reaction of 1 with CO2. As anticipated, the CO2 reaction showed an immediate transformation of 1 to multiple products (Scheme 1). The major product was identified as a tetrameric cluster {(PNCOONaP)Ni(CO)2}4 (2). Isotope labelling experiments with 13CO2 revealed that 2 contains CO molecules originated from CO2. The carbonate species, {(PNP)Ni}2-μ-CO3-κ2O,O (3), was also produced indicating that the reductive disproportionation of CO2 occurs with a nickel(0) species. In fact, a reaction product mixture also contains additional portions of {(PNP)Ni}2-μ-CO2-κ2C,O (4)14 and (PNP)Ni(CO) (5) as minor products.13 Therefore, the CO2 reaction of 1 involves multiple reaction pathways, vide infra.
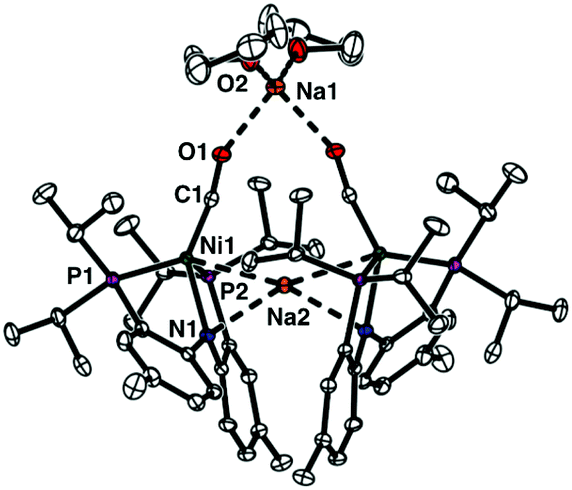 | ||
| Fig. 1 Displacement ellipsoid (50%) representations of {Na}{(PNP)Ni(CO)} (1) in dimeric assembly with two co-crystallized THF molecules. All hydrogen atoms are omitted for clarity. See the ESI† for details. | ||
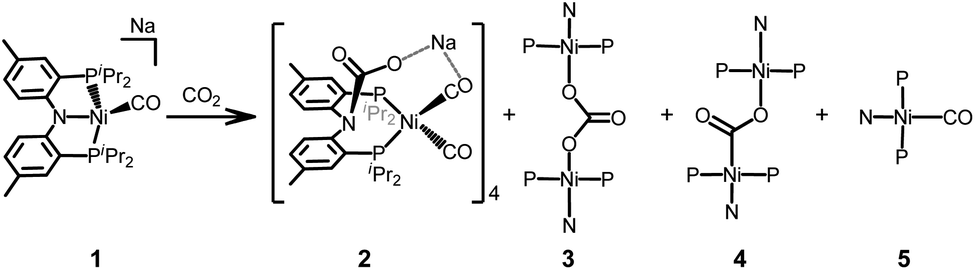 | ||
| Scheme 1 | ||
Results and discussion
Reaction of a nickel(0) monocarbonyl species with CO2(g)
By exposing CO2(g) to a degassed solution of {Na}{(PNP)Ni(CO)} (1) in THF at room temperature, an immediate reaction occurs (<1 min) revealing a color change from orange to dark green. This solution was found to contain several diamagnetic species according to the 31P NMR spectrum (see the ESI†). A major pentane-insoluble product was isolated in 35.5% yield as a light green solid, vide infra. The remaining pentane soluble portion contains two known diamagnetic products, {(PNP)Ni}2-μ-CO2-κ2C,O (4, 8.8%)14 and (PNHP)Ni(CO)2 (6, 2.8%)14,15 with several unidentified species according to the 31P NMR spectroscopic data (see the ESI†). In fact, there is an additional paramagnetic product (PNP)NiI(CO) (5).13 After the green species and 4 were removed from a product mixture, compound 5 was isolated as a green crystalline solid in >22% yield from the recrystallization of a cold diethyl ether solution at −35 °C. The identity of 5 was confirmed with IR (νCO 1931 cm−1) and XRD crystallographic data (see the ESI†).13The green unknown species exhibits a broad singlet at 35.5 ppm (ν1/2 = ∼900 Hz, C6D6) in the 31P NMR spectrum. Single crystals suitable for X-ray diffraction were obtained by slow evaporation of an ether solution of a green product. The solid-state structure reveals an unanticipated Td symmetric tetrameric nickel cluster, {(PNCOONaP)Ni(CO)2}4 (2) in which a CO2 molecule is incorporated into each central nitrogen atom of a PNP ligand (Fig. 2). The carbamate vibration in 2 detected at 1626 cm−1 also supports its structural data.16 In the nickel cluster, each sodium ion binds to four oxygen atoms from carbamates (O3, O3′ and O4′′) and CO (O1) to construct a tetrameric structure (dO1–Na1 = 2.455(6), dO3–Na1 = 2.243(6), and dO4–Na1 = 2.200(6) Å, Fig. 2). Two C–O bond distances for carbamate, dC3–O3 and dC3–O4 are 1.25(1) and 1.28(1) Å, respectively. The bond distance between Ni1 and N1 in 2 is significantly elongated (dNi1–N1 = 3.339(5) Å) compared with that of 1 (dNi1–N1 = 2.054(1) Å, Fig. 1). There are two CO ligands coordinated to each nickel center (dC1–O1 = 1.132(8) and dC2–O2 = 1.155(8) Å) along with two phosphorus atoms allowing for the formation of a four coordinate zero-valent nickel center.
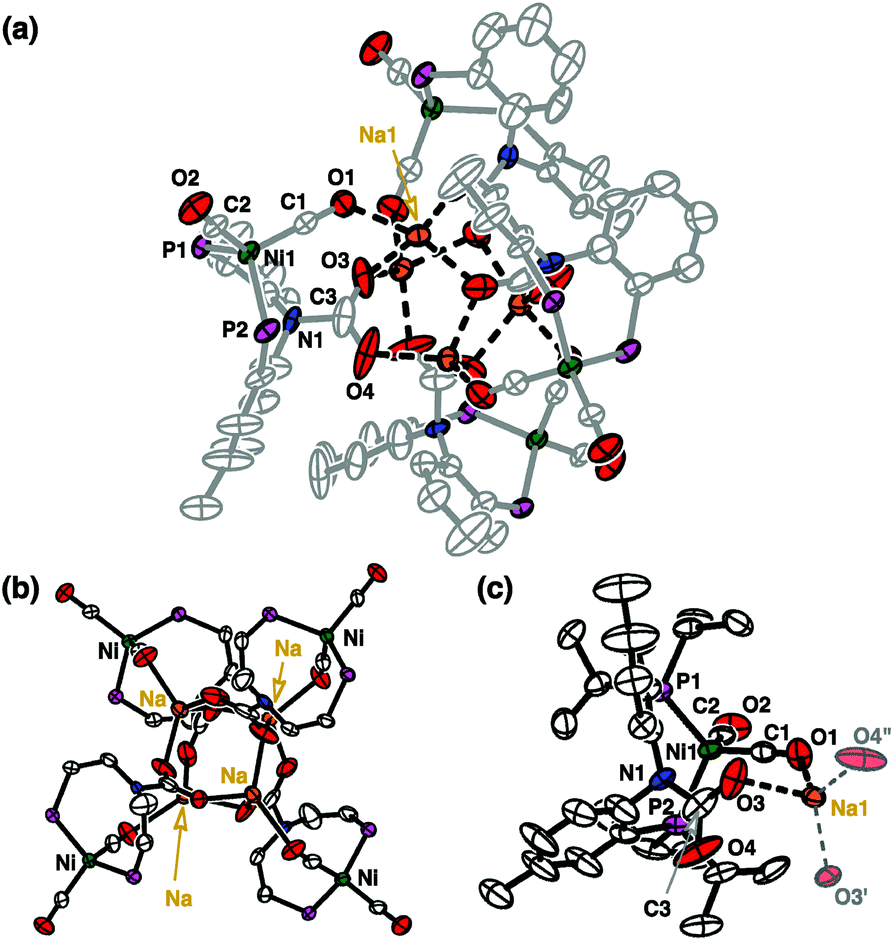 | ||
| Fig. 2 Displacement ellipsoid (50%) representations for (a) the cluster complex {(PNCOONaP)Ni(CO)2}4 (2), (b) the core structure of 2 with omission of ligand isopropyl and aromatic groups, and (c) a monomer unit of 2 composed of (PNCOOP)Ni(CO)2 and a sodium ion. All hydrogen atoms are omitted for clarity. See the ESI† for details. | ||
The nickel(0) center of 2 is reminiscent to the previously reported nickel(0) dicarbonyl species, (PNHP)Ni(CO)2 (6).14 The IR spectrum of 2 exhibits CO vibrations at 1976 and 1905 cm−1, which were observed at 1994 and 1933 cm−1 in 6.14
In order to comprehend the solution-state structure, the diffusion coefficient measurements of 2 with analogous compounds were conducted by pulse gradients spin echo nuclear magnetic resonance (PGSE-NMR) experiments.17 The diffusion coefficient for 2 was obtained as 4.877 × 10−10 m2 s−1 in a non-coordinating solvent, C6D6 at 25 °C. In comparison, the diffusion coefficients of a mononuclear complex (PNHP)Ni(CO)2 (6) and a dinuclear complex {(PNP)Ni}2-μ-CO2-κ2C,O (4) as reference compounds were also measured as 1.079 × 10−9 and 7.737 × 10−10 m2 s−1, respectively (Table 1). The hydrodynamic radius of 2 (rSolution = 7.366 Å) derived from the Stokes–Einstein equation (eqn (1)) is similar to the solid-state radius estimated from XRD data (rSolid = 8.8640 Å).18 Thus, the diffusion NMR spectroscopy experiments suggest that the tetrameric structure of 2 is maintained in a benzene solution.
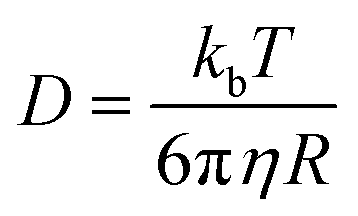 | (1) |
| Compound | Diffusion constant (×10−10 m2 s−1) | Hydrodynamic radius (Å) |
|---|---|---|
| 2 | 4.877 | 7.366 |
| 4 | 7.737 | 4.643 |
| 6 | 10.79 | 3.329 |
Origin of CO in the generation of {(PNCOONaP)Ni(CO)2}4 (2)
According to the structure of {(PNCOONaP)Ni(CO)2}4 (2), two CO molecules are coordinated to each nickel ion. One can anticipate that one of the CO ligands has originated from either CO2(g) or another Ni–CO species during the reaction. From the reaction of 1 with a CO/CO2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gas mixture, the yield of 2 was dramatically increased to 66.6% (see the Experimental section). This experiment suggests that the formation of 2 requires free CO. Yet, we cannot unfortunately differentiate two different CO sources. We then utilized 13C-labelled CO2(g) in the synthesis of 2 from the reaction of 1-12CO (see the Experimental section). The isolated 13C-labelled product 2-13CO2 revealed three augmented 13C-NMR signals at 165.31, 203.64 and 205.88 ppm (see the ESI†). The signal at 165.31 ppm corresponds to 13C of carbamate and those at 203.64 (t, J = 5.3 Hz) and 205.88 ppm represent two 13CO ligands in 2. The intensity of two carbonyl peaks is similar to each other indicating that labelled 13CO has been incorporated over two different positions at a nickel(0) center. However, the 12CO coordination in 2-13CO2 presumably remains since both carbonyl peak intensities are significantly lower than expected (see the ESI†). In fact, the infrared spectrum also supports that both 12CO (1976 and 1905 cm−1) and 13CO (1967sh and 1884 cm−1) are present in 2-13CO2. The vibration for a carbamate group is shifted to 1579 cm−1 from 1626 cm−1 (Δ(13CO2) = −47 cm−1, see the ESI†). Thus, the labelled product can be formulated as {(PN13COONaP)Ni(CO)x(13CO)2−x}4 (2-13CO2). This result indicates that new CO(g) might be generated from CO2 conversion and the corresponding CO(g) participates in the formation of 2.
:1) gas mixture, the yield of 2 was dramatically increased to 66.6% (see the Experimental section). This experiment suggests that the formation of 2 requires free CO. Yet, we cannot unfortunately differentiate two different CO sources. We then utilized 13C-labelled CO2(g) in the synthesis of 2 from the reaction of 1-12CO (see the Experimental section). The isolated 13C-labelled product 2-13CO2 revealed three augmented 13C-NMR signals at 165.31, 203.64 and 205.88 ppm (see the ESI†). The signal at 165.31 ppm corresponds to 13C of carbamate and those at 203.64 (t, J = 5.3 Hz) and 205.88 ppm represent two 13CO ligands in 2. The intensity of two carbonyl peaks is similar to each other indicating that labelled 13CO has been incorporated over two different positions at a nickel(0) center. However, the 12CO coordination in 2-13CO2 presumably remains since both carbonyl peak intensities are significantly lower than expected (see the ESI†). In fact, the infrared spectrum also supports that both 12CO (1976 and 1905 cm−1) and 13CO (1967sh and 1884 cm−1) are present in 2-13CO2. The vibration for a carbamate group is shifted to 1579 cm−1 from 1626 cm−1 (Δ(13CO2) = −47 cm−1, see the ESI†). Thus, the labelled product can be formulated as {(PN13COONaP)Ni(CO)x(13CO)2−x}4 (2-13CO2). This result indicates that new CO(g) might be generated from CO2 conversion and the corresponding CO(g) participates in the formation of 2.
Carbon monoxide can be generated through the reductive disproportionation of CO2, which gives CO and CO32− from two CO2 and two additional electrons, according to the literature.19 Nickel-mediated reductive disproportionation of CO2 was demonstrated with nickel(I) and nickel(0) species.5,10,11 Thus, the formation of carbonate can be expected. In order to confirm the generation of carbonate, a nickel carbonate species {(PNP)Ni}2-μ-CO3-κ2O,O (3) was independently prepared from the reaction of {(PNP)NiII(CO)}{BF4} with Na2CO3 (see the Experimental section). Compound 3 exhibiting a singlet signal at 30.6 ppm in the 31P NMR spectrum was fully characterized by various spectroscopic methods. The solid state structure of 3 confirms a dinuclear nickel bridging carbonate species (see Fig. 3). The presence of 3 (5.3%) in a reaction product mixture of {Na}{(PNP)Ni(CO)} (1) with CO2(g) was confirmed by 31P NMR spectroscopy (see the ESI†). Therefore, carbon monoxide along with CO32− might be produced from the reductive disproportionation of CO2.
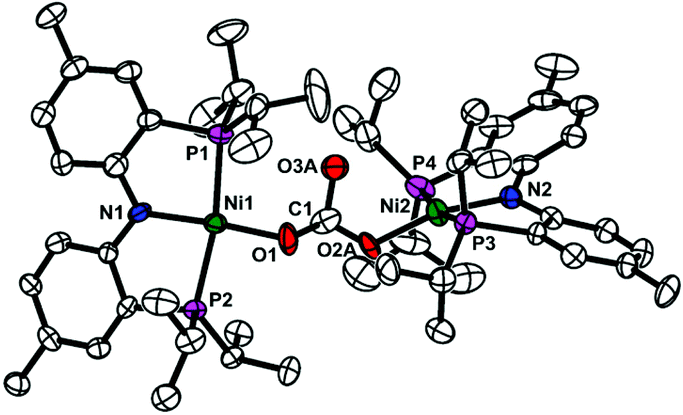 | ||
| Fig. 3 Displacement ellipsoid (50%) representations of {(PNP)Ni}2-μ-CO3-κ2O,O (3). A carbonate moiety was disordered over two distinctive positions. For clarity, only one component is shown. All hydrogen atoms are omitted for clarity. See the ESI† for details. | ||
After CO generation via reductive disproportionation, {(PNCOONaP)Ni(CO)2}4 (2) is produced by accepting both CO and CO2. Initially, the C–N bond between CO2 and an amide moiety in a PNP ligand can be formed. Further Ni(0)–N bond cleavage and CO coordination occur to produce 2. However, we cannot rule out the other pathway that the addition of CO2 to a nickel(0) center occurs to form (PNP)Ni–COONa14 followed by reductive elimination of a C–N bond. In an analogous PNP nickel system, C–N bond-forming reductive elimination reactions were reported.20 [RC(O)N(o-C6H4PPh2)2]Ni(CO)2 (R = Me or Et) can be generated from the reaction of (N(o-C6H4PPh2)2)NiCOR with addition of excess CO(g).20 However, this reaction is unlikely since the CO(g) addition to {(PNP)NiCOONa}2·(THF)14 does not give 2. Both CO(g) production mediated by Ni(0) and carbamate generation via nucleophilic addition of amide to CO2(g) suggest that the two major reactions are competitive.
Formation of {(PNP)Ni}2-μ-CO2-κ2C,O (4) and (PNP)Ni(CO) (5)
From the CO2 reaction, a dinuclear nickel carboxylate species {(PNP)Ni}2-μ-CO2-κ2C,O (4) was detected as a minor product (8.8%). According to a 13CO2 labelling experiment, {(PNP)Ni}2-μ-13CO2-κ2C,O (4-13CO2) was detected by its characteristic triplet of triplet signal at 192.40 ppm in the 13C NMR spectrum revealing that a CO2 moiety of 4 comes from CO2(g) (see the ESI†). This product can be generated from the direct addition of CO2 to a zero-valent nickel center followed by stabilization with a nickel(II) source (Scheme 2). The yield of 4 from the reaction of {Na}{(PNP)Ni(CO)} (1) with CO2(g) increases in the presence of a nickel(II) source such as {(PNP)Ni(CO)}{BF4} (36%) or 3 (26%).21 In these reactions, the cluster 2 was not generated (see the Experimental section). These experiments suggest another pathway of the CO2 reaction of 1 to generate a carboxylate species through a direct Ni–C bond formation. This reaction involves the elimination of a CO coordination from a nickel center. In fact, compound 4 can further react with CO. By exposing 4 toward CO, a monovalent nickel–CO adduct (PNP)Ni(CO) (5) was immediately produced in 87% yield, suggesting that the free CO(g) can be a directing agent to induce the inner-sphere electron transfer.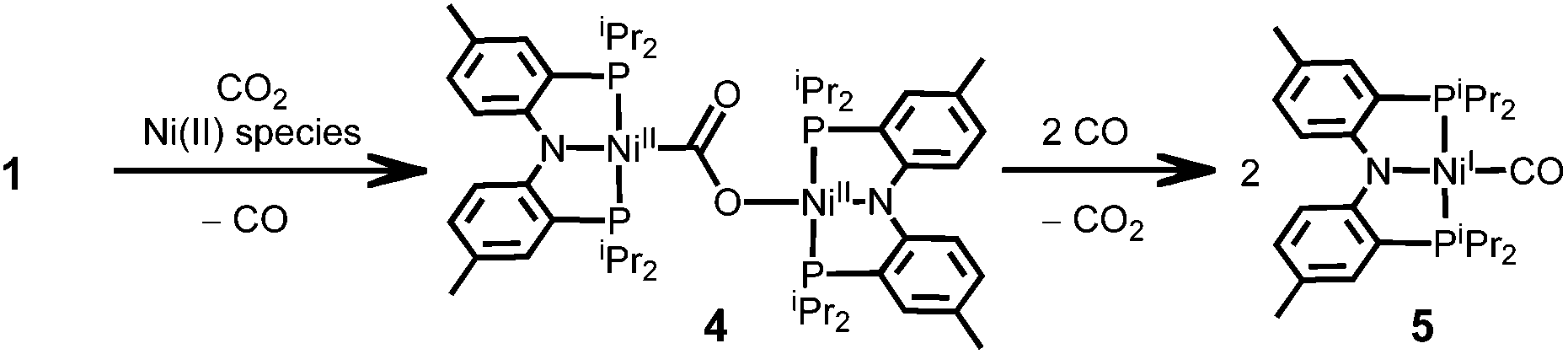 | ||
| Scheme 2 | ||
Conclusions
The reaction of a zerovalent nickel complex {Na}{(PNP)Ni(CO)} (1) with CO2(g) results in the formation of an unexpected cluster complex {(PNCOONaP)Ni(CO)2}4 (2) as a major product along with {(PNP)Ni}2-μ-CO3-κ2O,O (3), {(PNP)Ni}2-μ-CO2-κ2C,O (4), (PNP)Ni(CO) (5) and other byproducts. The XRD data of 2 clearly show a Td symmetric tetrameric nickel cluster structure, where a CO2 molecule is incorporated into an amide moiety of a PNP ligand. The cluster structure of 2 is maintained in its solution state in benzene supported by its small diffusion coefficient measured by diffusion NMR spectroscopy. According to 13CO2 labelling experiments, labelled carbon monoxide and carbamate moieties found in {(PN13COONaP)Ni(CO)x(13CO)2−x}4 (2-13CO2) originate from 13CO2. As a major CO production, reductive disproportionation of CO2 occurs to produce both CO and {(PNP)Ni}2(μ-CO3) (3). In fact, a direct CO2 oxidative reaction might occur with 1 to give {(PNP)Ni}2-μ-CO2-κ2C,O (4) as a minor reaction pathway. Therefore, the formation of 4 and 5 along with 2 and 3 indicates that the CO2 reaction of 1 involves multiple reaction pathways.Experimental
General consideration
All manipulations were carried out using standard Schlenk or glovebox techniques under a N2 atmosphere. Solvents were deoxygenated and dried by thoroughly sparging with Ar gas followed by passage through an activated alumina column. Solvents were tested with a standard purple solution of sodium benzophenone ketyl in tetrahydrofuran in order to confirm effective oxygen and moisture removal. CO2 gas (99.999% purity) was dried by storage in a column of activated 3-Å molecular sieves at a pressure of 20 bar. All reagents were purchased from commercial vendors and used without further purification unless otherwise stated. {Na}{(PNP)Ni(CO)} (1),13 {(PNP)Ni}2-μ-CO2-κ2C,O (4),14 (PNP)Ni(CO) (5),13 (PNHP)Ni(CO)2 (6),14 {(PNP)NiCOONa}2·THF14 and {(PNP)Ni(CO)}{BF4}13 were prepared according to literature procedures. Elemental analyses were carried out on a Thermo Scientific FLASH 2000 series instrument at the KAIST Research Analysis Center. Deuterated solvents were purchased from Euriso-top, degassed, and dried over activated 4-Å molecular sieves prior to use.Spectroscopic measurements
A Bruker AVHD-400 spectrometer was used to measure 1H and 31P NMR data. 13C NMR spectra were recorded on Bruker AVHD-400 and Agilent Technologies DD2 600 spectrometers. The chemical shifts for 1H NMR and 13C NMR spectra were quoted in parts per million (ppm) referenced to residual solvent peaks. 31P NMR spectra were decoupled by broad band proton decoupling and chemical shifts were quoted in parts per million (ppm) referenced to external phosphoric acid as 0.0 ppm. The following abbreviations are used to describe peak splitting patterns when appropriate: s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, dd = doublet of doublet, tt = triplet of triplet, bs = broad singlet. Coupling constants, J, were reported in hertz (Hz). Diffusion NMR experiments for 2, 4 and 6 were conducted using a Bruker AVANCE HD 800 spectrometer at Korea Basic Science Institute. The data were collected in C6D6 at 298K with the bipolar pulse pairs stimulated echo pulse sequence (ledbpgp2s). The pulse field gradient was incremented from 0 to 47 G cm−1 with 16 steps. UV-vis spectra were recorded with an Agilent Cary 60 UV-vis spectrophotometer using a 1 cm two-window quartz cell sealed with a screw-cap purchased from Hellma Analytics (117.100-QS). Infrared spectra were recorded using KBr pellets by using a Bruker VECTOR 33. Frequencies are given in reciprocal centimeters (cm−1) and only selected absorbances were reported.X-ray crystallography
The diffraction data of {Na}{(PNP)Ni(CO)} (1)13 were collected on an ADSC Quantum-210 detector at 2D SMC at the Pohang Accelerator Laboratory, Korea. Crystals of 1 suitable for X-ray diffraction were obtained by slow diffusion of pentane into a THF solution. A crystal of suitable size and quality was coated with Paratone-N oil and mounted on a Dual-Thickness MicroLoops LD purchased from MiTeGen. The data were collected with Si(111) double crystal monochromated synchrotron radiation (λ = 0.70000 Å) at 100 K. The ADSC Q210 ADX program22 was used for data collection and HKL3000sm (Ver. 703r)23 was used for cell refinement, reduction and absorption correction. The diffraction data of 2, 3, 5 and {(PNP)NiCOONa}2·THF14 were collected on a Bruker SMART APEX II instrument. Crystals of {(PNP)NiCOONa}2·(THF) suitable for X-ray diffraction were obtained by slow diffusion of pentane into a THF solution. The data were collected with graphite-monochromated MoKα radiation (λ = 0.71073 Å) under a stream of N2 (g) at 120 K. Cell parameters were determined and refined by the SMART program.24 Data reduction was performed using the SAINT software.25 An empirical absorption correction was applied using the SADABS program.26 The structures were solved by direct methods and all non-hydrogen atoms were subjected to anisotropic refinement by full-matrix least-squares on F2 by using the SHELXTL/PC package.27 Hydrogen atoms were placed at their geometrically calculated positions and refined riding on the corresponding carbon atoms with isotropic thermal parameters. A highly disordered ether solvent molecule for 2 could not be appropriately modeled and was removed using the PLATON program, SQUEEZE function.28510). IR (KBr pellet, cm−1): νCO 1976, 1905, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O 1626. Crystals of 2 suitable for X-ray diffraction were obtained by slow evaporation of an ether solution.
:1) and triphenylphosphine oxide (26 mg, 0.093 mmol) was added as an internal integration standard. The corresponding yields for 2 (0.022 mmol, 48%), 3 (0.0049 mmol, 5.3%), 4 (0.0077 mmol, 8.3%) and 6 (0.051 mmol, 2.8%) were determined by 31P NMR integration.
:1) was degassed by three freeze–pump–thaw cycles on the Schlenk line. 13CO2(g) was charged under ambient conditions resulting in an immediate color change from orange to dark green. After shaking for 10 min at room temperature, compounds 2, 3 and 4 were detected by using the 13C NMR spectrum (see the ESI†). 13C NMR (101 MHz, THF/benzene-d6) δ 164.92 (s) for 3 and 192.40 (tt, J = 26.2, 7.3 Hz) for 4. After additional shaking for 1 h at room temperature, all volatiles were removed under vacuum. The green solid residue was dissolved in 5 mL of toluene and stirred for 30 min at room temperature. The resulting solution was filtered through Celite, and all volatiles were removed under vacuum. The product, {(PN13COONaP)Ni(CO)x(13CO)2−x}4 (2-13CO2, 7.0 mg, 0.0029 mmol, 20%) was obtained as a light green solid after washing with pentane and drying under vacuum. 13C NMR (101 MHz, benzene-d6) δ 205.89 (s), 203.64 (t, J = 5.3 Hz), 165.31. IR (KBr pellet, cm−1): νCO 1976, 1905, ν13CO 1967sh, 1884, ν13CO 1579.
O 1626. Crystals of 2 suitable for X-ray diffraction were obtained by slow evaporation of an ether solution.
:1) and triphenylphosphine oxide (26 mg, 0.093 mmol) was added as an internal integration standard. The corresponding yields for 2 (0.022 mmol, 48%), 3 (0.0049 mmol, 5.3%), 4 (0.0077 mmol, 8.3%) and 6 (0.051 mmol, 2.8%) were determined by 31P NMR integration.
:1) was degassed by three freeze–pump–thaw cycles on the Schlenk line. 13CO2(g) was charged under ambient conditions resulting in an immediate color change from orange to dark green. After shaking for 10 min at room temperature, compounds 2, 3 and 4 were detected by using the 13C NMR spectrum (see the ESI†). 13C NMR (101 MHz, THF/benzene-d6) δ 164.92 (s) for 3 and 192.40 (tt, J = 26.2, 7.3 Hz) for 4. After additional shaking for 1 h at room temperature, all volatiles were removed under vacuum. The green solid residue was dissolved in 5 mL of toluene and stirred for 30 min at room temperature. The resulting solution was filtered through Celite, and all volatiles were removed under vacuum. The product, {(PN13COONaP)Ni(CO)x(13CO)2−x}4 (2-13CO2, 7.0 mg, 0.0029 mmol, 20%) was obtained as a light green solid after washing with pentane and drying under vacuum. 13C NMR (101 MHz, benzene-d6) δ 205.89 (s), 203.64 (t, J = 5.3 Hz), 165.31. IR (KBr pellet, cm−1): νCO 1976, 1905, ν13CO 1967sh, 1884, ν13CO 1579.
Acknowledgements
This work was supported by the Korea CCS R&D Center Grant funded by the Korea government (Ministry of Science, ICT & Future Planning; no. NRF-2014M1A8A1049353).Notes and references
- M. Aresta, Carbon dioxide as Chemical Feedstock, Wiley-VCH, Weinhein, Germany, 2010 CrossRef CAS PubMed; M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149 CrossRef CAS PubMed.
- D. J. Darensbourg, M. Y. Darensbourg, L. Y. Goh, M. Ludvig and P. Wiegreffe, J. Am. Chem. Soc., 1987, 109, 7539 CrossRef CAS; Y. Journaux, V. Lozan, J. Klingele and B. Kersting, Chem. Commun., 2006, 83 RSC; S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872 CrossRef PubMed; H.-W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225 CrossRef; G. T. Venkanna, S. Tammineni, H. D. Arman and Z. J. Tonzetich, Organometallics, 2013, 32, 4656 CrossRef PubMed; K. J. Jonasson and O. F. Wendt, Chem. – Eur. J., 2014, 20, 11894 CrossRef PubMed; T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672 CrossRef PubMed; M. Kreye, M. Freytag, P. G. Jones, P. G. Williard, W. H. Bernskoetter and M. D. Walter, Chem. Commun., 2015, 51, 2946 RSC; S. Murugesan, B. Stöger, M. Weil, L. F. Veiros and K. Kirchner, Organometallics, 2015, 34, 1364 CrossRef.
- B. C. Fullmer, H. Fan, M. Pink and K. G. Caulton, Inorg. Chem., 2008, 47, 1865 CrossRef CAS PubMed.
- B. Horn, C. Limberg, C. Herwig and B. Braun, Chem. Commun., 2013, 49, 10923 RSC.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636 RSC; M. Aresta and C. F. Nobile, J. Chem. Soc., Dalton Trans., 1977, 708 RSC.
- J. S. Anderson, V. M. Iluc and G. L. Hillhouse, Inorg. Chem., 2010, 49, 10203 CrossRef CAS PubMed.
- C. Bianchini, C. Mealli, A. Meli and M. Sabat, Inorg. Chem., 1984, 23, 2731 CrossRef CAS; N. Huang, X. Li, W. Xu and H. Sun, Inorg. Chim. Acta, 2013, 394, 446 CrossRef.
- Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458 RSC.
- C. H. Lee, D. S. Laitar, P. Mueller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 13802 CrossRef CAS PubMed.
- E. Y. Tsui and T. Agapie, Polyhedron, 2014, 84, 103 CrossRef CAS.
- D. Adhikari, S. Mossin, F. Basuli, B. R. Dible, M. Chipara, H. Fan, J. C. Huffman, K. Meyer and D. J. Mindiola, Inorg. Chem., 2008, 47, 10479 CrossRef CAS PubMed.
- C. Yoo, S. Oh, J. Kim and Y. Lee, Chem. Sci., 2014, 5, 3853 RSC.
- C. Yoo, J. Kim and Y. Lee, Organometallics, 2013, 32, 7195 CrossRef CAS.
- According to our observations, (PNHP)Ni(CO)2 (6) can be generated from 1 or 2 in the presence of a proton source such as water. Therefore, the yield of 6 varies from different experiments.
- D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857 CrossRef PubMed.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520 CrossRef CAS PubMed.
- r Solid (8.8640 Å) for 2 is derived from the molecular volume in XRD data. See the ESI† for details.
- G. R. Lee, J. M. Maher and N. J. Cooper, J. Am. Chem. Soc., 1987, 109, 2956 CrossRef CAS; K. K. Pandey, Coord. Chem. Rev., 1995, 140, 37 CrossRef; L. Contreras, M. Paneque, M. Sellin, E. Carmona, P. J. Pérez, E. Gutiérrez-Puebla, A. Monge and C. Ruiz, New J. Chem., 2005, 29, 109 RSC; N. W. Davies, A. S. P. Frey, M. G. Gardiner and J. Wang, Chem. Commun., 2006, 4853 RSC; A. R. Sadique, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2008, 47, 784 CrossRef PubMed; O. T. Summerscales, A. S. P. Frey, F. G. N. Cloke and P. B. Hitchcock, Chem. Commun., 2009, 198 Search PubMed; A.-C. Schmidt, A. V. Nizovtsev, A. Scheurer, F. W. Heinemann and K. Meyer, Chem. Commun., 2012, 48, 8634 RSC.
- L.-C. Liang, Y.-T. Hung, Y.-L. Huang, P.-S. Chien, P.-Y. Lee and W.-C. Chen, Organometallics, 2012, 31, 700 CrossRef CAS.
- Both divalent nickel species {(PNP)Ni(CO)}{BF4} and {(PNP)Ni}2-μ-CO3-κ2O,O (3) do not react with CO2 under the same conditions.
- A. J. Arvai and C. Nielsen, ADSC Quantum-210 ADX Program, Area Detector System Corporation, Poway, CA, USA, 1983 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, ed. C. W. Cater Jr. and R. M. Sweet, Academic Press, New York, 1997, Vol. 276, part A, p. 307 Search PubMed.
- SMART (version 5.0); data collection software, Bruker AXS, Inc., Madison, WI, 1998 Search PubMed.
- SAINT (version 5.0); data integration software, Bruker AXS, Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS: program for absorption correction with the Bruker SMART system, Universitat Göttingen, Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL (version 6.1), Bruker AXS, Inc., Madison, WI, 2000 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures, tables giving characterization data for 1–5 and X-ray crystallographic data. CCDC 1446704-1446707 and 1453323. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00011h |
| This journal is © the Partner Organisations 2016 |