
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect electrochemical synthesis of pentafluorophenyl esters via oxyl-radical-promoted nucleophilic aromatic substitution†
Edward G. V. Hilvano,
Min-Chieh Liang,
Jacob J. Piane and
Eric D. Nacsa *
*
The Pennsylvania State University, Department of Chemistry, University Park, PA 16802, USA. E-mail: nacsa@psu.edu
First published on 5th June 2025
Abstract
An electrochemical coupling between carboxylic acids and pentafluorophenol (PFP–OH) to access synthetically versatile pentafluorophenyl (PFP) esters has been developed. Novel reactivity of PFP–OH was turned on by modulating its oxidation state, leveraging both its native O-nucleophilicity and its latent, oxidation-induced C-electrophilicity to promote a unique cascade of nucleophilic aromatic and acyl substitutions. Its esterification with acids was thus achieved for the first time without exogenous dehydrating agents. The acidity of PFP–OH and the oxidizability of its conjugate base enabled its mild and selective activation via deprotonation–oxidation, readily affording PFP esters that are useful in many applications (peptide synthesis, chemical biology, etc.) and that contain redox-sensitive functional groups. Finally, we verified in a unified forum that an amino-acid-derived PFP ester can be converted into a range of acyl-substitution products while retaining key stereochemical information, and we demonstrated that PFP esters have excellent stability to hydrolysis, comparing favorably even to N-hydroxysuccinimidyl (NHS) esters.
Introduction
Nucleophilic acyl substitutions are indispensable synthetic transformations, providing access to a range of products such as amides, esters, thioesters, ketones, aldehydes, and other useful compounds.1–4 Since their development over 50 years ago, pentafluorophenyl (PFP) esters have emerged as leading forms of ‘active esters’5–9 designed to undergo these reactions. These acyl electrophiles contain good leaving groups (conjugate acid pKa = 4–10 in H2O)10–12 and thus react well with a broad range of nucleophiles to afford the corresponding acid-substitution products while requiring only a mild base to achieve high yields. Further notable classes of active esters include N-hydroxysuccinimidyl (NHS), N-hydroxybenzotriazolyl, and nitrophenyl esters, among others. Owing the empowering reactivity of PFP esters with nucleophiles, as well as their bench- and water stability (which is superior to that of NHS esters13–15 that are used for similar applications; see below), they have found use in a range of applications, including the synthesis of peptides,16–20 glycosides,21,22 materials,23–31 and pharmaceuticals,32–36 as chemical biology reagents,37–45 and for new synthetic methods46–55 (Fig. 1a). | ||
| Fig. 1 (a) Pentafluorophenyl (PFP) esters are versatile acyl electrophiles that have been used in a range of applications. (b) PFP esters are conventionally prepared using electrophilic dehydrating agents. This work avoids the handling of reactive electrophiles since they are electrochemically generated them in situ from PFP–OH, which must be used anyway to access the desired products. (c) Our reaction design involves the anodic activation of PFP–OH (3) as the corresponding oxyl radical (4), which activates F atoms to nucleophilic aromatic substitution by acid 2 (as its conjugate base, 5). Subsequently, resulting intermediate 6 acylates a second equivalent of PFP–OH (as its conjugate base, 7) to generate the desired PFP ester (1). | ||
PFP esters have exclusively been prepared, however, from the corresponding acids (2) and pentafluorophenol (PFP–OH, 3) using exogenous electrophilic dehydrating agents (Fig. 1b, left),16–50,56–58,59–79 and the native electrophilicity of the latter additives gives rise to operational hazards beyond those associated with essential starting materials 2 and 3. For instance, more-reactive variants that convert acids into acyl chlorides, such as SOCl2 and oxalyl chloride, are corrosive and acutely poisonous,80,81 and milder variants for direct coupling, like carbodiimides (DCC, EDC, etc.), are also toxins and dermal sensitizers.82–87
Cognizant of these challenges, we have developed a dehydrative electrochemical88–94 coupling of carboxylic acids with pentafluorophenol (3) to afford the corresponding PFP esters (1). No reactive electrophiles must be added or handled since they are all anodically generated in situ from PFP–OH (3), which must already be used for PFP ester synthesis (Fig. 1b, right). The only necessary additives (base, electrolyte, and solvent) are comparatively innocuous. As detailed below, this approach electrochemically modulates the oxidation state of PFP–OH to turn on otherwise-elusive reactivity. Moreover, the facile deprotonation of this reagent and subsequent oxidation of its conjugate base lead to mild conditions and an excellent scope for this new electrochemical method.
Design plan
Our reaction design (Fig. 1c) is initiated by deprotonation and single-electron anodic oxidation of PFP–OH (3, pKa = 20.1 for PFP–OH95 and Epox = +0.27 V vs. SCE for PFPO−, both in MeCN) to generate oxyl radical 4. The ortho- and para-C–F groups of this intermediate are activated (red shading on C) to otherwise-challenging nucleophilic aromatic substitution (SNAr) by carboxylate ion 5, owing to the exceptional electron-withdrawing character96 of the conjugated oxyl radical (purple shading) that has an underfilled octet. The resulting, putative O-aryl ester 6 (or a redox analog thereof) would then contain a good phenoxide leaving group (purple shading), making it susceptible to nucleophilic acyl substitution (SNAc, red shading on C) by PFP–O− (7). This step would afford desired PFP ester 1 and a quinone-type byproduct (not shown) after another anodic oxidation. The electrical circuit would be closed by cathodic proton reduction. Overall, this design uses the PFP–OH hydroxyl group in two ways: (1) as a latent umpolung electrophile, leading to the SNAr that activates the carboxylic acid, and (2) as a native nucleophile for SNAc, which forms the product.Results
Starting from this conceptual basis, we identified a simple protocol that efficiently converted model acid 8 and PFP–OH (3) into PFP ester 9 (Fig. 2, see scheme). Under optimized conditions, a mixture of 8 and 3, an organic base (N1,N1,N2,N2-tetramethylguanidine, TMG, 1.5 equiv.), and an electrolyte (Et4NOTs, 2 equiv.) in MeCN (0.05 M in 8) was electrolyzed at a constant cell potential of 3.0 V between a carbon-felt anode and a stainless-steel cathode in an undivided cell, affording PFP ester 9 in 90% yield. Optimal yields of 9 were obtained with a fivefold excess of PFP–OH, but similar or better yields were often obtained with a more-modest excess (3 equiv. of PFP–OH and 1 equiv. of TMG, see below and ESI†). | ||
| Fig. 2 Control experiments electrolyzed a mixture of acid 8 (0.5 mmol, 1 equiv.), PFP–OH (3, 5 equiv.), TMG (1.5 equiv.), and Et4NOTs (2 equiv.) in MeCN (0.05 M in 8) at a constant cell potential of 3.0 V between a carbon-felt anode and stainless-steel cathode. Yields were determined by 1H NMR analysis with an internal standard. Deviations are noted below the scheme. See ESI† for details. | ||
The impact of key reaction parameters is also shown in Fig. 2. Carbon anodes proved uniquely effective, and a lower-porosity material (graphite) was less efficient than C felt (58% yield, entry 1a). Replacing the stainless-steel cathode with C felt was counterproductive (34% yield, entry 1b). Only Pt proved comparable (87% yield, entry 1c), but it was not used thereafter owing to its much-higher cost. Tetraalkylammonium PF6 and BF4 electrolytes proved inferior to Et4NOTs (53–64% yields, entries 2a–c), although Et4NCl performed similarly (80% yield, entry 2d). Me4NOH could serve the dual role as electrolyte and base (80% yield without TMG, entry 2e), albeit in slightly reduced yield. Changes to the cell potential or switching to constant current gave lower yields (entries 3a–d), although a 15 mA current proved efficient (88% yield, entry 3c). The only solvent remotely comparable to MeCN was DMF (67% yield, entry 4a). DMSO (10% yield, entry 4b) represented the performance of most other solvents assessed. The process withstood appreciable amounts of water (5 & 10 equiv., giving 75% and 66% yields, respectively, entries 4c & d). The optimal base, TMG, could be exchanged for Et3N without issue in the model reaction (89% yield, entry 5a), but alternative organic bases such as DBU (55% yield, entry 5b) and especially inorganic bases, all of which were insoluble, were ineffective (9–15% yield, entries 5c–e). As mentioned above, a fivefold excess of PFP–OH (3) gave optimal yields when 1.5 equiv. TMG was used (compare the optimized 90% yield to entries 6a–e), but a threefold excess of PFP–OH with less TMG (1 equiv.) gave comparable results (87% yield, entry 7c). Almost all products obtained throughout this study employed one of these ratios (see below and ESI†). Finally, reaction mixtures were typically sparged with nitrogen and then electrolyzed under a nitrogen atmosphere. The reaction did not proceed under air (0% yield, entry 8a), and skipping the sparging step significantly lowered the efficiency, even if the electrolysis was performed under inert atmosphere (51% yield, entry 8b).
We then evaluated the range of carboxylic acids that could undergo this novel electrochemical PFP esterification. Functionalized aromatic and aliphatic acids were both broadly viable, as shown in Fig. 3. For example, the PFP ester of benzoic acid (10a) was obtained in 82% yield, and its para-halogenated (F, Cl, Br, and I) and CF3 analogs were formed in 70–83% yields (10b–f). Benzoic acids with cyano and methoxy groups at different positions were competent substrates, giving products 10g–k in 50–72% yields, as were 2,4,6-trimethoxybenzoic acid (product 10l, 51% yield) and pentafluorobenzoic acid (product 10m, 46% yield). PFP benzoates with nitrogen-containing para-azido (10n, 61% yield) and NHBoc (10o, 85% yield) groups were similarly effective. A range of extended aromatic and heteroaromatic acids were also tolerated, cleanly affording 2-naphthoic, 3-thienyl, 2- and 3-pyridyl, 6-quinolinyl PFP esters 10p and 11–14 in 72–81% yields.
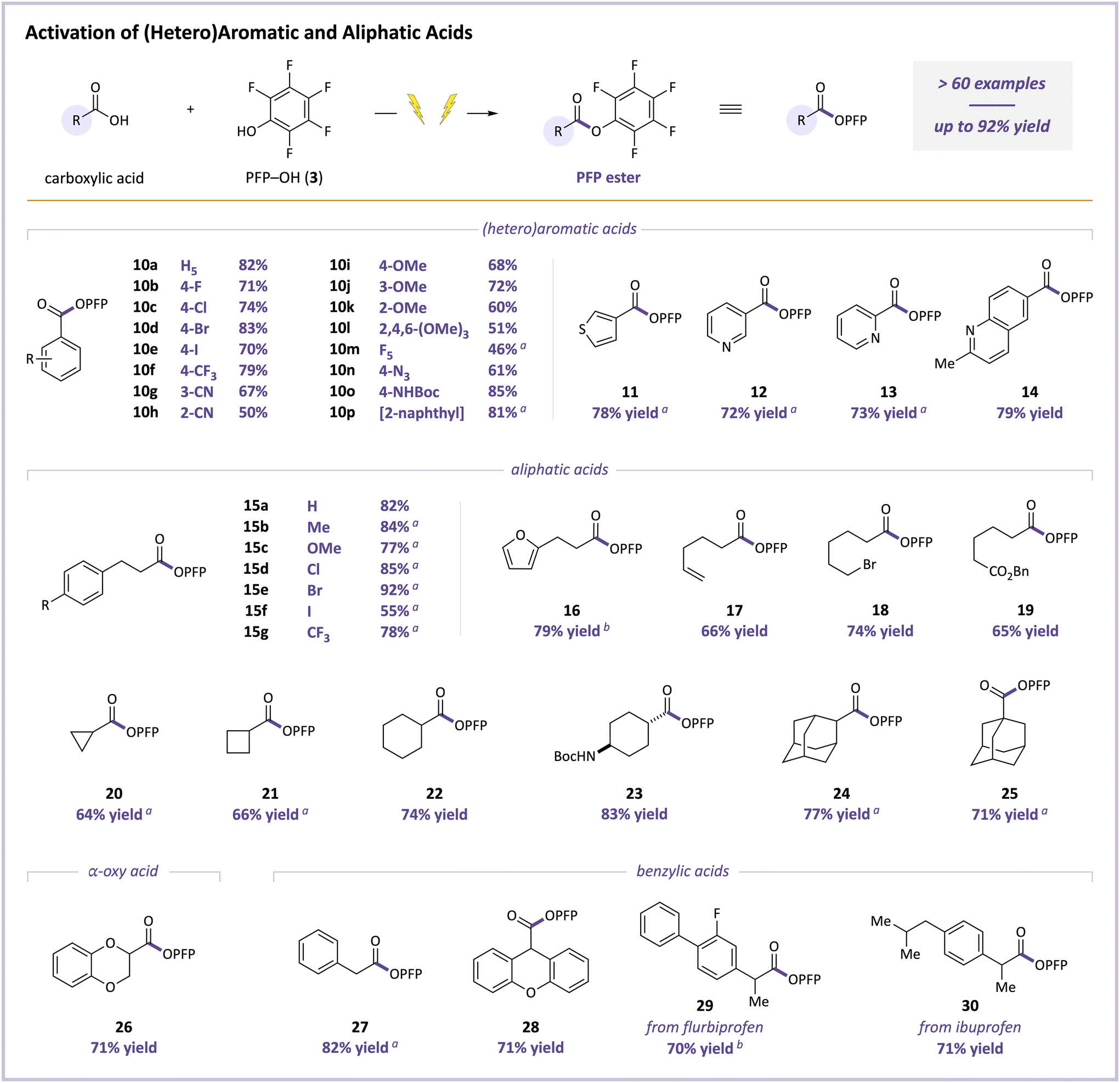 | ||
Fig. 3 Scope for PFP esterification of (hetero)aromatic and aliphatic acids. A mixture of acid (0.5 mmol, 1 equiv.), PFP–OH (3, 3–5 equiv.), TMG (1–1.5 equiv.), and Et4NOTs (2 equiv.) in MeCN (0.05 M in acid) were electrolyzed at a constant cell potential of 3.0 V between a carbon-felt anode and stainless-steel cathode. Yields of isolated products. See ESI† for details. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated as the corresponding N-benzylamide. bPerformed on a larger scale (2 mmol of acid). Isolated as the corresponding N-benzylamide. bPerformed on a larger scale (2 mmol of acid). | ||
In terms of aliphatic acids, 3-phenylpropionate product 15a was isolated in 82% yield, and its para-methyl, methoxy, chloro, bromo, iodo, and trifluoromethyl analogs 15b–g were obtained in 55–92% yields (with only iodo product 15f below 77% yield). Products containing furan, alkene, alkyl bromide, and benzyl ester groups (16–19, 65–79% yields) were also generated cleanly. PFP esters pendant from 3-, 4-, and 6-membered rings (20–22, 64–74% yields), a 4-NHBoc-substituted cyclohexane (23, 83% yield), and both secondary and tertiary adamantyl groups (24 & 25, 77% and 71% yields, respectively) were similarly prepared. Finally, an α-oxy acid and a variety of benzylic acids, which are potentially sensitive to oxidative decarboxylation,97,98 underwent efficient electrolysis, producing PFP esters 26–30 in 70–82% yields. The latter two examples employed the NSAIDs flurbiprofen and ibuprofen as substrates.
PFP esters derived from amino acids have proven particularly valuable as building blocks in peptide synthesis,16–20 materials chemistry,23–31 and chemical biology,37,38,41,42,45 so we also thoroughly verified whether these products were accessible using our electrochemical system. Encouragingly, an excellent range of these PFP esters were efficiently prepared (Fig. 4). Boc-protected β-amino product 31 was obtained in 75% yield. The majority of our efforts then focused on α-amino acids. Common Cbz, Boc, and Fmoc N-protecting groups were all well-tolerated on alanine, affording products 32–34 in 75–81% yields. The PFP ester of serine (35) bearing a free OH group was formed in 52% yield, and O-benzyl protection gave product 36 in higher 72% yield. With appropriate protecting groups, PFP esters of the functionalized amino acids cysteine (37, 52% yield), methionine (38, 88% yield), lysine (39, 74% yield), and arginine (40, 86% yield) were also cleanly isolated. A gram-scale preparation of arginine product 40 proceeded in nearly identical 81% yield. Functional aromatic-containing side chains were also well-tolerated, generating phenylalanine-, tyrosine-, tryptophan-, and histidine-derived products 41–45 in 55–72% yields. More-substituted α-amino acids including proline and unnatural building blocks afforded products 46–48 in 57–66% yields. Critically, excellent stereoretention was observed, as all non-racemic products had ≥96% ee, with most >99% ee. A dipeptide was converted to 49 (64% yield) without epimerization (20:1 dr), and the PFP ester of biotin (50), which is a useful reagent in chemical biology,39,40,43,44 was isolated in 65% yield. Throughout these studies, in cases where yields of PFP esters were modest, acid-derived byproducts were typically not observed. Instead, unproductive acid remained intact. Oligomeric PFP–OH-derived byproducts, which owing to their low polarity were readily removed by chromatography, accounted for the mass balance.
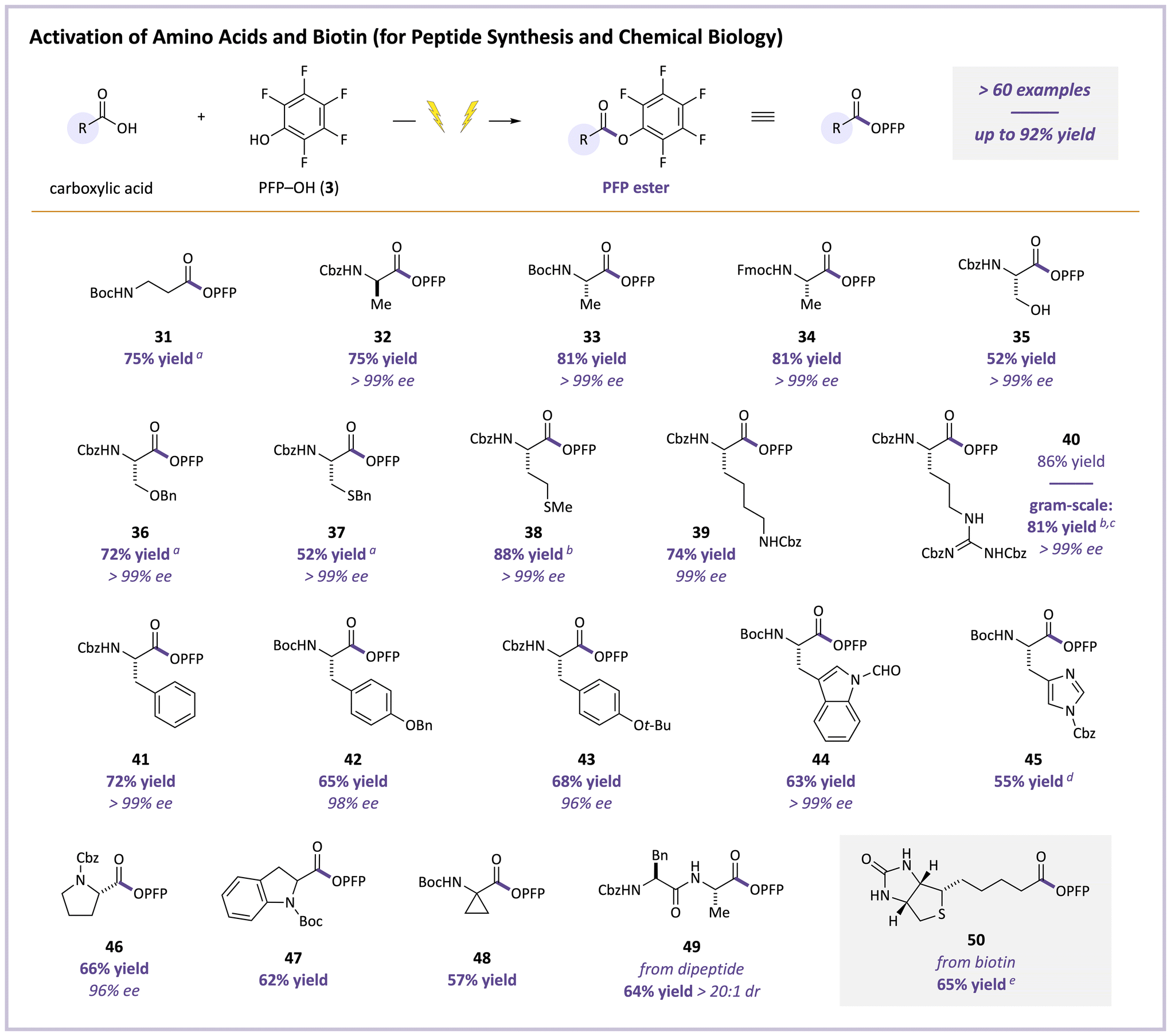 | ||
| Fig. 4 Scope for PFP esterification of amino acids and biotin. A mixture of acid (0.5 mmol, 1 equiv.), PFP–OH (3, 3–5 equiv.), TMG (1–1.5 equiv.), and Et4NOTs (2 equiv.) in MeCN (0.05 M in acid) were electrolyzed at a constant cell potential of 3.0 V between a carbon-felt anode and stainless-steel cathode. Yields of isolated products. See ESI† for details. aIsolated as the corresponding N-benylamide. bPerformed on a larger scale (2 mmol of acid). c3.5-V cell potential. dYield determined by 19F NMR. e3:1 DMF/MeCN solvent. | ||
Having established the broad scope of our electrochemical PFP ester synthesis, we completed our synthetic work by confirming the well-established versatility of these products in downstream synthetic transformations (Fig. 5a). Amino-acid-derived product 41 was prepared in 72% yield (see Fig. 4) and >99% ee. Treatment of this PFP ester with a range of nucleophiles under simple conditions (1.1 equiv. of nucleophile, 1 equiv. of Et3N, MeCN solvent) rapidly and efficiently produced the corresponding carboxylic-acid derivatives either without any or with only a minimal loss of enantiopurity. Specifically, O-alkyl and O-aryl esters 51 & 52 and S-alkyl and S-aryl thioesters 53 & 54 were isolated in 84–95% yields and 96–99% ees. N-Alkyl, N-aryl, and Weinreb amides 55–57 were similarly obtained in 92–98% yields and all in ≥99% ee. Lastly, using α-amino acid esters as the nucleophiles afforded dipeptides 58–60 in 70–92% yields and without epimerization (all ≥99:1 dr), which can be challenging in peptide synthesis.99–101 Although these outcomes are consistent with the well-established performance of PFP esters in a range of applications,16–55 these results showcase that these electrophiles are highly effective acyl surrogates (i.e., providing high yield and minimizing racemization) with a broad range of nucleophiles and without needing to re-optimize reaction conditions.
 | ||
| Fig. 5 (a) Synthetic transformations of amino-acid-derived PFP ester product 36. A range of O-, S-, and N-nucleophiles efficiently generated the corresponding alkyl and aryl ester, thioester, and amide products, including a selection of dipeptides, with minimal racemization. Conditions: a solution of PFP ester 36 (0.5 mmol, 1 equiv.), nucleophile (1.1 equiv.), and Et3N (1 equiv.) in MeCN (0.1 M in 36) were stirred at rt. Yields of purified products are reported. See ESI† for details. aPerformed with a larger excess of nucleophile (3 equiv.). (b & c) Stabilities of acyl electrophiles (acyl chloride, anhydride, NHS ester, and PFP ester) derived from cyclohexane carboxylic acid stored (b) under air and (c) in aqueous solution (0.1 M in 4:1 CD3CN/D2O), with % remaining measured by 1H NMR analysis. | ||
We also sought to compare the stability of amino-acid-derived PFP ester 41 to that of other acyl electrophiles, but we were unable to prepare any quantity of the acyl chloride despite reports of its synthesis.102–109 We therefore compared the stabilities of the acyl chloride, anhydride, NHS ester, and PFP ester derived from cyclohexane carboxylic acid stored as neat compounds under air (Fig. 5b) and in aqueous MeCN (Fig. 5c) by 1H NMR analysis. As expected, the acid chloride decomposed much more rapidly than all other electrophiles, with a half-life of ∼24 h stored under air and decomposing completely within 72 h. The half-life of the anhydride was ∼100 h under air, and it fully decomposed within 300 h, whereas neither NHS nor the PFP active esters suffered any detectable decomposition after 300 h. Starker differences were observed in aqueous solution. The acyl chloride fully decomposed within 15 minutes. The anhydride had a surprisingly long half-life of ∼140 h. The active esters again decomposed much more slowly, but the PFP ester remarkably proved ∼6-fold more stable than the NHS ester. The combined results across Fig. 5 therefore showcase both the synthetic versatility and practical robustness of these acyl electrophiles.
Electrochemical studies were then undertaken to account for the excellent chemoselectivity of this new electrochemical reaction. First, analysis of the reaction components by cyclic voltammetry (CV) revealed that phenoxide 7, the anion of PFP–OH (3), is the most oxidizable species in solution by a significant margin (Fig. 6a). Neither PFP–OH (Eoxp = + 1.58 V vs. SCE) nor model acid 8 (Eoxp > +2 V vs. SCE) were readily oxidized, which is consistent with the need for a base in the electrochemical reaction. The standard base employed throughout these studies (TMG, pKBH+ = 23.4 in MeCN,110 the standard reaction solvent) can deprotonate both the carboxylic acid (pKa = 23.5 for AcOH in MeCN)95 and PFP–OH (pKa = 20.1 in MeCN),95 although given their relative acidities and the stoichiometries employed (either 1:5:1.5 or 1:3:1 of carboxylic acid/PFP–OH/TMG), PFP–OH should be deprotonated to a much-greater extent than the carboxylic acid (this acid–base equilibrium also makes TMG oxidation unlikely, see ESI†). Even without accounting for this speciation, which would selectively activate PFP–OH to oxidation, PFP–O− (7, Eoxp = +0.27 V vs. SCE) proved more oxidizable than the carboxylate derived from acid 8 (61, Eoxp = +0.96 V vs. SCE) by 0.69 V. We postulate that this facile oxidation underpins the generality of the synthetic protocol, which did not lead to undesired oxidation of α-amino acids, benzylic acids, or dialkyl sulfides. Consistent with this scenario, the potential of the anode during preparative constant-cell-potential electrolysis underwent a modest anodic drift, but it stayed within a range of +0.64 V to +0.77 V vs. SCE (Fig. 6b). Presumably, the anode selectively oxidizes the PFP–OH/PFP–O−mixture present at any given time, but never reaches an energy that would destroy the carboxylate or other groups.111
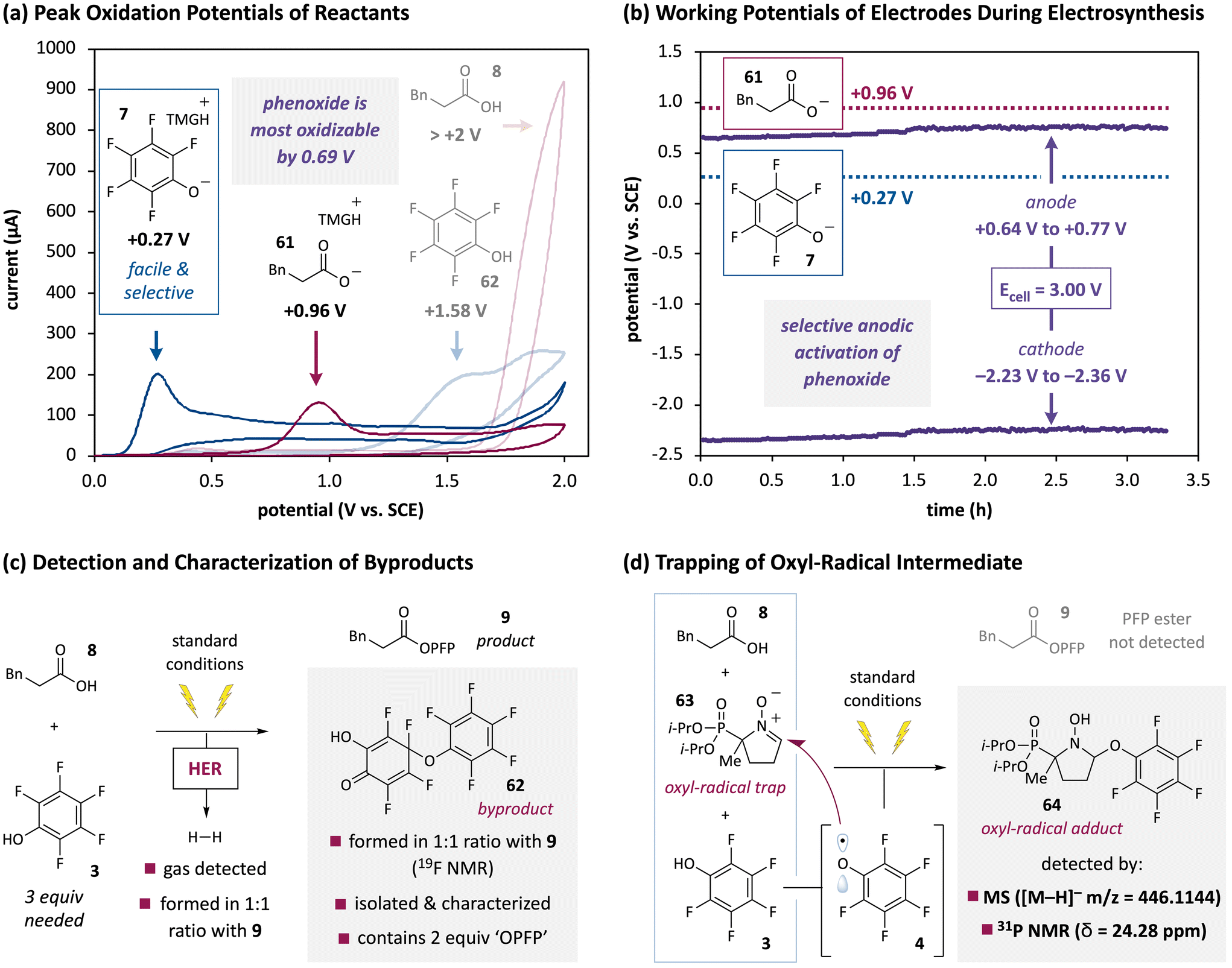 | ||
| Fig. 6 Preliminary mechanistic experiments. (a) CV analyses revealed that the conjugate base of PFP–OH (phenoxide 7) is oxidized at a significantly lower potential (+0.27 V vs. SCE) than any other possible major species in solution. (b) The potential of the anode varied from +0.64 V to +0.77 V vs. SCE during electrolysis, which can selectively engage phenoxide 7 while leaving carboxylates and other potentially oxidizable species intact. (c) Both H2 and 62, which contains two ‘OPFP’ units, were formed in equimolar ratios with PFP ester 9. (d) The model reaction forming PFP ester 9 did not proceed when adding radical trap 63, and oxyl-radical adduct 64 was detected by HRMS and 31P NMR. See ESI† for details. | ||
Finally, preliminary studies were performed to probe the reaction mechanism. As shown in Fig. 1c, we proposed that a net oxidation of the organic reactants would be enabled by cathodic hydrogen evolution. Indeed, preparative experiments were performed with a vent needle since the reaction vessel otherwise became noticeably pressurized and occasionally burst. A hydrogen detector confirmed the formation of this gas, and semiquantitative estimates indicated that it was produced in an equimolar amount with PFP ester 9 (Fig. 6c). Furthermore, when the reaction was monitored by 19F NMR, an organic byproduct was also formed in a ∼1:1 ratio with desired product 9. Careful isolation enabled its characterization and structural assignment as 62, which contains two ‘OPFP’ units. This byproduct explains why a threefold excess of PFP–OH is needed for the reaction to proceed (see Fig. 2). As discussed below, it also required a minor revision of our mechanistic hypothesis (Fig. 1c). On the other hand, the proposed single-electron oxidation to generate PFP–OH-derived oxyl radical 4, which would facilitate the proposed SNAr,96 was consistent with radical-trapping experiments (Fig. 6d). Addition of oxyl-radical trap 63112 to a standard preparative electrolysis completely prevented the formation of PFP ester 9, and oxyl-radical adduct 64 was detected both by HRMS and 31P NMR analysis (no adduct formation or substrate conversion of any kind occurred without electrolysis, see ESI†).
These observations led to the revised mechanistic proposal shown in Fig. 7. As initially suggested in Fig. 1c, deprotonation and single-electron anodic oxidation of PFP–OH (3) generate oxyl radical 4, the highlighted C–F bonds of which are activated to SNAr by carboxylate ion 5.96 Putative intermediate 6 then undergoes a further anodic oxidation and is trapped by a second equivalent of PFP–O− (7), generating closed-shell acyl electrophile 65. Finally, SNAc with a third and final equivalent of PFP–O− (7) affords PFP ester 1 and byproduct 62.
 | ||
| Fig. 7 Revised mechanistic hypothesis. Deprotonation and anodic oxidation of PFP–OH (3) generates oxyl radical 4, which undergoes SNAr with carboxylate 5. Resulting open-shell O-aryl ester 6 is oxidized further and trapped by a second equivalent of PFP–O− (7), producing acyl electrophile 65. SNAc by a third equivalent of PFP–O− (7) affords PFP ester 1 and byproduct 62. | ||
Conclusions
We have developed a novel electrochemical coupling of carboxylic acids with pentafluorophenol (PFP–OH) to access synthetically versatile pentafluorophenyl (PFP) esters. This system strategically modulates the oxidation state of the hydroxyl group in PFP–OH to turn on otherwise-elusive reactivity. By leveraging both the latent electrophilicity and the native nucleophilicity of this reagent, a unique SNAr/SNAc cascade ultimately generates the PFP ester product. As a result, this useful transformation can be accomplished for the first time without electrophilic dehydrating agents. Moreover, owing to the acid–base and electrochemical properties of PFP–OH that enabled its selective activation under mild conditions, an excellent range of PFP esters that are useful in a wide range of applications and that contain oxidation-sensitive functional groups were efficiently prepared. Finally, we confirmed that an amino-acid-derived PFP ester reliably affords a range of acyl-substitution products without any or with only minimal epimerization, and we demonstrated that PFP esters have excellent stability to hydrolysis, comparing favorably even to N-hydroxysuccinimidyl (NHS) esters.Author contributions
All authors were involved in the discovery and conception of the project. E. G. V. H., M.-C., L., and J. J. P. performed experiments and collected data. All authors analyzed data. E. D. N. prepared the manuscript and E. G. V. H. prepared the ESI.† All authors revised the manuscript.Data availability
Detailed experimental procedures, compound characterization data, preliminary mechanistic experiments, copies of NMR spectra (PDF).The datasets supporting this article have been uploaded as part of the ESI.†
Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors are grateful for financial support from The Pennsylvania State University Eberly College of Science. E. D. N. thanks the American Chemical Society Petroleum Research Fund for a Doctoral New Investigator Grant (Award 65252-DNI) and the National Science Foundation for a CAREER Award (CHE-2339405). Mass spectrometric analysis was performed at the Penn State Proteomics and Mass Spectrometry Core Facility, University Park, PA (RRID: CR_024462).References
- K. M. Lokanatha Rai, Carbonyl Compounds - Chemistry and Synthetic Applications, Notion Press Media, 2020 Search PubMed.
- Comprehensive Organic Synthesis, ed. P. Knochel, Elsevier, Amsterdam, 2nd edn, 2014, vol 6, pp. 296-597 Search PubMed.
- K. P. C. Vollhardt and N. E. Schore, in Organic Chemistry: Structure and Function, W. H. Freeman and Company, New York, NY, 5th edn, 2007, ch. 19, pp. 856–908 Search PubMed.
- J. D. Roberts and M. C. Caserio, Basic Principles of Organic Chemistry, W. A. Benjamin, Inc., New York, NY, USA, 1965 Search PubMed.
- J. Yang, H. Huang and J. Zhao, Active ester-based peptide bond formation and its application in peptide synthesis, Org. Chem. Front., 2023, 10, 1817–1846 RSC.
- J. Dussart-Gautheret, J. Deschamp, M. Monteil, O. Gager, T. Legigan, E. Migianu-Griffoni and M. Lecouvey, Formation of 1-Hydroxymethylene-1,1-bisphosphinates through the Addition of a Silylated Phosphonite on Various Trivalent Derivatives, J. Org. Chem., 2020, 85, 14559–14569 CrossRef CAS PubMed.
- M. Bodanszky and A. Bodanszky, in The Practice of Peptide Synthesis, Springer, Berlin, 2nd edn, 1994, pp. 96–107 Search PubMed.
- L. Kisfaludy, T. Mohacsi, M. Low and F. Drexler, Pentafluorophenyl acetate: a new, highly selective acetylating agent, J. Org. Chem., 1979, 44, 654–656 CrossRef CAS.
- R. B. Merrifield, Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- D. Stefanidis, S. Cho, S. Dhe-Paganon and W. P. Jencks, Structure-reactivity correlations for reactions of substituted phenolate anions with acetate and formate esters, J. Am. Chem. Soc., 1993, 115, 1650–1656 CrossRef CAS.
- D. G. McCarthy, A. F. Hegarty and B. J. Hathaway, N-hydroxy-compounds as acyl transfer agents. Part 1. Kinetics and mechanism of nucleophilic displacements on 1-hydroxybenzotriazole esters and crystal and molecular structure of 1-benzoyloxybenzotriazole, J. Chem. Soc., Perkin Trans. 2, 1977, 224–231 RSC.
- D. E. Ames and T. F. Grey, The synthesis of some N-hydroxyimides, J. Chem. Soc., 1955, 631–636 RSC.
- C. Ma, M. Chen, W. Chu, J. Tao, D. Kong, M. Zhang and W. Feng, A Practical and Total Synthesis of Pasireotide: Synthesis of Cyclic Hexapeptide via a Three-Component Condensation, Molecules, 2019, 24, 2185 CrossRef CAS PubMed.
- S. Asano, J. T. Patterson, T. Gaj and C. F. Barbas, 3rd, Site-selective labeling of a lysine residue in human serum albumin, Angew. Chem., Int. Ed., 2014, 53, 11783–11786 CrossRef CAS PubMed.
- G. W. Anderson, J. E. Zimmerman and F. M. Callahan, N-Hydroxysuccinimide Esters in Peptide Synthesis, J. Am. Chem. Soc., 1963, 85, 3039–3039 CrossRef CAS.
- A. Wu and H. Yamamoto, Super silyl-based stable protecting groups for both the C- and N-terminals of peptides: applied as effective hydrophobic tags in liquid-phase peptide synthesis, Chem. Sci., 2023, 14, 5051–5061 RSC.
- A. T. Ślósarczyk, R. Ramapanicker, T. Norberg and L. Baltzer, Mixed pentafluorophenyl and o-fluorophenyl esters of aliphatic dicarboxylic acids: efficient tools for peptide and protein conjugation, RSC Adv., 2012, 2, 908–914 RSC.
- C. A. G. N. Montalbetti and V. Falque, Amide bond formation and peptide coupling, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- V. N. Karel'skii, E. P. Krysin, A. A. Antonov and G. E. Rostovskaya, Use of pentafluorophenyl esters in the synthesis of intermediate fragments of ACTH, Chem. Nat. Compd., 1982, 18, 90–94 CrossRef.
- L. Kisfaludy, J. E. Roberts, R. H. Johnson, G. L. Mayers and J. Kovacs, Synthesis of N-carbobenzoxyamino acid and peptide pentafluorophenyl esters as intermediates in peptide synthesis, J. Org. Chem., 1970, 35, 3563–3565 CrossRef CAS PubMed.
- A. Imamura, A. Kimura, H. Ando, H. Ishida and M. Kiso, Extended applications of di-tert-butylsilylene-directed alpha-predominant galactosylation compatible with C2-participating groups toward the assembly of various glycosides, Chem. – Eur. J., 2006, 12, 8862–8870 CrossRef CAS PubMed.
- H. Ando, A. Imamura, M. Kiso and H. Ishida, Method of Alpha-Selective Glycosylation, European Patent, 1,626,053 A2, 2004 Search PubMed.
- J. J. Tarrio, R. Rodriguez, B. Fernandez, E. Quinoa and F. Freire, Dissymmetric Chiral Poly(diphenylacetylene)s: Secondary Structure Elucidation and Dynamic Luminescence, Angew. Chem., Int. Ed., 2022, 61, e202115070 CrossRef CAS PubMed.
- A. P. P. Kröger, J.-W. D. Paats, R. J. E. A. Boonen, N. M. Hamelmann and J. M. J. Paulusse, Pentafluorophenyl-based single-chain polymer nanoparticles as a versatile platform towards protein mimicry, Polym. Chem., 2020, 11, 6056–6065 RSC.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, A simple and efficient synthesis of functionalized cyclic carbonate monomers using a versatile pentafluorophenyl ester intermediate, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS PubMed.
- P. J. Roth, K. T. Wiss, R. Zentel and P. Theato, Synthesis of Reactive Telechelic Polymers Based on Pentafluorophenyl Esters, Macromolecules, 2008, 41, 8513–8519 CrossRef CAS.
- M. M. Dominguez, M. Wathier, M. W. Grinstaff and S. E. Schaus, Immobilized hydrogels for screening of molecular interactions, Anal. Chem., 2007, 79, 1064–1066 CrossRef CAS PubMed.
- M. W. Grinstafe, J. Butlin, M. A. Carnahan, K. R. D'Alessio and T. P. Hickey, Low-Swelling Hydrogel Sealants for Wound Repair, World Patent, 2007/001926 A2, 2007 Search PubMed.
- M. W. Grinstafe and M. A. Carnahan, Dendritic Polymers, Crosslinked Gels, and Their Uses as Ophthalmic Sealants and Lenses, World Patent, 2006/031358 A2, 2006 Search PubMed.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Synthesis of pentafluorophenyl(meth)acrylate polymers: New precursor polymers for the synthesis of multifunctional materials, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- M. Wathier, P. J. Jung, M. A. Carnahan, T. Kim and M. W. Grinstaff, Dendritic macromers as in situ polymerizing biomaterials for securing cataract incisions, J. Am. Chem. Soc., 2004, 126, 12744–12745 CrossRef CAS PubMed.
- M. Calvert, R. P. Sweeney, H. M. Chen, H. Bajwa, S. A. Nasseri, D. Habibi and S. G. Withers, Branched montbretin A mimics allow derivatisation and potent amylase inhibition, Org. Biomol. Chem., 2023, 21, 7977–7983 RSC.
- G. Bernardes, L. Couturier, J. Becher and E. Gil de Montes Rojas, Quinone Protected Forms and Conjugates, World Patent, 2023/227757 A1, 2023 Search PubMed.
- S. Nasim, S. Pei, F. K. Hagen, C. T. Jordan and P. A. Crooks, Melampomagnolide B: a new antileukemic sesquiterpene, Bioorg. Med. Chem., 2011, 19, 1515–1519 CrossRef CAS PubMed.
- A. Homma, H. Sato, T. Tamura, A. Okamachi, T. Emura, T. Ishizawa, T. Kato, T. Matsuura, S. Sato, Y. Higuchi, T. Watanabe, H. Kitamura, K. Asanuma, T. Yamazaki, M. Ikemi, H. Kitagawa, T. Morikawa, H. Ikeya, K. Maeda, K. Takahashi, K. Nohmi, N. Izutani, M. Kanda and R. Suzuki, Synthesis and optimization of hyaluronic acid-methotrexate conjugates to maximize benefit in the treatment of osteoarthritis, Bioorg. Med. Chem., 2010, 18, 1062–1075 CrossRef CAS PubMed.
- S. Lociuro, P. Tavecchia, R. Ciabatti and E. Restelli, Derivatives of Antibiotic GE2270 Factors C2A, D2, and E, US Patent, 6,008,225 A, 1999 Search PubMed.
- H. Ren, Q. Hu, J. Yang, X. Zhou, X. Liu, J. Tang, H. Hu, Y. Shen and Z. Zhou, Single-Molecule Dendritic MRI Nanoprobes Reveal the Size-Dependent Tumor Entrance, Adv. Healthcare Mater., 2023, 12, e2302210 CrossRef PubMed.
- H. Ren, Q. Hu, Y. Sun, X. Zhou, Y. Zhu, Q. Dong, L. Chen, J. Tang, H. Hu, Y. Shen and Z. Zhou, Surface chemistry mediates the tumor entrance of nanoparticles probed using single-molecule dual-imaging nanodots, Biomater. Sci., 2023, 11, 7051–7061 RSC.
- V. Waser, M. Mukherjee, R. Tachibana, N. V. Igareta and T. R. Ward, An Artificial [Fe4S4]-Containing Metalloenzyme for the Reduction of CO2 to Hydrocarbons, J. Am. Chem. Soc., 2023, 145, 14823–14830 CrossRef CAS PubMed.
- W. Wang, R. Tachibana, Z. Zou, D. Chen, X. Zhang, K. Lau, F. Pojer, T. R. Ward and X. Hu, Manganese Transfer Hydrogenases Based on the Biotin-Streptavidin Technology, Angew. Chem., Int. Ed., 2023, 62, e202311896 CrossRef CAS PubMed.
- D. Kang, S. Lee and J. Kim, Bioorthogonal Click and Release: A General, Rapid, Chemically Revertible Bioconjugation Strategy Employing Enamine N-oxides, Chem, 2022, 8, 2260–2277 CAS.
- M. Scherer, K. Fischer, F. Depoix, T. Fritz, R. Thiermann, K. Mohr and R. Zentel, Pentafluorophenyl Ester-based Polymersomes as Nanosized Drug-Delivery Vehicles, Macromol. Rapid Commun., 2016, 37, 60–66 CrossRef CAS PubMed.
- J. M. Chambers, L. M. Lindqvist, A. Webb, D. C. Huang, G. P. Savage and M. A. Rizzacasa, Synthesis of biotinylated episilvestrol: highly selective targeting of the translation factors eIF4AI/II, Org. Lett., 2013, 15, 1406–1409 CrossRef CAS PubMed.
- L. Dafik, V. Kalsani, A. K. Leung and K. Kumar, Fluorinated lipid constructs permit facile passage of molecular cargo into living cells, J. Am. Chem. Soc., 2009, 131, 12091–12093 CrossRef CAS PubMed.
- R. Kluger and A. Alagic, Chemical cross-linking and protein-protein interactions-a review with illustrative protocols, Bioorg. Chem., 2004, 32, 451–472 CrossRef CAS PubMed.
- H. K. Banovetz, K. L. Vickerman, C. M. David, M. Alkan and L. M. Stanley, Palladium-Catalyzed Intermolecular Alkene Carboacylation via Ester C-O Bond Activation, Org. Lett., 2021, 23, 3507–3512 CrossRef CAS PubMed.
- H. Li, Y. Hou, C. Liu, Z. Lai, L. Ning, R. Szostak, M. Szostak and J. An, Pentafluorophenyl Esters: Highly Chemoselective Ketyl Precursors for the Synthesis of α,α-Dideuterio Alcohols Using SmI2 and D2O as a Deuterium Source, Org. Lett., 2020, 22, 1249–1253 CrossRef CAS PubMed.
- J. Buchspies, D. J. Pyle, H. He and M. Szostak, Pd-Catalyzed Suzuki-Miyaura Cross-Coupling of Pentafluorophenyl Esters, Molecules, 2018, 23, 3134 CrossRef PubMed.
- S. Specklin and J. Cossy, Chemoselective synthesis of beta-ketophosphonates using lithiated alpha-(trimethylsilyl)methylphosphonate, J. Org. Chem., 2015, 80, 3302–3308 CrossRef CAS PubMed.
- B. G. Avitabile, C. A. Smith and D. B. Judd, Pentafluorophenyl sulfonate ester as a protecting group for the preparation of biaryl- and heterobiaryl sulfonate esters, Org. Lett., 2005, 7, 843–846 CrossRef CAS PubMed.
- M. Zhu, P. Wang, Q. Zhang, W. Tang and W. Zi, Diastereodivergent Aldol-Type Coupling of Alkoxyallenes with Pentafluorophenyl Esters Enabled by Synergistic Palladium/Chiral Lewis Base Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202207621 CrossRef CAS PubMed.
- Q. Zhang, M. Zhu and W. Zi, Synergizing palladium with Lewis base catalysis for stereodivergent coupling of 1,3-dienes with pentafluorophenyl acetates, Chem, 2022, 8, 2784–2796 CAS.
- K. J. Schwarz, C. M. Pearson, G. A. Cintron-Rosado, P. Liu and T. N. Snaddon, Traversing Steric Limitations by Cooperative Lewis Base/Palladium Catalysis: An Enantioselective Synthesis of alpha-Branched Esters Using 2-Substituted Allyl Electrophiles, Angew. Chem., Int. Ed., 2018, 57, 7800–7803 CrossRef CAS PubMed.
- L. Hutchings-Goetz, C. Yang and T. N. Snaddon, Enantioselective alpha-Allylation of Aryl Acetic Acid Esters via C1-Ammonium Enolate Nucleophiles: Identification of a Broadly Effective Palladium Catalyst for Electron-Deficient Electrophiles, ACS Catal., 2018, 8, 10537–10544 CrossRef CAS PubMed.
- K. J. Schwarz, J. L. Amos, J. C. Klein, D. T. Do and T. N. Snaddon, Uniting C1-Ammonium Enolates and Transition Metal Electrophiles via Cooperative Catalysis: The Direct Asymmetric alpha-Allylation of Aryl Acetic Acid Esters, J. Am. Chem. Soc., 2016, 138, 5214–5217 CrossRef CAS PubMed.
- H. Ando and A. Imamura, Method of Alpha-Selective Glycosylation, European Patent, 1,626,053 A2, 2004 Search PubMed.
- M. Adamczyk, J. R. Fishpaugh and K. J. Heuser, Preparation of succinimidyl and pentafluorophenyl active esters of 5- and 6-carboxyfluorescein, Bioconjugate Chem., 1997, 8, 253–255 CrossRef CAS PubMed.
- Despite significant progress in the activation of acids (see following reference), the poor nucleophilicity of PFP–OH in its neutral form may account for its complete absence as a reported nucleophile in any of these methods.
- J. Zhou, M. Paladino and D. G. Hall, Direct Boronic Acid Promoted Amidation of Carboxylic Acids with Poorly Nucleophilic Amines, Eur. J. Org. Chem., 2022, e202201050 CrossRef CAS.
- Y. Zheng, Y. Zhao, S. Tao, X. Li, X. Cheng, G. Jiang and X. Wan, Green Esterification of Carboxylic Acids Promoted by tert–Butyl Nitrite, Eur. J. Org. Chem., 2021, 2713–2718 CrossRef CAS.
- S. Nagahara, Y. Okada, Y. Kitano and K. Chiba, Biphasic electrochemical peptide synthesis, Chem. Sci., 2021, 12, 12911–12917 RSC.
- L. A. Wolzak, J. I. Vlugt, K. J. Berg, J. N. H. Reek, M. Tromp and T. J. Korstanje, Titanium–catalyzed esterification reactions: beyond Lewis acidity, ChemCatChem, 2020, 12, 5229–5235 CrossRef CAS.
- Handoko, S. Satishkumar, N. R. Panigrahi and P. S. Arora, Rational Design of an Organocatalyst for Peptide Bond Formation, J. Am. Chem. Soc., 2019, 141, 15977–15985 CrossRef CAS PubMed.
- Z. Liu, H. Noda, M. Shibasaki and N. Kumagai, Catalytic Oligopeptide Synthesis, Org. Lett., 2018, 20, 612–615 CrossRef CAS PubMed.
- H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Unique physicochemical and catalytic properties dictated by the B3NO2 ring system, Nat. Chem., 2017, 9, 571–577 CrossRef CAS PubMed.
- H. Lundberg, F. Tinnis and H. Adolfsson, Direct amide coupling of non-activated carboxylic acids and amines catalysed by zirconium(IV) chloride, Chem. – Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed.
- N. Gernigon, R. M. Al-Zoubi and D. G. Hall, Direct amidation of carboxylic acids catalyzed by ortho-iodo arylboronic acids: catalyst optimization, scope, and preliminary mechanistic study supporting a peculiar halogen acceleration effect, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed.
- H. Adolfsson, H. Lundberg and F. Tinnis, Titanium(IV) Isopropoxide as an Efficient Catalyst for Direct Amidation of Nonactivated Carboxylic Acids, Synlett, 2012, 2201–2204 CrossRef.
- R. M. Al-Zoubi, O. Marion and D. G. Hall, Direct and waste-free amidations and cycloadditions by organocatalytic activation of carboxylic acids at room temperature, Angew. Chem., Int. Ed., 2008, 47, 2876–2879 CrossRef CAS PubMed.
- R. K. Mylavarapu, K. Gcm, N. Kolla, R. Veeramalla, P. Koilkonda, A. Bhattacharya and R. Bandichhor, Boric Acid Catalyzed Amidation in the Synthesis of Active Pharmaceutical Ingredients, Org. Process Res. Dev., 2007, 11, 1065–1068 CrossRef CAS.
- T. Funatomi, K. Wakasugi, T. Misaki and Y. Tanabe, Pentafluorophenylammonium triflate (PFPAT): an efficient, practical, and cost-effective catalyst for esterification, thioesterification, transesterification, and macrolactone formation, Green Chem., 2006, 8, 1022–1027 RSC.
- K. Ishihara, M. Nakayama, S. Ohara and H. Yamamoto, Direct ester condensation from a 1:1 mixture of carboxylic acids and alcohols catalyzed by hafnium(IV) or zirconium(IV) salts, Tetrahedron, 2002, 58, 8179–8188 CrossRef CAS.
- K. Manabe, X. M. Sun and S. Kobayashi, Dehydration reactions in water. Surfactant-type Bronsted acid-catalyzed direct esterification of carboxylic acids with alcohols in an emulsion system, J. Am. Chem. Soc., 2001, 123, 10101–10102 CrossRef CAS PubMed.
- K. Wakasugi, T. Misaki, K. Yamada and Y. Tanabe, Diphenylammonium triflate (DPAT): efficient catalyst for esterification of carboxylic acids and for transesterification of carboxylic esters with nearly equimolar amounts of alcohols, Tetrahedron Lett., 2000, 41, 5249–5252 CrossRef CAS.
- K. Ishihara, S. Ohara and H. Yamamoto, Direct condensation of carboxylic acids with alcohols catalyzed by Hafnium(IV) salts, Science, 2000, 290, 1140–1142 CrossRef CAS PubMed.
- K. Ishihara and H. Yamamoto, Arylboron Compounds as Acid Catalysts in Organic Synthetic Transformations, Eur. J. Org. Chem., 1999, 527–538 CrossRef CAS.
- Y. Masaki, N. Tanaka and T. Miura, Mild Esterification and Transesterification of Carboxylic Acids Catalyzed by Tetracyanoethylene and Dicyanoketene Dimethyl Acetal, Chem. Lett., 1997, 26, 55–56 CrossRef.
- K. Ishihara, S. Ohara and H. Yamamoto, 3,4,5-Trifluorobenzeneboronic Acid as an Extremely Active Amidation Catalyst, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS PubMed.
- J. Otera, N. Danoh and H. Nozaki, Novel template effects of distannoxane catalysts in highly efficient transesterification and esterification, J. Org. Chem., 1991, 56, 5307–5311 CrossRef CAS.
- S. J. Barbee, J. J. Stone and R. J. Hilaski, Acute inhalation toxicology of oxalyl chloride, Am. Ind. Hyg. Assoc. J., 1995, 56, 74–76 CrossRef CAS PubMed.
- J. Pauluhn, Study for Acute Inhalation Toxicity in Rats in Accordance with OECD Guideline No. 403 (Exposure: 1 × 1 Hour), Bayer AG, Leverkusen, Germany, 1987 Search PubMed.
- M. Matsumoto, H. Ito, A. Tateishi, Y. Kobayashi, K. Satoh, K. Numata and H. Miyakawa, Effects of polycaprolactone degradation products on the water flea, Daphnia magna: Carbodiimide additives have acute and chronic toxicity, J. Appl. Toxicol., 2023, 43, 1840–1848 CrossRef CAS PubMed.
- I. Surh, M. Behl, S. A. Elmore and R. S. Chhabra, Comparative dermal toxicity of dicyclohexylcarbodiimide and diisopropylcarbodiimide in rodents, Cutaneous Ocul. Toxicol., 2012, 31, 177–187 CrossRef CAS PubMed.
- National Toxicology Program, NTP Report on the Toxicology Study of Diispropylcarbodiimide (CAS No. 693-13-0) in Genetically Modified (FVB Tg.AC Hemizygous) Mice and Carcinogenicity Study of Diispropylcarbodiimide in Genetically Modified [B6.129-Trp53tm1Brd (N5) Haploinsufficient] Mice (Dermal Studies), Report NTP GMM 10, 2007.
- National Toxicology Program, NTP Genetically Modified Model Report on the Toxicology Studies of Dicyclohexylcarbodiimide (CASRN 538-75-0) in F344/N Rats, B6C3F1 Mice, and Genetically Modified (FVB Tg.AC Hemizygous) Mice and Carcinogenicity Study of Dicyclohexylcarbodiimide in Genetically Modified [B6.129-Trp53tm1Brd (N5) Haploinsufficient] Mice (Dermal Studies), Report NTP GMM 09, 2007.
- National Toxicology Program, Toxicology and Carcinogenesis Studies of Diisopropylcarbodiimide in F344/N Rats and B6C3F1 Mice (Dermal Studies), Report NTP TR 523, 2007.
- A. B. Moshnikova, V. N. Afanasyev, O. V. Proussakova, S. Chernyshov, V. Gogvadze and I. P. Beletsky, Cytotoxic activity of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide is underlain by DNA interchain cross-linking, Cell. Mol. Life Sci., 2006, 63, 229–234 CrossRef CAS PubMed.
- M. C. Leech and K. Lam, A practical guide to electrosynthesis, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef PubMed.
- E. O. Bortnikov and S. N. Semenov, Unconventional approaches for organic electrosynthesis: Recent progress, Curr. Opin. Electrochem., 2022, 35, 101050 CrossRef CAS.
- J. C. Siu, N. Fu and S. Lin, Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Making electrochemistry easily accessible to the synthetic chemist, Green Chem., 2020, 22, 3358–3375 RSC.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, Organic electrosynthesis: from academia to industry, React. Chem. Eng., 2020, 5, 977–990 RSC.
- A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Electrifying Organic Synthesis, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Strengths of Acids in Acetonitrile, Eur. J. Org. Chem., 2021, 1407–1419 CrossRef.
- N. Y. Shin, E. Tsui, A. Reinhold, G. D. Scholes, M. J. Bird and R. R. Knowles, Radicals as Exceptional Electron-Withdrawing Groups: Nucleophilic Aromatic Substitution of Halophenols Via Homolysis-Enabled Electronic Activation, J. Am. Chem. Soc., 2022, 144, 21783–21790 CrossRef CAS PubMed.
- M. K. Abdel Latif, J. N. Spencer, B. E. Kidd, L. A. Madsen and J. M. Tanko, Electrochemical Oxidation of Aliphatic Carboxylates: Kinetics, Thermodynamics, Mechanism, and the Role of Hydrogen Bonding, ChemElectroChem, 2024, 11, e202300514 CrossRef CAS.
- J. Hioe and H. Zipse, Radical stability and its role in synthesis and catalysis, Org. Biomol. Chem., 2010, 8, 3609–3617 RSC.
- Y. Zhou, H. Li, Y. Huang, J. Li, G. Deng, G. Chen, Z. Xi and C. Zhou, Suppression of alpha-carbon racemization in peptide synthesis based on a thiol-labile amino protecting group, Nat. Commun., 2023, 14, 5324 CrossRef CAS PubMed.
- L. Ferrazzano, M. Catani, A. Cavazzini, G. Martelli, D. Corbisiero, P. Cantelmi, T. Fantoni, A. Mattellone, C. De Luca, S. Felletti, W. Cabri and A. Tolomelli, Sustainability in peptide chemistry: current synthesis and purification technologies and future challenges, Green Chem., 2022, 24, 975–1020 RSC.
- S. Carganico and A. M. Papini, in Amino Acids, Peptides and Proteins in Organic Chemistry: Building Blocks, Catalysis and Coupling Chemistry, ed. A. B. Hughes, Wiley–VCH Verlag GmbH & Co. KGaA, 2011, vol. 3, ch. 9, pp. 313–348 Search PubMed.
- N. Sugisawa, A. Ando and S. Fuse, Rapid and column-chromatography-free peptide chain elongation via a one-flow, three-component coupling approach, Chem. Sci., 2023, 14, 6986–6991 RSC.
- D. Jang and J.-G. Kim, A Convenient, One-Pot Procedure for the Preparation of Acyl and Sulfonyl Fluorides Using Cl3CCN, Ph3P, and TBAF(t-BuOH)4, Synlett, 2010, 3049–3052 CrossRef.
- M. P. Paradisi, G. P. Zecchini, I. Torrini and G. Lucente, Synthesis, stereochemistry and conformational properties of diastereomeric cyclic dipeptides containing tetrahydro–1,4−thiazine–3,5−dicarboxylic acid, J. Heterocycl. Chem., 2009, 27, 1661–1664 CrossRef.
- G. Evindar and R. A. Batey, Parallel synthesis of a library of benzoxazoles and benzothiazoles using ligand-accelerated copper-catalyzed cyclizations of ortho-halobenzanilides, J. Org. Chem., 2006, 71, 1802–1808 CrossRef CAS PubMed.
- U. Schmidt, U. Beutler and A. Lieberknecht, Total Synthesis of the Antitumor Antibiotic WF–3161, Angew. Chem., Int. Ed., 2003, 28, 333–334 CrossRef.
- K. Hirai, H. Koike, T. Ishiba, S. Ueda, I. Makino, H. Yamada, T. Ichihashi, Y. Mizushima, M. Ishikawa, Y. Ishihara, Y. Hara, H. Hirose, N. Shima and M. Doteuchi, Amino acid amides of 2-[(2-aminobenzyl)sulfinyl]benzimidazole as acid-stable prodrugs of potential inhibitors of H + K+ ATPase, Eur. J. Med. Chem., 1991, 26, 143–158 CrossRef CAS.
- U. Schmidt, M. Kroner and U. Beutler, Amino Acids and Peptides; 67.1 Easy Preparation and Use of Benzyloxycarbonyl Derivatives of Amino Acid Chlorides and α-Hydroxycarboxylic Acid Chlorides, Synthesis, 1988, 475–477 CrossRef CAS.
- O. Dangles, F. Guibe, G. Balavoine, S. Lavielle and A. Marquet, Selective cleavage of the allyl and (allyloxy)carbonyl groups through palladium-catalyzed hydrostannolysis with tributyltin hydride. Application to the selective protection-deprotection of amino acid derivatives and in peptide synthesis, J. Org. Chem., 1987, 52, 4984–4993 CrossRef CAS.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, On the Basicity of Organic Bases in Different Media, Eur. J. Org. Chem., 2019, 6735–6748 CrossRef CAS.
- D. Nicewicz, H. Roth and N. Romero, Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry, Synlett, 2015, 714–723 CrossRef.
- L. Zoia and D. S. Argyropoulos, Phenoxy radical detection using 31P NMR spin trapping, J. Phys. Org. Chem., 2009, 22, 1070–1077 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, electrochemical characterization data, copies of NMR spectra and copies of HPLC for stereochemical determinations. See DOI: https://doi.org/10.1039/d5ob00798d |
| This journal is © The Royal Society of Chemistry 2025 |