
Recent progress and perspectives on heteroatom doping of hematite photoanodes for photoelectrochemical water splitting
Juhyung
Park
,
Jihun
Kang
,
Sourav
Chaule
and
Ji-Hyun
Jang
 *
*
School of Energy and Chemical Engineering, Department of Energy Engineering, School of Carbon Neutrality, UNIST, Ulsan 44919, Republic of Korea. E-mail: clau@unist.ac.kr
First published on 16th October 2023
Abstract
Over the past few decades, extensive research on photoelectrochemical (PEC) water splitting has been conducted as a promising solution to meet the increasing demand for cleaner and renewable energy in a sustainable manner. Among the various photocatalysts, hematite (α-Fe2O3) has gained significant attention due to its advantageous characteristics, such as a high theoretical solar-to-hydrogen conversion efficiency value, a suitable band gap energy for visible light absorption, chemical stability, and low cost. However, the high PEC potential of α-Fe2O3 is hindered by several limitations, including band gap mismatch, short hole diffusion length, and low electrical conductivity. Several modifications are necessary to enhance the viability of α-Fe2O3 as an efficient photocatalyst for PEC water splitting. This review article primarily focuses on strategies aimed at improving PEC water oxidation performance, especially by addressing its poor transport behavior through heteroatom doping. In particular, we explore the co-doping approach involving unintentional Sn dopants, which are diffused from the fluorine-doped tin oxide substrate during high-temperature annealing.
1. Introduction
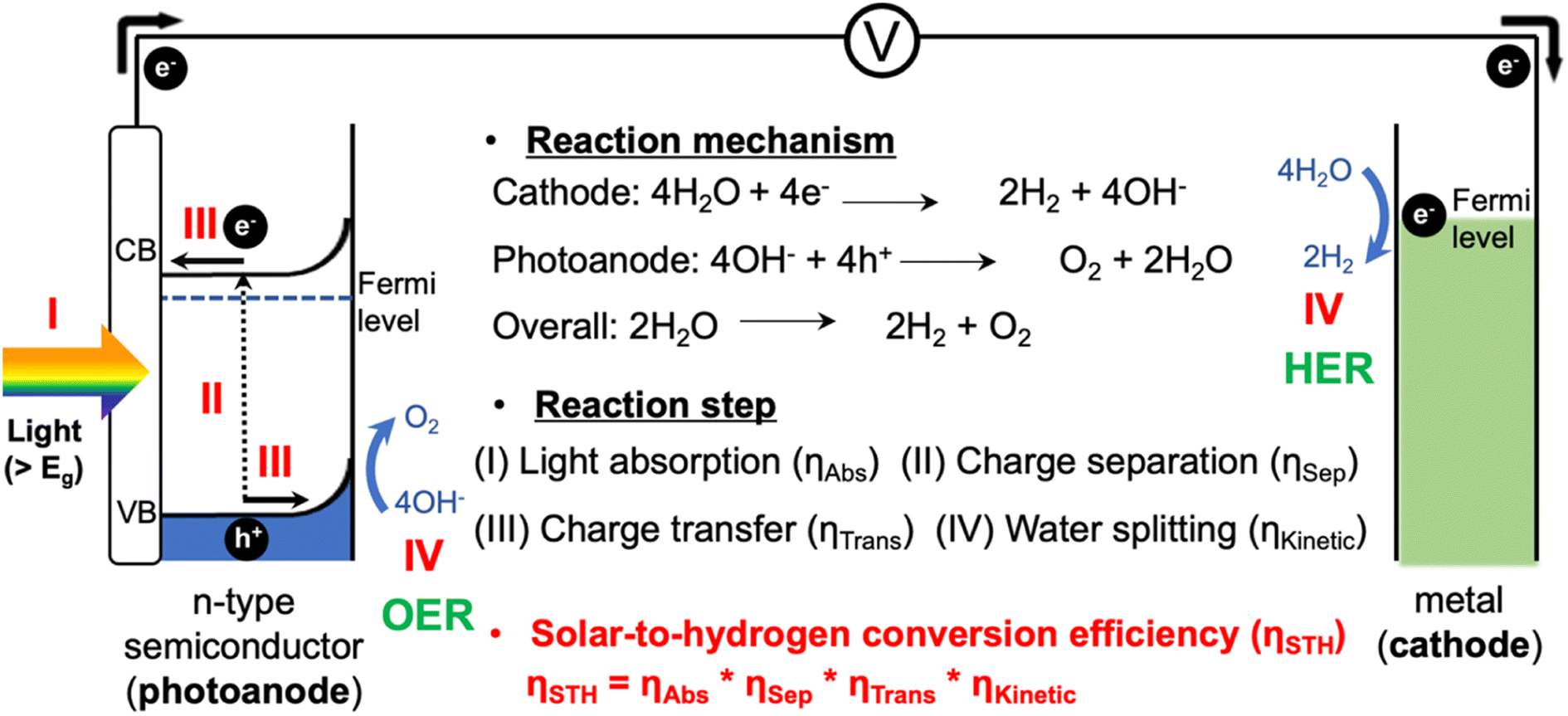
With the increasing global demand for energy, solar energy has received significant attention as a clean and infinite energy source. Since the development of the photoelectrochemical (PEC) water splitting technology by Fujishima and Honda in 1972, PEC water splitting has emerged as one of the most promising strategies for converting solar energy into hydrogen energy.1PEC systems, typically consisting of a photocathode and a photoanode, are capable of utilizing solar energy to split water into hydrogen and oxygen, providing a cleaner and more sustainable energy source (Fig. 1).2,3 These systems are scalable and adaptable to various energy demands, from small-scale applications to large-scale industrial usage. PEC systems also possess the potential for high energy conversion efficiencies as they directly convert sunlight into chemical energy utilizing semiconductor materials. However, there are still many hurdles for the commercialization of green hydrogen produced by PEC systems.4 Existing PEC systems face challenges in achieving high efficiency due to several factors related to semiconductor material properties and poor reaction kinetics.5–7 Finding photocatalysts that are highly efficient, durable, and low cost remains a challenge, and system complexity requires expertise in multiple disciplines. Addressing these challenges, including enhancing efficiency and reducing costs through research and development, is crucial for the wide application of PEC systems as a sustainable energy solution.
 | ||
| Fig. 1 Schematic illustration of the basic principle of the PEC water splitting system. | ||
In this review, we specifically discuss doping strategies of hematite (α-Fe2O3) photoanodes which are broadly used to improve their poor conductivity for efficient PEC water splitting systems. Particularly, we explore the effects of Sn doping caused by the thermal diffusion from the fluorine-doped tin oxide (FTO) bottom substrate under high-temperature annealing conditions. Additionally, we introduce the research trends of doping exhibiting the synergistic effects between unintentional dopant Sn and various other dopants in a multi-dopant system.
2. PEC background
2.1 Basic principle of PEC water oxidation
This review covers the simple photoelectrochemical (PEC) water splitting system, where a photoanode and a metal counter electrode are connected through an electric circuit, as illustrated in Fig. 1.8 The PEC water splitting process involves four key steps: light absorption, charge separation, charge transfer (injection), and surface reaction. During the light absorption step, the photoelectrode absorbs photons with energy higher than the band gap of the material (Eg), which generates photoexcited carriers (h+ and e−). Once these carriers are generated, it becomes essential to separate them promptly to avoid recombination. Specifically, the photogenerated carriers (e−/h+) migrate from the bulk region to the surface of the electrodes. Subsequently, they diffuse to the solid–electrolyte interface, initiating the water splitting reaction. At the surfaces of the photoanode and metal cathode, the oxygen evolution reaction (OER): 4OH− + 4h+ → O2 + 2H2O, and the hydrogen evolution reaction (HER): 4H2O + 4e− → 2H2 + 4OH− are initiated, respectively.9,10As per recent reports, a PEC system should have a solar-to-hydrogen conversion efficiency (STH) of over 10% to achieve the benchmark efficacy necessary to meet economic needs.11,12 Therefore, the overall STH conversion efficiency is the most practical parameter for evaluating the PEC water splitting performance, which is determined by considering these four steps, including the light absorption properties, charge separation and transfer efficiencies, and surface OER kinetics.13
Since these factors heavily rely on the intrinsic properties of the material, identifying the most suitable photocatalyst becomes crucial in achieving higher STH conversion efficiency.14
2.2 Hematite photoanode
An ideal photocatalyst must meet several requirements, including an appropriate bandgap, extended charge lifetime, excellent stability in aqueous systems, and affordability. Various n-type semiconductor materials, such as TiO2, ZnO, WO3, BiVO4, and α-Fe2O3, have been extensively investigated for their unique electrochemical properties, low cost, and high stability.15–23 Each one exhibits its own set of advantages and disadvantages as described in Table 1.| Advantage | Disadvantage | |
|---|---|---|
| Fe2O3 | Earth-abundance and low cost | Short hole diffusion length (2–4 nm) |
| Nontoxicity and good PEC stability | Low electron mobility (∼10−1 cm2 V−1 s−1) | |
| Narrow bandgap (2.0–2.2 eV) | Sluggish water oxidation kinetics | |
| WO3 | Stable in acid conditions | Large band gap of 2.6–3.0 eV |
| Moderate hole-diffusion length (∼150 nm) | Photo-corrosion during PEC reaction | |
| Good electron mobility (∼12 cm2 V−1 s−1) | Sluggish water oxidation kinetics | |
| ZnO | Environmentally friendly | Large bandgap of 3.2 eV |
| Electron lifetime exceeding 10 s | Fast charge recombination | |
| High electron mobility | Insufficient surface reactivity | |
| BiVO4 | Suitable bandgap of 2.4–2.5 eV | Poor electrical conductivity |
| Relatively high conduction band edge | Sluggish water oxidation kinetics | |
| Sluggish OER kinetics | Poor chemical stability | |
| TiO2 | Earth-abundance and high stability | Large bandgap of 3.0–3.2 eV |
| Suitable valence band edge | Low absorption for visible light | |
| Good electrical properties | Poor charge separation |
There are many challenges involved in synthesizing an ideal photocatalyst that can fulfill all the above-mentioned requirements. Therefore, to drive PEC technology towards commercial viability, it is imperative to explore a diverse range of approaches, including alloying, creating heterojunctions, and doping, to optimize the materials.27–29
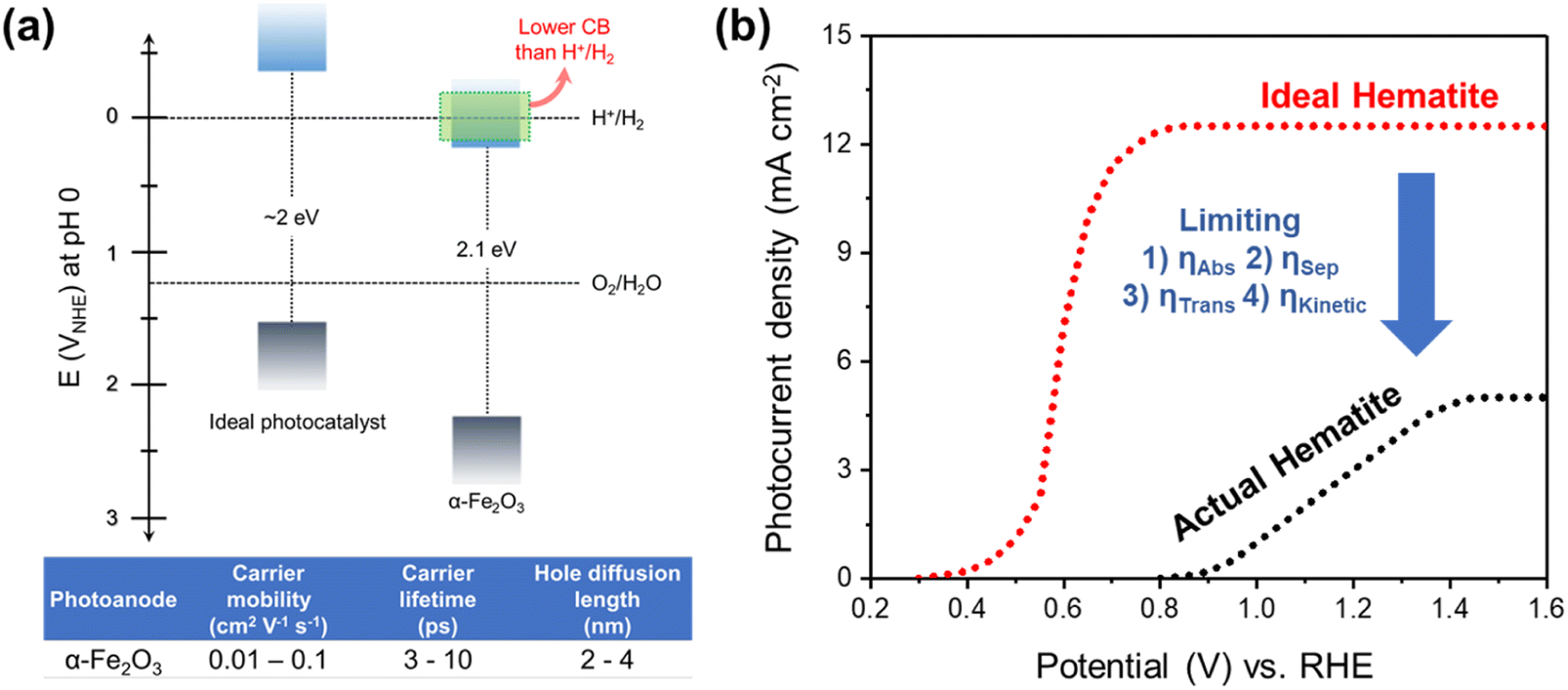
Hematite (α-Fe2O3) is one of the promising photoanode candidates for PEC water splitting as it possesses an ideal bandgap for a broad range of visible light absorption, high chemical stability in alkaline electrolytes, natural abundance, and non-toxicity.30–32 However, the practical STH conversion efficiency of hematite is far below the theoretical values (∼15%) due to its short hole diffusion length (2–4 nm), low electrical conductivity, sluggish water oxidation kinetics, and inappropriate band position for water oxidation and reduction (Fig. 2a).33,34 Note that hematite can achieve a theoretical maximum STH efficiency of 15%, which exceeds the benchmark efficiency of 10%. Despite how it may sound, there are several challenges involved in reaching the maximum theoretical STH efficiency.
 | ||
| Fig. 2 (a) Band edge positions and electronic properties of hematite. (b) PEC performance of the ideal hematite photoanode and actual hematite photoanode. | ||
Fig. 2b illustrates the differences in the performance of the ideal and actual hematite photoanode.35J–V curves represent the actual hematite photoanode with the anodic onset potential and lower photocurrent density compared to the ideal hematite photoanode primarily due to the sluggish charge transfer and poor separation efficiency of the actual hematite photoanode. Extensive research efforts have been directed towards multiple aspects, such as growing hematite on diverse substrates (e.g., metal, FTO, patterned FTO), applying various modifications, including constructing junctions, doping, nanostructuring, and interfacial engineering with overlayer and underlayer coating. These endeavors aim to enhance charge separation, transfer, and surface OER (oxygen evolution reaction) kinetics, as depicted in Fig. 3.
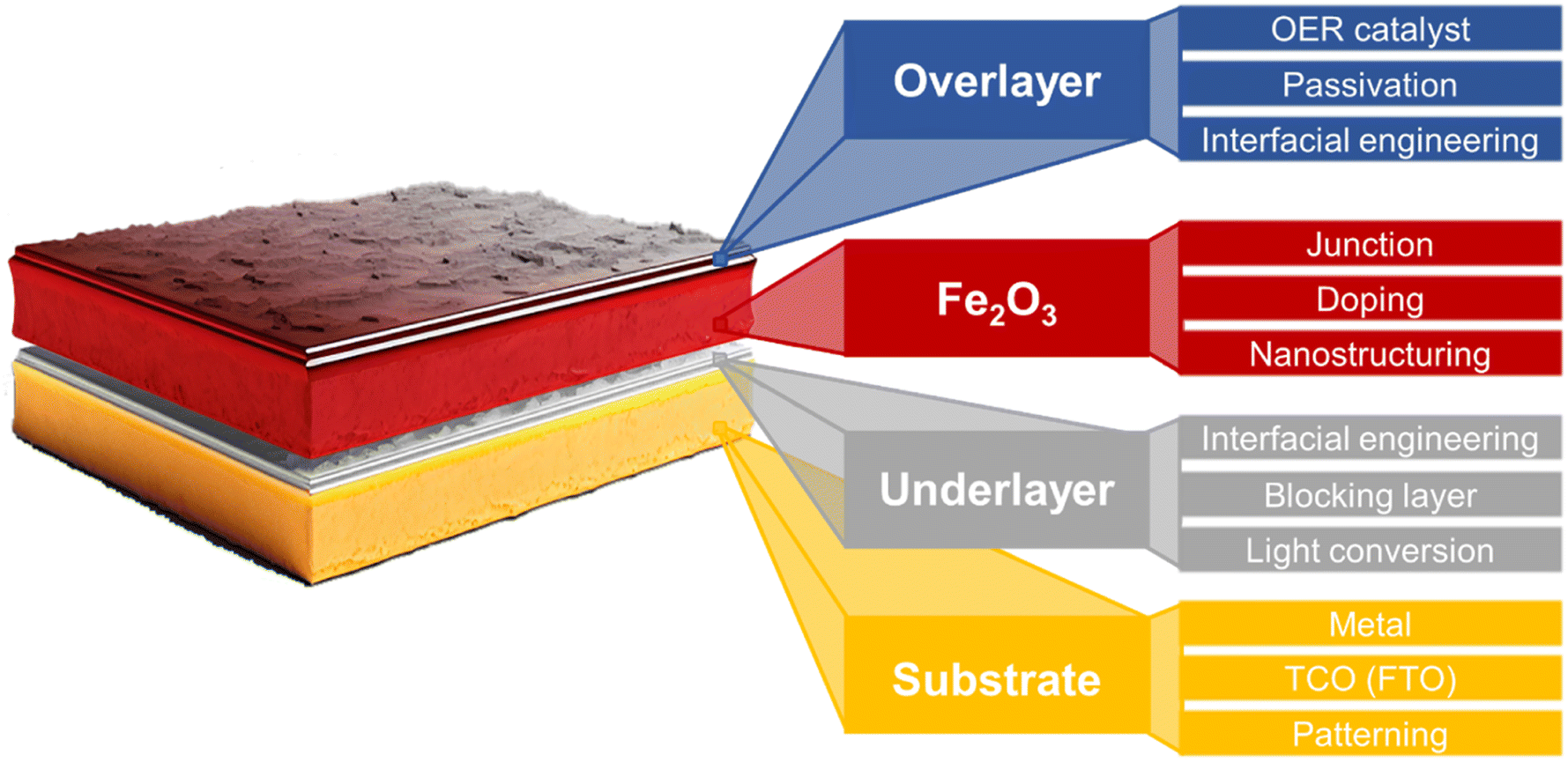 | ||
| Fig. 3 Schematic illustration of various strategies for enhancing PEC efficiencies of a hematite photoanode.35–43 | ||
This review primarily focuses on heteroatom-doped hematite on a fluorine-doped tin oxide (FTO) substrate and summarizes future perspectives that can lead to further advancements in this field. It offers valuable insights into the modification strategies employed and the progress achieved in terms of electronic and morphological changes through heteroatom doping. It also highlights the technical challenges associated with the α-Fe2O3 photoanode, which currently hinder the realization of its full potential in practical solar hydrogen production systems.
3. Heteroatom doping
3.1 Heteroatom doping and limitations
The primary aim of heteroatom doping is to modify the photocatalytic properties, such as bulk carrier concentration, conductivity, and charge separation efficacy of semiconductors, which is a widely utilized approach (Table 2). By introducing dopants into the semiconductor lattice, both the electronic and morphological properties can be enhanced to improve photocatalytic activity. In light of this, both metallic n-type and non-metallic n-type dopants, each possessing distinct photocatalytic potential, are commonly employed to enhance the performance of n-type α-Fe2O3.| Dopant | Photocurrent density (mA cm−2) | Potential vs. RHE | Electrolyte | Carrier density (1020 cm−3) | Year (ref.) |
|---|---|---|---|---|---|
| Ti | 1.25 | 1.23 | 1 M NaOH | — | 2016 (ref. 52) |
| Ti | 0.92 | 1.23 | 1 M KOH | — | 2016 (ref. 53) |
| P | 2.70 | 1.23 | 1 M NaOH | 1.01 | 2015 (ref. 54) |
| P | 1.48 | 1.23 | 1 M KOH | 5.80 | 2017 (ref. 55) |
| P/Ti | 2.50 | 1.23 | 1 M NaOH | 3.48 | 2023 (ref. 56) |
| P/Ru | 4.55 | 1.23 | 1 M KOH | — | 2023 (ref. 50) |
| Si | 2.20 | 1.23 | 1 M NaOH | — | 2006 (ref. 57) |
| Si/Ti | 2.44 | 1.23 | 1 M NaOH | — | 2016 (ref. 52) |
| Si/Ti | 2.36 | 1.23 | 1 M NaOH | — | 2020 (ref. 58) |
| Si/Ti | 3.00 | 1.23 | 1 M NaOH | 1.52 | 2022 (ref. 59) |
| Sn | 0.56 | 1.23 | 1 M NaOH | — | 2010 (ref. 60) |
| Sn | 1.24 | 1.23 | 1 M NaOH | 0.538 | 2011 (ref. 61) |
| Sn | 0.98 | 1.23 | 1 M NaOH | — | 2015 (ref. 62) |
| Sn | 1.38 | 1.23 | 1 M KOH | — | 2017 (ref. 63) |
| Sn/Be | 1.70 | 1.23 | 1 M NaOH | 0.803 | 2016 (ref. 64) |
| Sn/Si | 3.00 | 1.23 | 1 M NaOH | 2.90 | 2021 (ref. 65) |
| Ge | 1.40 | 1.23 | 1 M NaOH | 0.302 | 2014 (ref. 66) |
| Ge | 3.50 | 1.23 | 1 M NaOH | 3.10 | 2021 (ref. 67) |
| B | ∼0.1 | 1.23 | 1 M NaOH | — | 2018 (ref. 44) |
| B/Ti | 1.92 | 1.23 | 1 M NaOH | 8.50 | 2018 (ref. 44) |
| Mg | ∼0.5 | 1.23 | 1 M KOH | — | 2012 (ref. 47) |
| Mg | 0.763 | 1.23 | 1 M NaOH | 41.1 | 2022 (ref. 45) |
| Mg/P | 2.4 | 1.23 | 1 M NaOH | 2.97 | 2018 (ref. 46) |
| Al | ∼0 | 1.23 | 1 M NaOH | 0.02 | 2014 (ref. 68) |
| Al/Ti | 1.30 | 1.23 | 1 M NaOH | 1.70 | 2014 (ref. 68) |
| Zr | 1.50 | 1.23 | 1 M KOH | 5.88 | 2017 (ref. 69) |
| Ta | 1.93 | 1.23 | 1 M NaOH | — | 2020 (ref. 70) |
| Ta/Sn | 2.37 | 1.23 | 1 M NaOH | — | 2019 (ref. 71) |
| Pt | 2.19 | 1.23 | 1 M NaOH | 0.0277 | 2013 (ref. 72) |
| Pt | 2.71 | 1.23 | 1 M KOH | 2023 (ref. 51) | |
| Nb | 0.63 | 1.00 | 0.5 M Na2SO4 | 9.42 | 2016 (ref. 73) |
| Nb/Sn | 1.88 | 1.23 | 1 M KOH | 0.321 | 2019 (ref. 74) |
| Gd | ∼1.5 | 1.23 | 1 M KOH | 0.0217 | 2022 (ref. 32) |
Additionally, the incorporation of p-type dopants into metal-doped hematite is proposed as a solution to reduce the high charge recombination rate of metal-doped hematite.44,45 This strategy aims to create extra states situated above the valence band of hematite and establish a p–n junction at the surface, efficiently mitigating charge recombination by providing an additional driving force for charge separation.46,47
In addition to p-type and n-type doping using dopants of various oxidation numbers, trivalent dopants such as Al3+ and B3+ can act as donor levels in hematite, increasing carrier density and potentially enhancing its efficiency for photoelectrochemical water splitting.48 This is because trivalent dopants like Al3+ and B3+ create a higher positive charge density than the surrounding Fe atoms due to their smaller ionic size, even with the same ionic charge. Consequently, the surrounding oxygen atoms move toward the trivalent dopants because of the stronger coulombic attraction compared to Fe. This transformation in chemical bonding introduces lattice strain and ultimately reduces the overall lattice volume, which is beneficial for electron hopping.49
Several types of dopants play a crucial role in enhancing the PEC activities of hematite by addressing challenges such as low electron mobility (10−2 cm2 V−1 s−1) and low carrier density (1018 cm3), and reducing the overpotential for hematite water oxidation.30 Additionally, these dopants offer the ability to achieve diverse effects, including the control of morphological and optical properties.20 As mentioned above, various dopants exhibit diverse characteristics, and the doping methods are critical research areas for optimizing hematite's performance. Consequently, recent research has placed its focus on various doping techniques, encompassing atomic-level doping engineering.50,51 In this perspective article, we will delve specifically into the heteroatom doping method, particularly involving surface overlayers, among the array of available approaches.
Fig. 4a shows the X-ray photoelectron spectroscopy (XPS) depth profile for P/Fe of P,Ti-Fe2O3, which revealed evidence of P doping from the surface overlayer to the hematite lattice. Due to the P doping effect, P,Ti-Fe2O3 exhibited a shift of an O 1s peak towards higher binding energy, indicating the presence of oxygen atoms bonded with phosphorus (Fig. 4b). This led to an enhanced carrier density of P,Ti-Fe2O3 compared to that of Ti-Fe2O3 (Fig. 4c). The band structure analysis confirmed the band shift resulting from the injection of P (Fig. 4d) and revealed that the gradient P doping facilitated more efficient charge transport by creating an internal electric field (Fig. 4e). In J–V curves, P,Ti-Fe2O3 exhibited a photocurrent density of 2.5 mA cm−2 at 1.23 VRHE, which was a 94% improvement compared to that of a Ti-Fe2O3 photoanode as shown in Fig. 4f. The formation of a pore structure holds great importance in hematite, which greatly suffers from the very short hole diffusion length (2–4 nm) issue. This highlights the significance of the pore generation method. In addition to the alterations in electronic properties, the in situ P doping method created a mesoporous nanorod structure of P,Ti-Fe2O3 with much shortened diffusion pathways (Fig. 4h), different from the conventional wormlike structure of Ti-Fe2O3 (Fig. 4g). The pore generation occurred as a result of water evaporation in the P,Ti-FeOOH nanorod within the FePO4 hard template during the high-temperature dehydration process, which created vacancies.
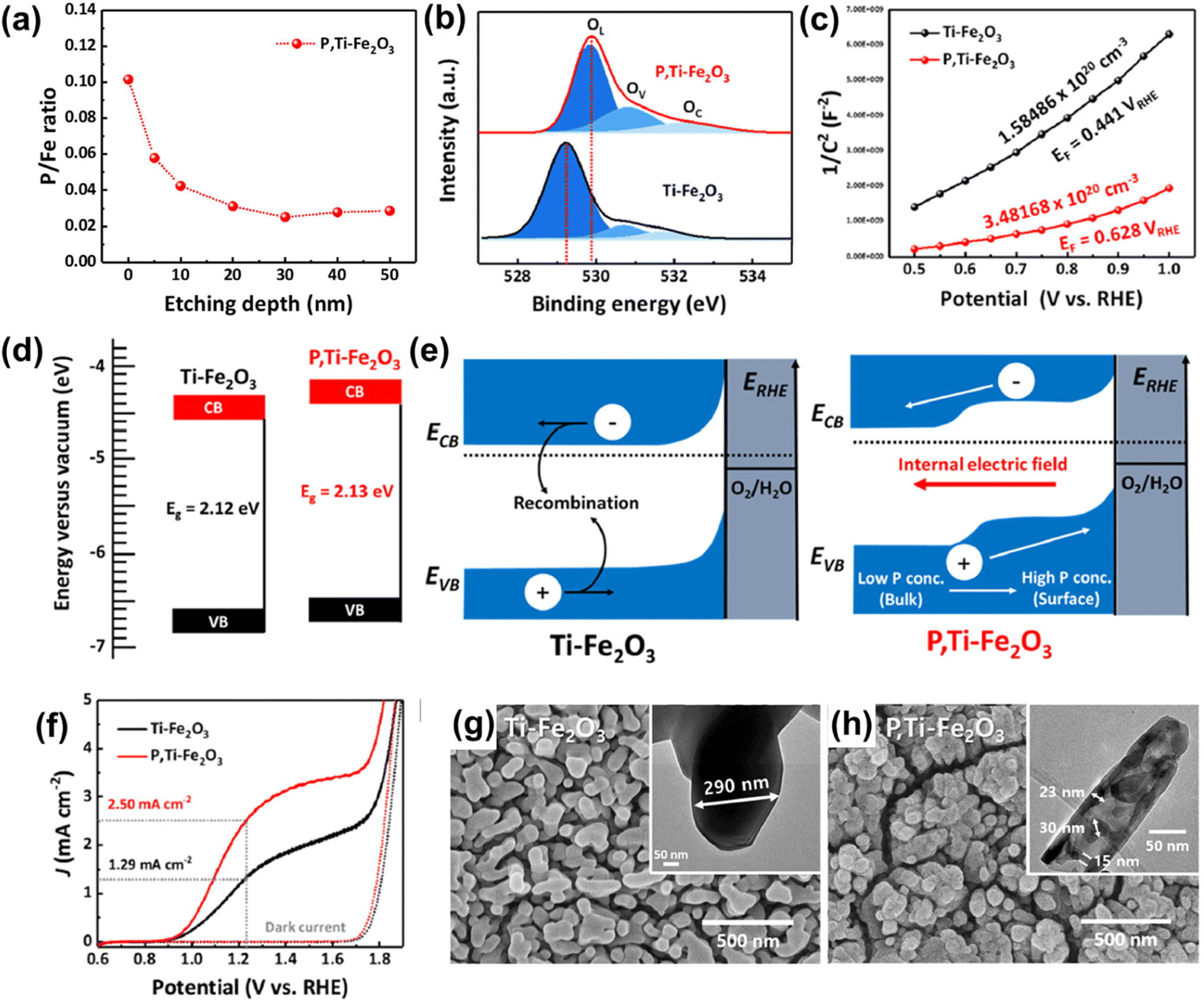 | ||
| Fig. 4 (a) XPS depth profiles for P/Fe of P,Ti-Fe2O3. (b) XPS O 1s spectra of Ti-Fe2O3 and P,Ti-Fe2O3. (c) Mott–Schottky plots at a frequency of 1 kHz. (d) Band diagrams and (e) band edge positions of Ti-Fe2O3 and P,Ti-Fe2O3 photoanodes. (f) J–V curves for Ti-Fe2O3 and P,Ti-Fe2O3 under 1 sun and dark conditions in a three-electrode system. SEM image of (g) Ti-Fe2O3, and (h) P,Ti-Fe2O3 (insets in (g) and (h) show TEM images of Ti-Fe2O3 and P,Ti-Fe2O3). Reproduced from ref. 56 with permission from Elsevier, Copyright 2023. | ||
The synthesis development of porous hematite, along with doping, has also offered a new opportunity to overcome the limited efficacy of the hematite photoanode. A porous structure improves solar energy conversion by (i) enhancing light scattering, (ii) facilitating an electron transfer pathway, and (iii) increasing the specific surface area. In 2016, Jang et al. reported a synthesis mechanism for porous hematite on an FTO surface.52 In the conventional hematite synthesis method, the high-temperature conditions caused Ti-FeOOH nanorods to coalesce, resulting in a worm-like, non-porous structure of Ti-Fe2O3 (Fig. 5b). On the other hand, to create a nanoporous hematite structure, they employed a Ti-doped amorphous SiOx overlayer on Ti-FeOOH. With the Ti-doped SiOx overlayer, the grain boundary motion appeared to occur inwards within each rod, accompanied by the evolution of HCl and H2O gases under the rigid SiOx overlayer through the dehydration process. Thus, Ti-doped SiOx overlayer coated hematite exhibited numerous pores (Ti-(SiOx/np-Fe2O3)) in the core and thick frames in the shell (Fig. 5c). The uniform distribution of Si atoms was confirmed through EDS mapping images (Fig. 5d) and XPS analysis (Fig. 5e). As a result of the nanoporous structure formation and the passivation effect of the SiOx overlayer on Ti-(SiOx/np-Fe2O3), the Ti-(SiOx/np-Fe2O3) photoanode exhibited an improved photocurrent density of 2.44 mA cm−2 at 1.23 VRHE (Fig. 5f). Through the development of doping methods using surface overlayer coating, not only heteroatom doping but also the fabrication of a nanoporous structure became easily achievable.
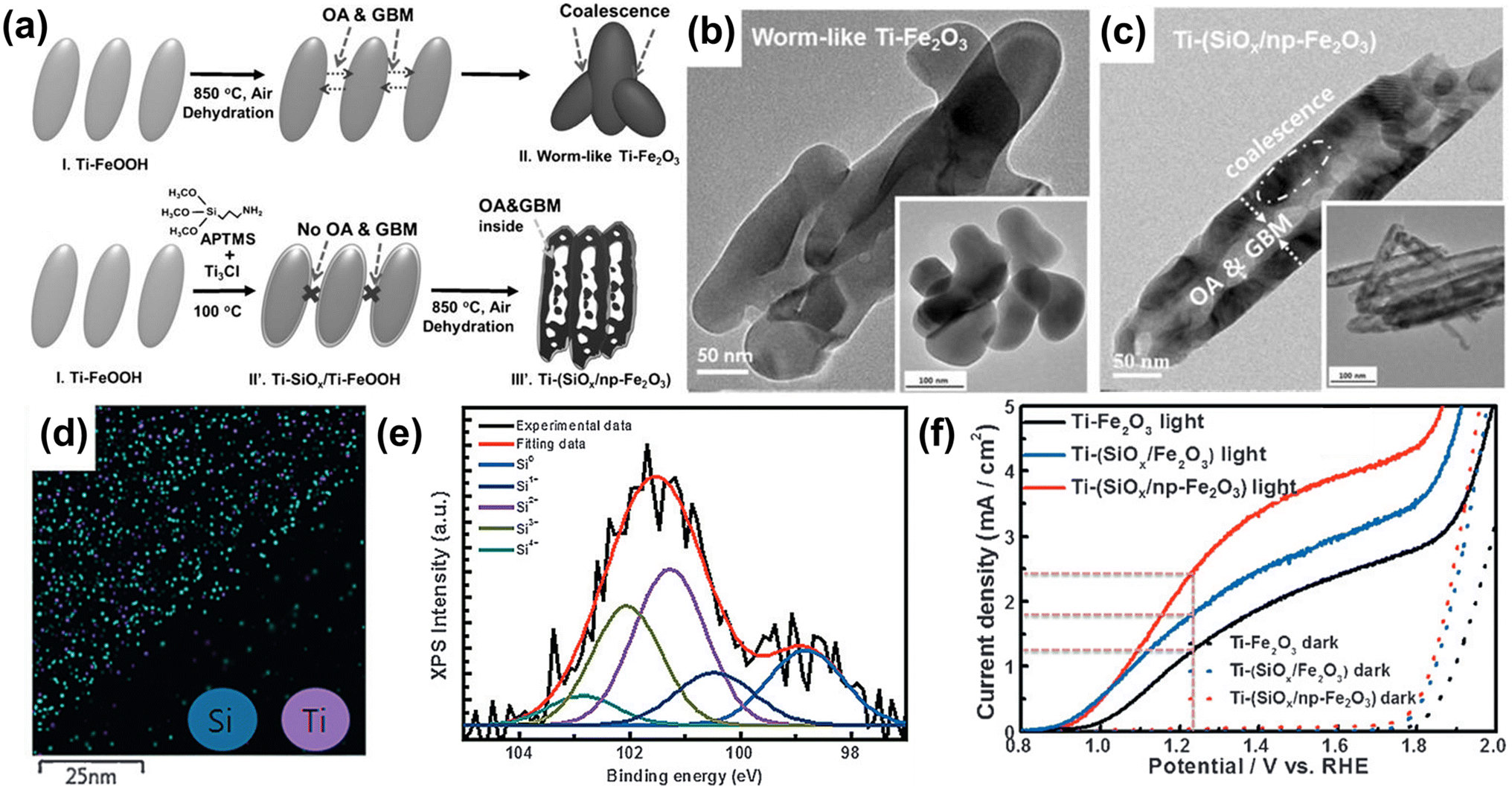 | ||
| Fig. 5 (a) The fabrication procedures for conventional worm-like Ti-Fe2O3 and Ti-(SiOx) layer-coated porous hematite (Ti-(SiOx/Fe2O3)). TEM image of (b) a worm-like Ti-Fe2O3 and (c) Ti-(SiOx/np-Fe2O3). (d) EDS mapping images of Si and Ti of Ti-(SiOx/np-Fe2O3). (e) XPS Si 2p spectra of Ti-(SiOx/np-Fe2O3). (f) J–V curves of Ti-Fe2O3, Ti-(SiOx/Fe2O3), and Ti-(SiOx/np-Fe2O3) under 1 sun and dark conditions with a three-electrode system. Reproduced from ref. 52 with permission from Wiley-VCH, Copyright 2016. | ||
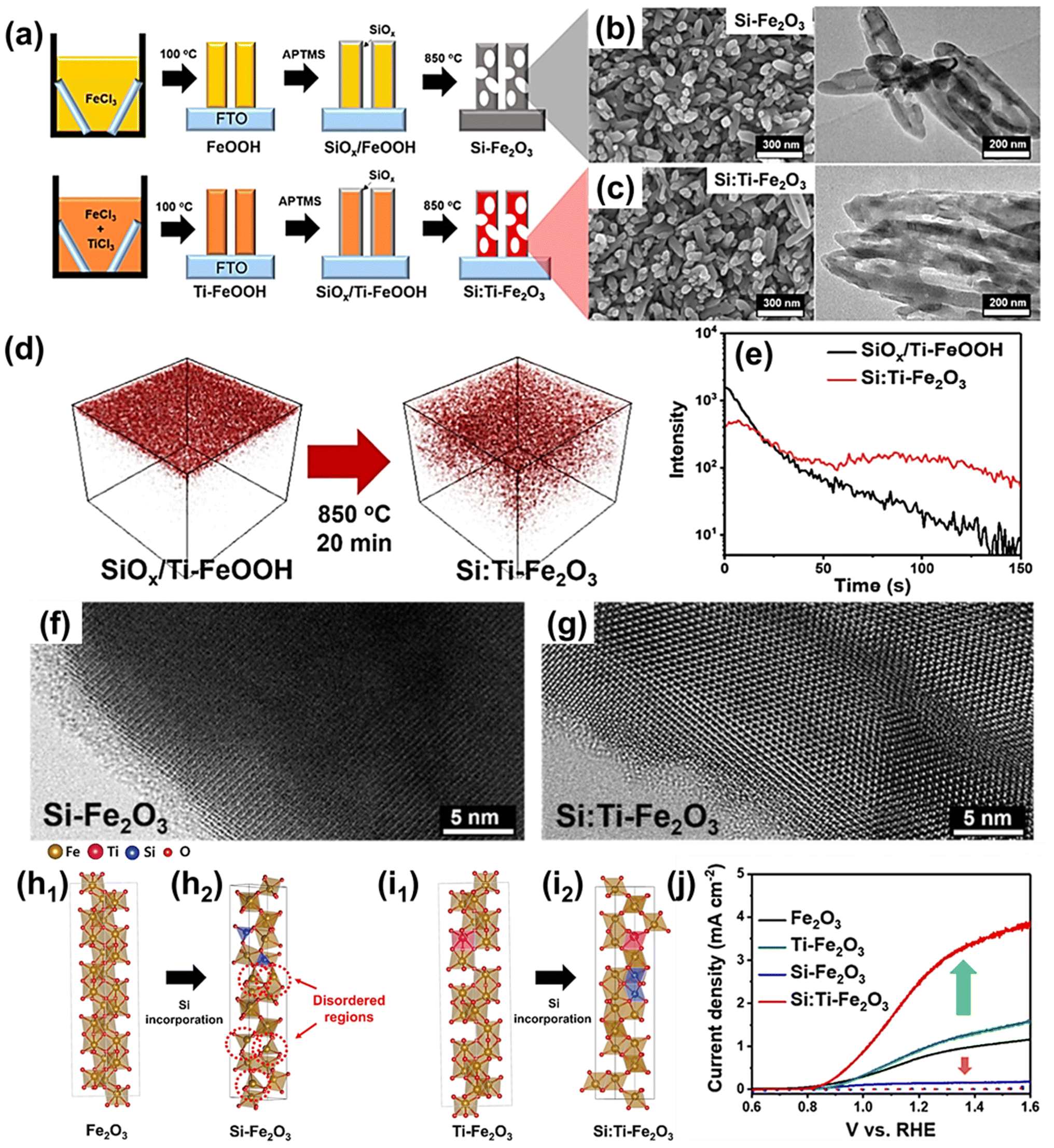 | ||
| Fig. 6 (a) Scheme for photoanode fabrication with the SiOx layer w/wo the host Ti-dopant. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images for (b) Si-Fe2O3 and (c) Si:Ti-Fe2O3. (d) Three-dimensional TOF-SIMS Si distribution before (SiOx/Ti-FeOOH film) and after (Si:Ti-Fe2O3 film) annealing and (e) TOF-SIMS depth profiles for Si distribution. High-magnification bright-field TEM images of (f) Si-Fe2O3 and (g) Si:Ti-Fe2O3 after etching the SiOx layer. (h) Atomic structures without host Ti-dopants for (h1) Fe2O3 and (h2) Si-Fe2O3. (i) Atomic structures with host Ti-dopant for (i1) Ti-Fe2O3 and (i2) Si:Ti-Fe2O3. (j) J–V curves of Fe2O3, Ti-Fe2O3, Si-Fe2O3, and Si:Ti-Fe2O3. Reproduced from ref. 59 with permission from American Chemical Society, Copyright 2022. | ||
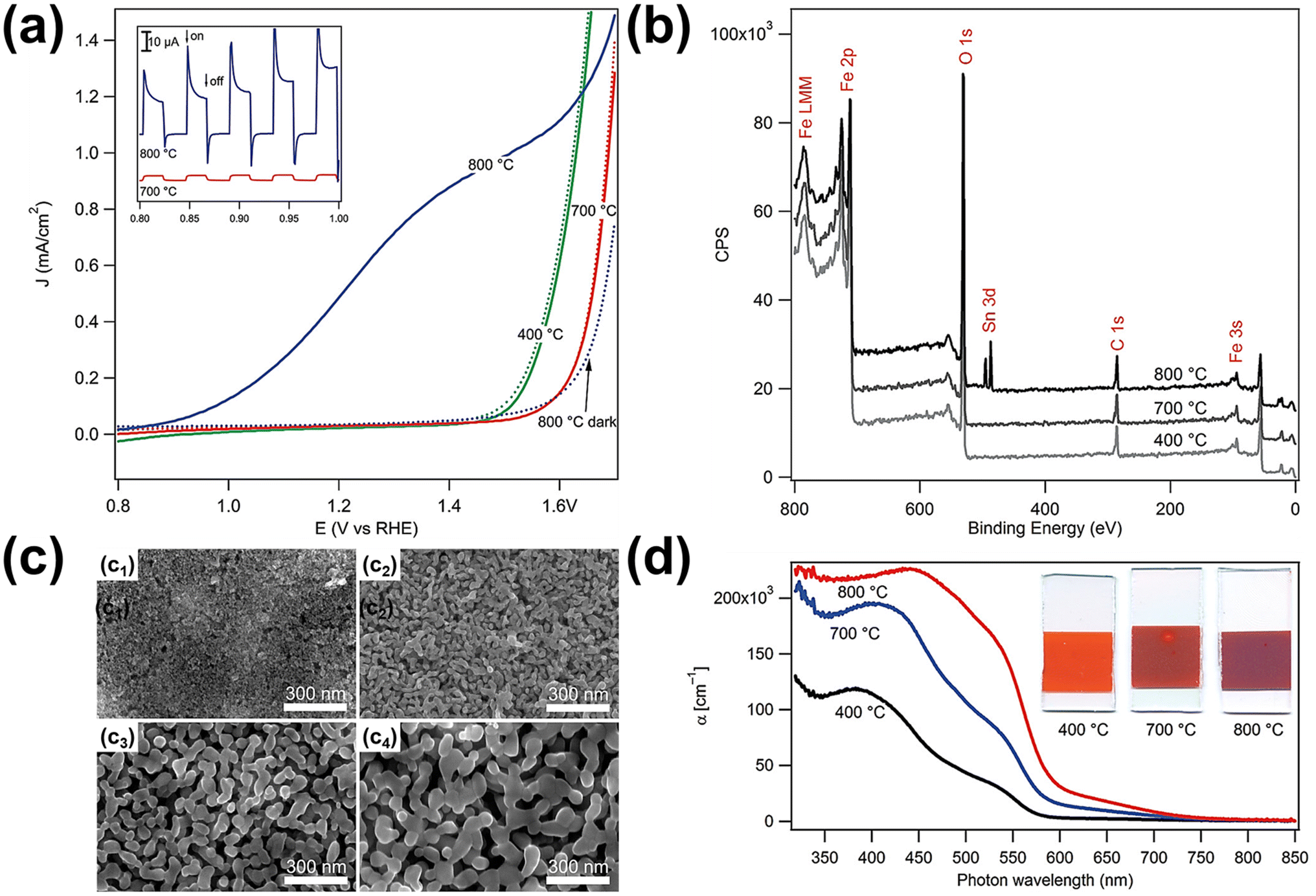 | ||
| Fig. 7 (a) Electrochemical water oxidation current vs. voltage curves of the photoanodes sintered at different temperatures in the dark (dotted curves) and under simulated solar light (solid curves, AM 1.5G 100 mW cm−2, spectrally corrected). The inset shows the photocurrent transient curves for the 700 and 800 °C samples created by chopping the light source during a current vs. voltage sweep. (b) X-ray photoelectron spectroscopy survey data for the electrodes sintered at different temperatures. (c) Scanning electron micrographs of mesoporous hematite films prepared on SiO2/F:SnO2 substrates after different heat treatments: (c1) as deposited with a porogen, (c2) after 10 h at 400 °C, and (c3) after 20 min at 700 °C and (c4) 800 °C. (d) Absorption coefficient as a function of wavelength and the typical appearance of the electrodes (25 mm height, all derived from the same parent substrate). Reproduced from ref. 60 with permission from American Chemical Society, Copyright 2010. | ||
A more detailed study of Sn diffusion from the FTO substrate into hematite was carried out by Annamalai et al., where they reported that Sn acted as an n-type dopant.62 Depending on the annealing time (at 800 °C for 2, 10, and 20 min), different amounts of Sn in the FTO substrate were diffused into the hematite lattice. A sudden decline in photocurrent was observed in hematite nanorods sintered for 20 min (Fig. 8a). This was attributed to the significant amount of diffused Sn content exceeding 3% in hematite, consequently causing the deformation of the FTO substrate and increasing resistivity due to changes in the stoichiometry of the FTO substrate. To investigate FTO deformation caused by excess Sn diffusion, the Fourier-transformed spectra of extended X-ray absorption fine structure (EXAFS) analysis were examined for the Sn K-edge of the FTO substrate (Fig. 8b). The intensity of the peak in the region of 0.8 to 2.0 Å, corresponding to short Sn–O bonding, followed the sequence 20 min < 0 min ≈ 2 min < 10 min, implying enhanced ordering with an increase in Sn diffusion up to 10 min of annealing. Due to a trade-off between Sn diffusion into the hematite lattice and loss of Sn in the FTO substrate, applying optimized annealing conditions with moderate Sn diffusion minimized the deformation of FTO, leading to high PEC activities. Based on these findings, they concluded that Sn doping had a positive impact, particularly when performed at an optimal sintering time of 10 minutes. This resulted in achieving the highest photocurrent density of 0.98 mA cm−2 at 1.23 VRHE. The study emphasized the significance of controlling the Sn diffusion from the FTO substrate, as it influenced various factors including charge recombination centers, structural disordering, and electrical conductivity of both the photoanode and the substrate.
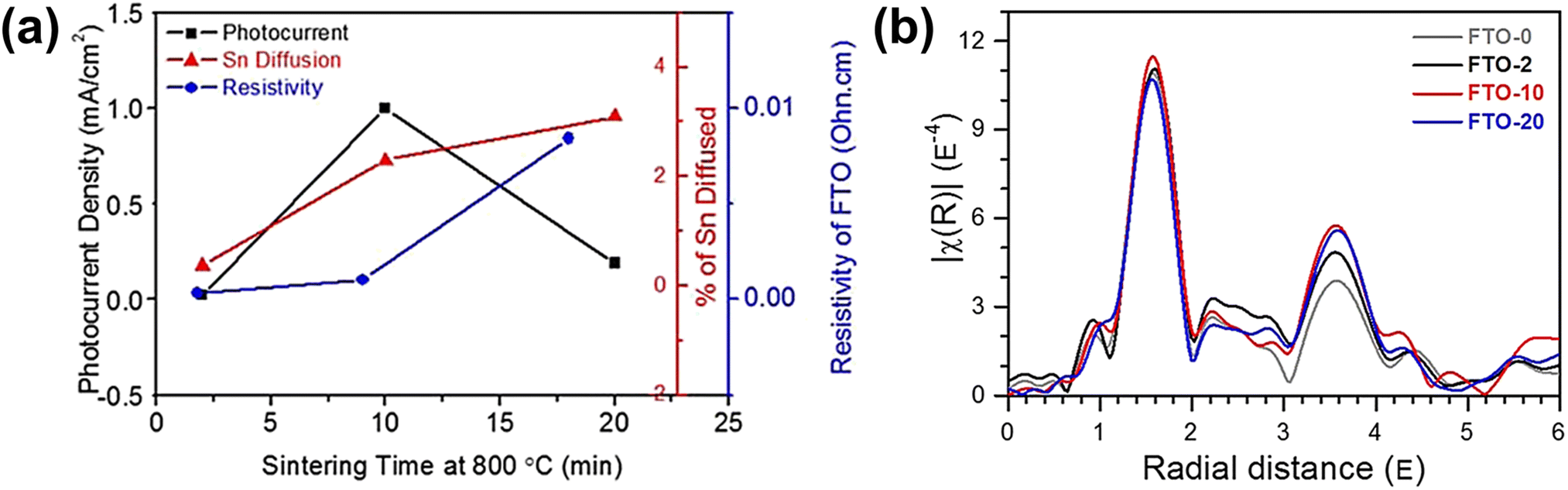 | ||
| Fig. 8 (a) Photocurrent density (mA cm−2), atomic percent of Sn diffusion of hematite photoanodes, and resistivity of the FTO substrate sintered at 800 °C for 2, 10, and 20 min. k3-weighted Fourier transforms for (b) Sn K-edge EXAFS of FTO substrates sintered at 800 °C for 0, 2, 10, and 20 min. Reproduced from ref. 62 with permission from American Chemical Society, Copyright 2015. | ||
Li et al. developed a method to control unintentionally diffused Sn and further enhance the Sn diffusion effects from the FTO substrate obtained by a bottom-up approach.63 They observed that while Sn enhances the PEC performance of hematite, the amount of Sn diffusing from the bottom is limited. Moreover, the high-temperature treatment required for Sn diffusion was found to cause structural deformation. To address these issues, they utilized a top-down method for Sn diffusion, decorating an additional Sn precursor on the surface of hematite to increase Sn concentration. They also encapsulated the hematite nanowires with silica through thermal diffusion. Compared to the normal hematite nanowire (Sn-Fe2O3) with unintentional Sn content diffused from the bottom FTO substrate, which also substantially shrank after high-temperature annealing, the one synthesized by drop-casting a Sn precursor and encapsulating it with a silica shell exhibited robust characteristics under the high-temperature annealing (800 °C) process (Fig. 9a). This resulted in a morphology-and-doping-engineered hematite nanowire (E-I-Sn-Fe2O3). By silica encapsulation and the addition of a Sn precursor before high-temperature annealing, E-I-Sn-Fe2O3 retained its morphology while improving the uniformity of dopant distribution along the nanowire growth axis as shown in XPS spectra and SIMS depth profiles in Fig. 9b and c. The high Sn concentration at the first 100 nm of E-I-Sn-Fe2O3 may arise from the uneven deposition of the Sn precursor onto the densely arranged NW arrays. They claimed that the enhanced photocurrent density and IPCE of the E-I-Sn-Fe2O3 are due to retaining the nanowire morphology and uniform distribution of Sn (Fig. 9d and e). This study presents the effects of intentional Sn doping on the morphology and doping uniformity to control unintentionally diffused Sn from the FTO substrate and assess its influence on the photoelectrochemical (PEC) performance of hematite photoanodes.
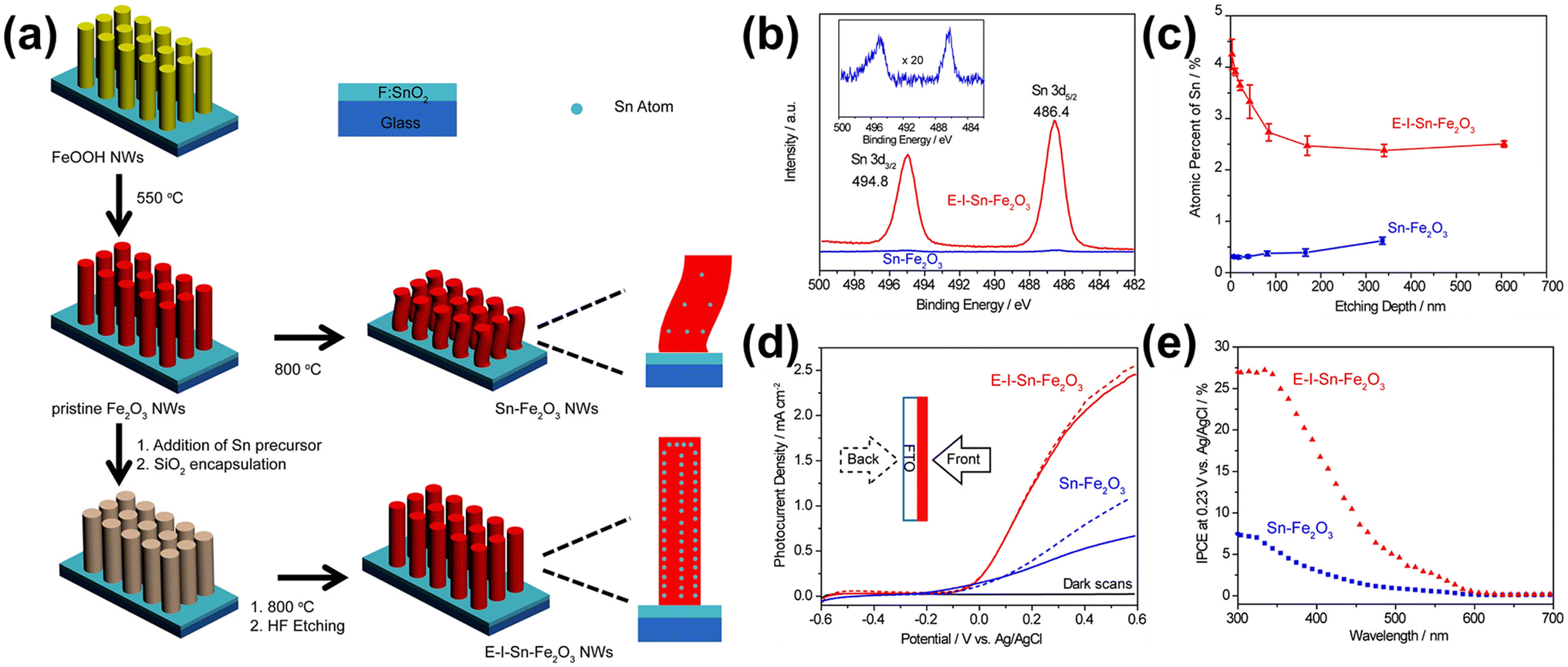 | ||
| Fig. 9 (a) Schematic illustration of the preparation of Sn-Fe2O3 NWs and E-I-Sn-Fe2O3 NWs. (b) XPS Sn 3d spectra of Sn-Fe2O3 and E-I-Sn-Fe2O3. Inset: magnified Sn 3d spectrum of Sn-Fe2O3. (c) SIMS depth profiles of Sn-Fe2O3 and E-I-Sn-Fe2O3. Error bars are evaluated based on parallel experiments. (d) J–V curves of Sn-Fe2O3 and E-I-Sn-Fe2O3 collected at 20 mV s−1 in a 1.0 M KOH aqueous electrolyte under one sun illumination (100 mW cm−2) and in the dark. The solid and dashed lines represent the data collected under the front (solid lines) and back (dashed lines) illuminations, respectively. (e) IPCE spectra of Sn-Fe2O3 and E-I-Sn-Fe2O3 collected at 0.23 V vs. Ag/AgCl. Reproduced from ref. 63 with permission from American Chemical Society, Copyright 2017. | ||
3.2 Multiple dopants (B, Si, Ti, and Ge) for controlling metal dopants of Fe2O3 (unintentionally diffused Sn)
The positive effect of Sn diffusion from the FTO substrate into the hematite lattice is described in the previous section. In more advanced co-doping systems, the role of Sn diffused from the FTO substrate may be quite different. There have been several reports that Sn doping can induce negative effects even under the doping limit when multiple heteroatoms are doped into the hematite lattice due to bad interaction among different atoms. Therefore, material design with theoretical validation should be carefully studied, so that material synthesis close to the designed system can be approached more scientifically. In this section, we introduce several reports on the hematite photoelectrochemical effects with controlled Sn content at the surface region where the recombination should be carefully considered to boost the OER properties.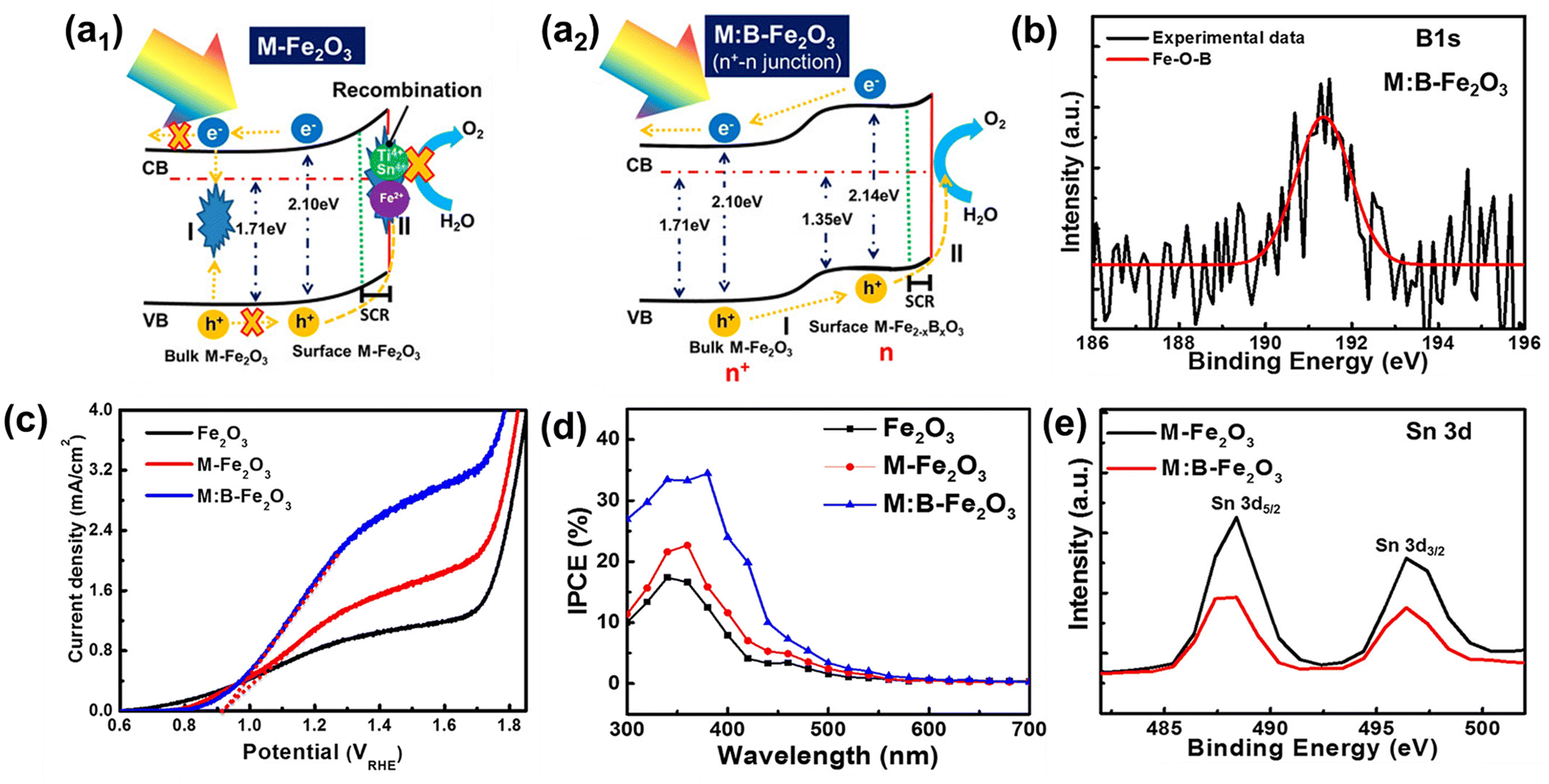 | ||
| Fig. 10 (a) Schematic illustration of an energy band diagram and charge transfer process of (a1) M-Fe2O3 and (a2) M:B-Fe2O3, (b) XPS B 1s spectra, (c) J–V curves, (d) IPCE, and (e) XPS Sn 3d spectra. Reproduced from ref. 44 with permission from American Chemical Society, Copyright 2018. | ||
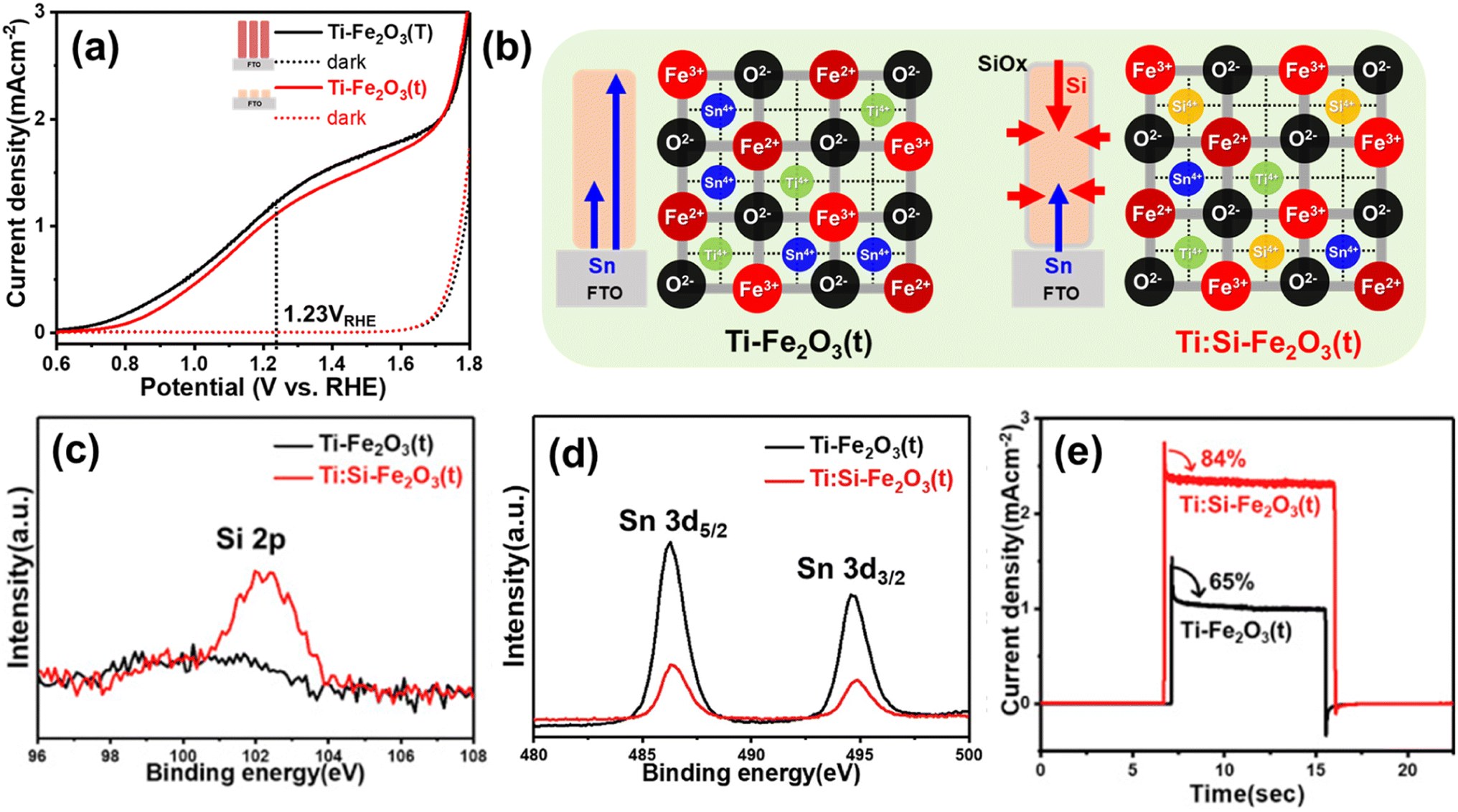 | ||
| Fig. 11 (a) J–V curves of Ti-Fe2O3(T) and Ti-Fe2O3(t) under 1 sun and dark conditions. (b) Schematic diagram of decreased metal dopants (Sn4+ and Ti4+) through non-metallic Si doping in Ti:Si-Fe2O3(t). XPS spectra of (c) Si 2p, and (d) Sn 3d. (e) Transient photocurrent density curves (0.001 point per s) measured at 1.23 VRHE of Ti-Fe2O3(t), and Ti:Si-Fe2O3(t). Reproduced from ref. 58 with permission from Elsevier, Copyright 2020. | ||
To further reveal these assumptions, they developed a two-step annealing process that enables systematic control of Si and Sn content.65 In the case of the conventional one-step annealed SiOx overlayer-coated hematite (A-1 step, Fig. 12a), the diffusion of Sn from the FTO substrate to the hematite surface was regulated by the thickness of the SiOx overlayer film during prolonged thermal energy exposure, as reported before.58 In contrast, a two-step annealing method (B-1 and B-2 steps, Fig. 12a) allowed precise control over the overall amount of Sn released from the substrate by managing the diffusion length of Sn towards the surface region. In the B-1 step, Sn doping occurred only in the bulk region due to the reduced diffusion length during a short annealing time. Subsequently, in the B-2 step, the thermal diffusion of Sn from the bottom was significantly hindered, resulting from the strong binding energy between the Sn atoms, which were pre-occupied by the diffusion process during the B-1 step, and the O atoms in the bulk region. As a result, the Sn content in the surface region was more controlled, allowing for intentional Si doping to readily take place at the surface region. The different concentrations of the dopant in Sn:Si-Fe2O3/1 (made by one-step annealing, high Sn and low Si) and Sn:Si-Fe2O3/2 (made by two-step annealing, low Sn and high Si) were confirmed by XPS depth profiles in Fig. 12b and c. The reduced surface charge trapped holes and enhanced charge-transfer efficiencies of Sn:Si-Fe2O3/2 compared to Sn:Si-Fe2O3/1 were confirmed using the Nyquist plot in Fig. 12d. In the result, Sn:Si-Fe2O3/2 illustrated a higher photocurrent density of 2.3 mA cm−2 at 1.23 VRHE due to the excellent OER activity of Si over Sn (Fig. 12e) at the surface region. The results provided a pathway for designing an optimal doping system with a targeted concentration of Si and Sn by manipulating the annealing process.
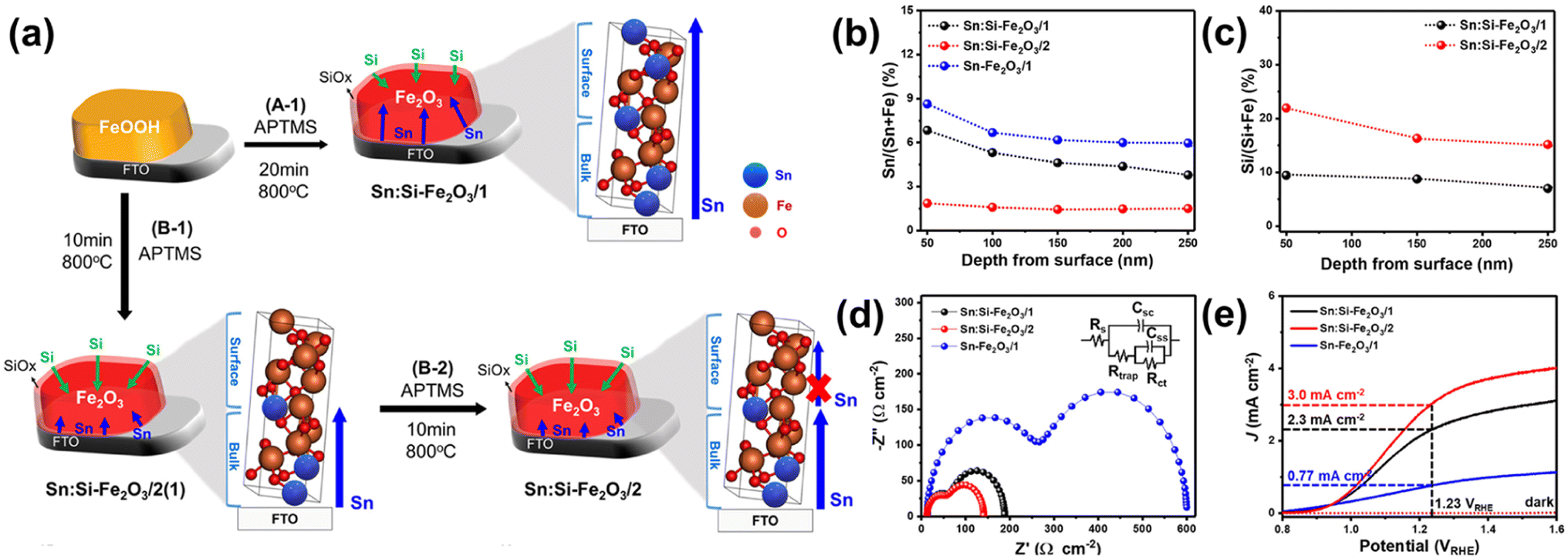 | ||
| Fig. 12 (a) Fabrication procedures of Sn:Si-Fe2O3/1 and Sn:Si-Fe2O3/2. (b) XPS depth profiles of Si and (c) Sn according to the depth from the surface of Sn:Si-Fe2O3/1–2. (d) EIS measurements at 1.23 VRHE, and (e) J–V curves of Sn:Si-Fe2O3/1–2, and Sn-Fe2O3/1. Reproduced from ref. 65 with permission from American Chemical Society, Copyright 2021. | ||
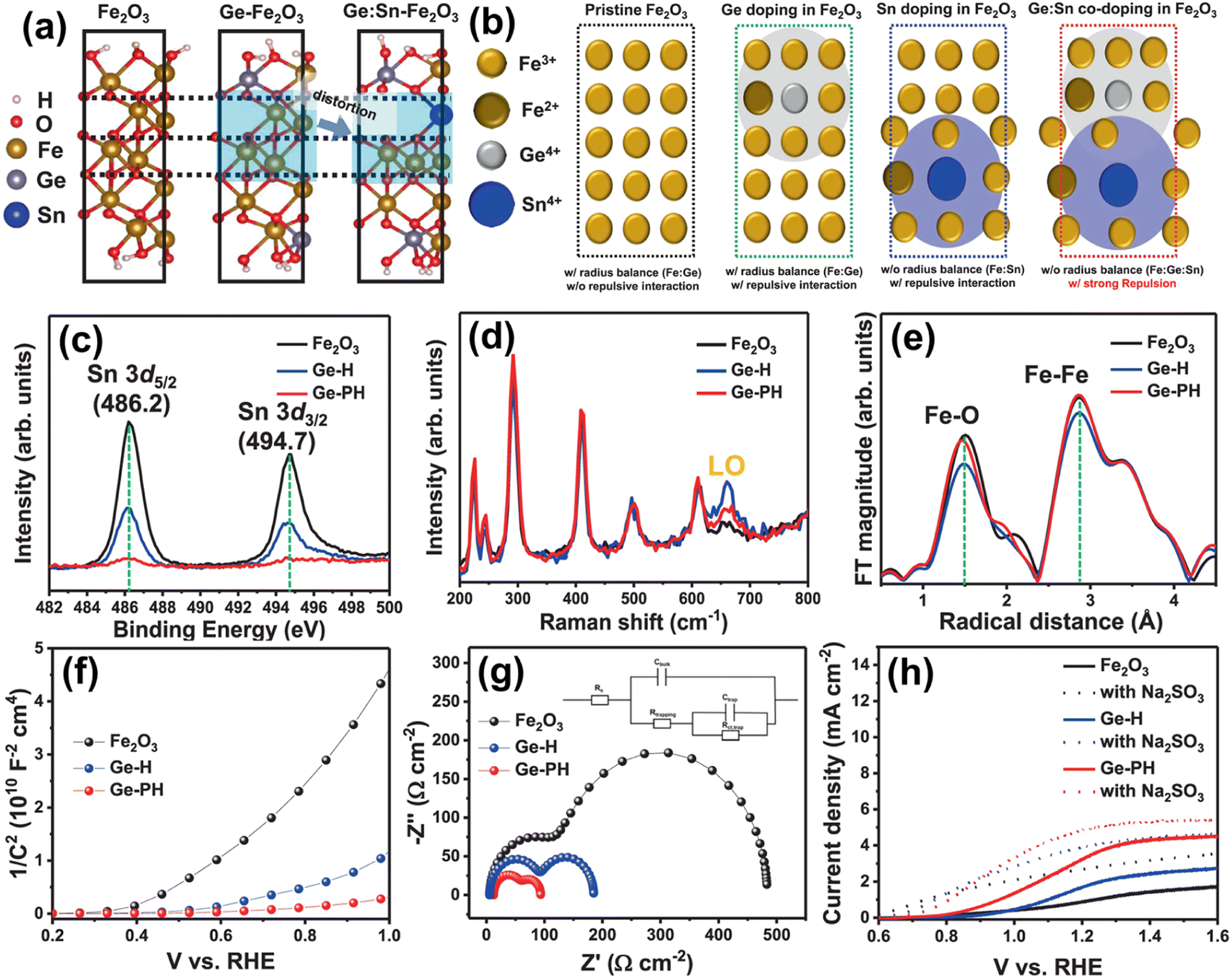 | ||
| Fig. 13 (a) DFT calculations of atomic arrangements of Fe2O3 with different dopant environments. (b) Atomic structure of Ge-doped Fe2O3 Sn-doped Fe2O3 and Ge:Sn co-doped Fe2O3. (c) XPS spectra of Sn. (d) Raman spectra. (e) Fourier transform of the EXAFS data at the Fe k-edge of the hematite nanostructures. (f) Mott–Schottky plots and (g) EIS measurements (with the circuit model). (h) J–V curves of Fe2O3, Ge-H, and Ge-PH under illumination in 1 M NaOH (solid line) and a solution mixture of 1 M NaOH and 0.5 M Na2SO3 (dashed lines). Reproduced from ref. 67 with permission from Springer Nature, Copyright 2021. | ||
In this perspective, we have explored various studies focusing on enhancing the morphological and electrical properties of hematite through heteroatom doping methods. To advance the co-doping system further, promising solutions have been proposed, focusing on the control of Sn diffusion from the substrate. Specifically, our investigation delved into the interactions between different dopants and Sn in the co-doped hematite photoanodes, which utilize the common FTO substrate.
Up to now, the highest efficiency of the hematite-based photoanode has been about 7 mA cm−2 at 1.23 VRHE, and the efficiency has been increasing at a fairly rapid rate.76 However, in order to reach the theoretical efficiency of about 12 mA cm−2 at 1.23 VRHE, in addition to the synergistic effect between the substrate and other multiple dopants covered in this review, further study on the various components and different aspects of doping such as single atomic-level engineering is required.50,51
4. Summary and outlook
This review focused on the effects of Sn diffusion from the FTO substrate and the strategies used to control Sn diffusion via multiple dopants. Prior research has shown that moderate levels of Sn doping can enhance the PEC efficiency of a hematite photoanode. However, when combined with multiple dopants in the hematite lattice, Sn doping might exhibit detrimental effects. Consequently, to establish a more systematic approach to the hematite co-doping system, it is essential to design a more sophisticated doping system by considering the positive or negative interaction between the dopant and unintentional dopant Sn necessarily originating from the experimental process.Efforts to improve solar energy conversion efficiency have focused on the individual efficiencies of sequential photocatalysis courses, including light harvesting, charge separation, and charge transport for solar fuel generation. Numerous studies have independently addressed these limitations, leading to significant improvements.
Nevertheless, to further boost efficiency and establish a cohesive system, it is imperative to concurrently consider the synergistic impacts of the various dopants and PEC components discussed in this review.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2019M1A2A2065612, NRF-2020R1A2C2014050, and NRF-2020R1A5A1019631).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- W. Yang, R. R. Prabhakar, J. Tan, S. D. Tilley and J. Moon, Chem. Soc. Rev., 2019, 48, 4979–5015 RSC.
- R.-T. Gao, N. T. Nguyen, T. Nakajima, J. He, X. Liu, X. Zhang, L. Wang and L. Wu, Sci. Adv., 2023, 9, eade4589 CrossRef PubMed.
- C. Avcıoğlu, S. Avcıoğlu, M. F. Bekheet and A. Gurlo, Mater. Today Energy, 2022, 24, 100936 CrossRef.
- Y. Yang, S. Niu, D. Han, T. Liu, G. Wang and Y. Li, Adv. Energy Mater., 2017, 7, 1700555 CrossRef.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- K. Kim, P. Thiyagarajan, H.-J. Ahn, S.-I. Kim and J.-H. Jang, Nanoscale, 2013, 5, 6254–6260 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- Y. Kuang, T. Yamada and K. Domen, Joule, 2017, 1, 290–305 CrossRef CAS.
- J. Seo, H. Nishiyama, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2018, 57, 8396–8415 CrossRef CAS PubMed.
- J. H. Kim and J. S. Lee, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed.
- X. T. Xu, L. Pan, X. Zhang, L. Wang and J. J. Zou, Adv. Sci., 2019, 6, 1801505 CrossRef PubMed.
- A. G. Tamirat, J. Rick, A. A. Dubale, W.-N. Su and B.-J. Hwang, Nanoscale Horiz., 2016, 1, 243–267 RSC.
- D. K. Lee, D. Lee, M. A. Lumley and K.-S. Choi, Chem. Soc. Rev., 2019, 48, 2126–2157 RSC.
- K.-Y. Yoon, H.-J. Ahn, M.-J. Kwak, S.-I. Kim, J. Park and J.-H. Jang, J. Mater. Chem. A, 2016, 4, 18730–18736 RSC.
- C. Jiang, S. J. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- K. Arifin, R. M. Yunus, L. J. Minggu and M. B. Kassim, Int. J. Hydrogen Energy, 2021, 46, 4998–5024 CrossRef CAS.
- H.-J. Ahn, M.-J. Kwak, J.-S. Lee, K.-Y. Yoon and J.-H. Jang, J. Mater. Chem. A, 2014, 2, 19999–20003 RSC.
- X. Liu, F. Wang and Q. Wang, Phys. Chem. Chem. Phys., 2012, 14, 7894–7911 RSC.
- P. Sharma, J. W. Jang and J. S. Lee, ChemCatChem, 2019, 11, 157–179 CrossRef CAS.
- P. Thiyagarajan, H. J. Ahn, J. S. Lee, J. C. Yoon and J. H. Jang, Small, 2013, 9, 2341–2347 CrossRef CAS PubMed.
- R.-T. Gao, D. He, L. Wu, K. Hu, X. Liu, Y. Su and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 6213–6218 CrossRef CAS PubMed.
- R. T. Gao and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 23094–23099 CrossRef CAS PubMed.
- H. Shi, H. Guo, S. Wang, G. Zhang, Y. Hu, W. Jiang and G. Liu, Energy Fuels, 2022, 36, 11404–11427 CrossRef CAS.
- D. K. Lee and K.-S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- G. Wang, Y. Yang, Y. Ling, H. Wang, X. Lu, Y.-C. Pu, J. Z. Zhang, Y. Tong and Y. Li, J. Mater. Chem. A, 2016, 4, 2849–2855 RSC.
- P. Liao, M. C. Toroker and E. A. Carter, Nano Lett., 2011, 11, 1775–1781 CrossRef CAS PubMed.
- Z. Zhou, P. Huo, L. Guo and O. V. Prezhdo, J. Phys. Chem. C, 2015, 119, 26303–26310 CrossRef CAS.
- A. Alarawi, V. Ramalingam and J.-H. He, Mater. Today Energy, 2019, 11, 1–23 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- T. W. Hamann, Dalton Trans., 2012, 41, 7830–7834 RSC.
- H. Chai, S. Wang, X. Wang, J. Ma and J. Jin, ACS Catal., 2022, 12, 3700–3709 CrossRef CAS.
- S. Shen, S. A. Lindley, X. Chen and J. Z. Zhang, Energy Environ. Sci., 2016, 9, 2744–2775 RSC.
- C. Li, Z. Luo, T. Wang and J. Gong, Adv. Mater., 2018, 30, 1707502 CrossRef PubMed.
- B. Iandolo, B. Wickman, I. Zorić and A. Hellman, J. Mater. Chem. A, 2015, 3, 16896–16912 RSC.
- J. Park, K.-Y. Yoon, M.-J. Kwak, J. Kang, S. Kim, S. Chaule, S.-J. Ha and J.-H. Jang, ACS Appl. Mater. Interfaces, 2023, 15, 9341–9349 CrossRef CAS PubMed.
- Y. Lin, G. Yuan, S. Sheehan, S. Zhou and D. Wang, Energy Environ. Sci., 2011, 4, 4862–4869 RSC.
- K. Kim, I.-H. Kim, K.-Y. Yoon, J. Lee and J.-H. Jang, J. Mater. Chem. A, 2015, 3, 7706–7709 RSC.
- T. Hisatomi, H. Dotan, M. Stefik, K. Sivula, A. Rothschild, M. Grätzel and N. Mathews, Adv. Mater., 2012, 24, 2699–2702 CrossRef CAS PubMed.
- L. Steier, I. Herraiz-Cardona, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, S. D. Tilley and M. Grätzel, Adv. Funct. Mater., 2014, 24, 7681–7688 CrossRef CAS.
- H.-J. Ahn, K.-Y. Yoon, M. Sung, H. Yoo, H. Ahn, B. H. Lee, J. Lee and J.-H. Jang, ACS Energy Lett., 2023, 8, 2595–2602 CrossRef CAS.
- C. H. Bak, K. Kim, K. Jung, J.-B. Kim and J.-H. Jang, J. Mater. Chem. A, 2014, 2, 17249–17252 RSC.
- H. J. Ahn, M. J. Kim, K. Kim, M. J. Kwak and J. H. Jang, Small, 2014, 10, 2325–2330 CrossRef CAS PubMed.
- H.-J. Ahn, K.-Y. Yoon, M.-J. Kwak, J. Park and J.-H. Jang, ACS Catal., 2018, 8, 11932–11939 CrossRef CAS.
- S. Wang, C. Meng, Y. Bai, Y. Wang, P. Liu, L. Pan, L. Zhang, Z. Yin and N. Tang, ACS Appl. Nano Mater., 2022, 5, 6781–6791 CrossRef CAS.
- F. Li, J. Li, F. Li, L. Gao, X. Long, Y. Hu, C. Wang, S. Wei, J. Jin and J. Ma, J. Mater. Chem. A, 2018, 6, 13412–13418 RSC.
- Y. Lin, Y. Xu, M. T. Mayer, Z. I. Simpson, G. McMahon, S. Zhou and D. Wang, J. Am. Chem. Soc., 2012, 134, 5508–5511 CrossRef CAS PubMed.
- A. Kleiman-Shwarsctein, M. N. Huda, A. Walsh, Y. Yan, G. D. Stucky, Y.-S. Hu, M. M. Al-Jassim and E. W. McFarland, Chem. Mater., 2010, 22, 510–517 CrossRef CAS.
- R. Rivera, H. P. Pinto, A. Stashans and L. Piedra, Phys. Scr., 2011, 85, 015602 CrossRef.
- R.-T. Gao, L. Liu, Y. Li, Y. Yang, J. He, X. Liu, X. Zhang, L. Wang and L. Wu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2300493120 CrossRef CAS PubMed.
- R.-T. Gao, J. Zhang, T. Nakajima, J. He, X. Liu, X. Zhang, L. Wang and L. Wu, Nat. Commun., 2023, 14, 2640 CrossRef CAS PubMed.
- H.-J. Ahn, K.-Y. Yoon, M.-J. Kwak and J.-H. Jang, Angew. Chem., Int. Ed., 2016, 55, 9922–9926 CrossRef CAS PubMed.
- Y. F. Xu, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, Adv. Funct. Mater., 2016, 26, 4414–4421 CrossRef CAS.
- Y. Zhang, S. Jiang, W. Song, P. Zhou, H. Ji, W. Ma, W. Hao, C. Chen and J. Zhao, Energy Environ. Sci., 2015, 8, 1231–1236 RSC.
- Z. Luo, C. Li, S. Liu, T. Wang and J. Gong, Chem. Sci., 2017, 8, 91–100 RSC.
- J. Kang, K.-Y. Yoon, J.-E. Lee, J. Park, S. Chaule and J.-H. Jang, Nano Energy, 2023, 107, 108090 CrossRef CAS.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed.
- J. Park, K.-Y. Yoon, T. Kim, H. Jang, M.-J. Kwak, J. Y. Kim and J.-H. Jang, Nano Energy, 2020, 76, 105089 CrossRef CAS.
- K.-Y. Yoon, J. Park, H. Lee, J. H. Seo, M.-J. Kwak, J. H. Lee and J.-H. Jang, ACS Catal., 2022, 12, 5112–5122 CrossRef CAS.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Gratzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS PubMed.
- Y. Ling, G. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119–2125 CrossRef CAS PubMed.
- A. Annamalai, A. Subramanian, U. Kang, H. Park, S. H. Choi and J. S. Jang, J. Phys. Chem. C, 2015, 119, 3810–3817 CrossRef CAS.
- M. Li, Y. Yang, Y. Ling, W. Qiu, F. Wang, T. Liu, Y. Song, X. Liu, P. Fang and Y. Tong, Nano Lett., 2017, 17, 2490–2495 CrossRef CAS PubMed.
- A. Annamalai, H. H. Lee, S. H. Choi, S. Y. Lee, E. Gracia-Espino, A. Subramanian, J. Park, K.-j. Kong and J. S. Jang, Sci. Rep., 2016, 6, 23183 CrossRef CAS PubMed.
- J. Park, K.-Y. Yoon, M.-J. Kwak, J.-E. Lee, J. Kang and J.-H. Jang, ACS Appl. Mater. Interfaces, 2021, 13, 54906–54915 CrossRef CAS PubMed.
- J. Liu, Y. Cai, Z. Tian, G. Ruan, Y. Ye, C. Liang and G. Shao, Nano Energy, 2014, 9, 282–290 CrossRef CAS.
- K.-Y. Yoon, J. Park, M. Jung, S.-G. Ji, H. Lee, J. H. Seo, M.-J. Kwak, S. Il Seok, J. H. Lee and J.-H. Jang, Nat. Commun., 2021, 12, 4309 CrossRef CAS PubMed.
- Z. Fu, T. Jiang, L. Zhang, B. Liu, D. Wang, L. Wang and T. Xie, J. Mater. Chem. A, 2014, 2, 13705–13712 RSC.
- C. Li, A. Li, Z. Luo, J. Zhang, X. Chang, Z. Huang, T. Wang and J. Gong, Angew. Chem., 2017, 129, 4214–4219 CrossRef.
- H. Zhang, D. Li, W. J. Byun, X. Wang, T. J. Shin, H. Y. Jeong, H. Han, C. Li and J. S. Lee, Nat. Commun., 2020, 11, 4622 CrossRef PubMed.
- H. Zhang, W. Y. Noh, F. Li, J. H. Kim, H. Y. Jeong and J. S. Lee, Adv. Funct. Mater., 2019, 29, 1805737 CrossRef.
- J. Y. Kim, G. Magesh, D. H. Youn, J.-W. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 2681 CrossRef PubMed.
- Y. Fu, C. L. Dong, W. Y. Lee, J. Chen, P. Guo, L. Zhao and S. Shen, ChemNanoMat, 2016, 2, 704–711 CrossRef CAS.
- H. Zhang, J. H. Park, W. J. Byun, M. H. Song and J. S. Lee, Chem. Sci., 2019, 10, 10436–10444 RSC.
- P. S. Shinde, A. Annamalai, J. H. Kim, S. H. Choi, J. S. Lee and J. S. Jang, Sol. Energy Mater. Sol. Cells, 2015, 141, 71–79 CrossRef CAS.
- D. Hansora, D. Cherian, R. Mehrotra, J.-W. Jang and J. S. Lee, Joule, 2023, 7, 884–919 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |