
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable materials for renewable energy storage in the thermal battery†
Samantha L.
Piper
 a,
Craig M.
Forsyth
a,
Mega
Kar
b,
Callum
Gassner
a,
R.
Vijayaraghavan
c,
S.
Mahadevan
c,
Karolina
Matuszek
*a,
Jennifer M.
Pringle
b and
Douglas R.
MacFarlane
*a
a,
Craig M.
Forsyth
a,
Mega
Kar
b,
Callum
Gassner
a,
R.
Vijayaraghavan
c,
S.
Mahadevan
c,
Karolina
Matuszek
*a,
Jennifer M.
Pringle
b and
Douglas R.
MacFarlane
*a
aSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: karolina.matuszek@monash.edu; douglas.macfarlane@monash.edu
bInstitute for Frontier Materials, Deakin University, Burwood Campus, Burwood, Victoria 3125, Australia
cCell for Industrial Safety and Risk Analysis, CLRI, 6000-20 Chennai, India
First published on 15th February 2023
Abstract
The “Thermal Battery” offers the possibility of an inexpensive renewable energy storage system, deployable at either distributed- or grid-scale. For high efficiency, a crucial component of this system is an effective phase change material (PCM) that melts within the intermediate temperature range (100–220 °C) and is inexpensive, stable and sustainable. Here, we report on five aliphatic dicarbamate PCMs. For the first time, we demonstrate high thermal stability of the two best performing materials to elucidate their critical properties including lifetimes in this application. These results are considered alongside the origins of the raw materials and the synthetic routes to provide a preliminary “whole picture” assessment of the sustainability of renewable energy systems employing these materials.
Sustainability spotlightThe global transition to the post-petroleum era is central to most of the UN's Sustainable Development Goals. New technology utilising phase change materials (PCMs) in Carnot Thermal Batteries can meaningfully contribute to this transition, offering an economical and efficient system for storing renewable energy. Crucial to this technology is inexpensive, stable and renewable PCMs that melt in the range of 100–220 °C. This work demonstrates the feasible application of dicarbamate PCMs, which are shown to meet these criteria through extensive stability tests and a sustainability and cost assessment. The proposed materials perform very well in these analyses, particularly when considered alongside existing technologies. |
1 Introduction
Inexpensive, efficient and sustainable energy storage technologies are vital for a global pivot away from fossil fuels. This pivot, in turn, is vital for the climate1 – and so the urgency of technological advancement in this field is clear. Thankfully, recent progress has been rapid; lithium ion batteries, for example, have more than tripled in energy density since the early 1990s and decreased considerably in price, allowing their widespread application from portable electronics to stationary grid-storage.2 However, lithium ion batteries cannot carry all of the energy storage burden – especially considering their reliance on materials that are not renewable or abundant and while their recyclability remains a very open question. Ongoing grid decarbonisation to meet greenhouse gas emission reduction targets will therefore require rapid uptake of multiple types of renewable energy storage systems.3In the context of solar and wind energy, “thermal batteries” – i.e. thermal energy storage (TES) technologies – can offer unique benefits including low cost, good reliability and high scalability. The term “thermal battery” broadly encompasses all energy storage systems where the energy is stored in the form of heat. An advanced concept of the thermal battery is one in which the thermal storage is integrated into a reversible heat pump/organic Rankine cycle system. Where the primary input is wind and PV solar power, and with electrical power as the primary output, this is often referred to as the “Carnot Battery” (Fig. 1).4,5
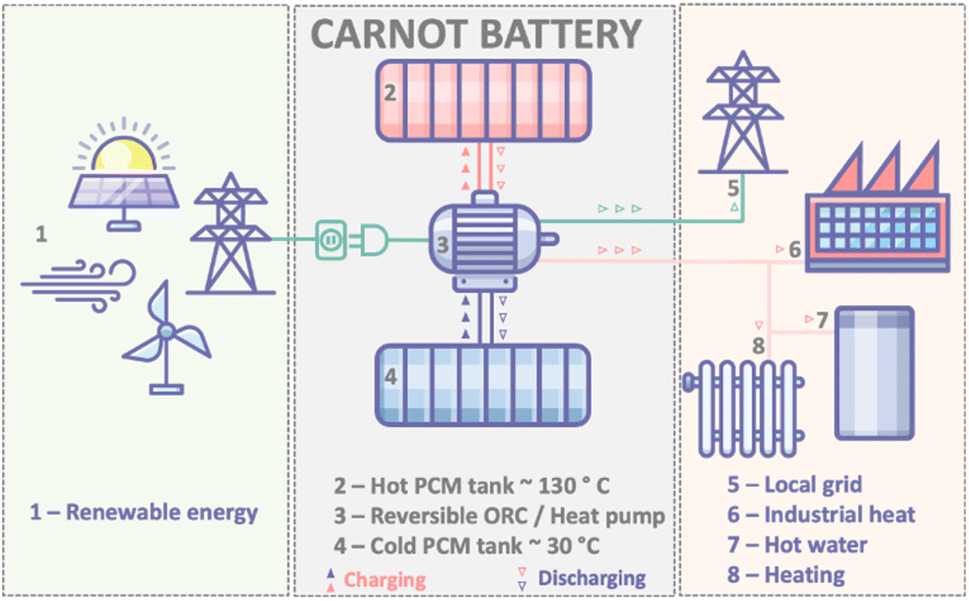 | ||
| Fig. 1 Schematic of a PCM based Carnot thermal battery. In the charge cycle, excess renewables are used in a heat pump to pump heat (red and blue upward arrows) from a cold reservoir to melt a phase change material in a hot reservoir. In the discharge cycle, the heat pump operates in the reverse mode as an organic Rankine cycle engine using the stored thermal energy (red and blue downward arrows) to generate electricity and release heat that can be used in a variety of applications. For optimum efficiency the PCM should store energy in the 100–220 °C range. | ||
In the charge mode of a Carnot battery, renewable energy drives a heat pump, with the effect of pumping thermal energy from a cold reservoir to a hot reservoir where the generated heat is stored in a thermal energy storage material. As is typical with heat pumps, the quantity of heat generated can be 2–3 times the electrical energy input to drive the pump. On discharge, the TES material transfers heat to a working fluid, which drives the heat pump in reverse mode (otherwise known as an organic Rankine cycle (ORC) engine), generating power and providing lower grade heat for various thermal energy applications, e.g. water heating. Thus far, power-to-power round trip efficiencies of 72% have been demonstrated for such a Carnot battery; notably this metric is for power output only and does not yet include the other uses of the low-grade heat that is output from the ORC. It is predicted that close to 100% round-trip energy ratio will be achievable once this new technology is optimised, including the additional waste heat applications.4,6,7
Crucial to optimising this technology is the choice of an appropriate TES material. Latent heat storage materials, otherwise known as phase change materials (PCMs), are particularly attractive for this application as they can store large amounts of energy within narrow temperature ranges. Most commonly PCMs utilise the melting transition, where the energy uptake on melting is determined by the heat of fusion, ΔHf, of the material. Gravimetric energy densities are therefore many times higher for PCMs than those of sensible thermal energy storage materials (i.e. those that do not exploit a phase change),7 while the ability of PCMs to transfer heat at constant temperatures also yields higher theoretical Carnot efficiencies.6
Intermediate melting temperature PCMs (i.e. with Tm = 100–220 °C) are particularly favourable for this application as high-efficiency power generation is achievable when coupled with ORC engines.8 Furthermore, the lower grade energy released from ORC operation at these temperatures can otherwise find use in a cascade of applications including industrial heat, space and water heating, etc.
Unfortunately, there is a limited choice of known PCMs that melt within this temperature range and meet the criteria required to offer a sustainable energy storage solution. There are two key reasons for this: firstly, naturally occurring high ΔHf materials (e.g. glycerol, fatty acids, etc.)9,10 usually melt at much lower temperatures (<100 °C), with the exception of sugar alcohols that suffer from extreme supercooling which impedes the energy release mechanism.11 PCMs that melt in the desired range are therefore typically synthetic,12–21 high density polyethylene (HDPE) being a classic example.22 Such synthetic PCMs thus carry environmental footprints derived from both raw material sourcing and synthesis. For HDPE, this “fossil-carbon backpack” is heavy.
Secondly, thermal stability is challenging to achieve when operating at these elevated temperatures, however this is rarely the subject of detailed studies in the literature; nevertheless, it is a crucial property. While thermal cycling data on proposed PCMs is sometimes reported, it is common to include only 100 cycles, without isothermal holding at the upper temperature limit. This can result in misleading claims of viable new PCMs – even the best material regarding all other thermal and physical properties will not perform well when subjected to lifecycle assessment analyses, if it lacks sufficient thermal stability for a long-life application.23
In this work, we report five aliphatic dicarbamate PCMs (Fig. 2), two of which are novel, and using these materials as exemplars, seek to answer the following questions:
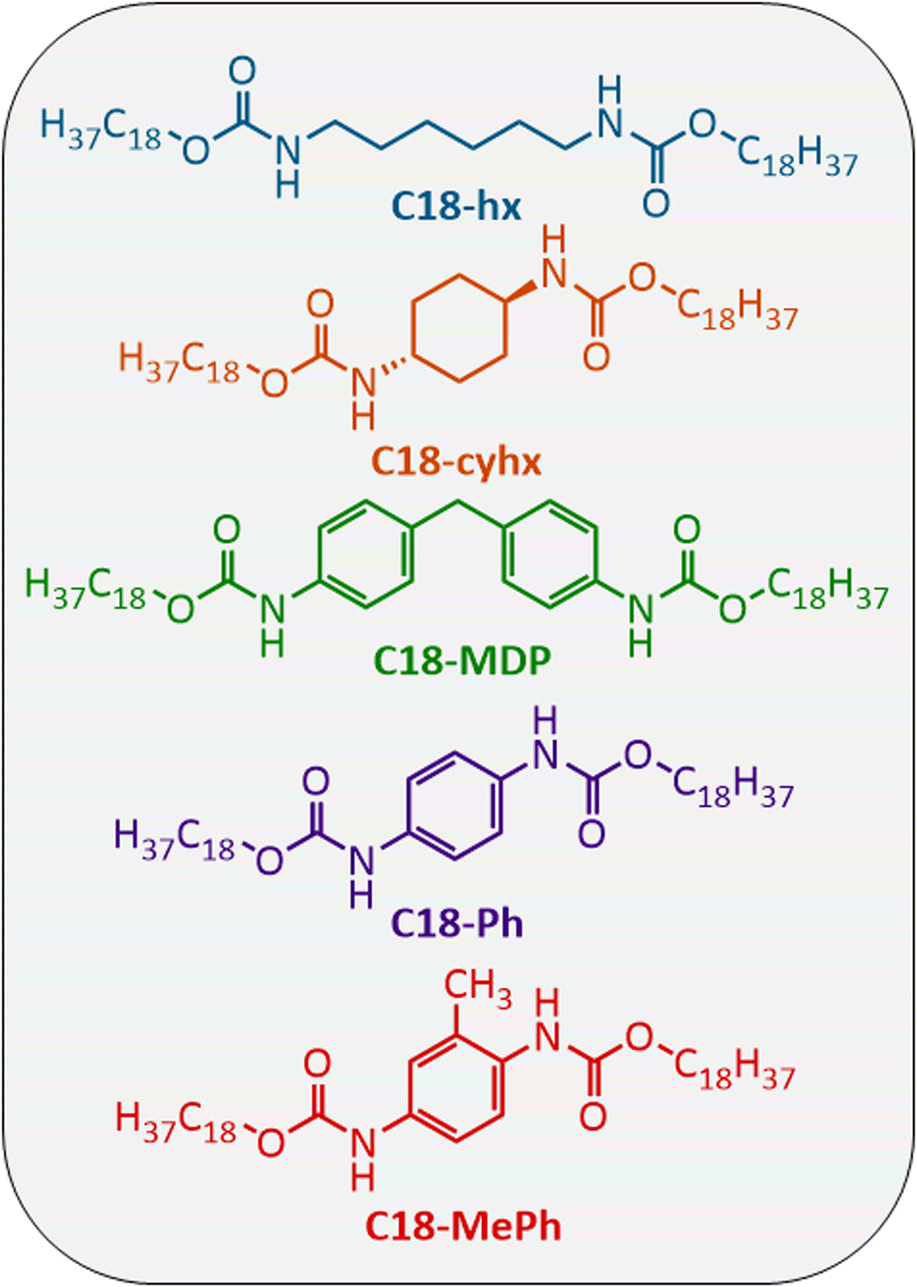 | ||
| Fig. 2 Chemical structures of the five dicarbamates studied here for use as intermediate temperature PCMs. | ||
1. How is extended thermal stability best studied in the laboratory?
2. How thermally stable does a PCM have to be to offer promise for practical application?
These PCMs are chosen as their structural features should conceivably mimic the synergy of hydrogen bonding and van der Waals interactions that is found in many naturally occurring high ΔHf materials (e.g. fatty acids9 and amines24). The functionalisation of fatty acids to overcome their typical drawbacks for PCM applications (i.e. corrosivity, sublimation tendencies) has demonstrated success in the literature previously. Examples of PCMs derived from fatty acids include fatty ureas,25 esters,26–29 thioureas,30 carbonates31 and amides.17,18,32,33 These examples typically have melting temperatures below the intermediate temperature range (Tm < 100 °C), with a notable exception being the fatty diamides and dicarbamates derived from stearic acid.17,18,34 The structures targeted here incorporate carbamate functionalities capable of forming intermolecular hydrogen bonds, with long aliphatic chains derived from stearic acid. Alongside their hydrogen bonding ability, the use of thermally stable carbamate groups should assist in targeting this property, which is particularly important given the high melting (and therefore operating) temperatures of these materials.
We begin by discussing the thermal properties of the materials, and correlate these properties to solid-state packing arrangements and interactions by studying the crystal structures of the two best performers. We then describe a series of experiments and develop novel analysis methods that probe the thermal stability of the materials to elucidate their possible lifetimes in application. In the final section we propose an approach to the assessment of the sustainability of these materials in renewable energy storage systems by considering the origins of the raw materials and the synthetic routes, alongside the thermal stability results. In a recent publication, Aftab et al.34 reported a study that includes some of these compounds as part of an investigation into the effect of chain length and functionality of a large series of materials, including photo-absorbing composites, although not on the in-depth thermal stability and sustainability aspects discussed here.
2 Results and discussion
2.1. Thermal properties of dicarbamate PCMs
Amongst the various properties that determine the viability of a given material to act as a PCM, Tm and ΔHf are chief in dictating application and efficiency. As these two properties are not inherently linked to chemical functionalities in the way that others often are (for example, flammability or chemical stability), but arise instead from less predictable factors (such as changes in molecular arrangements and intermolecular interactions35), the optimisation of these properties is a common first step in the design of new PCMs. The melting points and enthalpies of fusion of the five dicarbamates studied here, as determined by differential scanning calorimetry (DSC, full experimental details are provided in the ESI†), are detailed in Table 1 (curves are shown in Fig. 3).| T m onset/°C (±2 °C) | T m peak/°C (±2 °C) | ΔHf/J g−1 (±5%) | |
|---|---|---|---|
| a For simplicity, the thermal properties reported here for C18-MePh correspond to those of the highest stability polymorph, which forms when a ramp rate of 1 °C min−1 is used. A ramp rate of 10 °C min−1 was used for the determination of ΔHf and Tm for the other four materials as thermal properties were found to be independent of the ramp rate. | |||
| C18-hx | 119 | 121 | 200 |
| C18-cyhx | 156 | 158 | 68 |
| C18-MDP | 129 | 131 | 147 |
| C18-Ph | 149 | 151 | 135 |
| C18-MePh | 97a | 103a | 140 |
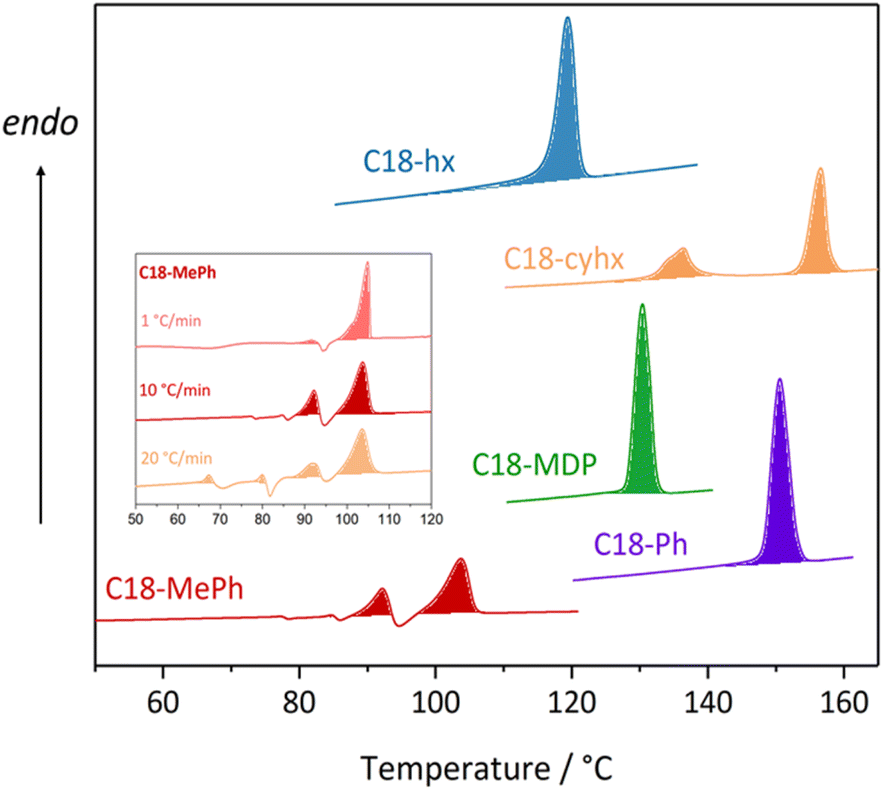 | ||
| Fig. 3 DSC traces showing the second heating cycle for the five dicarbamates, at a ramp rate of 10 °C min−1. The inset shows the DSC curves of the second heating cycle of C18-MePh at ramp rates of 1 °C min−1, 10 °C min−1, and 20 °C min−1, as the features of the traces differ at different ramp rates for this material. | ||
Despite the similarities in their fundamental chemical structures and functionalities, there is substantial variation in the melting properties exhibited by the five materials. C18-hx has the highest enthalpy of fusion among the series, melting with a ΔHf of 200 J g−1 at 119 °C (here we define Tm as the onset of the melting transition – refer to ESI† for further discussion on this topic), making it a promising candidate for PCM applications. An impressive enthalpy of fusion is also observed for C18-MDP, melting with ΔHf = 147 J g−1 at 129 °C. C18-Ph shows similar promise for PCM applications, melting with an enthalpy of fusion of 135 J g−1 at 149 °C. Importantly, these materials display minimal supercooling, with onset of crystallisation temperatures less than 5 °C below the onset of melting temperatures. Low supercooling, which is regularly observed for alkanes and their derivates due to the good nucleating ability of the n-alkyl substituents,36 is beneficial for PCM applications as it allows the efficient release of energy at temperatures close to the storing range.
The thermal behaviours of C18-cyhx and C18-MePh differ to the simple melting/crystallisation described above, with both materials appearing to undergo solid-state transitions prior to melting. In the DSC curve of C18-cyhx, a reversible solid–solid transition is observed at 134 °C (ΔH = 30 J g−1) prior to a melting transition of 68 J g−1 at 156 °C. The thermal behaviour of C18-MePh is yet more complex, with various endo- and exothermic transitions appearing on the heating cycle that vary depending on the ramp rate used (inset, Fig. 3). These transitions are attributed to the melting and crystallisation of various polymorphs of this material. Notably, when heated at a ramp rate of 1 °C min−1, only one melting transition is observed at 103 °C, which has a high enthalpy of fusion of 140 J g−1. Given that this slow ramp rate best resembles the temperature conditions of application, this material may be considered a promising material for the intermediate temperature PCM application.
2.2. Crystal structures of C18-hx and C18-MDP
The molecular origins of the thermal properties described in the previous section are important to understand, as insights will aid the design of future PCMs. Nevertheless, structure–property relationships that govern melting properties – particularly ΔHf – are often elusive. Much of our research is focussed on elucidating these relationships; we are currently conducting a detailed study on the five dicarbamates reported here and hope to publish a full report of these results in the near future. Here, with the use of synchrotron X-ray crystallography, we report the crystal structures of the two best performing (highest ΔHf) materials, C18-hx and C18-MDP, which are shown in Fig. 4 (and are confirmed to be representative of the bulk samples through powder X-ray diffraction; calculated and experimental patterns are shown in Fig. S9 and S10†).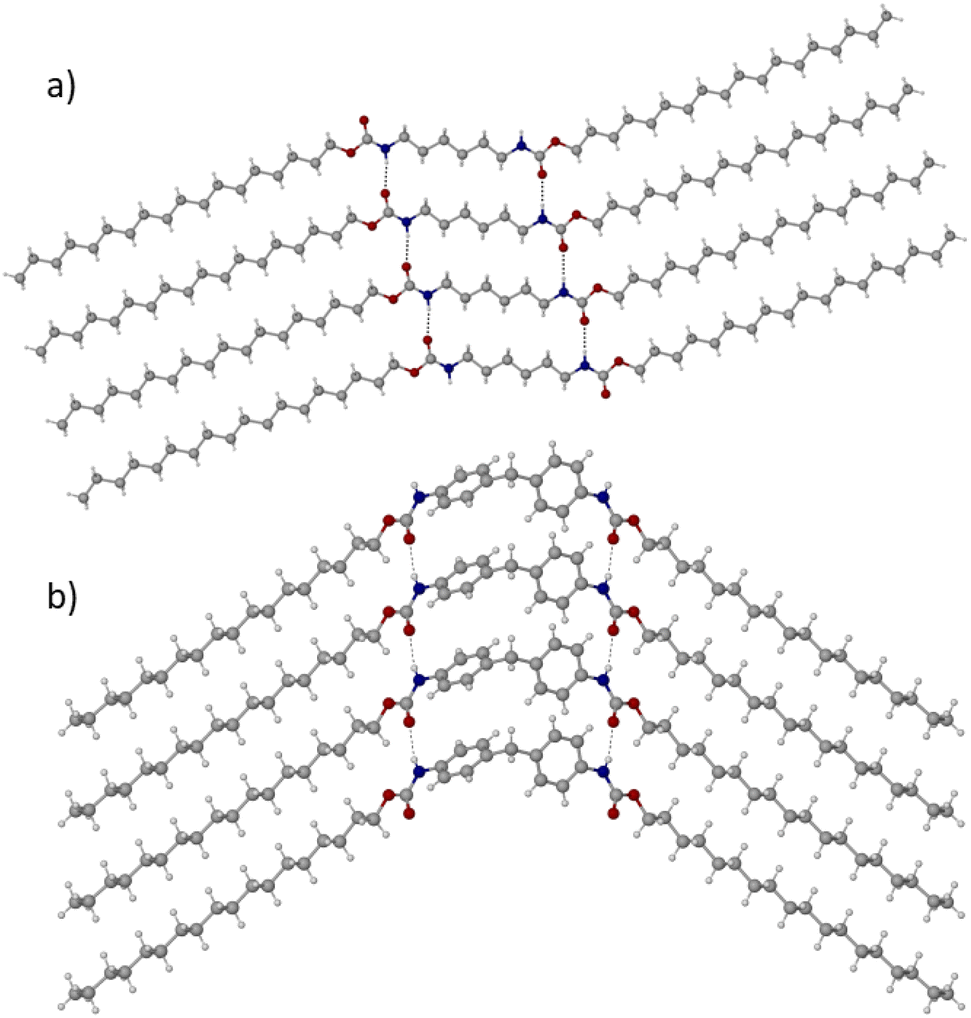 | ||
| Fig. 4 Crystallographically determined structures of C18-hx (a) and C18-MDP (b). Four molecules are shown to demonstrate the ladder motif formed by the intermolecular hydrogen bonds (represented as broken black lines). | ||
C18-hx crystallises in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, with 0.5 molecules in the asymmetric unit. The molecule is near-planar, with a slight twist between the planes of the hydrocarbon chains and the plane of the hexyl core group (24.5°). Within the plane, the molecule exists in a Z conformation, with a 22° angle between the external hydrocarbon chains and the core hexyl chain (Fig. 4a).
space group, with 0.5 molecules in the asymmetric unit. The molecule is near-planar, with a slight twist between the planes of the hydrocarbon chains and the plane of the hexyl core group (24.5°). Within the plane, the molecule exists in a Z conformation, with a 22° angle between the external hydrocarbon chains and the core hexyl chain (Fig. 4a).
C18-MDP crystallises in the monoclinic P21/m space group, with 0.5 molecules in the asymmetric unit. Due to the tetrahedral geometry of the methylene bridge in the MDP core group, the molecule exists in a bent geometry, with an angle between the core group centroid and the centroids of the external alkyl chains of 101.3°. The two phenylene groups of the MDP core are tilted relative to each other with an angle between planes of 83.8° (Fig. 4b).
In both structures, each molecule participates in four intermolecular hydrogen bonding interactions – two where the N–H groups of the urethane functionalities are acting as proton donors to a carbonyl oxygen of a urethane group from a neighbouring molecule, and two where the carbonyl oxygens are acting as proton acceptors in the reverse scenario. The hydrogen bonds are more linear and marginally shorter in the structure of C18-hx compared to those in C18-MDP (∠(N–H⋯O) = 175.1(19)°, d(N⋯O) = 2.880(2) Å and ∠(N–H⋯O) = 161(3)°, d(N⋯O) = 2.909(4) Å respectively). It is well-known in the literature that hydrogen bond length and linearity correlate with hydrogen bond strength, with shorter and more linear H-bonds typically being the strongest.37,38 Hydrogen bond strength, in turn, is known to have beneficial contributions to increasing ΔHf.12,14,19,39 The more favourable geometry of the H-bonds in C18-hx may therefore contribute to the higher ΔHf observed for this material.
In both structures, the hydrogen bonding motifs form a ladder-like structure, resulting in sheets of molecules that are bound by hydrogen bonding interactions between the urethane groups and van der Waals interactions between hydrocarbon chains of neighbouring molecules (Fig. 4). In both structures, these hydrocarbon chains pack with triclinic symmetry of the methylene subcells (commonly referred to as β-polymorphic symmetry in waxes and related aliphatic compounds40,41). This packing arrangement is typically accompanied by low free-energies and subsequently high enthalpies of fusion,42 explaining the high ΔHf of the two materials.
In the following section, we elucidate the possible lifetimes of C18-hx and C18-MDP in the thermal energy storage application by probing the thermal stability of the materials.
3 Determining PCM viability for thermal energy storage applications
It is crucial that the material not only exhibits the desired thermal properties (e.g. an appropriate Tm and high ΔHf), but also retains these properties over multiple heating/cooling cycles. In reality, a good PCM should be able to operate over many years without degradation. Various accelerated testing protocols may be used to demonstrate, and probe, the thermal stability in this context. Here we chose to proceed with C18-hx and C18-MDP, as these two materials exhibit high enthalpies of fusion, with the additional benefit of being derived from inexpensive starting materials (as discussed in Section 5).3.1 Thermal reversibility of carbamate bonds
The thermal stability of the carbamate bond has been extensively studied as it is similar to the polyurethane linkage.43 While decomposition routes resulting in the generation of amines, olefins, and CO2 are possible at high temperatures, the main degradation pathway between 150 °C and 200 °C is dissociation into the starting alcohol and isocyanate.44 The point of equilibrium for the reversible reaction is therefore somewhere in the region of 150 °C, but is dependent on the nature of the alcohol and diisocyanate involved. To elucidate this temperature for C18-hx and C18-MDP, which is an important property as the thermal dissociation of the carbamate bonds is a clear first step towards decomposition, we studied the compounds with variable temperature FTIR (Fig. 5). A peak corresponding to N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO stretching (∼2275 cm−1) is first observed at 155 °C for C18-MDP and 160 °C for C18-hx, confirming that thermal dissociation of the carbamate bonds occurs in this region of temperature.
CO stretching (∼2275 cm−1) is first observed at 155 °C for C18-MDP and 160 °C for C18-hx, confirming that thermal dissociation of the carbamate bonds occurs in this region of temperature.
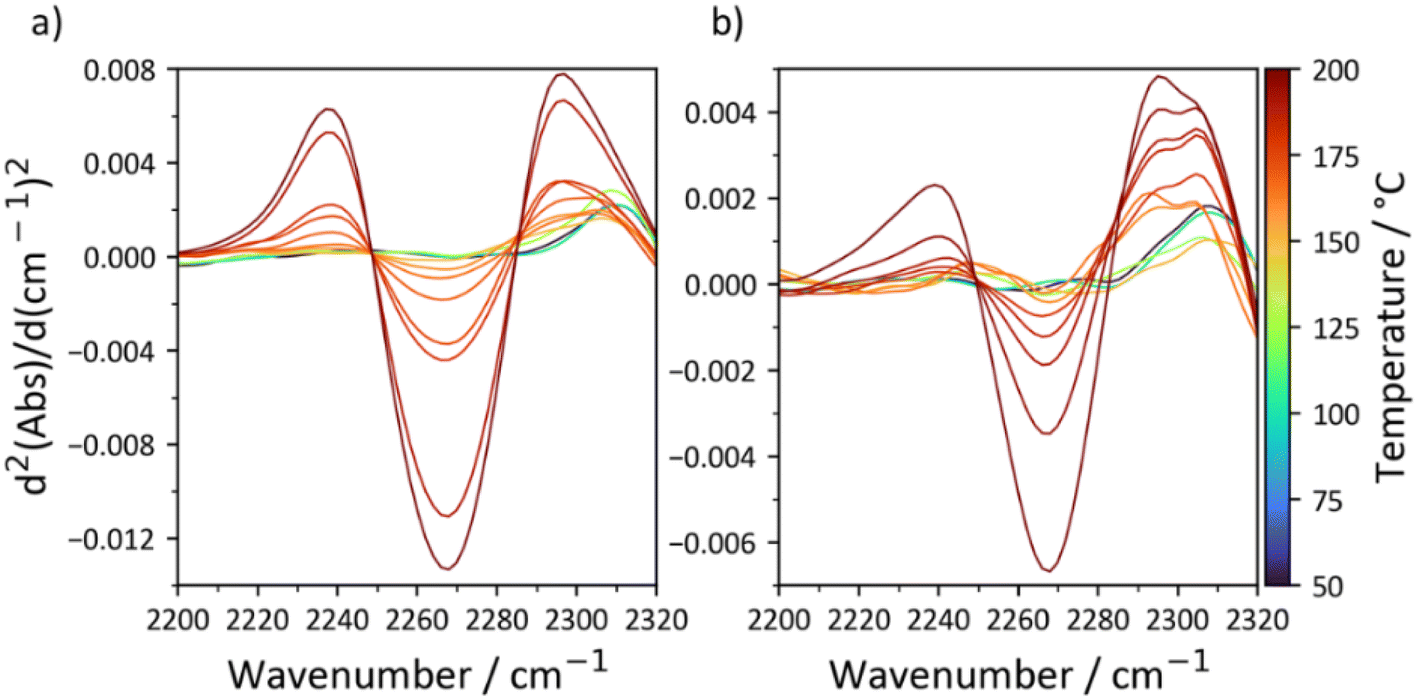 | ||
| Fig. 5 Second derivative IR spectra of C18-MDP (a) and C18-hx (b) in the 2200 cm−1–2320 cm−1 region. The isocyanate NCO stretch is observed at ∼2275 cm−1. | ||
In a closed vessel and in the absence of any nucleophilic impurities, dissociation in the molten state is unproblematic for the PCM application as the urethane bond will reform upon cooling. Nonetheless, to verify this and verify that no other decomposition reactions are involved, the following sections describe extensive thermal stability testing regimes that probe the thermal stability and upper temperature operating limits of C18-hx and C18-MDP.
3.2 Thermogravimetric analysis
A first estimation of an upper operating temperature limit can be obtained from thermogravimetric analysis (TGA),45 as shown in Fig. 6a. However, significant mass loss can take place well below these temperatures when the heating rate is slowed or isothermal heating is used.45–47 A more rigorous TGA method involves holding samples at various isothermal temperatures and measuring the time taken for 1% mass loss to occur, which is denoted t0.99;46,47 typical data is shown in Fig. 6b and c (blue axes and data points). As this value decreases exponentially with increasing temperature, due to the Arrhenius relationship between reaction rate and temperature (k = Ae−Ea/RT), the applicability of this relationship can be confirmed or otherwise in a ln(time) vs. 1/T plot as shown in Fig. 6b and c (wine coloured axes and data points) for C18-hx and C18-MDP. | ||
| Fig. 6 (a) TGA curves of C18-hx, C18-cyhx, C18-MDP, C18-Ph and C18-MePh at a 10 °C min−1 heating rate. The inset shows the expanded temperature range where decomposition begins. (b) and (c) Show the isothermal TGA time taken for 1% mass loss to occur (top and right axes in blue) and Arrhenius plots of this data with linear fits (wine coloured axes and data points) for C18-hx and C18-MDP respectively. | ||
The linear trend of ln(time) vs. 1/T allows the extrapolation of thermal stability information to lower temperatures, where the time taken to observe degradation would be too long to be feasibly tested by TGA methods. Additionally, this fit allows determination of the activation energy of decomposition reactions, Ea; in this case 108 kJ mol−1 and 118 kJ mol−1 for C18-hx and C18-MDP respectively. The higher activation energy of the decomposition reaction of C18-MDP suggests greater thermal stability for this material, as indicated directly by the TGA curves in Fig. 6a.
Using the linear relationships established here, if we are to assume that the two PCMs will operate at a maximum temperature of Tm + 10 °C (i.e. 129 °C for C18-hx and 139 °C for C18-MDP), extrapolating the time taken for 1% degradation to occur to these temperatures gives values of 20 days for C18-hx and 62 days for C18-MDP. Given that the fraction of time spent at these maximum temperatures during application (i.e. in the charged state) would likely be ∼25%, an estimate of lifetime can be obtained.
To carry out this estimation we assume that end of life corresponds to 20% degradation, and that the rate of the decomposition reaction is independent of the extent of the reaction. This yields a lifetime of 4 years for C18-hx, and a particularly promising lifetime of 13 years for C18-MDP. Note, of course, that the “degradation” involved here is chiefly mass loss of volatile products, and that the piercing of the sample pan and the constant flow of nitrogen over the sample in the standard TGA test would be expected to increase the extent of decomposition by constantly removing the volatile products. In application, the PCMs would be confined in closed systems similar to those of the long-term thermal stability tests described in the next section. Hence these are lower bounds on lifetime. For example, though this extrapolation suggests 1% thermal degradation of C18-hx after 12 days at 135 °C, we found no evidence of degradation in a sample after being held at this temperature for 40 days when contained within a capped vial in a low moisture atmosphere. This method allows for a rapid first estimation of possible lifetime, which should then be supported by more long-term studies, as presented below.
3.3 Extended thermal stability studies
As the first step in extended thermal stability studies, we found that there were no significant changes to the thermal properties of either C18-hx or C18-MDP after >500 heating/cooling cycles in the DSC (ramp rate 10 °C min−1) to a maximum temperature of Tm + 10 °C.To better understand the resistance of these materials to thermal degradation, we next held the samples in their molten state at various temperatures above Tm. Initially, samples were held at 135 °C in silicone-capped vials in a low moisture atmosphere. When no changes to thermal properties were observed for either material after 40 days at this temperature, as determined by DSC, we replicated the conditions with fresh samples at 150 °C. Samples were held at this temperature for over three months, with small samples frequently extracted throughout this time period for property testing by DSC. The results of these DSC tests are shown in Fig. 7.
 | ||
| Fig. 7 Thermal properties (ΔHf and Tm) for samples of C18-MDP (a and b) and C18-hx (d and e) after being held at 150 °C for various periods of time, as determined by DSC. (c and f) DSC scans of C18-MDP and C18-hx before and after being held for extended periods at 150 °C. | ||
The decrease in enthalpy of fusion and melting point of C18-hx after 100 days at 150 °C suggests degradation has occurred in the sample, as decomposition products act as impurities that are known to lower melting points and decrease enthalpies of fusion.48 Notably, there are also two additional peaks on the heating curve of C18-hx at 59 °C (3 J g−1) and 68 °C (10 J g−1) (Fig. 7c), likely attributed to the melting of some decomposition products. The first of these two new peaks can be attributed to the melting of free stearyl alcohol (Tm = 58–59 °C), suggesting that the equilibrium point for the thermal dissociation reaction of the carbamate bond of C18-hx is around 150 °C (consistent with the stability observed at 135 °C, from the previous tests described above). This result is not problematic given that the testing was done at a temperature more than 30 °C above the melting point of the PCM – this temperature condition would not be routinely reached in application. It is likely that nucleophilic impurities, e.g. water from the atmosphere, hindered the reformation of some of the original carbamate bonds on cooling.
On the other hand, the consistency in Tm and ΔHf of C18-MDP over the 100 day period suggests negligible decomposition has occurred, supporting the TGA experiments that indicate high thermal stability for this material. This may suggest that the components of C18-MDP are less susceptible to side reactions than those of C18-hx once the carbamate bond is dissociated.
3.4 Accelerated rate calorimetry and flashpoint testing
While sufficient thermal stability is important to ensure a long lifetime in application, it is also important to ensure that a thermal degradation event would not be catastrophic. Accelerated rate calorimetry (ARC) is a useful technique for evaluating the safety of materials at elevated temperatures. This technique measures the self-heating and any adiabatic pressure increase of a system in response to temperature and time. The results from ARC studies on C18-hx and C18-MDP show that there is no exothermic event and no self-heating or “thermal runaway” in either an air or nitrogen atmosphere up to 400 °C (Fig. S11–S14†), indicating that both materials are quite safe operating in this temperature range. In both materials, a small but gradual increase in pressure is observed with increasing temperature, resulting in a maximum pressure of ∼4.6 bar for both materials at 400 °C. Notably this pressure generation does not begin until around 250 °C, which is well above the operating temperature of both materials.Further supporting the operational safety of the materials, a flash point was not detected for C18-hx until 280–290 °C, more than 150 °C higher than the expected upper temperature limit for this PCM in application. Flash point testing was only conducted on this material as very large sample sizes are required for the test, and the aliphatic core group of C18-hx suggested that it may be the more susceptible of the two materials to flammability and thus the most important to investigate.
4 Sustainability assessment
In the development of sustainable energy systems it is important that lifecycle assessment (LCA) indicates an overall positive outcome after assessing any environmental cost of raw material sourcing and synthesis. While detailed lifecycle assessment is outside the scope of this paper, we can make some comments regarding the possible outcomes of such analyses by considering the starting materials and synthetic route, as well as expected outcomes when the materials eventually reach the end of their first life.4.1 Raw materials and synthetic route
There are several important ways in which the synthetic route used here align with the Principles of Green Chemistry,49 which are fundamental to the outcome of any LCA.50 The PCMs can be formed in solvent- and catalyst-free conditions, without the need for further isolation or purification. Furthermore, no by-products are formed as the reaction has an atom efficiency of 100%, and ∼70% of the mass of the final products are derived from renewable stearyl alcohol (76% in C18-hx). The synthetic route also has low energy consumption, as the compounds form quickly once the reaction vessel is elevated to a temperature above the Tm of both starting materials (∼60 °C in all cases).A potential drawback of this synthetic route is the use of diisocyanate starting materials, which are problematic due to the use of phosgene in their synthesis. Lifecycle assessment analyses have suggested that present day isocyanate production results in 6.5 kg CO2 (ref. 51) per kg of aliphatic diisocyanate and 2.76 kg CO2 per kg of MDI.52 Given the proportion of these isocyanates in the PCMs, CO2 emissions as a direct result of these isocyanates is 1541 kg per tonne of C18-hx and 872 kg per tonne of C18-MDP. Considering these on the basis of lifetime energy stored and delivered, conservatively assuming an operating life of 5 years, the CO2 emissions from using these isocyanates is 0.014 kg kW−1 h−1 for C18-hx and 0.012 kg kW−1 h−1 for C18-MDP (see ESI† for calculations). This is in stark contrast to the ∼1 kg of CO2 emitted per kW h from coal-fired power, and impressive when considered alongside the ∼0.029 kg kW−1 h−1 emitted from the manufacture and use of lithium ion batteries (assuming a battery lifetime of 2500 cycles).53
However, it is notable that there is significant research underway in developing more sustainable synthetic routes to polyurethanes, the findings from which will be applicable here. For example, efforts are being directed toward the sustainable production of diisocyanates; pentamethylene diisocyanate has been reported as the first bio-based crosslinker.54 This is particularly relevant in the context of C18-hx due to the structural similarity to the hexamethylene diisocyanate (HDI) used in its synthesis. Similar difunctionalised materials have shown minimal variation in thermal properties when the length of the central alkane group linking the functionalities is altered beyond Cn > 2.18 It would therefore be expected that replacing the hexane core group in C18-hx with a pentane core group by use of this more sustainable diisocyanate would yield a bio-based compound with similar thermal properties to C18-hx. Regarding the aromatic core groups, earlier this year BASF reported the first greenhouse gas neutral aromatic isocyanate55 – a methylene diisocyanate (MDI), which is the diisocyanate used in the synthesis of C18-MDP.
Other routes to sustainable polyurethanes are phosgene-free, or phosgene- and isocyanate-free.56 Phosgene-free routes typically involve the reaction of a diamine with environmentally-benign dimethyl carbonate, followed by catalytic decomposition of the subsequent dicarbamate to yield the corresponding diisocyanate. This has been achieved for both MDI57 and HDI.58
Of the synthetic routes to polyurethanes that bypass the use of diisocyanates altogether, some involve using CO2 as a reactant, inviting the possibility of another route to carbon-negative production processes in the future. For example, the incorporation of CO2 into epoxides to form cyclic organic carbonates as intermediates for urethane formation can result in 100% bio-based polyurethanes.59 This is a favourable route as the chemical transformation from epoxide to carbonate is 100% atom efficient, and as carbonates are of broad utility, developing effective and sustainable catalysts for this transformation is of substantial interest to many researchers.60
Overall, one can expect that the CO2 emissions burden of isocyanate production calculated above might be substantially reduced in the future as these sustainable pathways become industrialised.
The above calculations and discussion do not yet consider the possible benefits of using stearyl alcohol, which is the major component of the PCMs. Stearyl alcohol is widely considered to be a sustainable material, as it is derived from biorenewable stearic acid, which is found in vegetable oils and animal fats. Notably, at the present time, there is a carbon-footprint associated with using stearyl alcohol despite its biorenewable origins – for example, one LCA study suggests a carbon-footprint of cetearyl alcohol (a cetyl/stearyl alcohol mixture) of 11.9 kg CO2 per kg.61 In this case, a palm plantation provides the crop source and land-use is responsible for 64% of the emissions associated with the production process. The authors describe the value as an over-estimation due to drastic initial assumptions, including that plantation occurs on undegraded soil and does not account for the fact that land-clearing only occurs once. The value decreases to 1.9 kg CO2 per kg cetearyl alcohol if meadows are used for the initial plantation. In either land-use case, this can actually have a carbon-storage effect when considering the CO2 removed by the crop over time. Notably, the above values also assume non-renewable energy is used for refining and transport. To eliminate the need for further refining of cetearyl alcohol to isolate pure stearyl alcohol, we synthesised C18-hx with a mixture of cetyl and stearyl alcohol (i.e. C16/C18-hx) finding only a small decrease in the ΔHf of the material when compared to C18-hx (186 J g−1) and slightly lower Tm (116 °C).
There are other notable avenues for stearyl alcohol production – sunflower oil has a much higher stearic acid content than palm kernel oil (18% compared to 1–3%) with some mutants having stearic acid contents as high as 37%.62 The remainder of the oil is largely oleic acid which can be converted to stearic acid through hydrogenation; a process already required in the production of fatty alcohols from fatty acids.63 Sunflower oil production also has a lower global warming effect than palm oil and uses significantly less water.64 Furthermore, researchers have demonstrated the valorisation of low-cost feedstock waste in producing C18 fatty acids; waste cooking oil and fat has been shown to be a viable raw material for the production of stearic acid by introduction of lipase enzymes.65 The otherwise dumping of this waste can have profound environmental impacts, as well as energy and economic costs which have been estimated to be ∼3 kW h kg−1 and 0.49 USD per kg.66,67
Of course, the use of stearyl alcohol produced from any of the sources above in the PCM represents a conversion and sequestering of CO2 in the material for the duration of its lifetime. For stearyl alcohol, this represents a negative carbon contribution of −2.1 kg CO2 per kg PCM.
Other factors that are outside the scope of this paper to meaningfully calculate, such as energy use during synthesis and transport, are consistent regardless of the origins of the materials used. Notably, CO2 emissions from the synthesis can be considered negligible.
4.2 Re-use/recycling prospects at the “end of first life”
Re-use or recycling instead of disposal as the final phase in LCA will drastically improve system performance.68 The end of life – or end of first-life – of C18-hx and C18-MDP is assumed here to be the point in time at which the efficiency of the system is reduced to 80% of its initial performance (75% is commonly used for lithium ion batteries). At this point, at least 80% of the PCM should not have decomposed and purification by recrystallisation in ethanol could be used to regenerate the pristine material for ongoing use. This is a key benefit of using materials that can be solubilised – the purification procedure is more challenging, if achievable at all, for materials that cannot be easily solubilised (e.g. HDPE).Processing of the remaining (≤20%) decomposed sample will depend on the decomposition products; some insights can be gained here by review of the available literature. Prenveille et al.69 studied dicarbamates analogous to C18-MDP and C18-hx with shorter alkyl chains as model compounds when investigating the reactivity of urethanes at high temperatures. In line with our observations of C18-MDP being more thermally stable than C18-hx, the authors reported more side-product formation in the HDI-derived samples than the MDI samples after annealing at temperatures between 190 °C and 220 °C. In the hexane samples, decomposition products were identified to be mostly allophanates (which form when an isocyanate reacts with a urethane), with some isocyanaurates (which form either by reaction of an isocyanate with an allophanate, or via the cyclic trimerisation of three isocyanates). Allophanates are favourable decomposition products here as their formation is reversible – thermal treatment can be used to regenerate the urethane and isocyanate group,70 which can be re-used in synthesis to regenerate the PCM.
Interestingly, the authors reported seeing only trace decomposition products in MDI samples after the same annealing process, which were reported to be uretdiones and ureas. As with allophanates, uretdiones, which are dimers of isocyanates, are favourable decomposition products here as they thermally dissociate to reform the isocyanates at elevated (>150 °C) temperatures, or at moderate temperatures in the presence of (usually phosphine) catalysts.71 In fact, uretdiones are of commercial value in the blocked isocyanate industry.69 Urea containing decomposition products may find commercial value elsewhere, or are generally considered to be biodegradable and likely to break down in the environment.
Thus, there is clearly immense potential for re-using the bulk of the material at the end of its first life in application – ∼80% should be recoverable by simple recrystallisation methods, while a significant proportion of the remaining 20% can be regenerated to starting materials through facile dissociation reactions. Repeated recycling in this way extends the effective lifetime of the materials by a factor of approximately four and is consistent with the principles of a circular economy.
5 Cost analysis
While these PCMs offer many benefits over conventional power generation and energy storage technologies (high biorenewable content, no rare earth minerals, etc.) – they must also be competitive in the market. Here we have performed a simple cost analysis for the preparation of these materials, and estimate USD 453 per tonne for C18-hx and USD 342 per tonne for C18-MDP (Table S4†). This results in a cost of USD 7.9 per kW h of stored energy per cycle for C18-hx and USD 8.3 per kW h for C18-MDP. Notably, this is a fraction of the cost per kW h that lithium ion batteries offer (USD132 per kW h in 2021 (ref. 72)). Importantly, the stored thermal energy is versatile; either used directly for heating applications (which constitute 50% of total global energy consumption73) or for electricity generation in the Carnot battery. Notably, this cost analysis is for initial synthesis from raw materials, and does not attempt to incorporate the further economic and energy benefits of recycling the materials at end of first life as discussed in the previous section.6 Conclusions
The thermal battery holds the promise of providing an inexpensive energy storage solution on both a distributed- and grid-scale. However, its cost efficiency and sustainability is critically dependent on the source, performance, and price of the phase change material involved. In this work we have described a family of PCMs that are readily and atom efficiently produced from carbamate chemistry and are based on sustainable, or potentially sustainable, materials. The high biorenewable content of the PCMs presents the opportunity for carbon-negative syntheses in the near future. Particularly, two of the materials – C18-hx and C18-MDP – show very high enthalpies of fusion and suitable melting points for the thermal battery application.On the complex question of the stability in operation, we propose several approaches to studying this property, including various thermal analysis experiments and variable temperature FTIR spectroscopic methods. The two materials that represent the most promising PCMs show very high thermal stability; in particular, these studies suggest an operating life of 10+ years for C18-MDP. Promisingly, the products formed upon eventual decomposition are expected to be mostly adducts that can be converted back to the starting materials by simple heating methods. The ability to re-purify the PCMs at the end of their first life by recrystallisation methods could increase their overall lifetime by a factor of approximately four.
At the fundamental level, synchrotron X-ray crystallography was used here to study the intermolecular interactions and solid-state packing arrangements of the two materials with the most favourable thermal properties, C18-hx and C18-MDP. The reported structures demonstrate the effects of subtle differences between the structures on their melting properties. These structure–property relationships are important to understand, as their optimisation is a clear step towards the development of next-generation PCMs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the Australian Research Council through its Linkage Project scheme (LP190100522) and collaborating organisation Energy Storage Pty Ltd. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.References
- International Energy Agency (IEA), World Energy Outlook 2019, 2019 Search PubMed.
- M. S. Ziegler and J. E. Trancik, Energy Environ. Sci., 2021, 14, 1635–1651 RSC.
- International Renewable Energy Agency, Innovation Outlook: Thermal Energy Storage, Abu Dhabi, 2020 Search PubMed.
- O. Dumont, A. Charalampidis and V. Lemort, 18th International Refrigeration and Air Conditioning Conference at Purdue, 2021 Search PubMed.
- D. Steger, C. Regensburger, B. Eppinger, S. Will, J. Karl and E. Schlücker, Energy, 2020, 208, 118216 CrossRef CAS.
- R. B. Peterson, Energy, 2011, 36, 6098–6109 CrossRef.
- O. Dumont, G. F. Frate, A. Pillai, S. Lecompte and V. Lemort, J. Energy Storage, 2020, 32, 101756 CrossRef.
- K. Matuszek, M. Kar, J. Pringle and D. Macfarlane, Chem. Rev., 2023, 123, 491–514 CrossRef CAS PubMed.
- Y. Yuan, N. Zhang, W. Tao, X. Cao and Y. He, Renew. Sustain. Energy Rev., 2014, 29, 482–498 CrossRef CAS.
- O. Okogeri and V. N. Stathopoulos, Int. J. Thermofluids, 2021, 10, 100081 CrossRef CAS.
- S. Tomassetti, A. Aquilanti, P. F. Muciaccia, G. Coccia, C. Mankel, E. A. Koenders and G. Di Nicola, J. Energy Storage, 2022, 55, 105456 CrossRef.
- K. Matuszek, R. Vijayaraghavan, M. Kar and D. R. MacFarlane, Cryst. Growth Des., 2019, 20, 1285–1291 CrossRef.
- K. Matuszek, R. Vijayaraghavan, C. M. Forsyth, S. Mahadevan, M. Kar and D. R. MacFarlane, ChemSusChem, 2020, 13, 159–164 CrossRef CAS.
- S. L. Piper, C. M. Forsyth, M. Kar, D. MacFarlane, K. Matuszek and J. M. Pringle, Mater. Adv., 2021, 2, 7650–7661 RSC.
- K. Matuszek, R. Vijayaraghavan, M. Kar, S. Mahadevan and D. R. MacFarlane, ChemSusChem, 2020, 14, 2757–2762 CrossRef.
- S. L. Piper, M. Kar, D. R. MacFarlane, K. Matuszek and J. M. Pringle, Green Chem., 2022, 24, 102–117 RSC.
- M. C. Floros, K. L. C. Kaller, K. D. Poopalam and S. S. Narine, Sol. Energy, 2016, 139, 23–28 CrossRef CAS.
- K. D. Poopalam, L. Raghunanan, L. Bouzidi, S. K. Yeong and S. S. Narine, Sol. Energy Mater. Sol. Cells, 2019, 201, 110056 CrossRef CAS.
- R. D. McGillicuddy, S. Thapa, M. B. Wenny, M. I. Gonzalez and J. A. Mason, J. Am. Chem. Soc., 2020, 142, 19170–19180 CrossRef CAS PubMed.
- H. Zhang, W. Xu, J. Liu, M. Li and B. Yang, J. Mol. Liq., 2019, 282, 474–483 CrossRef CAS.
- H. Zhang, M. Li and B. Yang, J. Phys. Chem. C, 2018, 122, 2467–2474 CrossRef CAS.
- H. Weingrill, K. Resch-Fauster, T. Lucyshyn and C. Zauner, Polym. Test., 2019, 76, 433–442 CrossRef CAS.
- M. Kenisarin and K. Mahkamov, Renew. Sustain. Energy Rev., 2007, 11, 1913–1965 CrossRef CAS.
- T. Chen, H. Sun, P. Mu, Z. Zhu, J. An, W. Liang and A. Li, Sol. Energy Mater. Sol. Cells, 2020, 206, 110340 CrossRef CAS.
- N. Gökşen Tosun, A. Çetin and C. Alkan, Int. J. Energy Res., 2022, 46, 20600–20610 CrossRef.
- A. A. Aydın, Sol. Energy Mater. Sol. Cells, 2013, 113, 44–51 CrossRef.
- A. A. Aydın and A. Aydın, Sol. Energy Mater. Sol. Cells, 2012, 96, 93–100 CrossRef.
- Ö. Gök, J. Energy Storage, 2022, 56, 105859 CrossRef.
- M. C. Floros and S. S. Narine, Mater. Lett., 2014, 137, 252–255 CrossRef CAS.
- C. Alkan, Y. Tek and D. Kahraman, Turk. J. Chem., 2011, 35, 769–777 CAS.
- J. A. Kenar, Sol. Energy Mater. Sol. Cells, 2010, 94, 1697–1703 CrossRef CAS.
- G. Canik and C. Alkan, Sol. Energy, 2010, 84, 666–672 CrossRef CAS.
- C. Alkan, G. Canik, H. Dünya and A. Sarı, Sol. Energy Mater. Sol. Cells, 2011, 95, 1203–1207 CrossRef CAS.
- W. Aftab, J. Shi, M. Qin, Z. Liang, F. Xiong, A. Usman, S. Han and R. Zou, Energy Storage Mater., 2022, 52, 284–290 CrossRef.
- H. Mehling, Sol. Energy, 2013, 95, 290–299 CrossRef CAS.
- J. A. Noël, L. M. LeBlanc, D. S. Patterson, L. Kreplak, M. D. Fleischauer, E. R. Johnson and M. A. White, J. Phys. Chem. B, 2019, 123, 7043–7054 CrossRef PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- J. A. Gerlt, M. M. Kreevoy, W. Cleland and P. A. Frey, Chem. Biol., 1997, 4, 259–267 CrossRef CAS PubMed.
- T. Inagaki and T. Ishida, J. Phys. Chem. C, 2016, 120, 7903–7915 CrossRef CAS.
- K. Larsson, Acta Chem. Scand., 1966, 20, 20 Search PubMed.
- K. Sato, Chem. Eng. Sci., 2001, 56, 2255–2265 CrossRef CAS.
- C. Himawan, V. Starov and A. Stapley, Adv. Colloid Interface Sci., 2006, 122, 3–33 CrossRef CAS PubMed.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- T. Hentschel and H. Münstedt, Polymer, 2001, 42, 3195–3203 CrossRef CAS.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef CAS.
- K. J. Baranyai, G. B. Deacon, D. R. MacFarlane, J. M. Pringle and J. L. Scott, Aust. J. Chem., 2004, 57, 145 CrossRef CAS.
- T. J. Wooster, K. M. Johanson, K. J. Fraser, D. R. MacFarlane and J. L. Scott, Green Chem., 2006, 8, 691–696 RSC.
- J. Sohns, B. Seifert and E. Hahne, Int. J. Thermophys., 1981, 2, 71–87 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Frontiers, 1998, 640, 1998 Search PubMed.
- T. E. Swarr, D. Cespi, J. Fava and P. Nuss, Application of Lifecycle Assessment to Green Chemistry Objectives, in Handbook of Green Chemistry, ed. E. S. Beach and S. Kundu, John Wiley & Sons, Ltd, vol. 10, 2017, p. 1 Search PubMed.
- Cefic – The European Chemical Industry Council, Aliphatic Isocyanates for Polyurethane Products, 2014 Search PubMed.
- PlasticsEurope, Toluene Diisocyanate (TDI) & Methylenediphenyl Diisocyanate, MDI, 2021 Search PubMed.
- H. E. Melin, Analysis of the Climate Impact of Lithium-Ion Batteries and How to Measure it, Circular Energy Storage, 2019 Search PubMed.
- W. Li, H. Li, C. Wu, B. Han, P. Ouyang and K. Chen, Chem. Eng. J., 2021, 425, 131527 CrossRef CAS.
- BASF, BASF Expands Portfolio of Climate Friendly Products Introducing the First Isocyanate Not Carrying a CO2 Backpack, https://www.basf.com/global/en/media/news-releases/2022/02/p-22-132.html, accessed September 2022 Search PubMed.
- H. Khatoon, S. Iqbal, M. Irfan, A. Darda and N. K. Rawat, Prog. Org. Coat., 2021, 154, 106124 CrossRef CAS.
- N. Lucas, A. P. Amrute, K. Palraj, G. Shanbhag, A. Vinu and S. Halligudi, J. Mol. Catal. A: Chem., 2008, 295, 29–33 CrossRef CAS.
- D.-L. Sun, J.-Y. Luo, R.-Y. Wen, J.-R. Deng and Z.-S. Chao, J. Hazard. Mater., 2014, 266, 167–173 CrossRef CAS PubMed.
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238–1254 CrossRef CAS PubMed.
- Y. Fan, M. Tiffner, J. Schörgenhumer, R. Robiette, M. Waser and S. R. Kass, J. Org. Chem., 2018, 83, 9991–10000 CrossRef CAS PubMed.
- J. Guilbot, S. Kerverdo, A. Milius, R. Escola and F. Pomrehn, Green Chem., 2013, 15, 3337 RSC.
- S. Anushree, M. André, D. Guillaume and F. Frédéric, Agron. Sustainable Dev., 2017, 37, 18 CrossRef.
- L. Giraldo, G. Camargo, J. Tirano and J. C. Moreno-Piraján, E-J. Chem., 2010, 7, 1138–1147 CrossRef CAS.
- J. H. Schmidt, J. Cleaner Prod., 2015, 87, 130–138 CrossRef.
- S. Kumar and S. Negi, 3 Biotech, 2015, 5, 847–851 CrossRef PubMed.
- F. F. de Albuquerque Landi, C. Fabiani, B. Castellani, F. Cotana and A. L. Pisello, Waste Manag., 2022, 138, 219–233 CrossRef PubMed.
- M. Ripa, C. Buonaurio, S. Mellino, G. Fiorentino and S. Ulgiati, Int. J. Perform. Eng., 2014, 10, 347 Search PubMed.
- X. Tian, J. Xie, M. Xu, Y. Wang and Y. Liu, Waste Manag., 2022, 145, 72–82 CrossRef PubMed.
- T. Prenveille, C. Garreau, M. Matner, D. Dijkstra, W. Oppermann and D. Johannsmann, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 621–629 CrossRef CAS.
- A. Lapprand, F. Boisson, F. Delolme, F. Méchin and J.-P. Pascault, Polym. Degrad. Stab., 2005, 90, 363–373 CrossRef CAS.
- E. Querat, L. Tighzert, J. Pascault and K. Dušek, Appl. Macromol. Chem. Phys. Angew Makromol. Chem., 1996, 242, 1–36 CrossRef CAS.
- BloombergNEF, https://about.bnef.com/blog/battery-pack-prices-fall-to-an-average-of-132-kwh-but-rising-commodity-prices-start-to-bite/, accessed September 2022.
- International Energy Agency (IEA), Renewables, Paris, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2220003 and 2220004. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2su00111j |
| This journal is © The Royal Society of Chemistry 2023 |