
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomass components toward H2 and value-added products by sunlight-driven photocatalysis with electronically integrated Auδ−–TiO2: concurrent utilization of electrons and holes†
Himanshu
Bajpai
 ab,
Inderjeet
Chauhan
ab,
Kranti N.
Salgaonkar
ab,
Nitin B.
Mhamane
ab and
Chinnakonda S.
Gopinath
*ab
ab,
Inderjeet
Chauhan
ab,
Kranti N.
Salgaonkar
ab,
Nitin B.
Mhamane
ab and
Chinnakonda S.
Gopinath
*ab
aCatalysis and Inorganic Chemistry Division, CSIR – National Chemical Laboratory, Dr Homi Bhabha Road, Pune 411 008, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201 002, India. E-mail: cs.gopinath@ncl.res.in
First published on 17th February 2023
Abstract
While the progress of photocatalytic overall water splitting is hindered mainly by sluggish oxygen evolution kinetics, photocatalytic hydrogen generation using biomass components, such as glycerol as a sacrificial reagent, is a prudent way to efficient hydrogen production. Can we also utilize holes effectively to oxidize the sacrificial agent to value-added products (VAPs)? Towards answering this question of concurrent utilization of electrons and holes, a successful attempt has been made. Herein, we report a facile photo-deposition method to prepare well-dispersed plasmonic Au integrated with P25–TiO2. XPS studies show the nature of gold to be electron-rich or anionic, demonstrating strong electronic integration with titania and possible charge transfer from oxygen-vacancy sites to gold, which in turn helps the simultaneous production of large amounts of H2 (18 mmol h−1 g−1) and VAPs (glycolaldehyde, dihydroxyacetone, and formic acid) formation from glycerol. Elemental mapping and HRTEM images demonstrate a uniform distribution of Au on TiO2, leading to strong Au–TiO2 nano-heterojunctions, and hence a high photocatalytic activity. H2 yield decreases by 4 and 18-fold with glucose and cellulose, respectively, compared to glycerol. A systematic photocatalytic study was carried out under aerobic and anaerobic conditions to understand the glycerol oxidation products. In addition to H2, 4–10% of glycerol in 5 h was oxidized to VAPs in direct sunlight underscoring the high activity associated with Au@TiO2. Further, the C–C cleavage of glycerol also occurs to a significant extent. Hence, significantly prolonged irradiation is expected to result in improved results with longer-chain biomass components to VAPs. It is worth exploring the current approach with other metal-integrated semiconductors with different redox potentials and various biomass components towards H2 and VAP formation.
Sustainability spotlightPresent research is towards addressing the circular economy by converting biomass component molecules to value added products, namely glycolaldehyde, dihydroxyacetone and formic acid, along with hydrogen by direct sunlight driven photocatalysis. While conventional heterogeneous catalysis of biomass conversion requires high temperature/pressure, present method achieves the same at ambient conditions with solar energy. Further CO2 is almost completely avoided in the present work by not producing the same in the reaction and by employing solar energy for reaction. Further solar energy is effectively stored in the chemical bonds of value added products. Therefore, this research contributes to the circular economy. Present work aligns with the goals 7 of “Affordable and Clean Energy” 12 of “Responsible Consumption and Production” and 13 of “Climate Action” in the UN's Sustainable Development Goals. |
Introduction
Fossil fuels for global energy are like glucose to the human body, which move the entire world but are also the main cause of CO2 emission and hence contribute to climate change; however, unlike fossil fuels, the global energy demand can be at least partially fulfilled by renewable energy sources, e.g., solar and wind energy.1 Hydrogen as a fuel is ideal for high gravimetric energy (120 MJ kg−1) compared to any other fossil fuel and produces no harmful by-products, except water, when subjected to combustion. However, in the present scenario, >95% of hydrogen is produced by fossil fuels, such as steam reforming of methane/naphtha, resulting in greenhouse gases as by-products and causing global warming. To deal with global warming and the future energy crisis while considering limited fossil fuel availability, combining solar energy and biomass/biowaste can lead to sustainable and green energy by solar water splitting (SWS).2–4 Can we also concurrently convert biomass to value-added products (VAPs), so that the whole process could be considered green? We are partially successful in our efforts to convert biomass to H2 and VAPs.SWS at zero potential can fulfill the future energy demand; however, the main drawback is its low efficiency. It has been 50 years since the first report of SWS appeared in 1972, and we are still far away from finding an efficient photocatalytic system of hydrogen generation from SWS.5 Certain factors hamper efficiency, such as (1) sluggish oxygen evolution kinetics, (2) high rate of electron–hole recombination, and (3) 3–5 orders of difference in time scales of photophysical and photochemical processes.6–8
Since biomass components, such as cellulose and glucose, have very low economic value and glycerol is a by-product of biodiesel industries; it is worth employing them as feedstock and/or sacrificial agent. It is also known that the production of biodiesel and hence glycerol generation is also increasing every year by 10%. The conversion of glycerol into VAPs is a point of attraction in the present era, as its under-utilization is posing a threat to the biodiesel industry; glycerol availability at a very low price should be exploited commercially.9–11 Glycerol could be used in photocatalytic water splitting not only to increase H2 production but also as a sacrificial reagent to consume holes for partial oxidation to VAPs.12–15 The same concept can be extended to other biomass components for their conversion to VAPs.
Plasmonic metals (Au, Ag, Cu) integrated with semiconductors are known to enhance charge separation and work as a photosensitizer due to the surface plasmon resonance (SPR) effect, via either the direct electron transfer or resonant energy transfer.16–18 Since TiO2 is a well-known stable semiconductor, the large bandgap and high electron–hole recombination result in low efficiency. Plasmonic metals, especially gold shows significantly different absorption spectra with different particle sizes and morphologies and can be integrated with TiO2 to utilize visible light in direct sunlight.19–22 However, in SWS, the overall kinetics is mainly hindered due to the four-electron O2 evolution process, therefore, to enhance the overall kinetics, a sacrificial reagent,23 such as methanol, triethanolamine, sulphide–sulphite is normally employed, which consumes holes rapidly and minimizes electron–hole recombination. From the term sacrificial, it is clear that these chemicals increase hydrogen production; however, can we prudently utilize holes, so that hole consumption leads to the conversion of sacrificial agents to VAPs too? Hence, a versatile catalyst is required to meet the dual purpose of photocatalytic H2 production as well as oxidation of low-value sacrificial agent (biomass component) specifically into VAPs.24–27
Zang et al.28 employed Au/TiO2 system for photo-reforming of ethanol; they obtained 7 mmol h−1 g−1 of H2 generation with formaldehyde and carbon dioxide as by-products but did not quantify the by-products. Wang et al.29 attempted photocatalytic H2 generation using Ag2O/TiO2 system, however, the reported H2 yield was 0.3 mmol h−1 g−1, and there was no VAP reported. For the Cu/TiO2 system, several researchers have attempted photo-reforming of glycerol as a model sacrificial reagent. Trang et al.30 reported 16 mmol h−1 g−1 H2 yield under direct sunlight; however, there are no VAPs reported. With 1.6 mmol h−1 g−1 of H2, Montini et al. produced CO2, CH4, glycolaldehyde, and di-hydroxy acetone by converting glycerol into VAPs.31 In the majority of the literature, authors mostly paid attention to H2 production and not much on the selectivity of the oxidation products; however, in the present study, we exploited the large-scale and concurrent utilization of electrons and holes toward hydrogen and VAPs, respectively.
Herein we report the Au@TiO2 composite, prepared via the facile photo deposition method, by taking advantage of the positive zeta potential of TiO2. Elemental mapping and X-ray photoelectron spectroscopy (XPS) was employed to understand the distribution and electronic environment of the Au@TiO2, respectively. The photocatalytic hydrogen evolution reaction (HER) was carried out with Au@TiO2 in aqueous glycerol, which also showed high glycerol conversion and selectively to three major VAPs. HER studies were also carried out with glucose and cellulose. 1H NMR was employed to identify and quantify the oxidation products.
Experimental section
Synthesis of Au/TiO2 composite
Electronically integrated and highly-dispersed Au@TiO2 was prepared by a modified procedure of controlled photochemical deposition. 200 mg P25–TiO2 was dispersed in 100 ml of water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (60:40) solution in a 250 ml quartz round bottom (RB) flask; after stirring for 10 min. 3.08 mg of the Au-precursor (HauCl4·3H2O) was added to prepare 0.5 wt% Au-containing TiO2. Other compositions were also prepared in a similar manner with the required amount of Au-precursor. The solution was subjected to N2 purging in the dark for 30 min. with stirring for uniform interaction of [AuCl4]−1 with TiO2. It is very likely that gold-center is attracted to oxygen vacancy sites. This solution was subjected to UV lamp irradiation for 1 h. After irradiation, the solution was centrifuged and washed with hot water (338 K) for several times, and then dried in the oven at 343 K. Different wt% of Au with 0.1, 0.5, and 1% with respect to TiO2 were also synthesized and they are denoted as 0.1Au@TiO2, 0.5Au@TiO2 and 1Au@TiO2, respectively.
:methanol (60:40) solution in a 250 ml quartz round bottom (RB) flask; after stirring for 10 min. 3.08 mg of the Au-precursor (HauCl4·3H2O) was added to prepare 0.5 wt% Au-containing TiO2. Other compositions were also prepared in a similar manner with the required amount of Au-precursor. The solution was subjected to N2 purging in the dark for 30 min. with stirring for uniform interaction of [AuCl4]−1 with TiO2. It is very likely that gold-center is attracted to oxygen vacancy sites. This solution was subjected to UV lamp irradiation for 1 h. After irradiation, the solution was centrifuged and washed with hot water (338 K) for several times, and then dried in the oven at 343 K. Different wt% of Au with 0.1, 0.5, and 1% with respect to TiO2 were also synthesized and they are denoted as 0.1Au@TiO2, 0.5Au@TiO2 and 1Au@TiO2, respectively.
Photochemical hydrogen production measurement
For photocatalytic hydrogen production, 3 mg of any Au@TiO2 catalyst was taken in a 50 ml borosilicate RB flask, with 20 ml of different percentages of aqueous glycerol (0.14, 0.6, and 1.4 M glycerol), and subjected to 10 min of N2 purging. Then, the photocatalyst was irradiated with light under one sun condition (100 mW cm−2 with a 450 W Newport solar simulator fitted with a 1.5 AM filter) or direct sunlight (average light intensity was 60 mW cm−2). Hydrogen was the only gas-phase product, and it was confirmed and quantified by GC (Agilent 7890A) analysis.Photochemical glycerol oxidation measurement
To measure the glycerol oxidation products, 5 mg of the catalyst was initially dispersed in 5 ml of 0.05 M glycerol aqueous solution, and then kept under nitrogen or oxygen purging for 30 min, depending on the type of the reaction carried out subsequently. It was realized from the initial measurements with the same glycerol concentration (as that for the above-mentioned H2 production) for oxidation, the conversion was low and this made the NMR analysis also difficult for the product identification. In view of this difficulty, a few other catalyst:glycerol ratios were also evaluated and optimized to the maximum glycerol conversion along with the ease of product identification. The maximum glycerol conversion to value-added products was observed with a 5 mg catalyst in a 5 ml solution of 50 mM glycerol. The solution was kept under nitrogen or oxygen purging for 30 min, depending on the type of reaction carried out subsequently, after that, the catalyst-glycerol suspension was irradiated under one sun condition or direct sunlight. All oxidized products of glycerol were quantified using Agilent GC (7890A) and 1H NMR. Liquid product identification and quantification were carried out by NMR (Bruker AV 500), with 1 mmol KHP (potassium hydrogen phthalate) as the internal standard.where Cx,Ix, Ical, Ncal, Nx, Ccal are the concentration of the product, integral area of the product, integral area of the standard, number of protons from the standard and the product, and concentration of the standard, respectively. Various liquid products were measured by NMR and hydrogen through GC, and the reproducibility was within a 5% error margin.
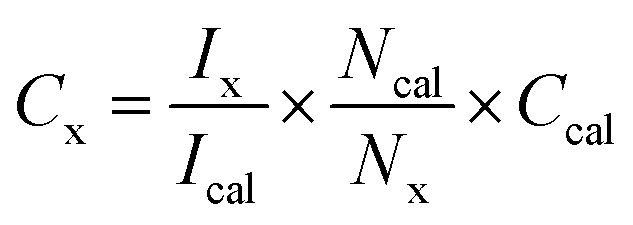
Materials characterization
Diffuse reflectance UV-Vis measurement was carried out on a Shimadzu spectrophotometer (model-UV2550) with spectral grade BaSO4 as the reference material. Powder X-ray diffraction (XRD) data were collected on a Pan analytic X'pert pro dual goniometer diffractometer using Cu-Kα (1.5418 Å) radiation with a Ni filter. HRTEM analysis of the material was conducted on a JEOL JEM F-200 HTEM instrument operating at 200 kV equipped with EDX (energy dispersive X-ray analysis). XPS analysis was carried out on an instrument from Thermo Fisher Scientific Instruments, UK with monochromatic Al Kα as the X-ray source with 6 mA beam current and operating at 12 kV. All the spectra were calibrated, with respect to adventitious carbon observed at 284.9 eV. Glycerol product quantification was carried out with Bruker AV 500, with NS-32, DS-4, SW-24.9934 ppm with water suppression sequence using Bruker Top Spin. Many characterization tests were repeated using at least three sets of samples/materials prepared in different batches. An Agilent GC (7890A) was used for gas analysis with a molecular sieve-coated packed column using TCD as a detector. Zeta potential measurements were performed on a Brookhaven Instruments NanoBrook series zeta potential analyser.Results and discussions
UV-visible absorption by Au@TiO2
UV-visible reflectance spectrum was measured and converted to absorption. UV-Vis spectra were recorded on all Au@TiO2 photocatalysts and the results are shown in Fig. 1, along with parent TiO2. SPR (surface plasmon resonance) absorption features were observed on all Au@TiO2 materials at 545 nm, irrespective of Au content.32 While 0.1Au@TiO2 showed low-absorption intensity, the other two compositions show comparable absorption behaviour with an increase of SPR intensity, associated with higher absorption ability in the visible region compared to 0.1Au@TiO2. It is also to be noted that the colour associated with the photocatalyst appeared to be increasingly becoming deep purple (inset image of Fig. 1). It is likely that the deep purple colour associated with 1Au@TiO2 could be preventing more light absorption, compared to 0.5Au@TiO2, which resulted in low photocatalytic hydrogen evolution, and the same is discussed in the later section.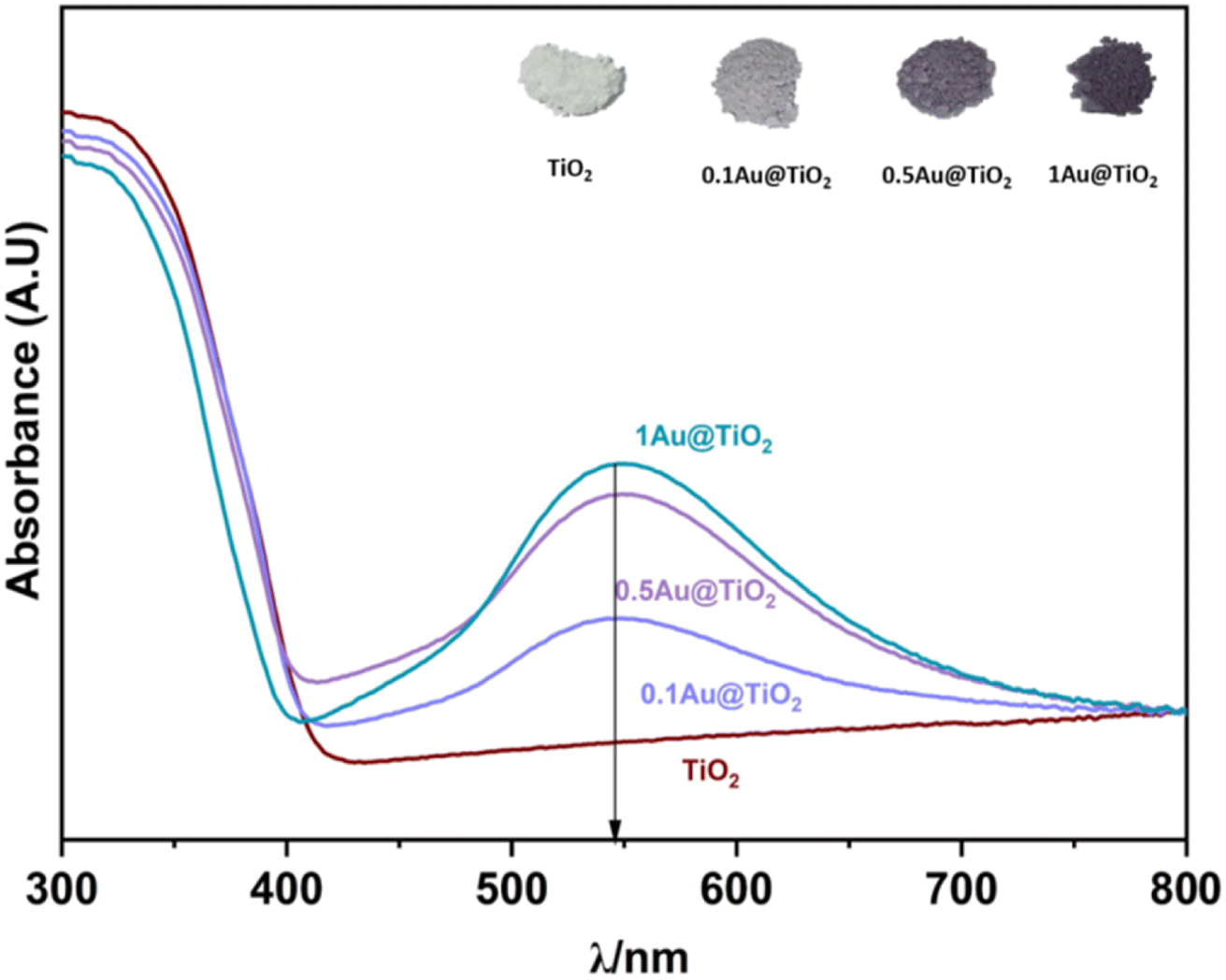 | ||
| Fig. 1 UV-visible absorption spectra of TiO2, 0.1Au@TiO2, 0.5Au@TiO2, and 1Au@TiO2, inset shows the image of all photocatalysts. SPR feature was observed at 545 nm for all xAu@TiO2. | ||
Solar hydrogen production
Solar hydrogen production was measured under one sun (100 mW cm−2) and direct sunlight in Pune, Maharashtra, India, from February to May with an average light intensity of 60 mW cm−2. It is to be noted that sunlight power fluctuates between 52 and 70 mW cm−2 depending on the time of the day, cloud cover, etc. However, for practical purposes, we considered 60 mW cm−2 as the average solar power. 60–65 mW cm−2 power was observed for at least 50% of the total reaction time. Solar hydrogen evolution measurement was carried out systematically under different reaction conditions. During hydrogen evolution measurement, 3 mg of the catalyst was used with 20 ml solution and kept under irradiation without stirring. It is to be noted that a uniform suspension, without settling of any particles, was observed with the catalyst in an aqueous glycerol solution, and hence the experiments were carried out without stirring (see photos shown in Fig. S1 in ESI†). In Fig. 2a, different weight percent loadings of Au on TiO2 were subjected to hydrogen evolution under direct sunlight with 0.6 M glycerol as a sacrificial reagent. TiO2 gave 0.86 mmol h−1 g−1 of hydrogen, after loading with Au (xAu@TiO2), and absorption of composite shifted to the visible light range due to the SPR effect of Au, as shown in UV-Vis absorption spectra (Fig. 1), indicating the strong absorption at 545 nm resulting in higher H2 production. With 0.1 wt% of Au loading, hydrogen evolution was observed to be 7.4 mmol h−1 g−1, which is 8-fold higher than that of bare TiO2.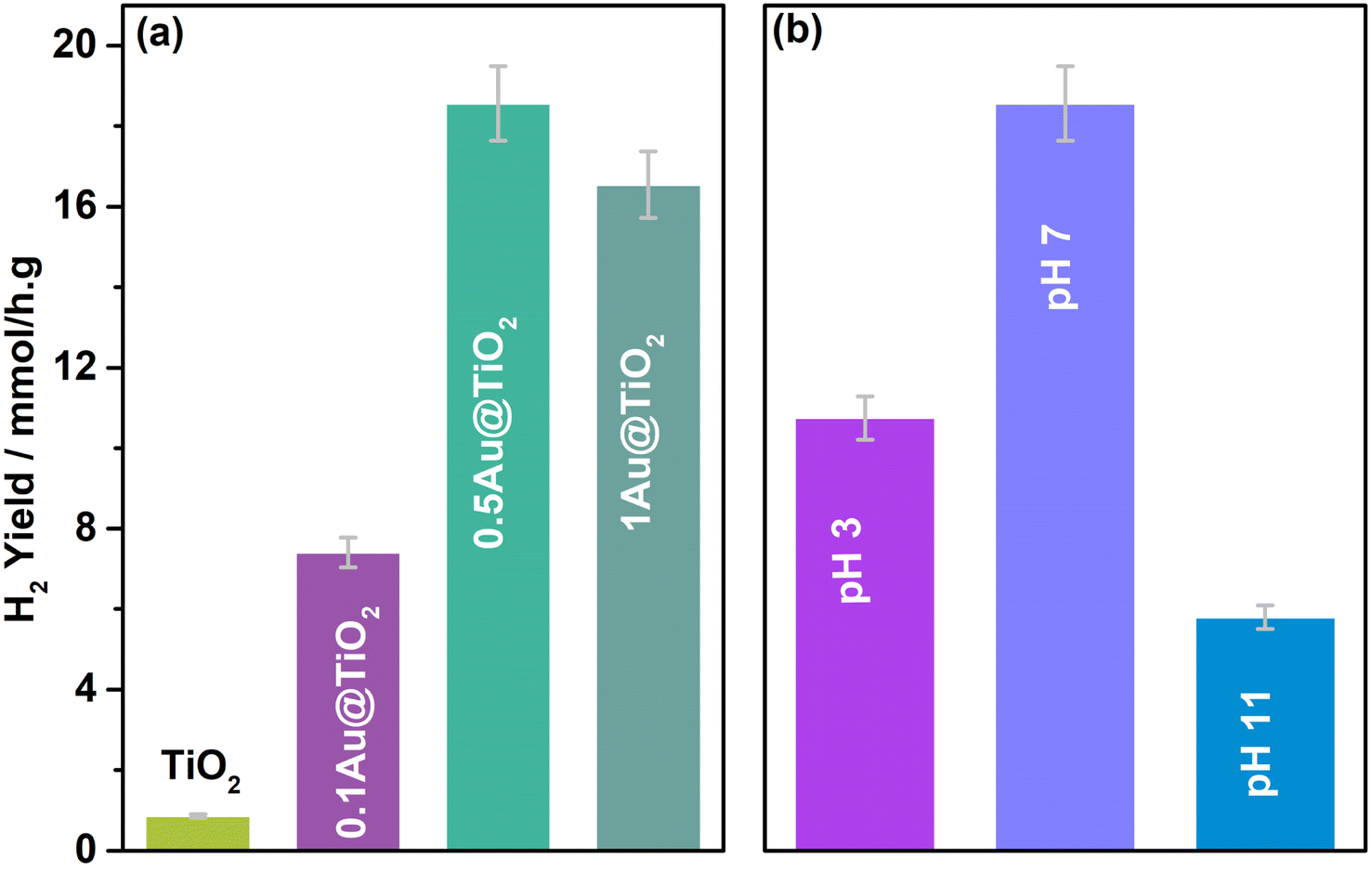 | ||
| Fig. 2 (a) Photocatalytic hydrogen yield observed for (a) different wt% of Au on TiO2 and (b) 0.5Au@TiO2 at different pH conditions; to maintain the pH, 0.01 M HCl and NaOH were used as acid and base, respectively. 5% glycerol was used as a sacrificial reagent under direct sunlight with an average light intensity of 60 mW cm−2. | ||
While increasing the weight% of Au from 0.1 to 1, 0.5% exhibited the highest hydrogen production (18 mmol h−1 g−1) than the 1% Au-containing (16.5 mmol h−1 g−1) composite under direct sunlight. It is to be noted that comparable SPR feature intensity was observed for 0.5 and 1 wt% of Au@TiO2, reflecting a similar observation as in solar hydrogen production. These observations hint that the Au-dispersion remains the same for 0.5 and 1 wt% of Au. In fact, a marginally lower H2 yield was observed with 1 wt% Au, indicating that there could be other changes. This is supported by a significant change observed with Au 4f binding energy and the same is explained later in the XPS section. Some of the top results reported for H2 production with Au and Cu-based systems from literature are compiled in Table 1. Results obtained from the current studies are superior to the results reported earlier with hydrogen and VAPs production, as shown in Table 1; also, direct sunlight irradiation shows significantly better results than with one sun condition. Reported higher hydrogen production, than that from this study, is invariably observed under UV irradiation; needless to say that significant UV light absorption by titania too contributes to the total activity. While glycerol was used as a sacrificial agent by a few groups for hydrogen generation, no other VAPs of glycerol were observed or reported in the literature. This factor makes the current work far superior in terms of the concurrent utilization of holes and electron charge carriers for versatile light energy to chemical energy conversion to H2 as well as VAPs.
| Catalyst | Light source and power | H2 (mmol h−1 g−1) | Reaction conditions | VAPs | Ref. |
|---|---|---|---|---|---|
| 1.5% Au/TiO2 P25 | UV light | 28.3 | 10% glycerol in water (6.5 mg in 20 ml solution) | NA | 11 |
| 1% Au/TiO2 P25 | One sun (UV-Vis) | 0.307 | Water:methanol = 3:1 (20 mg catalyst in 40 ml solution) |
NA | 16 |
| 5% Au/TiO2 hierarchical spheres | UV-Vis (100 mW cm−2) | 8.06 | 30 vol% methanol in water (50 mg catalyst in 100 ml solution) | NA | 36 |
| 0.5% Au/TiO2 nanorods | UV light | 29 | 10% glycerol in water (6.5 mg catalyst in 20 ml solution) | NA | 37 |
| 2% Au/TiO2 | UV light | 30.3 | 10% glycerol in water (6.5 mg catalyst in 20 ml solution) | NA | 38 |
| 0.3% AuTiO2 | Simulated light (21.1 mW cm−2) | 0.2 | 1% TEOA with water (250 mg with continuous flow of nitrogen) | NA | 39 |
| 1% Au/TiO2 anatase | Solar lamp (10.7 mW cm−2) | 2.55 | 5% glycerol in water (50 mg in 50 ml solution) | NA | 40 |
| Cu2O/Bi2O3/TiO2 | UV-Vis | 6.2 | 5% glycerol | NA | 41 |
| Cu2O/TiO2 | 400 W metal halide lamp | 4.7 | 5 v/v% glycerol/water (100 mg catalyst in 200 ml) with pH 10 | NA | 42 |
| Cu2O/TiO2 | SB-110P/F lamp (100 W), 365 nm | 15.3 | 5 v/v% glycerol/water (2.5 mg catalyst in 25 ml) | NA | 43 |
| CuOx–PtO2/TiO | 300 W xenon lamp | 1.3 | 10% glycerol (50 mg catalyst in 100 ml) | NA | 44 |
| 3% Cu/TiO2 | 100 mW cm−2 | 0.05 | 1% acetic acid (25 mg in 25 ml) | CH4, CO2 | 45 |
| 0.5% Au@TiO2 | One sun condition (100 mW cm−2) | 15 | 5% (0.6 M) glycerol in water (3 mg in 20 ml) | Dihydroxyacetone, glycolaldehyde, formic acid | Current work |
| 0.5% Au@TiO2 | Direct sunlight (60 mW cm−2) | 18 | 5% (0.6 M) glycerol in water (3 mg in 20 ml) | Dihydroxyacetone, glycolaldehyde, formic acid | Current work |
As shown in the structural characterization studies in the following sections, the highest hydrogen generation activity observed with 0.5Au@TiO2 is attributed to an atom-like sub-nanometre Au cluster dispersion over TiO2, as discussed in the later sections. Furthermore, photocatalytic hydrogen evolution measurements at different pH values were carried out with 0.5Au@TiO2 catalyst with 5% glycerol under direct sunlight, and the results are shown in Fig. 2b. The pH change was achieved with the required amount of dilute (0.01 M) HCl and NaOH under acidic and alkaline conditions, respectively, and pH change was ensured using a pH meter (Mettler Toledo seven compact S210). Interestingly and critically, the highest amount of hydrogen was observed at neutral pH; hydrogen production was found to decrease on either increasing or decreasing the pH to basic or acidic conditions. In the acidic pH, zeta potential is close to the zero-point charge, which results in significant agglomeration of TiO2 particles and hence a decrease in photocatalytic activity was observed, however, under basic pH conditions, due to the abundantly-available hydroxyl groups, the dissociation of Ti–OH is decreased drastically. The zeta potentials measured for all three pH conditions are shown in Fig. S2 (in ESI†) fully supporting this result. These factors are known to hinder the photocatalytic activity, and it was suggested from the kinetic model by Estahbanati et al.46 At neutral pH (pH = 7) good dispersion of the catalyst in the water/glycerol was observed. Even after a few days, we did not observe any settling of the catalyst (Fig. S1†) but a highly-dispersed uniform suspension was maintained, which is also an interesting observation in the present work. In general, with powdered catalyst, due to the stirring, significant scattering of the light occurs and the same reduces the light absorption by the catalyst; if there is no stirring, the catalyst would settle at the bottom of the reactor and also reduces the photoactivity of the composite; this problem could be overcome by making the catalyst in a thin film form (as in Si PV) or should be dispersed in the solution throughout the reaction.6,47
Three different glycerol concentrations (1, 5, and 10%) were employed to understand the effect of sacrificial reagent concentration; activity was measured periodically with 0.5Au@TiO2 in 20 ml aqueous glycerol solution under one sun condition and the results are shown in Fig. 3a. An interesting trend in results was observed, and the following points deserved to be highlighted. (1) 5% (0.6 M) glycerol containing aqueous solution showed a constant H2 yield of 15 mmol h−1 g−1 as a function of irradiation time; among the three different glycerol concentrations (1, 5, and 10%), indeed, 5% showed the best performance. (2) While 10% (1.4 M) glycerol concentration also showed a constant H2 yield of 11 mmol h−1 g−1, it is lower than that at 5% glycerol concentration. (3) In contrast to the above two points, 1% (0.14 M) glycerol-containing solution showed the lowest H2 yield, although initially, it showed 7 mmol h−1 g−1 yield, H2 yield reaches a plateau at later hours. The influence of the sacrificial agent concentration on hydrogen generation has been addressed significantly in the literature.33–35 Dependence of the hydrogen generation has been demonstrated with different concentrations of the reactant. Beyond a threshold concentration of the sacrificial agent, active sites are saturated with those molecules and the same decreases the photoactivity. In the present case, the viscosity factor of glycerol also adds to the diffusion issues, which decreases the rate at 10% glycerol concentrations. Generally, the observed results indicated that an optimum glycerol concentration was required to achieve the best performance in the present experiments; except hydrogen, no other gaseous product was observed. For the oxidation product of the glycerol, due to the high concentration of glycerol employed for the results shown in Fig. 2 and 3, it was challenging to identify and quantify the product by NMR. Another set of the study was carried out to explore the products of glycerol oxidation at low glycerol concentrations, which will be discussed later.
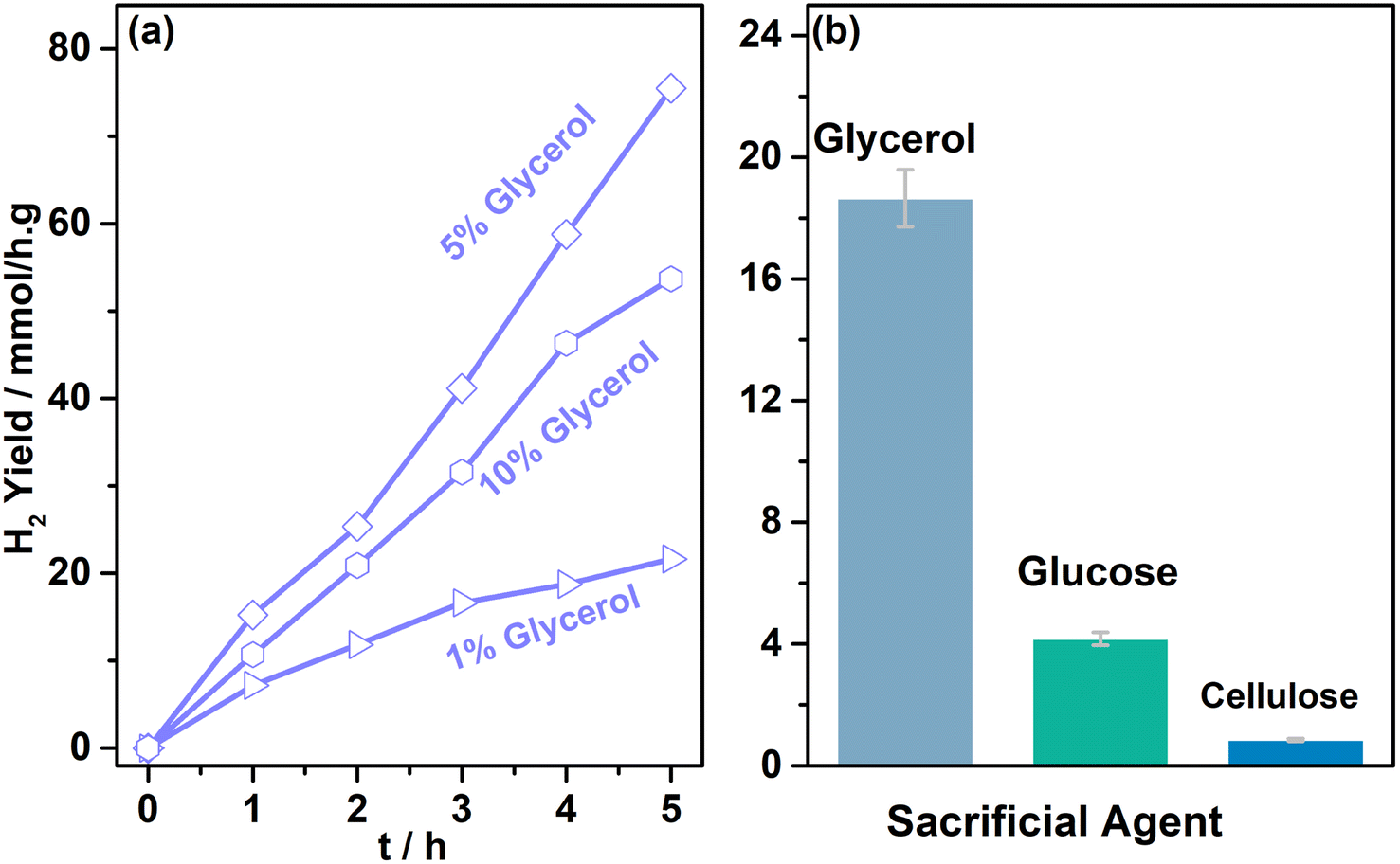 | ||
| Fig. 3 (a) Photocatalytic hydrogen yield observed with 0.5 wt% of Au over TiO2 with different concentrations of glycerol under one sun condition (100 mW cm−2, AM 1.5 filter). (b) Photocatalytic hydrogen yield observed with different biomass components as sacrificial reagents using 0.5 wt% Au on TiO2 under direct sunlight with an average intensity of 60 mW cm−2. | ||
In addition to glycerol, glucose, and cellulose were also evaluated as sacrificial agents; it is known that they all have similar building blocks, except for chain lengths. The average H2 yield obtained is shown in Fig. 3b. 20 ml of 0.35 M glucose solution was employed with 3 mg of the catalyst (0.5Au@TiO2), this is mainly to maintain the similar number of carbon atoms as that of 5% glycerol solution. 4.1 mmol h−1 g−1 H2 yield was observed under direct sunlight with glucose, while glycerol solution exhibited 18 mmol h−1 g−1. A four-fold decrease in H2 yield with glucose is attributed primarily to the 6-carbon unit; more difficult C–C cleavage is the leading/silent reaction also occurs, which consumes a significant number of electrons and protons and slows down the overall kinetics. However, the result was still significant, and, in fact, it opens a new window to the utilization of biomass components for photocatalytic hydrogen production. One more reason for the lower activity with glucose, compared to that with glycerol as a sacrificial reagent, is due to the settling of the catalyst in the bottom of the RB flask; due to this, the light absorption capacity decreases and results in lower photoactivity. In the case of cellulose, 200 mg of cellulose was taken in 20 ml water with 3 mg catalyst and subjected to direct sunlight irradiation for 5 h. Because cellulose is not soluble in water, activity decreased drastically to 0.8 mmol h−1 g−1. To improve and produce an efficient sacrificial reagent for photocatalytic hydrogen production, the solubility of cellulose needs to be increased but without employing harsh basic or acidic conditions. Dispersed cellulose particles in the solution would tend to scatter the light, rather than light absorption, which also results in lower photoactivity.6,48–50 Ideally a systematic approach from waste biomass components to hydrogen and any VAP would be more appealing.51,52 Our efforts to identify any oxidation products of glycerol from the results shown in Fig. 1 and 2 was not successful, due to high glycerol and low catalyst content. Hence the conditions were modified to enhance the oxidized product concentration and the observed results are shown and discussed in the following sections.
Glycerol oxidation by direct solar photocatalysis
Glycerol oxidation products were measured from systematic photocatalytic experiments. 5 mg of 0.5Au@TiO2 with 5 ml of 0.05 M glycerol was used for photocatalytic glycerol oxidation. These experiments were carried out in aerobic (purged with O2), anaerobic (purged with N2), and dry air conditions. Representative product analysis data obtained using NMR are shown in Fig. S3 in ESI.†Fig. 4 shows the data on photocatalytic glycerol oxidation carried out under the three conditions mentioned above, and the products observed along with hydrogen under (a) direct sunlight, and (b) under one sun condition. Three major liquid products, namely, glycolaldehyde, dihydroxyacetone, and formic acid, were observed under both reaction conditions. In anaerobic conditions under direct sunlight, 0.32, 0.12, and 0.17 M g−1 of glycolaldehyde, dihydroxyacetone, and formic acid were observed, respectively, in 5 h; along with the liquid products, 32 mmol g−1 of hydrogen was observed in 5 h reaction time. When purging was carried out using dry air, the amount of oxidation product was also found to be increased; 0.49, 0.19, 0.13 M g−1 of glycolaldehyde, dihydroxyacetone, and formic acid, respectively, were observed along with 23 mmol g−1 hydrogen in 5 h; 2.8 mmol g−1 of CO2 was also observed in the gaseous products. In the case of the aerobic condition, the overall product formation rate increased twice that of anaerobic conditions, along with glycerol conversion. Significant over-oxidation to CO2 (7.5 mmol g−1) was also observed, and hydrogen generation decreased to 1.9 mmol g−1. This indicates that in the presence of oxygen, oxidation of glycerol also occurs instead of water reduction. Expectedly, oxygen helps in the oxidation of the glycerol, through superoxo/peroxo radicals by electron consumption or the oxygen reduction reaction, this in turn decreases the hydrogen production.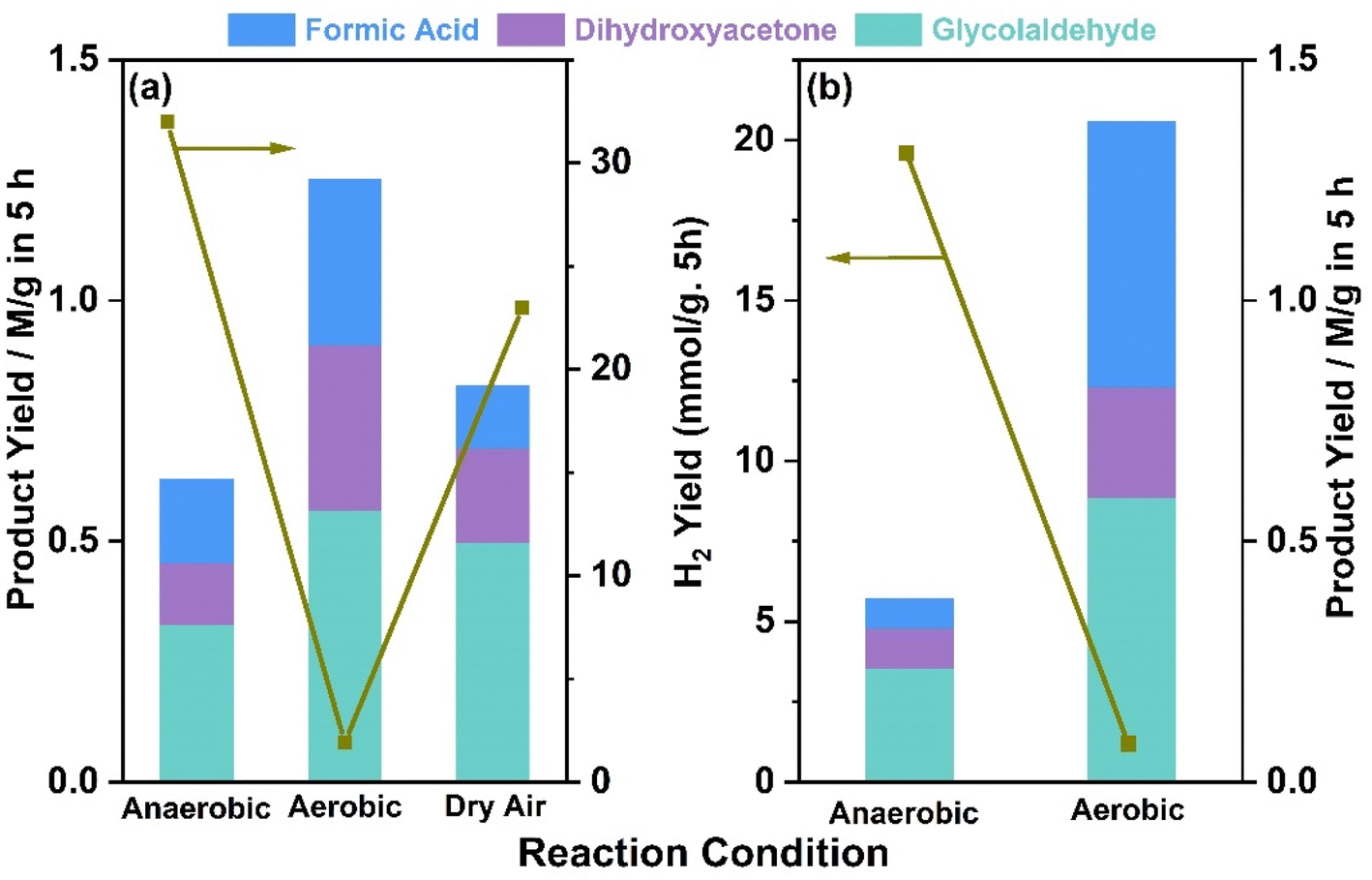 | ||
| Fig. 4 Photocatalytic glycerol oxidation with 0.5%Au@TiO2 under (a) direct sunlight with an average intensity of 60 mW cm−2, and (b) one sun condition (100 mW cm−2). All product concentrations were for five hours of the reaction time in both panels. | ||
Under one sun condition, similar liquid, and gaseous products were observed with the same trend. However, over-oxidation was comparatively lower with a smaller amount of CO2 production (5.3 mmol g−1) under one sun condition than under direct sunlight. An interesting observation to be noted is the significantly large amount of formic acid and smaller CO2 under one sun irradiation and aerobic conditions, compared to direct sunlight irradiation. A careful comparison of both results revealed that while glycolaldehyde was observed at the same level, formic acid was observed to increase at the cost of dihydroxyacetone under one sun condition. It is also to be noted that the temperature of the solution and the catalyst increases to around 333 K under direct sunlight, while the same was observed to be 320 K under one sun condition, carried out under laboratory ambient conditions. Hence, the overall increase in the reaction rate in direct sunlight may be partly attributed to an enhancement in temperature. Controlled experiments were also carried out at 333 K under one sun condition to ensure the temperature effect in direct sunlight. The results observed at 333 K are equivalent to those of sunlight conditions within the experimental accuracy. This further confirmed the influence of temperature under both natural and simulated light conditions. More power (100 mW cm−2) under one sun condition leads to the decomposition of dihydroxyacetone to formic acid. More experiments under a controlled atmosphere are required to ascertain the extent of the influence of the temperature and illumination power. In addition, the advantage of higher temperature and lower light power with direct sunlight may be utilized in a prudent manner. These experiments suggest that with regulated oxygen content, aqueous glycerol can be a very efficient reactant to generate VAPs along with hydrogen. A simple back calculation by accounting for all the carbon-containing products, including CO2, originating from the oxidation of glycerol, leads to glycerol conversion levels between 6 and 13% in 5 h with 52% selectivity for glycolaldehyde under anaerobic conditions. High TON values of 1280 and 920 were observed with 0.5Au@TiO2 for hydrogen under anaerobic and dry air conditions, respectively, while high TON values (68–113) were observed for VAPs under aerobic conditions (Tables S4–S6 in ESI†). A marginal decrease in hydrogen production by 1Au@TiO2 than that by 0.5Au@TiO2 suggested the higher activity associated with Au-clusters with the latter. We attribute this to the high dispersion of gold and electronic integration with TiO2 with the latter. By changing the batch to a continuous flow type with controlled oxygen or dry air, the higher activity would be possible by minimizing CO2. More experiments in this direction are desired. The present results also demonstrate the cleavage of C–C bonds occurring from glycerol all the way to formic acid and then to CO2, and Fig. S7† shows a plausible pathway for glycerol oxidation to VAPs. Hence, there are possibilities existing to fine-tune the reaction conditions to maximize the value-added liquid oxidation products with hydrogen production. As formic acid is expected to undergo fast oxidation to CO2, a product separation stage and recirculation of unreacted glycerol would be a good idea for achieving higher glycerol conversion to the same VAPs.
Structural analysis and elemental mapping
XRD, HRTEM, and elemental mapping methods were employed to understand the structure and morphology of the photocatalysts. XRD results of TiO2 and xAu@TiO2 are shown in Fig. S8.† As expected, anatase and rutile features were observed in both photocatalysts. However, after gold integration with TiO2, no difference is discernible in the XRD pattern up to 1 wt% of Au integrated TiO2 compared to virgin TiO2; indeed, 0.5 and 1 wt% of Au loading is too small and this could be one reason for the absence of any gold features in the XRD results. However, it rules out the clustering of gold atoms to big particle size(s). This naturally leads to the question of the status of gold in xAu@TiO2 the photocatalysts.To understand the structural and/or integration aspects of Au and its distribution over TiO2, HRTEM analysis was carried out and the representative results are shown in Fig. S9.† TiO2 particles with an average size of 20 ± 1 nm were observed, which is typical for P25 TiO2. However, after careful investigations of multiple batches of 0.5Au@TiO2, we did not observe any Au particle over TiO2. Nonetheless, it is essential to understand the role of gold to understand the SPR features at 545 nm, as shown in Fig. 1, and very high photocatalytic activity was observed for hydrogen generation and glycerol oxidation under solar irradiation conditions. To investigate this phenomenon, we carried out elemental mapping, and the results are shown in Fig. 5. Elemental mapping gives an idea about the small Au-cluster formation on TiO2, and demonstrates the homogeneous distribution of Au on TiO2 particles. Ti and O mapping shown in Fig. 5 panels follow the expected trend In their distribution. However, in the case of Au (yellow), a very fine and homogeneous dispersion of Au on TiO2 particles was observed. It is to be noted that the atomic weight of gold is more than four times that of titanium, and gold content in terms of the atomic percent is about 0.12–0.14% in 0.5Au@TiO2. Although many earlier studies have reported Au@TiO2 such a fine distribution has not been shown; uniform distribution is shown, probably, for the first time, with a photo deposition method. It is to be noted that the zeta potential of TiO2 P25 is positive, and hence negatively charged metal-ion precursor could be loaded over TiO2. During nitrogen purging in the dark, the negative charge [AuCl4]−1 is expected to be uniformly deposited over TiO2 upon UV irradiation, and [AuCl4]−1 gets reduced to Au with uniform dispersion.53 We also speculate the oxygen vacancies on the titania surface could be a possible driving force for a very high distribution. Under irradiation conditions, pre-deposited gold precursor ions get homogenously reduced over TiO2. Indeed this leads to a highly integrated xAu@TiO2. The lower the amount of gold deposited, the better the distribution observed. The low atom percent of Au and its high dispersion on titania are the reasons that its features could not be detected in XRD, while UV-Vis absorption shows the characteristic SPR feature. The specific surface area of Au on TiO2 is likely to be higher, which results in high hydrogen production as well as glycerol conversion to VAPs. A number of earlier studies indicated the electronic and structural integration of the catalyst components leading to a facile charge transfer between semiconductors and the metal towards efficient photocatalytic activity, which was also analyzed by XPS and discussed in the next section.
 | ||
| Fig. 5 STEM image of 0.5%Au@TiO2 and corresponding elemental distribution for Ti (green), Au (yellow), and oxygen (red) at scale bar of (a–d) 30 nm. A very uniform distribution of Au over TiO2 nanoparticles could be observed at high resolution. | ||
X-ray photoelectron spectroscopy studies
XPS provides information on the electronic interaction between various components as well as the surface composition of composite materials. To understand the electronic interaction between TiO2 and Au, XPS studies were carried out and the results are shown in Fig. 6. The deconvolution of Ti 2p core level shows that Ti was present in two different states for Ti3+ (457.45 ± 0.05 eV) and Ti4+(458.6 ± 0.05 eV); however, Ti4+ was predominantly available (92%), while only 8% of Ti3+ was available on the virgin TiO2 surface.54 However, with 0.5Au@TiO2, there was a decrease in the Ti3+ content by 50% (to 4%) as that of pristine TiO2, and this is attributed to the deposition/integration of gold with those sites. As metal atoms are known to be electrophilic, there is a driving force to deposit gold in Ti3+ or oxygen vacancy sites. This integration also leads to charge transfer from TiO2 to Au and makes the composite electronically integrated. In Fig. 6b, O 1s spectra of TiO2 and 0.5Au@TiO2 are shown. Three different oxygen species observed at 529.75 ± 0.05 eV (red trace), 530.45 ± 0.05 eV (blue trace), ∼531.8 eV (green trace) are attributed to lattice oxygen of TiO2, oxygen associated with Ti3+, and the hydroxyl group, respectively. While there was no change in the hydroxyl group intensity was observed before and after Au-integration, the percentage of oxygen associated with Ti3+ decreased marginally in the O 1s spectra. This reiterates that gold cluster deposition occurs selectively at the defect sites.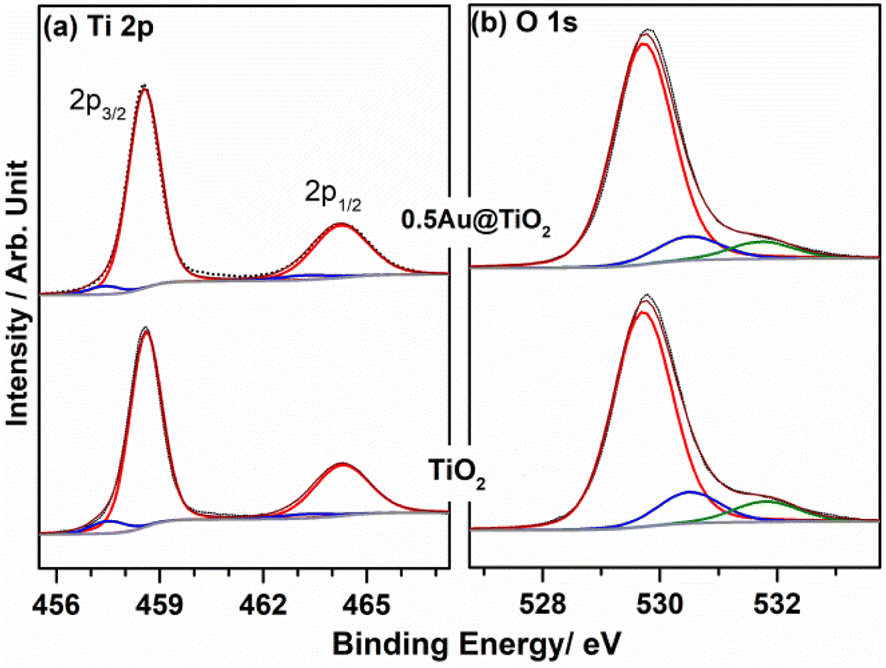 | ||
| Fig. 6 (a) Ti 2p, and (b) O 1s core level spectra of TiO2 P25 and 0.5Au@TiO2. Ti3+ and Ti4+ are given in blue and red traces, respectively. Red, blue, and green traces in O 1s corresponds to TiO2 lattice oxygen, O-associated with Ti3+ and OH group, respectively. | ||
Fig. 7 shows the Au 4f spectrum of 0.5Au@TiO2. Generally, bulk and metallic gold species appear at 84 ± 0.1 eV for the Au 4f7/2 core level in the XPS spectrum.54–56 However, interestingly, there was a large negative shift of 0.9 eV was observed with the Au 4f core levels of 0.5Au@TiO2. Almost 1 eV shift to low BE suggests a large charge transfer from titania to gold. This observation could be possibly due to a combination of the following reasons. (a) As mentioned in the discussion of Ti 2p and O 1s, Au species gets integrated into the defect site of TiO2, which is fully supported by the observation of a 50% decrease in Ti3+ content in 0.5Au@TiO2, compared to that in virgin TiO2. These XPS results also point toward an electronic integration between Au and TiO2, and helping to create metal-semiconductor nano-Schottky or heterojunctions. Such junctions are essential for electron–hole pair separation, which in turn increase the utilization of the charge carriers through redox reactions. (b) Small clusters of gold are expected to behave differently than nanoclusters or bulk gold. Elemental mapping indicates the extensive distribution of small gold clusters. Due to the small size of such clusters, gold is likely deposited uniformly on the interior and exterior surfaces of the pores of titania. (c) Small clusters are expected to be electrophilic and prefer to bind at sites richer in electrons. This leads to a preferential interaction between defect sites of titania and small gold clusters. Combining the above factors makes the gold cluster behave more like anionic gold. However, a significant reduction in BE shift was observed with 1Au@TiO2 (0.6 eV at 83.4 eV compared to that with metallic gold at 84 eV) and shows a reduction in anionic character compared to that with 0.5Au@TiO2 (Fig. 7b). Although the gold content was doubled, hydrogen generation was observed to be marginally lower, suggesting that the state of gold does matter for the best performance. This synergy between the metal and semiconductor helps toward the charge separation, which in turn helps high photocatalytic glycerol oxidation to VAPS and hydrogen evolution.
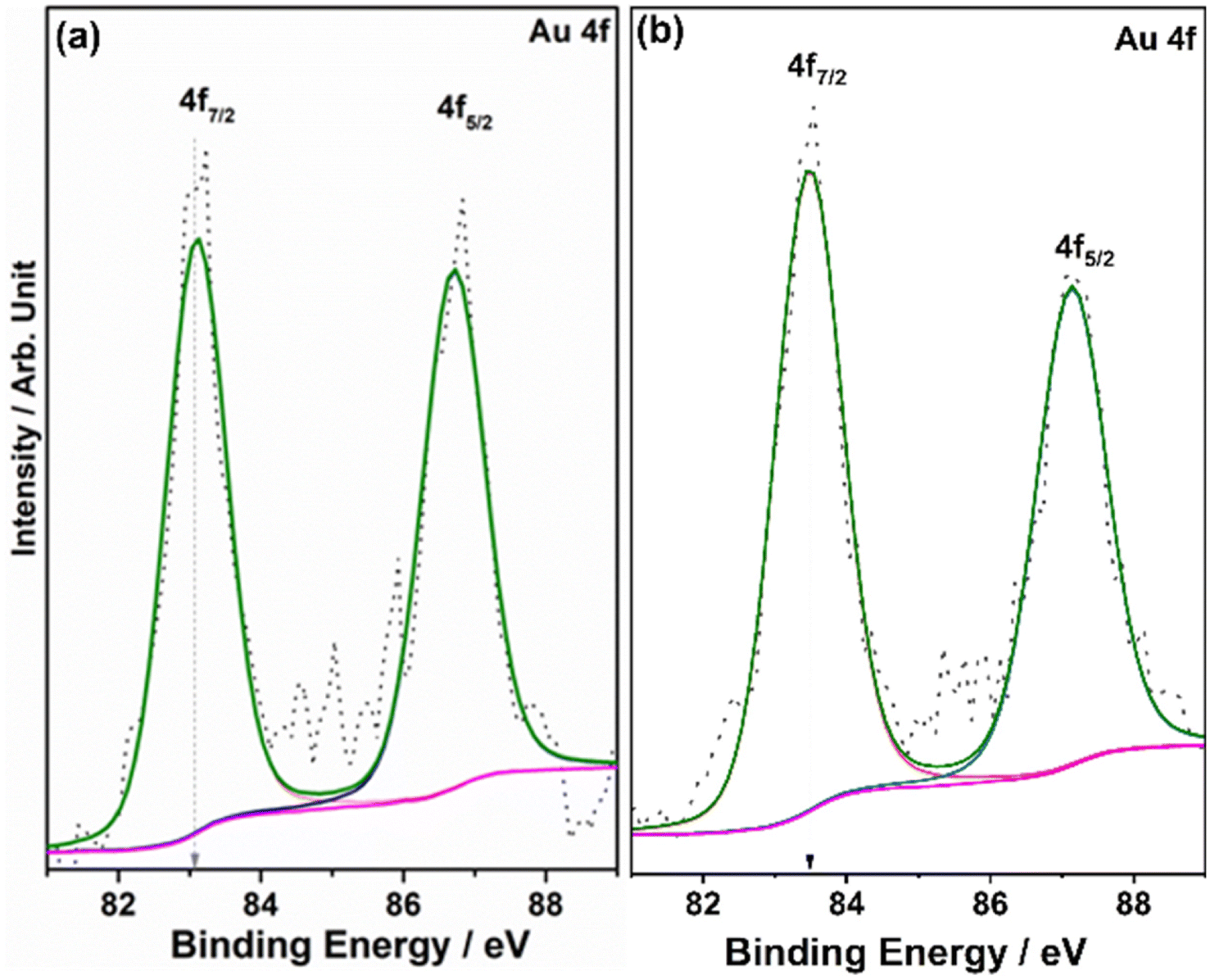 | ||
| Fig. 7 XPS spectra for Au 4f core levels for (a) 0.5Au@TiO2 and (b) 1Au@TiO2. Compared to the standard metallic gold 4f7/2 feature at 84.0 eV, the appearance of the same at 83.1 and 83.4 eV for 0.5Au@TiO2 and 1Au@TiO2, respectively, underscores the difference in electronic integration between Au and TiO2, and the extent of the anionic character of gold. | ||
Mechanism
The integration of the plasmonic metal with semiconductors can lead to efficient photocatalytic activity. Based on the experimental data obtained from various studies and discussed in the earlier sections for Au@TiO2, a possible reaction mechanism is proposed in Fig. 8. TiO2 exhibits a bandgap of 3.2 eV, which absorb only 4–5% of the UV light present in the solar spectrum, the electron and hole separation is, indeed, one of the main issues for the low efficiency of photocatalytic activity with virgin TiO2. Small and highly-dispersed gold cluster integration enhanced its light absorption capacity to the visible region, amplifying its electron–hole generation via the LSPR effect of gold particles, which is evident from the UV-Vis spectra. Also, the electronically integrated anionic Au-clusters with TiO2 suggest a negative shift of the Fermi level of the composite, which is more facile for charge separation and enhances hydrogen generation 20 times, compared to that in virgin TiO2.57,58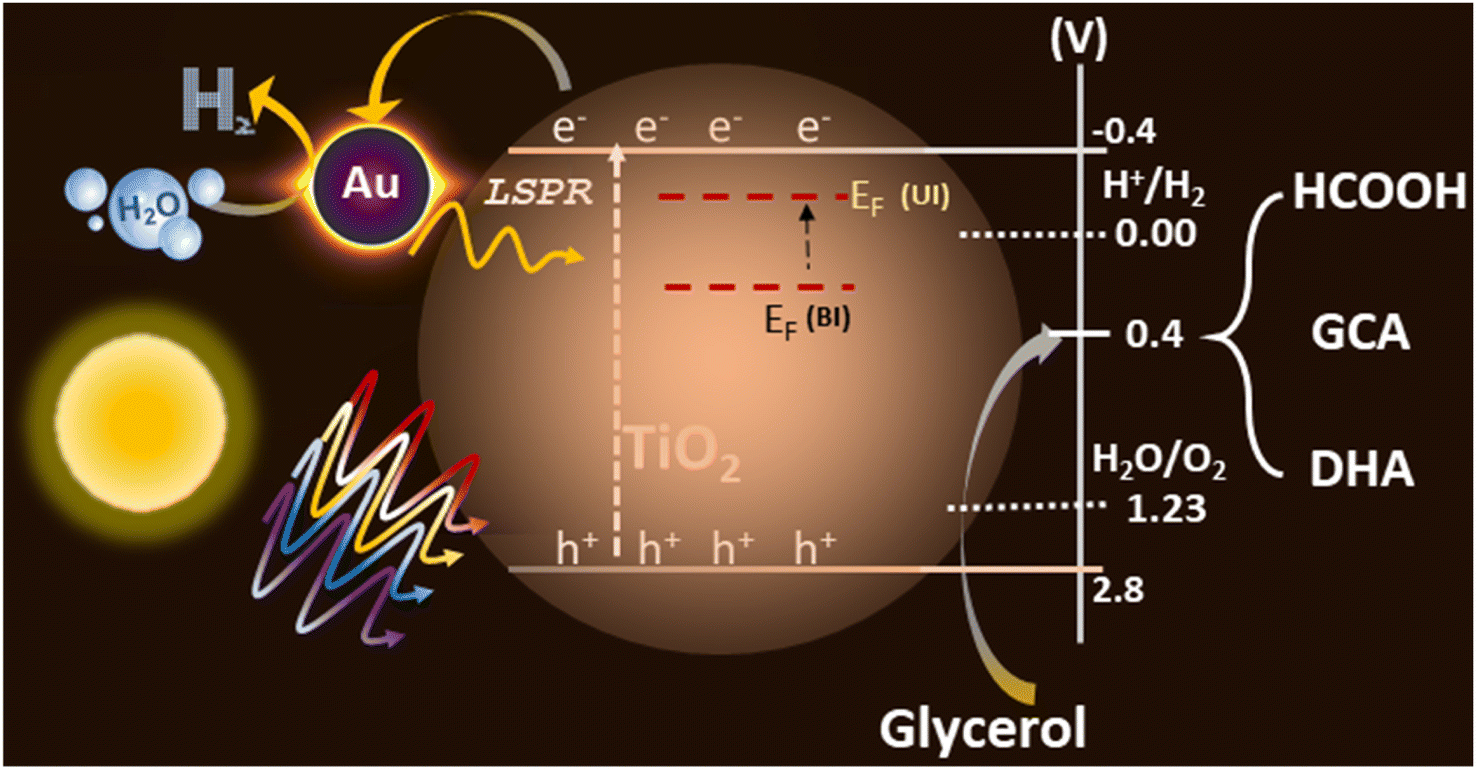 | ||
| Fig. 8 Possible mechanism for plasmon-induced Au@TiO2 toward photocatalytic hydrogen generation and glycerol oxidation towards the value-added products. EF levels shown are indicative and not to the scale; however, an upward shift under illumination occurs due to electron storage in gold clusters. Abbreviations: EF(UI) Fermi level under illumination, EF(BI) Fermi level before illumination or fresh catalyst, GCA (glycolaldehyde), DHA (dihydroxyacetone). | ||
Photocatalysis involves both photophysical and photochemical processes. It is well-known that the time scale of photophysical (≤ ns) and photochemical (≥ μs) processes are very different and it is necessary to address this issue.6 By selectively storing electrons in gold, utilization of holes, which are also known as the minority charge carriers, for oxidation increases to a large extent. Basically, the charge separation enhances both the reduction and oxidation aspects of the photocatalysis and hence an overall increase in the activity is observed. Further, the integration of Au helps in the photophysical process via the LSPR effect and as an electron sink for efficient charge separation, this leads to an enhanced redox process and underscores the dual functional character of the gold clusters with light absorption as well as catalysis. XPS results show that gold is present as anionic gold, which also proves the electron transfer is occurring from TiO2 to gold, where water reduction is taking place. However, the oxidation process occurs via holes present in the valence band of TiO2, from glycerol to VAPs (Fig. S7†). Glycerol and glucose require low oxidation potential (0.4 V vs. RHE with Pt/C and 0.4 vs. RHE with Au, respectively) than water,59,60 while VB of TiO2 is around 2.7 V with high oxidation potential. The potential required for both reduction and oxidation is met with the above arrangement, which helps in the large utilization of holes, resulting in low charge recombination and high photocatalytic activity. The large dispersion of atom-like gold clusters observed in STEM results (Fig. 5) supports the effective charge carrier separation as well as its utilization. Further, by depositing gold at the oxygen vacancy sites, not only electronic integration was achieved, but charge recombination was also minimized. These factors increased the charge utilization and improved the redox activity towards H2 and VAP formation. A significantly high formic acid formation underscores the high potential associated with holes and helps achieve C–C cleavage of glycerol components. This particular aspect should be utilized to tackle other biomass components too.
Conclusions
We synthesized xAu@TiO2 (x = 0.1, 0.5, and 1 wt%) composites via facile photo-deposition, with small Au clusters uniformly deposited and electronically integrated on TiO2. 0.5Au@TiO2 exhibits maximum hydrogen yield in direct sunlight with glycerol as a sacrificial agent compared to other biomass components. This study focused on two aspects; (1) novel integration of gold with TiO2 and its large distribution due to small clusters, which increases the effective charge transfer between semiconductor to metal and reduces the electron–hole recombination. (2) Glycerol is not only a promising sacrificial reagent but could be oxidized to value-added products under aerobic conditions. This aspect helps in the utilization of photogenerated electrons and holes to hydrogen and value-added products of glycerol, respectively. Glucose and cellulose were also shown to exhibit hydrogen generation activity, and it is suggested to explore them more with different catalyst systems to find a potential solution for hydrogen and value-added products. We believe the low activity with glucose/cellulose, compared to glycerol, is due to the consumption of charge carriers in breaking C–C bonds in the initial hours. Hence, prolonging the reaction for a longer time in sunlight is expected to produce VAPs and more hydrogen. Alternately, 1.5 and 2 sun conditions could be employed to perform the glycerol and glucose oxidation reactions, which is expected to increase the temperature further, in addition to light power. More work in this direction is likely to improve the activity in the desired direction of the biomass components to VAPS and hydrogen.The preparation method can be utilized to prepare different noble metal integrated TiO2 composites, a combination of SPR active metal and another transition metal in the form of alloy or bimetal co-catalysts is likely to provide more options for tuning the redox potential and hence the possibility of selectively producing one value-added product at a time and hence product separation step could be avoided. A combination of such co-catalysts with different semiconductors could also be evaluated for partial oxidation of biomass components by photocatalysis.61 With an ideal catalyst, one may be able to achieve maximum C–C cleavage towards making 100% of C1-oxygenates, such as formic acid and formaldehyde. For maximum light absorption, thin-film and powder form with proper dispersion have the potential for large-scale application.6,47,62 Recent work by Domen et al. on a 100 m2 photocatalyst panel for water splitting in sunlight indicates the possibility to circulate a thin layer of suspension over large area devices for the maximum absorption of sunlight to maximize the reactivity.63 Less expensive system should be more practical and sustainable for large-scale synthesis, but earlier literature reports did not address the identification and quantification of the oxidized VAPs produced during hydrogen evolution. In fact, we suggest the practitioners of photocatalysis move from employing a mere sacrificial agent to generating VAPs, as this makes an attractive proposition.
It has been 50 years since the announcement of the first proof of concept of water splitting by Fujishima and Honda;5 however, photocatalytic hydrogen evolution research and technology is still at the infantry level. We believe that a small step suggested in the present manuscript could significantly impact these strategies and can potentially make the photocatalytic hydrogen evolution and glycerol oxidation process by photocatalysis attractive.
Conflicts of interest
There are no conflicts or competing interests to declare.Acknowledgements
HB and NBM thank UGC, New Delhi for research fellowships; IC and KNS thank INSPIRE and Maharashtra state government for research fellowships, respectively. We acknowledge the financial support from CSIR, New Delhi, through the HCP-44 project.References
- J. Gong, C. Li and M. R. Wasielewski, Chem. Soc. Rev., 2019, 48, 1862–1864 RSC.
- S. Rajaambal, K. Sivaranjani and C. S. Gopinath, J. Chem. Sci., 2015, 127, 33–47 CrossRef CAS.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- (a) C. S. Gopinath and N. Nalajala, J. Mater. Chem. A, 2021, 9, 1353–1371 RSC; (b) S. Rajaambal, K. Sivaranjani and C. S. Gopinath, Dalton Trans., 2014, 43, 12546–12554 RSC.
- K. K. Patra, P. A. Bharad, V. Jain and C. S. Gopinath, J. Mater. Chem. A, 2019, 7, 3179–3189 RSC.
- N. Fajrina and M. Tahir, Int. J. Hydrogen Energy, 2019, 44, 540–577 CrossRef CAS.
- M. R. Karimi Estahbanati, M. Feilizadeh, F. Attar and M. C. Iliuta, React. Chem. Eng., 2021, 6, 197–219 RSC.
- D. Lv, Y. Lei, D. Zhang, X. Song, Y.-W. Li, J. W. H. Niemantsverdriet, W. Hao, Y. Deng and R. Su, J. Phys. Chem. C, 2020, 124, 20320–20327 CrossRef CAS.
- R. Deas, S. Pearce, K. Goss, Q. Wang, W.-T. Chen and G. I. N. Waterhouse, Appl. Catal., A, 2020, 602, 117706 CrossRef CAS.
- G. Dodekatos and H. Tüysüz, Catal. Sci. Technol., 2016, 6, 7307–7315 RSC.
- H. Sakurai, M. Kiuchi, C. Heck and T. Jin, Chem. Commun., 2016, 52, 13612–13615 RSC.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- B.-H. Wu, W.-T. Liu, T.-Y. Chen, T.-P. Perng, J.-H. Huang and L.-J. Chen, Nano Energy, 2016, 27, 412–419 CrossRef CAS.
- A. A. Melvin, K. Illath, T. Das, T. Raja, S. Bhattacharyya and C. S. Gopinath, Nanoscale, 2015, 7, 13477–13488 RSC.
- (a) P. Devaraji and C. S. Gopinath, Int. J. Hydrogen Energy, 2018, 43, 601–613 CrossRef CAS; (b) P. Devaraji, N. K. Sathu and C. S. Gopinath, ACS Catal., 2014, 4, 2844–2853 CrossRef CAS.
- (a) K. K. Patra and C. S. Gopinath, J. Phys. Chem. C, 2018, 122, 1206–1214 CrossRef CAS; (b) K. K. Patra, B. Bhuskute and C. S. Gopinath, Sci. Rep., 2017, 7, 6515 CrossRef PubMed; (c) K. K. Patra and C. S. Gopinath, ChemCatChem, 2016, 8, 3294–3301 CrossRef CAS.
- F. J. Méndez, A. González-Millán and J. A. García-Macedo, Int. J. Hydrogen Energy, 2019, 44, 14945–14954 CrossRef.
- X. Shi, Z. Lou, P. Zhang, M. Fujitsuka and T. Majima, ACS Appl. Mater. Interfaces, 2016, 8, 31738–31745 CrossRef CAS.
- R. Fiorenza, M. Bellardita, L. D'Urso, G. Compagnini, L. Palmisano and S. Scirè, Catalysts, 2016, 6, 121 CrossRef.
- (a) J. B. Priebe, J. Radnik, A. J. J. Lennox, M.-M. Pohl, M. Karnahl, D. Hollmann, K. Grabow, U. Bentrup, H. Junge, M. Beller and A. Brückner, ACS Catal., 2015, 5, 2137–2148 CrossRef CAS; (b) P. A. Bharad, K. Sivaranjani and C. S. Gopinath, Nanoscale, 2015, 7, 11206–11215 RSC.
- H. Bajpai, K. K. Patra, R. Ranjan, N. Nalajala, K. P. Reddy and C. S. Gopinath, ACS Appl. Mater. Interfaces, 2020, 12, 30420–30430 CrossRef CAS PubMed.
- A. Villa, G. M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499–4502 CrossRef CAS.
- A. V. Puga, A. Forneli, H. García and A. Corma, Adv. Funct. Mater., 2014, 24, 241–248 CrossRef CAS.
- (a) E. Taboada, I. Angurell and J. Llorca, J. Photochem. Photobiol., A, 2014, 281, 35–39 CrossRef CAS; (b) N. K. Sathu, P. Devaraji and C. S. Gopinath, J. Nanosci. Nanotechnol., 2016, 16, 9203–9208 CrossRef CAS.
- B. Zhou, J. Song, Z. Zhang, Z. Jiang, P. Zhang and B. Han, Green Chem., 2017, 19, 1075–1081 RSC.
- X. Zhang, L. Luo, R. Yun, M. Pu, B. Zhang and X. Xiang, ACS Sustainable Chem. Eng., 2019, 7, 13856–13864 CrossRef CAS.
- C. Wang, X. Cai, Y. Chen, Z. Cheng, X. Luo, S. Mo, L. Jia, R. Shu, P. Lin, Z. Yang, S. Sun, E. Pu and Y. Shen, Int. J. Hydrogen Energy, 2017, 42, 17063–17074 CrossRef CAS.
- T. N. Q. Trang, L. T. N. Tu, T. V. Man, M. Mathesh, N. D. Nam and V. T. H. Thu, Composites, Part B, 2019, 174, 106969 CrossRef CAS.
- T. Montini, V. Gombac, L. Sordelli, J. J. Delgado, X. Chen, G. Adami and P. Fornasiero, ChemCatChem, 2011, 3, 574–577 CrossRef CAS.
- Y. Yang, F. Li, J. Chen, J. Fan and Q. Xiang, ChemSusChem, 2020, 13, 1979–1985 CrossRef CAS PubMed.
- L. S. Al-Mazroai, M. Bowker, P. Davies, A. Dickinson, J. Greaves, D. James and L. Millard, Catal. Today, 2007, 122, 46–50 CrossRef CAS.
- N. Strataki, V. Bekiari, D. I. Kondarides and P. Lianos, Appl. Catal., B, 2007, 77, 184–189 CrossRef CAS.
- Y. Li, G. Lu and S. Li, Appl. Catal., A, 2001, 214, 179–185 CrossRef CAS.
- H. Hu, D. Qian, P. Lin, Z. Ding and C. Cui, Int. J. Hydrogen Energy, 2020, 45, 629–639 CrossRef CAS.
- A. G. Dosado, W.-T. Chen, A. Chan, D. Sun-Waterhouse and G. I. N. Waterhouse, J. Catal., 2015, 330, 238–254 CrossRef CAS.
- W.-T. Chen, A. Chan, D. Sun-Waterhouse, J. Llorca, H. Idriss and G. I. N. Waterhouse, J. Catal., 2018, 367, 27–42 CrossRef CAS.
- P. Jiménez-Calvo, V. Caps, M. N. Ghazzal, C. Colbeau-Justin and V. Keller, Nano Energy, 2020, 75, 104888 CrossRef.
- S. A. Balsamo, S. Sciré, M. Condorelli and R. Fiorenza, J. Interdiscip. Multidiscip. Res., 2022, 5, 92–104 CAS.
- A. K. R. Police, S. V. P. Vattikuti, K. K. Mandari, M. Chennaiahgari, P. S. M. Venkata, D. K. Valluri and C. Byon, Ceram. Int., 2018, 44, 11783–11791 CrossRef CAS.
- M. O. Segovia-Guzmán, M. Román-Aguirre, J. Y. Verde-Gomez, V. H. Collins-Martínez, G. Zaragoza-Galán and V. H. Ramos-Sánchez, Catal. Today, 2020, 349, 88–97 CrossRef.
- I. Majeed, M. A. Nadeem, A. Badshah, F. K. Kanodarwala, H. Ali, M. A. Khan, J. A. Stride and M. A. Nadeem, Catal. Sci. Technol., 2017, 7, 677–686 RSC.
- M. Jung, J. N. Hart, D. Boensch, J. Scott, Y. H. Ng and R. Amal, Appl. Catal., A, 2016, 518, 221–230 CrossRef CAS.
- M. Imizcoz and A. V. Puga, Catal. Sci. Technol., 2019, 9, 1098–1102 RSC.
- M. R. Karimi Estahbanati, N. Mahinpey, M. Feilizadeh, F. Attar and M. C. Iliuta, Int. J. Hydrogen Energy, 2019, 44, 32030–32041 CrossRef CAS.
- N. Nalajala, K. K. Patra, P. A. Bharad and C. S. Gopinath, RSC Adv., 2019, 9, 6094–6100 RSC.
- B. Tudu, N. Nalajala, K. P. Reddy, P. Saikia and C. S. Gopinath, ACS Sustainable Chem. Eng., 2021, 9, 13915–13925 CrossRef CAS.
- B. Tudu, N. Nalajala, P. Saikia and C. S. Gopinath, Sol. RRL, 2020, 4, 1900557 CrossRef CAS.
- B. Tudu, N. Nalajala, K. P. Reddy, P. Saikia and C. S. Gopinath, ACS Appl. Mater. Interfaces, 2019, 11, 32869–32878 CrossRef CAS PubMed.
- H. Kasap, D. S. Achilleos, A. Huang and E. Reisner, J. Am. Chem. Soc., 2018, 140, 11604–11607 CrossRef CAS PubMed.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- H. Wu, W. Jiang, L. Shi, R. Li, L. Huang and C. Li, iScience, 2022, 25, 103572 CrossRef CAS PubMed.
- K. Sivaranjani and C. S. Gopinath, J. Mater. Chem., 2011, 21, 2639–2647 RSC.
- K. Roy, C. P. Vinod and C. S. Gopinath, J. Phys. Chem. C, 2013, 117, 4717–4726 CrossRef CAS.
- A. A. Melvin, P. A. Bharad, K. Illath, M. P. Lawrence and C. S. Gopinath, ChemistrySelect, 2016, 1, 917–923 CrossRef CAS.
- L. Cheng, D. Zhang, Y. Liao, F. Li, H. Zhang and Q. Xiang, J. Colloid Interface Sci., 2019, 555, 94–103 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- M. Simões, S. Baranton and C. Coutanceau, Appl. Catal., B, 2010, 93, 354–362 CrossRef.
- N. Schloegel, G. K. H. Wiberg and M. Arenz, Electrochim. Acta, 2022, 410, 140023 CrossRef.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- N. Nalajala, K. N. Salgaonkar, I. Chauhan, S. P. Mekala and C. S. Gopinath, ACS Appl. Energy Mater., 2021, 4, 13347–13360 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Contains photographs for the dispersion of the catalyst suspension after 5 h of irradiation (S1), zeta potential (S2), NMR spectra (S3), glycerol oxidation product analysis (S4–S6), plausible reaction pathway of glycerol to VAPs (S7), X-ray diffraction (S8), and HRTEM images (S9). See DOI: https://doi.org/10.1039/d2su00145d |
| This journal is © The Royal Society of Chemistry 2023 |