
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surfactant stabilization of vanadium iron oxide derived from Prussian blue analog for lithium-ion battery electrodes†
Behnoosh
Bornamehr
ab,
Hiba
El Gaidi
ab,
Stefanie
Arnold
 ab,
Emmanuel
Pameté
a and
Volker
Presser
*abc
ab,
Emmanuel
Pameté
a and
Volker
Presser
*abc
aINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science & Engineering, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
cSaarene – Saarland Center for Energy Materials and Sustainability, Campus C4 2, 66123 Saarbrücken, Germany
First published on 27th July 2023
Abstract
Due to their high energy density, Li-ion batteries have become indispensable for energy storage in many technical devices. Prussian blue and its analogs are a versatile family of materials. Apart from their direct use as an alkali-ion battery electrode, they are a promising source for templating other compounds due to the presence of carbon, nitrogen, and metallic elements in their structure, ease of synthesis, and high tunability. In this study, homogeneous iron vanadate derivatization from iron vanadium Prussian blue was successfully carried out using an energy efficient infrared furnace utilizing CO2 gas. Iron-vanadate is an inherently unstable electrode material if cycled at low potentials vs. Li/Li+. Several parameters were optimized to achieve a stable electrochemical performance of this derivative, and the effect of surfactants, such as tannic acid, sodium dodecylbenzene sulfonate, and polyvinylpyrrolidone were shown with their role in the morphology and electrochemical performance. While stabilizing the performance, we demonstrate that the type and order of addition of these surfactants are fundamental for a successful coating formation, otherwise they can hinder the formation of PBA, which has not been reported previously. Step-by-step, we illustrate how to prepare self-standing electrodes for Li-ion battery cells without using an organic solvent or a fluorine-containing binder while stabilizing the electrochemical performance. A 400 mA h g−1 capacity at the specific current of 250 mA g−1 was achieved after 150 cycles while maintaining a Coulombic efficiency of 99.2% over an extended potential range of 0.01–3.50 V vs. Li/Li+.
1. Introduction
Li-ion batteries are considered the key technology for portable energy storage, and their market is growing with the energy transition. To meet the increasing requirements for improved power and energy densities and their environmental impact, continuous research is conducted for next-generation battery materials that are more sustainable and offer enhanced electrochemical performance.1 Many alternatives to commercial battery materials can be synthesized by derivatization from Prussian blue and its analogs (PBAs). While easy and cheap to synthesize, PBA-derived metal-vanadates have remained unexplored, although they can offer high capacities.2So far, work on the iron–vanadium (FeV) PBA in aqueous media has shown that the material decomposes and cannot offer a stable capacity. Coating the particles with a more stable PBA, namely with the CuFe, has been reported to create a barrier from the dissolution of FeV, thereby improving the stability.3 To the best of our knowledge, there are no reports on the derivatization from the FeV. For example, PBAs are used in energy storage and conversion as cathodes for commercial Na-ion batteries.4 Due to their inherent porous structure arising from the cyanide ligands in the lattice, they have an advantage in ion mass transport.5 In parallel, they also are a great precursor for self-templating as they contain metallic precursor elements in their structure and cyanide ligands that can decompose to nitrogen-doped carbon during derivatization. Additionally, PBAs can be prepared in aqueous media at room temperature, are highly tunable in morphology and chemistry, and can be converted to a variety of compounds at temperatures of 300–500 °C.6,7
Target derivative compounds for alkali-ion storage are mainly metal oxides, sulfides, and phosphides due to their high storage capacity.8,9 Mixed metal compounds can offer high capacities due to a higher lithiation degree while distributing the volumetric expansion over multiple steps that otherwise cause pulverization in single metal compounds.10 Among the mixed vanadium oxides, iron vanadate incorporates low-cost iron in its structure, a rock-forming element with the highest abundance among transition metals. The first report on iron-vanadate as an electrode in Li-ion batteries in Fe2VO4 spinel structure was reported with an initial capacity of 250 mA h g−1 at 250 mA g−1.11 Further studies in potassium-ion batteries elucidated that Fe2VO4 undergoes conversion reactions that break down the mixed metal oxide into vanadium oxide and iron oxide. A capacity loss accompanies this transition if the particles are not confined or protected.12 Vanadium-iron oxide, compared to the current commercial battery electrode materials, also bears the advantage of easy recovery after its end of life.13 Therefore, the dissolution of vanadium during cycling is directly related to its solubility.
Using PBAs for mixed metal oxide derivatization provides multiple advantages from a sustainability point of view. First, the synthesis can be done in pH-neutral aqueous media, avoiding the hazards of organic or low-pH components in preparation and waste management. Second, the synthesis is done at room temperature without autoclaves, heating energy, or pressure, reducing the carbon footprint and energy usage compared to other methods, such as hydrothermal or solid-state synthesis.1 Third, the synthesis can be scaled to larger volumes due to simplicity. For example, the PBA synthesis, particle coating, and attachment to carbon nanotubes (CNTs) can be done in one step. Additionally, derivatization from PBAs to mixed metal oxides usually requires low energy, employing temperatures lower than 500 °C for a duration of 2 h.14
In this work, we show how to derive homogeneous iron vanadium mixed metal oxide by templating the FeV PBA via a low-energy heat treatment and how to prepare self-standing electrodes without using a binder or organic solvents. Multiple parameters in the synthesis, processing, and electrode preparation are then improved to stabilize the material's performance, and their role is investigated. We analyze the effect of different surfactants on the electrochemical stability, namely tannic acid, sodium dodecylbenzene sulfonate, and polyvinylpyrrolidone. The role of surfactant types and their addition to different metal salts are explained following their effect on salt solubility.
2. Experimental
2.1. Synthesis of the FeV
FeV was prepared on CNTs from an aqueous solution with 658.58 mg of potassium ferricyanide (K3Fe(CN)6, 99.0%, Sigma Aldrich), 326 mg of vanadyl sulfate (VOSO4, 97.0%, Sigma Aldrich), and 50 mg of CNTs (NC7000, Nanocyl).CNTs served as a network provider and substrate for the FeV particles to form a self-standing electrode. All aqueous solutions were prepared with ultrapure water (ρ > 18.18 MΩ cm). The formation of the FeV was done by dropwise addition of 100 mL of an aqueous solution of 10 mM vanadyl sulfate at a rate of 40 mL h−1 to 100 mL aqueous 10 mM solution of potassium ferricyanide mixed with 50 mg CNT powder under magnetic stirring. The mixture remained overnight to rest. Multiple rounds of centrifugation and washing in ultrapure water and ethanol obtained the FeV powder after drying at 80 °C overnight.
2.2. Synthesis of FeV–PVP/SDBS
FeV–PVP/SDBS1 was prepared by dropwise addition at a 40 mL h−1 rate of 100 mL aqueous solution of vanadyl sulfate mixed with 1 g polyvinylpyrrolidone (PVP) or sodium dodecylbenzene sulfonate (SDBS) to a 100 mL aqueous solution of potassium ferricyanide mixed with 50 mg CNTs as a powder under magnetic stirring.FeV–PVP/SDBS2 was prepared by dropwise addition at a 40 mL h−1 rate of 100 mL aqueous solution of potassium ferricyanide powder mixed with 1 g PVP or SDBS to a 100 mL aqueous solution of vanadyl sulfate mixed with 50 mg CNTs under magnetic stirring.
2.3. Oxidation of the FeV
The oxidation of the material was done by heat treatment in an infrared furnace (Behr infrared furnace IRF 10) under 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 sccm Ar/CO2 flow. After 30 min of purging, the furnace was heated to 400 °C for 1000 s and held for 1 h. The FeV oxidized powder was obtained after cooling in the furnace to room temperature for 1000 s, and the product was labeled FVO. The samples with PVP or SDBS were denoted similarly after heat treatment: FVO–PVP1 or FVO–PVP2 and FVO–SDBS1 or FVO–SDBS2.
:50 sccm Ar/CO2 flow. After 30 min of purging, the furnace was heated to 400 °C for 1000 s and held for 1 h. The FeV oxidized powder was obtained after cooling in the furnace to room temperature for 1000 s, and the product was labeled FVO. The samples with PVP or SDBS were denoted similarly after heat treatment: FVO–PVP1 or FVO–PVP2 and FVO–SDBS1 or FVO–SDBS2.
2.4. Material characterization
Scanning electron microscopy (SEM) was carried out to characterize the morphology using a ZEISS GEMINI 500 employing an acceleration voltage of 1 kV. Transmission electron microscopy (TEM) was carried out using a 2100F system (JEOL) at a voltage of 200 kV for imaging. For SEM, samples were mounted on an aluminum stub fixed with double-sided copper tape and analyzed without a conductive sputter coating. For TEM, a copper grid coated with lacey carbon was used as the sample holder, and the powder was dispersed in ethanol via ultrasonic bath and then dried on the copper grid drop by drop.To analyze the present phases in the material, X-ray diffraction (XRD) was carried out using a D8 Discover diffractometer (BRUKER AXS) with a copper source (Cu-Kα, 40 kV, 40 mA), a Göbel mirror, and a 1 mm point focus. About 10 mg of as-synthesized powder was used. Each angular range of 20° 2θ was covered by a two-dimensional VANTEC detector and recorded for 2000 s. Measurement was carried out on a full range of 20–80° 2θ. Before the measurement, a NIST 1976b corundum standard calibration and peak position adjustment were conducted. The powder samples were fixed by pressing on a glass sample holder with a depth of 0.5 mm. All scans went through background subtraction and were normalized between 0 and 100.
Raman spectroscopy was conducted with a Renishaw inVia Raman microscope employing an Nd-YAG laser and 633 nm excitation wavelength with a power of 87 μW at the focal point of the sample with a numeric aperture of 0.75. For each sample, spectra from 10 points were recorded with 20 s exposure time and accumulated 20 times. The powder samples were placed on microscope glass slides. Spectra were treated by cosmic ray removal and normalized. The system was calibrated before and after the measurement with a silicon standard.
Thermogravimetric analysis (TGA) on powder materials was performed by a Netzsch TG-209-1 Libra system. Alumina cups cleaned by acid and base washing were used for the measurement, and the change in mass was calibrated to the cup mass. At least 10 mg of as-synthesized powder was used. The mass change was recorded by temperature increase to 800 °C under a heating rate of 5 °C min−1 in an inert environment (Ar 99.999%).
Inductively coupled plasma optical emission spectroscopy (ICP OES) on the residual solution after centrifugation was carried out by Horiba Jobin Yvon Ultima 2. A conical atomizer with a pressure of 2.67 bar and a flow of 0.76 L min−1 was used with wavelengths of λ = 258.207 nm for iron and λ = 309.311 nm for vanadium. The solutions were thinned to 1/100 of their concentration by ultra-pure water.
2.5. Electrode preparation
Sample powders were mixed with different CNT ratios from 5 mass% to 20 mass% of total electrode mass. For FeV mixed with CNT, 30 mg of pure FeV was mixed with CNT, then sonicated for 10 min in EtOH and vacuum filtered on a polytetrafluorethylene (PTFE) filter (Merck). For FeV grown on CNT, the as-synthesized powder was sonicated and filtered after oxidation. The electrodes were then dried overnight at 80 °C and cold rolled in two steps from 150 μm to 120 μm and from 120 μm to 100 μm by a pressure-controlled electric hot rolling press (HR01, MTI). 10 mm discs were punched and used as negative electrodes for electrochemical characterization without further binder use or slurry preparation.2.6. Electrochemical characterization
Electrodes were tested against a lithium chip (11 mm) in CR2032 coin cells' two-electrode configuration. For electrolyte, 1 M LiPF6 or 2 M LiTFSI salt in a mixture of ethylene carbonate and dimethyl carbonate (EC:DMC, 1:1 by volume, Sigma, battery grade) was used. For separation between the electrodes, Celgard 2325 (18 mm) and Whatman GF/F glass fiber (18 mm) were used. All cells were rested for 6 h prior to electrochemical testing.
All electrochemical measurements were carried out in a potential window of 0.01–3.50 V vs. Li/Li+. Galvanostatic cycling with potential limitation (GCPL) measurement was carried out with an Arbin battery cycler. Cyclic voltammetry (CV) at various rates and electrochemical impedance spectroscopy (EIS) in the frequency range from 100 kHz to 1 mHz was carried out via a BioLogic VMP-300 potentiostat/galvanostat. All tests were done in a climate chamber with a regulated temperature of 25 ± 1 °C. Specific capacity from GCPL tests was calculated separately for each cell per electrode mass (FVO + CNT composite mass) with an average of 1.75 ± 0.3 mg. Discharge capacity was divided by charge capacity in each cycle to calculate the Coulombic efficiency.
3. Results and discussion
3.1. Synthesis of FeV and FVO
A schematic representation of the synthesis method is shown in Fig. 1. As the vanadium precursor, vanadyl sulfate was used, which is reported as the recycling product from hydrometallurgical separation from iron.13 The synthesis, assembly, and stabilization were carried out in one step by adding the precursors to each other (Fig. 1A). Through room temperature coprecipitation without using acidic media, FeV was prepared (Fig. 1B). Heat treatment was done via infrared radiation, which significantly reduces the heating and cooling time compared to conventional tube furnaces with resistive heating (Fig. 1C). Electrode preparation was carried out by direct filtration of the composite suspension in ethanol (Fig. 1D).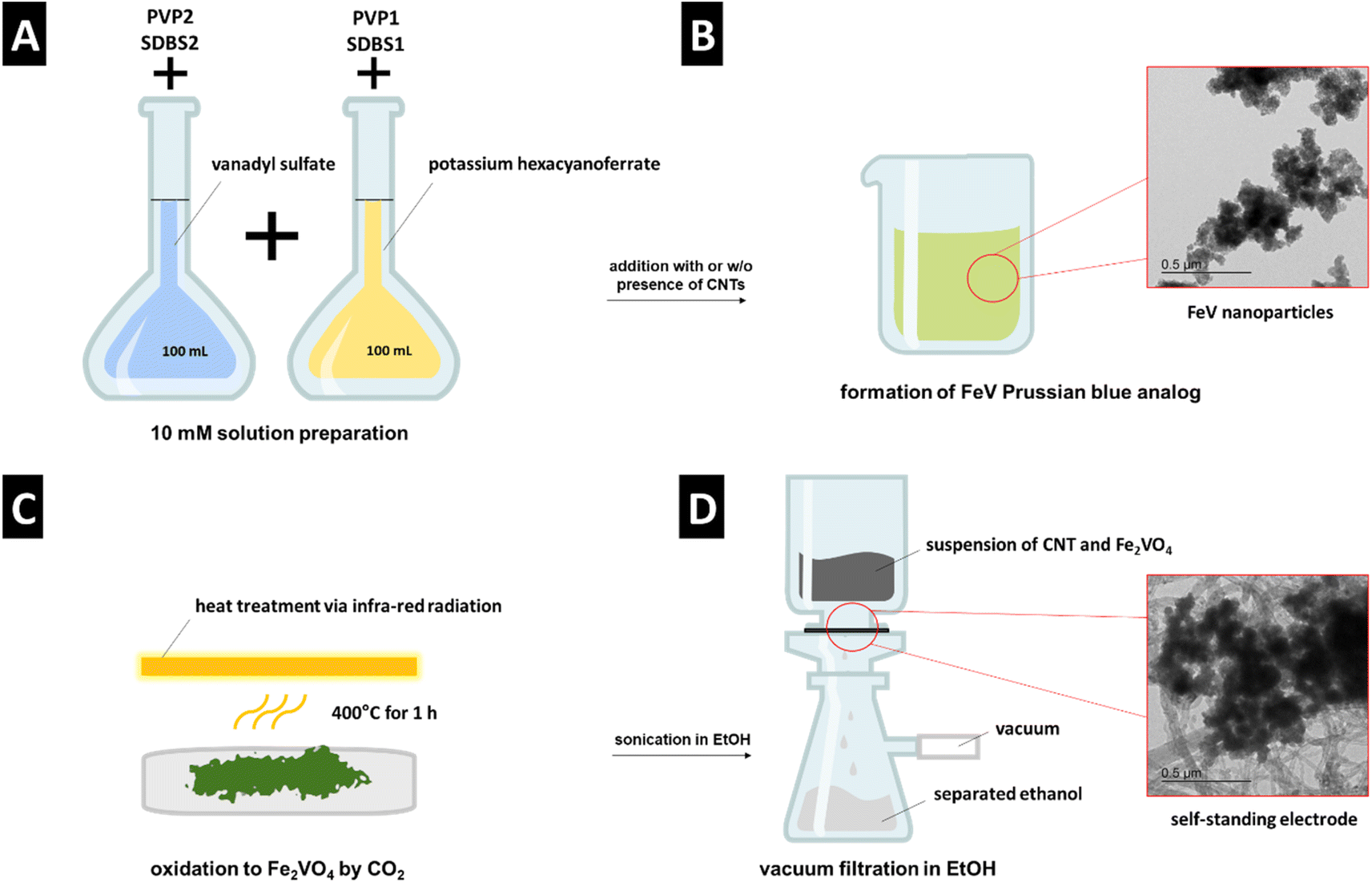 | ||
| Fig. 1 Schematic representation of (A) and (B) FeV synthesis, (C) heat treatment to FVO, (D) electrode preparation. | ||
The morphology of as-synthesized FeV is shown in Fig. 2A and B. The structure comprises typical nanoparticles in the size range of ∼4–10 nm with high aggregation which was retained after oxidation (Fig. 2C and D). Formation of FeV Prussian blue,15 similar to Thurnball blue, was successful with a unit of M(III)–C–N–M′(II) that accommodates four CN-groups in a primitive unit (Fig. 2E). The X-ray diffractogram data agree with iron vanadium cyanide V1.5Fe(CN)6 reflections in a cubic lattice with a space group of F(0) and a lattice parameter of 10.13 Å, and similar to other PBAs with strong reflections at ∼25° 2θ and ∼35° 2θ.3,16 After successful derivatization to the mixed metal oxide of Fe2VO4, crystallization in cubic lattice space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and a lattice parameter of 8.42 Å took place. Conversion to mixed metal oxide was only successful under CO2 flow which has a less oxidizing nature compared to oxygen.17–19 This is in line with early works on the solid-state synthesis of mixed iron–vanadium oxide which is highly dependent on the reducing atmosphere and a high control of CO:CO2 is needed for the formation of different iron–vanadium oxides.20,21 Here, instead of single metal oxide formation heating to 1000–1200 °C for Fe2VO4 formation, the iron–vanadium oxide directly can be derived at a low temperature of 400 °C from its parent PBA.
m and a lattice parameter of 8.42 Å took place. Conversion to mixed metal oxide was only successful under CO2 flow which has a less oxidizing nature compared to oxygen.17–19 This is in line with early works on the solid-state synthesis of mixed iron–vanadium oxide which is highly dependent on the reducing atmosphere and a high control of CO:CO2 is needed for the formation of different iron–vanadium oxides.20,21 Here, instead of single metal oxide formation heating to 1000–1200 °C for Fe2VO4 formation, the iron–vanadium oxide directly can be derived at a low temperature of 400 °C from its parent PBA.
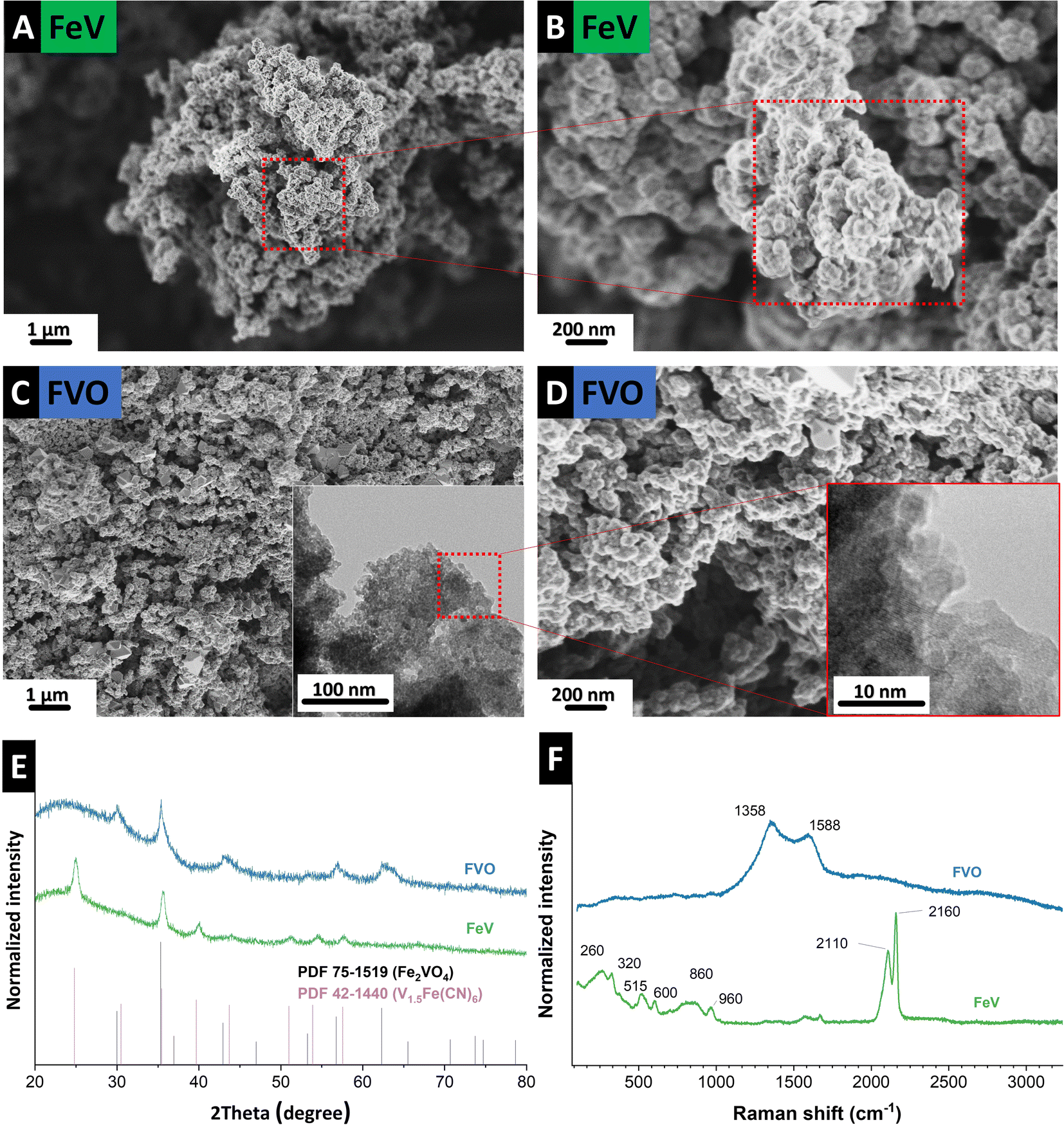 | ||
| Fig. 2 Scanning electron micrographs of (A) and (B) FeV, (C) and (D) FVO particles after heat treatment, (E) X-ray diffractograms, and (F) Raman spectra of the FeV and FVO particles. | ||
While in the derivatization of mixed metal oxides from PBAs, usually oxygen or synthetic air is used,22 using synthetic air for FeV oxidation, even when mixed with 50 sccm Ar, did not result in the formation of homogeneous mixed metal oxide. Fig. S1, ESI† shows the images taken from the resulting powders after oxidation under synthetic air. Inhomogeneity in color at the low stream and upstream of the furnace, yellow (Fig. S1B, ESI†) instead of the black color of mixed vanadium/iron oxide shows that the presence of oxygen gas complicates homogeneous derivatization of mixed metal oxide and could also explain the lack of reports on derivatization from vanadium PBAs.
Raman spectroscopy confirms the formation of PBA and oxide derivatization of FVO under CO2. Before the heat treatment, successful FeV formation is observed (Fig. 2F) with ν(CN) bands at 2110 cm−1 and 2160 cm−1, representing the typical cyanide ligand stretching vibration in the A1g and E1g modes, respectively.16,23 Lower frequency bands correspond to bonding with metallic atoms, such as Fe–C stretching in the 450–620 cm−1 spectral window. The band at 960 cm−1 that is outside the M–C and M–CN–M vibrations can be an indicator of V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the vanadyl group as vanadium–oxygen bands are usually observed in this region.3,24,25 After heat treatment, v(CN) bands are absent indicating full derivatization of the precursor. At the same time, D-mode and G-mode appear at 1358 cm−1 and 1588 cm−1, respectively, due to the formation of disordered carbon from the presence of CN in the FeV.
O in the vanadyl group as vanadium–oxygen bands are usually observed in this region.3,24,25 After heat treatment, v(CN) bands are absent indicating full derivatization of the precursor. At the same time, D-mode and G-mode appear at 1358 cm−1 and 1588 cm−1, respectively, due to the formation of disordered carbon from the presence of CN in the FeV.
Successfully derived mixed metal oxide from FeV was filtered as a self-standing electrode by sonicating the FVO and CNTs mixture. The surface of these self-standing electrodes (Fig. S2, ESI†) was characterized by SEM. Further analysis of the material morphology by SEM (Fig. 3A and B) showed a homogeneous distribution of the FVO in the CNT network and the retained morphology of the FeV template during the heat treatment. The crystallinity of the derived components was approved by TEM with d-spacing measurements of 4.9 Å and 2.8 Å, indexed with (111) and (220) plane distances of Fe2VO4, respectively (Fig. 3C and D).
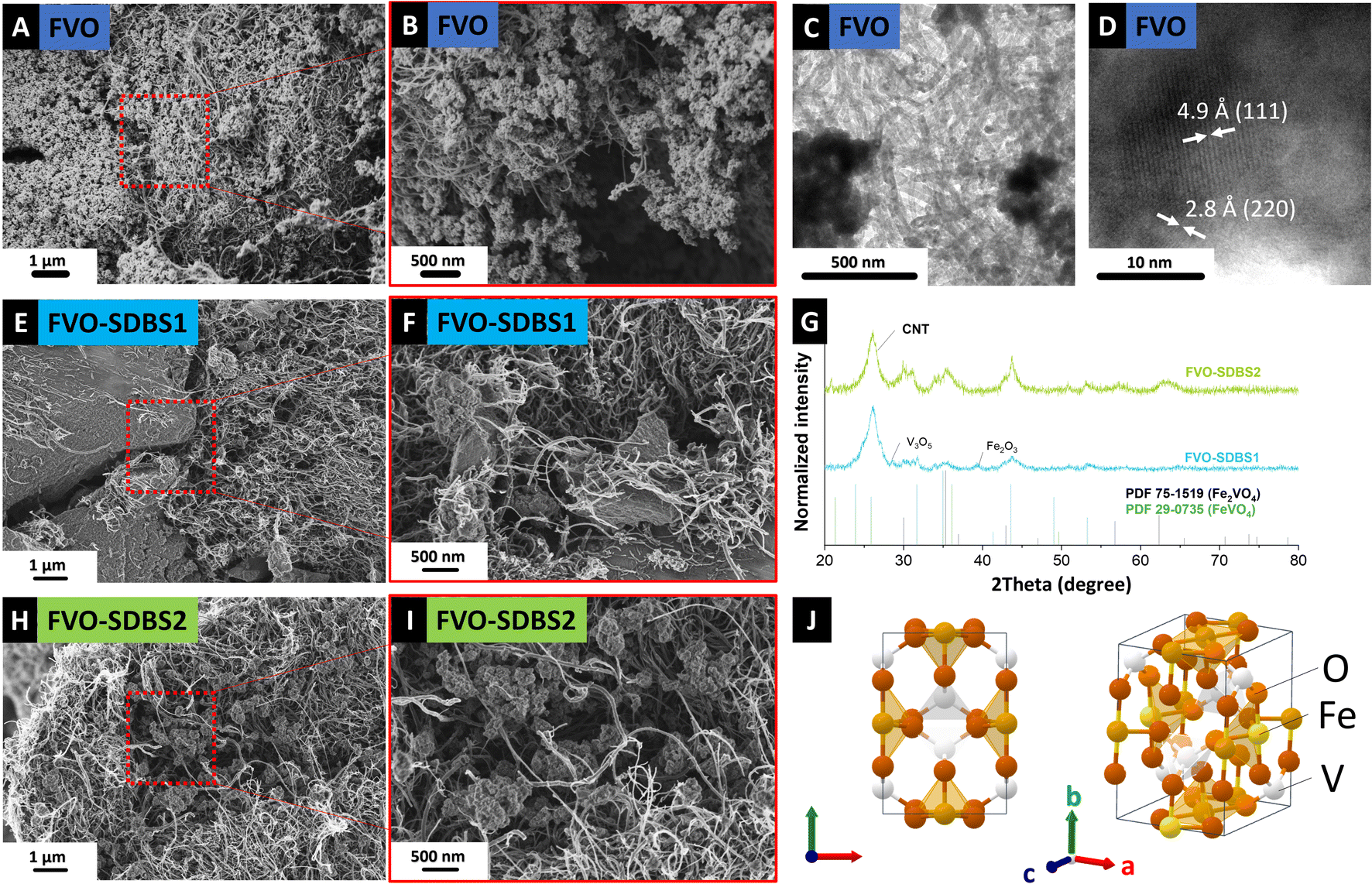 | ||
| Fig. 3 (A) and (B) Scanning electron micrographs and (C) and (D) transmission electron micrographs of FeV grown in situ on CNT after oxidation, (E) and (F) scanning electron micrographs of FeV with SDBS added to hexacyanoferrate after oxidation, (G) X-ray diffractogram of FVO–SDBS1 and FVO-SDBS-2, (H) and (I) scanning electron micrographs of FeV with SDBS added to vanadyl sulfate after oxidation and (J) crystal structure of FeVO4. | ||
Fig. S3, ESI,† shows the material synthesis in the presence of tannic acid (TA). Using catechols such as polydopamine and tannins has been reported as a successful strategy to coat Prussian blue derivatives (PBDs) with a carbon coating.26 Data from the XRD and Raman characterization after the synthesis indicate a possible FeV formation but with broad reflections and weak bands (Fig. S3C and D, ESI†). Inhomogeneous structure and loss of the nanoparticles after oxidation show that tannic acid does not form a coating while retaining the material morphology (Fig. S3B, ESI†). This is assumed to be due to the strong bonding of the catechols to the iron–cyanide complexes and affecting the PBA formation and disturbing subsequent derivatization.26
Therefore, other surfactants, such as PVP and SDBS, were used to form a coating with weaker bonding to the precursor salts. SDBS and PVP have been reported as additives that successfully tune the particle size or enable structure engineering of the materials by selectively covering particle edges and faces.27–30 To evaluate the effect of these surfactants on the PBA synthesis, the remaining concentration of Fe and V in the centrifugation supernatant after material synthesis was analyzed by ICP (Table 1). For the synthesis of FeV, concentrations of 402 mg L−1 iron and 51 mg L−1 vanadium were recorded. This corresponds to a Fe/V ratio of 7.8 that was observed with a yellow solution color, which aligns with the remaining potassium ferricyanide. In all samples with surfactant addition, a higher concentration of the metals was detected compared to the blank FeV. This is the highest for PVP1, showing that adding PVP to the ferricyanide solution (PVP1) complicates the FeV formation the most. In contrast, a successful particle formation has been observed so far in studies on PB31 and other PBAs.29
| FeV | FeV–PVP1 | FeV–SDBS1 | FeV–PVP2 | FeV–SDBS2 | |
|---|---|---|---|---|---|
| Fe (mg L−1) | 402.3 ± 6.9 | 532.8 ± 11.3 | 381.0 ± 6.2 | 472.2 ± 6.3 | 341.2 ± 6.7 |
| V (mg L−1) | 51.3 ± 1.1 | 205.4 ± 3.5 | 72.1 ± 0.6 | 175.3 ± 2.6 | 78.2 ± 0.9 |
| Fe/V ratio | 7.8 | 2.6 | 5.3 | 2.7 | 4.4 |
ICP results (Table 1) show for FeV–PVP1, the present iron concentration increases to 532.8 mg L−1 and vanadium to 205.4 mg L−1. It has been reported that PVP forms a strong bond with metal ions through their imide unit32 and SDBS forms micelles of M(DBS)x29 (M = metal) in aqueous solutions. Adding the PVP to vanadyl sulfate solution (PVP2) shows a lower concentration of remaining metals in the solution, with 472.2 mg L−1 iron and 175.3 mg L−1 vanadium. Still, for PVP, both metals are stabilized in the solvents. This aligns with the green color of the solution after centrifugation resembling the remaining FeV dissolved in the solution (Fig. S4A, ESI†).
The dependence of the FeV on the surfactant addition was higher for SDBS, with the Fe/V ratio in the solution changing drastically from 5.3 for SDBS1 to 4.4 for SDBS2. For SDBS1, 381.0 mg L−1 iron and 72.1 mg L−1 vanadium were detected, and for SDBS2 the concentration was less than for SDBS1 with 341.2 mg L−1 iron and 78.2 mg L−1 vanadium. This shows that SDBS, similar to PVP, impedes the FeV formation on CNTs. Compared to the blank FeV, in the presence of SDBS, bonding is stronger to vanadium, but less iron has remained in the solution.33 When SDBS is first added to iron cyanide, more iron remains in the solution (SDBS1), and when added to the vanadyl sulfate, more vanadium is observed (SDBS2). For both samples with SDBS, FeV was successfully synthesized on CNTs (Fig. S4 and S5, ESI†) and retained its nanostructure morphology after oxidation (Fig. 3E, F and H, I).
Further characterization indicated a lack of substantial FeV formation in the presence of PVP as well as metal oxides. This includes a lack of FVO particles on the CNTs (Fig. S6A–C, ESI†), XRD reflections, and Raman bands after the treatment (Fig. S6E and F, ESI†). Additional TGA analysis under Ar showed a similar mass loss of 11% at 800 °C compared to FVO (Fig. S6D, ESI†).
For samples treated with SDBS, a lower intensity of the XRD reflections was observed FVO–SDBS1 sample compared to FVO–SDBS2 after oxidation, in line with the ICP results on the dissolved metals in the supernatant (Fig. 3G). Similar to FVO, reflections of Fe2VO4 were observed in FVO–SDBS1 and FVO–SDBS2. Additionally, the formation of FeVO4 was visible. In FVO–SDBS1, minor peaks of vanadium oxide V3O5 and iron oxide Fe2O3 were also detected, showing a decrease in the phase homogeneity compared to pristine FVO. The formation of FeVO4 is assumed to be due to the lost Fe ions in the supernatant and the single metal oxides from defective FeV lattice formation because of the M(DBS) micelle formation.
The formation of single metal oxides can, however, increase the electrochemical stability. These oxides are thermodynamically more stable than the mixed oxide of iron and vanadium and less prone to chemical dissociation during cycling.12 Moreover, the mixed metal oxide of FeVO4 provides V5+ and Fe3+ in addition to the V4+ and Fe2+ in Fe2VO4 (ref. 34) and therefore contributes to more stable cycling by multi-valence redox reactions35 and causes less mechanical stressing of the particles. FeVO4 crystallizes in a spinel structure36 with an orthorhombic lattice and is reported as electrochemically active for lithium storage.37
3.2. Electrochemical performance
LiPF6 and LiTFSI were used as electrolyte salts and improved performance by using LiTFSI was observed similarly as reported in the literature.38 For the cell with LiPF6, capacity starts with a value of 580 mA h g−1, followed by a short activation in the first 20 cycles, reaching 615 mA h g−1. A low scattering of the Coulombic efficiencies was recorded in a 95–96% range in the first 40 cycles. After the initial 10 cycles, capacity begins to fall, and the Coulombic efficiency shows a high scattering in parallel, which indicates side reactions.39 The initial capacity increase appears likely because of material activation and access to more electrochemically active particles.40 However, with the lack of a protective layer, when the electrolyte accesses the FVO particles, they are continuously etched and disintegrated, resulting in capacity fading. This is a common pitfall for mixed metal components, especially materials that undergo conversion reactions and do not return to the initial chemistry, such as iron–vanadium oxide.41 Post-mortem analysis of the electrode material observed the morphology change during the process. Transmission electron micrographs show that the electrode stays intact regarding FVO on CNTs, but the FVO particle aggregates swell and become larger after cycling (Fig. S7A, ESI†).It has been shown in the literature that vanadium is prone to dissolution and electrolyte etching both in aqueous3 and organic media38,42 and results in a capacity loss in vanadium-based compounds. Iron–vanadium oxide is also prone to irreversible cycling after undergoing conversion reactions.11 This mixed metal oxide is usually broken into vanadium oxide and iron oxide, which are more thermodynamically stable.12,41 However, stability studies on different electrolytes and their molarities indicate a larger electrolyte molecule size, such as in the case of LiTFSI, and higher molarities may impede the fast dissolution of vanadium ions by acting as a diffusion barrier at the surface of electrode particles.38Fig. 4B shows the stability of the as-grown FeV cycled in 2 M LiTFSI in EC/DMC 1:1 (by volume). The initial capacity reaches 700 mA h g−1 after 10 cycles and then stabilizes at around 370 mA h g−1 after a decrease in capacity observed over 100 cycles. Compared to 1 M LiPF6, the Coulombic efficiencies show less scattering as well. To see if the change of electrolyte salt affects the lithiation mechanism, the electrochemical signatures in measured potential curves were compared. Respective charge and discharge curves in Fig. 4C and D show similar electrochemical thermodynamics of the sample cycled in 1 M LiFP6 and 2 M LiTFSI, only with the extension of the curves to higher capacities and better retention of the redox processes. This shows that the change in the electrolyte does not introduce new redox reactions but only increases the materials by hampering its etching by the electrolyte. Although the effect of vanadium dissolution is mitigated here, the capacity fading and Coulombic efficiency scattering indicate the instability of iron vanadate after 100 cycles of conversion, as the FVO particles are not protected by any coating.
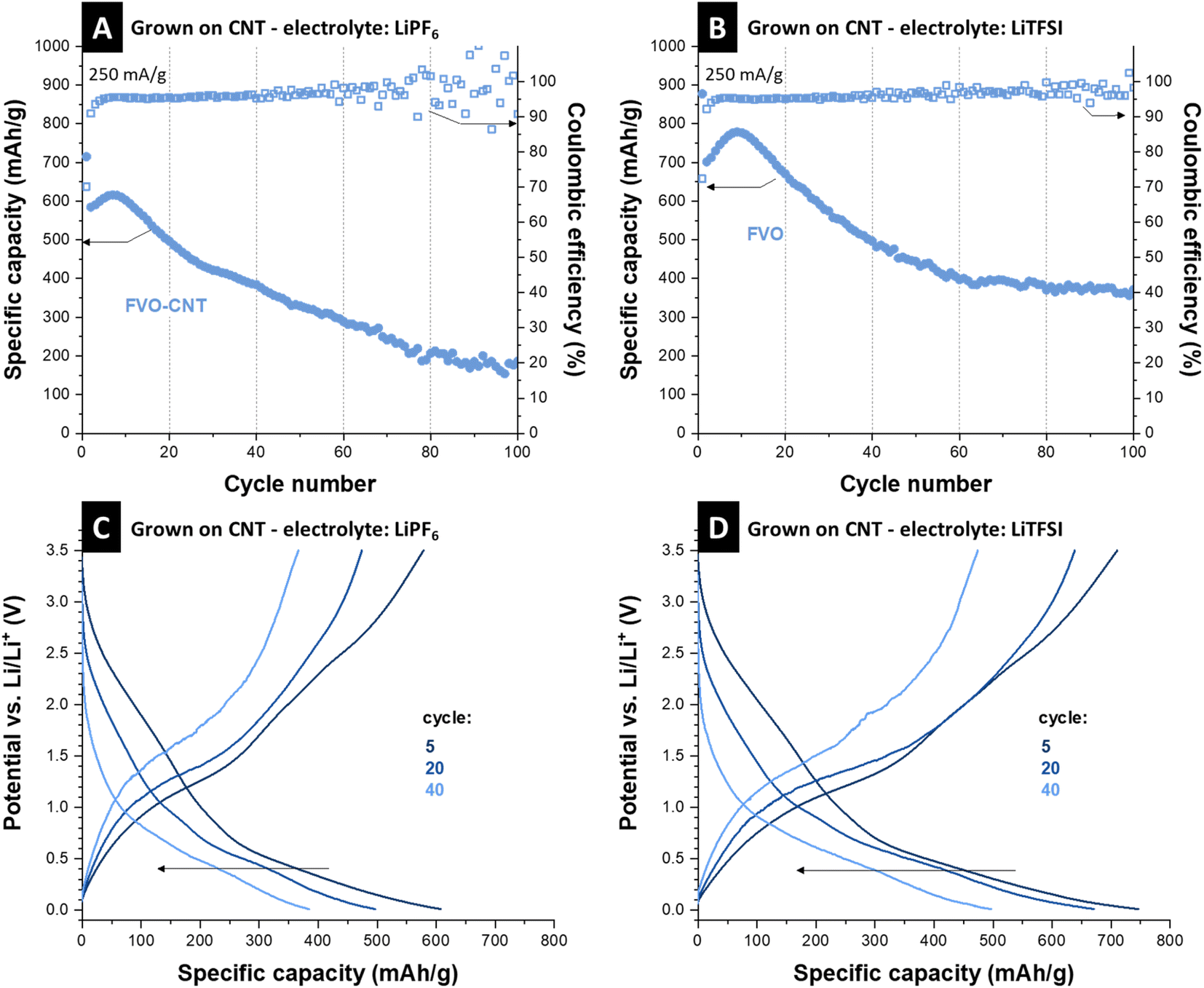 | ||
| Fig. 4 Stability performances of FVO electrodes at the specific current of 250 mA g−1 in (A) 1 M LiPF6 in EC/DMC, and (B) 2 M LiTFSI in EC/DMC with their Coulombic efficiencies, and (C) and (D) their respective charge–discharge curves at cycles 5, 20, and 40. | ||
Caging of the conversion type materials or formation of composites with electrochemically stable components can decrease capacity fading.43 Here, SDBS is used to form a stable coating on the material. For both FVO–SDBS1 and FVO–SDBS2, an improved cycling behavior is observed compared to the FVO material (Fig. 5A and B). FVO–SDBS2 shows a similar cycling behavior to FVO but with slower kinetics. The initial capacity of FVO–SDBS2 with 520 mA h g−1 is lower than FVO with 700 mA h g−1. This is a result of two combined effects: first, the loss of vanadium due to dissolution when SDBS is added, and second, a lower amount of active material in the total electrode mass due to the presence of SDBS coating, which is not redox active but increases the electrode mass. This capacity then increases to 560 mA h g−1 with further cycling and subsequently decreases, similar to FVO, accompanied by a scattering of the values for the Coulombic efficiency. In general, the use of SDBS surfactant, when added to vanadyl sulfate (SDBS2), delays the material deterioration by the electrolyte but is ineffective in stabilizing the cycling for longer cycles. TGA curves showed the incorporated SDBS coating is about 11 mass% for FVO–SDBS2 and 50 mass% for FVO–SDBS1 (Fig. S8A, ESI†). To elucidate the electrochemical behavior of FVO–SDBS1 and FVO–SDBS2 materials under dynamic and non-destructive conditions, electrochemical impedance spectroscopy analysis was conducted. Fig. S8B, ESI,† presents the Nyquist plots obtained from this analysis along with the corresponding equivalent circuit. Both materials exhibit two distinct semi-circles in the high and middle-frequency regions. The high-frequency semi-circle can be ascribed to the bulk electrolyte behavior, originating from the impedance of a layer that forms on the interface between the electrode and electrolyte.44 This layer corresponds to the combined carbon layer formed by the SDBS coating and solid electrolyte interphase (SEI), and its corresponding resistance and capacitance are annotated by EEI (electrode–electrolyte interphase). The higher REEI for FVO–SDBS2 indicates a thicker coating layer. With 103.2 Ω, this is about 90% more resistive than that of FVO–SDBS1 (Table S1, ESI†) since the SDBS coating is less for FVO–SDBS2, this resistance results from the higher etching of the sample as observed in the stability results. The mid-frequency semi-circle corresponds to the electrode polarization (Rct), which is linked to the kinetics of the electrochemical reaction occurring during the charge/discharge process.45 With 177.5 Ω for FVO–SDBS1 and 207.6 Ω for FVO–SDBS2, slightly more favorable kinetics are present for FVO–SDBS1. A great difference is observed in the values of ESR for the samples. The equivalent series resistance (ESR) of the cell made with the FVO–SDBS1 electrode is lower than that obtained with the device with FVO–SDBS2 materials with 10.8 Ω for FVO–SDBS1 and 52.3 Ω for FVO–SDBS2. Accordingly, the lower ESR value of the FVO–SDBS1 cell is due to the coating, which ultimately provides better electrode–electrolyte contact.45 The lower Rct and Warburg resistances for the FVO–SDBS1 cell correlate to the better charge transfer process and diffusional effects of lithium-ion on the host FVO–SDBS1 than the FVO–SDBS2 material.
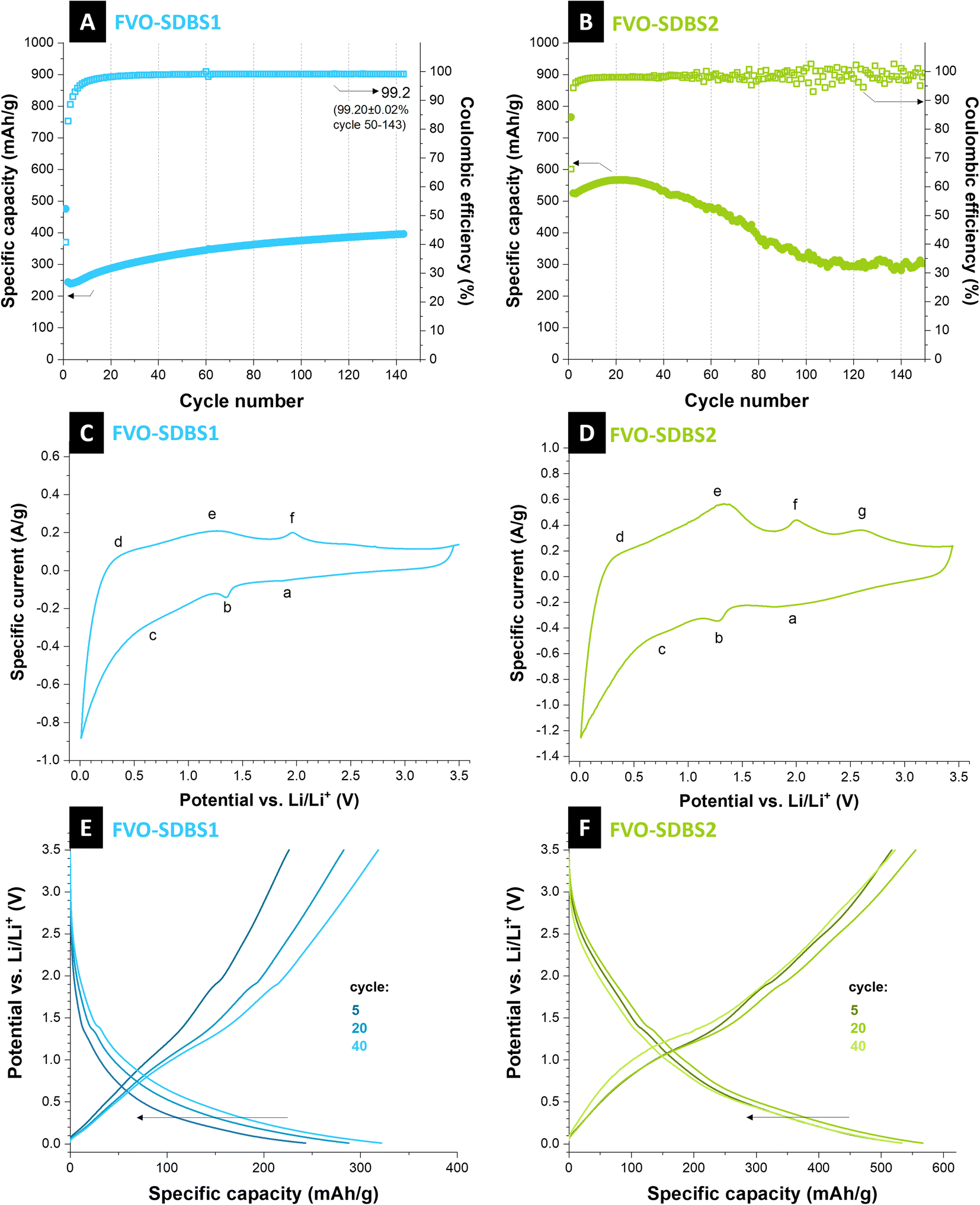 | ||
| Fig. 5 (A) and (B) Stability of FVO–SDBS1 and FVO–SDBS2 at the specific current of 250 mA g−1, respectively. (C) and (D) The respective cyclic voltammetry at the scanning rate of 0.1 mV s−1, and (E) and (F) the respective galvanostatic charge–discharge curves. | ||
FVO–SDBS1 shows a different cycling behavior than previous materials. The capacity increase does not end after 10–20 cycles and the materials continuously increase in capacity with more mass accessed by the electrolyte and also no subsequent fading is observed. However, the general capacity values are lower than previous materials. This confirms the lower initial capacity values. The capacity is 250 mA h g−1 initially and gradually increases to 400 mA h g−1 after 140 cycles while delivering a Coulombic efficiency of 99.2%. In contrast to the previous samples, no capacity fading follows after the initial increase. This shows SDBS coats the active material effectively when added to iron cyanide. As the charge and discharge curves remain the same during cycling and show that the redox processes are not changed, the increase in the capacity can be attributed to the accessibility of more active material during cycling and lithiation. The rate handling of FVO–SDBS1 also shows improved capacity stability compared to the FVO electrode. A high capacity of 740 mA h g−1 was achieved under 50 mA g−1 for FVO–SDBS1. However, similar to FVO, the material was not capable of delivering storage capacity at higher rates, such as 2500 mA g−1 and 5000 mA g−1 which shows the high dependency of both materials on diffusion (Fig. S9A, ESI†).
Cyclic voltammetry at different rates was carried out for FVO–SDBS1. From specific current values at maximum potential, 0.5 V, and 2.75 V, b-values were calculated (Fig. S10A, ESI†). The b-values ranged from 0.64 to 0.71, indicative of a behavior between diffusion-controlled (b = 0.5) and non-diffusion-controlled (b = 1.0) charge storage (Fig. S10B and C, ESI†).46
Cyclic voltammograms of FVO–SDBS1 and FVO–SDBS2 showed similar peaks. However, they were more pronounced in the FVO–SDBS2 with a larger integrated area under the cyclic voltammograms area, arising from the higher ratio of the active material in this sample (Fig. 5C and D). Three pairs of current peaks can be indexed, showing multiple electron electrochemical redox reactions. We see oxidation peaks for FVO–SDBS2 at 0.3 V, 1.3 V, 2.0 V, and 2.6 V vs. Li/Li+. These are less distinct in FVO–SDBS1, indicating lower electrochemical accessibility of the active material caused by the SDBS coating. The sharp oxidation peak at 0.30 V vs. Li/Li+ and the respective reduction peak at 0.01 V vs. Li/Li+ displays the Li-intercalation into ordered graphite structure (graphitic layer) of the carbon coating and the MWCNTs and the Li-deintercalation from LiC6 (LiC6 → C6 + Li+ + e−).47 Further, the peak at 1.30 V vs. Li/Li+ can be attributed to the Li ions extraction in the inner channels of the CNTs. Finally, the very weak peak at 2.25 V characterizes the extraction of Li ions bonded to surface functional groups.48
Anodic peaks at 1.30 V and 2.00 V vs. Li/Li+ can also be assigned to the multi-step oxidation of Fe0 to Fe3+ and the de-lithiation of vanadium oxide (or vanadyl groups).41 Peaks obtained in the cathodic scan located at 0.80 V, 1.30 V, and 1.80 V vs. Li/Li+ can be associated with nano stoichiometric Li+-intercalated compounds and further conversion formation of iron oxide and vanadium oxide, while lithiation followed by the multi-step reduction of iron back to metallic iron.12,41 The obtained reduction and oxidation peaks from cyclic voltammetry agree with the galvanostatic discharge and charge profiles tested at a specific current of 250 mA g−1 in a voltage range between 0.01 V and 3.50 V vs. Li/Li+ as shown in Fig. 5E and F.
As the redox peaks in the cyclic voltammogram are broad and small, in the charge and discharge profiles, no flat plateaus, but rather gradual slopes are observed for different redox reactions with a weak plateau at ∼1.5 V vs. Li/Li+, similar to previous reports on FeVO4 (ref. 37) and Fe2VO4 (ref. 40). Studies on mixed iron–vanadium with similarly shaped cyclic voltammograms show this results from the formation of LixVOy and metallic Fe through multi-step reduction.40 Further post-mortem analysis after discharge and washing shows that the electrode retains its morphology, and the network of CNTs remains (Fig. S11A, ESI†). XRD analysis shows that the main formed phase after cycling is FeVO4 in an orthorhombic lattice, same as the initial phase present in the electrode with the strongest line at 34° 2θ corresponding to the (112) plane. Additional present reflections can be indexed with Fe2O3 (Fig. S11B, ESI†).
Table 2 shows the electrochemical performance of comparable studies for lithium-ion storage with values similar to those obtained in this study. Achieved capacity values outperform Fe0.12V2O5 nanowire arrays on a Ti foil that are self-standing electrodes similar to this study.49 Our results are comparable to Fe2VO4 nanowires achieved by Tao et al.,40 although they used phosphorous additive, which is a high-capacity alloying element and contributes to the total material capacity and increases the conductivity. So far, the present work offers the largest potential window of ∼3.5 V among other studies while providing stable capacity from conversion reactions. In contrast, other studies rely on limiting the voltage window to reduce capacity fading.11,49 With the coating from SDBS surfactant in the present study, stability results outperform all other iron vanadium reports so far. However, the achieved stable capacity is lower than that of the initial (∼700–800 mA h g−1) due to the high mass of SDBS incorporation. Among comparable studies, the capacity values achieved by Luo et al.41 are the highest, comparable to our initial values before stabilization. In their research, surface treatment in ethanol was carried out to form stable porous mixed metal oxide. However, the stability of this treatment faded during cycling. As our surface treatment shows great stability in the current study, an improved conductivity may increase the capacity for iron vanadium oxide further.40 Therefore, decreasing the SDBS coating mass or substituting it with similar surfactants that result in a more conductive layer can further enhance the performance compared to other studies on iron–vanadium oxide.
| Material/reference | AM composition | Potential (V vs. Li/Li+) | Electrode composition | Electrolyte | Capacity | Cycles/capacity retention vs. the first cycle |
|---|---|---|---|---|---|---|
| Fe2VO4 porous microparticles/41 | Fe2VO4 | 0.01–2.50 | 70% AM/20% super P/10% sodium alginate | 1 M LiPF6 in EC/DMC (1:1 by volume) |
799 mA h g−1 at 500 mA g−1 | 500/78% |
| Fe2VO4/11 | Fe2VO4 | 1.00–2.00 | 70% AM/20% acetylene black/10% PTFE | 1 M LiPF6 in EC/DMC (1:1 by volume) |
136 mA h g−1 at 5 C | 80/91% |
| Fe2VO4 carbon mesoporous nanowires/40 | P-Fe2VO4 | 0.01–3.00 | 70% AM/20% super P/10% sodium alginate | 1 M LiPF6 in EC/DMC (1:1 by volume, 5% FEC additive) |
486 mA h g−1 at 5000 mA g−1 | 250/109% |
| Iron vanadate nanowire arrays/49 | Fe0.12V2O5@Ti foil | 2.00–3.60 | 100% AM | 1 M LiPF6 in EC/DMC (1:1 by volume) |
278 mA h g−1 at 30 mA g−1 | 100/82% |
| Iron vanadate nanowire arrays/49 | Fe0.12V2O5@Ti foil | 1.00–4.00 | 100% AM | 1 M LiPF6 in EC/DMC (1:1 by volume) |
382 mA h g−1 at 30 mA g−1 | 50/81% |
| This work (FVO–SDBS1) | FeVO 4 and Fe 2 VO 4 on CNT | 0.01–3.50 | 100% AM |
2 M LiTFSI in EC/DMC (1:1 by volume) |
400 mA h g− 1 at 250 mA g− 1 | 150/160% |
4. Conclusions and outlook
Derivatization of mixed vanadium-iron oxide from its respective FeV Prussian blue analog (PBA) was reported for the first time. FeV formation, assembly on CNT, and coating were in one step at room temperature. Water-based salt of vanadyl sulfate was used for vanadium incorporation, as this salt has been reported as the product of vanadium recovery from iron–vanadium waste. Heat treatment was carried out under CO2 flow with infrared radiation. Electrodes were prepared by suspension in ethanol and subsequent filtration to decrease the needed heating energy and use of toxic material in the electrode preparation. We use different surfactants to analyze their effect on the electrochemical stability. TA, PVP, and SDBS surfactants have been reported as effective coating agents for morphology design regardless of the order in which the surfactants are added.We show that for PBAs with higher solubility, such as FeV, the type, and order of the surfactants' addition can directly impede material crystallization and PBA formation. SDBS was shown to be an effective surfactant to stabilize the capacity of FeV only when added to hexacyanoferrate by forming a protective layer. However, both TA and PVP are unsuitable for coating the FeV and stabilizing its derivative. After stabilizing the bare FVO using an SDBS coating, a capacity of 400 mA h g−1 at a rate of 250 mA g−1 after 150 cycles were achieved with a Coulombic efficiency of 99.2%.
Since the coating mass was 50% of the electrode, the capacity can be further increased by optimizing the type and mass of the surfactant to decrease the coating thickness. This work can facilitate the path to derivatization of other complex mixed metal oxides (and other mixed metal components) from their soluble and tricky PBA and show how to stabilize their performance for electrochemical energy storage.
Author contributions
BB: conceptualization, methodology, investigation, data curation, validation, visualization, writing – original draft. HEG: methodology, investigation, data curation, writing – original draft. STA: methodology, validation, writing – original draft. EP: methodology, investigation, data curation, validation, writing – original draft. VP: validation, funding acquisition, writing – review and editing.Conflicts of interest
The authors have no conflict of interest.Acknowledgements
This work was part of the electrospun vanadium oxide and sulfide-carbon hybrids project (HEROES-4-Li-Na-batteries) by the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft), project number 452180147. The authors thank Andrea Jung (INM) for carrying out ICP-OES and Samantha Husmann (formerly INM) for her comments on the work. The work of HEG was part of the École Européenne d'Ingénieurs en Génie des Matériaux (EEIGM) carried out at Saarland University. E. P. acknowledges the financial support from the Alexander von Humboldt Foundation.References
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- D. Xia, H. Gao, M. Li, F. Gong and M. Li, Energy Storage Mater., 2021, 35, 169–191 CrossRef.
- T.-U. Choi, G. Baek, S. G. Lee and J.-H. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 24817–24826 CrossRef CAS PubMed.
- C. Q. Lim and Z.-K. Tan, ACS Appl. Energy Mater., 2021, 4, 6214–6220 CrossRef CAS.
- M. Hu, N. L. Torad and Y. Yamauchi, Eur. J. Inorg. Chem., 2012, 2012, 4795–4799 CrossRef CAS.
- C. Xuan, J. Zhang, J. Wang and D. Wang, Chem.–Asian J., 2020, 15, 958–972 CrossRef CAS PubMed.
- B. Bornamehr, V. Presser, A. J. G. Zarbin, Y. Yamauchi and S. Husmann, J. Mater. Chem. A, 2023, 11, 10473–10492 RSC.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502175 CrossRef.
- T. Yang, D. Xia, Z. Wang and Y. Chen, Mater. Lett., 2009, 63, 5–7 CrossRef CAS.
- C. Yang, F. Lv, Y. Zhang, J. Wen, K. Dong, H. Su, F. Lai, G. Qian, W. Wang and A. Hilger, Adv. Energy Mater., 2019, 9, 1902674 CrossRef CAS.
- M. Tavakoli and D. Dreisinger, Hydrometallurgy, 2014, 141, 17–23 CrossRef CAS.
- J. Chen, L. Wei, A. Mahmood, Z. Pei, Z. Zhou, X. Chen and Y. Chen, Energy Storage Mater., 2020, 25, 585–612 CrossRef.
- S. F. Kettle, E. Diana, E. M. Marchese, E. Boccaleri and P. L. Stanghellini, J. Raman Spectrosc., 2011, 42, 2006–2014 CrossRef CAS.
- B. Bornamehr, V. Presser and S. Husmann, ACS Omega, 2022, 7, 38674–38685 CrossRef CAS PubMed.
- A. K. Boehm, S. Husmann, M. Besch, O. Janka, V. Presser and M. Gallei, ACS Appl. Mater. Interfaces, 2021, 13, 61166–61179 CrossRef CAS PubMed.
- B. Krüner, J. Lee, N. Jäckel, A. Tolosa and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 9104–9115 CrossRef PubMed.
- A. Tolosa, B. Krüner, S. Fleischmann, N. Jäckel, M. Zeiger, M. Aslan, I. Grobelsek and V. Presser, J. Mater. Chem. A, 2016, 4, 16003–16016 RSC.
- M. Wakihara, Y. Shimizu and T. Katsura, J. Solid State Chem., 1971, 3, 478–483 CrossRef CAS.
- D. Wang, F. Li and B. Yan, J. Alloys Compd., 2019, 797, 1050–1058 CrossRef CAS.
- C.-H. Chuang, L.-Y. Hsiao, M.-H. Yeh, Y.-C. Wang, S.-C. Chang, L.-D. Tsai and K.-C. Ho, ACS Appl. Energy Mater., 2020, 3, 11752–11762 CrossRef CAS.
- G. Moretti and C. Gervais, J. Raman Spectrosc., 2018, 49, 1198–1204 CrossRef CAS.
- R. Fehrmann, S. Boghosian, G. Papatheodorou, K. Nielsen, R. W. Berg and N. Bjerrum, Inorg. Chem., 1989, 28, 1847–1853 CrossRef CAS.
- N. A. Gvozdik and K. J. Stevenson, Electrochim. Acta, 2021, 383, 138300 CrossRef CAS.
- S.-J. Park, S. S. Shin, J. H. Jo, C. H. Jung, H. Park, Y.-I. Park, H.-J. Kim and J.-H. Lee, J. Hazard. Mater., 2023, 442, 129967 CrossRef CAS PubMed.
- M. Hu, A. A. Belik, M. Imura, K. Mibu, Y. Tsujimoto and Y. Yamauchi, Chem. Mater., 2012, 24, 2698–2707 CrossRef CAS.
- Q. Zhang, T. Zhang, J. Ge and Y. Yin, Nano Lett., 2008, 8, 2867–2871 CrossRef CAS PubMed.
- L. Liu, Z. Hu, L. Sun, G. Gao and X. Liu, RSC Adv., 2015, 5, 36575–36581 RSC.
- J. Nai, J. Zhang and X. W. D. Lou, Chem, 2018, 4, 1967–1982 CAS.
- T. Uemura and S. Kitagawa, J. Am. Chem. Soc., 2003, 125, 7814–7815 CrossRef CAS PubMed.
- M. Hu, S. Furukawa, R. Ohtani, H. Sukegawa, Y. Nemoto, J. Reboul, S. Kitagawa and Y. Yamauchi, Angew. Chem., Int. Ed., 2012, 51, 984–988 CrossRef CAS PubMed.
- A. Hemmatifar, N. Ozbek, C. Halliday and T. A. Hatton, ChemSusChem, 2020, 13, 3865–3874 CrossRef CAS PubMed.
- A. Wold, D. Rogers, R. Arnott and N. Menyuk, J. Appl. Phys., 1962, 33, 1208–1209 CrossRef CAS.
- Z. Gan, L. Chen, F. Xiong, X. Cai, W. Cui, X. Sang, Q. An and L. Wu, Chem. Commun., 2021, 57, 3615–3618 RSC.
- Y. Oka, T. Yao, N. Yamamoto, Y. Ueda, S. Kawasaki, M. Azuma and M. Takano, J. Solid State Chem., 1996, 123, 54–59 CrossRef CAS.
- N. Yan, Y. Xu, H. Li and W. Chen, Mater. Lett., 2016, 165, 223–226 CrossRef CAS.
- Y. Maeyoshi, K. Yoshii, M. Shikano and H. Sakaebe, ACS Appl. Energy Mater., 2021, 4, 13627–13635 CrossRef CAS.
- B. Tratnik, N. Van de Velde, I. Jerman, G. Kapun, E. Tchernychova, M. Tomšič, A. Jamnik, B. Genorio, A. Vizintin and R. Dominko, ACS Appl. Energy Mater., 2022, 5(9), 10667–10679 CrossRef CAS PubMed.
- Y. Tao, N. Yang, C. Liang, D. Huang, P. Wang, F. Cao, Y. Luo and H. Chen, ChemElectroChem, 2020, 7, 2395–2403 CrossRef CAS.
- Y. Luo, D. Huang, C. Liang, P. Wang, K. Han, B. Wu, F. Cao, L. Mai and H. Chen, Small, 2019, 15, 1804706 CrossRef PubMed.
- L. Li, Z. Li, A. Yoshimura, C. Sun, T. Wang, Y. Chen, Z. Chen, A. Littlejohn, Y. Xiang and P. Hundekar, Nat. Commun., 2019, 10, 1764 CrossRef PubMed.
- Z.-W. Zhao, T. Wen, K. Liang, Y.-F. Jiang, X. Zhou, C.-C. Shen and A.-W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 3757–3765 CrossRef CAS PubMed.
- N. Ogihara, S. Kawauchi, C. Okuda, Y. Itou, Y. Takeuchi and Y. Ukyo, J. Electrochem. Soc., 2012, 159, A1034 CrossRef CAS.
- M. Gaberscek, J. Moskon, B. Erjavec, R. Dominko and J. Jamnik, Electrochem. Solid-State Lett., 2008, 11, A170 CrossRef CAS.
- J. Liu, J. Wang, C. Xu, H. Jiang, C. Li, L. Zhang, J. Lin and Z. X. Shen, Adv. Sci., 2018, 5, 1700322 CrossRef PubMed.
- R. Paul, V. Etacheri, V. G. Pol, J. Hu and T. S. Fisher, RSC Adv., 2016, 6, 79734–79744 RSC.
- X. X. Wang, J. N. Wang, H. Chang and Y. F. Zhang, Adv. Funct. Mater., 2007, 17, 3613–3618 CrossRef CAS.
- Y. Cao, D. Fang, R. Liu, M. Jiang, H. Zhang, G. Li, Z. Luo, X. Liu, J. Xu and W. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 27685–27693 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Digital images of iron–vanadium oxide self-standing electrode and synthesis solutions, additional characterization of FeV and FVO including SEM, XRD, Raman, TGA, and TEM. Additional electrochemical characterizations. See DOI: https://doi.org/10.1039/d3se00854a |
| This journal is © The Royal Society of Chemistry 2023 |