
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of fully renewable, furfural-derived building blocks via formal Diels–Alder cycloaddition of atypical addends†
Răzvan C.
Cioc‡
*a,
Eva
Harsevoort‡
a,
Martin
Lutz
b and
Pieter C. A.
Bruijnincx
 *a
*a
aOrganic Chemistry and Catalysis, Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl; r.c.cioc@uu.nl
bDr M. Lutz, Structural Biochemistry, Bijvoet Centre for Biomolecular Research, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands
First published on 31st August 2023
Abstract
Diels–Alder (DA) cycloaddition of furanics is emerging as a key transformation in circular chemistry, providing access to highly versatile, biobased platform molecules. Further development of this technology into viable industrial applications faces major challenges, a notorious one being the lack of reactivity of the most readily available furans, i.e. the furfural derivatives. Herein we describe the remarkably-facile intramolecular DA reaction of allyl acetals of different furfurals to efficiently afford formal DA adducts with the atypical, unreactive dienophile allyl alcohol. Our methodology gives access to unprecedented oxanorbornene derivatives in high chemo-, regio- and stereoselectivity, which can be readily diversified into valuable products. This offers the potential of scalable production of renewable chemical building blocks from cheap, bioderived platform molecules.
Introduction
Circularity and sustainability considerations require routes to be developed to produce ‘drop-in’ chemical commodities as well as novel, functional alternatives from renewable resources rather than fossil feedstock. For this, the Diels–Alder [4 + 2] cycloaddition (DA) of biobased furanics has emerged as a particularly useful synthetic tool,1 as it allows fully atom-efficient conversion of bioderived feedstocks into highly useful products (i.e. bearing the ubiquitous 6-membered carbocyclic motif). Indeed, in addition to the usual synthetic targets of the century-old DA chemistry (natural products,2–4 bioactive compounds,5 drug delivery systems and biomaterials,6,7 responsive polymers,8–10etc.), furan DA chemistry is now increasingly part of synthesis routes towards various renewable commodity and specialty chemicals, for potential application as (novel) monomers, adhesives, curing agents, plasticizers, lubricants, coatings, etc.1,11–17To ensure both overall sustainability and economic viability, such furan DA conversion technology needs to be green and efficient and based on the most readily available biobased furanics to ensure the necessary redox- and atom-efficiency. Meeting these prerequisites in full is a challenge. First of all, there is an unfortunate mismatch between those furans most directly accessible by biorefining and currently most abundantly produced and their DA reactivity: formyl-furans (furfural and derivatives) are essentially unreactive dienes.18–21 Furthermore, the reactive dienophiles of choice, e.g. maleic anhydride (and derivatives thereof), are only available from biomass at max 80% theoretical carbon yield, impacting the carbon footprint of the process (Fig. 1B).22 Indeed, the current scope of furan DA reactions is dominated by the so-called ‘normal electron demand’ cycloadditions of electron-rich furans and electron-deficient dienophiles, both activated inputs;1,12 very few examples with alternative substitution patterns of the addends are available in the literature.18,23 More so, adducts originating from the combination of electronically-mismatched pairs (i.e. electron-poor furan/electron-rich dienophile, both deactivated inputs) are absent altogether; this area of the oxanorbornene chemical space is essentially empty. Next, furan DA cycloadditions are typically equilibrium-limited, disallowing high product yields. In addition, the adduct is typically thermally labile, which limits the options and operating window for downstream processing. Finally, controlling the regiochemistry (and sometimes stereochemistry) of unsymmetrical addend coupling is highly challenging, as is the subsequent purification of the major isomer from the resulting mixture.24
 | ||
| Fig. 1 Furan DA cycloaddition: current paradigm and design of novel route to new scaffolds. | ||
Herein we report a novel strategy that addresses all these issues and enables the direct, selective and efficient production of versatile DA adducts of furfural by exploiting an atypical dienophile, allyl alcohol. As part of our prior work on sustainable furan DA chemistry, we noted that the intramolecular DA cycloaddition with the bifunctional furfuryl acrylate25 failed due to a highly unfavorable equilibrium between the most stable conformation of this molecule (i.e. linear) and the necessary, reactive conformer (folded), originating among other factors from the large dipole moment of the latter. As intramolecular DA cycloaddition does offer the potential to overcome most of the challenges noted above, we did pursue this approach further and hypothesized that tethering the dienophile to the furan via an ether linkage instead would improve the conformational flexibility and thus allow the DA reaction. However, since the olefinic bond would no longer be electronically-activated, we anticipated a more difficult internal cycloaddition. Intramolecular furan DA reactions are fairly common and, encouragingly, have been reported for both activated as well as unactivated tethered dienophiles (mainly amine-based, but also (thio)ether or alkyl linkages).26 Notably, all previously disclosed examples involve complex precursors, tediously synthesized and structurally incompatible with our aim, i.e. the scalable production to renewable chemical building blocks from cheap, bioderived platform molecules.
Results and discussion
Precursor design and reaction optimization
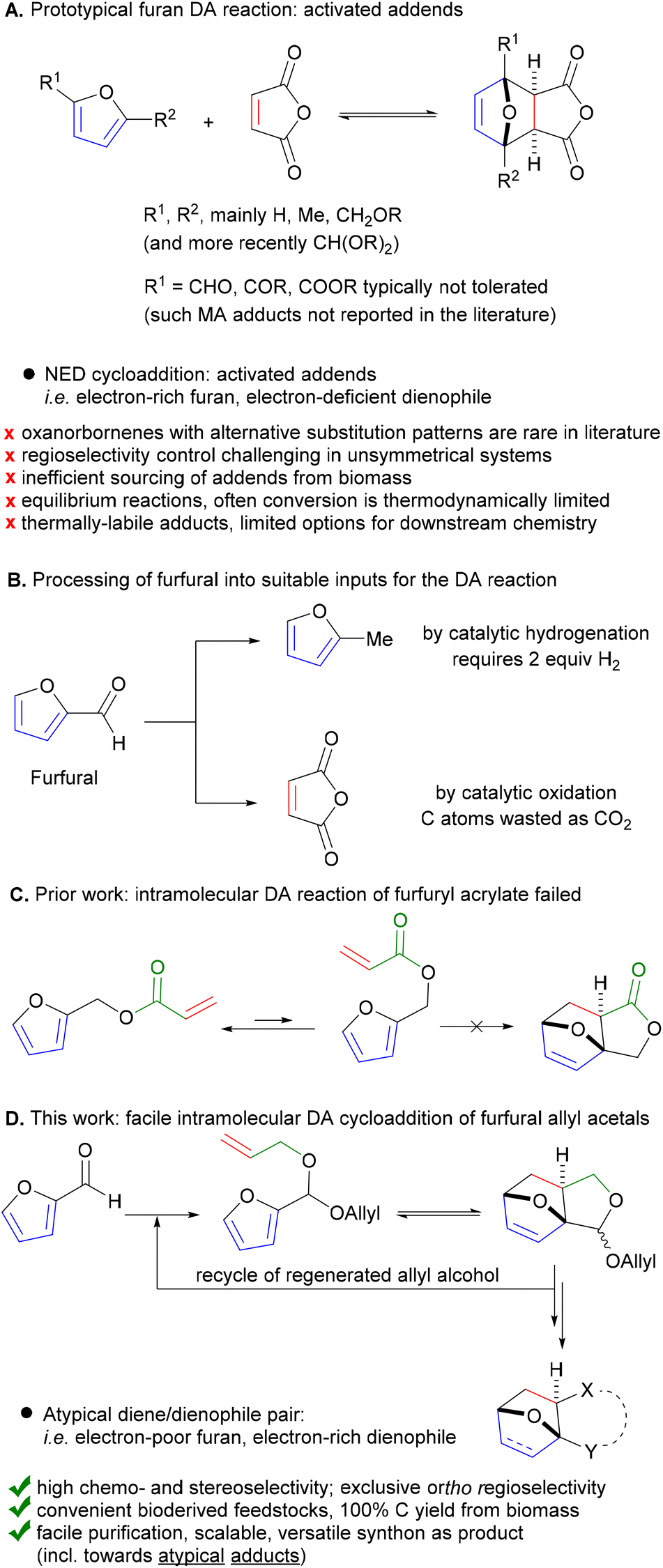
Promisingly, a small variation on the initial acrylate attempt already showed some DA cycloaddition activity: the furfuryl allyl ether 2a gave some of the corresponding adduct 3a (neat, 80 °C, 24 h), albeit in a low isolated yield of 11% and low chemoselectivity (33%) (Scheme 1). While modest, this activity did suggest that enhancing the population of the reactive conformer is a more important contributor to the feasibility of the reaction than activation of the olefin by an electron-withdrawing substituent is. Accordingly, we shifted focus to diallyl acetal 2b, a furfural derivative, which we anticipated to be a superior DA substrate due to enhanced conformational flexibility, favorable statistics and potentially the Thorpe–Ingold effect; moreover, the direct use of 2b is highly desirable from a redox-economy point of view. Gratifyingly, the formation of 3b under similar conditions turned out to be significantly more exergonic (K ≈ 10 vs. K < 1 for 3a); in addition, furfural 1b and acetal 2b are less prone to degradation as compared to 1a and 2a. Together, this allowed for the isolation of the adduct in a much-improved yield (66%) and selectivity.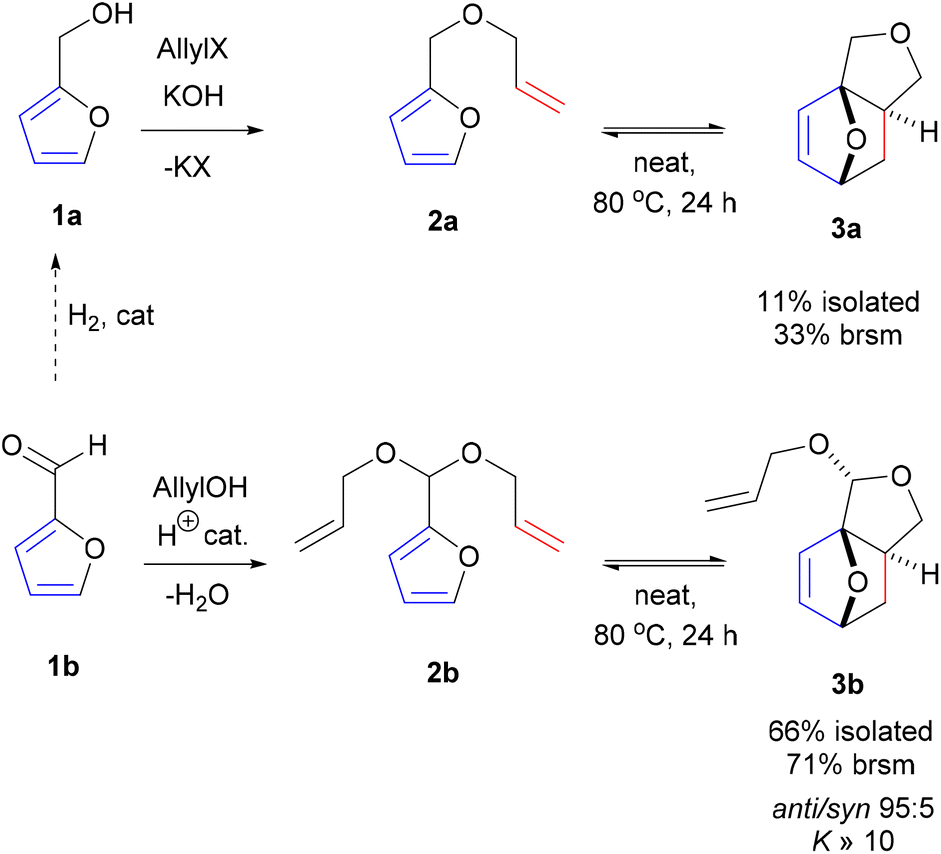 | ||
| Scheme 1 Intramolecular DA cycloadditions with furfural allyl ether 2a/furfural diallyl acetal 2b; brsm = based on recovered starting material. | ||
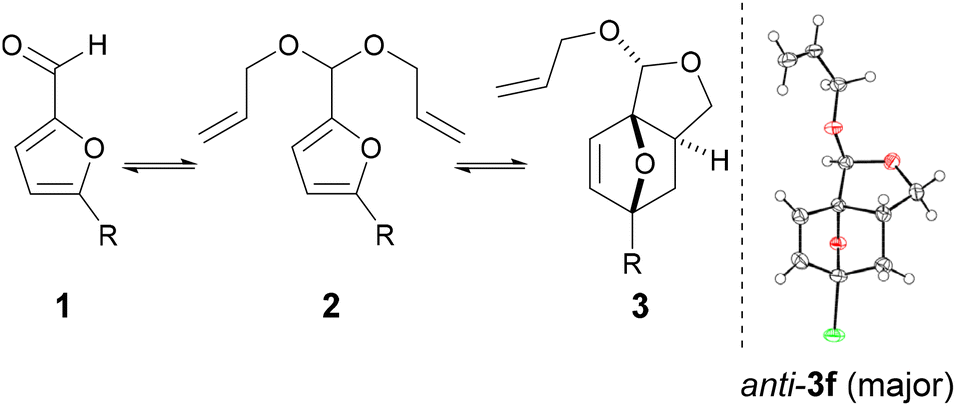
With the intention to simplify the process as much as possible, we performed the acetalization and the cycloaddition in one single step. Thus, with excess allyl alcohol (10 equiv.), catalytic trifluoroacetic acid (TFA) (10 mol%) and in the presence of 3 Å molecular sieves, both the acetalization and the cycloaddition equilibria are pushed towards 3b, which was produced in 75–80% yield after 24 h at 100 °C. The reaction was 100% exo-stereoselective due to the high preference for cis disposition of the fused C5–C6 rings; the diastereoisomeric ratio of the resulting anti- and syn-acetal pair was also very high, at ∼95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. Monitoring the reaction in time showed the transformation to be selective, both at low as well as high furfural conversions (Fig. 2) and to be near to equilibrium after 24 h. Note that the DA reaction proceeds with thermal activation alone and TFA does not play a catalytic role here in this reaction. Indeed, furfural diallyl acetal conversion to the DA adduct in the absence of TFA was comparable.
:5. Monitoring the reaction in time showed the transformation to be selective, both at low as well as high furfural conversions (Fig. 2) and to be near to equilibrium after 24 h. Note that the DA reaction proceeds with thermal activation alone and TFA does not play a catalytic role here in this reaction. Indeed, furfural diallyl acetal conversion to the DA adduct in the absence of TFA was comparable.
 | ||
| Fig. 2 Profile of the one-pot acetalization/DA in time (20 equiv. allyl alcohol, TFA 10 mol%, 3 Å MS, 100 °C). | ||
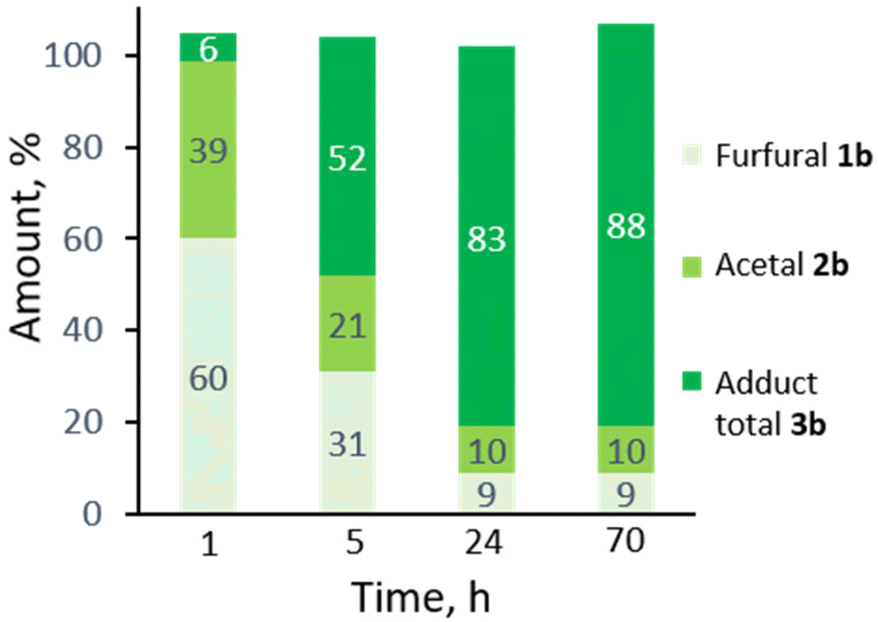
While 3b (oil at ambient temperature) can be separated from unconverted furfural and 2b by chromatography (and presumably by vacuum distillation), we envisaged that a more convenient purification by crystallization would be possible after deprotection of the acetal group (which also offers the additional advantage of further valorization of the combined diastereoisomeric acetal pair). Acid hydrolysis of 3b towards 4b was facile (HCl 2 equiv., H2O, 50 °C) and highly selective; 4b, a crystalline solid (see Scheme 2 for the X-ray crystal structure) could be isolated in highly pure form by precipitation. While 4b is exclusively anti in the solid state, in solution the hemiacetal diastereoisomeric structures are in a solvent-dependent equilibrium; this suggest that the open hydroxy-aldehyde form (i.e. the formal adduct of furfural and ally alcohol), is accessible for chemical derivatization, see Scheme 3 below and the ESI for more details.†
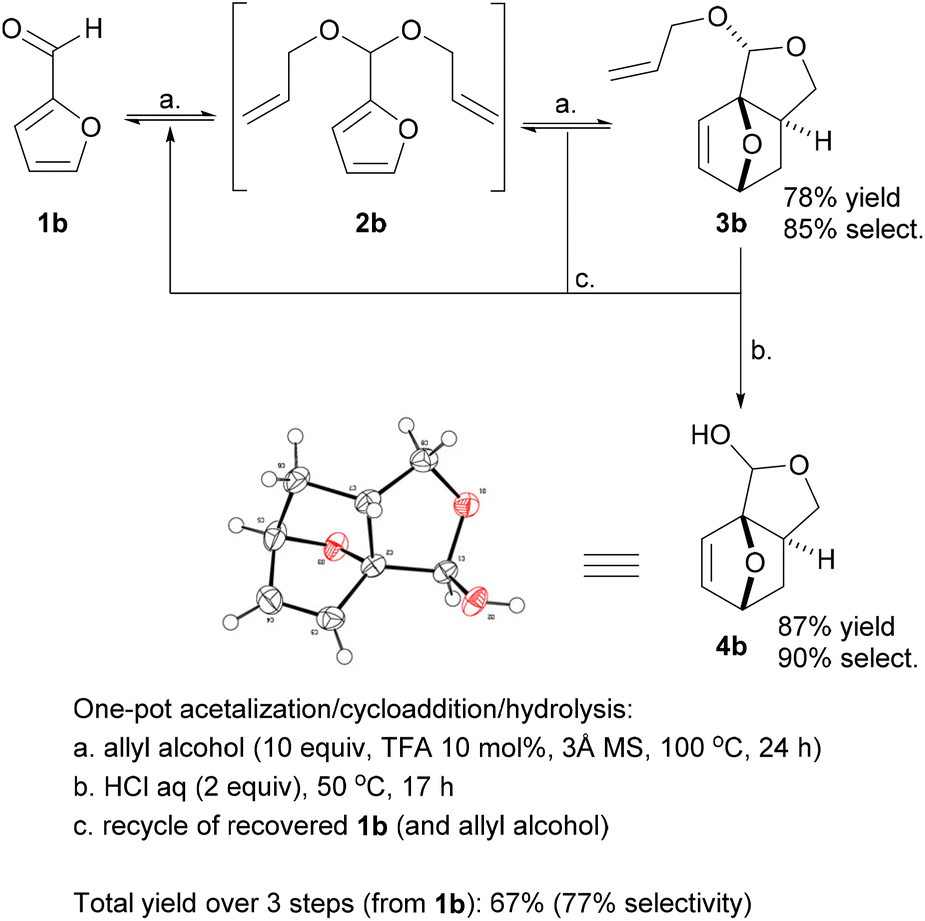 | ||
| Scheme 2 Selective one-pot furfural acetalization/DA cycloaddition/deprotection towards tricyclic hemiacetal 4b; XRD structure of isolated solid anti-4b; note that in solution, the anti and syn hemiacetal forms are in a solvent-dependent equilibrium. | ||
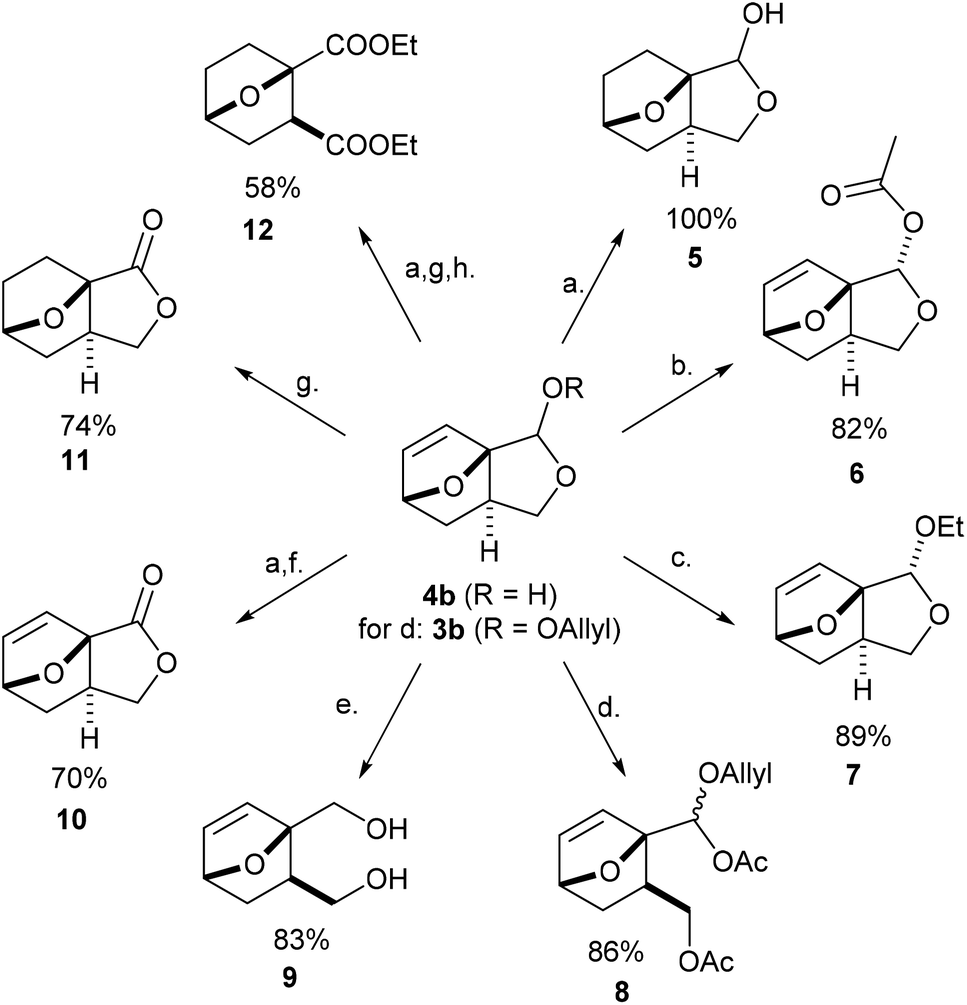 | ||
| Scheme 3 Functionalization of the novel DA adducts into value-added products; unless otherwise stated, 4b is the starting point: (a) Pd/C (0.5 wt%), H2, EtOAc, rt, 22 h; (b) AcCl, TEA, DCM, rt, 5 h; (c) Amberlyst-H cat., EtOH, 30 °C, 20 h; (d) MeSO3H, Ac2O, 50 °C, 2 h, using 3b as starting material (R = Allyl); (e) NaBH4, MeOH, rt, 2 h; (f) CuBr (5 mol%), TEMPO (5 mol%), bipy, NMI, MeCN, rt, 2 d; (g) Shvo's cat. (0.05 mol%), acetone, 50 °C, 4 d; (h) (1) EtOH, HCl, rt, 21 h; (2) NaClO2, NaClO, TEMPO cat., rt, 48 h, (3) SOCl2, EtOH, rt, 21 h. | ||
Deprotection also converts residual 2b to furfural, which can be recovered for recycling; similarly, (the excess) allyl alcohol can also be readily recovered at either stage of the synthesis by distillation. As there was no indication of side reactions involving allyl alcohol (according to GC and NMR analyses), this component can in principle be recovered in entirety; only one equivalent is incorporated in the final product 4b.
This synthesis proved very robust and scale-up to >100 mmol was facile. The acetalization/DA reaction produced adduct 3b in 78% yield, with 85% selectivity and the remainder almost fully accounted for with 6% unconverted 1b and 8% residual 2b. The acid hydrolysis of this crude reaction mixture proceeded with approx. 90% selectivity, and a very high isolated yield of the pure hemiacetal 4b (87%).
Reaction scope
With this promising synthetic route in hand, the intramolecular DA cycloaddition of the acetals of other readily available biobased 5-substituted furfurals was studied (Table 1). Notably, under similar reaction conditions, the ratio of 3:2 (calculated by NMR analysis of the crude reaction mixtures) was in all examples around 10 or higher; this suggests that the thermodynamics of the reaction is not negatively impacted by the substitution at the furan 5-position. Exceptionally, for R = Br, this ratio is at least 19:1; this positive impact of a halogen substituent on the intramolecular furan DA, the ‘halogen-effect’,27–29 has been observed before. This reaction was somewhat less diastereoselective, however, with a ratio of anti-3f to syn-3f of 83:17. For all the other examples, the minor syn-3 diastereoisomer accounted for ≤7%; resolution of the diastereoisomers was in all cases possible by chromatography. Notably, single crystal X-ray structure determination of the major 3f isomer allowed the unambiguous assignment of the configuration of the acetal centers in the series (Table 1). In terms of the reaction's overall performance, adducts 3e (R = CH2OEt) and 3f (R = Br) were obtained in comparable yields to the title example 3b, whereas for R = Me and R = CH2OH the less efficient acetalization of 1 limited the yields to somewhat lower values.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Furan | 1, % | 2, % | Yield 3, % |
dr
3
, anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :syn :syn
|
| General procedure: 1 and allyl alcohol (10 equiv.), TFA (10 mol%), 3 Å MS, 100 °C, 24 h; residual 1 and 2, yield of 3 and dr of 3 refer to crude values determined by quantitative 1H-NMR analysis; isolated yields of purified anti-3 in brackets.a 12% of syn-3f also isolated. | ||||||
| 1 | H | 1b | 8% | 7% | 69% (53%) | 96:4 |
| 2 | Me | 1c | 26% | 5% | 54% (41%) | 95:5 |
| 3 | CH2OH | 1d | 22% | 5% | 47% (31%) | 96:4 |
| 4 | CH2OEt | 1e | 11% | 6% | 72% (60%) | 93:7 |
| 5 | Br | 1f | 7% | <4% | 76% (56%)a | 83:17 |
Follow-up chemistry
With easy access to gram quantities of 4b, we explored the synthetic potential of this novel DA adduct. The scaffold proved highly versatile and selective functionalization of the unsaturated hemiacetal into various added-value products was facile, as summarized in Scheme 3. Catalytic hydrogenation of the olefinic bond proceeded quantitatively on Pd/C, without compromising the reduction-labile hemiacetal group; this conversion is important in preventing side reactions resulting from the retro-DA channel (at temperatures >70 °C and/or in the presence of activators).30,31 Next, the free OH group underwent facile ester- and etherification, giving 6 and 7 in high yields under mild conditions (preserving the anti-stereochemistry). These fragrant molecules show structural similarities to aroma compounds like camphor and eucalyptol and might accordingly find use in the fragrances industry. Ensuing, the masked aldehyde functionality in 4b (or 3b) can be exploited in producing carbonyl derivatives upon the opening of the fused C5 ring, for instance the α-allyloxyalkyl acetate 8 (by treatment of 3b with MeSO3H in Ac2O) and oxime 13 (see Scheme 4). Alternatively, the hemiacetal ring can also be opened reductively with NaBH4, leading to diol 9, a bifunctional compound with potential use in polymer applications. Otherwise, going up the oxidation scale can be performed aerobically with the Cu/TEMPO catalytic system, producing the unsaturated tricyclic lactone 10;32 oxidation by transfer hydrogenation failed on 4b but proved feasible with 5 as substrate (with Shvo's catalyst), affording the saturated analogue 11 in 74% isolated yield. This lactone served as precursor for the straightforward synthesis of diester 12, a molecular analogue of diethyl o-phthalate, and thus a potential bioderived functional replacement of this industrially-important chemical commodity. Thus, this novel furan DA platform allows straightforward access to arene analogues and, more generally, to highly functional molecular scaffolds endowed with reactivity for various applications, such as cross-linking agents, polyesters/urethanes, plasticizers, etc. Preliminary studies show that 4b (and 3b) are reluctant to aromatization under standard conditions (e.g. with MeSO3H/Ac2O), something which requires further study.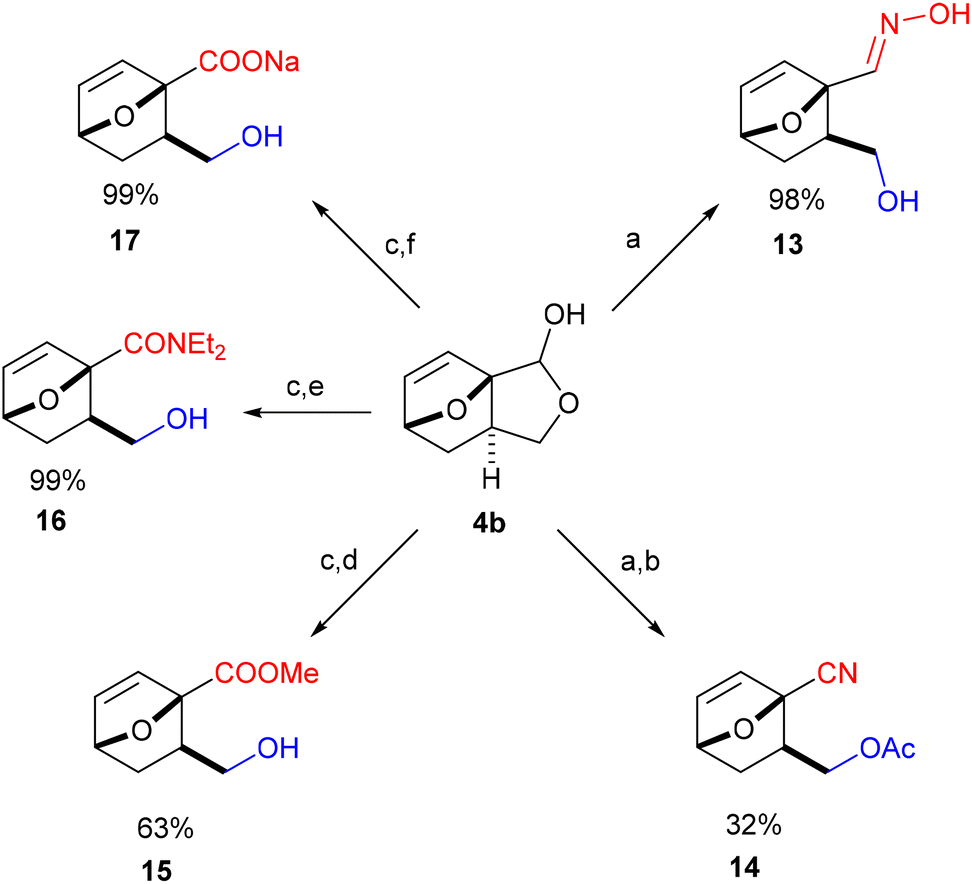 | ||
| Scheme 4 Divergent access of atypical furan DA adducts starting from platform molecule 4b: (a) NH2OH·HCl, NaHCO3, water, rt, 17 h; (b) DMAP (10 mol%), Ac2O, NEt3, CH2Cl2, rt, 6 d; (c) CuBr (5 mol%), TEMPO (5 mol%), bipy, NMI, MeCN, rt, 2 d; (d) AmberlystH, MeOH, rt, 2 h; (e) Et2NH, rt, 18 h; (f) NaOH, MeOH, rt. | ||
Finally, by simple functional group interconversion, key products 4b and 10 can be elaborated into a variety of atypical oxanorbornenes, such as oxime 13, nitrile 14, ester 15, amide 16 and carboxylate salt 17 (Scheme 4). Notably, together with many of the molecules in Scheme 3, these products are formally DA adducts of allyl alcohol, a dienophile that is not reactive enough to undergo direct cycloaddition reactions with furan and even less so with deactivated derivatives such as furfural (oxime), 2-furonitrile or methyl 2-furoate, as required here. Thus, our methodology is ideal for the rapid, efficient and divergent synthesis of (electronically-) unusual furan DA adducts, populating previously unexplored areas of the oxanorbornene chemical space (see also Fig. 1). In addition, this synthetic strategy allows for complete control over the regio- and (relative) stereochemistry of the products, which is an important advantage over other (DA-based) routes. In this respect, the ortho/exo configuration obtained here is most challenging, as it is severely disfavored sterically.25
Reaction mechanism
DFT calculations allowed for a better understanding of the factors contributing to the success of this reaction (B3LYP/6-311+g(d), GD3BJ dispersion, in methanol (SMD)) (Fig. 3). In accordance with experiment, anti-3b was found to be more stable than syn-3b (by approx. 3 kcal mol−1); only the pathway leading to the major isomer was investigated. As hypothesized and in contrast to furfuryl acrylate, the difference between the reactive conformation and the ground state is small for both furfuryl allyl ether 2a and furfuryl diallyl acetal 2b, 1.3 and 1.1 kcal mol−1, respectively. This leads to thermally accessible kinetic barriers for the cycloaddition of 27.3 and 25.9 kcal mol−1, respectively, in good agreement with the experimental observations (reactions proceed at 80–100 °C). Importantly, the cycloaddition with 2b is confirmed to be significantly more exergonic (by approx. 3 kcal mol−1) than the reaction with furfuryl allyl ether 2a. This difference is indeed reflected in the experiments: in the studied temperature range (80–100 °C), the equilibrium was on the adduct side in the case of the acetal (K ≈ 10) whereas the reaction with the ether was endergonic. Finally, the DFT study allows us to rule out the intermolecular pathway: the direct cycloaddition between furfural and allyl alcohol is both considerably slower and endergonic relative to its intramolecular equivalent, as expected for this electronically-mismatched pair of addends. The strategy reported here thus provides a welcome alternative to still formally obtain the attractive, renewable adducts of this combination.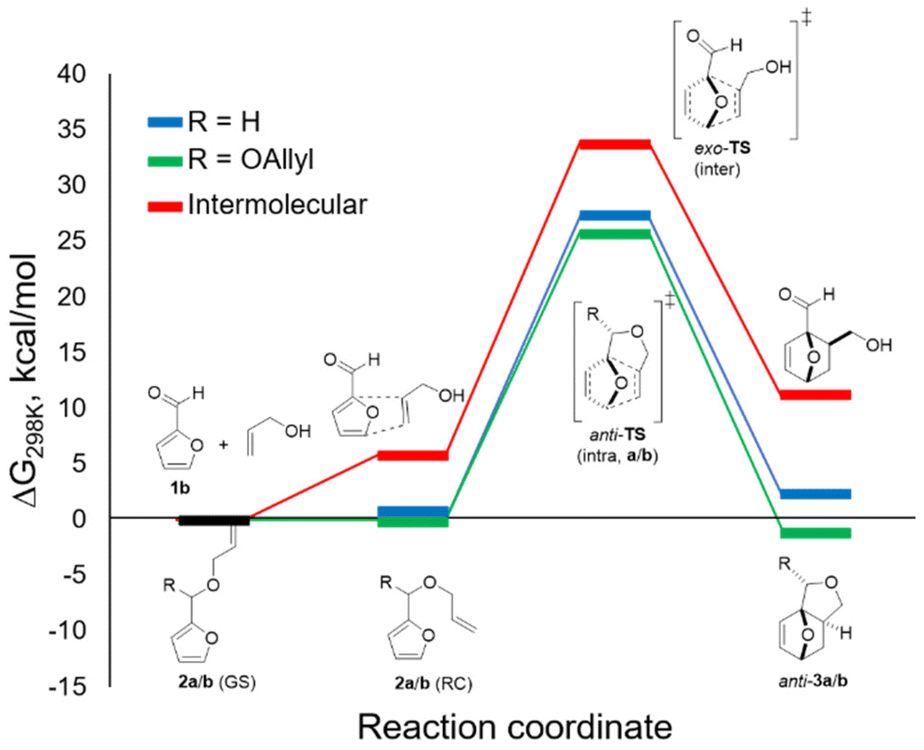 | ||
| Fig. 3 DFT mechanism (calculated with B3LYP at 6-311+g(d) and GD3BJ dispersion, in methanol (SMD)); only the paths leading to the anti-3 adducts were calculated; blue = furfuryl allyl ether 2a; green: furfural diallyl acetal 2b; red: intermolecular reaction between furfural and allyl alcohol; GS = ground state, RC = reactive conformation. | ||
Conclusions
The remarkably selective furan Diels–Alder cycloaddition of furfural allyl acetals provides ready access to a highly versatile molecular scaffold and thus to a range of renewable chemical building blocks. Contrary to many related (intermolecular) [4 + 2] cycloadditions, the cyclization of furfural diallyl acetal is both kinetically and thermodynamically feasible at convenient operating temperatures (80–100 °C). Importantly, the route provides straightforward access to various atypical furan adducts that are not accessible via the intermolecular coupling of the unreactive DA precursors (i.e. furfural and allyl alcohol). The high selectivity (including regio- and stereo-), operational simplicity and facile product purification allow for a scalable synthesis; in addition, the renewable origin of the two precursors and full carbon retention in the product, combined with the straightforward diversification of the tricyclic scaffold via atom-economical transformations make this chemistry highly attractive for the sustainable preparation of bioderived chemical products, both drop-in and new. Finally, given the high synthetic utility of furan DA reactions and the versatility of our novel adduct platform, we strongly believe this chemistry can also find application in other fields, such as drug discovery, artificial flavor development, and natural product synthesis.Author contributions
R. C. C. and P. C. A. B. conceived the research, and directed and supervised the project. R. C. C. and E. H. designed and executed the experiments and analysed the data. M. L. performed the single crystal structure determination. R. C. C. and P. C. A. B. acquired funding for the project. R. C. C., E. H. and P. C. A. B. wrote the paper and all authors commented on different versions of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research (NWO OCENW.XS4.250). Dr L. Witteman, Dr T. N. Ran and Dr J. T. B. H. Jastrzebski (Utrecht University) are acknowledged for technical assistance. We thank Dr J. Sastre Torano (Utrecht University) for performing the ESI-MS measurements. The X-ray diffractometer has been financed by NWO. The computational work was carried out on the Dutch national e-infrastructure with the support of the SURF Foundation.References
- F. A. Kucherov, L. V. Romashov, G. M. Averochkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2021, 9, 3011–3042 CrossRef CAS.
- P. Vogel, J. Cossy, J. Plumet and O. Arjona, Tetrahedron, 1999, 55, 13521–13642 CrossRef CAS.
- S. Roscales and J. Plumet, Heterocycles, 2015, 90, 741–810 CrossRef CAS PubMed.
- S. Bur and A. Padwa, in Methods and Applications of Cycloaddition Reactions in Organic Syntheses, ed. N. Nishiwaki, John Wiley & Sons, Inc, 1st edn, 2014, pp. 355–406 Search PubMed.
- C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477–499 CrossRef CAS PubMed.
- M. Gregoritza and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 97, 438–453 CrossRef CAS PubMed.
- T. Elschner, F. Obst and T. Heinze, Macromol. Biosci., 2018, 18, 1800258 CrossRef PubMed.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61–75 CrossRef CAS.
- M. Vauthier, L. Jierry, J. C. Oliveira, L. Hassouna, V. Roucoules and F. B. Gall, Adv. Funct. Mater., 2019, 1806765, 1–16 Search PubMed.
- Y.-L. Liu and T.-W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- R. F. A. Gomes and J. M. J. M. Ravasco, ChemSusChem, 2021, 14, 3047–3053 CrossRef PubMed.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736–2748 CrossRef CAS PubMed.
- S. Dutta and N. S. Bhat, Biomass Convers. Biorefin., 2023, 13, 541–554 CrossRef.
- H. Zhang, M. Jiang, Y. Wu, L. Li, Z. Wang, R. Wang and G. Zhou, Green Chem., 2021, 23, 2437–2448 RSC.
- H. Zhang, M. Jiang, Y. Wu, L. Li, Z. Wang, R. Wang and G. Zhou, ACS Sustainable Chem. Eng., 2021, 9, 6799–6809 CrossRef CAS.
- C. Plass, N. Adebar, R. Hiessl, J. Kleber, A. Grimm, A. Langsch, R. Otter, A. Liese and H. Gröger, Eur. J. Org. Chem., 2021, 6086–6096 CrossRef CAS.
- R. C. Cioc, M. Lutz, E. A. Pidko, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Green Chem., 2021, 23, 367–373 RSC.
- I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. De Oliveira Vigier, R. Wischert and F. Jérôme, Angew. Chem., Int. Ed., 2018, 57, 10510–10514 CrossRef CAS PubMed.
- F. A. Kucherov, K. I. Galkin, E. G. Gordeev and V. P. Ananikov, Green Chem., 2017, 19, 4858–4864 RSC.
- G. Averochkin, E. Gordeev, M. Skorobogatko, F. Kucherov and V. P. Ananikov, ChemSusChem, 2021, 14, 3110–3123 CrossRef CAS PubMed.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. Martin Alonso, R. Mariscal and M. López Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- R. C. Cioc, T. J. Smak, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Green Chem., 2021, 23, 5503–5510 RSC.
- K. I. Galkin and V. P. Ananikov, Int. J. Mol. Sci., 2021, 22, 11856 CrossRef CAS PubMed.
- C. S. Lancefield, B. Fölker, R. C. Cioc, K. Stanciakova, R. E. Bulo, M. Lutz, M. Crockatt and P. C. A. Bruijnincx, Angew. Chem., Int. Ed., 2020, 59, 23480–23484 CrossRef CAS PubMed.
- F. Lewis and L. Harwood, in Targets in Heterocyclic Systems, 2012, pp. 1–30 Search PubMed.
- S. N. Pieniazek and K. N. Houk, Angew. Chem., Int. Ed., 2006, 45, 1442–1445 CrossRef CAS PubMed.
- R. L. Rae, J. M. Zurek, M. J. Paterson and M. W. P. Bebbington, Org. Biomol. Chem., 2013, 11, 7946–7952 RSC.
- A. Padwa, K. R. Crawford, C. S. Straub, S. N. Pieniazek and K. N. Houk, J. Org. Chem., 2006, 71, 5432–5439 CrossRef CAS PubMed.
- S. Thiyagarajan, H. C. Genuino, M. Śliwa, J. C. Van Der Waal, E. De Jong, J. Van Haveren, B. M. Weckhuysen, P. C. A. Bruijnincx and D. S. Van Es, ChemSusChem, 2015, 8, 3052–3056 CrossRef CAS PubMed.
- S. Thiyagarajan, H. C. Genuino, J. C. Van Der Waal, E. De Jong, B. M. Weckhuysen, J. Van Haveren, P. C. A. Bruijnincx and D. S. Van Es, Angew. Chem., Int. Ed., 2016, 55, 1368–1371 CrossRef CAS PubMed.
- J. M. Hoover, J. E. Steves and S. S. Stahl, Nat. Protoc., 2012, 7, 1161–1166 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2126534 and 2126535. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc02357e |
| ‡ Equal first authorship contribution. |
| This journal is © The Royal Society of Chemistry 2023 |