
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of cisplatin-binding proteins by competitive cysteinome profiling†
Xianghe
Wang
a,
Yihai
Zhang
a and
Chu
Wang
 *ab
*ab
aSynthetic and Functional Biomolecules Center, Beijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing, China. E-mail: chuwang@pku.edu.cn
bPeking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China
First published on 1st August 2023
Abstract
Cisplatin is a widely used cancer metallodrug that induces cytotoxicity by targeting DNA and chelating cysteines in proteins. Here we applied a competitive activity-based protein profiling strategy to identify cisplatin-binding cysteines in cancer proteomes. A novel cisplatin target, MetAP1, was identified and functionally validated to contribute to cisplatin's cytotoxicity.
As an anti-cancer drug discovered in 1965, cis-dichlorodiamineplatinum(II) (cisplatin) is widely used in the treatment of testicular and ovarian cancer. According to statistics, over 50% of cancer patients have been treated with cisplatin in different treatment stages.1 When cisplatin enters cells, two chloride ions are removed by hydrolysis and the divalent platinum ions can attack adenines or guanines on DNA.2 As the cytotoxicity of cisplatin is generally believed to be mainly caused by the formation of Pt-DNA adducts,3 many research studies have focused on the interaction between cisplatin and DNA in recent years.4,5 Nevertheless, it has been estimated that only about 1% of the intracellular platinum can bind to DNA,6 and because of the unique structure of the diammonium platinum dihydrate, other intracellular nucleophilic substances can also be attacked extensively, including RNA, reduced cysteine side chains on proteins, thiol-containing ligands (e.g. glutathione), etc.7,8
Several protein targets of cisplatin have been biochemically characterized that may contribute to its cytotoxicity and drug resistance.9 For example, the human copper transport protein (Atox1) was shown to be potentially relevant to cisplatin resistance,10 and the interaction of cisplatin with human superoxide dismutase (SOD1) could cause cytotoxicity.11 In addition, proteomic techniques such as Multi-dimensional Protein Identification Technology (MudPIT) and Laser Ablation Inductively Coupled Plasma Mass Spectrometry (LA-ICP-MS) have been implemented for large-scale identification of cisplatin-binding proteins,12,13 however, they are limited in detecting targets of low abundance. While clickable cisplatin analogue probes have been developed to label and enrich cisplatin-binding proteins in cells,14,15 introduction of the bioorthogonal group may change the physical and chemical properties of cisplatin so that certain bona fide targets are missed.
Activity-based protein profiling (ABPP) is a powerful chemoproteomic strategy that can be applied to identify the protein targets of small molecules in complex biological systems.16,17 In particular, a site-specific and quantitative version of ABPP, isoTOP-ABPP, has been developed for global profiling of functional cysteines in proteomes.18 When it is operated in a competitive manner, the strategy can be used not only to identify cysteines that are sensitive to electrophilic metabolites and covalent drugs,19–22 but also to profile key metal-binding sites for zinc and iron–sulfur clusters.23,24
In the current study, we were inspired to apply this competitive ABPP strategy to globally profile cisplatin-binding cysteines in proteomes. Firstly, we verified by in-gel fluorescence that cisplatin could compete with the labeling of a cysteine-reactive and alkyne-functionalized iodoacetamide probe (IAyne) in living cells (ESI,† Fig. S1). We then performed a competitive rdTOP-ABPP25 experiment to identify cisplatin-binding sites (Fig. 1a). Briefly, proteomes were obtained from normal and cisplatin-treated MCF-7 cells, labeled by the IAyne probe for 1 h, and then conjugated with an acid-cleavable biotin tag by Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC).26,27 After enrichment by streptavidin and on-bead digestion by trypsin, the probe-adducted peptides from the normal and cisplatin-treated samples were isotopically labeled by light and heavy dimethylation reagents, respectively, prior to combination. The IAyne-adducted peptides were released by acid cleavage and analyzed using LC-MS/MS. After quantitation by CIMAGE2.0,28 we detected and quantified a total of 1947 peptides that belong to 1115 proteins in all three replicates with high confidence (Fig. 1b).
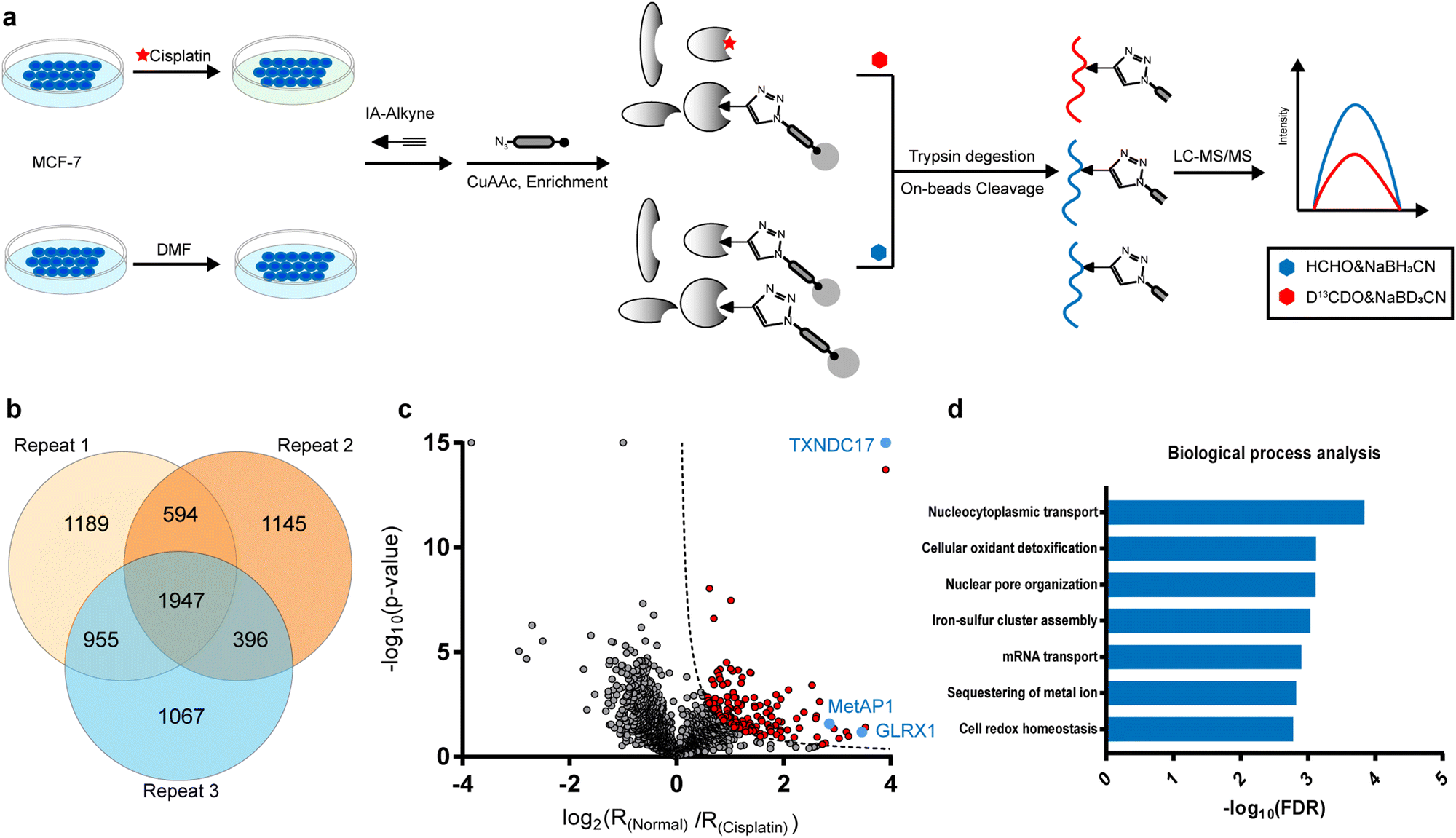 | ||
Fig. 1 Quantitative profiling of cisplatin-binding cysteines by rdTOP-ABPP. (a) The scheme of quantitative profiling of cisplatin-binding cysteines by rdTOP-ABPP. Equal numbers of MCF-7 cells were treated with N,N-dimethylformamide (DMF) and 300 μM cisplatin, respectively, for 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h. The whole proteomes were labeled with the cysteine-reactive IAyne probe and then subjected to the rdTOP-ABPP procedures. (b) Venn diagram showing the number of cisplatin-binding peptides quantified from three biological replicates. (c) Volcano plot of the rdTOP-ABPP ratios for each peptide quantified in the cisplatin-treated group as compared to those in the normal cells. Highlighted in red are cisplatin-targeted cysteines with high confidence in unique peptides. Highlighted in blue are the targets that are biochemically verified in the current study. (d) Gene ontology analysis of the identified cisplatin-binding proteins in terms of biological processes. h. The whole proteomes were labeled with the cysteine-reactive IAyne probe and then subjected to the rdTOP-ABPP procedures. (b) Venn diagram showing the number of cisplatin-binding peptides quantified from three biological replicates. (c) Volcano plot of the rdTOP-ABPP ratios for each peptide quantified in the cisplatin-treated group as compared to those in the normal cells. Highlighted in red are cisplatin-targeted cysteines with high confidence in unique peptides. Highlighted in blue are the targets that are biochemically verified in the current study. (d) Gene ontology analysis of the identified cisplatin-binding proteins in terms of biological processes. | ||
In light of the IAyne competition by cisplatin, a credible cisplatin-binding cysteine should yield a light/heavy (“normal/cisplatin”) ratio higher than 1. We analyzed, for every cysteine quantified, the statistical difference (p value) among all three replicates using a Student's t-test and drew a volcano plot (Fig. 1c). After applying the cutoff of −log10(p value) × log2(ratio) >1.5, we obtained 125 cysteines from 107 proteins as strong candidates for cisplatin-binding targets (Fig. 1c).
According to the gene ontology (GO) analysis, the 109 cisplatin-binding proteins are enriched in the biological process of cellular oxidant detoxification (Fig. 1d), which are exemplified by glutaredoxin-1 (GLRX1), glutaredoxin-related protein 5 (GLRX5), thioredoxin (TXN) and thioredoxin domain-containing protein 17(TXNDC17). This is consistent with previous reports that cisplatin induces ROS by disturbing oxidoreductases.29 Interestingly, another enriched pathway is the nucleocytoplasmic transport (Fig. 1d), which is supported by the presence of multiple proteins that are functionally involved in the assembly and maintenance of nuclear pore complexes, including nuclear pore glycoprotein p62 (NUP62), nucleoporin NUP35 (NUP35), nuclear pore complex protein NUP205 (NUP205), nuclear pore complex protein Nup98-Nup96 (NUP98), etc. These data suggest that cisplatin may enter a nucleus by interacting with a nuclear pore complex30 and affect its function.
Satisfyingly, several proteins identified in our work have been previously reported as cisplatin-binding proteins (Table 1). Our rdTOP-ABPP experiments additionally revealed the detailed cisplatin-binding sites in them. For example, rdTOP-ABPP identified C23/C26 and C43/C46 as cisplatin-sensitive cysteines in GLRX1 and TXNDC17, respectively, and we therefore set out to validate them biochemically. For each target, we first recombinantly expressed and purified the protein and showed by in-gel fluorescence that cisplatin could dose-dependently inhibit IAyne's labeling (Fig. 2a and d). We then constructed the corresponding cysteine mutants (C23A and C26A for GLRX1, C43A and C46A for TXNDC17) and validated by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) that the mutants have a much reduced platinum(II) signal as compared to their wild-type protein (Fig. 2b and e). Finally, we incubated the wild-type protein with cisplatin and analyzed the complex using LC-MS/MS. Representative MS/MS spectra supports that the platinum(II) ion is coordinated by the two identified cysteines in each protein in a bidentate mode, as previously reported10,31 (Fig. 2c and f). These results unambiguously confirmed that cisplatin binds these two proteins through the functional cysteine sites as profiled by the rdTOP-ABPP experiments.
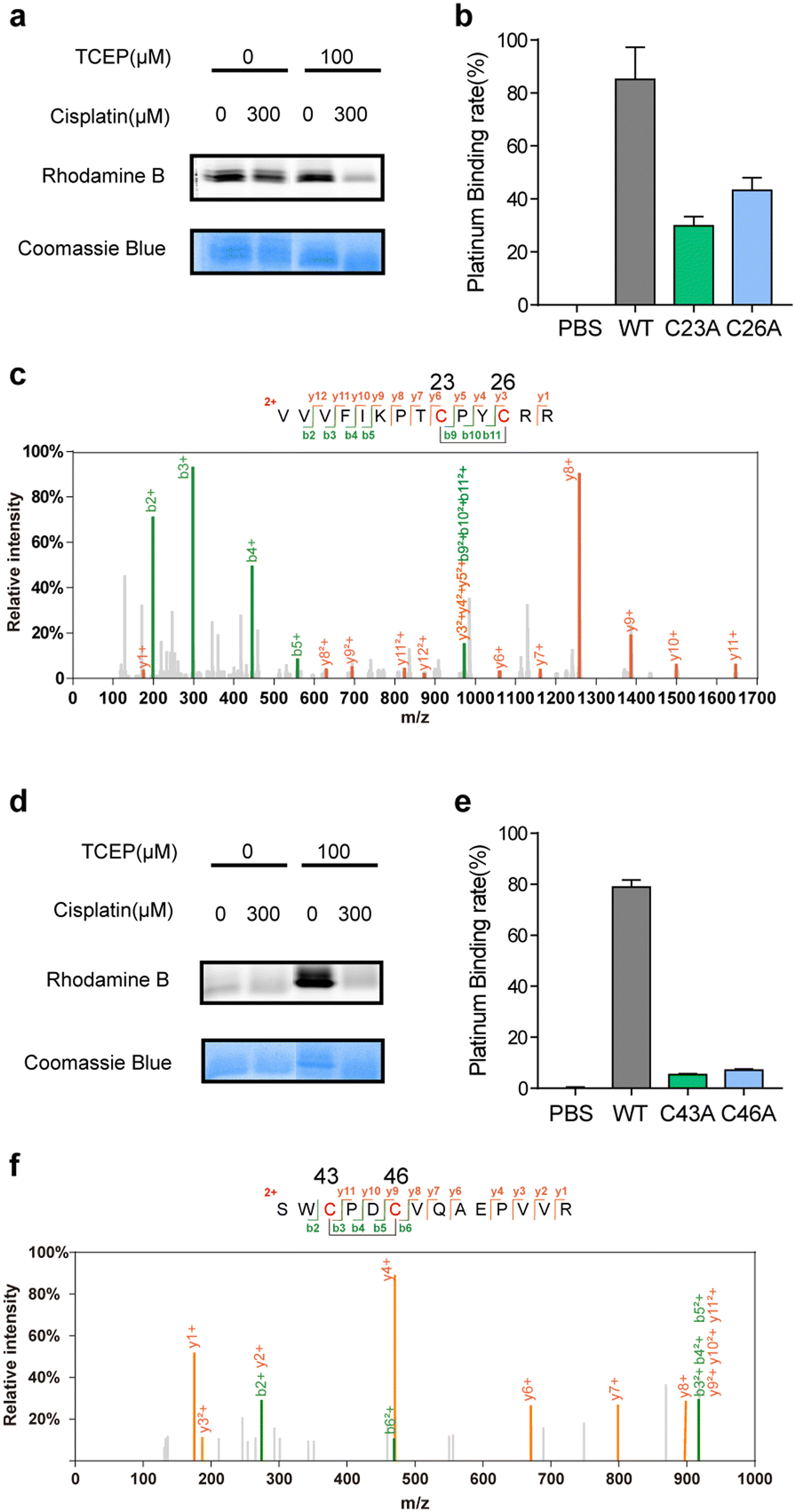 | ||
| Fig. 2 Verification of cisplatin-binding sites in human GLRX1 and TXNDC17. (a) Cisplatin competes with IAyne's labeling on the purified and reduced GLRX1 as displayed by in-gel fluorescence. The concentration of cisplatin is 300 μM. (b) Wild-type (WT) GLRX1 shows much higher cisplatin-binding signals than the corresponding cysteine mutants (C23A and C26A) as measured by ICP-MS. (c) Representative MS/MS spectrum supporting that the Cys23 and Cys26 of GLRX1 are bridged by cisplatin. (d) Cisplatin competes with IAyne's labeling on the purified and reduced TXNDC17 as displayed by in-gel fluorescence. The concentration of cisplatin is 300 μM. (e) Wild-type (WT) TXNDC17 shows much higher cisplatin-binding signals than the corresponding cysteine mutants (C43A and C46A) as measured by ICP-MS. (f) Representative MS/MS spectrum supporting that the Cys43 and Cys46 of TXNDC17 are bridged by cisplatin. | ||
In addition to GLRX1 and Trip14, a novel target of cisplatin, methionine aminopeptidases 1 (MetAP1), has attracted our attention. In eukaryotic cells, there are two types of methionine aminopeptidases (MetAP1 and MetAP2) and they function to co-translationally remove the N-terminal methionine from nascent proteins. In Saccharomyces cerevisiae, the loss of either enzyme will lead to a phenotype of slow growth, while the simultaneous loss is lethal.36 Recently, human MetAP2 has been identified as the target of a tumor-inhibiting natural product fumagillin.37 However, neither protein is known to have cisplatin-binding activity. Cys14 of MetAP1 was identified in our rdTOP-ABPP data with a ratio of 7.2 and a p value of 0.0263. This specific cysteine belongs to a zinc finger motif in the N-terminal domain of MetAP1, which also contains Cys9, Cys36 and Cys40. It plays an important role in regulating the enzyme's activity.38 Loss of function of the zinc finger disturbs the interaction between MetAP1 and its metal cofactors.39
We recombinantly expressed and purified the wild-type MetAP1 and the corresponding mutant C14S (ESI,† Fig. S3). The concentration-dependent competition between cisplatin and IAyne can be clearly observed for the wild-type MetAP1 by in-gel fluorescence (Fig. 3a). ICP-MS analysis shows MetAP1 binds cisplatin with a molar ratio of 1:0.47, while that of the C14S mutant is significantly lower to 1:0.11 (Fig. 3b). Unfortunately, we were unable to find a high-quality MS/MS spectrum for the cisplatin-bridged peptide probably due to the fact that the sequence (VCETDGCSSEAK) contains a high percentage of acidic amino acids and is not friendly for detection under the positive-ion mode of LC-MS/MS.
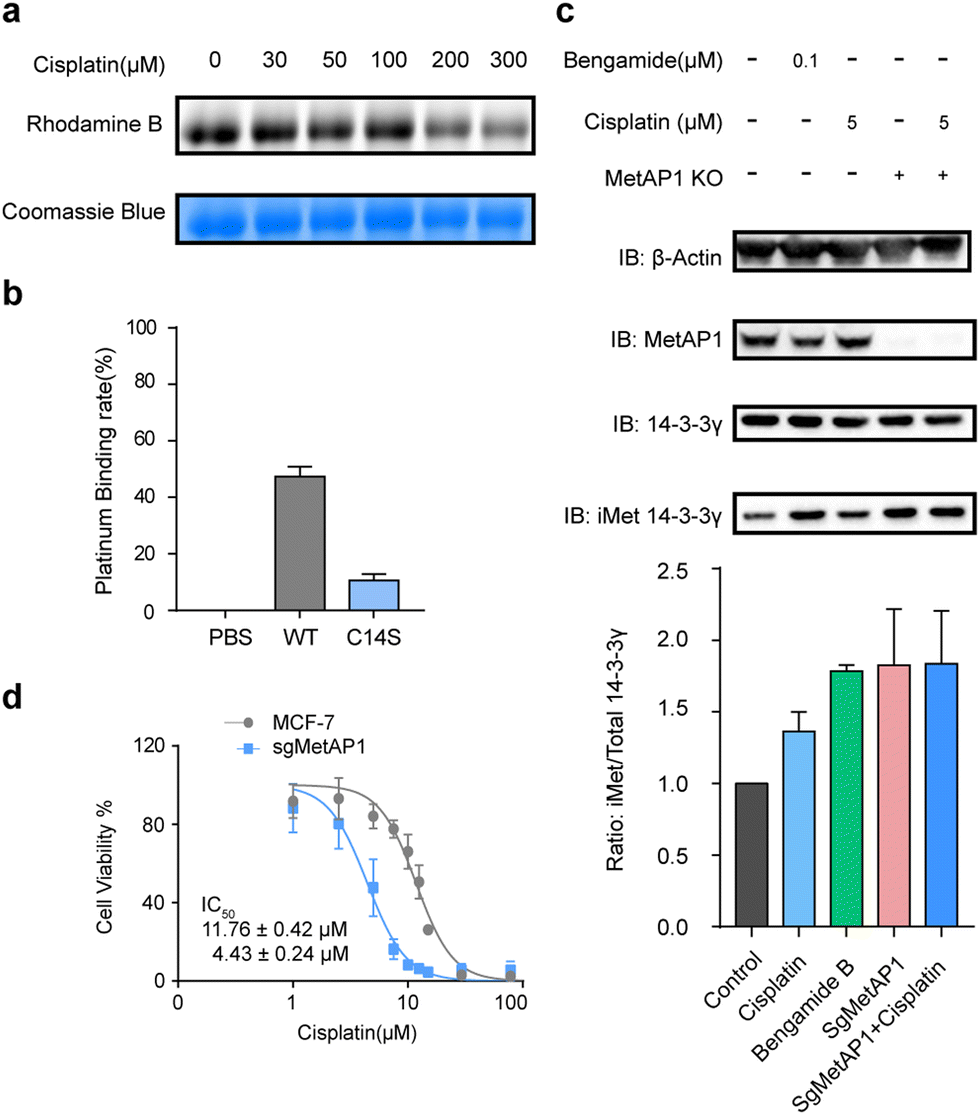 | ||
| Fig. 3 Cisplatin binds and inhibits human MetAP1. (a) Cisplatin competes with IAyne on MetAP1 dose dependently. (b) MetAP1 is a cisplatin-binding protein as measured by ICP-MS, whose binding site contains Cys14. (c) Inhibition of iMet processing of 14-3-3γ by cisplatin in human cell lines. The levels of iMet 14-3-3γ and the total 14-3-3γ were immunoblotted (top) and quantified by ImageJ (bottom). (d) Knockout of MetAP1 sensitized cells to cisplatin. Dose-dependent cell death induced by cisplatin was measured by MTT. Results are from three independent experiments. *p < 0.05, **P < 0.01, ***P < 0.001. Statistical differences were determined by a two-sided Student's t-test. | ||
We next explored the effect of cisplatin on the activity of purified MetAP1. Using an established MetAP1 activity assay39 that quantifies the release of Met from the heptapeptide MAHAIHY in the presence of zinc as the metal cofactor, we observed that cisplatin could not affect the activity-purified MetAP1 (ESI,† Fig. S4). The result is not completely surprising as the residual zinc ions in the assay buffer may block the binding of cisplatin by occupying the sites of the zinc finger motif. Also, the cisplatin-binding site is far away from the enzyme's catalytic center (within the domain spanning from His203 to Glu358) both in the primary sequence and 3D structure. Considering that the zinc finger motifs of MetAP1 play an important role of regulating MetAP1's activity in vivo,40 we suspected that cisplatin might affect the activity of MetAP1 in living cells. To test this hypothesis, we set up an assay to evaluate the effect of cisplatin on cellular MetAP1 activity by probing the degree of N-terminal initiator methionine (iMet) retention of the known MetAP1/2 substrate, 14-3-3γ (i.e. iMet-14-3-3γ).41 A commonly used inhibitor of MetAP1/2, bengamide B, was used to treat cells as a reference and to further rule out the possible interference from MetAP2, MetAP1 was knocked out by CRISPR-Cas9 (MetAP1-KO) to set the background activity of MetAP2. The results showed that both cisplatin and bengamide B could inhibit the activity of intracellular MetAP1, which looks to be the dominant MetAP activity in MCF-7 cells (Fig. 3c). Finally, the cell viability assays demonstrated that knocking out MetAP1 significantly increased the sensitivity of cells toward cisplatin treatment (Fig. 3d), suggesting that MetAP1 partially contributes to protect cells from cisplatin-induced death.
In summary, we report the application of competitive cysteinome profiling to identify cisplatin-binding cysteines in MCF-7 proteomes. Among the targets of cisplatin, we not only verified its binding sites in GLRX1 and TXNDC17, but also functionally characterized a novel target, MetAP1, in terms of cisplatin binding on the enzyme activity and cellular toxicity. While the former provided strong evidence that the thioredoxin and glutathione systems would be disturbed by cisplatin to impact the intracellular ROS level, the latter suggests that MetAP1 could serve as a potential target for improving cytotoxicity of cisplatin to avoid tumor resistance. Considering that the competitive labeling was performed in cell lysates and changes in cysteine ratios are not all directly caused by cisplatin, the resulting proteomic data by ABPP should be carefully interpreted. Further biochemical experiments are advised to confirm the targets as cisplatin-binding proteins before functional assay are applied. In addition, whether these targets are functionally impactful in vivo remain to be explored. Nevertheless, the ABPP-based chemoproteomic technology proves to be an enabling tool to systemically study protein-metal/metallodrug interactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Computing Platform of the Center for Life Science for supporting the proteomics data analysis and Analytical Instrumentation Center of College of Chemistry and Molecular Engineering, Peking University for ICP-MS analysis. This work is supported by the National Natural Science Foundation of China (No. 21925701 and No. 92153301) and the National Key R&D Program of China (2022YFA1304700) to C. W.References
- S. Ghosh, Bioorg. Chem., 2019, 88, 102955 CrossRef PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- S. R. Zhang, X. M. Zhong, H. Yuan, Y. Guo, D. F. Song, F. Qi, Z. Z. Zhu, X. Y. Wang and Z. J. Guo, Chem. Sci., 2020, 11, 3829–3835 RSC.
- S. G. Chaney, S. L. Campbell, B. Temple, E. Bassett, Y. B. Wu and M. Faldu, J. Inorg. Biochem., 2004, 98, 1551–1559 CrossRef CAS PubMed.
- H. M. Moon, J. S. Park, I. B. Lee, Y. I. Kang, H. J. Jung, D. An, Y. Shin, M. J. Kim, H. I. Kim, J. J. Song, J. Kim, N. K. Lee and S. C. Hong, Nucleic Acids Res., 2021, 49, 12035–12047 CrossRef CAS PubMed.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 CrossRef CAS PubMed.
- B. Michalke, J. Trace Elem. Med. Biol., 2010, 24, 69–77 CrossRef CAS PubMed.
- R. N. Bose, S. K. Ghosh and S. Moghaddas, J. Inorg. Biochem., 1997, 65, 199–205 CrossRef CAS PubMed.
- F. Arnesano and G. Natile, Semin. Cancer Biol., 2021, 76, 173–188 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, J. Am. Chem. Soc., 2009, 131, 14196–14197 CrossRef CAS PubMed.
- L. Banci, I. Bertini, O. Blazevits, V. Calderone, F. Cantini, J. F. Mao, A. Trapananti, M. Vieru, I. Amori, M. Cozzolino and M. T. Carri, J. Am. Chem. Soc., 2012, 134, 7009–7014 CrossRef CAS PubMed.
- J. Will, W. S. Sheldrick and D. Wolters, J. Biol. Inorg. Chem., 2008, 13, 421–434 CrossRef CAS PubMed.
- E. Moreno-Gordaliza, D. Esteban-Fernandez, C. Giesen, K. Lehmann, A. Lazaro, A. Tejedor, C. Scheler, B. Canas, N. Jakubowski, M. W. Linscheid and M. M. Gomez-Gomez, J. Anal. At. Spectrom., 2012, 27, 1474–1483 RSC.
- R. M. Cunningham and V. J. DeRose, ACS Chem. Biol., 2017, 12, 2737–2745 CrossRef CAS PubMed.
- X. Yuan, W. Zhang, Y. He, J. Yuan, D. Song, H. Chen, W. Qin, X. Qian, H. Yu and Z. Guo, Metallomics, 2020, 12, 1834–1840 CrossRef CAS PubMed.
- M. J. Niphakis and B. F. Cravatt, Annu. Rev. Biochem., 2014, 83, 341–377 CrossRef CAS PubMed.
- C. Wang and N. Chen, Acta Chim. Sin., 2015, 73, 657–668 CrossRef CAS.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–795 CrossRef CAS PubMed.
- C. Wang, E. Weerapana, M. M. Blewett and B. F. Cravatt, Nat. Methods, 2014, 11, 79–85 CrossRef CAS PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. Gonzalez-Paez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- R. Sun, L. Fu, K. Liu, C. Tian, Y. Yang, K. A. Tallman, N. A. Porter, D. C. Liebler and J. Yang, Mol. Cell. Proteomics, 2017, 16, 1789–1800 CrossRef CAS PubMed.
- D. W. Bak and E. Weerapana, Nat. Chem. Biol., 2023, 19, 356–366 CrossRef CAS PubMed.
- N. J. Pace and E. Weerapana, ACS Chem. Biol., 2014, 9, 258–265 CrossRef CAS PubMed.
- F. Yang, J. Gao, J. Che, G. Jia and C. Wang, Anal. Chem., 2018, 90, 9576–9582 CrossRef CAS PubMed.
- J. Szychowski, A. Mahdavi, J. J. Hodas, J. D. Bagert, J. T. Ngo, P. Landgraf, D. C. Dieterich, E. M. Schuman and D. A. Tirrell, J. Am. Chem. Soc., 2010, 132, 18351–18360 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- J. J. Gao, Y. Liu, F. Yang, X. M. Chen, B. F. Cravatt and C. Wang, J. Proteome Res., 2021, 20, 4893–4900 CrossRef CAS PubMed.
- S. Mirzaei, K. Hushmandi, A. Zabolian, H. Saleki, S. M. R. Torabi, A. Ranjbar, S. SeyedSaleh, S. O. Sharifzadeh, H. Khan, M. Ashrafizadeh, A. Zarrabi and K. S. Ahn, Molecules, 2021, 26, 2382 CrossRef CAS PubMed.
- Y. Kinoshita, T. Kalir, J. Rahaman, P. Dottino and D. S. Kohtz, Am. J. Pathol., 2012, 180, 375–389 CrossRef CAS PubMed.
- L. Messori and A. Merlino, Inorg. Chem., 2014, 53, 3929–3931 CrossRef CAS PubMed.
- I. Moraleja, E. Moreno-Gordaliza, M. L. Mena and M. M. Gomez-Gomez, Talanta, 2014, 120, 433–442 CrossRef CAS PubMed.
- S. Prast-Nielsen, M. Cebula, I. Pader and E. S. Arner, Free Radical Biol. Med., 2010, 49, 1765–1778 CrossRef CAS PubMed.
- E. S. Arner, H. Nakamura, T. Sasada, J. Yodoi, A. Holmgren and G. Spyrou, Free Radical Biol. Med., 2001, 31, 1170–1178 CrossRef CAS PubMed.
- L. J. Li, W. Guo, K. Wu, Y. Zhao, Q. Luo, Q. W. Zhang, J. A. Liu, S. X. Xiong and F. Y. Wang, Rapid Commun. Mass Spectrom., 2016, 30, 168–172 CrossRef CAS PubMed.
- S. G. Bernier, N. Taghizadeh, C. D. Thompson, W. F. Westlin and G. Hannig, J. Cell. Biochem., 2005, 95, 1191–1203 CrossRef CAS PubMed.
- X. Hu, A. Addlagatta, J. Lu, B. W. Matthews and J. O. Liu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18148–18153 CrossRef CAS PubMed.
- S. Zuo, Q. Guo and C. Ling, Mol. Gen. Genet., 1995, 246, 247–253 CrossRef CAS PubMed.
- A. Weiss, C. C. Murdoch, K. A. Edmonds, M. R. Jordan, A. J. Monteith, Y. R. Perera, A. M. Rodriguez Nassif, A. M. Petoletti, W. N. Beavers, M. J. Munneke, S. L. Drury, E. S. Krystofiak, K. Thalluri, H. Wu, A. R. S. Kruse, R. D. DiMarchi, R. M. Caprioli, J. M. Spraggins, W. J. Chazin, D. P. Giedroc and E. P. Skaar, Cell, 2022, 185, 2148–2163 CrossRef CAS PubMed.
- J. A. Vetro and Y. H. Chang, J. Cell. Biochem., 2002, 85, 678–688 CrossRef CAS PubMed.
- V. Jonckheere, D. Fijalkowska and P. Van Damme, Mol. Cell. Proteomics, 2018, 17, 694–708 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00042g |
| This journal is © The Royal Society of Chemistry 2023 |