
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical probes and methods for the study of protein arginine methylation
Tyler
Brown
,
Terry
Nguyen
,
Bo
Zhou
 and
Y. George
Zheng
*
and
Y. George
Zheng
*
Department of Pharmaceutical and Biomedical Sciences, University of Georgia, Athens, GA 30602, USA. E-mail: yzheng@uga.edu; Fax: +1-(706) 542-5358; Tel: +1-(706) 542-0277
First published on 28th July 2023
Abstract
Protein arginine methylation is a widespread post-translational modification (PTM) in eukaryotic cells. This chemical modification in proteins functionally modulates diverse cellular processes from signal transduction, gene expression, and DNA damage repair to RNA splicing. The chemistry of arginine methylation entails the transfer of the methyl group from S-adenosyl-L-methionine (AdoMet, SAM) onto a guanidino nitrogen atom of an arginine residue of a target protein. This reaction is catalyzed by about 10 members of protein arginine methyltransferases (PRMTs). With impacts on a variety of cellular processes, aberrant expression and activity of PRMTs have been shown in many disease conditions. Particularly in oncology, PRMTs are commonly overexpressed in many cancerous tissues and positively correlated with tumor initiation, development and progression. As such, targeting PRMTs is increasingly recognized as an appealing therapeutic strategy for new drug discovery. In the past decade, a great deal of research efforts has been invested in illuminating PRMT functions in diseases and developing chemical probes for the mechanistic study of PRMTs in biological systems. In this review, we provide a brief developmental history of arginine methylation along with some key updates in arginine methylation research, with a particular emphasis on the chemical aspects of arginine methylation. We highlight the research endeavors for the development and application of chemical approaches and chemical tools for the study of functions of PRMTs and arginine methylation in regulating biology and disease.
1. Protein methylation
Post-translational modifications (PTMs) of proteins represent a fundamental molecular mechanism for the modulation of physicochemical properties and biological functions of cellular proteins, ranging from stability to conformation, localization, enzymatic activity, and biomolecular interactions. So far, more than 400 different types of PTM markers have been identified in eukaryotic cells.1,2 Based on an analysis of the dbPTM database,2 methylation, mainly arginine methylation and lysine methylation, is listed as the fifth most frequent and ubiquitous PTM marker, following phosphorylation, acetylation, ubiquitination, and succinylation. Examination of the PhosphoSite database demonstrates that protein methylation occurs widely in the human proteome.3 Especially, arginine is the most frequently methylated residue in human proteins as compared to other methylated residues, including lysine, histidine, cysteine, glutamine, glutamate, and aspartate.4–10 In the PhosphoSite database, more than 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 arginine methylation sites are reported, with a ranging distribution across 4207 human proteins; in comparison, 5212 lysine methylation sites are found across 2814 proteins (Fig. 1). These numbers were recently expanded in an updated study by Shechter and coworkers to 15386 methylarginine sites amongst 5255 proteins.11 Almost certainly, these studies do not represent the full scope of arginine and lysine methylation in the human proteome as we would anticipate that more methylated substrates and sites will be uncovered and added to the PTM databases from future proteomic screenings.
000 arginine methylation sites are reported, with a ranging distribution across 4207 human proteins; in comparison, 5212 lysine methylation sites are found across 2814 proteins (Fig. 1). These numbers were recently expanded in an updated study by Shechter and coworkers to 15386 methylarginine sites amongst 5255 proteins.11 Almost certainly, these studies do not represent the full scope of arginine and lysine methylation in the human proteome as we would anticipate that more methylated substrates and sites will be uncovered and added to the PTM databases from future proteomic screenings.
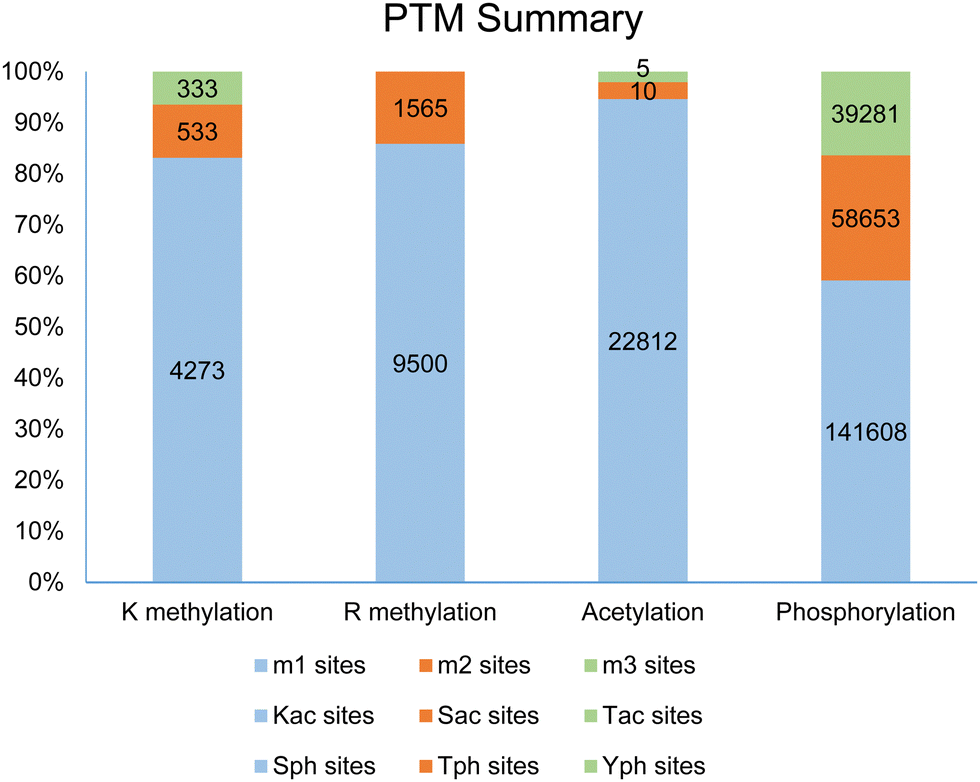 | ||
| Fig. 1 A survey of protein methylation, acetylation, and phosphorylation data in the PhosphoSite database. For lysine methylation, a total of 2814 methylated proteins containing 5212 methylation sites are curated, and the numbers of identified monomethyllysine, dimethyllysine and trimethyllysine sites are 4273, 533, and 333, respectively (data procured April 19, 2021)). For arginine methylation, a total of 4207 methylated proteins containing 11153 methylation sites are curated, and the numbers of identified monomethylarginine and dimethylarginine sites are 9500 and 1565, respectively. For protein acetylation, predominant acetylation sites are on lysine residues, with a small number of acetylations occurring on serine and threonine residues. Protein phosphorylation occurs mostly on serine residues, followed by threonine and tyrosine residues. The numbers of identified phosphorylation sites are much larger than those of methylation and acetylation; one reason accounting for this issue could be that protein phosphorylation is much more extensively studied than the other PTMs. | ||
Methylation of arginine and lysine residues on proteins exists in different stoichiometric states which are distinguished by the number of methyl groups covalently attached to the side chain amine of lysine or the side chain guanidine of arginine. The methylation of the ε-amino group of lysine residues can exist in three states: monomethyllysine (MMK), dimethyllysine (DMK), or trimethyllysine (TMK). Methylation of arginine residues can exist as NG-monomethylarginine (MMA, Rme1) or dimethylarginine (DMA, Rme2). In the DMA states, the two methyl groups can be bonded either to the same terminal ω-nitrogen of the guanidino group or to each of the two ω-nitrogens, resulting in either asymmetric NG,NG-dimethylarginine (ADMA, Rme2a) or symmetric NG,N′G-dimethylarginine (SDMA, Rme2s), respectively. Investigation of proteomic profiling studies shows that the numbers of monomethylarginine sites are much greater than dimethylarginine sites (e.g., 9500 MMA sites compared to 1565 DMA sites in the PhosphoSite database, Fig. 1).6,11–14 Similarly, the numbers of lysine methylation sites follows the order of monomethyllysine > dimethyllysine > trimethyllysine (e.g., 4273 MMK sites, 533 DMK sites, and 333 TMK sites in the PhosphoSite database).6,9 Existence of such a large number of monomethylarginine and monomethyllysine residues can be a reflection of the relatively low processivity of protein arginine N-methyltransferases (PRMTs) and protein lysine N-methyltransferases (PKMTs).15–18 At first glance, an interpretation of this phenotype can be that monomethylated arginine or lysine residues serve as an intermediate along the production path toward the formation of di- or tri-methylated residues. Nevertheless, studies also have shown that monomethylated states can exist as independent entities and may possess distinctive roles in protein regulation. A well-known example of functional differences between methylation states is that monomethyllysine 4 of histone H3 (H3K4me1) is often associated with enhancers in chromatin while H3K4me3 is correlated with active promoters.19,20 Also, unimodal H3K4me1 centered on translational start sites is a characteristic feature of the poised epigenetic state in embryonic stem cells and germ cells.21 For arginine methylation, a recent study shows that MMA proteins have ontological characteristics distinct from ADMA and SDMA proteins.11
Notably, the site diversity of a PTM in proteins can be different from its occurring abundance. This is practically witnessed in the case of arginine methylation. As aforementioned and supported by numerous MS-based proteomic screening studies, a much larger number of MMA sites are mapped out as compared to ADMA and SDMA sites in cellular proteins.4,6,9,12,22 On the other hand, the global abundance of MMA, ADMA, and SDMA in cells and tissues seem to be divergent from their methylation site diversity. The traditional measurement of the abundance of methylated arginines in proteins is based on spectrometric quantitation of individual amino acid peaks eluted from liquid chromatographic separation of digested proteins acquired either chemically or enzymatically. In an early study by Allfrey and coworkers, chromatographic analysis of amino acid hydrolysates showed that ADMA is the main methylated arginine derivative found in non-histone nuclear proteins in rat liver, while MMA is present only in trace amounts.5 Using protein hydrolysis and high-performance liquid chromatography, Bulau et al. found that 0.6–0.7% of total arginine residues in mouse heart and kidney protein hydrolysates are methylated, with ADMA being the dominant form (83% of total methylated residues) and SDMA occurring minorly (17% of total methylated residues, while MMA abundance was low and not detected.23 In that study, crude kidney extracts were shown to exhibit a significantly higher level of free ADMA and SDMA (0.037 nmol mg−1 and 0.033 nmol mg−1 protein, respectively), supporting that the kidney may be the main route for clearance of free ADMA and SDMA from the body.24 Chromatographic analysis by Clarke and coworkers showed that 0.45% of the total arginine residues in the proteins isolated from MEF cells are methylated, with an abundance distribution of 0.012% MMA, 0.4% ADMA, and 0.034% SDMA.25 Recently, nuclear magnetic resonance spectroscopy was applied to determine the global abundance of arginine methylation, proclaimed having a better accuracy than antibody enrichment and mass spectrometry.26 Testing of nine human cell lines showed that ADMA was the dominant methylarginine species in proteins (1–3.4% of total arginine residues), while SDMA/MMA levels were significantly lower: ∼10% of ADMA. Maron et al.11 used direct-injection mass spectrometry with the high-resolution Orbitrap Fusion Lumos to characterize the relative abundances of Rme2s and Rme2a. With that method, they found that Rme2a residues were approximately 20× higher than Rme2s in A549 lung adenocarcinoma cells and that Rme2s residues were ∼25% of the total Rme2 in the Xenopus cell-free egg extract. Altogether, the cellular ADMA abundance is higher than that of SDMA and MMA.
The difference between the diversity and the abundance of methylarginine implies that although a large number of monomethylated arginine (MMA) sites are found to exist in cellular proteins by MS/MS mapping, their abundance level likely is quite low as compared to ADMA. This is supported by the overall low abundance of MMA observed in the chromatographic analysis of protein hydrolysates. Regardless of the global methylation abundance, we also emphasize the need for quantitative studies for measuring the abundance ratios of different arginine methylation states in specifically methylated proteins. This is necessary and essential as differentially methylated states of individual proteins can be quite divergent from the global abundance ratios and may also vary according to cellular contexts. For example, in myelin basic protein (MBP), 2% of arginine residues are MMA and 1.5% are SDMA, while ADMA is not detectable.23,27 From the perspective of metabolic effects, the high abundance of ADMA residues in proteins seems to be in good alignment with the long observed physiological effect of the end products of protein arginine methylation: it is the free ADMA molecule (not MMA or SDMA) that is abundant in plasma and acts as a major regulator of NO synthesis and endothelial functions.28,29
2. Protein arginine methyltransferases (PRMTs)
Although it seems to be commonplace today that arginine methylation is one of the most prominent PTMs and is functionally important for eukaryotic organisms, the field has taken many years to get into shape. A particular delay is that it took more than 30 years for scientists to discover the genes that encode protein arginine methyltransferases since the initial discovery of protein arginine methylation in the 1960s. The methylation of the guanidinium group of arginine residues in proteins was first recognized in 1967 by Paik and Kim who studied the methylation of calf thymus nuclear histones using radioactive S-adenosyl-L-methionine (AdoMet, SAM) that was labeled with C-14 on the methyl group to be transferred: 14C-SAM. Following acidic hydrolysis, paper chromatography, and radioactive ninhydrin detection, two unknown methylated amino acid peaks near arginine were found.30 A year later, Paik and Kim determined that one of the methylated amino acid peaks near arginine in calf thymus nuclear histones was MMA (evidenced by observed 14C-methylurea).31 In that paper, Paik and Kim also partially purified an enzyme from the calf thymus named “protein methylase I” that was capable of methylating histones. Friedman et al.32 in 1969 observed methylated arginines from acid-hydrolyzed proteins in the nuclei of rat liver cells, and Nakajima et al.33 in 1971 isolated and identified NG-monomethyl, NG,NG-dimethyl- and NG,N'G-dimethylarginines from the protein hydrolysates of bovine brains, which provides direct evidence for the existence of arginine methylation in vivo. Ever since the discovery of arginine methylation, scientists have been fervently looking for the enzymes that catalyze arginine methylation.34 For example, Kim and coworkers have partially purified two arginine methyltransferases from calf brain: one with particular substrate specificity for histones, and the other for myelin basic protein.35 The histone-specific arginine methyltransferase was found later to methylate the heterogeneous nuclear ribonucleoprotein particle (hnRNP) protein A1, producing both MMA and ADMA.36 However, prior to 1996, the molecular identities of the protein arginine methyltransferases were not established, and no genes encoding for arginine methyltransferases had been found.37A breakthrough in the arginine methylation field occurred in 1996 when the groups of Clarke and Herschman identified and cloned the first protein arginine methyltransferase gene in rats that was named PRMT1.38 In this paper, the authors found that the immediate early gene product TIS21/BTG1 interacts with PRMT1 and enhances its methyltransferase activity, which provided the first example of PRMT activity regulation. Later on, Clarke and coworkers showed that PRMT1 contributed to the majority of arginine methylation in mammalian cells.39 At about the same time as mammalian PRMT1 discovery in 1996, the first PRMT enzyme in the yeast Saccharomyces cerevisiae was discovered independently by the groups of Clarke and Herschman37 and by the Silver group,40 which was named RMT1 or Hmt1p by the authors. RMT1/Hmt1p is homologously similar to mammalian PRMT1 and has a strong activity in methylating the poly(A) + -RNA-binding proteins hnRNAP A1 and Npl3p. Interestingly, yeast Hmt1p/RMT1 is not essential for normal cell growth, but cells missing Hmt1p and also bearing mutations in the mRNA-binding proteins Npl3p or Cbp80p can no longer survive.41
Through sequence homology search followed by biochemical activity validation, nine PRMT members have been discovered consecutively to exist in mammalian cells, all of which belong to the class I of SAM-dependent methyltransferases and bind SAM with a characteristic Rossmann fold.42–47 Based on their end-methylation products, PRMT1, -2, -3, -4, -6, and -8 are grouped into type I enzymes that catalyze methylation of arginine residue to produce MMA and can further methylate MMA to produce ADMA (Fig. 2).38,44,48–52 PRMT4 is also known as coactivator-associated arginine methyltransferase 1 (CARM1). PRMT5 and PRMT9 are grouped into type II enzymes that produce MMA and SDMA.53–55 PRMT7 is grouped as a type III enzyme that only catalyzes the conversion of an arginine residue to MMA.56,57
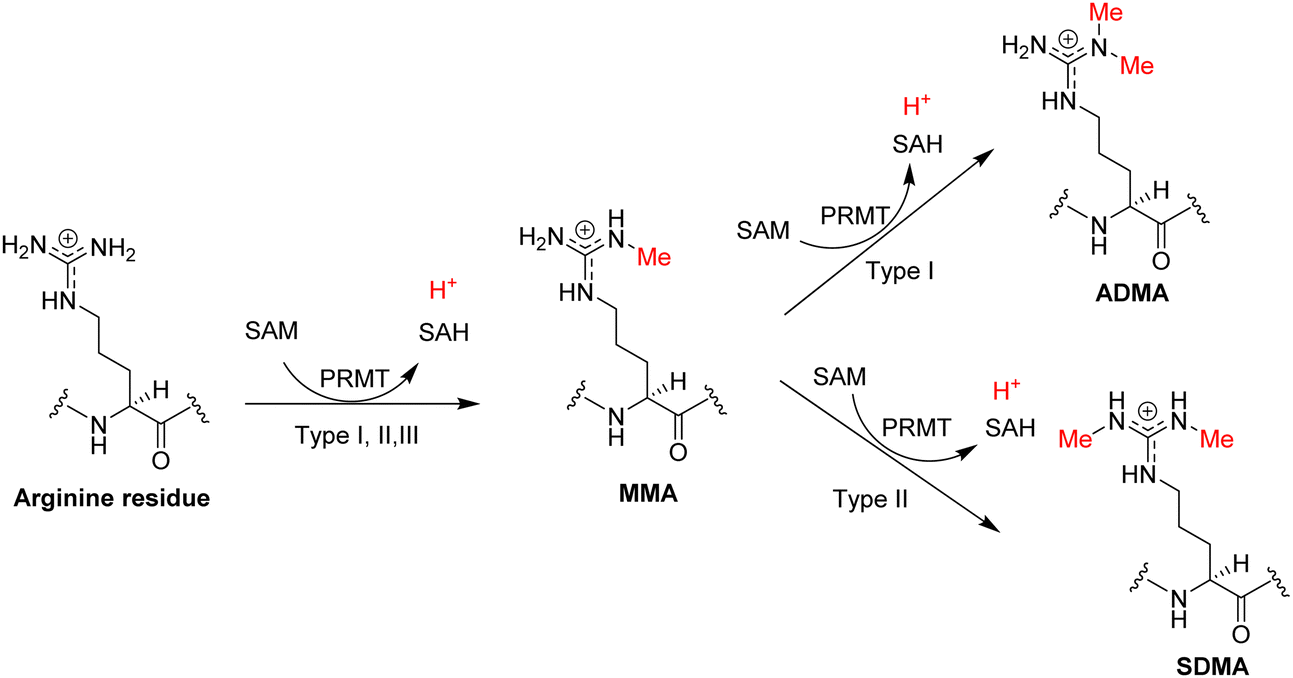 | ||
| Fig. 2 Arginine methylation reaction. The PRMT proteins are generally classified into three types according to their catalyzed methylarginine products: MMA, ADMA, and SDMA. | ||
Among the nine PRMT members, PRMT1 makes a dominant contribution to the arginine methylation levels in eukaryotic cells.25,58 In a Rat1 cell lysate model, immunodepletion of PRMT1 almost completely abolishes total protein methylation signals.39 Knockout of the PRMT1 gene in MEF cells leads to a 52–58% loss of the normal steady-state levels of ADMA.25,59 Treatment of A549 cells with the PRMT1 inhibitor MS023 reduced the ADMA level by ∼50%, as examined by western blotting.11 Protein extracts from spleens of mice showed a 78–86% reduction in ADMA after MS023 treatment.14 Another study using NMR quantification revealed that MS023 reduced the ADMA level in HeLa cells by 68%.26 In agreement with the predominant role of PRMT1 in mammalian cells, a study in Saccharomyces cerevisiae showed that Hmt1p accounts for 89% of ADMA and 66% of MMA in proteins, demonstrating its predominant role for arginine methylation in yeasts.37 NMR analysis of methylarginines in S. cerevisiae showed that deletion of Hmt1p nearly completely abolishes ADMA and MMA levels (SDMA is undetectable in that study).26 Taking into account the leading contribution of PRMT1 in arginine methylation which is more abundant than all the other types of protein methylations, we may draw the conclusion that PRMT1 is the dominant methyltransferase for total protein methylations in eukaryotic organisms.
PRMT5 accounts for the vast majority of the type-II arginine methylase activity and knockout of PRMT5 in MEF cells abolished ∼95% of SDMA signals.60 In A549 cells, inhibition of PRMT5 with inhibitor GSK591 abolished SDMA signals down to below the threshold of detection.11 An NMR study showed that GSK591 reduces the SDMA and MMA levels in HeLa cells by 72%.26 In comparison to PRMT5, the other type-II member PRMT9 only plays a very minor role in SDMA formation.60 In MEF cells, PRMT9 is only responsible for a small percentage of SDMA production (∼5%), including the methylation of the splicing factor SF3B2 (SAP145).55,60
Except for the nine classical PRMT members, two other proteins are reported of possessing arginine methylation activity. NADH:ubiquinone oxidoreductase complex assembly factor 7 (NDUFAF7, Q7L592) was found to symmetrically dimethylate a highly conserved Arg-85 in NADH: ubiquinone oxidoreductase core subunit S2 (NDUFS2), which is a subunit of mitochondrial complex I.61–63 Methylation of Arg-85 in NDUFS2 results in the release of NDUFAF7 and promotes the protein–protein interaction with NDUFS7 for pre-assembly of active complex I.62 Superimposition of the PRMT5 crystal structure with mitochondrial dysfunction protein A (MidA), an ortholog of NDUFAF7 in Dictyostelium discoideum, revealed certain structural similarities and identified conserved residues for SAH binding.64 We superposed the AlphaFold structure of NDUFAF7 with the human PRMT5 structure (PDB ID: 5FA5) and observed good overlap in the Rossmann fold domain (Fig. 3). Docking of SAH into the NDUFAF7 structure showed interactions of the purine ring with F217, the ribose portion with E156, as well as hydrogen bonding interactions of the carboxylate with T89 and the ammonium group with G124. We also docked a prototypical arginine residue (Ac-R-NHMe) into NDUFAF7. Within the arginine-binding pocket, the guanidino group was observed to form interactions with E216, D219, and D330, with E216 and D219 taking the form of a pseudo-double-E loop typically present in PRMTs. The guanidino group also formed a pi–cation interaction with F87. This interaction is an important structural feature as NDUFAF7 has been reported to have the same type-II activity as PRMT5, which contains a conserved phenylalanine residue in the arginine binding pocket that has been shown to be important for the selectivity of SDMA over ADMA formation.65
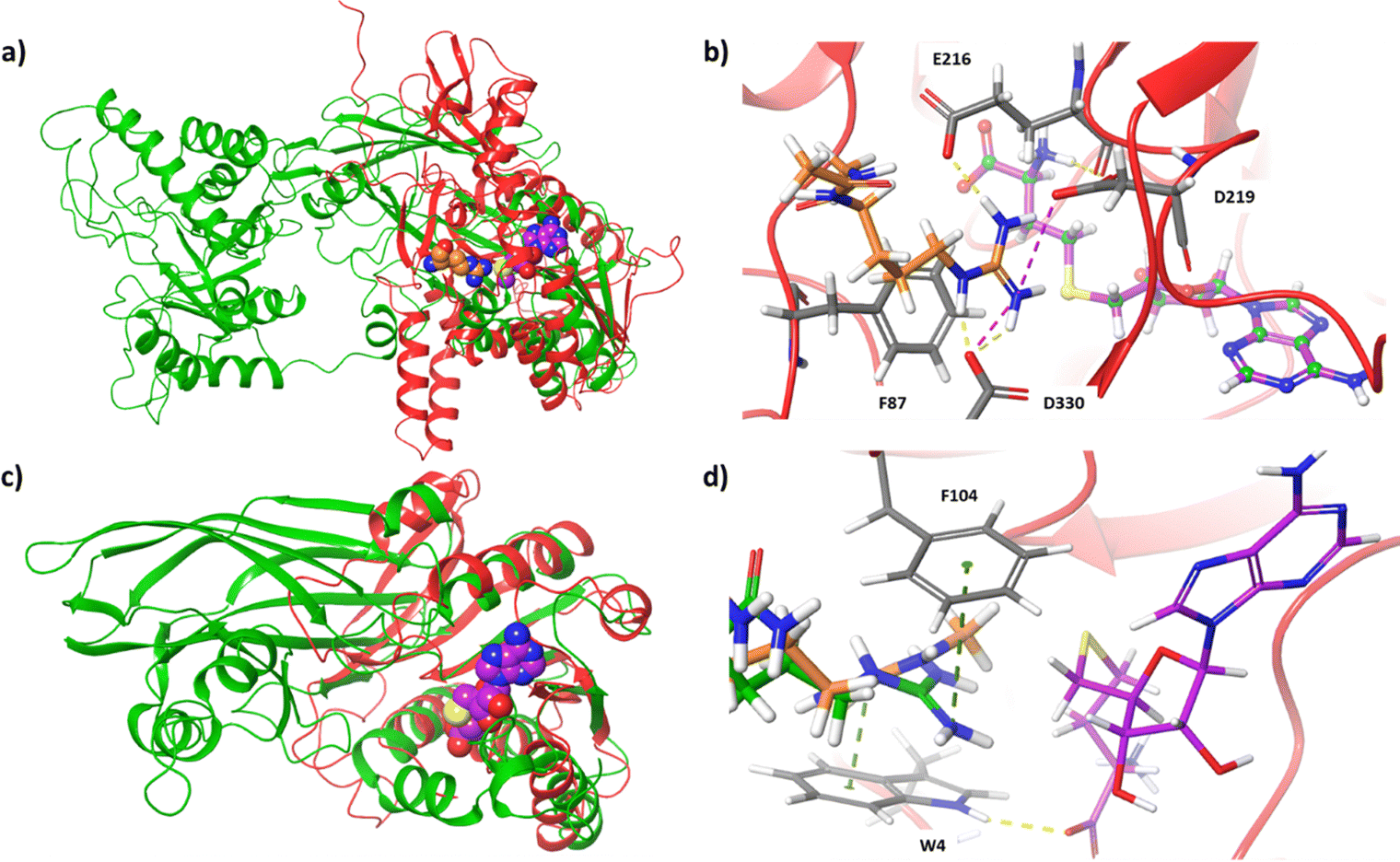 | ||
| Fig. 3 Structural analysis of NDUFAF7 and Mettl23. (a) Structural overlay of PRMT5 (PDB ID: 5FA5, green) with NDUFAF7 (AlphaFold, red). The crystal structure ligands MTA (purple) and arginine (orange) demonstrate the active site. (b) Docked pose of arginine (orange) and SAH (purple) within NDUFAF7 active site. (c) Structural overlay of PRMT1 (PDB ID: 6NT2, green) with Mettl23 (AlphaFold, red). The crystal structure ligand, SAH (purple), determines the active site. (d) Docked poses of a tripeptide containing arginine (green), MMA (orange), and SAH (purple) within the active site of Mettl23. | ||
METTL23 is a class I methyltransferase member that was reported to catalyze the asymmetric dimethylation of histone H3 Arg-17 (H3R17me2a) in mouse oocytes.66 METTL23-catalyzed H3R17me2a was demonstrated to support histone H3 deposition and DNA demethylation for paternal reprogramming. The protein alignment of PRMT1-8 with METTL23 reveals some sequence similarities in the methyltransferase core.66 However, METTL23 (Q86XA0) is only 199 amino acids long, which is much shorter than the catalytic domain of typical PRMTs (∼310 residues). Therefore, it is doubtful whether METTL23 possesses authentic enzymatic activity of an arginine methyltransferase. We modeled the AlphaFold structure of METTL23 using PRMT1 (PDB ID: 6NT2). A good overlap is clearly seen within the Rossmann fold domain of PRMT1 (Fig. 3). With a size two-thirds of typical type-I PRMTs, METTL23 lacks the β-barrel domain. Docking of SAH into the Mettl23 structure adopts a pose similar to SAH in the PRMT1 crystal structure. Upon first glance at the pocket, no double-E loop motif is observed. We docked a tripeptide derived from H3, Ac-APR-NHMe, into the structure of METTL23. Interestingly, the arginine residue does dock with a suitable orientation within the pocket; however, rather than interacting with two acidic residues, the guanidino group interacts with D101 and is also sandwiched between W4 and F104 to form pi–cation interactions. Investigation of the predicted arginine binding pose and protein surface also provides some insight into how this enzyme may produce ADMA over SDMA. The pocket itself around the guanidino group is asymmetrical, with more space closer to the sulfur of SAH and less space approaching D101. When bound, the guanidino group appears to be closely associated with D101 through ionic interaction, with the nitrogen to be methylated pointing towards SAH with a distance of approximately 4 Å. After methylation, the steric bulk of the methylated terminal nitrogen has increased; therefore, the monomethylated arginine substrate would bind to METTL23 in such a way that the less bulky terminal nitrogen is pointed towards D101 (away from SAM) to place the bulkier terminal nitrogen into the more open portion of the pocket (towards SAM). It will be interesting to see how substrate specificity and protein–protein interactions of METTL23 are achieved.
3. Biological functions of arginine methylation
Given the large abundance and widespread nature of arginine methylation in the eukaryotic proteome, it is unsurprising that PRMTs and arginine methylation regulate diverse biological processes. Knockout of PRMT1 or PRMT5 was proved to be fatal in new mice, demonstrating that these genes are essential for the organism's development. Quite a few comprehensive review articles have been dedicated to discussing the regulatory functions of PRMTs in normal cellular physiology and in disease processes, and readers are referred to these articles for detailed coverage.67–79 This article is geared more toward providing an account of chemical approaches and tools in the study of arginine methylation and thus does not tend to discuss the biological functions of arginine methylation.A striking feature of arginine methylation is that the majority of arginine methylated substrates are nucleic acid-binding proteins and thereby are involved in RNA or DNA-participated processes.73 This feature somehow is expected because the arginine side chain is nitrogen-rich and positively charged, which is an ideal functional group for interaction with nucleic acids that contain phosphate and heteroatomic purines and pyrimidines. Indeed, arginine residues are abundantly enriched in nucleic acid-binding proteins.80
Accumulating amounts of preclinical and clinical evidence points out the intimate connection of PRMTs with multiple human disease states, especially cancer.43,81–84 Generally, PRMT expression and arginine methylation are upregulated in cancer, the reason for which is unknown. We notice that one round of arginine methylation releases one proton ion (Fig. 2), indicating that increased PRMT expression and activity could provide a proton source to acidify the intracellular milieu. Proton-forming biochemical reactions have been proposed as a molecular mechanism to counteract excessive alkaline stress in tumorous cells and chronic inflammation. Therefore, upregulation of arginine methylation may play a functional role in maintaining cellular acid–base homeostasis.85 Future studies are warranted to address this hypothesis.
4. Biochemical assays of arginine methylation
To address PRMT functions in physiology and pathology, many efforts have been made to develop versatile or specific assay tools for PRMTs.86–89 The study of arginine methylation depends on sensitive and quantitative assays for PRMT activity detection and quantitation. The investigation of arginine methylation in protein samples extracted from lysed cells or animal tissues mainly is achieved by western blotting, total MS, or MS/MS analysis of digested peptides. On the other hand, in vitro assays of measuring the enzymatic activities of purified PRMT proteins can be designed with greater diversity and flexibility. These assays are important for characterizing the activities of different forms of PRMTs and for quantitatively testing and screening PRMT inhibitors. We highlight herein the reported biochemical assays for PRMT activity measurement (Fig. 4).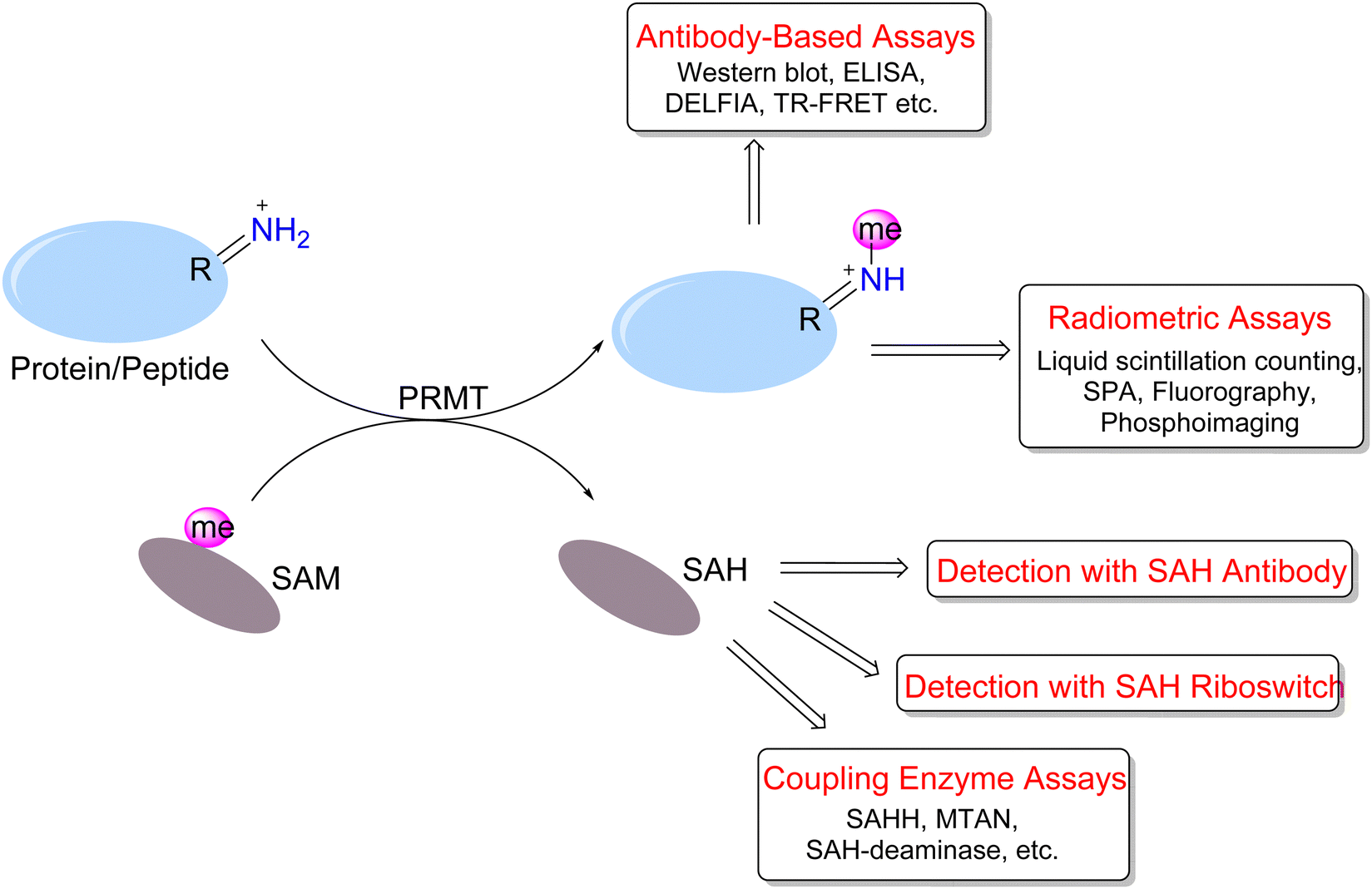 | ||
| Fig. 4 Different forms of biochemical assays for PRMT activity measurement. | ||
A number of biochemical assays have been designed for PRMTs and other methyltransferases (Fig. 4).90–98 The radiometric assays represent a gold standard for quantitating in vitro methyltransferase activities of PRMTs owing to their superior sensitivity and protocol straightforwardness.99 The principle is that PRMTs transfer the radioisotope-labeled methyl group from [3H]-SAM or [14C]-SAM to their peptide or protein substrates, and methylated products are detected based on their incorporated radiation amounts. The common protocol is that reaction mixtures are resolved on an SDS-PAGE gel and then methylated proteins/peptide products are visualized and quantitated using fluorography or phosphorimaging.100–106 Albeit having the advantage of visualization and sensitivity, the gel imaging assay takes overnight or longer for films to be adequately sensitized for image production.
For the detection of methylated substrates using liquid scintillation counting, different approaches have been developed to separate the products from unreacted SAM in the reaction mixture. Glass fiber or phosphocellulose filter paper is commonly used for affinity isolation of positively-charged peptides, for example, histones or RGG-rich peptides.103,107–120 These materials are quite cheap and have large surface areas suited for bulk peptide binding. The Hevel group developed a pipette chromatography method (ZipTip assay) for peptide product isolation which has the advantage of easy operation and fast assay speed and is particularly useful for analyzing small volumes of reaction.121 To circumvent the washing step in radiometric assays for throughput improvement, an ideal method is the scintillation proximity assay (SPA) in which scintillation signals are generated from the micrometer proximity of biotinylated substrates to streptavidin-coated scintillant microsphere beads (either FlashPlates or streptavidin-coated microscopic beads).109,122–133 SAM molecules in the bulk solution do not need to be removed because they fall out of the SPA distance and do not produce any scintillation signals.
Immunosorbent assays are widely used for detection and quantitation of protein arginine methylation. The quality of the methylarginine-specific antibodies has to be ensured. A typical format of these assays is the enzyme-linked immunosorbent assay (ELISA),100,134,135 in which methylated substrates are adsorbed onto microplate wells and incubated with a primary antibody that is further probed with horseradish peroxidase (HRP)-labeled secondary antibody. After washing out the unbound antibodies, chemiluminescence is developed from the HRP. The dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA)101,136–138 is similar to the ELISA, except that the antibody is labeled with a lanthanide probe instead of HRP. The lanthanide dissociates from the antibody by the addition of an enhancement cocktail and exhibits amplified fluorescence when excited at 340 nm. The lanthanide fluorophore features a large Stokes shift, a long decay time and a narrow emission spectrum, which together minimizes the interference of background fluorescence. Other assay methods involving lanthanide labels are homogeneous (no-wash) technologies that include time-resolved fluorescence resonance energy transfer (TR-FRET)139–142 and AlphaLISA.139,143 In the TR-FRET assay, energy is transferred from the donor europium chelate to the acceptor fluorophore within the FRET distance (∼10 nm). For AlphaLISA, donor beads convert ambient oxygen to singlet oxygen when excited at 680 nm, which can diffuse about 200 nm in solution. Then, the europium in the acceptor bead within this range receives energy and emits fluorescence.
In addition to the direct detection of methylated proteins/peptides, the production of SAH or the consumption of SAM provides a means for methylation measurement. Graves et al.144 employed anti-SAH antibodies and fluorescein–SAH conjugate tracers to measure SAH generated as a result of methyltransferase activity. The produced SAH competes with the tracer in the antibody/tracer complex. The release of the tracer results in a decrease in fluorescence polarization. The antibody had more than a 150-fold preference for binding SAH relative to SAM. The limit of detection was approximately 5 nM (0.15 pmol) SAH in the presence of 3 μM SAM. Time- and enzyme-concentration-dependent decreases in fluorescence polarization were observed in the catechol-O-methyltransferase (COMT) assay. In principle, this method is applicable to PRMT activity measurement.
Detection of SAM and SAH can be achieved by riboswitches (RNA aptamers) that selectively bind to SAM or SAH and discriminate against each other.145,146 The Hammond group developed a series of RNA aptamers that selectively bind SAH and trigger fluorescence biosensing.147 Its use in enzyme activity measurement was demonstrated for DNA and protein methyltransferases. Scientists at BellBrook Labs have developed an AptaFluor® SAH methyltransferase TR-FRET assay that is based on RNA aptamer binding of SAH. The riboswitch binds SAH with nanomolar affinity and exquisite selectivity. They split the riboswitch into two halves: one half is labeled with a terbium chelate and the other with DyLight® 650. SAH binding induces the assembly of a trimeric complex which generates a TR-FRET signal. This mix-and-read assay is robust, sensitive, histone acetyltransferase (HAT) compatible, and could be applied to different methyltransferases.
Quite a few assays have also been developed to detect the side product SAH through enzyme-coupled reactions. Such enzyme-driven coupling assays attempt to convert SAH into derivatives possessing colorimetric, fluorescence, or luminescence properties. In the SAH hydrolase (SAHH)-coupled assay, SAH is hydrolyzed into adenosine and homocysteine, the latter of which subsequently reacts with the fluorogenic probes ThioGlo or CPM, generating blue fluorescence.128,148–150 Scientists at BellBrook Labs used adenosine kinase to generate AMP from adenosine, and the formed AMP is detected by a competitive fluorescence polarization immunoassay.151,152
In another approach, SAH is converted into adenine by SAH/5′-methylthioadenosine nucleosidase (SAHN/MTAN) and then to hypoxanthine by adenine deaminase (ADA). The methylation process is monitored by the absorbance difference of adenine and hypoxanthine at 265 nm.153 G-Biosciences further developed an assay kit (Cat. # 786-430) that introduces Xanthine oxidase converting hypoxanthine into urate and hydrogen peroxide (H2O2), the latter of which is spectroscopically measured at 510 nm with the colorimetric reagent 3,5-dichloro-2-hydroxybenzenesulfonic acid (DHBS). Luo and coworkers implemented a biochemical strategy by adding coupling enzymes that convert adenine to AMP to ATP so that luciferase-linked bioluminescence can be generated.154 The luciferase coupling assay is featured for its ultra-sensitivity and quantitative linear response to SAH. The same assay format was also designed for characterizing DNA methyltransferases.155
The Shechter group has developed a simple coupling assay for SAH measurement using the SAH-deaminase TM0936.156 This enzyme converts SAH to S-inosyl-L-homocysteine (SIH), the process of which can be monitored by absorbance at 263 nm. The utility of TM0936 for several methyltransferases including PRMTs was demonstrated in 96-well plate format with excellent high-throughput performance. Furthermore, the authors substituted SAM with 8-aza-SAM as the methyl donor. Both 8-aza-SAM and 8-aza-SAH are fluorescent at 360 nm, but the end product 8-aza-SIH upon the action of TM0936 is a poor fluorophore. Thus, the loss of fluorescence emission at 360 nm was a quantitative indicator of the methyltransferase reaction. Compared to other coupling methylation assays, this method only utilizes a single coupling substance and thus is experimentally more straightforward.
There are other types of assay approaches that are important for elucidating protein arginine methylation on specific substrates or in the proteomic scope. The classic method of amino acid hydrolysis and the subsequent analysis of methylated amino acids using different chromatography approaches play a critical role in analyzing types and levels of arginine methylation in different protein and peptide samples.98 Furthermore, mass spectrometry techniques (MS and MS/MS) have become more and more sensitive and accurate and find significant use in distinguishing and quantifying MMA, SDMA, and ADMA markers in cellular proteins.157
In addition to the above-mentioned methods for studying steady-state reactions of arginine methylation, a few biochemical probes and assays were designed to investigate the pre-steady state of arginine methylation. PRMT1 is reported to methylate glycine and arginine-rich (GAR) motifs and histone H4 at the R3 site.158 Based on this, the Zheng laboratory designed and synthesized fluorescent probes for PRMT1 using a fluorescently labeled GAR-rich peptide (R4-FL) and a labeled H4 peptide.159 These fluorescent peptides are used as chemical probes to examine the substrate binding and methyltransferase activity of PRMT1. In their work, fluorescein was selected as a reporter group because it has long absorption and emission wavelengths and its photophysical properties are sensitive to the local micro-environment. Both fluorescence intensity and anisotropy can be used to measure the binding of R4-FL and H4-FL with PRMT1. These two probes are effective in measuring the association constants of PRMT1 with its nascent substrates through competitive binding assays and were shown to effectively manifest enzyme–substrate interactions. Of particular interest is that the fluorescence intensity of R4-FL or H4-FL is sensitive to the progression of their binding with and methylation by PRMT1, supporting that it is a useful fluorescent probe to investigate the transient kinetic mechanism of PRMT catalysis. Indeed, based on the stopped-flow fluorescence data, the authors concluded that PRMT1 catalyzes H4 methylation via a multiple-step process including an ultra-fast substrate-binding step, then a modestly fast formation of the ternary PRMT1 complex, and the methyl transfer is the rate-limiting step for arginine methylation.160 Later on, they combined the stopped-flow data of H4-FL fluorescence and intrinsic tryptophan fluorescence of PRMT1 and were able to generate a transient kinetic model of PRMT1 catalysis.16 Further, they applied the stopped-flow fluorescence approach as a homogeneous, continuous assay for PRMT1 inhibitor detection and characterization.161 Except for quantitatively determining the potency (IC50) of PRMT1 inhibitors, the approach also distinguishes between different modes of inhibition: i.e., cofactor-competitive, substrate-competitive, and mixed-type.
5. Small molecule covalent PRMT inhibitors
The pathological significance of PRMTs has stimulated growing enthusiasm for designing and screening chemical inhibitors to target individual PRMT members for therapeutic development. A number of small-molecule, reversible organic inhibitors have been developed to target PRMTs, and there are several extensive reviews published discussing the attributes and uses of PRMT inhibitors.71,76,95,162 Readers are referred to these references for more information. It is necessary to highlight that PRMT inhibitors not only have therapeutic potential, but also have significant value to basic research in PRMT biology: potent, selective, cell-active compounds with well-defined mechanisms of action are powerful chemical genetic tools to investigate biological roles and mechanisms of individual PRMT enzymes.Reported PRMT inhibitors can be classified into SAM competitive,163 substrate competitive, and allosteric inhibitors, based on the binding mode of these compounds. So far, there is no clear evidence of which type of PRMT inhibitors will have more preferential benefit as disease therapeutics. Taking into account the micromolar to millimolar concentrations of SAM inside cells and the submicromolar potency of SAM and SAH in PRMT binding, nascent PRMT proteins in cells likely exist in SAM- or SAH-bound forms. In this regard, inhibitors that have substrate competitive and SAM uncompetitive nature may have a mechanistic advantage to prohibit PRMT activity because they bind to SAM/SAH complexed PRMT forms directly. On the other hand, SAM-competitive inhibitors may have to compete with endogenous SAM and SAH for PRMT binding, and thus their potencies could be influenced by SAM/SAH concentration variations under varying biological contexts. Allosteric inhibitors of PRMTs may have the advantage of targeting both free PRMT forms and SAM/SAH bound forms.
Apart from the mainstay reversible binding inhibitors, covalent ligands have been made for PRMT inhibition. An early example of such inhibitors was investigated by Dillon et al. in 2012.164 The discovery was made through a competitive activity-based protein profiling (ABPP) assay,165 whereby a reactive cysteine residue C101 close to the SAM-binding pocket of PRMT1 was selected to be the target residue for covalent bond formation.166 The assay utilized an activity-based fluorescence polarization approach with an AlexaFluor-maleimide probe (maleimide–AF488) as a reporter for targeting PRMT1 and a C101A mutant was used as a control. The authors found that a significantly stronger signal was observed with the wild-type PRMT1 compared with the C101A mutant after incubating with the probe. Screening of 16000 compounds was conducted and yielded two inhibitors: CID 5380390 and CID 2818500 (Fig. 5). Both inhibitors contain a nitroalkene moiety that was believed to serve as an electrophile to react with Cys101 that is only conserved in PRMT1 and PRMT8. A full mechanism of action study and full biochemical characterization of these compounds has not yet been done; however, due to their small size, it is possible that they could serve as a scaffold for the future development of more specific covalent PRMT1 inhibitors.
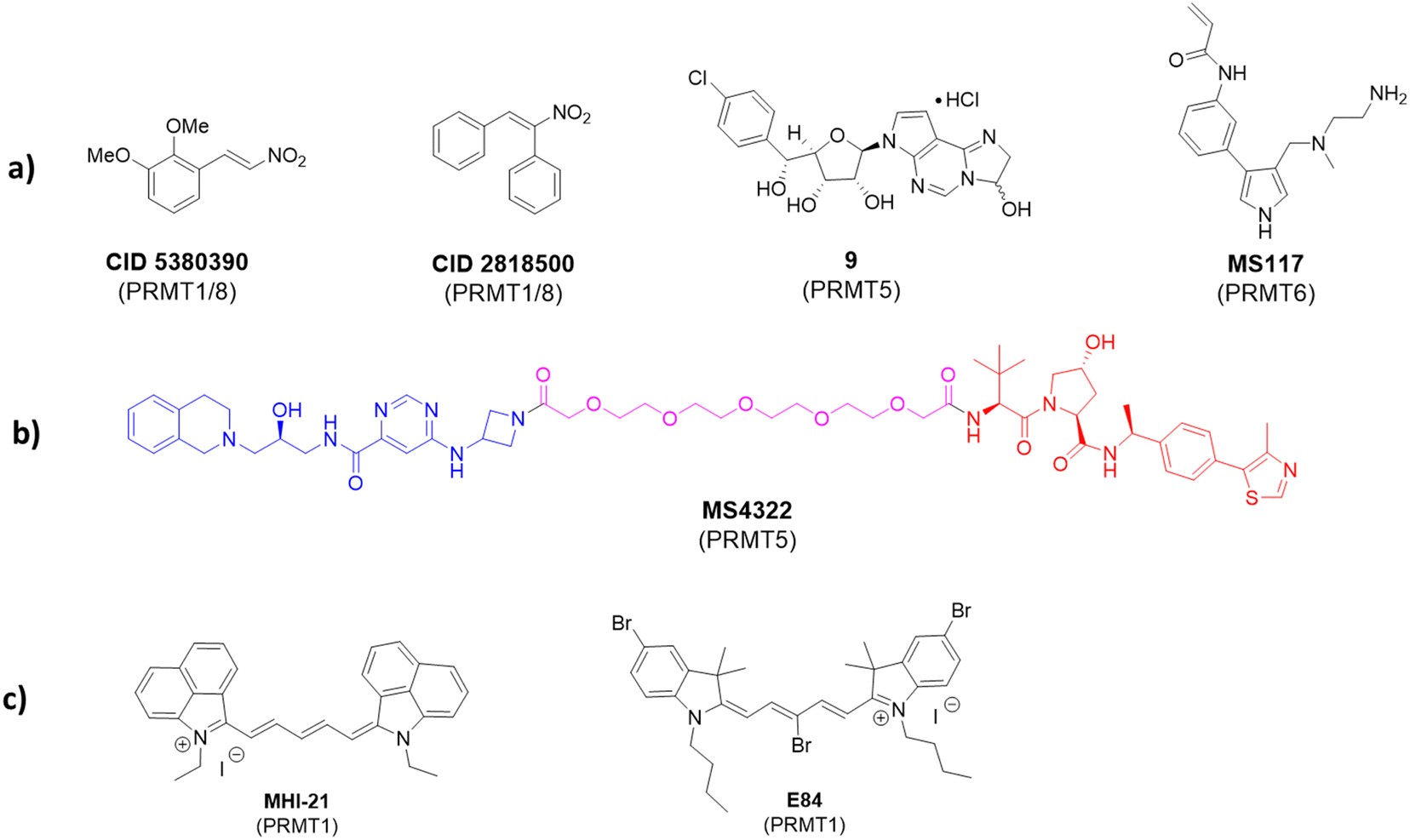 | ||
| Fig. 5 Chemical probes of PRMTs with different properties. (a) Covalent inhibitors designed for PRMTs. Red color highlights reactive groups for covalent reaction with PRMTs. (b) A degrader of PRMT5 containing PRMT5 ligand (blue), linker (purple), and VHL E3 Ligase ligand (red). (c) Long-wavelength fluorescent compounds that inhibit PRMTs. | ||
The approach of utilizing a reactive cysteine residue is common in covalent inhibitor design and is not unique to PRMT1 design. Within PRMT5, there is also a reactive cysteine close to the SAM pocket (C449) near the N6 amino group on the purine ring. This residue is unique to PRMT5, whereas in other PRMTs, the homologous residue is a serine. Lin et al. envisioned this as a unique opportunity to develop covalent inhibitors of PRMT5.167 In their approach, the authors designed an analog based on the previously developed LLY-283 as a starting point. A derivative containing a hemiaminal cyclized between N1 and N6 of the adenine ring (compound 9, Fig. 5) was synthesized. This hemiaminal was believed to open to provide an aldehyde under physiological conditions that would then be able to react with C449. Both the hemiaminal and the free aldehyde were assessed in biochemical tests and showed comparable IC50 values of 11 nM and 19.5 nM, respectively.167 In addition, the koff rate was measured for the hemiaminal in both wild-type and C449S mutant, with the wild-type having a significantly slower off-rate. Typically, IC50 values are good measurements for reversible inhibition; however, the potency of a covalent inhibitor is time-dependent. Thus, potency is better understood for irreversible inhibition through measurement of kinact/KI, where kinact is the rate constant of enzyme inactivation and KI is the inhibitory constant. For the described compound, kinact/KI was found to be 1.2 × 105 M−1 min−1. Additionally, the authors co-crystallized the free aldehyde with PRMT5:MEP50 (PDB ID: 6K1S) which showed C449 at a distance within a bond length of the molecule. Interestingly, the authors reported that based on the associated dihedral angles between the covalent inhibitor and C449, elimination had also occurred, which resulted in a trans double bond between the sulfur of C449 and N6 of the adenine ring.
Lately, a covalent inhibitor of PRMT6 was developed by Shen et al.168 This compound (MS117) was designed as an analog of the previously described compound MS023. PRMT6 contains a unique cysteine residue (C50) that was identified to be accessible via substitution of the phenyl ring of MS023. The authors designed MS117 by replacing the para-isopropoxy substituent of the phenyl ring with a meta-acrylamide to serve as the electrophile for the covalent linkage.168 Biochemical analysis provided an IC50 value of MS117 to be 18 nM with a kinact/KI value of 8.3 × 104 M−1 s−1. The covalent bond formation was confirmed by mass-spectrometry and a crystal structure was obtained (PDB ID: 6P7I) that established a covalent bond between C50 and the acrylamide group. MS117 showed moderate selectivity for PRMT6 against other type-I PRMTs. In cells, it was found to significantly reduce levels of asymmetrically demethylated H3R2, a marker produced by PRMT6 alone, while only having moderate effects on other markers produced by PRMT1, -3, and -4 without having significant cellular toxicity in MCF-7, PNT2, and HEK293T cells. By comparing the low selectivity of MS023 and the higher selectivity of MS117 for PRMT6 among type-I PRMT isoforms, the introduction of a target-specific covalency speaks for a doable strategy for engendering selectivity enhancement for nonselective reversible inhibitors.
6. PRMT degraders
With the growing popularity of proteolysis-targeting chimeras (PROTACs), it stands to reason that PRMTs would eventually be targeted with the PROTAC strategy. The first set of PRMT degraders that include MS4322 (Fig. 5) was developed by Shen et al. in 2020.169 This PROTAC molecule was designed by linking the known PRMT5 inhibitor EPZ015666 to the von Hippel-Lindau E3 ligase (VHL) ligand with the goal of ubiquitinating and ultimately degrading intracellular levels of PRMT5. Indeed, MS4322 was demonstrated to reduce cellular levels of PRMT5 and in turn SDMA levels in proteins in a PRMT5-, VHL E3 ligase-, and proteasome-dependent manner in five different cancer cell lines after treatment. The compound also exhibited a reduction in proliferation of these cell lines. MS4322 represents a new targeting strategy for PRMTs and has opened the door for future development of other PROTACs to study the cellular effects of PRMT degradation. The pharmacological significance of PROTAC probes and their distinction from standard PRMT inhibitors await to be fully investigated in cellular and preclinical models.7. PRMT imaging probes
Fluorescent probes that alter fluorescence intensity upon binding with a target enzyme are excellent probes to image its expression level and distribution in cells and tissues. In 2012, we studied a series of trimethine cyanine derivatives containing indolium, benz[e]indolium, or benz[c,d]indolium heterocyclic moieties and evaluated their inhibition for the enzymatic activity of PRMT1.170 The compound MHI-21 demonstrated an inhibitory effect on PRMT1 with an IC50 value of 4.1 μM (Fig. 5(c)). The steady-state enzymatic measurements demonstrated that MHI-21 is a noncompetitive inhibitor with regard to both H4 and SAM substrates. This compound was found to cross the plasma membrane and cause chromatin dysfunction in HeLa cells. Of uniqueness, MHI-21 has the marked feature of carbocyanine compounds with its spectral absorption and fluorescence emission in the near-infrared region. Measured in methanol, MHI-21 has its absorption maximum at 758 nm and its emission maximum at 774 nm. This optical property was explored for the microscopic imaging of PRMT1 in cells. Treatment of HeLa cells with MHI-21 led to bright near-infrared fluorescence imaging of HeLa cells as observed under confocal microscopy using a 633 nm laser for fluorescence excitation and a 715 to 785 nm long pass for emission. The compound was found to be highly concentrated around- and in the nucleus, with a weak level in the cytoplasmic vesicles.Later, we studied more cyanine compounds as PRMT inhibitors.171 Among them, a pentamethine compound E-84 (Fig. 5(c)) showed inhibition of PRMT1 with the potency in the low micromolar level and certain degrees of specificity for PRMT1 over CARM1, PRMT5, and PRMT8 ranging from 6- to 25-fold. Molecular docking suggested that E-84 acts by partially occupying the cofactor binding site and blocking the proximity of the cofactor and substrate arginine. E-84 is not only a potent PRMT1 inhibitor but a strong tool to label PRMT1.172 E84 fluoresces brightly when it binds to PRMT1 and excitation with a red laser. Based on this spectral property, we used E-84 as an optical probe to study PRMT1-regulated functions in living cells.172 We found that the 293T cells transfected with plasmids expressing either isoforms of PRMT1 (V1 or V2) have approximately sevenfold higher fluorescence signals than the 293T cells transfected with the vector plasmid. Therefore, fluorescence intensity was correlated with the PRMT1 expression level. There were no apparent negative effects on proliferation of labeled cells under a low concentration of E-84. Interestingly, using E-84, we were able to identify and sort two populations of PRMT1 expression out of the long-term hematopoietic stem cells (LT-HSCs). The two populations of cells displayed little difference in the proliferation rate. However, PRMT1-low LSK cells were able to repopulate peripheral blood with multiple lineages, whereas the PRMT1-low LSK cells failed to reconstitute multilineages. Thus, E84 is a useful molecular tool to probe the role of PRMT1 in hematopoiesis and leukemogenesis. Developing E84 and other fluorogenic probes to label PRMTs provides a convenient approach superior to antibodies for studying cell epigenetic status and differentiation state changes induced by PRMTs.
8. Peptide- and peptoid-based chemical probes
PRMTs can recognize and methylate peptides as efficiently as full-length proteins. Therefore, short peptide substrates are an excellent starting point for the design of specific chemical probes to study PRMTs. This is important because the use of SAM-analogs and other small molecule inhibitors for targeting PRMTs could be compromised by the off-target issue.95 The Thompson group was among the first to explore peptide-based chemical scaffolds for PRMT inhibition. In 2011, they reported a histone H4 tail peptide bearing a chloroacetamidine moiety named C21 that acts as an irreversible PRMT1 inhibitor (Fig. 6(a)).173,174 The chloroacetamidine warhead is structurally similar to the guanidino group of the arginine residue and could covalently bond with a nucleophilic group like the sulfur in the cysteine side chain. It was proposed that the C21 probe targets Cys101 of PRMT1, which is only conserved in PRMT1 and PRMT8. Taking advantage that C21 irreversibly inactivates PRMT1, the Thompson group further implemented the probe for activity-based protein profiling by conjugating C21 with reporter tags such as biotin (Fig. 6(b), B-C21) or fluorescein (Fig. 6(c), F-C21). Although F-C21 labeled recombinant PRMT1, the fluorescent probe could not detect cellular PRMT1 due to low levels of active protein. B-C21 showed higher sensitivity and detected as little as 25 ng of PRMT1 and isolated PRMT1 from cell extracts. Using the probe, they found that the activity of PRMT1 is negatively regulated spatially and temporally by estrogen.173 Consistent with the property of chloroacetamidine-H4 probes, the Knuckley group later made and tested a chloroacetamidine-containing unnatural peptide with an alkyne group on one end (PI-16yne, Fig. 6(d)), and in vitro assays showed that PI-16yne selectively inhibited and labeled PRMT1 over PRMT5 at concentrsations <25 μM.175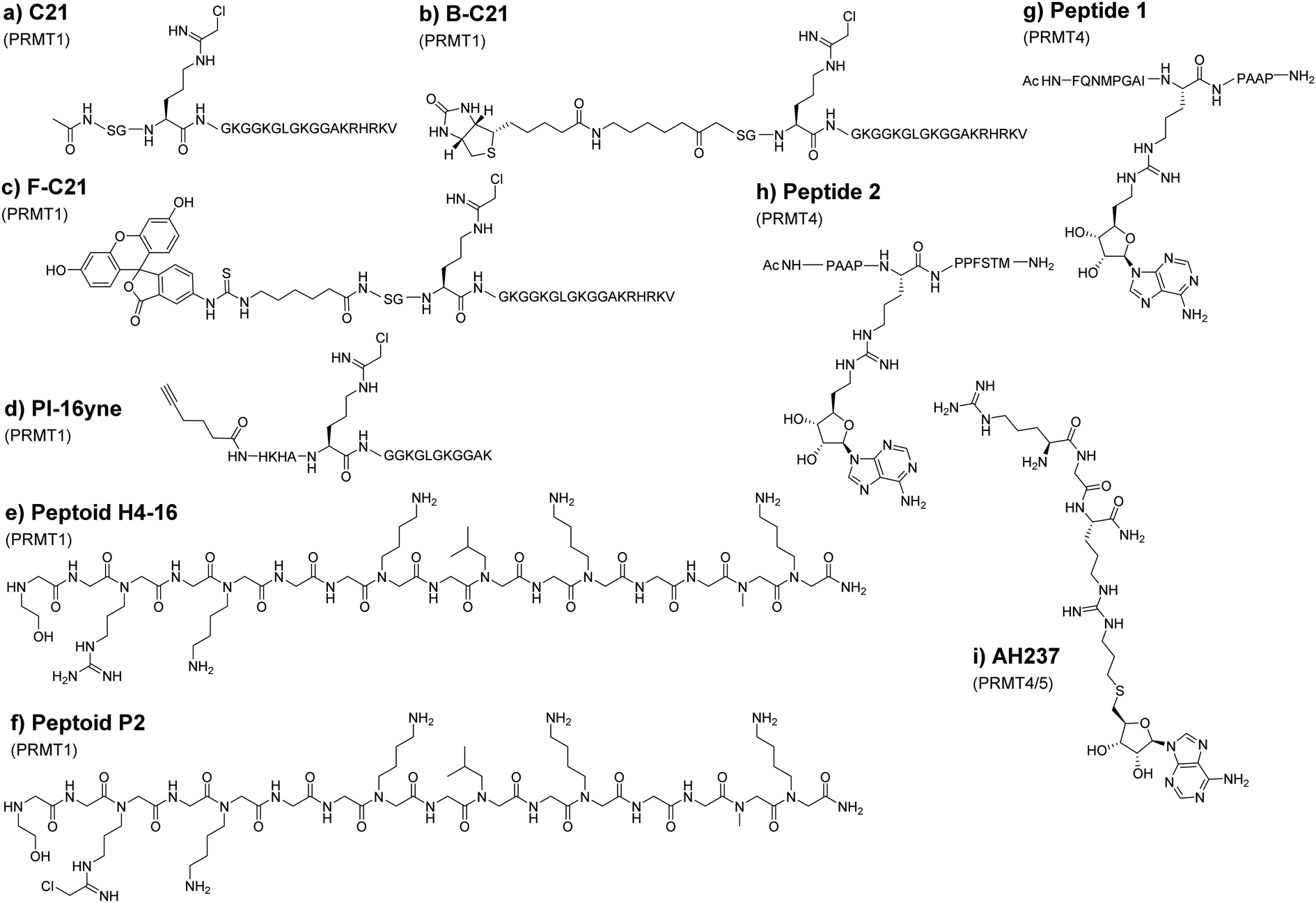 | ||
| Fig. 6 Peptide and peptoid probes of PRMTs. (a) C21 peptide which contains the chloracetamidine moiety synthesized by Obianyo (b) B-C21 displays a biotin conjugated onto C21. (c) F-C21 is C21 peptide with fluorescein bound to the N-terminal tail. (d) PI-16yne peptide probe for PRMT1 labeling. (e) Peptoid H4-16 was among the several peptoids by Mann and coworkers that demonstrated the potential of peptoid inhibitors. (f) Peptoid P2 by DuBose displayed the improvement of the peptoid inhibitors through the chloracetamidine moiety. (g) and (h) Peptides 1 and 2 were among a few peptide-adenosine probes that established the potential of these bisubstrate inhibitors. (i) AH237 synthesized by Al-Hamashi was a bisubstrate inhibitor that had the best IC50 value among the other peptide-adenosine probes. | ||
Though the peptide probes show amenable selectivity, their utility is limited due to their susceptibility to proteolysis. Adopting peptide mimetic structures such as peptoids can circumvent this issue to a degree. The structural difference between peptoids and peptides is that the side chain of peptoids is located on the nitrogen of the amide backbone rather than on the α-carbon in natural amino acids, thus making peptoids more resistant against proteolysis.176 For this reason, Knuckley and coworkers explored histone H4 peptoid structures as an alternative probe design. The initial design includes several peptoid analogs of the histone H4 tail which all showed weak inhibitor activities against PRMT1 with IC50 values in the high micromolar range. Nevertheless, the H4-16 peptoid (Fig. 6(e)) clearly exhibits a certain degree of selectivity towards PRMT1 over PRMT5.177 To improve the potency, the chloroacetamidine warhead that had been used in previous studies was conjugated onto the peptoid. This substituent greatly improves the IC50 as well as its selectivity much like in the previous study.178 Notably, the positive charge at the N-terminus of the H4 sequence provides additional binding stabilization and inhibition that was drawn from the IC50 value of different peptoids. Further tests showed that the H4 peptoid-chloroacetamidine compound (peptide P2, Fig. 6(f)) causes apoptosis and autophagy in cancerous cells while leaving healthy cells relatively unharmed. However, the mechanism by which the inhibitor discriminates between cancerous cells and healthy cells was not delineated. This would suggest that there is still much to be discovered with regard to peptoid-based probes but exemplifies a great promise in optimizing peptoids for use as selective probes for various PRMT isozymes.
9. Peptide-adenosine bisubstrate chemical probes
Linking the substrate peptide sequence and a portion of the SAM structure generates potential bisubstrate inhibitors of PRMTs. Such compounds with bivalent binding modes may have enhanced potency over peptide inhibitors or SAM analogs alone. In 2016, van Haren and coworkers developed a peptide that mimics the transition state formed between substrates during methyltransferase catalysis.179 This probe consists of a peptide fragment (peptides 1 and 2, Fig. 6(g), (h)) from the poly A-binding protein, a highly targeted protein for CARM1 methylation, in which alkyl adenosine was conjugated to the guanidine of arginine on the peptide. The probes were found to inhibit CARM1 in sub-micromolar concentrations and were used by the group in the structural investigation of CARM1. In an effort to improve upon these probes, Huang and coworkers used shorter tripeptides that were then conjugated to adenosine.180 The best inhibitor, AH237 (Fig. 6(i)), reported by the group achieved an IC50 value of 2.8 nM for CARM1 as compared to an IC50 value of 5.9 μM for the major type I enzyme PRMT1. AH237 showed its strongest potency for PRMT5, with an IC50 value of 0.42 nM. It remains intriguing to see how these bisubstrate probes perform in cellular tests.10. Cofactor-based PRMT probes
SAM is the major methyl donor and plays an essential role in biological methylation reactions. These methylation reactions are catalyzed by methyltransferases. To date, several efforts have been made to develop SAM analogs with SAM's methyl moiety replaced by other alkyl substituents as a chemical probe to investigate the methylation reactions. For example, 5′-(diaminobutyric acid)-N-iodoethyl-5′-deoxyadenosine ammonium hydrochloride (AAI) was developed as a synthetic cofactor for SAM-dependent DNA methyltransferases (Fig. 7(a)).181 Inspired by this, Thompson and coworkers reported the first use of AAI as an alternative cosubstrate of PRMT1 and identified that AAI can be used to generate methyltransferase inhibitors. An aziridinium ion is a key intermediate in this reaction process. After reacting with an arginine 3 residue on the histone H4 peptide, a bisubstrate adduct is generated which serves as an inhibitor of PRMT1. In principle, when appropriate AAI modifications were applied such as alkyne or azide functionalization, the ability of a PRMT to catalyze the transfer of AAI to the substrate peptide could be used to identify PRMT substrates and provide potent and selective inhibitors for other SAM-dependent methyltransferases.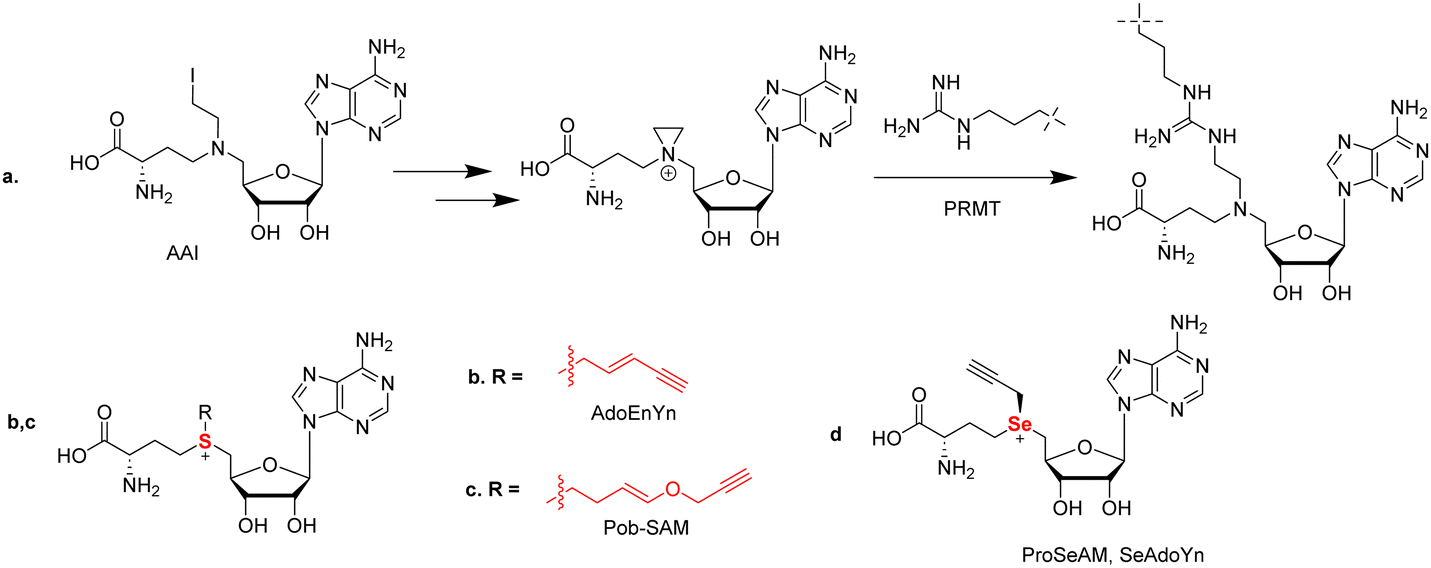 | ||
| Fig. 7 (a) Conjugation reaction of AAI with the target arginine residue in the substrate; (b) structure of AdoEnYn; (c) structure of 4-propargyloxy-but-2-enyl SAM (Pob-SAM); (d) structure of propargylic Se-adenosyl-L-selenomethionine (ProSeAM, SeAdoYn). | ||
The copper-catalyzed azide–alkyne cycloaddition (CuAAC) has been a highlight of click chemistry since its inception by Sharpless and Meldal in 2002.182,183 This reaction entails the cyclization of a terminal alkyne and an azide to form a 1,4-disubstuted 1,2,3-triazole promoted by a Cu(I) catalyst. The reaction is highly chemospecific and is tolerable in aqueous media. Such high specificity and the ability to be conducted in water has in turn opened the door to a wide degree of biorthogonal approaches ultimately leading to both Sharpless and Meldal, as well as Bertozzi for her work in biorthogonal click chemistry, sharing the 2022 Nobel Prize in Chemistry. With such a useful nature, it is no surprise that probes utilizing click chemistry have been developed towards PRMTs.
In PRMT catalysis, the sulfonium of SAM acts as a methyl donor during the methylation reaction. It is simple to hypothesize that chemical modification of the methyl group of SAM to different alkyl groups (e.g. a propargyl group) could potentially generate different alkyl donors through enzymatic reaction. The first modified SAM-analogs developed were sulfonium-β-sp/sp2-alkyl SAM analogs toward native protein methyltransferases (PMTs) as cofactor surrogates in 2010 by the Weinhold group.184 In their report, they designed the SAM-based cofactor 5'-[(S)-[(3S)-3-amino-3-carboxypropyl](E)-pent-2-en-4-ynylsulfonio]-5′-deoxyadenosine (AdoEnYn) by replacing the methyl group of SAM with a pent-2-en-4-ynyl side chain (Fig. 7(b)). They explained that the double bond can compensate unfavorable steric effects within the SN2-like transition state by conjugative stabilization, making the structure more stable.184 The terminal alkyne is used for further modifications through bioorthogonal CuAAC click chemistry. They found that this probe is active for the mammalian ASH2-MLL complex and can be applied to identify novel MTase substrates and define the methylated proteome through conjunction with protein arrays.184 However, other PMTs cannot accept this probe due to steric hindrance by the pent-2-en-4-ynyl side chain.87,185 To expand the capability of SAM analogs as chemical biology probes to label target PMTs, a set of internal alkene-containing SAM analogs were synthesized by Luo's group and evaluated with several recombinant PMTs (Fig. 7(c)).186 These bulkier SAM analogs were found to be poor co-substrates for the wild-type PMTs; nevertheless, alkyl transfer activity was gained for a G9a Y1154A mutant that relieved some steric hindrances.186
In 2011, Binda and coworkers reduced the size of the side chain by using propargyl-SAM as a cofactor that was accepted by H3K9 lysine methyltransferase SETDB1. The propargyl moiety was observed to be transferred to an isolated H3 substrate; however, the propargyl-SAM probe is unstable at physiological pH and will decompose via an allene intermediate. Other methyltransferases tested by this assay including PRMT1 are unable to accept the probe in in vitro assays. Taking this observed limitation into context, this probe has been limited to use as a relatively specific reagent for the study of SETDB1.187
To address the stability issue, the Luo and Weinhold group developed a SAM surrogate, propargylic Se-adenosyl-L-selenomethionine (ProSeAM, SeAdoYn) by replacing the sulfur of propargyl SAM with selenium (Fig. 7(d)).188,189 This method could strongly enhance the stability of the propargylic cofactor caused by the reduction of the acidity of the methylene adjacent to the center. Due to the less steric hindrance in the active site and the stronger activation of the cofactor surrogate, ProSeAM can be utilized by multiple native PMTs. The Weinhold group found that it was active toward G9a, Set 7/9, and PRMT1, but in the Luo's work, they found only G9a, but not Set7/9 and PRMT1 are active toward ProSeAM. They reasoned that these differences may be due to the sensitive antibody-coupled horseradish peroxidase assay used under Weinhold's conditions.
The Luo's laboratory then aimed at developing SAM analog cofactors that are inert toward native PRMTs but can be recognized by engineered PRMTs. They developed a “bump and hole” strategy to analyze the process of methylation reaction.190,191Via this strategy, they reported 4-propargyloxy-but-2-enyl SAM (Pob-SAM) that showed binding with a PRMT1 Y39F-M48G double mutant and was successfully able to label H4 in vitro. Labeling of protein substrates was also achieved in HEK293T cells that expressed the mutated PRMT1.87 Given the sequence homology among type-I PRMTs, it might be possible to apply the same mutations for other PRMT members in order to generate their respective bioorthogonal probes.
While ProSeAM is a promiscuous binder of methyltransferases, it has been utilized in the development of inhibitors. Using an azide-containing biotin, the Sodeoka group performed a CuAAC reaction with propargylated substrates from cell lysate. Together with the SILAC technique, they were able to identify an inhibitor based on the structure of chaetocin (a lysine methyltransferase inhibitor) that selectively inhibits non-histone methylation.192,193 Further analysis showed an enrichment of protein substrates that align with PRMT1 and upon further testing, it was observed that the compound functions as a modest PRMT1 inhibitor with an IC50 value of 71.7 μM.192,193 While further medicinal chemistry modifications to the structure could produce a more selective inhibitor, this work does highlight the utility of ProSeAM in analyzing the methylome and identifying PRMT substrates and potential inhibitors.
11. Photoaffinity probes
Most strategies for chemical probe design involve non-covalent interactions. In the design and use of photoaffinity labeling, a covalent bond can be formed between the probe and the protein of interest using only light as a trigger. For methyltransferases, it has been observed that UV irradiation is able to initiate chemical cross-linking of [3H]-SAM with its binding proteins thus providing initial evidence to demonstrate the methyltransferase activity of an enzyme in question; this has also been applied to PRMTs.39,194–196 The mechanism behind SAM crosslinking was first proposed in 1984 by Yu.197 In that work, Yu describes the crosslinking of SAM to catechol O-methyltransferase (COMT) and reports that the entire SAM molecule is attached to the enzyme rather than just a single methyl group. A study reported by Takata in 1992 found SAM linked to a tyrosine residue in rat guanidinoacetate methyltransferase (rGAMT); however, they stated that the residue type may not matter in terms of crosslinking and that the reactive site may only need to be a good radical acceptor.198 Van Loon postulated that the crosslinking is derived from two possibilities; a radical pathway that proceeds through the formation of a radical at the C8 position of the purine ring that reacts with the closest radical acceptor (Fig. 8(a)), or the less likely pathway that SAM is instead not photoactivated and reacts with a photoactivated residue within the binding pocket.198 In 2017, Bera and coworkers published their work through which some semblance of initiation of the reaction mechanism could be theorized.199 From their study, a radical is most likely to be formed at the C8 position of purine rings due to retention of the aromaticity of the 6-membered ring. When they performed reactions with adenine they highlight that while the 2-substituted product is lower energy than the 8-substituted product, the transition state of the C8 substitution is much lower energy than the C2 substituted transition state in both the gas phase and the condensed phase.199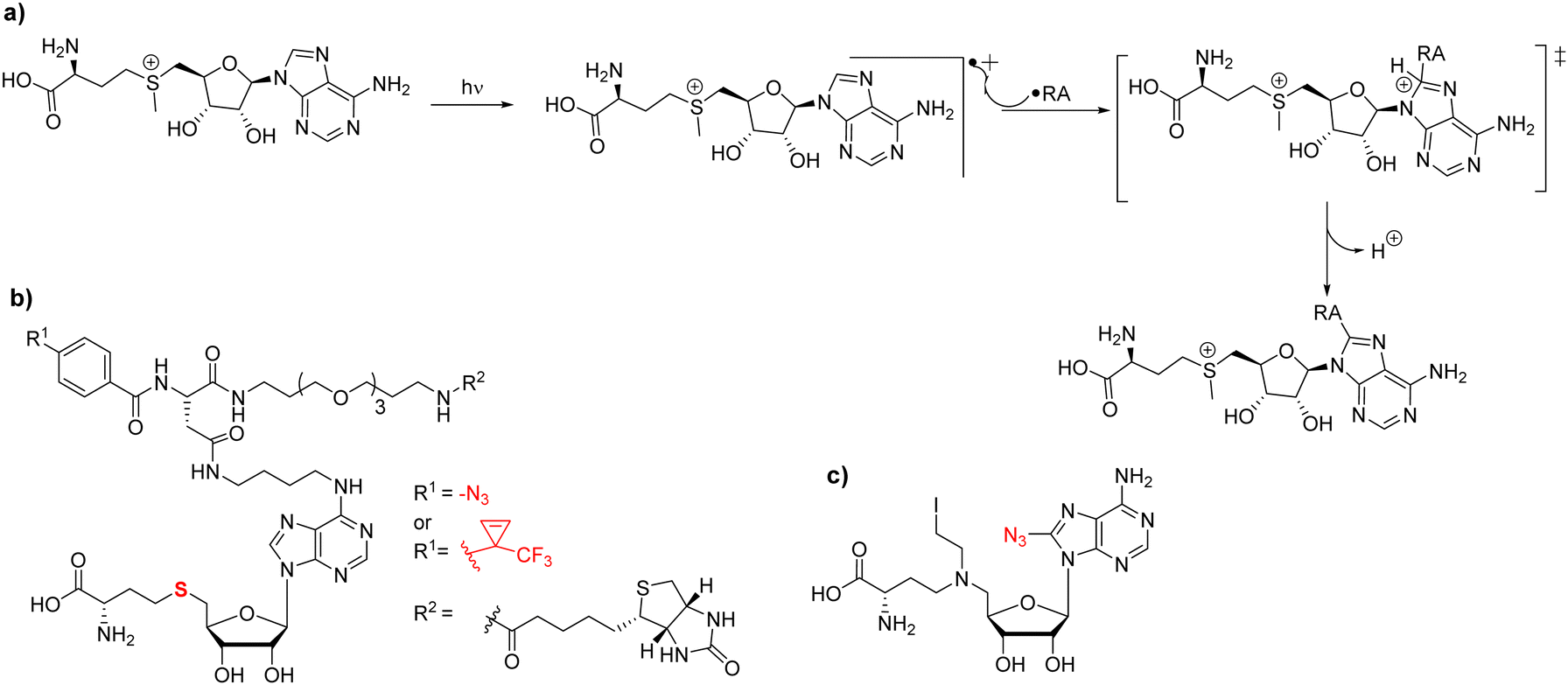 | ||
| Fig. 8 Photoaffinity probes for PRMT labeling. (a) Generalized proposed reaction mechanism whereby UV irradiation of SAM produces an excited radical cation state on the purine ring. This radical cation will subsequently react with a radical acceptor (RA) within a SAM-binding protein to form a covalent linkage between SAM and the protein. (b) SAH-based capture compounds developed by the Weinhold group targeting M. TaqI. (c) 8-Azido-5′-(diaminobutyric acid)-N-iodoethyl-5′-deoxyadenosine. | ||
As a part of the development of photochemical probes, one strategy is the utilization of capture compound mass spectrometry (CCMS) as a tool to investigate the protein–ligand interactions in medicinal chemistry.200 A capture compound contains three main groups: a selectivity function for specific and reversible binding to target protein(s), a photoreactive group that upon irradiation with UV light will form a covalent linkage to a group in close proximity (i.e. the target protein), and a sorting function to isolate the covalently captured proteins. Ideally, the photoreactive groups require low-intensity UV wavelengths to initiate crosslinking reactions thereby resulting in minimal damage to the structure of the nucleic acid and/or the protein. Because of its a good binding affinity and chemical stability, SAH is often used as a selectivity function in photoaffinity probes to capture SAM-dependent MTases.201 A SAH-based photolabeling probe was designed by the Weinhold group (Fig. 8(b)).202 SAH was modified by using linkers at either the N6 or C8 position of the purine ring that contained different photo-crosslinking groups (azide or diazirinine), terminating in a biotin group as a sorting function. In their experiments, the probes were successfully applied to the isolation of the DNA MTase M. Taql and subsequent determination of the dissociation constant (KD) for MTase-cofactor complexes. Currently, photoaffinity probes have been primarily developed for DNA methyltransferases and most recently lysine methyltransferases, leaving an obvious gap in the field of arginine methyltransferases.203 In 2016, analogs of this design were further generated by the Cravatt group where alterations were made to either the photoaffinity group and/or the reporter in an effort to chemically profile the human proteome-wide methyltransferases.204 Using the compounds, the authors were able to identify at least 40 known or predicted methyltransferases with high confidence indicated by consistent enrichment across multiple experiments. PRMT1, -3, -4, and -6 were all a part of the high-confidence group. PRMT5 and PRMT8 were also observed; however, their enrichments were less consistent across experiments and were thus deemed a moderate confidence target. These differences in enrichment are possibly due to binding affinity variations of the probe to individual PRMT members. Conceivably, more extensive modifications to these probes could be a viable method of tuning their selectivity for different members of the family.
In 2011, Mai reported an azide-bearing SAM analog, 8-azido-5′-(diaminobutyric acid)-N-iodoethyl-5′-deoxyadenosine which serves as a biochemical tool to identify sites and substrates of the methylation reaction (Fig. 8(c)).205 This probe was designed as a nitrogen mustard that at physiological pH would cyclize to form a reactive aziridinium. This aziridinium would then be brought into close proximity to the substrate arginine via both molecules binding to the associated PRMT. This will allow for the guanidino group to attack the aziridinium resulting in a covalently linked substrate to the SAM analog. The associated azido group was linked to a biotin group in a later work from the same group via click chemistry, effectively producing a means of labeling the substrates in an enzyme-dependent fashion.206 In principle, the utility of this compound may not be just for substrate identification, but also could be able to photoactively label SAM-dependent methyltransferases. In particular, it has been previously shown that the 8-azidoadenine moiety can be irradiated with light to generate a reactive nitrene that would subsequently form a covalent linkage to bound enzyme.207,208
12. Arginine analogs for site-specific installation of methylarginine marks
In studying how specific arginine methylation type and location affect intracellular processes, a particular interest is in the recognition of methylated arginine residues by reader or effector protein modalities. Using solid-phase peptide synthesis, it is convenient to introduce MMA, ADMA, and SDMA residues in peptides and apply them to study how methylarginines are recognized by proteins downstream of the methylation. However, this strategy is not applicable for studying methylarginine–protein interactions in vivo. In biological samples, it is difficult to control the level and degree of methylation at a specific arginine residue as a PRMT often produces both MMA and ADMA/SDMA at more than one residue on the substrate. Therefore, it is technically challenging to assess how one particular mark on a given protein alters the binding of another protein. To overcome this problem, Le and coworkers designed a series of arginine analogs to install specific methylarginine marks in a site-specific manner (Fig. 9).209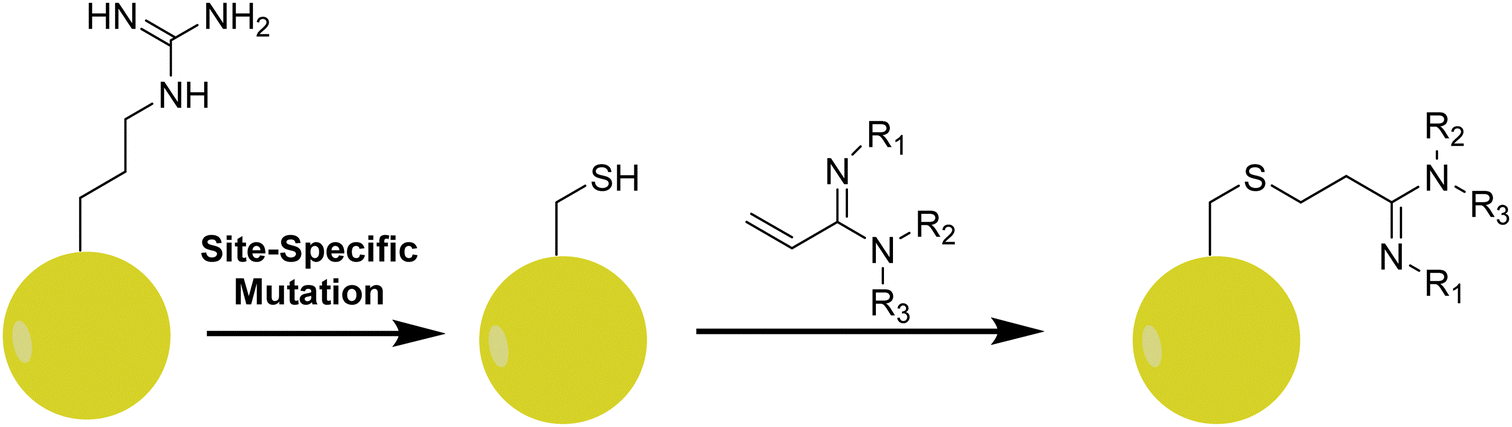 | ||
| Fig. 9 Incorporation of methylarginine mimics into proteins. After a site-specific mutation of an arginine residue to cysteine, a target protein is then reacted with an acrylamidine analog to generate the methylarginine analog. R1 = R2 = R3 = H (Arg); R1 = Me, R2 = R3 = H (MMA); R1 = H, R2 = R3 = Me (ADMA); R1 = R2 = Me, R3 = H (SDMA). | ||
These analogs were designed using an amidine functional group to mimic the guanidino group of arginine that would be linked to a protein containing an arginine to cysteine mutation (in this case, recombinant Xenopus H4R3C and H3R2C). The amidine functional group was synthesized in two steps starting from either 3-hydroxypropionitrile (in the case of MMA and ADMA) or 3-hydroxy-N-methylpropanamide (in the case of SDMA). Subsequent halogenation and elimination afforded α,β-unsaturated methylated amidines which easily underwent conjugate addition with the free cysteine residues of the recombinant proteins. The authors utilized the effector protein TDRD3 (a protein known to recognize H4R3Me2a) along with arginine analog-modified peptides as a proof of concept study. Using isothermal calorimetry (ITC), a 15-mer peptide of H4 showed an ∼1.6-fold increase in binding affinity for TDRD3 when it contained an ADMA mark at R3 as opposed to unmethylated arginine. This was further supported in a fluorescence polarization assay using His6-MBP-TDRD3 and a tetramethylrhodamine-tagged peptide. Competitive peptides were added to antagonize the fluorescence, and the H4R3Me2a peptide was observed to show the most potency with the arginine analog (H4R3C-ADMAA) demonstrated comparable activity. The unmethylated H4R3 and its associated analog displayed no affinity for TDRD3 in this assay. Lastly, the authors were able to modify histone proteins with the arginine analogs and were able to reconstitute nucleosomes containing these marks. This opens the door to the potential of utilizing these analogs in the assessment of the functionality of the methylarginines pertaining to protein–protein interactions as a whole, rather than peptide–protein interactions.209
While cysteine-directed strategies like what is described above are a great avenue for accessing a protein with a singular modification, the use of protein semisynthesis has opened the door to the generation of full-length histones bearing multiple modifications. The semisynthetic method utilizes either synthetic protein fragments alone (native chemical ligation) or in conjunction with recombinant protein fragments (expressed protein ligation) whereby a fragment containing a C-terminal thioester reacts with an N-terminal cysteine to produce a native peptide bond.90,210 Whereas there is a lack of cysteine residues in histones, the advent of chemical desulfurization has greatly improved ligation techniques in the semisynthesis of proteins. Further, sulfur-containing derivatives of amino acids can be utilized to direct the ligation of modified residues at sites adjacent to residues other than just cysteine.210
13. Molecular reporters that recognize methylated arginines
Early work on the study of arginine and lysine recognition stems from the investigation of pi–cation interactions. Ngola and coworkers found that adding anions onto the aromatic structures found on cyclophanes induces a dipole moment that increases its affinity towards cations (Fig. 10(a)).211 Gamal-Eldin and Macartney later found that such interactions could be achieved without aromaticity. In their investigation, cucurbit[n]uril could recognize arginine and lysine, where the compound had a higher affinity towards trimethyllysine over lysine and a slight preference for SDMA over ADMA (Fig. 10(b)).212 Rather than pi–cation interactions, the authors suggested that the mechanism for the compound's affinity for the residues was through hydrophobic and van der Waals interactions.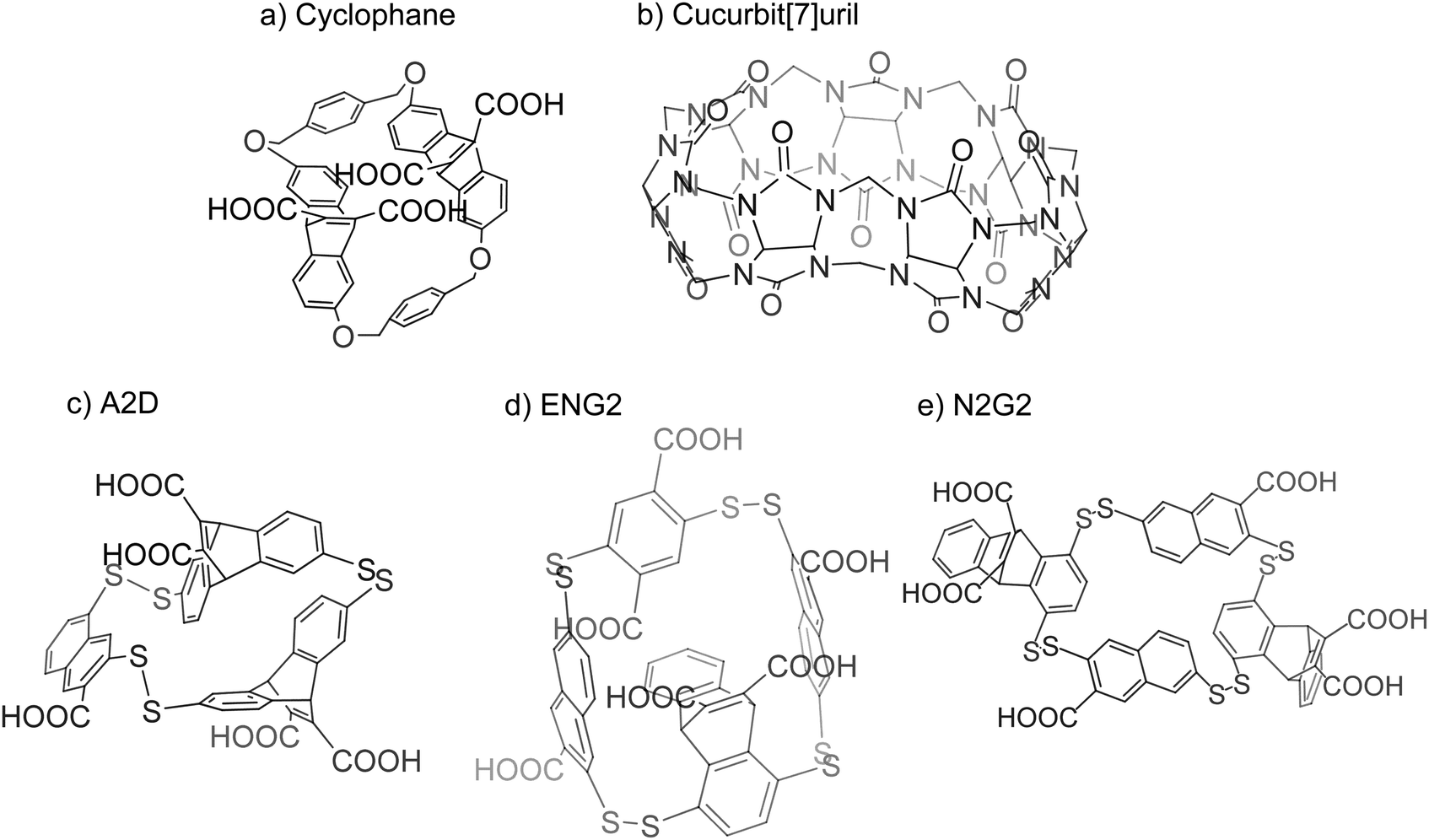 | ||
| Fig. 10 Synthetic receptors for methylarginine recognition. (a) Cyclophanes used by Dougherty's group led to the discovery that anionic substituents would increase an aromatic structure's affinity to cations. (b) Cucurbit[7]uril demonstrated that aromaticity was not required for molecular reporters of arginine and lysine. (c) The Waters group used dynamic combinatorial chemistry to discover new molecular reporters for methylated states of arginine. A2D was found to have selective interactions with ADMA. (d) and (e) The Waters group improved their synthetic receptors with synthesizing ENG2 and N2G2. ENG2 had specificity for dimethylated arginine over trimethylated lysine but could not distinguish between SDMA and ADMA. N2G2, however, was able to selectively interact with ADMA rather than SDMA or trimethylated lysine. | ||
Further study on these reporters is resourced on dynamic combinatorial chemistry in which molecules act as building blocks for a probe. These compounds would be placed in a solution and allowed to react with each other reversibly and form an equilibrium of various products. Upon the introduction of the target molecule, the equilibrium of the products begins to favor a final compound that interacts with the target molecule and is stabilized by it. The result is the discovery of a polymer that has a strong affinity for the target.213 The Waters group took advantage of this technique to screen for synthetic receptors that would mimic protein binding pockets for dimethylated arginine. In using various methylated states of arginine and lysine as a target molecule, the authors described a synthetic receptor, A2D, which displays specificity for ADMA over SDMA (Fig. 10(c)).214 It was suggested that the size difference between ADMA and SDMA may play a role in this specificity as the pocket may only be able to accommodate one methyliminium group. This synthetic receptor, however, did not show any selectivity with regard to trimethyllysine. The group furthered this investigation with two more synthetic receptors found through dynamic combinatorial chemistry, ENG2 and N2G2 (Fig. 10(d) and (e)).215 ENG2 seems to prefer dimethylarginine over dimethyllysine but offers no selectivity between SDMA and ADMA. For N2G2, only ADMA has an affinity towards the synthetic receptor. It was believed that the higher desolvation cost of SDMA discourages its binding while trimethyllysine is excluded due to steric hindrance as the pocket promotes the binding of planar molecules such as the guanidino group found on arginine side chains.216 Mullins and coworkers used these synthetic receptors to develop a protocol to interrogate methylation states of lysine and arginine. They created a dynamic combinatorial library consisting of subunits of the previously discovered synthetic receptors and allowed the solution to equilibrate before adding in a modified target peptide. In the presence of the altered peptide, the subunits will form a known product that has an affinity for the modifications and will then be found in higher abundance in the solution when compared to the solution without the target peptide. This difference can be observed through HPLC to determine the modification present on the peptide as the synthetic receptor has previously been tested for its affinity towards designated modifications.217
The described molecular reporters represent a start in discriminating between various modifications of lysine and arginine. While the main challenge being tackled revolves around the specificity of the reporters, other issues still remain to be resolved. With the exception of cucurbit[n]uril, the presence of multiple carboxylic groups on the reporters may discourage cell permeability, limiting these reporters to in vitro assays. Another limitation concerning the reporters is that of the disulfide bond found in A2D, ENG2, and N2G2. Although these compounds seem promising in terms of their selectivity, the disulfide bond proves to be fragile under reductive intracellular environments, further limiting their use for cellular assays.
14. Arginine selective probes
In the study of protein structure and interaction, a commonly met issue is how to investigate the properties of key pockets, such as enzyme active sites. Residue-specific labeling of target proteins has been a valuable tool to solve this issue. The big challenge of arginine-specific labeling is how to control exact reaction sites, especially overcoming competitive reactions with lysine residues that are more abundant and reactive.Glycation is an important nonenzymatic modification of proteins. In this process, the nucleophilic amino group of arginine or lysine reacts with carbonyl groups of the sugar to form a Schiff base product by the Maillard reaction. Based on the process of glycation, several chemical probes have been developed by using these dicarbonyl moieties to allow possible arginine-specific conjugation. Methylglyoxal (MGO) belongs to a family of α-oxo aldehydes and has been observed to participate in glycation to form stable modifications known as advanced glycation end products (AGEs) (Fig. 11(a)).218 Because of its capability to react with arginine and lysine, it has been studied for its use as chemical probes, although this has not been a fruitful endeavor as MGO-based chemical probes have been found to be nonselective in their activity as well as their lack of fluorescence.219
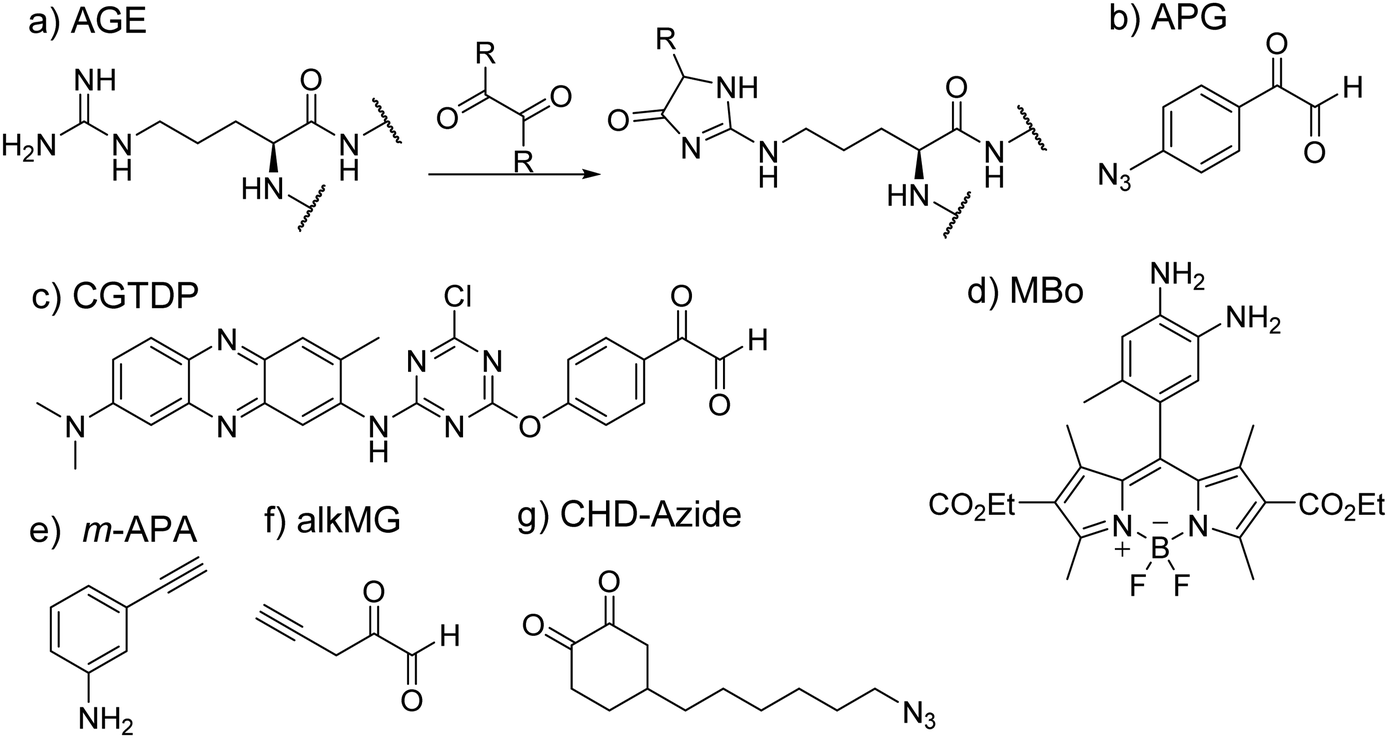 | ||
| Fig. 11 Arginine targeting probes. (a) The generic reaction between the arginine guanidino group and the dicarbonyl group. (b) APG probe. (c) CGTDP in which neutral red acted as a polarity-sensitive fluorophore. (d) Mbo as a probe for histone glycation. (e) Ray and coworker targeted the Schiff base formed in the glycation of arginine and used m-APA to react with this intermediate to probe modified residues. (f) alkMG took advantage of arginine glycation and was then visualized through CuAAC addition with either biotin azide or TAMRA azide. (g) CHD-azide underwent the click reaction with biotin-PEG4-alkyne for enrichment and identification of arginine modified proteins. | ||
A direct probe involving MGO was synthesized by the Johannsen group in which an alkyne was conjugated onto MGO (i.e. alkMG, Fig. 11(f)).220,221 This allowed for the enrichment of proteins that have been modified by MGO through CuAAC addition of biotin azide.221 The probe can permeate cell membranes and modify proteins sensitive to α-oxoaldehydes. A relevant technique developed by Ray et al. is the use of an aniline analog as a probe for MGO-based AGEs (Fig. 11(e)). The group thought to discern the level of glycation in histones given that histones are in high abundance and have long half-lives.222 Aniline was found to react with the Schiff base formed as a result of the reaction between glyoxals and arginine or lysine before the cyclization of the AGEs. The alkyne conjugated onto the aniline probe allowed for labeling and visualization of the probe through click chemistry with TAMRA azide or enrichment with biotin azide (Fig. 11(b)).223 However, this probe was found to be more sensitive to glyoxal rather than MGO. To address this challenge and specifically target MGO, Spiegel and coworkers developed a fluorescent probe that would conjugate with MGO over other dicarbonyls.224 The probe, MBo (Fig. 11(d)), is based on a scaffold that was originally used in biochemical assays involving nitric oxide. However, the authors found that the shorter half-life of nitric oxide would not interfere with the probe's ability to detect MGO modifications. Such investigations led to the discovery that glycation of histones was found mainly in histone H3 and that the effect of glycation caused improperly formed chromatin.225
Cyclohexane-1,2-dione as a common dicarbonyl reagent is an effective compound to track the reactive arginine residues in proteins. In 2018, Chowdhury and coworkers developed an arginine-selective probe, cyclohexanedione-azide (CHD-Azide), for identification and enrichment of reactive arginine residues in proteins (Fig. 11(g)).226 Click chemistry and the biotin–avidin affinity chromatography were used to enrich labeled peptides. This probe was tested by using several standard peptides with arginine residues at different positions in the sequence. Two selective arginine residues, Arg-39 and Arg-33, were identified as the most reactive arginine residues present in RNase A.
Phenylglyoxal and p-hydroxyphenylglyoxal have been used as common α-dicarbonyl reagents to selectively modify arginine residues. Based on phenylglyoxal, a polarity-sensitive fluorescent probe, 3-(4-chloro-6-p-glyoxal-phenoxy-1,3,5-triazinylamino)-7-(dimethylamino)-2-methylphenazine (CGTDP) was developed by Ma's group (Fig. 11(c)).227 In this work, the authors conjugated a phenylglyoxal group with a neutral red moiety. With an analytical long-wavelength characteristic and sensitive emission shift, Neutral red is an excellent polarity-sensitive fluorophore. This probe was applied to detect the polarity and conformational changes of the active site of creatine kinase. Dawson developed a class of arginine-specific reagents by using a commercially available reagent, the p-azidophenylglyoxal (APG) molecule (Fig. 11(b)).228 Therein, the phenylglyoxal group of APG can target arginine residues and subsequently be reacted with alkynyl fluorophores such as Biotin-PEG4-alkyne and DBCO-TAMRA reagents with CuAAC chemistry. Given that methylation alters the chemical reactivity of the guanidino group of arginine residues, reactive dicarbonyl reagents have the potential to distinguish arginine residues in different methylation states. There may be future work possible with the concept of MGO-based probes as it was suggested that dimethylated arginine may not be reactive with MGO.229 This offers an opportunity for further development of dicarbonyl based probes to examine the arginine methylation activity of proteins by PRMTs.
15. Summary
The field of protein methylation, especially arginine and lysine methylation, has boomed significantly since mid-1990s. The major driving force for such a big advancement is the increased recognition of the physiological and pathological significance of protein methylation in biology and disease as well as the advancement of modern genetic and molecular technologies that enable its functional investigations.34 Since the discovery of the first PRMT gene, PRMTs have been demonstrated to be essential cellular enzymes that play important regulatory roles in diverse biological pathways. Evidence of the causative relationship between PRMT deregulation and disease pathogenesis is amounting. In this account, we have discussed different kinds of chemical methods and probes designed to interrogate PRMT mechanisms and analyze the processes regulated by arginine methylation. These chemical biology tools have affected many aspects of protein methylation research and empowered our functional investigation of PRMT biology.Research in the PRMT field is constantly evolving. Challenges and opportunities are both present. To elicit more open discussion, we list a few questions that we regard deserve substantive studies henceforth. First, an interesting observation is that lysine methylation has gained much greater attention than arginine methylation in the area of histone modification and chromatin biology. Time will tell whether arginine methylation and lysine methylation are equally important in chromatin and gene regulation. Admittedly, a technical limitation of histone arginine methylation is the lack of specific antibodies that recognize different arginine methylation marks on histones for ChIP-seq examination. Second, contrary to the fact that the abundance of arginine methylation is higher than that of lysine methylation in cellular proteins, the number of PRMT genes/proteins is much smaller than that of PKMT genes/proteins. What is the evolutionary etiology for such a phenomenon? It might also be possible that some unknown PRMT enzymes wait to be discovered. In relevance, a trimethylarginine marker has been found to exist in proteins;230 however, the structural features of trimethylarginine and the enzymes that catalyze arginine trimethylation are not elucidated. Third, arginine methylation marks in proteins are found to be relatively stable.26 Nevertheless, some weak-activity arginine demethylases have been reported. It remains to elaborate whether these arginine demethylases regulate dynamic changes in arginine methylation levels in different biological contexts and whether arginine demethylation is of importance to in vivo physiology and pathophysiology. Last but not least, most PRMT research thus far is concentrated on mammalian cells and organisms. Arginine methylation in other types of organisms (e.g. protozoan parasites) is much less studied.231 Given that knockout of PRMT1 or PRMT5 causes lethal defects in mammalian organisms,74 there is a promise that PRMTs could be a druggable target in anti-parasitic therapeutics development. Research is needed to elucidate functions of PRMTs in pathogenic microorganisms as well as to develop selective inhibitors for pathogen-unique PRMT enzymes. We envision that the landscape of PRMT research will look very different from nowadays in the foreseeable future. Through this course, chemical approaches and tools will be continuously renovated and brought into practice.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Institutes of Health for supporting this work (grant #R01GM126154, 1R35GM149230, and 1R21AI158176). We apologize to those colleagues whose valuable work could not be cited due to space constraints.References
- G. A. Khoury, R. C. Baliban and C. A. Floudas, Sci. Rep., 2011, 1, 1–5 CrossRef PubMed.
- S. Ramazi and J. Zahiri, Database, 2021, 2021, 1–20 CrossRef PubMed.
- P. V. Hornbeck, B. Zhang, B. Murray, J. M. Kornhauser, V. Latham and E. Skrzypek, Nucleic Acids Res., 2015, 43, D512–520 CrossRef CAS PubMed.
- M. Bremang, A. Cuomo, A. M. Agresta, M. Stugiewicz, V. Spadotto and T. Bonaldi, Mol. BioSyst., 2013, 9, 2231–2247 RSC.
- L. C. Boffa, J. Karn, G. Vidali and V. G. Allfrey, Biochem. Biophys. Res. Commun., 1977, 74, 969–976 CrossRef CAS PubMed.
- A. Guo, H. Gu, J. Zhou, D. Mulhern, Y. Wang, K. A. Lee, V. Yang, M. Aguiar, J. Kornhauser, X. Jia, J. Ren, S. A. Beausoleil, J. C. Silva, V. Vemulapalli, M. T. Bedford and M. J. Comb, Mol. Cell. Proteomics, 2014, 13, 372–387 CrossRef CAS PubMed.
- K. Wang, M. Dong, J. Mao, Y. Wang, Y. Jin, M. Ye and H. Zou, Anal. Chem., 2016, 88, 11319–11327 CrossRef CAS PubMed.
- M. Zhang, J. Y. Xu, H. Hu, B. C. Ye and M. Tan, Proteomics, 2018, 18, 1700300 CrossRef PubMed.
- A. W. Wilkinson, J. Diep, S. B. Dai, S. Liu, Y. S. Ooi, D. Song, T. M. Li, J. R. Horton, X. Zhang, C. Liu, D. V. Trivedi, K. M. Ruppel, J. G. Vilches-Moure, K. M. Casey, J. Mak, T. Cowan, J. E. Elias, C. M. Nagamine, J. A. Spudich, X. D. Cheng, J. E. Carette and O. Gozani, Nature, 2019, 565, 372–376 CrossRef CAS PubMed.
- J. Murn and Y. Shi, Nat. Rev. Mol. Cell Biol., 2017, 18, 517–527 CrossRef CAS PubMed.
- M. I. Maron, S. M. Lehman, S. Gayatri, J. D. DeAngelo, S. Hegde, B. M. Lorton, Y. Sun, D. L. Bai, S. Sidoli, V. Gupta, M. R. Marunde, J. R. Bone, Z. W. Sun, M. T. Bedford, J. Shabanowitz, H. Chen, D. F. Hunt and D. Shechter, iScience, 2021, 24, 102971 CrossRef CAS PubMed.
- D. Musiani, J. Bok, E. Massignani, L. Wu, T. Tabaglio, M. R. Ippolito, A. Cuomo, U. Ozbek, H. Zorgati, U. Ghoshdastider, R. C. Robinson, E. Guccione and T. Bonaldi, Sci. Signaling, 2019, 12, eaat8388 CrossRef CAS PubMed.
- K. Lott, J. Li, J. C. Fisk, H. Wang, J. M. Aletta, J. Qu and L. K. Read, J. Proteomics, 2013, 91, 210–225 CrossRef CAS PubMed.
- J. Y. Fong, L. Pignata, P. A. Goy, K. C. Kawabata, S. C. Lee, C. M. Koh, D. Musiani, E. Massignani, A. G. Kotini, A. Penson, C. M. Wun, Y. Shen, M. Schwarz, D. H. Low, A. Rialdi, M. Ki, H. Wollmann, S. Mzoughi, F. Gay, C. Thompson, T. Hart, O. Barbash, G. M. Luciani, M. M. Szewczyk, B. J. Wouters, R. Delwel, E. P. Papapetrou, D. Barsyte-Lovejoy, C. H. Arrowsmith, M. D. Minden, J. Jin, A. Melnick, T. Bonaldi, O. Abdel-Wahab and E. Guccione, Cancer Cell, 2019, 36, 194–209 e199 CrossRef CAS PubMed.
- J. J. Hamey, R. J. Separovich and M. R. Wilkins, J. Proteome Res., 2018, 17, 3485–3491 CrossRef CAS PubMed.
- H. Hu, C. Luo and Y. G. Zheng, J. Biol. Chem., 2016, 291, 26722–26738 CrossRef CAS PubMed.
- M. Wang, R. M. Xu and P. R. Thompson, Biochemistry, 2013, 52, 5430–5440 CrossRef CAS PubMed.
- F. Frederiks, M. Tzouros, G. Oudgenoeg, T. van Welsem, M. Fornerod, J. Krijgsveld and F. van Leeuwen, Nat. Struct. Mol. Biol., 2008, 15, 550–557 CrossRef CAS PubMed.
- A. Barski, S. Cuddapah, K. Cui, T. Y. Roh, D. E. Schones, Z. Wang, G. Wei, I. Chepelev and K. Zhao, Cell, 2007, 129, 823–837 CrossRef CAS PubMed.
- N. D. Heintzman, R. K. Stuart, G. Hon, Y. Fu, C. W. Ching, R. D. Hawkins, L. O. Barrera, S. Van Calcar, C. Qu, K. A. Ching, W. Wang, Z. Weng, R. D. Green, G. E. Crawford and B. Ren, Nat. Genet., 2007, 39, 311–318 CrossRef CAS PubMed.
- S. Bae and B. J. Lesch, Front. Cell. Dev. Biol., 2020, 8, 289 CrossRef PubMed.
- S. C. Larsen, K. B. Sylvestersen, A. Mund, D. Lyon, M. Mullari, M. V. Madsen, J. A. Daniel, L. J. Jensen and M. L. Nielsen, Sci. Signaling, 2016, 9, rs9 CrossRef PubMed.
- P. Bulau, D. Zakrzewicz, K. Kitowska, B. Wardega, J. Kreuder and O. Eickelberg, Biotechniques, 2006, 40, 305–310 CrossRef CAS PubMed.
- P. Vallance and J. Leiper, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 1023–1030 CrossRef CAS PubMed.
- S. Dhar, V. Vemulapalli, A. N. Patananan, G. L. Huang, A. Di Lorenzo, S. Richard, M. J. Comb, A. Guo, S. G. Clarke and M. T. Bedford, Sci. Rep., 2013, 3, 1311 CrossRef PubMed.
- F. Zhang, J. Kerbl-Knapp, M. J. Rodriguez Colman, A. Meinitzer, T. Macher, N. Vujic, S. Fasching, E. Jany-Luig, M. Korbelius, K. B. Kuentzel, M. Mack, A. Akhmetshina, A. Pirchheim, M. Paar, B. Rinner, G. Horl, E. Steyrer, U. Stelzl, B. Burgering, T. Eisenberg, B. Pertschy, D. Kratky and T. Madl, Cells Rep. Methods, 2021, 1, 100016 CrossRef CAS PubMed.
- G. E. Deibler and R. E. Martenson, J. Biol. Chem., 1973, 248, 2387–2391 CrossRef CAS PubMed.
- M. D. Fulton, T. Brown and Y. G. Zheng, Int. J. Mol. Sci., 2019, 20, 3322 CrossRef CAS PubMed.
- R. J. Nijveldt, T. Teerlink, C. van Guldener, H. A. Prins, A. A. van Lambalgen, C. D. Stehouwer, J. A. Rauwerda and P. A. van Leeuwen, Nephrol., Dial., Transplant., 2003, 18, 2542–2550 CrossRef CAS PubMed.
- W. K. Paik and S. Kim, Biochem. Biophys. Res. Commun., 1967, 29, 14–20 CrossRef CAS PubMed.
- W. K. Paik and S. Kim, J. Biol. Chem., 1968, 243, 2108–2114 CrossRef CAS PubMed.
- M. Friedman, K. H. Shull and E. Farber, Biochem. Biophys. Res. Commun., 1969, 34, 857–864 CrossRef CAS PubMed.
- T. Nakajima, Y. Matsuoka and Y. Kakimoto, Biochim. Biophys. Acta, 1971, 230, 212–222 CrossRef CAS PubMed.
- W. K. Paik, D. C. Paik and S. Kim, Trends Biochem. Sci., 2007, 32, 146–152 CrossRef CAS PubMed.
- S. K. Ghosh, W. K. Paik and S. Kim, J. Biol. Chem., 1988, 263, 19024–19033 CrossRef CAS PubMed.
- R. Rajpurohit, S. O. Lee, J. O. Park, W. K. Paik and S. Kim, J. Biol. Chem., 1994, 269, 1075–1082 CrossRef CAS PubMed.
- J. D. Gary, W. J. Lin, M. C. Yang, H. R. Herschman and S. Clarke, J. Biol. Chem., 1996, 271, 12585–12594 CrossRef CAS PubMed.
- W. J. Lin, J. D. Gary, M. C. Yang, S. Clarke and H. R. Herschman, J. Biol. Chem., 1996, 271, 15034–15044 CrossRef CAS PubMed.
- J. Tang, A. Frankel, R. J. Cook, S. Kim, W. K. Paik, K. R. Williams, S. Clarke and H. R. Herschman, J. Biol. Chem., 2000, 275, 7723–7730 CrossRef CAS PubMed.
- M. F. Henry and P. A. Silver, Mol. Cell. Biol., 1996, 16, 3668–3678 CrossRef CAS PubMed.
- A. E. McBride, V. H. Weiss, H. K. Kim, J. M. Hogle and P. A. Silver, J. Biol. Chem., 2000, 275, 3128–3136 CrossRef CAS PubMed.
- H. L. Schubert, R. M. Blumenthal and X. Cheng, Trends Biochem. Sci., 2003, 28, 329–335 CrossRef CAS PubMed.
- Y. Yang and M. T. Bedford, Nat. Rev. Cancer, 2013, 13, 37–50 CrossRef CAS PubMed.
- S. K. Tewary, Y. G. Zheng and M. C. Ho, Cell. Mol. Life Sci., 2019, 76, 2917–2932 CrossRef CAS PubMed.
- M. T. Bedford and S. G. Clarke, Mol. Cell, 2009, 33, 1–13 CrossRef CAS PubMed.
- Y. Morales, T. Cáceres, K. May and J. M. Hevel, Arch. Biochem. Biophys., 2016, 590, 138–152 CrossRef CAS PubMed.
- J. Fuhrmann, K. W. Clancy and P. R. Thompson, Chem. Rev., 2015, 115, 5413–5461 CrossRef CAS PubMed.
- V. Cura and J. Cavarelli, Life, 2021, 11, 1263 CrossRef CAS PubMed.
- A. Frankel and S. Clarke, J. Biol. Chem., 2000, 275, 32974–32982 CrossRef CAS PubMed.
- D. Chen, H. Ma, H. Hong, S. S. Koh, S. M. Huang, B. T. Schurter, D. W. Aswad and M. R. Stallcup, Science, 1999, 284, 2174–2177 CrossRef CAS PubMed.
- A. Frankel, N. Yadav, J. H. Lee, T. L. Branscombe, S. Clarke and M. T. Bedford, J. Biol. Chem., 2002, 277, 3537–3543 CrossRef CAS PubMed.
- J. Lee, J. Sayegh, J. Daniel, S. Clarke and M. T. Bedford, J. Biol. Chem., 2005, 280, 32890–32896 CrossRef CAS PubMed.
- A. Hadjikyriacou and S. G. Clarke, Biochemistry, 2017, 56, 2612–2626 CrossRef CAS PubMed.
- M. Wang, J. Fuhrmann and P. R. Thompson, Biochemistry, 2014, 53, 7884–7892 CrossRef CAS PubMed.
- Y. Yang, A. Hadjikyriacou, Z. Xia, S. Gayatri, D. Kim, C. Zurita-Lopez, R. Kelly, A. Guo, W. Li, S. G. Clarke and M. T. Bedford, Nat. Commun., 2015, 6, 6428 CrossRef CAS PubMed.
- C. I. Zurita-Lopez, T. Sandberg, R. Kelly and S. G. Clarke, J. Biol. Chem., 2012, 287, 7859–7870 CrossRef CAS PubMed.
- Y. Feng, A. Hadjikyriacou and S. G. Clarke, J. Biol. Chem., 2014, 289, 32604–32616 CrossRef CAS PubMed.
- N. Stopa, J. E. Krebs and D. Shechter, Cell. Mol. Life Sci., 2015, 72, 2041–2059 CrossRef CAS PubMed.
- M. R. Pawlak, C. A. Scherer, J. Chen, M. J. Roshon and H. E. Ruley, Mol. Cell. Biol., 2000, 20, 4859–4869 CrossRef CAS PubMed.
- A. Hadjikyriacou, Y. Yang, A. Espejo, M. T. Bedford and S. G. Clarke, J. Biol. Chem., 2015, 290, 16723–16743 CrossRef CAS PubMed.
- O. Zurita Rendon, L. Silva Neiva, F. Sasarman and E. A. Shoubridge, Hum. Mol. Genet., 2014, 23, 5159–5170 CrossRef PubMed.
- V. F. Rhein, J. Carroll, S. Ding, I. M. Fearnley and J. E. Walker, J. Biol. Chem., 2013, 288, 33016–33026 CrossRef CAS PubMed.
- J. Carroll, S. Ding, I. M. Fearnley and J. E. Walker, J. Biol. Chem., 2013, 288, 24799–24808 CrossRef CAS PubMed.
- U. F. S. Hameed, O. Sanislav, S. T. Lay, S. J. Annesley, C. Jobichen, P. R. Fisher, K. Swaminathan and S. T. Arold, Cell Rep., 2018, 24, 1996–2004 CrossRef PubMed.
- L. Sun, M. Wang, Z. Lv, N. Yang, Y. Liu, S. Bao, W. Gong and R. M. Xu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20538–20543 CrossRef CAS PubMed.
- Y. Hatanaka, T. Tsusaka, N. Shimizu, K. Morita, T. Suzuki, S. Machida, M. Satoh, A. Honda, M. Hirose, S. Kamimura, N. Ogonuki, T. Nakamura, K. Inoue, Y. Hosoi, N. Dohmae, T. Nakano, H. Kurumizaka, K. Matsumoto, Y. Shinkai and A. Ogura, Cell Rep., 2017, 20, 2756–2765 CrossRef CAS PubMed.
- E. Guccione and S. Richard, Nat. Rev. Mol. Cell Biol., 2019, 20, 642–657 CrossRef CAS PubMed.
- C. Peng and C. C. L. Wong, Expert Rev. Proteomics, 2017, 14, 157–170 CrossRef CAS PubMed.
- R. S. Blanc and S. Richard, Mol. Cell, 2017, 65, 8–24 CrossRef CAS PubMed.
- M. T. Bedford, J. Cell Sci., 2007, 120, 4243–4246 CrossRef CAS PubMed.
- Q. Wu, M. Schapira, C. H. Arrowsmith and D. Barsyte-Lovejoy, Nat. Rev. Drug Discovery, 2021, 20, 509–530 CrossRef CAS PubMed.
- E. Guccione and S. Richard, Nat. Rev. Mol. Cell Biol., 2019, 20, 642–657 CrossRef CAS PubMed.
- B. M. Lorton and D. Shechter, Cell. Mol. Life Sci., 2019, 76, 2933–2956 CrossRef CAS PubMed.
- R. S. Blanc and S. Richard, Mol. Cell, 2017, 65, 8–24 CrossRef CAS PubMed.
- M. T. Bedford and S. Richard, Mol. Cell, 2005, 18, 263–272 CrossRef CAS PubMed.
- J. Jarrold and C. C. Davies, Trends Mol. Med., 2019, 25, 993–1009 CrossRef CAS PubMed.
- D. Levy, Cell. Mol. Life Sci., 2019, 76, 2873–2883 CrossRef CAS PubMed.
- X. Zhao, Z. Chong, Y. Chen, X. L. Zheng, Q. F. Wang and Y. Li, J. Biol. Chem., 2022, 102517 CrossRef CAS PubMed.
- A. Di Lorenzo and M. T. Bedford, FEBS Lett., 2011, 585, 2024–2031 CrossRef CAS PubMed.
- S. Gardini, S. Cheli, S. Baroni, G. Di Lascio, G. Mangiavacchi, N. Micheletti, C. L. Monaco, L. Savini, D. Alocci, S. Mangani and N. Niccolai, PLoS One, 2016, 11, e0148174 CrossRef PubMed.
- S. M. Greenblatt, F. Liu and S. D. Nimer, Exp. Hematol., 2016, 44, 435–441 CrossRef CAS PubMed.
- Y. Z. Yang and M. T. Bedford, Nat. Rev. Cancer, 2013, 13, 37–50 CrossRef CAS PubMed.
- H. Wei, R. Mundade, K. C. Lange and T. Lu, Cell Cycle, 2014, 13, 32–41 CrossRef CAS PubMed.
- H. Kim and Z. A. Ronai, Cell Stress, 2020, 4, 199–215 CrossRef CAS PubMed.
- H. Sun, Y. Zhou, M. F. Skaro, Y. Wu, Z. Qu, F. Mao, S. Zhao and Y. Xu, Cancer Res., 2020, 80, 1143–1155 CrossRef CAS PubMed.
- A. Spannhoff, R. Heinke, I. Bauer, P. Trojer, E. Metzger, R. Gust, R. Schule, G. Brosch, W. Sippl and M. Jung, J. Med. Chem., 2007, 50, 2319–2325 CrossRef CAS PubMed.
- R. Wang, W. H. Zheng, H. Q. Yu, H. T. Deng and M. K. Luo, J. Am. Chem. Soc., 2011, 133, 7648–7651 CrossRef CAS PubMed.
- R. Ragno, S. Simeoni, S. Castellano, C. Vicidomini, A. Mai, A. Caroli, A. Tramontano, C. Bonaccini, P. Trojer, I. Bauer, G. Brosch and G. Sbardella, J. Med. Chem., 2007, 50, 1241–1253 CrossRef CAS PubMed.
- A. S. M. Li, F. Li, M. S. Eram, A. Bolotokova, C. C. Dela Sena and M. Vedadi, Methods, 2020, 175, 30–43 CrossRef CAS PubMed.
- K. K. Li, C. Luo, D. Wang, H. Jiang and Y. G. Zheng, Med. Res. Rev., 2012, 32, 815–867 CrossRef CAS PubMed.
- W. L. Wooderchak, Z. S. Zhou and J. Hevel, Curr. Protoc. Toxicol., 2008, 4.26.1–4.26.12 Search PubMed.
- H. Zeng and W. Xu, in Epigenetic Technological Applications, ed. Y. G. Zheng, Academic Press, San Diego, 2015, pp.333–361 Search PubMed.
- M. Luo, ACS Chem. Biol., 2012, 7, 443–463 CrossRef CAS PubMed.
- A. Spannhoff, A.-T. Hauser, R. Heinke, W. Sippl and M. Jung, ChemMedChem, 2009, 4, 1568–1582 CrossRef CAS PubMed.
- H. Hu, K. Qian, M. C. Ho and Y. G. Zheng, Expert Opin. Invest. Drugs, 2016, 25, 335–358 CrossRef CAS PubMed.
- K. Y. Horiuchi, M. M. Eason, J. J. Ferry, J. L. Planck, C. P. Walsh, R. F. Smith, K. T. Howitz and H. Ma, Assay Drug Dev. Technol., 2013, 11, 227–236 CrossRef CAS PubMed.
- D. Cheng, V. Vemulapalli and M. T. Bedford, Methods Enzymol., 2012, 512, 71–92 CAS.
- T. M. Lakowski, C. Zurita-Lopez, S. G. Clarke and A. Frankel, Anal. Biochem., 2010, 397, 1–11 CrossRef CAS PubMed.
- S. M. Carlson and O. Gozani, J. Mol. Biol., 2014, 426, 3350–3362 CrossRef CAS PubMed.
- D. Cheng, N. Yadav, R. W. King, M. S. Swanson, E. J. Weinstein and M. T. Bedford, J. Biol. Chem., 2004, 279, 23892–23899 CrossRef CAS PubMed.
- E.-M. Bissinger, R. Heinke, A. Spannhoff, A. Eberlin, E. Metzger, V. Cura, P. Hassenboehler, J. Cavarelli, R. Schüle, M. T. Bedford, W. Sippl and M. Jung, Bioorg. Med. Chem., 2011, 19, 3717–3731 CrossRef CAS PubMed.
- D. Cheng, S. Valente, S. Castellano, G. Sbardella, R. Di Santo, R. Costi, M. T. Bedford and A. Mai, J. Med. Chem., 2011, 54, 4928–4932 CrossRef CAS PubMed.
- S. Castellano, C. Milite, R. Ragno, S. Simeoni, A. Mai, V. Limongelli, E. Novellino, I. Bauer, G. Brosch, A. Spannhoff, D. Cheng, M. T. Bedford and G. Sbardella, ChemMedChem, 2010, 5, 398–414 CrossRef CAS PubMed.
- K. Bonham, S. Hemmers, Y.-H. Lim, D. M. Hill, M. G. Finn and K. A. Mowen, FEBS J., 2010, 277, 2096–2108 CrossRef CAS PubMed.
- J. Dowden, W. Hong, R. V. Parry, R. A. Pike and S. G. Ward, Bioorg. Med. Chem. Lett., 2010, 20, 2103–2105 CrossRef CAS PubMed.
- J. Dowden, R. A. Pike, R. V. Parry, W. Hong, U. A. Muhsen and S. G. Ward, Org. Biomol. Chem., 2011, 9, 7814–7821 RSC.
- S. H. Sinha, E. A. Owens, Y. Feng, Y. Yang, Y. Xie, Y. Tu, M. Henary and Y. G. Zheng, Eur. J. Med. Chem., 2012, 54, 647–659 CrossRef CAS PubMed.
- L. Yan, C. Yan, K. Qian, H. Su, S. A. Kofsky-Wofford, W.-C. Lee, X. Zhao, M.-C. Ho, I. Ivanov and Y. G. Zheng, J. Med. Chem., 2014, 57, 2611–2622 CrossRef CAS PubMed.
- F. Liu, F. Li, A. Ma, E. Dobrovetsky, A. Dong, C. Gao, I. Korboukh, J. Liu, D. Smil, P. J. Brown, S. V. Frye, C. H. Arrowsmith, M. Schapira, M. Vedadi and J. Jin, J. Med. Chem., 2013, 56, 2110–2124 CrossRef CAS PubMed.
- M. Allan, S. Manku, E. Therrien, N. Nguyen, S. Styhler, M.-F. Robert, A.-C. Goulet, A. J. Petschner, G. Rahil, A. Robert MacLeod, R. Déziel, J. M. Besterman, H. Nguyen and A. Wahhab, Bioorg. Med. Chem. Lett., 2009, 19, 1218–1223 CrossRef CAS PubMed.
- A. V. Purandare, Z. Chen, T. Huynh, S. Pang, J. Geng, W. Vaccaro, M. A. Poss, J. Oconnell, K. Nowak and L. Jayaraman, Bioorg. Med. Chem. Lett., 2008, 18, 4438–4441 CrossRef CAS PubMed.
- T. Huynh, Z. Chen, S. Pang, J. Geng, T. Bandiera, S. Bindi, P. Vianello, F. Roletto, S. Thieffine, A. Galvani, W. Vaccaro, M. A. Poss, G. L. Trainor, M. V. Lorenzi, M. Gottardis, L. Jayaraman and A. V. Purandare, Bioorg. Med. Chem. Lett., 2009, 19, 2924–2927 CrossRef CAS PubMed.
- E. Therrien, G. Larouche, S. Manku, M. Allan, N. Nguyen, S. Styhler, M.-F. Robert, A.-C. Goulet, J. M. Besterman, H. Nguyen and A. Wahhab, Bioorg. Med. Chem. Lett., 2009, 19, 6725–6732 CrossRef CAS PubMed.
- H. Wan, T. Huynh, S. Pang, J. Geng, W. Vaccaro, M. A. Poss, G. L. Trainor, M. V. Lorenzi, M. Gottardis, L. Jayaraman and A. V. Purandare, Bioorg. Med. Chem. Lett., 2009, 19, 5063–5066 CrossRef CAS PubMed.
- J. S. Sack, S. Thieffine, T. Bandiera, M. Fasolini, G. J. Duke, L. Jayaraman, K. F. Kish, H. E. Klei, A. V. Purandare, P. Rosettani, S. Troiani, D. Xie and J. A. Bertrand, Biochem. J., 2011, 436, 331–339 CrossRef CAS PubMed.
- L. Alinari, K. V. Mahasenan, F. Yan, V. Karkhanis, J.-H. Chung, E. M. Smith, C. Quinion, P. L. Smith, L. Kim, J. T. Patton, R. Lapalombella, B. Yu, Y. Wu, S. Roy, A. De Leo, S. Pileri, C. Agostinelli, L. Ayers, J. E. Bradner, S. Chen-Kiang, O. Elemento, T. Motiwala, S. Majumder, J. C. Byrd, S. Jacob, S. Sif, C. Li and R. A. Baiocchi, Blood, 2015, 125, 2530–2543 CrossRef CAS PubMed.
- R. A. Baiocchi, C. Li, H. Lai and S. Sif, WO2014145214A2, 2014.
- B. R. Selvi, K. Batta, A. H. Kishore, K. Mantelingu, R. A. Varier, K. Balasubramanyam, S. K. Pradhan, D. Dasgupta, S. Sriram, S. Agrawal and T. K. Kundu, J. Biol. Chem., 2010, 285, 7143–7152 CrossRef CAS PubMed.
- Y. Feng, M. Li, B. Wang and Y. G. Zheng, J. Med. Chem., 2010, 53, 6028–6039 CrossRef CAS PubMed.
- J. Wang, L. Chen, S. H. Sinha, Z. Liang, H. Chai, S. Muniyan, Y.-W. Chou, C. Yang, L. Yan, Y. Feng, K. Kathy Li, M.-F. Lin, H. Jiang, Y. George Zheng and C. Luo, J. Med. Chem., 2012, 55, 7978–7987 CrossRef CAS PubMed.
- B. B. Suh-Lailam and J. M. Hevel, Anal. Biochem., 2010, 398, 218–224 CrossRef CAS PubMed.
- J. Wu, N. Xie, Y. Feng and Y. G. Zheng, J. Biomol. Screening, 2012, 17, 237–244 CrossRef CAS PubMed.
- P. Rathert, X. Cheng and A. Jeltsch, BioTechniques, 2007, 43, 602 CrossRef CAS PubMed , 604, 606 passim.
- A. Dhayalan, E. Dimitrova, P. Rathert and A. Jeltsch, J. Biomol. Screening, 2009, 14, 1129–1133 CrossRef CAS PubMed.
- J. Wu, N. Xie, Y. Feng and Y. G. Zheng, J. Biomol. Screening, 2012, 17, 237–244 CrossRef CAS PubMed.
- G. Ibanez, D. Shum, G. Blum, B. Bhinder, C. Radu, C. Antczak, M. Luo and H. Djaballah, Comb. Chem. High Throughput Screening, 2012, 15, 359–371 CrossRef CAS PubMed.
- H. Hu, E. A. Owens, H. Su, L. Yan, A. Levitz, X. Zhao, M. Henary and Y. G. Zheng, J. Med. Chem., 2015, 58, 1228–1243 CrossRef CAS PubMed.
- A. Siarheyeva, G. Senisterra, A. Allali-Hassani, A. Dong, E. Dobrovetsky, G. A. Wasney, I. Chau, R. Marcellus, T. Hajian, F. Liu, I. Korboukh, D. Smil, Y. Bolshan, J. Min, H. Wu, H. Zeng, P. Loppnau, G. Poda, C. Griffin, A. Aman, P. J. Brown, J. Jin, R. Al-awar, C. H. Arrowsmith, M. Schapira and M. Vedadi, Structure, 2012, 20, 1425–1435 CrossRef CAS PubMed.
- H. Ü. Kaniskan, M. M. Szewczyk, Z. Yu, M. S. Eram, X. Yang, K. Schmidt, X. Luo, M. Dai, F. He, I. Zang, Y. Lin, S. Kennedy, F. Li, E. Dobrovetsky, A. Dong, D. Smil, S.-J. Min, M. Landon, J. Lin-Jones, X.-P. Huang, B. L. Roth, M. Schapira, P. Atadja, D. Barsyte-Lovejoy, C. H. Arrowsmith, P. J. Brown, K. Zhao, J. Jin and M. Vedadi, Angew. Chem., Int. Ed., 2015, 54, 5166–5170 CrossRef CAS PubMed.
- W. Zheng, G. Ibáñez, H. Wu, G. Blum, H. Zeng, A. Dong, F. Li, T. Hajian, A. Allali-Hassani, M. F. Amaya, A. Siarheyeva, W. Yu, P. J. Brown, M. Schapira, M. Vedadi, J. Min and M. Luo, J. Am. Chem. Soc., 2012, 134, 18004–18014 CrossRef CAS PubMed.
- L. H. Mitchell, A. E. Drew, S. A. Ribich, N. Rioux, K. K. Swinger, S. L. Jacques, T. Lingaraj, P. A. Boriack-Sjodin, N. J. Waters, T. J. Wigle, O. Moradei, L. Jin, T. Riera, M. Porter-Scott, M. P. Moyer, J. J. Smith, R. Chesworth and R. A. Copeland, ACS Med. Chem. Lett., 2015, 6, 655–659 CrossRef CAS PubMed.
- M. S. Eram, Y. Shen, M. M. Szewczyk, H. Wu, G. Senisterra, F. Li, K. V. Butler, H. Ü. Kaniskan, B. A. Speed, C. dela Seña, A. Dong, H. Zeng, M. Schapira, P. J. Brown, C. H. Arrowsmith, D. Barsyte-Lovejoy, J. Liu, M. Vedadi and J. Jin, ACS Chem. Biol., 2015, 11, 772–781 CrossRef PubMed.
- D. Smil, M. S. Eram, F. Li, S. Kennedy, M. M. Szewczyk, P. J. Brown, D. Barsyte-Lovejoy, C. H. Arrowsmith, M. Vedadi and M. Schapira, ACS Med. Chem. Lett., 2015, 6, 408–412 CrossRef CAS PubMed.
- N. Fontán, P. García-Domínguez, R. Álvarez and Á. R. de Lera, Bioorg. Med. Chem., 2013, 21, 2056–2067 CrossRef PubMed.
- M. van Haren, L. Q. van Ufford, E. E. Moret and N. I. Martin, Org. Biomol. Chem., 2015, 13, 549–560 RSC.
- A. Spannhoff, R. Heinke, I. Bauer, P. Trojer, E. Metzger, R. Gust, R. Schüle, G. Brosch, W. Sippl and M. Jung, J. Med. Chem., 2007, 50, 2319–2325 CrossRef CAS PubMed.
- R. Heinke, A. Spannhoff, R. Meier, P. Trojer, I. Bauer, M. Jung and W. Sippl, ChemMedChem, 2009, 4, 69–77 CrossRef CAS PubMed.
- A. Spannhoff, R. Machmur, R. Heinke, P. Trojer, I. Bauer, G. Brosch, R. Schüle, W. Hanefeld, W. Sippl and M. Jung, Bioorg. Med. Chem. Lett., 2007, 17, 4150–4153 CrossRef CAS PubMed.
- Y. Xie, R. Zhou, F. Lian, Y. Liu, L. Chen, Z. Shi, N. Zhang, M. Zheng, B. Shen, H. Jiang, Z. Liang and C. Luo, Org. Biomol. Chem., 2014, 12, 9665–9673 RSC.
- E. Chan-Penebre, K. G. Kuplast, C. R. Majer, P. A. Boriack-Sjodin, T. J. Wigle, L. D. Johnston, N. Rioux, M. J. Munchhof, L. Jin, S. L. Jacques, K. A. West, T. Lingaraj, K. Stickland, S. A. Ribich, A. Raimondi, M. P. Scott, N. J. Waters, R. M. Pollock, J. J. Smith, O. Barbash, M. Pappalardi, T. F. Ho, K. Nurse, K. P. Oza, K. T. Gallagher, R. Kruger, M. P. Moyer, R. A. Copeland, R. Chesworth and K. W. Duncan, Nat. Chem. Biol., 2015, 11, 432–437 CrossRef CAS PubMed.
- N. Rioux, K. W. Duncan, R. J. Lantz, X. Miao, E. Chan-Penebre, M. P. Moyer, M. J. Munchhof, R. A. Copeland, R. Chesworth and N. J. Waters, Xenobiotica, 2015, 1–10 Search PubMed.
- K. W. Duncan, N. Rioux, P. A. Boriack-Sjodin, M. J. Munchhof, L. A. Reiter, C. R. Majer, L. Jin, L. D. Johnston, E. Chan-Penebre, K. G. Kuplast, M. Porter Scott, R. M. Pollock, N. J. Waters, J. J. Smith, M. P. Moyer, R. A. Copeland and R. Chesworth, ACS Med. Chem. Lett., 2015, 7, 162–166 CrossRef PubMed.
- X.-R. Yu, Y. Tang, W.-J. Wang, S. Ji, S. Ma, L. Zhong, C.-H. Zhang, J. Yang, X.-A. Wu, Z.-Y. Fu, L.-L. Li and S.-Y. Yang, Bioorg. Med. Chem. Lett., 2015, 25, 5449–5453 CrossRef CAS PubMed.
- T. L. Graves, Y. Zhang and J. E. Scott, Anal. Biochem., 2008, 373, 296–306 CrossRef CAS PubMed.
- J. X. Wang and R. R. Breaker, Biochem. Cell Biol., 2008, 86, 157–168 CrossRef CAS PubMed.
- R. T. Batey, Wiley Interdiscip. Rev.: RNA, 2011, 2, 299–311 CrossRef CAS PubMed.
- Y. Su, S. F. Hickey, S. G. Keyser and M. C. Hammond, J. Am. Chem. Soc., 2016, 138, 7040–7047 CrossRef CAS PubMed.
- E. Collazo, J. F. Couture, S. Bulfer and R. C. Trievel, Anal. Biochem., 2005, 342, 86–92 CrossRef CAS PubMed.
- R. Wang, G. Ibanez, K. Islam, W. Zheng, G. Blum, C. Sengelaub and M. Luo, Mol. BioSyst., 2011, 7, 2970–2981 RSC.
- A. Allali-Hassani, G. A. Wasney, A. Siarheyeva, T. Hajian, C. H. Arrowsmith and M. Vedadi, J. Biomol. Screening, 2012, 17, 71–84 CrossRef CAS PubMed.
- M. Kumar, T. Zielinski and R. G. Lowery, Assay Drug Dev. Technol., 2015, 13, 200–209 CrossRef CAS PubMed.
- T. A. Klink, M. Staeben, K. Twesten, A. L. Kopp, M. Kumar, R. S. Dunn, C. A. Pinchard, K. M. Kleman-Leyer, M. Klumpp and R. G. Lowery, J. Biomol. Screening, 2012, 17, 59–70 CrossRef CAS PubMed.
- K. M. Dorgan, W. L. Wooderchak, D. P. Wynn, E. L. Karschner, J. F. Alfaro, Y. Cui, Z. S. Zhou and J. M. Hevel, Anal. Biochem., 2006, 350, 249–255 CrossRef CAS PubMed.
- G. Ibanez, J. L. McBean, Y. M. Astudillo and M. Luo, Anal. Biochem., 2010, 401, 203–210 CrossRef CAS PubMed.
- I. Hemeon, J. A. Gutierrez, M. C. Ho and V. L. Schramm, Anal. Chem., 2011, 83, 4996–5004 CrossRef CAS PubMed.
- E. S. Burgos, R. O. Walters, D. M. Huffman and D. Shechter, Chem. Sci., 2017, 8, 6601–6612 RSC.
- H. Wang, R. M. Straubinger, J. M. Aletta, J. Cao, X. Duan, H. Yu and J. Qu, J. Am. Soc. Mass Spectrom., 2009, 20, 507–519 CrossRef CAS PubMed.
- M. T. Bedford and S. Richard, Mol. Cell, 2005, 18, 263–272 CrossRef CAS PubMed.
- Y. Feng, N. Xie, J. Wu, C. Yang and Y. G. Zheng, Biochem. Biophys. Res. Commun., 2009, 379, 567–572 CrossRef CAS PubMed.
- Y. Feng, N. Xie, M. Y. Jin, M. R. Stahley, J. T. Stivers and Y. G. Zheng, Biochemistry, 2011, 50, 7033–7044 CrossRef CAS PubMed.
- K. Qian, H. Hu, H. Xu and Y. G. Zheng, Signal Transduction Targeted Ther., 2018, 3, 6 CrossRef PubMed.
- D. Dilworth and D. Barsyte-Lovejoy, Cell. Mol. Life Sci., 2019, 76, 2967–2985 CrossRef CAS PubMed.
- R. Ferreira de Freitas, D. Ivanochko and M. Schapira, Molecules, 2019, 24, 4492 CrossRef CAS PubMed.
- M. B. Dillon, D. A. Bachovchin, S. J. Brown, M. G. Finn, H. Rosen, B. F. Cravatt and K. A. Mowen, ACS Chem. Biol., 2012, 7, 1198–1204 CrossRef CAS PubMed.
- D. K. Nomura, M. M. Dix and B. F. Cravatt, Nat. Rev. Cancer, 2010, 10, 630–638 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–799 CrossRef CAS PubMed.
- H. Lin, M. Wang, Y. W. Zhang, S. Tong, R. A. Leal, R. Shetty, K. Vaddi and J. I. Luengo, ACS Med. Chem. Lett., 2019, 10, 1033–1038 CrossRef PubMed.
- Y. Shen, F. Li, M. M. Szewczyk, L. Halabelian, K. S. Park, I. Chau, A. Dong, H. Zeng, H. Chen, F. Meng, D. Barsyte-Lovejoy, C. H. Arrowsmith, P. J. Brown, J. Liu, M. Vedadi and J. Jin, J. Med. Chem., 2020, 63, 5477–5487 CrossRef CAS PubMed.
- Y. Shen, G. Gao, X. Yu, H. Kim, L. Wang, L. Xie, M. Schwarz, X. Chen, E. Guccione, J. Liu, M. T. Bedford and J. Jin, J. Med. Chem., 2020, 63, 9977–9989 CrossRef CAS PubMed.
- S. H. Sinha, E. A. Owens, Y. Feng, Y. Yang, Y. Xie, Y. Tu, M. Henary and Y. G. Zheng, Eur. J. Med. Chem., 2012, 54, 647–659 CrossRef CAS PubMed.
- H. Hu, E. A. Owens, H. Su, L. Yan, A. Levitz, X. Zhao, M. Henary and Y. G. Zheng, J. Med. Chem., 2015, 58, 1228–1243 CrossRef CAS PubMed.
- H. Su, C. W. Sun, S. M. Liu, X. He, H. Hu, K. M. Pawlik, T. M. Townes, X. Han, C. A. Klug, M. Henary, Y. Chen, L. Li, Y. G. Zheng and X. Zhao, Blood Adv., 2018, 2, 2829–2836 CrossRef CAS PubMed.
- O. Obianyo, C. P. Causey, J. E. Jones and P. R. Thompson, ACS Chem. Biol., 2011, 6, 1127–1135 CrossRef CAS PubMed.
- K. L. Bicker, O. Obianyo, H. L. Rust and P. R. Thompson, Mol. BioSyst., 2011, 7, 48–51 RSC.
- S. A. Mann, A. Salsburg, C. P. Causey and B. Knuckley, Bioorg. Med. Chem., 2019, 27, 224–229 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- S. A. Mann, M. K. DeMart, B. May, C. P. Causey and B. Knuckley, Biochem. J., 2020, 477, 2971–2980 CrossRef CAS PubMed.
- M. B. DuBose, T. Sartawi, T. M. Sawatzky, C. P. Causey, F. K. Rehman and B. Knuckley, J. Biol. Chem., 2022, 298, 102205 CrossRef CAS PubMed.
- M. J. van Haren, N. Marechal, N. Troffer-Charlier, A. Cianciulli, G. Sbardella, J. Cavarelli and N. I. Martin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3625–3630 CrossRef CAS PubMed.
- A. A. Al-Hamashi, D. Chen, Y. Deng, G. Dong and R. Huang, Acta Pharm. Sin. B, 2021, 11, 2709–2718 CrossRef CAS PubMed.
- C. S. Zhang, R. L. Weller, J. S. Thorson and S. R. Rajski, J. Am. Chem. Soc., 2006, 128, 2760–2761 CrossRef CAS PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- W. Peters, S. Willnow, M. Duisken, H. Kleine, T. Macherey, K. E. Duncan, D. W. Litchfield, B. Luscher and E. Weinhold, Angew. Chem., Int. Ed., 2010, 49, 5170–5173 CrossRef CAS PubMed.
- K. Islam, W. H. Zheng, H. Q. Yu, H. T. Deng and M. K. Luo, ACS Chem. Biol., 2011, 6, 679–684 CrossRef CAS PubMed.
- R. Wang, G. Ibanez, K. Islam, W. H. Zheng, G. Blum, C. Sengelaub and M. K. Luo, Mol. BioSyst., 2011, 7, 2970–2981 RSC.
- O. Binda, M. Boyce, J. S. Rush, K. K. Palaniappan, C. R. Bertozzi and O. Gozani, ChemBioChem, 2011, 12, 330–334 CrossRef CAS PubMed.
- S. Willnow, M. Martin, B. Luscher and E. Weinhold, ChemBioChem, 2012, 13, 1167–1173 CrossRef CAS PubMed.
- I. R. Bothwell, K. Islam, Y. L. Chen, W. H. Zheng, G. Blum, H. T. Deng and M. K. Luo, J. Am. Chem. Soc., 2012, 134, 14905–14912 CrossRef CAS PubMed.
- K. Islam, Cell Chem. Biol., 2018, 25, 1171–1184 CrossRef CAS PubMed.
- R. Wang and M. K. Luo, Curr. Opin. Chem. Biol., 2013, 17, 729–737 CrossRef CAS PubMed.
- Y. Sohtome, T. Shimazu, J. Barjau, S. Fujishiro, M. Akakabe, N. Terayama, K. Dodo, A. Ito, M. Yoshida, Y. Shinkai and M. Sodeoka, Chem. Commun., 2018, 54, 9202–9205 RSC.
- Y. Sohtome, T. Shimazu, Y. Shinkai and M. Sodeoka, Acc. Chem. Res., 2021, 54, 3818–3827 CrossRef CAS PubMed.
- B. P. Pollack, S. V. Kotenko, W. He, L. S. Izotova, B. L. Barnoski and S. Pestka, J. Biol. Chem., 1999, 274, 31531–31542 CrossRef CAS PubMed.
- J. H. Lee, J. R. Cook, B. P. Pollack, T. G. Kinzy, D. Norris and S. Pestka, Biochem. Biophys. Res. Commun., 2000, 274, 105–111 CrossRef CAS PubMed.
- X. Zhang and X. D. Cheng, Structure, 2003, 11, 509–520 CrossRef CAS PubMed.
- P. H. Yu, Can. J. Biochem. Cell Biol., 1984, 62, 964–969 CrossRef CAS PubMed.
- Y. Takata and M. Fujioka, Biochemistry, 1992, 31, 4369–4374 CrossRef CAS PubMed.
- P. P. Bera, T. Stein, M. Head-Gordon and T. J. Lee, Astrobiology, 2017, 17, 771–785 CrossRef CAS PubMed.
- H. Koster, D. P. Little, P. Luan, R. Muller, S. M. Siddiqi, S. Marappan and P. Yip, Assay Drug Dev. Technol., 2007, 5, 381–390 CrossRef PubMed.
- T. Deguchi and J. Barchas, J. Biol. Chem., 1971, 246, 3175–3181 CrossRef CAS PubMed.
- C. Dalhoff, M. Huben, T. Lenz, P. Poot, E. Nordhoff, H. Koster and E. Weinhold, ChemBioChem, 2010, 11, 256–265 CrossRef CAS PubMed.
- M. N. Maas, J. C. J. Hintzen and J. Mecinovic, Chem. Commun., 2022, 58, 7216–7231 RSC.
- B. D. Horning, R. M. Suciu, D. A. Ghadiri, O. A. Ulanovskaya, M. L. Matthews, K. M. Lum, K. M. Backus, S. J. Brown, H. Rosen and B. F. Cravatt, J. Am. Chem. Soc., 2016, 138, 13335–13343 CrossRef CAS PubMed.
- V. Mai and L. R. Comstock, J. Org. Chem., 2011, 76, 10319–10324 CrossRef CAS PubMed.
- S. J. Hymbaugh, L. M. Pecor, C. M. Tracy and L. R. Comstock, ChemBioChem, 2019, 20, 379–384 CrossRef CAS PubMed.
- S. Valenzuela, J. Pizarro, A. M. Sandino, M. Vasquez, J. Fernandez, O. Hernandez, J. Patton and E. Spencer, J. Virol., 1991, 65, 3964–3967 CrossRef CAS PubMed.
- D. Polshakov, S. Rai, R. M. Wilson, E. T. Mack, M. Vogel, J. A. Krause, G. Burdzinski and M. S. Platz, Biochemistry, 2005, 44, 11241–11253 CrossRef CAS PubMed.
- D. D. Le, A. T. Cortesi, S. A. Myers, A. L. Burlingame and D. G. Fujimori, J. Am. Chem. Soc., 2013, 135, 2879–2882 CrossRef CAS PubMed.
- M. Holt and T. Muir, Annu. Rev. Biochem., 2015, 84, 265–290 CrossRef CAS PubMed.
- S. M. Ngola, P. C. Kearney, S. Mecozzi, K. Russell and D. A. Dougherty, J. Am. Chem. Soc., 1999, 121, 1192–1201 CrossRef CAS.
- M. A. Gamal-Eldin and D. H. Macartney, Org. Biomol. Chem., 2013, 11, 488–495 RSC.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- L. I. James, J. E. Beaver, N. W. Rice and M. L. Waters, J. Am. Chem. Soc., 2013, 135, 6450–6455 CrossRef CAS PubMed.
- A. G. Mullins, N. K. Pinkin, J. A. Hardin and M. L. Waters, Angew. Chem., Int. Ed., 2019, 58, 5282–5285 CrossRef CAS PubMed.
- J. E. Beaver and M. L. Waters, ACS Chem. Biol., 2016, 11, 643–653 CrossRef CAS PubMed.
- A. G. Mullins, L. E. St. Louis and M. L. Waters, Chem. Commun., 2020, 56, 3947–3950 RSC.
- P. J. Thornalley, Mol. Aspects Med., 1993, 14, 287–371 CrossRef CAS PubMed.
- D. Scumaci, E. Olivo, C. V. Fiumara, M. La Chimia, M. T. De Angelis, S. Mauro, G. Costa, F. A. Ambrosio, S. Alcaro, V. Agosti, F. S. Costanzo and G. Cuda, Cells, 2020, 9, 1968 CrossRef CAS PubMed.
- C. Sibbersen, J. Palmfeldt, J. Hansen, N. Gregersen, K. A. Jorgensen and M. Johannsen, Chem. Commun., 2013, 49, 4012–4014 RSC.
- C. Sibbersen, A. M. S. Oxvig, S. B. Olesen, C. B. Nielsen, J. J. Galligan, K. A. Jorgensen, J. Palmfeldt and M. Johannsen, ACS Chem. Biol., 2018, 13, 3294–3305 CrossRef CAS PubMed.
- D. M. Ray, E. Q. Jennings, I. Maksimovic, X. Chai, J. J. Galligan, Y. David and Q. Zheng, ACS Chem. Biol., 2022, 17, 756–761 CrossRef CAS PubMed.
- Y. Chen, W. Qin, Z. Li, Z. Guo, Y. Liu, T. Lan and C. Wang, Future Med. Chem., 2019, 11, 2979–2987 CrossRef CAS PubMed.
- T. Wang, E. F. Douglass, Jr., K. J. Fitzgerald and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 12429–12433 CrossRef CAS PubMed.
- Q. Zheng, N. D. Omans, R. Leicher, A. Osunsade, A. S. Agustinus, E. Finkin-Groner, H. D’Ambrosio, B. Liu, S. Chandarlapaty, S. Liu and Y. David, Nat. Commun., 2019, 10, 1289 CrossRef PubMed.
- M. S. K. Wanigasekara, X. Huang, J. K. Chakrabarty, A. Bugarin and S. M. Chowdhury, ACS Omega, 2018, 3, 14229–14235 CrossRef CAS PubMed.
- S. J. Wang, X. C. Wang, W. Shi, K. Wang and H. M. Ma, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 415–422 CrossRef CAS PubMed.
- D. A. Thompson, R. Ng and P. E. Dawson, J. Pept. Sci., 2016, 22, 311–319 CrossRef CAS PubMed.
- F. O. Fackelmayer, Trends Biochem. Sci., 2005, 30, 666–671 CrossRef CAS PubMed.
- N. Bagwan, H. H. El Ali and A. Lundby, Sci. Rep., 2021, 11, 2184 CrossRef CAS PubMed.
- M. A. Rodriguez, Parasitology, 2022, 149, 427–435 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |