
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the effect of partial B-site Al3+–Mg2+ dual substitution on optoelectronic, surface, and photocatalytic properties of BaTaO2N†
Mirabbos
Hojamberdiev
 *ab,
Ronald
Vargas
cd,
Zukhra C.
Kadirova
ef,
Katsuya
Teshima
bg and
Martin
Lerch
a
*ab,
Ronald
Vargas
cd,
Zukhra C.
Kadirova
ef,
Katsuya
Teshima
bg and
Martin
Lerch
a
aInstitut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: khujamberdiev@tu-berlin.de; hmirabbos@gmail.com
bDepartment of Materials Chemistry, Shinshu University, Nagano 380-8553, Japan
cInstituto Tecnológico de Chascomús (INTECH) – Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET)/Universidad Nacional de San Martín (UNSAM), Avenida Intendente Marino, Km 8,2, (B7130IWA), Chascomús, Provincia de Buenos Aires, Argentina
dEscuela de Bio y Nanotecnologías, Universidad Nacional de San Martín (UNSAM), Avenida Intendente Marino, Km 8,2, (B7130IWA), Chascomús, Provincia de Buenos Aires, Argentina
eDepartment of Inorganic Chemistry, National University of Uzbekistan, 100174 Tashkent, Uzbekistan
fUzbekistan-Japan Innovation Center of Youth, University Street 2B, 100095 Tashkent, Uzbekistan
gResearch Initiative for Supra-Materials, Shinshu University, 4-17-1 Wakasato, Nagano 380-8553, Japan
First published on 3rd August 2022
Abstract
BaTaO2N is appraised to be one of the few promising 600 nm-class photocatalysts for solar water splitting. However, the presence of structural defects and low charge separation limits its photocatalytic activity. Compared with mono substitution, dual substitution can be more effective in engineering the structural defects and improving the photocatalytic activity if foreign ions are suitably selected. In this work, we involve a dual-substitution approach to partially substitute Al3+ and/or Mg2+ for Ta5+ in BaTaO2N. By maintaining the maximum concentration of Al3+–Mg2+ dual substitution at 5%, the effect of the Al3+–Mg2+ cosubstituent ratio on the optoelectronic, surface, and photocatalytic properties of BaTaO2N is investigated. The Al3+–Mg2+ dual substitution leads to the shift of optical absorption edge toward shorter wavelengths, increasing the optical bandgap energy of BaTaO2N. This effect is more pronounced in the samples with a higher concentration of Mg2+ due to the replacement of N3− by a large number of O2− to compensate charge balance. The initial reaction rates for the evolution of O2 and H2 reveal the improvement in the photocatalytic activity of BaTaO2N due to the partial Al3+–Mg2+ dual substitution. Higher O2 evolution is observed in the samples with a higher concentration of Mg2+, while the H2 evolution rate significantly relies on the increased concentration of Al3+. According to the density functional theory (DFT) calculations, the effective masses of electrons become slightly lower than that of pristine BaTaO2N after partial Al3+–Mg2+ (co)substitution, while a contrary tendency is observed for the effective masses of holes. The calculated positions of the valence band maximum and conduction band minimum are aligned with respect to the normal hydrogen electrode (NHE), and partial Al3+–Mg2+ (co)substituted BaTaO2N photocatalysts can be promising candidates for visible-light-induced water splitting.
1. Introduction
As a carbon-free chemical process to generate green hydrogen, solar water splitting depends on various important photochemical and photophysical properties of photocatalytic materials. Particularly, band structure, optical absorption, charge density, charge mobility, charge separation, defect density, surface structure, particle size, crystallinity, etc. can be modulated by (co)substituting cations and anions in the crystal structures of host photocatalytic materials by foreign ions to improve their solar water-splitting efficiency.1,2Many efforts have so far been made to enhance the water-splitting efficiency of various oxide and non-oxide photocatalytic materials by a cosubstitution. For instance, an apparent quantum yield (AQY) of 3.2% under visible light (420–800 nm) was reached by partial substitution of In3+ for Bi3+ and Mo6+ for V5+ in the host lattice of scheelite m-BiVO4 due to uplifting the conduction band edge position above the proton reduction potential (0 VRHE at pH = 7).3 Compared with pristine BiVO4, 0.5W–2Mo–BiVO4 exhibited a significant increase in the donor concentration (ND = 1.03 × 1028 m−3), lifetime (τD = 3.8 s), and incident photon conversion efficiency (IPCE = 41%), a decrease in the flat band potential (Vfb = 0.35 VRHE) and space charge layer thickness, and nearly ten times higher H2 evolution.4 A photocurrent density of 1.97 mA cm−2 at 1.23 VRHE and a relatively low onset potential of 0.68 VRHE were obtained by a simultaneous introduction of Sn and Mo in α-Fe2O3, where the former accelerated charge separation by improving conductivity and the latter induced high density of surface trapping states, leading to the inhibition of charge recombination kinetics in surface states.5 A significant increase in the concentration of (co)doped ions decreases the efficiency. By applying a computational method, Smart et al.6 recently found a doping clustering, which traps free-electron polarons and severely lowers the carrier concentration with respect to the doping concentration, to be responsible for the doping bottleneck in α-Fe2O3, and proposed a codoping with dopants having low binding energies for clustering, such as Sn–Ti, as a solution. The formation of oxygen vacancies in SrTiO3, which act as electron–hole recombination centers, was suppressed by Rh–La7 and La–Al8 codoping, resulting in apparent quantum yields of 1.1% at 420 nm and 78.43% under 365 nm, respectively. Layered perovskite Sr2TiO4 was activated by La/N codoping, using 0.5 wt% Rh/Cr2O3 as a cocatalyst, for overall water splitting under visible light due to the contribution of N and La for uplifting the valence band edge position and charge balancing, respectively.9 Wide-band-gap photocatalysts, such as NaTaO3,10 BaTa2O6,11 and KTaO3,12 were activated for H2 evolution under visible light irradiation by introducing the Ir/La codopants. Along with La as a charge balancer, doped Ir could form relatively shallow impurity levels, and the H2 evolution could proceed by electron transition from the impurity levels formed by Ir3+ to the conduction band. A negative shift in the onset potential of photoelectrochemical water splitting from about 0.8 VRHE (for pristine Ta3N5) to 0.55 VRHE under AM 1.5 G-simulated sunlight was observed for a Mg–Zr cosubstituted Ta3N5 photoanode because of the change in the bandgap potential.13 Very recently, Mg–Zr-codoped single-crystalline Ta3N5 exhibited 45-times greater photocatalytic water-reduction activity than undoped Ta3N5 and an outstanding apparent quantum yield (AQY) of 0.54% at 420 nm during the photocatalytic H2 evolution reaction due to the simultaneously well-regulated defect species and surface properties.14
As a promising candidate for visible-light-driven water splitting because of its excellent visible light absorption up to 660 nm, narrow band gap, sufficient valence band potential for water oxidation, good stability, and nontoxicity,15 perovskite BaTaO2N has gained significant research interest. Apparent quantum yields (AQY) of 0.06%,16 2.1%,17 6.8%,18 and 11.9%19 at 420 nm were progressively achieved for photocatalytic H2 and O2 evolution over BaTaO2N, respectively, and an incident photon-to-current efficiency of ≈43% at 1.23 VRHE was obtained in photoelectrochemical water oxidation over BaTaO2N.20 In addition to other important strategies applied, such as flux growth,21 particle morphology- and size-controlled synthesis,22 time-retrenched synthesis,23 thin-film fabrication,24 facet-controlled synthesis,25,26 solid–solution,16,27 surface modification,19 tensile uniaxial strain,28 and particle transfer method,29 the A-site or B-site substitution or partial substitution of atoms with different radii or valences have been proven to be effective in tailoring the surface local structure and anion ordering and modulating the optical, electronic, surface, and photocatalytic properties of BaTaO2N without altering its perovskite structure. Substituents with different valences act as either electron donors or acceptors and change the carrier concentration when introduced into the host lattice.30 Although divalent (Mg2+, Ca2+, Sr2+, and Zn2+), trivalent (Al3+, Ga3+, and Sc3+), tetravalent (Ti4+ and Zr4+), and hexavalent (Mo6+ and W6+) cations were singly introduced into the BaTaO2N lattice to improve its photocatalytic and photoelectrochemical water splitting efficiency,17,28,31–35 a dual-substitution effect on water splitting efficiency of BaTaO2N has not been explored yet.
Many outstanding works, including the above-mentioned ones, on the enhancement of water splitting efficiency of various photocatalytic materials by a dual substitution and our recent work,32 where 5% Mg2+ and 5% Al3+ independently promoted the photocatalytic sacrificial O2 and H2 evolution over BaTaO2N under visible light, respectively, inspired us to further explore the impact of a partial dual substitution on water splitting efficiency of BaTaO2N. In this study, by maintaining the maximum concentration of partial Al3+–Mg2+ dual substitution at 5%, the effect of the Al3+–Mg2+ cosubstituent ratio on the optoelectronic, surface, and photocatalytic properties of BaTaO2N is studied. By linking the materials characterization results to the evaluated photocatalytic activity, the contribution of the partial Al3+–Mg2+ dual substitution is discussed and insights into the possible underlying mechanisms are gained.
2. Experimental
2.1. Synthesis
Pristine and Al3+–Mg2+ cosubstituted BaTaO2N powders were synthesized by a solid-state reaction route. BaCO3 (99.99%, chemPUR), Ta2O5 (99%, Alfa Aeser), and Al2O3 (99.99%, Merck) or MgCO3 (>99%, Merck) as cosubstituent sources were first mixed manually in a stoichiometric ratio. Then, the well-homogenized mixture was placed in a platinum crucible, heated at 950 °C for 6 h using a localized NH3 delivery system (12.5 L h−1), with a heating rate of 500 °C h−1 and a natural cooling rate. The synthesized BaTaO2N (BTON) powders: with no substituent, 5% Al3+, 5% Mg2+, 2.5% Al3+ + 2.5% Mg2+, 3.5% Al3+ + 1.5% Mg2+, and 1.5% Al3+ + 3.5% Mg2+ were labeled as BTON1, BTON2, BTON3, BTON4, BTON5, and BTON6, respectively.2.2. Characterization
The crystal structure was determined by X-ray diffraction (XRD; PANalytical X’Pert PRO) analysis using Cu-Kα radiation (Bragg–Brentano geometry). The microstructure was examined by scanning electron microscopy (SEM; GeminiSEM 500 NanoVP, Carl Zeiss). The elemental content was analyzed by means of energy-dispersive X-ray spectroscopy (EDX; DSM 982 GEMINI, Carl Zeiss, with a Bruker Quantax XFlash® 6|60) and inductively coupled plasma-optical emission spectrometry (SPS5510, SII Nanotechnology Inc.). The UV-Vis diffuse reflectance spectra were measured using an Evolution 220 UV/Vis spectrometer (Thermo Fisher Scientific). The surface chemical composition and states of elements were analyzed using a PHI Quantera II scanning X-ray photoelectron microprobe (XPS; ULVAC-PHI, Inc.) with monochromatic Al-Kα radiation. The XPS profiles were fitted using a Gaussian–Lorentzian function, and the peak positions were normalized by positioning the C 1s peak at 284.5 eV.2.3. Photocatalytic activity tests
The photocatalytic activity of pristine and (co)substituted BaTaO2N samples was evaluated by comparing their H2 and O2 evolution promoted by Pt (0.5 wt%) and CoOx (2 wt% Co) cocatalysts, respectively. The cocatalysts were loaded according to the processes reported elsewhere.32 The H2 and O2 evolution half-reactions were separately carried out in a Pyrex® side-irradiation-type reactor connected to a closed gas circulation and evacuation system. A 300 W Xe arc lamp (Cermax-PE300BF, PerkinElmer) with a UV-cutoff filter (L42, HOYA) and a cold mirror (CM-1, Optline) was used as the visible-light source, and the irradiance of visible light was 200 mW cm−2. The quantity of evolved gases was analyzed by using a gas chromatograph (GC-8A, TCD, Ar gas carrier, Shimadzu), which was directly connected to the reactor. For the O2-evolution half-reaction, 100 mg of CoOx-loaded photocatalyst, 300 mL of 10 mM AgNO3 (sacrificial electron scavenger), and 200 mg of La2O3 (pH buffer) were used, while 100 mg of Pt-loaded photocatalyst and 300 mL of 10 vol% aqueous methanol solution were used for the H2-evolution half-reaction.2.4. Computational methods
Density functional theory (DFT) simulations were performed within Vienna ab initio simulation package (VASP)36,37 in the projector augmented waves (PAW) scheme. The pristine BaTaO2N cell (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, No. 221, Z = 1) was created based on the experimental structural data.38 Several types of substitution have been considered in the present work. A 2 × 1 × 1 supercell was used to simulate a 50 at% substitution content of Al or Mg, i.e., one substituent atom was substituted for Ta atom within the 10-atomic supercell. To model the cosubstitution contents of 25 at% Al and 25 at%. Mg, one Al and one Mg atom were substituted for two Ta atoms within the 20-atomic 2 × 2 × 1 supercell. Simulation of 50 at% substitution, where one quarter is Al and three quarters is Mg, and vice versa were modeled within a 2 × 2 × 2 supercell with 40 atoms. All simulated configurations with the corresponding chemical compositions are listed in Table S1 (ESI†). The generalized gradient approximation (GGA) of the exchange-correlation potential in the PBE form39 was adopted during geometry optimization. For the 2 × 1 × 1 and 2 × 2 × 1 supercells, Γ-centered 6 × 12 × 12 and 6 × 12 × 12 k-point grids were used, respectively. In the case of the 2 × 2 × 2 supercell, the Monkhorst–Pack k-point mesh 6 × 6 × 6 was applied.32 The cut-off energy was 400 eV for all models. The optimization continued until the residual forces on the atoms became less than 0.5 meV Å−1. The totally relaxed pristine and (co)substituted models were adopted for further calculations of the density of states (DOS) and band structures. To achieve the best agreement between experimental and theoretical results, the screened Coulomb hybrid HSE12s exchange–correlation functional was employed.40 During the calculation of the DOS structure, Γ-centered 6 × 12 × 12, 4 × 4 × 8, and 3 × 3 × 3 k-point grids in the case of (co)substituted models within the 2 × 1 × 1, 2 × 2 × 1, and 2 × 2 × 2 supercells were applied, respectively. The Gaussian smearing method was used for the electronic structure calculations. The effective masses of electrons and holes were calculated using the Sumo-bandstats program through the parabolic fitting to the conduction band minimum (CBM) and valence band maximum (VBM).41 The band edge positions were calculated according to the empirical formula:42–45
m space group, No. 221, Z = 1) was created based on the experimental structural data.38 Several types of substitution have been considered in the present work. A 2 × 1 × 1 supercell was used to simulate a 50 at% substitution content of Al or Mg, i.e., one substituent atom was substituted for Ta atom within the 10-atomic supercell. To model the cosubstitution contents of 25 at% Al and 25 at%. Mg, one Al and one Mg atom were substituted for two Ta atoms within the 20-atomic 2 × 2 × 1 supercell. Simulation of 50 at% substitution, where one quarter is Al and three quarters is Mg, and vice versa were modeled within a 2 × 2 × 2 supercell with 40 atoms. All simulated configurations with the corresponding chemical compositions are listed in Table S1 (ESI†). The generalized gradient approximation (GGA) of the exchange-correlation potential in the PBE form39 was adopted during geometry optimization. For the 2 × 1 × 1 and 2 × 2 × 1 supercells, Γ-centered 6 × 12 × 12 and 6 × 12 × 12 k-point grids were used, respectively. In the case of the 2 × 2 × 2 supercell, the Monkhorst–Pack k-point mesh 6 × 6 × 6 was applied.32 The cut-off energy was 400 eV for all models. The optimization continued until the residual forces on the atoms became less than 0.5 meV Å−1. The totally relaxed pristine and (co)substituted models were adopted for further calculations of the density of states (DOS) and band structures. To achieve the best agreement between experimental and theoretical results, the screened Coulomb hybrid HSE12s exchange–correlation functional was employed.40 During the calculation of the DOS structure, Γ-centered 6 × 12 × 12, 4 × 4 × 8, and 3 × 3 × 3 k-point grids in the case of (co)substituted models within the 2 × 1 × 1, 2 × 2 × 1, and 2 × 2 × 2 supercells were applied, respectively. The Gaussian smearing method was used for the electronic structure calculations. The effective masses of electrons and holes were calculated using the Sumo-bandstats program through the parabolic fitting to the conduction band minimum (CBM) and valence band maximum (VBM).41 The band edge positions were calculated according to the empirical formula:42–45| ECB = χ − Ee – 0.5 Eg | (1) |
| EVB = ECB − Eg | (2) |
3. Results and discussion
Since doping can also influence the formation of a secondary phase depending on its concentration,48 the crystal structure and purity of the synthesized samples were first analyzed by X-ray powder diffraction (XRD) analysis. The XRD patterns of pristine and (co)substituted samples are shown in Fig. 1. All reflections in the XRD patterns are identified as a single-phase cubic perovskite BaTaO2N with the space group Pmm (ICDD PDF 01-084-1748), and no reflections assignable to impurity phases are detected. In the perovskite structure of BaTaO2N, Ba cations with a larger ionic radius occupy A site and has a 12-fold coordination, while Ta cations with a smaller ionic radius occupy a B site and have a 6-fold coordination at the octahedral sites. Since the ionic radii of Al3+ (53.5 pm) and Mg2+ (72 pm) substituents are closer to that of Ta5+ (64 pm) than that of Ba2+ (161 pm), both are expected to be (co)substituted for Ta5+. Apparently, the magnified XRD patterns indicate a slight alteration in the 2θ angle position of the 110 reflection toward higher or lower 2θ angles when Ta5+ is partially substituted by Al3+ (BTON2) or Mg2+ (BTON3) in the octahedral coordination, indicating the lattice volume contraction or expansion, respectively.32 As the ionic radius of Al3+ is smaller than that of Ta5+, while the ionic radius of Mg2+ is larger than that of Ta5+, the 2θ angle position of the 110 reflections of the Al3+–Mg2+ dual-substituted samples (BTON4-6) is near to that of pristine BaTaO2N (BTON1). Also, the concurrent substitution of smaller O2− (126 pm) for larger N3− (157 pm) to compensate charge balance in the Al3+–Mg2+ dual-substitution for Ta5+ may have lessened the lattice expansion.13,32 An opposite trend was observed when oxygen occupying the substitutional and interstitial lattice sites of K2La2Ti3O10 was intentionally substituted by nitrogen.49 As the (co)substitution concentration is strictly controlled at 5%, no significant distortion in the crystal structure of BaTaO2N is observed.
 | ||
| Fig. 1 X-Ray diffraction patterns of BTON1 (a), BTON2 (b), BTON3 (c), BTON4 (d), BTON5 (e), and BTON6 (f). | ||
The photocatalytic water splitting activity of photocatalysts is greatly influenced by particle morphology, size, porosity, exposed facets, etc. Recently, it was found that the CoOx cocatalyst could function more effectively in photoluminescence quenching and generating greater band bending on the {010} facet in dual-faceted BiVO4 with respect to the {110} facet.50 Also, the BaTaO2N crystals with well-developed {111} facets25 and coexposed anisotropic {100} and {110} facets26 showed a significantly enhanced photocatalytic activity for H2 evolution in comparison to the BaTaO2N crystals with only {100} facets. Thus, the microstructures of pristine and (co)substituted samples were also analyzed, and the SEM images are shown in Fig. 2. Pristine BaTaO2N (BTON1) has irregular particles with an average size of 247 nm, and some particles contain pores (Fig. 2a). Evidently, the particle morphology and size were affected by changing the Al3+–Mg2+ cosubstituent ratio. Namely, introducing 5% Al3+ (BTON2) significantly reduced the number of small particles, and large bulky particles with an average size of 338 nm and surface pores were formed (Fig. 2b). On one hand, these surface pores can, in principle, provide a large surface area that is beneficial for the photocatalytic water splitting reactions and also a greater number of dangling bonds that can act as nucleation centers for cocatalyst particles.22 On the other hand, they can also affect both charge transport within the photocatalyst and mass transfer of reactants and products, impacting the overall reaction kinetics.51 In Fig. 2c, the particle size was drastically decreased to an average size of 87 nm, and the particles became more joined with an intimate contact and without any surface pores after substituting 5% Mg2+ for Ta5+ (BTON3). When 2.5% Al3+ and 2.5% Mg2+ (BTON4) were equally cosubstituted for Ta5+ (BTON4), the particles again became larger (285 nm) without a clear outline (Fig. 2d). When the Al3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg2+ cosubstituent ratio was set to 3.5%:1.5% (BTON5), larger and denser crystals with idiomorphic shapes appeared along with smaller irregular particles with surface pores (Fig. 2e). In contrast, when the Al3+:Mg2+ cosubstituent ratio was adjusted to 1.5%:3.5% (BTON6), the average particle size was reduced to 93 nm (Fig. 2f). This indicates that the total interfacial free energy and kinetic factors were more substantially influenced by Mg2+ than Al3+. In the previous study,32 the partial substitution of Mg2+ for Ta5+ in BaTaO2N similarly reduced the number of plate-like particles and led to the formation of particles with idiomorphic shapes in comparison to other substituents. Furthermore, the solid-state reaction here induced the formation of more irregular particles despite Al3+–Mg2+ dual substitution in comparison to the flux method applied previously to synthesize BaTaO2N particles,23,32,52 which may lead to the different photocatalytic activity. The EDX spectra of pristine and (co)substituted samples shown in Fig. S1 (ESI†) reveal the presence of Ba, Ta, O, N, Al, and Mg elements. The (co)substituent contents estimated by EDX and ICP-OES data are about 4.84% Al, 4.91% Mg, 2.42% Al + 2.38% Mg, 3.47% Al + 1.52% Mg, and 1.48% Al + 3.51% Mg for BTON2, BTON3, BTON4, BTON5, and BTON6, respectively, which are close to the nominal compositions of (co)substituents.
:Mg2+ cosubstituent ratio was set to 3.5%:1.5% (BTON5), larger and denser crystals with idiomorphic shapes appeared along with smaller irregular particles with surface pores (Fig. 2e). In contrast, when the Al3+:Mg2+ cosubstituent ratio was adjusted to 1.5%:3.5% (BTON6), the average particle size was reduced to 93 nm (Fig. 2f). This indicates that the total interfacial free energy and kinetic factors were more substantially influenced by Mg2+ than Al3+. In the previous study,32 the partial substitution of Mg2+ for Ta5+ in BaTaO2N similarly reduced the number of plate-like particles and led to the formation of particles with idiomorphic shapes in comparison to other substituents. Furthermore, the solid-state reaction here induced the formation of more irregular particles despite Al3+–Mg2+ dual substitution in comparison to the flux method applied previously to synthesize BaTaO2N particles,23,32,52 which may lead to the different photocatalytic activity. The EDX spectra of pristine and (co)substituted samples shown in Fig. S1 (ESI†) reveal the presence of Ba, Ta, O, N, Al, and Mg elements. The (co)substituent contents estimated by EDX and ICP-OES data are about 4.84% Al, 4.91% Mg, 2.42% Al + 2.38% Mg, 3.47% Al + 1.52% Mg, and 1.48% Al + 3.51% Mg for BTON2, BTON3, BTON4, BTON5, and BTON6, respectively, which are close to the nominal compositions of (co)substituents.
 | ||
| Fig. 2 SEM images of BTON1 (a), BTON2 (b), BTON3 (c), BTON4 (d), BTON5 (e), and BTON6 (f). | ||
Fig. 3 shows the UV-Vis diffuse reflectance spectra of pristine and (co)substituted samples. Pristine BaTaO2N (BTON1) has an optical absorption edge at 665 nm, corresponding to the optical bandgap energy of 1.86 eV. Obviously, the Al3+–Mg2+ cosubstituent ratio influenced visible-light absorption of BaTaO2N. That is, the optical absorption edges of (co)substituted samples shifted toward shorter wavelengths, resulting in the optical bandgap energies of 1.90, 2.01, 1.96, 1.93, and 1.99 eV for BTON2, BTON3, BTON4, BTON5, and BTON6, respectively. Interestingly, increasing the concentration of Mg2+ led to a greater shift toward shorter wavelengths in comparison to that of Al3+. This is due to the substitution of more N3− by O2− to compensate charge balance in the Mg2+-to-Ta5+ substitution.13,32,34 The valence band of BaTaO2N consists of hybridized N 2p and O 2p orbitals, and its position is affected by the N/O ratio as the N 2p orbitals are higher in energy than the O 2p orbitals. This is also reflected by the powder color of the synthesized samples shown in the insets of Fig. 3, where the higher the Mg2+ concentration, the brighter the powder color is. Although the Al3+–Mg2+ dual substitution induced a considerable blue-shift in light absorption, which is not beneficial in solar energy conversion, it is still advantageous in reducing the surface and bulk defects because of the suppression of Ta5+ reduction by Al3+/Mg2+ (co)substitution and altering the band edge positions with respect to water splitting potentials.53
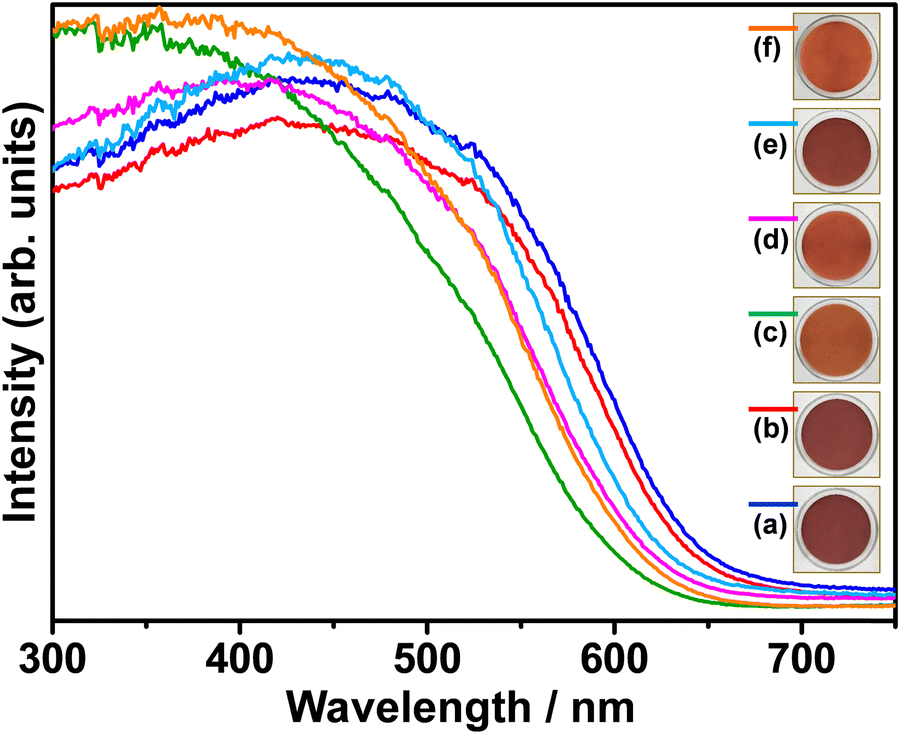 | ||
| Fig. 3 UV-Vis diffuse reflectance spectra of BTON1 (a), BTON2 (b), BTON3 (c), BTON4 (d), BTON5 (e), and BTON6 (f). | ||
To probe the surface chemical composition and oxidation state of elements, X-ray photoelectron spectroscopy (XPS) measurements were conducted. In the XPS core-level spectra of Ta 4f in Fig. 4, the overlapping peaks of the Ta 4f5/2 and Ta 4f7/2 states of the Ta5+ species bonded to N3− and O2− can be deconvoluted into four different peaks centered at the binding energies of 25.70–26.16, 23.67–24.14, 27.07–27.11, and 25.01–26.09 eV, respectively.54 Although there is no direct correlation between the intensities of the Ta(N) and Ta(O) peaks and Al3+–Mg2+ (co)substitution ratio, a slightly higher intensity in the Ta(O) peak can be observed in comparison to that of the Ta(N) peak when the Al3+:Mg2+ (co)substituent ratio is decreased because a large number of N3− were substituted by O2− to compensate charge balance in the Mg2+-to-Ta5+ substitution.13,32,34 No peaks associated with reduced tantalum species were noted as the partial substitution of Al3+ or/and Mg2+ suppressed the reduction of Ta5+.53 This leads to the reduction in the surface and bulk defects that can improve charge separation and photocatalytic activity.
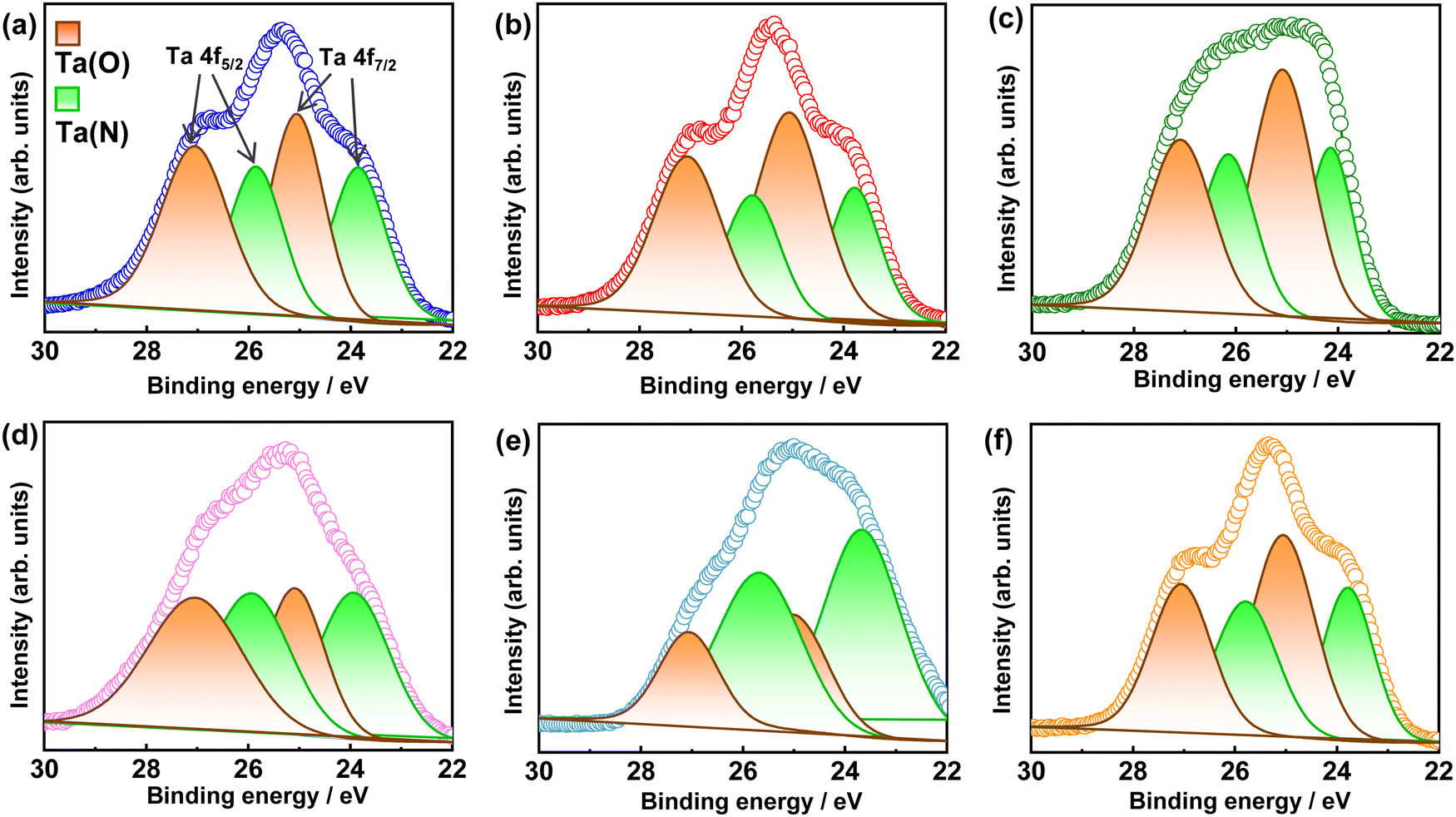 | ||
| Fig. 4 Ta 4f high-resolution X-ray photoelectron spectra of BTON1 (a), BTON2 (b), BTON3 (c), BTON4 (d), BTON5 (e), and BTON6 (f). | ||
Density functional theory (DFT) simulations were further involved to understand the electronic structures of pristine and (co)substituted BaTaO2N models. First, the structural parameters of pristine and (co)substituted BaTaO2N models were predicted by DFT-PBE. As shown in Table S1 (ESI†), the structural parameters of pristine BaTaO2N model were found to be in good agreement with the experimental data reported previously.32,40 It can be noted that Al substitution leads to a contraction of lattice constants due to a smaller ionic radius of Al3+ (53.5 pm) than that of Ta5+ (64 pm). An opposite trend is observed in Mg substitution because the ionic radius of Mg2+ (72 pm) is larger than that of Ta5+. The cosubstituted models can be characterized in the same manner depending on the Al and Mg contents. This is consistent with the X-ray diffraction data presented earlier.
Next, the effect of Al3+–Mg2+ (co)substitution at the Ta site in BaTaO2N on electronic band structures was studied by DFT-HSE12s. As shown in Fig. 5, the estimated bandgap energy of pristine BaTaO2N is 1.49 eV (direct-type), which is slightly lower than the experimentally32,55 and theoretically56 obtained bandgap values due to the well-known underestimation. The calculation results reveal that the (co)substitution of Al and/or Mg at the Ta site in BaTaO2N can generate acceptor states above the valence band maximum, shifting the valence band upward (Fig. 5). According to the density of states (DOS) plots, the distribution patterns of the atomic orbitals seem to be unchanged (Fig. S2, ESI†). The major contribution to the valence band comes from occupied O p and N p states, while the conduction band consists of empty Ta d states. The dependence of the bandgap value on Al or/and Mg contents in pristine and (co)substituted BaTaO2N models is shown in Fig. S3 (ESI†). Apparently, Al substitution results in a narrower bandgap value in comparison to Mg substitution. Meantime, three cosubstituted models are characterized by a significant decrease in the band gap with an increase in the Al content, which is beneficial to absorb a significant fraction of visible light. Among the three cosubstituted models, the BaTa0.5Al0.375Mg0.125O2N model has the narrowest band gap (1.36 eV).
 | ||
| Fig. 5 Electronic band structures of (a) BTON,32 (b) BTON:Al (50 at%), (c) BTON:Mg (50 at%), (d) BTON:Al:Mg (25:25 at%), (e) BTON:Al:Mg (37.5:12.5 at%), and (f) BTON:Al:Mg (12.5:37.5 at%). The Fermi level is set at 0 eV. | ||
The effective masses of electrons 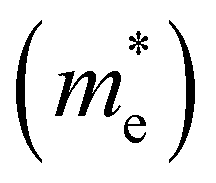 and holes
and holes 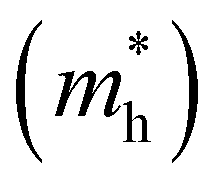 were also estimated along specific directions (Table S2, ESI†). It is known that lower effective masses of charge carriers indicate their higher mobility, which is important for enhancing the photocatalytic activity.45 In the case of pristine BaTaO2N, the effective masses of electrons and holes are comparable and low. With Al and/or Mg (co)substitution, the effective masses of electrons become slightly lower than that in pristine BaTaO2N, suggesting an improvement in the reduction ability of the (co)substituted BaTaO2N models. A contrary tendency can be observed for the effective masses of holes, which increased three or more times as compared with that of pristine BaTaO2N. The lowest effective masses of holes are noted for the BaTa0.5Al0.375Mg0.125O2N model, which may exhibit its stronger oxidizing ability among (co)substituted compounds. Suitable redox potentials are also known as one of the major criteria for developing high-efficiency visible-light-active photocatalysts.45,46 As shown in Table S3 (ESI†), the calculated positions of the valence band maximum and conduction band minimum are aligned with respect to the normal hydrogen electrode (NHE), and Al3+–Mg2+ (co)substituted BaTaO2N photocatalysts can be promising candidates for visible-light-induced water splitting.
were also estimated along specific directions (Table S2, ESI†). It is known that lower effective masses of charge carriers indicate their higher mobility, which is important for enhancing the photocatalytic activity.45 In the case of pristine BaTaO2N, the effective masses of electrons and holes are comparable and low. With Al and/or Mg (co)substitution, the effective masses of electrons become slightly lower than that in pristine BaTaO2N, suggesting an improvement in the reduction ability of the (co)substituted BaTaO2N models. A contrary tendency can be observed for the effective masses of holes, which increased three or more times as compared with that of pristine BaTaO2N. The lowest effective masses of holes are noted for the BaTa0.5Al0.375Mg0.125O2N model, which may exhibit its stronger oxidizing ability among (co)substituted compounds. Suitable redox potentials are also known as one of the major criteria for developing high-efficiency visible-light-active photocatalysts.45,46 As shown in Table S3 (ESI†), the calculated positions of the valence band maximum and conduction band minimum are aligned with respect to the normal hydrogen electrode (NHE), and Al3+–Mg2+ (co)substituted BaTaO2N photocatalysts can be promising candidates for visible-light-induced water splitting.
The effect of Al3+–Mg2+ dual substitution on visible-light-induced photocatalytic activity of BaTaO2N was investigated. The half-reaction time courses for the photocatalytic H2 and O2 evolution over pristine and (co)substituted BaTaO2N samples are shown in Fig. 6. As shown in Fig. 6a, the quantity of evolved O2 gradually increases in the following order within 5 hours of the photocatalytic reaction: 124.05 μmol < 171.4 μmol < 238.2 μmol < 271.5 μmol < 324.7 μmol < 406.2 μmol for BTON1, BTON2, BTON3, BTON5, BTON4, and BTON6, respectively. Clearly, compared with pristine BaTaO2N (BTON1) and mono-substituted samples (BTON2 and BTON3), cosubstituted samples exhibit higher O2 evolution, and the highest O2 evolution (406.2 μmol) is observed for BTON6 with 1.5% Al3+ + 3.5% Mg2+ cosubstituents. It is argued that due to the decrement in the surface and bulk defects as a result of the partial replacement of Ta5+ by a higher number of Mg2+ in the Al3+–Mg2+ dual substitution and altering the valence band position with respect to water oxidation potential. As shown in Fig. 6b, the highest quantity of evolved H2 is obtained for BTON5 (75.4 μmol) followed by BTON4 (60.5 μmol), BTON6 (51.9 μmol), BTON2 (45.4 μmol), BTON3 (31.7 μmol), and BTON1 (17.3 μmol), respectively.
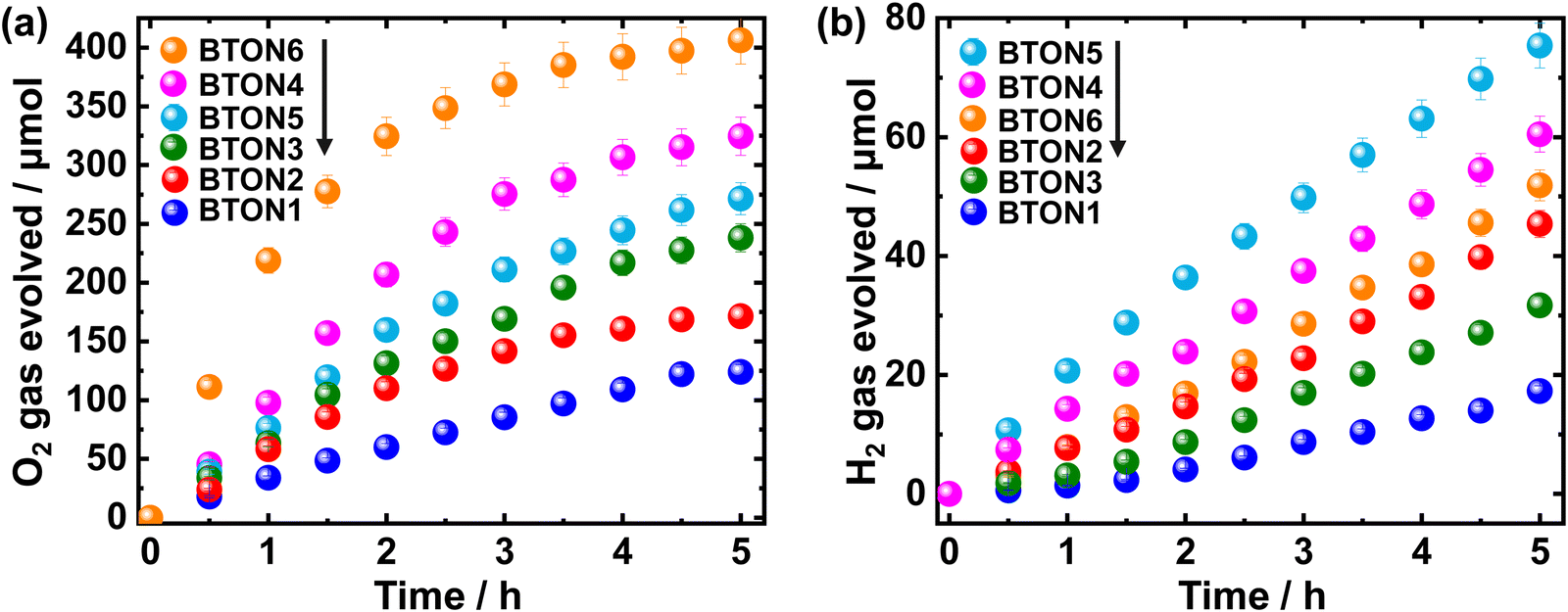 | ||
| Fig. 6 Reaction time courses for photocatalytic O2 (a) and H2 (b) evolution over BTON1, BTON2, BTON3, BTON4, BTON5, and BTON6 loaded with CoOx and Pt nanoparticles as O2 and H2 evolution cocatalysts under visible light irradiation. | ||
Kisch and Bahnemann57 suggested that the comparison of photocatalyst performance must be done using the kinetic parameters extracted from experimental measurements performed using the same types of light source and reactor. Then, it is convenient to estimate the reaction rate in the initial stages as the respective slope of the O2 and H2 evolved vs. irradiation time plots. For the O2 evolution, the reaction rates were estimated to be 178.66, 102.64, 79.33, 66.65, 55.89, and 31.44 μmol h−1 for BTON6, BTON4, BTON5, BTON3, BTON2, and BTON1, respectively. For the H2 evolution, 18.94, 12.81, 8.34, 7.35, 3.93, and 1.78 μmol h−1 for BTON5, BTON4, BTON6, BTON2, BTON3, and BTON1, respectively. Clearly, BaTaO2N modified with 1.48% Al + 3.51% Mg generated the highest quantity of O2 (178.66 μmol h−1) with an apparent quantum yield of 0.18% at 420 nm, and BaTaO2N modified with 3.47% Al + 1.52% Mg produced the highest quantity of H2 (18.94 μmol h−1) with an apparent quantum yield of 0.64% at 420 nm. Considering that the variation of the specific surface area between the photocatalysts does not have a greater impact,58 the trend observed in the initial reaction rate can be considered as evidence that reflects the improvement of the surface reactions due to the modification of the BaTaO2N photocatalyst with Al3+–Mg2+ dual substitution. In our recent work,32 the photocatalytic reaction rate of the cation-modified BaTaO2N was correlated with the energy difference of the adsorbed intermediates, where the photocatalytic evolution of H2 and O2 was significantly enhanced using Al- and Mg-modified BaTaO2N photocatalysts, respectively. Thus, it is argued that in the case of dual substitution of BaTaO2N with Al3+ and Mg2+, the photocatalytic performance to form H2 can be favored in the photocatalyst with a higher proportion of Al3+. Regarding the evolution of O2, it was reported that the modification with Mg2+ presented a higher reaction rate than the undoped BTON and the Al-doped BTON.32 The latter suggests that the highest reactivity for the evolution of O2 can be achieved with the photocatalyst having the highest percentage of Mg2+. Therefore, the Al3+–Mg2+ dual substitution can modulate the photocatalytic activity of BaTaO2N.
The Al3+–Mg2+ dual substitution in BaTaO2N leads to the improvement in the kinetics of photocatalytic processes as a result of efficient electron transfer and the reduction of recombination processes. The interrelation of both phenomena (electron transfer and recombination) in BaTaO2N photocatalysts has been discussed in previous works,13,20,23 and the optoelectronic properties were presented to be responsible for the changes in the photocatalytic behavior. For instance, the codoping of Ta3N5 with Mg and Zr13 and the modification of BaTaO2N with Ca and cobalt oxide17 significantly affected the optoelectronic properties in such a way that the co-doped photocatalysts could exhibit the lower onset potentials and higher photocatalytic activity for photoelectrochemical water splitting. Modification of BaTaO2N and LaTiO2N with Zn and Ca has also been shown to significantly influence carrier density, to shift the band edge position, and to improve the yield of photo-redox reactions.33 Similar results have been reported in BaTaO2N with various dopants.31,34
For Al3+–Mg2+-(co)substituted BaTaO2N, the photocatalytic performance observed for the evolution of O2 and H2 (Fig. 6), the changes in visible light absorption (Fig. 3), and DFT calculations (Fig. 5) support the alteration in the electronic states of the photocatalyst with Al3+ and/or Mg2+. Therefore, the difference in the dynamics of charge carriers affected the photocatalytic performance of pristine and (co)substituted photocatalysts. The relevance of DFT calculations to detect band structure effects that correlate with photocatalytic activity is convenient. Using the DFT calculations, Ni et al.59 estimated the changes in the electronic structure of ZnSe by co-substitution of Sb at Se sites and Sc or Y at Zn sites, suggesting the importance of the effect of strong Coulombic interactions. It was also noted that the dual substitution resulted in a reduced bandgap, absorption in visible light, and energetic position of the bands relative to the redox potentials of water. By applying the DFT calculations, the role of F and N in the co-doped TiO2 was studied,60 indicating that the co-substitution of foreign atoms affects the band structure and provides new pathways for the appearance of different physicochemical processes (e.g., enhanced adsorption of reagents, formation of new bonds, changes in the band structure, etc.) that provide improvements to the photocatalytic activity. As the dynamics of charge carriers is defined by the electronic structure, the DFT results presented here for BaTaO2N cosubstituted with Al3+ and Mg2+ (Fig. 5) allow to assertively detect those changes in the electronic band structure that could have effects and/or be related to the performance of the photocatalysts. Further, the effect of Al3+–Mg2+ dual substitution on surface property (water and methanol adsorption) of BaTaO2N is also theoretically explored.
Along with other factors, the adsorption of water molecules and formed intermediates on the photocatalyst surface has a strong influence on photocatalytic activity. In our recent work, the experimental photocatalytic reaction rates of pristine and cation-doped BaTaO2N surfaces terminated with TaO6, TaN6, and TaO4N2 were well presented using the adsorption energies of intermediates (H* for H2 evolution and HO* and O* for O2 evolution) estimated by molecular dynamics calculations.32 Another study has shown that the nickel modification can improve the adsorption of water molecules on anatase-TiO2, rutile-TiO2, and ZnO photoanodes, enhancing their photoelectrochemical performance.61 Particularly, Adsorption Locator for modelling has been broadly used for evaluating the adsorption interaction or non-bonded energies of organic and water molecules for various applications, including biomolecule/surface interactions,62 adsorption of SiF4 and HF gaseous molecules at the molecular level,63 next-generation protein-based biosensor surfaces,64 catalyst/adsorbents for oil recovery and viscosity reduction process,65 drug delivery tool in biological systems,66etc. Here, the influence of the Al3+–Mg2+ dual substitution on the adsorption of water molecules on the BaTaO2N(110) surfaces was also explored by combined Molecular Dynamics and Monte-Carlo computer simulation. The Forcite and Adsorption Locator modules in BIOVIA Materials Studio 2017 software67 was used to determine the most favorable adsorption sites and to evaluate the adsorption energy of water molecules on the BaTaO2N(110) surfaces at different concentrations of the Al3+ and Mg2+ (co)substituents (Table S5, ESI†). The simulation data reveal that the adsorption energies of water molecules increase linearly by the Al3+-for-Ta5+ substitution on the BaTaO2N(110) surface. The simultaneous adsorption of water and methanol molecules is higher than the adsorption of only water molecules (Fig. S4, ESI†), which increases depending on the Al3+ content. The differential adsorption (dEads/dNi) of water molecules in water or water-methanol systems on the BaTaO2N(110) surfaces have similar values (2.3–2.7 kcal mol−1). The methanol molecules interact more strongly with Al3+–Mg2+-cosubstituted surfaces (4.95–6.14 kcal mol−1). Fig. 7 shows close contacts between water molecules and Al3+–, Mg2+– and Al3+–Mg2+-(co)substituted BaTaO2N(110) surfaces. However, methanol molecules tend to interact better with magnesium atoms on the BaTaO2N(110) surface. Thus, compared with Al3+– or Mg2+ substitution, the Al3+–Mg2+ dual substitution can improve the adsorption of water and methanol molecules on the surface of BaTaO2N, enhancing its photocatalytic activity.
 | ||
| Fig. 7 Water and methanol molecules and field density distribution on the BaTaO2N(110) surfaces with 5% Al (a), 2.5% Al and 2.5% Mg (b), and 5% Mg (c). Atoms: grey – hydrogen, red – oxygen, blue – nitrogen, yellow – magnesium, pink – aluminum, light blue – tantalum, green – barium; Isosurface: red – methanol, green – water. | ||
4. Conclusions
In summary, the partial Al3+–Mg2+ dual substitution (5%) was applied to engineer structural defects and to modulate optoelectronic, surface, and photocatalytic activity of BaTaO2N. The optical absorption edge of BaTaO2N was shifted to shorter wavelengths after (co)substitution of Al3+ and/or Mg2+ for Ta5+, leading to the increase in the optical bandgap energy. This effect was more pronounced in the samples with higher content of Mg2+ because a large number of O2− were substituted for N3− to compensate charge balance. Similarly, a partial substitution of Mg2+ for Ta5+ affected the morphology of BaTaO2N particles in comparison to Al3+, reducing the average particle size significantly. The initial reaction rates for the evolution of O2 and H2 revealed the improvement in the photocatalytic performance of BaTaO2N photocatalysts due to Al3+–Mg2+ dual substitution. Particularly, BaTaO2N modified with 1.48% Al + 3.51% Mg generated the highest quantity of O2 (178.66 μmol h−1) and exhibited an apparent quantum yield of 0.18% at 420 nm, while BaTaO2N modified with 3.47% Al + 1.52% Mg produced the highest quantity of H2 (18.94 μmol h−1) and exhibited an apparent quantum yield of 0.64% at 420 nm. This enhancement in the photocatalytic O2 and H2 evolution over Al3+–Mg2+-(co)substituted BaTaO2N photocatalysts can be related to the changes in the defect density, dynamics of charge carriers, electronic band structure, improvement in water and methanol adsorption, and favorable shift in the band energy levels with respect to water reduction and oxidation potentials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Ina Remy-Speckmann, Dipl. Phys. Christoph Fahrenson, Ms Reiko Shiozawa, and Dr Aleksei G. Krasnov for their kind assistance in XRD, SEM-EDX and XPS analyses and DFT calculations, respectively. This project received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement no. 793882.References
- Q. Wang and K. Domen, Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- T. Takata and K. Domen, Defect engineering of photocatalysts by doping of aliovalent metal cations for efficient water splitting, J. Phys. Chem. C, 2009, 113, 19386–19388 CrossRef CAS.
- W. J. Jo, H. J. Kang, K.-J. Kong, Y. S. Lee, H. Park, Y. Lee, T. Buonassisi, K. K. Gleason and J. S. Lee, Phase transition-induced band edge engineering of BiVO4 to split pure water under visible light, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13774–13778 CrossRef CAS PubMed.
- M. Tayebi and B.-K. Lee, The effects of W/Mo-co-doped BiVO4 photoanodes for improving photoelectrochemical water splitting performance, Catal. Today, 2021, 361, 183–190 CrossRef CAS.
- J. Xiao, B. Du, S. Hu, J. Zhong, X. Chen, Y. Zhang, D. Cai, S.-F. Zhou and G. Zhan, Simultaneously Enhanced Charge Separation and Transfer in Cocatalyst-Free Hematite Photoanode by Mo/Sn Codoping, ACS Appl. Energy Mater., 2021, 4, 10368–10379 CrossRef CAS.
- T. J. Smart, V. U. Baltazar, M. Chen, B. Yao, K. Mayford, F. Bridges, Y. Li and Y. Ping, Doping Bottleneck in Hematite: Multipole Clustering by Small Polarons, Chem. Mater., 2021, 33, 4390–4398 CrossRef CAS.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Core/Shell Structured La- and Rh-Codoped SrTiO3 as a Hydrogen Evolution Photocatalyst in Z-Scheme Overall Water Splitting under Visible Light Irradiation, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- Y. Qin, F. Fang, Z. Xie, H. Lin, K. Zhang, X. Yu and K. Chang, La,Al-Codoped SrTiO3 as a Photocatalyst in Overall Water Splitting: Significant Surface Engineering Effects on Defect Engineering, ACS Catal., 2021, 11, 11429–11439 CrossRef CAS.
- X. Sun, Y. Mi, F. Jiao and X. Xu, Activating Layered Perovskite Compound Sr2TiO4 via La/N Codoping for Visible Light Photocatalytic Water Splitting, ACS Catal., 2018, 8, 3209–3221 CrossRef CAS.
- A. Iwase, K. Saito and A. Kudo, Sensitization of NaMO3 (M: Nb and Ta) Photocatalysts with Wide Band Gaps to Visible Light by Ir Doping, Bull. Chem. Soc. Jpn., 2009, 82, 514–518 CrossRef CAS.
- A. Iwase and A. Kudo, Development of Ir and La-codoped BaTa2O6 photocatalysts using visible light up to 640 nm as an H2-evolving photocatalyst for Z-schematic water splitting, Chem. Commun., 2017, 53, 6156–6159 RSC.
- A. Iwase and H. Misono, Development of visible-light-responsive Ir and La-codoped KTaO3 photocatalysts for water splitting, Chem. Commun., 2021, 57, 10331–10334 RSC.
- J. Seo, T. Takata, M. Nakabayashi, T. Hisatomi, N. Shibata, T. Minegishi and K. Domen, Mg−Zr Cosubstituted Ta3N5 Photoanode for Lower-Onset-Potential Solar-Driven Photoelectrochemical Water Splitting, J. Am. Chem. Soc., 2015, 137, 12780–12783 CrossRef CAS PubMed.
- J. Xiao, J. J. M. Vequizo, T. Hisatomi, J. Rabeah, M. Nakabayashi, Z. Wang, Q. Xiao, H. Li, Z. Pan, M. Krause, N. Yin, G. Smith, N. Shibata, A. Brückner, A. Yamakata, T. Takata and K. Domen, Simultaneously Tuning the Defects and Surface Properties of Ta3N5 Nanoparticles by Mg−Zr Codoping for Significantly Accelerated Photocatalytic H2 Evolution, J. Am. Chem. Soc., 2021, 143, 10059–10064 CrossRef CAS PubMed.
- M. Higashi, R. Abe, K. Teramura, T. Takata, B. Ohtani and K. Domen, Two step water splitting into H2 and O2 under visible light by ATaO2N (A = Ca, Sr, Ba) and WO3 with IO3−/I− shuttle redox mediator, Chem. Phys. Lett., 2008, 452, 120–123 CrossRef CAS.
- K. Maeda and K. Domen, Water Oxidation Using a Particulate BaZrO3-BaTaO2N Solid-Solution Photocatalyst That Operates under a Wide Range of Visible Light, Angew. Chem., Int. Ed., 2012, 51, 9865–9869 CrossRef CAS PubMed.
- S. Wei, G. Zhang and X. Xu, Activating BaTaO2N by Ca modifications and cobalt oxide for visible light photocatalytic water oxidation reactions, Appl. Catal., B, 2018, 237, 373–381 CrossRef CAS.
- Z. Wang, Y. Luo, T. Hisatomi, J. J. M. Vequizo, S. Suzuki, S. Chen, M. Nakabayashi, L. Lin, Z. Pan, N. Kariya, A. Yamakata, N. Shibata, T. Takata, K. Teshima and K. Domen, Sequential cocatalyst decoration on BaTaO2N towards highly-active Z-scheme water splitting, Nat. Commun., 2021, 12, 1005 CrossRef CAS PubMed.
- S. Jadhav, S. Hasegawa, T. Hisatomi, Z. Wang, J. Seo, T. Higashi, M. Katayama, T. Minegishi, T. Takata, J. M. Peralta-Hernández, O. S. Torres and K. Domen, Efficient photocatalytic oxygen evolution using BaTaO2N obtained from nitridation of perovskite-type oxide, J. Mater. Chem. A, 2020, 8, 1127–1130 RSC.
- J. Seo, M. Nakabayashi, T. Hisatomi, N. Shibata, T. Minegishi and K. Domen, Solar-Driven Water Splitting over a BaTaO2N Photoanode Enhanced by Annealing in Argon, ACS Appl. Energy Mater., 2019, 2, 5777–5784 CrossRef CAS.
- M. Hojamberdiev, K. Yubuta, J. J. M. Vequizo, A. Yamakata, S. Oishi, K. Domen and K. Teshima, NH3-Assisted Flux Growth of Cube-like BaTaO2N Submicron Crystals in a Completely Ionized Nonaqueous High-Temperature Solution and Their Water Splitting Activity, Cryst. Growth Des., 2015, 15, 4663–4671 CrossRef CAS.
- M. Hojamberdiev, K. Kawashima, T. Hisatomi, M. Katayama, M. Hasegawa, K. Domen and K. Teshima, Distinguishing the effects of altered morphology and size on the visible light-induced water oxidation activity and photoelectrochemical performance of BaTaO2N crystal structures, Faraday Discuss., 2019, 215, 227–241 RSC.
- M. Hojamberdiev, J. M. Mora-Hernandez, R. Vargas, A. Yamakata, K. Yubuta, E. M. Heppke, L. M. Torres-Martínez, K. Teshima and M. Lerch, Time-Retrenched Synthesis of BaTaO2N by Localizing an NH3 Delivery System for Visible-Light-Driven Photoelectrochemical Water Oxidation at Neutral pH: Solid-State Reaction or Flux Method?, ACS Appl. Energy Mater., 2021, 4, 9315–9327 CrossRef CAS.
- K. Teshima, Y. Hara, K. Yubuta, S. Oishi, K. Domen and M. Hojamberdiev, Application of Flux Method to the Fabrication of Ba5Ta4O15, Sr5Ta4O15, Sr2Ta2O7, and BaTaO2N Polycrystalline Films on Ta Substrates, Cryst. Growth Des., 2017, 17, 1583–1588 CrossRef CAS.
- Y. Luo, Z. Wang, T. Yamada, K. Yubuta, S. Suzuki, T. Hisatomi, K. Domen and K. Teshima, Platy BaTaO2N Crystals Fabricated from K2CO3–KCl Binary Flux for Photocatalytic H2 Evolution, ACS Appl. Energy Mater., 2020, 3, 10669–10675 CrossRef CAS.
- Y. Luo, S. Suzuki, Z. Wang, K. Yubuta, J. J. M. Vequizo, A. Yamakata, H. Shiiba, T. Hisatomi, K. Domen and K. Teshima, Construction of Spatial Charge Separation Facets on BaTaO2N Crystals by Flux Growth Approach for Visible-Light-Driven H2 Production, ACS Appl. Mater. Interfaces, 2019, 11, 22264–22271 CrossRef CAS PubMed.
- K. Hibino, M. Yashima, T. Oshima, K. Fujii and K. Maeda, Structures, electron density and characterization of novel photocatalysts, (BaTaO2N)1−x(SrWO2N)x solid solutions, Dalton Trans., 2017, 46, 14947–14956 RSC.
- Z. Lan, T. Vegge and I. E. Castelli, Theoretical Insight on Anion Ordering, Strain, and Doping Engineering of the Oxygen Evolution Reaction in BaTaO2N, Chem. Mater., 2021, 33, 3297–3303 CrossRef CAS.
- K. Ueda, T. Minegishi, J. Clune, M. Nakabayashi, T. Hisatomi, H. Nishiyama, M. Katayama, N. Shibata, J. Kubota, T. Yamada and K. Domen, Photoelectrochemical Oxidation of Water Using BaTaO2N Photoanodes Prepared by Particle Transfer Method, J. Am. Chem. Soc., 2015, 137, 2227–2230 CrossRef CAS PubMed.
- T. Takata and K. Domen, Defect Engineering of Photocatalysts by Doping of Aliovalent Metal Cations for Efficient Water Splitting, J. Phys. Chem. C, 2009, 113, 19386–19388 CrossRef CAS.
- M. Higashi, Y. Yamanaka, O. Tomita and R. Abe, Fabrication of Cation-Doped BaTaO2N Photoanodes for Efficient Photoelectrochemical Water Splitting Under Visible Light Irradiation, APL Mater., 2015, 3, 104418 CrossRef.
- M. Hojamberdiev, R. Vargas, Z. C. Kadirova, K. Kato, H. Sena, A. G. Krasnov, A. Yamakata, K. Teshima and M. Lerch, Unfolding the Role of B Site-Selective Doping of Aliovalent Cations on Enhancing Sacrificial Visible Light-Induced Photocatalytic H2 and O2 Evolution over BaTaO2N, ACS Catal., 2022, 12, 1403–1414 CrossRef CAS.
- Y. Bao, H. Zou, N. Yang, G. Li and F. Zhang, Synthesis of perovskite BaTaO2N with low defect by Zn doping for boosted photocatalytic water reduction, J. Energy Chem., 2021, 63, 358–363 CrossRef.
- H. Zhang, S. Wei and X. Xu, Mg modified BaTaO2N as an efficient visible-light-active photocatalyst for water oxidation, J. Catal., 2020, 383, 135–143 CrossRef CAS.
- K. Maeda, D. Lu and K. Domen, Oxidation of Water under Visible-Light Irradiation over Modified BaTaO2N Photocatalysts Promoted by Tungsten Species, Angew. Chem., Int. Ed., 2013, 52, 6488–6491 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- F. Pors, R. Marchand, Y. Laurent, P. Bacher and G. Roult, Etude structurale des perovskites oxyazotees BaTaO2N et BaNbO2N. Structural study of BaTaO2N and BaNbO2N oxynitrided perovskites, Mater. Res. Bull., 1988, 23, 1447–1450 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. E. Moussa, P. A. Schultz and J. R. Chelikowsky, Analysis of the Heyd-Scuseria-Ernzerhof density functional parameter space, J. Chem. Phys., 2012, 136, 1–11 CrossRef PubMed.
- A. M. Ganose, A. J. Jackson and D. O. Scanlon, Sumo: Command-Line Tools for Plotting and Analysis of Periodic Ab Initio Calculations, J. Open Source Softw., 2018, 3, 717 CrossRef.
- M. Hojamberdiev, E. Zahedi, E. Nurlaela, K. Kawashima, K. Yubuta, M. Nakayama, H. Wagata, T. Minegishi, K. Domen and K. Teshima, The Cross-Substitution Effect of Tantalum on the Visible-Light-Driven Water Oxidation Activity of BaNbO2N Crystals Grown Directly by an NH3-Assisted Flux Method, J. Mater. Chem. A, 2016, 4, 12807–12817 RSC.
- Z. Ma, Z. Yi, J. Sun and K. Wu, Electronic and Photocatalytic Properties of Ag3PC4VI (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, S, Se): A Systemic Hybrid DFT Study, J. Phys. Chem. C, 2012, 116, 25074–25080 CrossRef CAS.
O, S, Se): A Systemic Hybrid DFT Study, J. Phys. Chem. C, 2012, 116, 25074–25080 CrossRef CAS. - A. G. Krasnov, M. S. Napalkov, M. I. Vlasov, M. S. Koroleva, I. R. Shein and I. V. Piir, Photocatalytic Properties of Bi2−xTi2O7−1.5x (x = 0, 0.5) Pyrochlores: Hybrid DFT Calculations and Experimental Study, Inorg. Chem., 2020, 59, 12385–12396 CrossRef CAS PubMed.
- M. A. Butler and D. S. Ginley, Prediction of Flatband Potentials at Semiconductor-Electrolyte Interfaces from Atomic Electronegativities, J. Electrochem. Soc., 1978, 125, 228–232 CrossRef CAS.
- M. Benčina, M. Valant and M. Ben, Bi2Ti2O7-Based Pyrochlore Nanoparticles and Their Superior Photocatalytic Activity under Visible Light, J. Am. Ceram. Soc., 2018, 101, 82–90 CrossRef.
- H. Sameie, A. A. Sabbagh Alvani, N. Naseri, S. Du and F. Rosei, First-Principles Study on ZnV2O6 and Zn2V2O7: Two New Photoanode Candidates for Photoelectrochemical Water Oxidation, Ceram. Int., 2018, 44, 6607–6613 CrossRef CAS.
- K. Kawashima, M. Hojamberdiev, H. Wagata, M. Nakayama, K. Yubuta, S. Oishi, K. Domen and K. Teshima, Amount of tungsten dopant influencing the photocatalytic water oxidation activity of LaTiO2N crystals grown directly by an NH3-assisted flux method, Catal. Sci. Technol., 2016, 6, 5389–5396 RSC.
- K. Kawashima, M. Hojamberdiev, H. Wagata, K. Yubuta, K. Domen and K. Teshima, Protonated Oxide, Nitrided, and Reoxidized K2La2Ti3O10 Crystals: Visible-Light-Induced Photocatalytic Water Oxidation and Fabrication of Their Nanosheets, ACS Sustainable Chem. Eng., 2017, 5, 232–240 CrossRef CAS.
- Z. Xie, H. L. Tan, H. Wu, R. Amal, J. Scott and Y. H. Ng, Facet-dependent Spatial Charge Separation with Rational Co-catalyst Deposition on BiVO4, Mater. Today Energy, 2022, 26, 100986 CrossRef CAS.
- M. Zbiri, C. M. Aitchison, R. S. Sprick, A. I. Cooper and A. A. Y. Guilbert, Probing Dynamics of Water Mass Transfer in Organic Porous Photocatalyst Water-Splitting Materials by Neutron Spectroscopy, Chem. Mater., 2021, 33, 1363–1372 CrossRef CAS PubMed.
- M. Hojamberdiev and K. Kawashima, Exploring flux-grown transition metal oxynitride perovskites for photocatalytic water oxidation: A minireview, Energy Rep., 2020, 6, 13–24 CrossRef.
- J. Seo, D. Ishizuka, T. Hisatomi, T. Takata and K. Domen, Effect of Mg2+ substitution on the photocatalytic water splitting activity of LaMgxNb1–xO1+3xN2–3x, J. Mater. Chem. A, 2021, 9, 8655–8662 RSC.
- C. Wang, T. Hisatomi, T. Minegishi, Q. Wang, M. Zhong, M. Katayama, J. Kubota and K. Domen, Synthesis of Nanostructured BaTaO2N Thin Films as Photoanodes for Solar Water Splitting, J. Phys. Chem. C, 2016, 120, 15758–15764 CrossRef CAS.
- K. Kawashima, M. Hojamberdiev, K. Yubuta, K. Domen and K. Teshima, Synthesis and Visible-Light-Induced Sacrificial Photocatalytic Water Oxidation of Quinary Oxynitride BaNb0.5Ta0.5O2N Crystals, J. Energy Chem., 2018, 27, 1415–1421 CrossRef.
- X. Xu and H. Jiang, First-Principles Investigation on Anion Order, Electronic Structure and Dielectric Properties of BaTaO2N, J. Mater. Chem. A, 2019, 7, 14583–14591 RSC.
- H. Kisch and D. Bahnemann, Best practice in photocatalysis: Comparing rates or apparent quantum yields, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef CAS PubMed.
- F. Wu, G. Liu and X. Xu, Efficient photocatalytic oxygen production over Ca-modified LaTiO2N, J. Catal., 2017, 346, 10–20 CrossRef CAS.
- C. Ni, C. Fu, B. Wang, H. Yuan and H. Chen, Charge-compensated codoped pseudohexagonal zinc selenide nanosheets towards enhanced visible-light-driven photocatalytic water splitting for hydrogen production, Int. J. Hydrogen Energy, 2021, 46, 34305–34317 CrossRef CAS.
- A. Miyoshi, A. Kuwabara and K. Maeda, Effects of Nitrogen/Fluorine Codoping on Photocatalytic Rutile TiO2 Crystal Studied by First-Principles Calculations, Inorg. Chem., 2021, 60, 2381–2389 CrossRef CAS PubMed.
- M. Hojamberdiev, R. Vargas, V. S. Bhati, D. Torres, Z. C. Kadirova and M. Kumar, Unraveling the photoelectrochemical behavior of Ni-modified ZnO and TiO2 thin films fabricated by RF magnetron sputtering, J. Electroanal. Chem., 2021, 882, 115009 CrossRef CAS.
- S. Galvez-Martinez, E. Escamilla-Roa, M. P. Zorzano and E. Mateo-Marti, Defects on a pyrite (100) surface produce chemical evolution of glycine under inert conditions: experimental and theoretical approaches, Phys. Chem. Chem. Phys., 2019, 21, 24535–24542 RSC.
- M. Khnifira, A. Mahsoune, M. E. Belghiti, L. Khamar, M. Sadiq, M. Abdennouri and N. Barka, HF and SiF4 adsorption on carbon graphite (111) surface in aqueous medium: A combined DFT and MD simulation approach, Mater. Today: Proc., 2021, 37, 3987–3993 CAS.
- J. S. Cross, Y. Kubota, A. Chatterjee, S. Unni, T. Ikoma and M. Tagaya, Interfacial Modeling of Fibrinogen Adsorption onto LiNbO3 Single Crystal–Single Domain Surfaces, Int. J. Mol. Sci., 2021, 22, 5946 CrossRef CAS PubMed.
- T. Montoya, A. Amrollahi, G. Vitale, N. Hosseinpour and N. N. Nassar, Size Effects of NiO Nanoparticles on the Competitive Adsorption of Quinolin-65 and Violanthrone-79: Implications for Oil Upgrading and Recovery, ACS Appl. Nano Mater., 2020, 3, 5311–5326 CrossRef CAS.
- E. A. Abdullah, Theoretical study of a single-walled carbon nanotube and a cellulose biofiber as 5-fluorouracil anti-cancer drug carriers, Eur. J. Chem., 2022, 13, 69–77 CrossRef CAS.
- S. Sharma, P. Kumar and R. Chandra, Applications of BIOVIA Materials Studio, LAMMPS, and GROMACS in Various Fields of Science and Engineering, in Molecular Dynamics Simulation of Nanocomposites Using BIOVIA Materials Studio, Lammps and Gromacs, ed. S. Sharma, 2019, pp. 329–341 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00611a |
| This journal is © The Royal Society of Chemistry 2022 |