
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMassive amplification of photoluminescence and exceptional water stability of MAPbBr3 nanocrystals through core–shell nanostructure formation in a self-defence mechanism†
Shovon
Chatterjee
 ,
Tanmoy
Khan
,
Arghya
Sen
,
Nilimesh
Das
* and
Pratik
Sen
*
,
Tanmoy
Khan
,
Arghya
Sen
,
Nilimesh
Das
* and
Pratik
Sen
*
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur – 208 016, UP, India. E-mail: psen@iitk.ac.in; nilimesh@iitk.ac.in; Fax: +91 51 2259 6806; Tel: +91 51 2259 6312
First published on 26th July 2022
Abstract
Vulnerability to atmospheric conditions and their associated toxicity limit the practical/industrial use of perovskites despite their tremendous promise in optoelectronics. This study utilized an ionic liquid-like solvent to synthesize methylammonium lead bromide (MAPbBr3) nanocrystals (NCs). The synthesized NCs showed a moderate photoluminescence quantum yield (PLQY) of ∼19% and high environmental stability of at least six months. Further, the entire visible range was tuned through the anion-exchange method. More interestingly, the synthesized NCs formed a core–shell structure in a unique self-defence mechanism in the presence of water, which was proposed to be MAPbBr3@lead laurate. This core–shell structure was found to be beneficial for (a) preventing further degradation of the NC, and making it water stable (at least for two months), (b) improving the PLQY by surface modification inducing a massive five-fold amplification, and (c) restricting the anion-exchange reaction. Moreover, these unique properties were achieved without any special control. Moreover, the successful synthesis of other MAPbX3 (X = Cl, I) demonstrated its potential applicability.
1. Introduction
Halide perovskites are considered a potential game changer in the field of light-emitting diodes, solar cells, photodetectors, and bioimaging because of their unique optical and electronic properties.1–3 Despite the tremendous promise, their practical/industrial applicability is limited because of their vulnerability to atmospheric conditions, and due to toxicity related to the perovskite materials and their synthesis.1,4,5 This study utilized an ionic liquid-like solvent that could largely reduce synthesis-related toxicity, minimize structural defects, and protect the perovskite nanocrystal from degradation by water through a self-defence mechanism.The most widely used precursor solvents for perovskite synthesis are hazardous and toxic dimethylformamide (DMF) along with its homologous dimethylacetamide (DMAC), N-methyl-2-pyrrolidone (NMP), and skin-penetrating dimethyl sulfoxide (DMSO). Another critical problem with these solvents is their high coordinating ability with the perovskite surface, which eventually facilitates the degradation of the perovskite structure and the formation of surface defects.6–9 The use of less hazardous and non-coordinating organic solvents, like acetonitrile (ACN), for this purpose has been attempted in recent times.9,10 A recent breakthrough was achieved using ionic liquids (ILs) (like carboxylic acid-methylamine, imidazole-based ionic liquid, and so on) as the precursor medium.11,12 The first report came in 2015 by Moore et al. describing the synthesis of a hybrid lead halide perovskite (MAPbI3) thin film using methylammonium formate ionic liquid.13 Consequent studies have proved the massive potential of such an approach.11,14–17 A few studies have also been undertaken to venture into the fundamental roles of the ILs in controlling the perovskite nanocrystals’ (NCs) crystallization, nucleation, growth, interface modification, and overall power conversion efficiency (PCE).11,12,18,19 The use of such solvents serves many purposes; including (i) these can solubilize a vast series of materials;11 (ii) by forming active intermediates, it provides an opportunity to control the crystal-growth process;11,18,20 (iii) by interacting with uncoordinated lead, it might provide a much-needed enhancement in the PLQY;21 (iv) these liquids are considered environmentally friendly;11–14 (v) the synthesis of perovskites can be done in a simple one-step reaction.22 A recent perspective has predicted ILs to be the next reformer in the field of perovskite research considering these facts.11 Although the use of ILs in the synthesis process of the perovskite systems has emerged with high potential, the research is still in its infancy, and huge effort is still necessary. The use of ILs in the synthesis of perovskites is still being investigated for the improvement of solar cells.
In this context, we prepared lead halide perovskite NCs employing a lauric acid-based IL-like solvent system to address some of the issues related to perovskite NCs’ efficiency, stability, and synthetic toxicity. We were inspired by a few facts, as follows. (i) The only report on the synthesis of hybrid lead halide perovskite NCs with ILs was reported in 2020 by Hoang et al., in which they successfully synthesized luminescent MAPbBr3 nanocrystals.22 A systematic study involving the carboxylic acids of different carbon chain lengths from C1 to C4 suggested that the emissive property increased as the chain length of the acid increased. They achieved a moderately high quantum yield (50%) and even demonstrated water stability of up to 1 h. It can be assumed that a further increment in the chain length of carboxylic acid might increase the emissive property along with the environmental stability due to an effective capping. But, the ILs composed of long-chain carboxylic acids have never been employed in perovskite synthesis. (ii) In previous reports, all the organic acids used were short chains that lacked a hydrophobic character. From our knowledge, we know that lauric acid (LA)-based solvents show a high hydrophobic character. Consequently, we expect that the capping with LA might introduce high moisture and environmental stability to the prepared NCs. (iii) Furthermore, LA can act as a surface capping ligand to provides stability to the NCs, and this can nullify the destructive consequences of solvent coordination. (iv) LA induces confinement in contact with water, which may be a key to size tunability in NC preparation in the future.23 (v) LA is a harmless natural fatty acid and it is the major component of coconut oil.
This report demonstrates a single-step synthesis of MAPbBr3 NCs in lauric acid media. It showed a moderate photoluminescence quantum yield (PLQY) of ∼19%. In the presence of water, the formation of a core–shell structure by a unique self-defence mechanism rendered high water stability (at least up to two months) and a five-fold amplification of the PLQY to ∼96%. Further, the successful synthesis of other MAPbX3 compounds (X = Cl, I) demonstrated the generality of the procedure.
2. Experimental
Materials
Lauric acid or dodecanoic acid (Aldrich, 98%), methylamine (TCI, 40% solution in methanol), lead bromide (Sigma Aldrich, 99%), lead chloride (Sigma Aldrich, 99%), lead iodide (Sigma Aldrich, 99%), and oleylamine (TCI Chemicals, 60%) were used as received. Hydrochloric acid (Merck, 37% in water), hydrobromic acid (Alfa-Aesar, 48% water), hydroiodic acid (TCI, 57% in water), and hexane (TCI) were used without further purification.Preparation of the reaction medium
The reaction medium was prepared by adding 2 mL methylamine solution in 2 g LA at 60 °C. The mixture was stirred for 30 min under the same conditions. High temperature was required to liquefy LA (melting point 43.2 °C). This IL-like medium was then used to synthesize the perovskite NCs.Synthesis of CH3NH3PbBr3 NCs
We prepared the MAPbBr3 NCs by the direct addition of 200 mg of PbBr2 salt to 3 mL of the solvent medium at 60 °C (see Scheme 1). After the formation of bright green-luminescent MAPbBr3 NCs, we divided this into two parts (∼1.5 mL each containing the MAPbBr3 NC suspension in the solvent medium). Both parts were centrifuged (40 °C, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 577 g, 60 min), and the supernatants were discarded. Then, 1 mL of hexane was added to each part and sonicated (25 °C, 53 MHz, 10 min) followed by centrifugation (40 °C, 12577 g, 15 min). This process was repeated five times to discard the remaining medium. Then the MAPbBr3 residue from each part was redispersed in 5 mL of hexane (room temperature) and used for further studies. In this procedure, we observed that 200 mg of PbBr2 salt yielded about 249 mg of dry MAPbBr3 powder, that part obviously capped with lauric acid. Like previous reports of perovskite synthesis in IL medium,13,22 our synthesis was also a single-step process and did not require any anti-solvent treatment in the synthesis procedure. The reusability of the medium was at least 10 times, checked through preparing MAPbBr3 NCs in the recollected medium (see Fig. S3 and Section S2 of the ESI† for details), making it a superior choice.
577 g, 60 min), and the supernatants were discarded. Then, 1 mL of hexane was added to each part and sonicated (25 °C, 53 MHz, 10 min) followed by centrifugation (40 °C, 12577 g, 15 min). This process was repeated five times to discard the remaining medium. Then the MAPbBr3 residue from each part was redispersed in 5 mL of hexane (room temperature) and used for further studies. In this procedure, we observed that 200 mg of PbBr2 salt yielded about 249 mg of dry MAPbBr3 powder, that part obviously capped with lauric acid. Like previous reports of perovskite synthesis in IL medium,13,22 our synthesis was also a single-step process and did not require any anti-solvent treatment in the synthesis procedure. The reusability of the medium was at least 10 times, checked through preparing MAPbBr3 NCs in the recollected medium (see Fig. S3 and Section S2 of the ESI† for details), making it a superior choice.
 | ||
| Scheme 1 Schematic representation of the synthesis of MAPbBr3 NCs. The solvent medium was composed of lauric acid (LA) and methylamine (MA). Real images of this medium in daylight and under UV (365 nm) are shown (leftmost image of the scheme). MAPbBr3 NCs were instantly formed by the addition of solid PbBr2 to this medium. Real images of the synthesized MAPbBr3 NCs in daylight and under UV (365 nm) are shown (rightmost image). A strong green PL of MAPbBr3 NCs was observed under UV. | ||
Synthesis of MAPbBr3@lead laurate core–shell NCs
MAPbBr3@lead laurate core–shell NCs were prepared by the direct addition of a hexane dispersion of purified MAPbBr3 NCs to distilled water. A visible colour change from deep yellow to white indicated the formation of lead laurate shell on the MAPbBr3 NC core (see later for the detailed discussion). The MAPbBr3@lead laurate core/shell NPs were then extracted in hexane for further characterization (see the Video in the ESI†).Synthesis of CH3NH3PbCl3 and CH3NH3PbI3 NCs
MAPbCl3 and MAPbI3 NCs were prepared by the direct addition of 200 mg of salts (lead chloride and lead iodide) to 3 ml of prepared solvent medium. Immediately after the addition of the salts, MAPbCl3 (white) and MAPbI3 (black) NCs were formed, which were then purified by the similar procedure as MAPbBr3 NCs.Synthesis of oleylammonium halide (OAmX) salts
OAmX salts were prepared by the previous procedure reported by Dutta et al.24 In short, 10 ml of oleylamine (OAm) was taken along with the required amount of hydrohalic acid (For OAmCl 1 mL of HCl, for OAmBr 1.28 mL of HBr and for OAmI 1.5 mL of HI) in a 25 mL two-neck round-bottomed flask. The resulting reaction mixture was heated at 120 °C for 2 h in a nitrogen atmosphere. The reaction temperature was then increased to 150 °C and further heated for 30 min. The prepared OAmCl salt concentration was measured to be 0.85 mmol mL−1, whereas the concentrations of OAmBr and OAmI salts were 0.51 mmol mL−1 and 0.34 mmol mL−1, respectively. The yields of the synthesized salts are listed in Table S1 of the ESI.† The synthesized OAmX salts were characterized by 1H and 13C NMR (see Fig. S4–S6 and Section S3 of the ESI†). The hexane diluted solutions (0.3 M OAmX) were then used for the anion-exchange reactions.Microscopic characterizations
High-resolution transmission electron microscopy (HRTEM) images were collected on a Titan G2 60–300 system. Samples for HRTEM were prepared by casting one drop of the much-diluted suspension of NCs after 10 min of sonication on to a copper grid (Ted Pella). The grid was dried overnight before HRTEM analysis. The field emission scanning electron microscopy (FESEM) images were collected by NOVA a NANOSEM 450 system in the secondary electron mode.Powder X-ray diffraction (PXRD)
X-Ray diffraction patterns were collected on a PANalytical Xpert Powder diffractometer with Cu-Kα as the incident radiation (λ = 0.154 nm) in the 2θ range between 5° to 50° with steps of 0.01°. For sample preparation, we used the NC suspension in hexane. This NC dispersion was then drop-cast on a glass slide and kept in the open air for complete solvent evaporation.X-Ray photoelectron spectroscopy (XPS)
XPS data were collected using a PHI 5000 Versa Prob II instrument. Samples were prepared by the same process as for the powder XRD analysis on a very thin glass slide.NMR measurements
NMR spectra were recorded on a Bruker Avance 400 system (1H, 400 MHz; 13C, 100 MHz). The NMR spectra were recorded by dissolving the ionic liquid medium or OAmX salts into CDCl3.Absorption, emission, and PL-lifetime measurements
The absorption and emission spectra were collected on a commercial UV-visible spectrophotometer (UV-2450, Shimadzu, Japan) and spectrofluorometer (FluoroMax4, Jobin-Yvon, USA), respectively. PL transients were recorded using a commercial time correlated single photon counting (TCSPC) setup (LifeSpecII, Edinburgh Instruments, UK) with excitation by a 405 nm diode laser (EPL-405, Edinburgh Instruments, UK) at the magic angle condition. The full width at half maxima (FWHM) of the instrument response function (IRF) was 120 ps. PL transients were fitted with the sum of two exponentials along with the deconvolution by the IRF. The PLQY of the NCs was calculated using coumarin 152A in acetonitrile as the reference (see Fig. S7 and Section S4 of the ESI† for details). The prepared hexane suspension of MAPbBr3 NCs was diluted enough (up to an absorbance value of ∼0.3 at 400 nm) for the spectroscopic studies.3. Results and discussion
The solvent medium for the synthesis of perovskite NCs was prepared through the addition of methylamine and hydrophobic LA. The NMR spectrum of this medium is depicted in Fig. S1 of the ESI.† All the detected protons could be assigned to the corresponding chemical shifts of the cation and anion, which confirmed the formation of an IL-like medium. The synthesized IL-like medium showed no weight loss till 100 °C, as confirmed by the thermogravimetric analysis (TGA) (see Fig. S2 and Section S1 of the ESI†).3.1. Characterisation of MAPbBr3 NCs
The purified MAPbBr3 NCs exhibited a pure cubic crystal structure, as evident from the PXRD pattern (see Fig. S8 and Section S5 of the ESI†).5 X-Ray photoelectron spectroscopy (XPS) confirmed the presence of lead (Pb) and bromine (Br) on the surface of the NCs (see Fig. S11 and Section S6 of the ESI†). Further XPS analysis revealed that the synthesized NCs had a bromine-rich surface with a bromine to lead ratio of 3.13, which was very much in line with the previous reports.2,5 Various microscopic characterizations were done to build a clear idea about the microstructure of the synthesized MAPbBr3 NCs. The FESEM images showed that the purified NCs had a cubic morphology (see Fig. S14 and Section S7 of the ESI†). The transmission electron microscopy (TEM) images of the NCs revealed that the NCs were mainly rectangular, but some square shapes were also observed (Fig. 1a and Fig. S15 and Section S7 of the ESI†). The TEM images also suggested the formation of large NCs, sized >100 nm. A bright-field image of a single NC (Fig. 1b) along with selected area electron diffraction (SAED) further verified the formation of crystalline NC in the cubic phase5 (Fig. 1c). From the HRTEM image of the synthesized NCs, the crystal planes could be seen clearly (see Fig. 1d). Fourier filtration of the selected area from Fig. 1d produced Fig. 1e and f, where we could observe the (200) and (110) crystal planes with d-spacings of 0.291 nm and 0.396 nm, respectively. The high-angle annular dark-field (HAADF) image of the NCs further confirmed the high crystallinity of the rectangular-shaped NCs (see Fig. 1g). Fig. 1h shows the energy-dispersive spectroscopy (EDS) elemental mapping of the MAPbBr3 NCs from the TEM analysis, which showed a high density of lead (Pb), bromine (Br), and carbon (C) on the surface. The EDS distribution mapping of every individual element is also included in Fig. 1i. The EDS analysis showed that the NCs exhibited a bromine-rich surface with a bromine to lead ratio of 3.41. Interestingly, EDS analysis also revealed that the NC surface had a very high density of carbon atoms, which could be because of the presence of lauric acid as the capping ligand. | ||
| Fig. 1 Microscopic characterization of the synthesized MAPbBr3 NCs. (a) TEM image of the synthesized MAPbBr3 NCs. (b) TEM image of a single MAPbBr3 nanocrystal. (c) Selected area electron diffraction (SAED) of a single MAPbBr3 NC. (d) High-resolution TEM (HRTEM) image of synthesized MAPbBr3 NCs. (e and f) Fourier filtered images showing the (200) and (110) crystal planes. The inset contains the fast Fourier transformed (FFT) pattern. (g) High-angle annular dark-field (HAADF) image of the MAPbBr3 NCs. (h) EDS elemental mapping of the MAPbBr3 NCs. (i) Mapping of the induvial elements (Pb, C, O, and Br). | ||
3.2. Optical properties of the MAPbBr3 NCs
Steady-state and time-resolved PL studies were carried out to explore the optical properties of the synthesized MAPbBr3 NCs. These MAPbBr3 NCs showed broad absorption spectra with an excitonic band centred at 521 nm and a very strong PL centred at 525 nm (see Fig. 2a). The PLQY was 19% and the average PL lifetime was 13.6 ns (61% 9.8 ns and 39% 18.9 ns) (see Fig. 2b). The 9.8 ns and 18.9 ns time components were assigned to the radiative excitonic recombination and surface trap-assisted recombination process, respectively.5,25,26 The radiative rate constant (kr) was calculated to be 1.4 × 107 s−1, whereas, the non-radiative rate constant (knr) was 5.9 × 107 s−1 (see Section S9 of the ESI†). The PL intensity of the MAPbBr3 NCs dropped by 63% after six months in ambient conditions, indicating its moderate PL stability (see Fig. S18 and Section S8 of the ESI†). The PXRD pattern remained almost unaffected (see Fig. S9 and Section S8 of the ESI†), indicating its phase purity in ambient conditions. However, we could observe the appearance of some diffraction peaks at lower angles, suggesting the transformation of NC to lead laurate.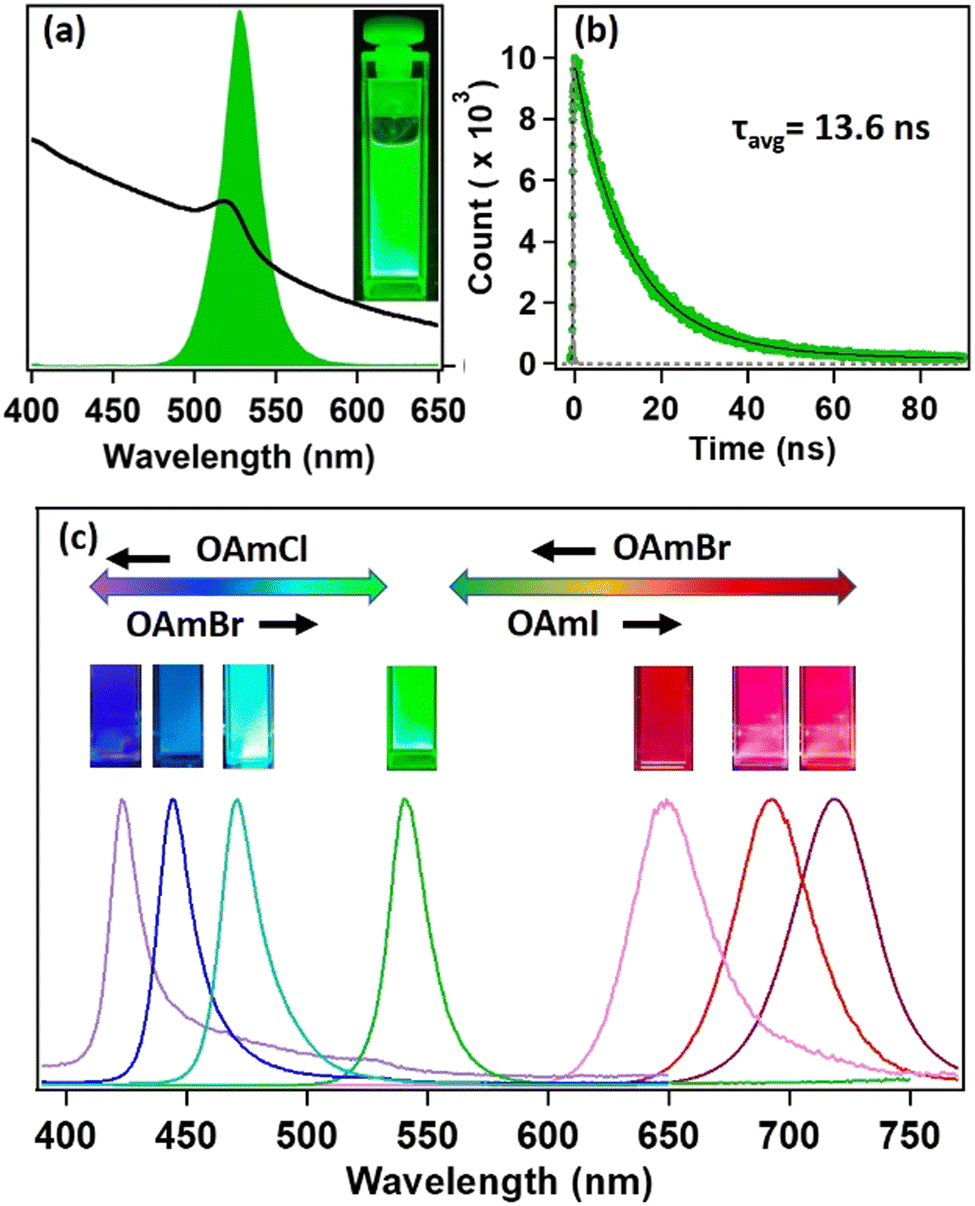 | ||
| Fig. 2 Optical properties of MAPbBr3 NCs. (a) Steady-state absorption (black) and photoluminescence (PL) spectra (green) of MAPbBr3 NCs. (b) PL transient of MAPbBr3 NCs upon exciting at 405 nm. (c) Tuning the PL through anion exchange by oleylammonium halides (OAmX; X = Cl, Br, I). | ||
3.3. Tuning of PL through anion exchange
Hybrid mixed lead halide perovskite NCs were synthesized from MAPbBr3 through the anion-exchange method in hexane at 25 °C, in order to investigate tuning the PL over the entire visible region. We used oleylammoium halides (OAmX), as the halide source for this tuning. The successive addition of OAmCl to the emissive MAPbBr3 NCs gradually shifted the PL towards the blue region, indicating the replacement of the bromide ions by chloride ions in the perovskite lattice (see Fig. 2c). The PL maximum reached 421 nm at the saturation point. This indicated the formation of MAPbCl3 NCs by replacing almost all the bromide ions of MAPbBr3 NCs. The addition of OAmBr to MAPbCl3 NCs again shifted the PL maxima towards the green region, indicating the reversibility of the process. The addition of OAmI to MAPbBr3 showed a similar phenomenon and shifted the PL maximum up to 721 nm, indicating the formation of MAPbI3 NCs (see Fig. 2c). The change in halide composition was also monitored through XPS (see Fig. S13 and Section S6 of the ESI†).3.4. Water stability of MAPbBr3 NC through core–shell formation
Being highly water-immiscible, LA is expected to safeguard MAPbBr3 NC from water when it is used as a capping ligand. The hydrophobic environment provided by the long alkyl chain of LA should not allow water molecules to go beyond the ligand environment and thus should prevent the water molecules from interacting with the perovskite structure and increase the water stability. Keeping this in mind, we dispersed our synthesized MAPbBr3 NCs into water. As expected, due to the hydrophobic character of the LA ligands, the NCs were unable to form a suspension in the water, instead tending to float on the water surface even after vigorous shaking (see the Video provided in the ESI†). The NCs showed a very stable intense green photoluminescence (see Fig. 3a) in water, though the colour of the NCs changed from deep yellow to white gradually. This incredible observation motivated us to perform a time-dependent PXRD study. The result showed an incremental decrease in the perovskite peaks with a concomitant appearance of new peaks in the low diffracting region (see Fig. 3b). The PXRD of these new peaks showed a high similarity with lead laurate (Ref. Code: 00-009-0712). The perovskite peaks fully disappeared after 30 min (see Fig. 3b). However, the strong photoluminescence band at 528 nm remained persistent, confirming the presence of MAPbBr3 perovskite NCs (see Fig. 3a). Interestingly, the PL intensity of the 528 nm band underwent a 5-fold increase to reach a near-unity PLQY (96%) (see Fig. S20 and Section S8 of the ESI†) in 30 min – representing a significant improvement over the previous report.22 We propose the formation of a lead laurate shell over MAPbBr3 NC as the possible mechanism for this observation. The masking of perovskite peaks in the PXRD may be because of the lead laurate shell thickness. The time-resolved study of this core–shell type NCs showed an average lifetime of 24 ns, which was almost twice that of the parent MAPbBr3 NCs (see Fig. 3c). The increment in average lifetime also signified the sufficient attenuation of the non-radiative pathways in the core–shell-type structure. The radiative and non-radiative rate constants were calculated to be 3.9 × 107 s−1 and 2.1 × 106 s−1, respectively (see Section S9 of the ESI†). XPS study of this samples revealed that the surfaces of the particles were mostly composed of carbon and oxygen with a minimal amount of lead (79% C, 16% O, and 5% Pb) (see Fig. S11 and Section S6 of the ESI†). The comparative XPS survey spectra of both MAPbBr3 NCs and MAPbBr3@lead laurate further gave a clear picture of the carbon- and oxygen-rich surface in the core–shell system, while for MAPbBr3 NCs, lead and bromine were sufficiently present on the surface. The high-resolution XPS of Pb-4f of MAPbBr3@lead laurate displayed a shift of the binding energy by 0.3 eV (143.4 eV for 4f5/2 and 138.5 eV for 4f7/2) towards higher energy than for the MAPbBr3 NCs (143.1 eV for 4f5/2 and 138.2 eV for 4f7/2) (see Fig. S11b and Section S6 of the ESI†). This signified the higher electronegative environment around the lead on the surface of the core–shell system. The O-1s binding energy also showed a 0.6 eV shift, which could also be attributed to the same fact (see Fig. S11c and Section S6 of the ESI†). This observation confirmed the formation of Pb-O bonds in the shell region. A TEM image of these core–shell-type NCs is shown in Fig. 3d, which shows they did not have a proper shape. A magnified image of this structure further revealed the formation of the lead laurate shell structure with a thickness of 5–8 nm (see Fig. 3e). The SAED pattern further confirmed the presence of crystalline MAPbBr3 NCs within the core of the structure (see Fig. 3f). The Fourier filtration results from the selected area of the HRTEM image (core part) of MAPbBr3@lead laurate (Fig. 3g) are presented in Fig. 3h and i, where (111) and (200) crystal planes with d-spacings of 0.37 nm and 0.30 nm, respectively, could be seen. The bright-field HAADF image (see Fig. 3j) further confirmed the formation of the core–shell structure. The EDS elemental mapping of the core–shell structure clearly showed that the core was lead and bromine rich, whereas the shell was carbon rich, which was a clear indication of lead laurate shell formation (see Fig. 3k). The EDS analysis from FESEM also allowed concluding the same (see Fig. S17 and Section S7 of the ESI†). However, the full disappearance of the perovskite peak in the PXRD also suggested that a large percentage of NCs had possibly been converted to lead laurate. However, the PL study supported that MAPbBr3 NCs remained in the system. TEM analysis further revealed that these NCs remained stable in the aqueous environment by forming the core–shell structure.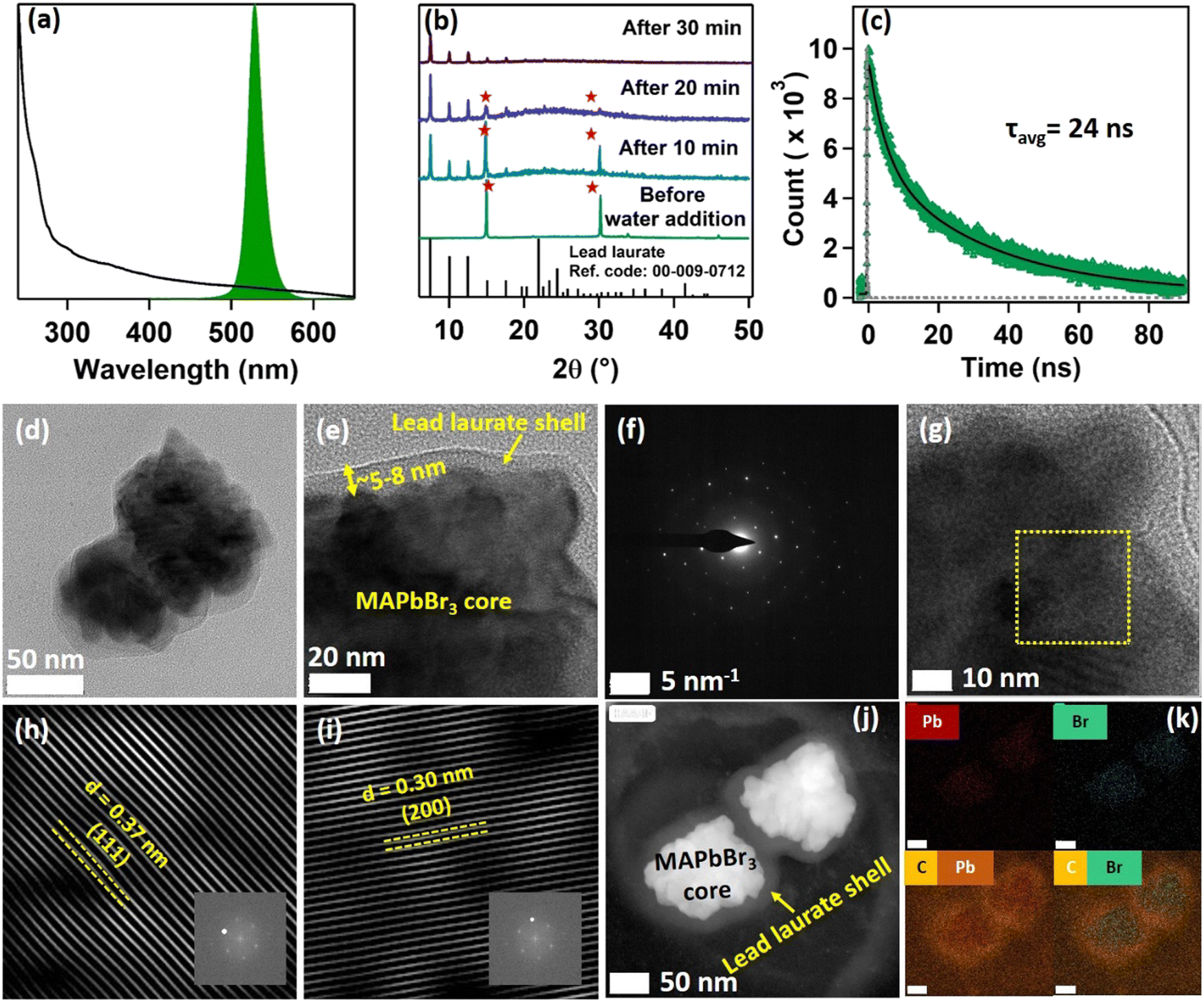 | ||
| Fig. 3 Characterization of MAPbBr3@lead laurate core–shell NCs. (a) Steady-state absorption (black) and PL (green) spectra. (b) Time-dependent change in the PXRD pattern of lauric acid-capped MAPbBr3 NCs in water. Red stars denote the perovskite peaks. (c) PL transient of MAPbBr3@lead laurate core–shell NCs upon exciting at 405 nm. (d) TEM image of MAPbBr3@lead laurate core–shell NCs. (e) Magnified TEM image of MAPbBr3@lead laurate core–shell NCs. (f) SAED pattern of MAPbBr3@lead laurate core–shell NCs. (g) HRTEM image of MAPbBr3@lead laurate core–shell NCs. (h–i) Fourier filtration of the selected area showing (111) and (200) crystal planes. Inset containing the FFT pattern from the selected area of the figure (g). (j) HAADF image of the MAPbBr3@lead laurate core–shell NCs. (k) EDS elemental mapping of MAPbBr3@lead laurate core–shell NCs (Pb, Br, C, and Pb together, C and Br together). The scale bar is 50 nm. | ||
After the self-growth of the lead laurate shell around the MAPbBr3 NC core, the system showed extreme stability in the water as the formed lead laurate shell was itself hydrophobic. The PL intensity remained almost unaffected even after 60 days in the water (see Fig. 4a and b). The photographic images of the floating MAPbBr3@lead laurate core–shell system in water under UV light showed a virtually unaffected PL intensity as seen with the bare eye (see Fig. 4b). Such observations proved that the NCs had water stability. To note, the only available report of MAPbBr3 NCs synthesized in a green medium displayed a much lower water stability (up to 1 h).22 The photostability and thermal stability were also checked for this core–shell system by monitoring its PL. This core–shell system retained almost 50% of its original PL intensity after irradiating continuously with UV light (365 nm, 6W) (see Fig. S22a of the ESI†). With the increase in the temperature, the PL intensity decreased monotonously and became almost non-luminescent around 70 °C, showing only moderate thermal stability (see Fig. S22b of the ESI†).
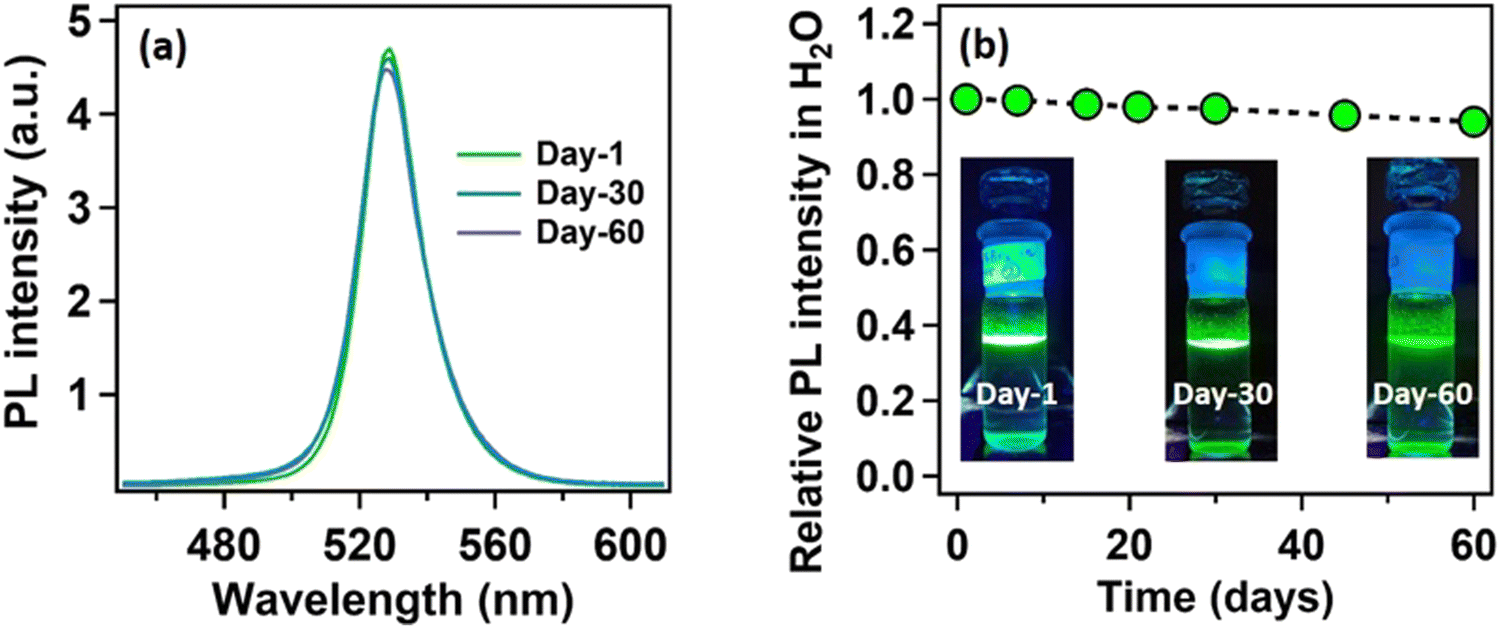 | ||
| Fig. 4 Water stability of the MAPbBr3@lead laurate core–shell NC system. (a) Comparative PL spectra of the MAPbBr3@lead laurate core–shell NC system in water, recorded on day-1, day-30, and day-60 upon exciting at 400 nm. (b) Relative change in PL intensity with the progress in days of the MAPbBr3@lead laurate core–shell NC system in water. Real images of MAPbBr3@lead laurate core–shell NC system in water under UV (365 nm) for day-1, day-30, and day-60 are given in the inset. | ||
This MAPbBr3@lead laurate core–shell structure did not respond to the anion exchange reaction (see Fig. S21 and Section S8 of the ESI†). The thick 5–8 nm lead laurate shell completely inhibited the anion-exchange process. This observation is important as the facile anion-exchange process in the perovskite NCs might inhibit its use in white light-emitting diodes (WLEDs).27–29 Earlier, Li et al. attempted to block the anion-exchange process by coating CsPbBr3 NC with SiO2, but failed.30 However in the present case, the anion-exchange reaction was ceased completely. This indicated that the shell thickness might act as an effective shield for the anion-exchange process. Recently, Imran et al. reported control over the anion-exchange reaction in CsPbX3 NC by using an amphiphilic polymer micelle and fabricating a stable WLED.29 We believe that our greener approach of synthesizing the MAPbBr3@lead laurate core–shell structure will play a crucial role in fabricating stable WLEDs in the near future. Moreover, such water stability and suppression of the anion exchangeability of MAPbBr3@lead laurate core–shell NCs were achieved without any special control or the extra perturbation generally needed to protect perovskite NCs from water.30,31 This surprising and positive effect inspired us to gain some insight into the mechanism of the process.
3.5. Mechanism of the core–shell formation through a self-defence mechanism
To gain an insight into the improved water stability and the formation of a lead laurate shell on lauric acid-capped MAPbBr3 NCs, we propose a plausible mechanism involving the surface dissolution process of the NCs and the formation of a lead laurate shell by the surface capping ligands in a self-defence mechanism. From the results, it was evident that the lauric acid-capped MAPbBr3 NC formed a shell while in contact with water. Recently, a very little amount of aqueous treatments have been found to be beneficial to improve the surface properties of the hybrid lead halide perovskite NCs and their water stability through the formation of core–shell structures.32,33 Even a tiny amount of water degrades the surface of the NCs on a small scale to produce the constituent cations and anions, which come into play in the water part.33 However, with LA being hydrophobic, the penetration of water molecules to the surface of the NCs is highly unlikely, while the formation of a lead laurate shell indicates the removal of lead ions (Pb2+) from the perovskite lattice site, which is the only source of the Pb2+ ions. This strongly suggests that water is somehow interacting with the MAPbBr3 NC surface by overcoming the effectual barrier of the LA. We propose that, due to the dynamic nature of ligand binding,34,35 it is quite possible that a small fraction of dynamically bound lauric acid may be slightly dislocated from the NC surface, leaving the NC surface to become uncoordinated and allowing a tiny few water molecules to interact. This small fraction of water is ideal and sufficient to dissolve a limited surface layer of the MAPbBr3 NCs. Different constituent cations and anions, like methylammonium (CH3NH3+), divalent lead (Pb2+), and bromide (Br−), can come into the water part and the divalent lead (Pb2+) forms the lead laurate shell by reacting with laurate anions.This small-scale water-assisted degradation of MAPbBr3 NC is a boon and beneficial in three ways: (1) within 30 min, an effective lead laurate shell has formed around the MAPbBr3 NC core that protects the NC from any further degradation by the water; (2) the formation of the shell replenishes the surface of the MAPbBr3 NC in a way that reduces the surface-related trap states and increases the radiative recombination rate, which ultimately increases the PLQY and average PL lifetime of the core–shell perovskite NC and makes it water stable; (3) the hydrophobic nature of LA itself acts as a controller to optimize the interaction between water and the perovskite surface. The reaction scheme on the surface of MAPbBr3 NCs is proposed to be
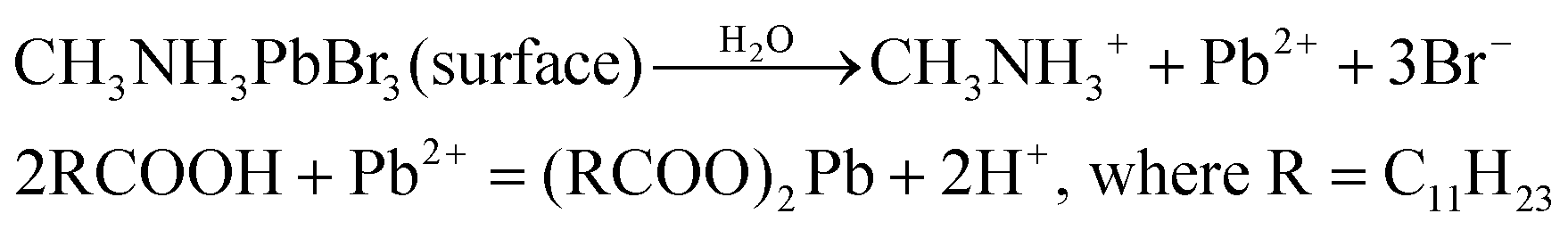
According to this reaction scheme, the medium should be acidic after the lead laurate shell formation and should also contain excess bromide ions. To confirm this, we measured the pH of the water after MAPbBr3 NC treatment. The water become acidic (pH 5.21) as expected after the formation of the lead laurate shell, whereas the pH of the water that was used for the experiment was neutral (pH 6.79). Further, we measured the bromide ion level of water as 200 ppm after the formation of the lead laurate shell by ion chromatography (which was below the detection limit before the MAPbBr3 NC treatment). These two observations further established our proposed mechanism, which is depicted in Scheme 2.
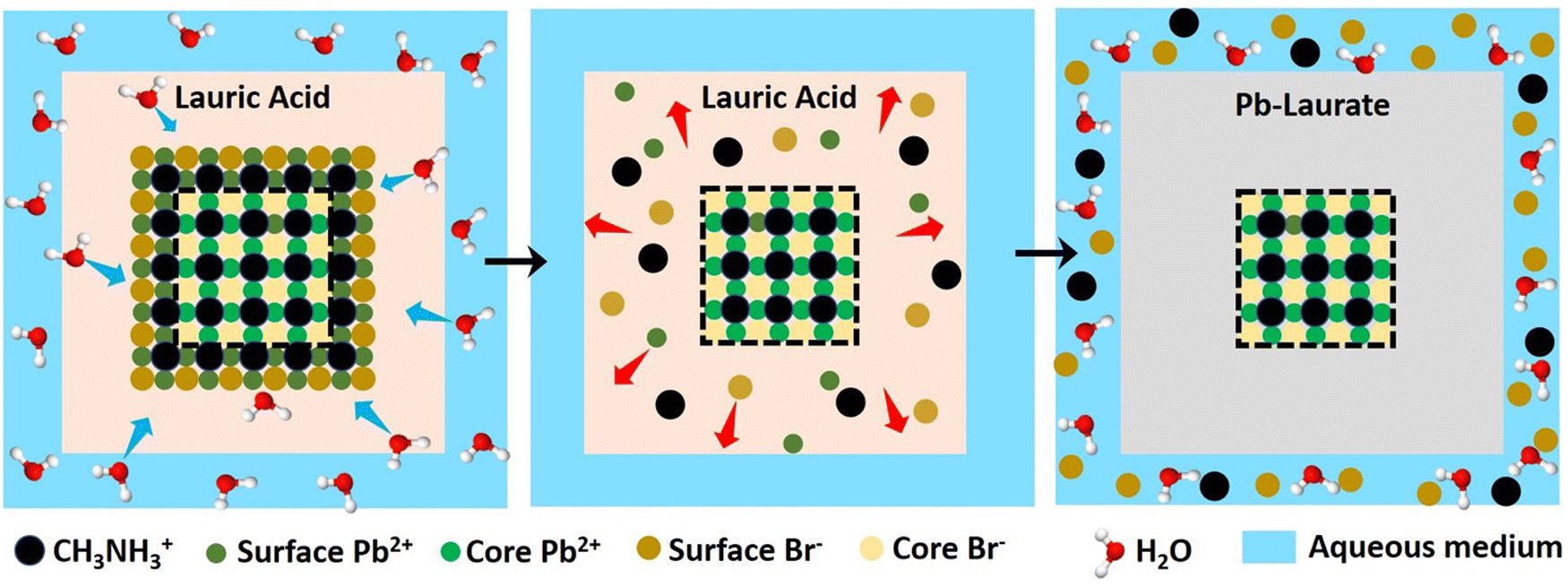 | ||
| Scheme 2 Schematic representation of the proposed mechanism of lead laurate shell formation around the MAPbBr3 NC in the presence of water. | ||
3.6. Synthesis of other MAPbX3 (X = Cl, I) NCs
To demonstrate the further applicability of the ionic liquid-like medium, we synthesized other MAPbX3 compounds (X = Cl, I). The purified MAPbCl3 and MAPbI3 NCs showed pure cubic and tetragonal crystalline phases, respectively (see Fig. S10 and Section S5 of the ESI†). For MAPbI3, the lower diffraction angle peaks were probably due to partial surface degradation to the lead laurate by the atmospheric moisture (see Fig. S10 and Section S5 of the ESI†). Detailed XPS analysis revealed a halide-rich surface in both MAPbCl3 (chlorine to lead ratio of 3:1) and MAPbI3 NCs (iodine to lead ratio of 3:4) (see Fig. S12 and Section S6 of the ESI†). However, high-resolution XPS showed two types of lead were present in the case of MAPbI3 NCs (see Fig. S12b and Section S6 of the ESI†). This may arise due to the partial surface degradation to lead laurate, which was in line with the PXRD analysis. FESEM analysis showed the formation of cubic to cuboid MAPbCl3 NCs sized ∼100–150 nm (see Fig. S16a and Section S7 of the ESI†); however, that of MAPbI3 NCs showed the formation of some irregular-shaped particles of ∼100 nm (see Fig. S16b and Section S7 of the ESI†). The MAPbCl3 NCs had a strong excitonic peak at 403 nm and a very weak PL at around 409 nm (see Fig. S19a and Section S8 of the ESI†). The MAPbI3 NCs showed a broad absorption and PL centred at 741 nm (see Fig. S19b and Section S8 of the ESI†). The time-resolved PL of MAPbCl3 and MAPbI3 NCs showed average lifetimes of 1.6 ns and 23.2 ns, respectively (see Fig. S19c and Section S8 of the ESI†).
4. Conclusions and future aspects
In conclusion, this report demonstrates the synthesis of MAPbBr3 NCs in lauric acid media, which were converted to MAPbBr3@lead laurate core–shell NCs in the presence of water. The MAPbBr3@lead laurate core–shell NCs showed a high PLQY and environmental stability. The main highlights of this work are depicted in Scheme 3 and are also summarized as follows: (i) we successfully synthesized green-luminescent MAPbBr3 NCs in lauric acid having a moderate PLQY of ∼19% in a single-step synthetic approach and observed a good environmental stability of up to 6 months. (ii) The PL over the entire visible range was tuned by synthesizing MAPbX3 NCs (where X is Cl, Br, I, and a mixture of either Cl and Br or Br and I) through the anion-exchange method. (iii) The NCs formed a core–shell structure (MAPbBr3@lead laurate) in a unique self-defence mechanism in the presence of water. First, owing to its hydrophobic nature, the shell safeguards the perovskite from further degradation by water and makes it water stable (for at least two months). Second, through surface modification, a five-fold increase in the PLQY and two-fold increase in the PL lifetime were achieved. Third, It prevents the anion-exchange process. To our delight, all these positive effects were achieved without any special control or extra perturbation. Further, the universality of the process was demonstrated by synthesizing MAPbCl3 and MAPbI3 NCs.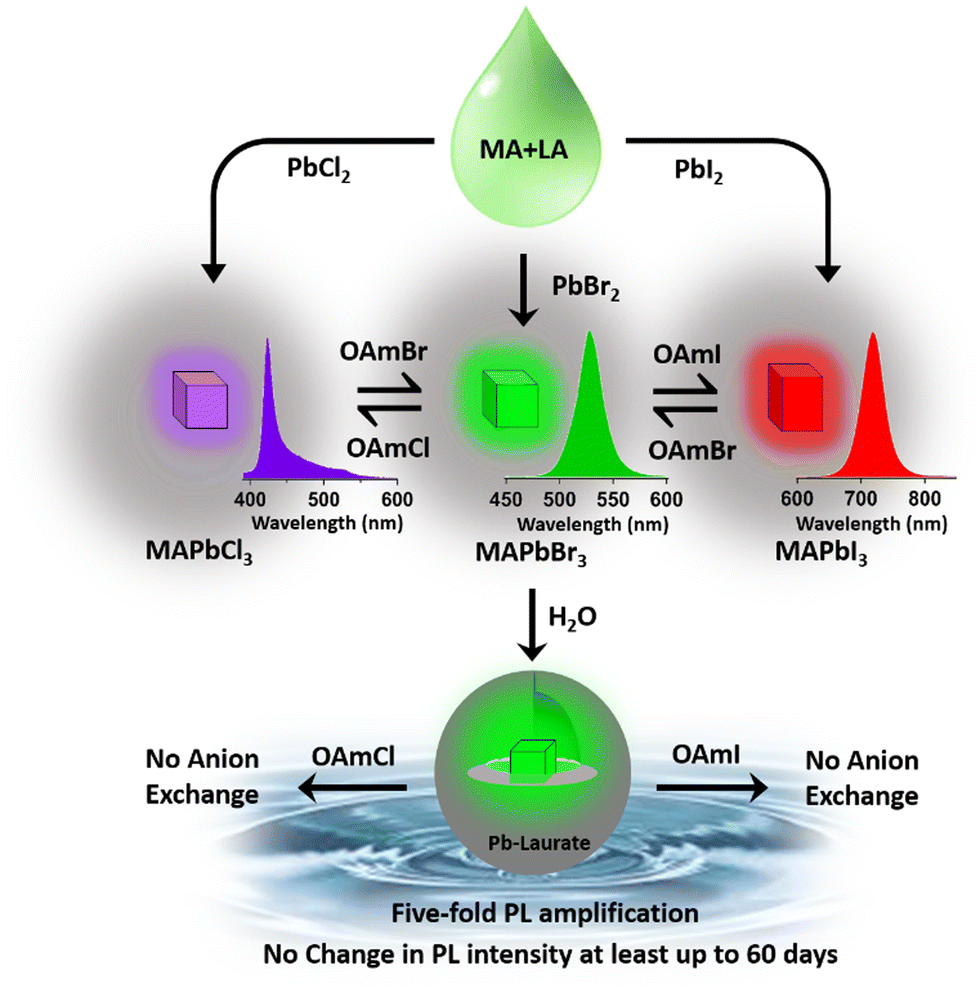 | ||
| Scheme 3 Schematic representation of the main highlights of the present work. Lauric acid and methyl amine (LA + MA) formed the solvent media, to which the addition of PbBr2 produced MAPbBr3 NCs without the use of any hazardous solvent. The entire visible range was tuned through anion exchange. The addition of water to MAPbBr3 NCs produced a core–shell structure in a unique self-defence mechanism that made the NCs water stable (for at least 60 days) with a five-fold increase in PLQY. The inhibition of anion exchange in the core–shell structure was also highlighted. | ||
Abbreviations
| MAPbBr3 | Methylammonium lead bromide |
| NC | Nanocrystal |
| PLQY | Photoluminescence quantum yield |
| MAPbBr3@lead | Methylammonium lead bromide core in lead laurate shell |
| LA | Lauric acid |
| OAmX | Oleylammonium halide salts |
Conflicts of interest
Authors declare no conflict of interests.Acknowledgements
SC and AS acknowledge IIT Kanpur for providing fellowships. TK and ND thank Prime Minister Research Fellowship (PMRF) and Council of Scientific and Industrial Research (CSIR), Government of India, respectively, for providing graduate fellowships. PS thanks IIT Kanpur for infrastructure and financial support.References
- Y. Dong, Y. Zhao, S. Zhang, Y. Dai, L. Liu, Y. Li and Q. Chen, J. Mater. Chem. A, 2018, 6, 21729–21746 RSC.
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X. G. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C. J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed.
- N. K. Tailor, M. Abdi-Jalebi, V. Gupta, H. Hu, M. I. Dar, G. Li and S. Satapathi, J. Mater. Chem. A, 2020, 8, 21356–21386 RSC.
- R. Vidal, J. A. Alberola-Borràs, S. N. Habisreutinger, J. L. Gimeno-Molina, D. T. Moore, T. H. Schloemer, I. Mora-Seró, J. J. Berry and J. M. Luther, Nat. Sustain., 2021, 4, 277–285 CrossRef.
- S. Chatterjee, M. Ghosal, K. Tiwari and P. Sen, J. Phys. Chem. Lett., 2021, 12, 546–551 CrossRef CAS PubMed.
- F. Zhang, S. Huang, P. Wang, X. Chen, S. Zhao, Y. Dong and H. Zhong, Chem. Mater., 2017, 29, 3793–3799 CrossRef CAS.
- J. C. Hamill, J. Schwartz and Y. L. Loo, ACS Energy Lett., 2018, 3, 92–97 CrossRef CAS.
- J. Stevenson, B. Sorenson, V. H. Subramaniam, J. Raiford, P. P. Khlyabich, Y. L. Loo and P. Clancy, Chem. Mater., 2017, 29, 2435–2444 CrossRef CAS.
- K. L. Gardner, J. G. Tait, T. Merckx, W. Qiu, U. W. Paetzold, L. Kootstra, M. Jaysankar, R. Gehlhaar, D. Cheyns, P. Heremans and J. Poortmans, Adv. Energy Mater., 2016, 6, 1–8 Search PubMed.
- Y. Hassan, O. J. Ashton, J. H. Park, G. Li, N. Sakai, B. Wenger, A. A. Haghighirad, N. K. Noel, M. H. Song, B. R. Lee, R. H. Friend and H. J. Snaith, J. Am. Chem. Soc., 2019, 141, 1269–1279 CrossRef CAS PubMed.
- T. Niu, L. Chao, W. Gao, C. Ran, L. Song, Y. Chen, L. Fu and W. Huang, ACS Energy Lett., 2021, 6, 1453–1479 CrossRef CAS.
- M. T. Hoang, F. Ünlü, W. Martens, J. Bell, S. Mathur and H. Wang, Green Chem., 2021, 23, 5302–5336 RSC.
- D. T. Moore, K. W. Tan, H. Sai, K. P. Barteau, U. Wiesner and L. A. Estroff, Chem. Mater., 2015, 27, 3197–3199 CrossRef CAS.
- S. Öz, J. Burschka, E. Jung, R. Bhattacharjee, T. Fischer, A. Mettenbörger, H. Wang and S. Mathur, Nano Energy, 2018, 51, 632–638 CrossRef.
- D. Li, L. Chao, C. Chen, X. Ran, Y. Wang, T. Niu, S. Lv, H. Wu, Y. Xia, C. Ran, L. Song, S. Chen, Y. Chen and W. Huang, Nano Lett., 2020, 20, 5799–5806 CrossRef CAS PubMed.
- L. Liu, Z. Tang, C. Xin, S. Zhu, S. An, N. Wang, L. Fan, C. Wei, Q. Huang, G. Hou, Y. Zhao, Y. Ding and X. Zhang, ACS Appl. Energy Mater., 2018, 1, 2730–2739 CrossRef CAS.
- D. Liu, Z. Shao, J. Gui, M. Chen, M. Liu, G. Cui, S. Pang and Y. Zhou, Chem. Commun., 2019, 55, 11059–11062 RSC.
- J. Y. Seo, T. Matsui, J. Luo, J. P. Correa-Baena, F. Giordano, M. Saliba, K. Schenk, A. Ummadisingu, K. Domanski, M. Hadadian, A. Hagfeldt, S. M. Zakeeruddin, U. Steiner, M. Grätzel and A. Abate, Adv. Energy Mater., 2016, 6, 1–6 Search PubMed.
- M. Li, C. Zhao, Z. K. Wang, C. C. Zhang, H. K. H. Lee, A. Pockett, J. Barbé, W. C. Tsoi, Y. G. Yang, M. J. Carnie, X. Y. Gao, W. X. Yang, J. R. Durrant, L. S. Liao and S. M. Jain, Adv. Energy Mater., 2018, 8, 1–8 Search PubMed.
- Y. Xia, C. Ran, Y. Chen, Q. Li, N. Jiang, C. Li, Y. Pan, T. Li, J. P. Wang and W. Huang, J. Mater. Chem. A, 2017, 5, 3193–3202 RSC.
- D. Chakraborty, N. Preeyanka, A. Akhuli and M. Sarkar, J. Phys. Chem. C, 2021, 125, 26652–26660 CrossRef CAS.
- M. T. Hoang, N. D. Pham, Y. Yang, V. T. Tiong, C. Zhang, K. Gui, H. Chen, J. Chang, J. Wang, D. Golberg, J. Bell and H. Wang, Green Chem., 2020, 22, 3433–3440 RSC.
- B. H. Morrow, P. H. Koenig and J. K. Shen, J. Chem. Phys., 2012, 137, 19 CrossRef PubMed.
- A. Dutta, R. K. Behera, P. Pal, S. Baitalik and N. Pradhan, Angew. Chem., Int. Ed., 2019, 58, 5552–5556 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X. G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- S. Paul and A. Samanta, ACS Energy Lett., 2020, 5, 64–69 CrossRef CAS.
- M. Imran, B. T. Mai, L. Goldoni, M. Cirignano, H. B. Jalali, F. Di Stasio, T. Pellegrino and L. Manna, ACS Energy Lett., 2021, 6, 2844–2853 CrossRef CAS PubMed.
- X. K. Liu, W. Xu, S. Bai, Y. Jin, J. Wang, R. H. Friend and F. Gao, Nat. Mater., 2021, 20, 10–21 CrossRef CAS PubMed.
- M. Bidikoudi, E. Fresta and R. D. Costa, Chem. Commun., 2018, 54, 8150–8169 RSC.
- M. Li, X. Zhang and P. Yang, Nanoscale, 2021, 13, 3860–3867 RSC.
- G. H. Ahmed, J. Yin, O. M. Bakr and O. F. Mohammed, ACS Energy Lett., 2021, 6, 1340–1357 CrossRef CAS.
- K. K. Liu, Q. Liu, D. W. Yang, Y. C. Liang, L. Z. Sui, J. Y. Wei, G. W. Xue, W. B. Zhao, X. Y. Wu, L. Dong and C. X. Shan, Light: Sci. Appl., 2020, 9, 1 CrossRef PubMed.
- Y. Liu, F. Li, Q. Liu and Z. Xia, Chem. Mater., 2018, 30, 6922–6929 CrossRef CAS.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- S. Chatterjee, P. Dey, N. Das, K. Tiwari, T. Maiti and P. Sen, Chem. – Eur. J., 2020, 26, 1506–1510 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD pattern of MAPbBr3 NCs, XPS data of MAPbBr3 NCs and MAPbBr3@lead laurate, FESEM data of MAPbBr3 NCs and MAPbBr3@lead laurate, the stability information of MAPbBr3 NCs, the anion exchange related data of MAPbBr3@lead laurate system and video of water stability of MAPbBr3@lead laurate NC. See DOI: https://doi.org/10.1039/d2ma00684g |
| This journal is © The Royal Society of Chemistry 2022 |