
Development of a continuous flow synthesis of FGIN-1-27 enabled by in-line 19F NMR analyses and optimization algorithms†
N.
Vasudevan
a,
Ehu C.
Aka
a,
Elvina
Barré
a,
Eric
Wimmer
b,
Daniel
Cortés-Borda
c,
Patrick
Giraudeau
 a,
Jonathan
Farjon
a,
Mireia
Rodriguez-Zubiri
a and
François-Xavier
Felpin
*a
a,
Jonathan
Farjon
a,
Mireia
Rodriguez-Zubiri
a and
François-Xavier
Felpin
*a
aUniversité de Nantes, CNRS, CEISAM UMR 6230, 2 rue de la Houssinière, 44322 Nantes, France. E-mail: fx.felpin@univ-nantes.fr; Tel: +33-025-112-5422 Web: http://felpin.univ-nantes.fr/
bInstitut de Recherches Servier, 125 Chemin de Ronde, 78290 Croissy sur Seine, France
cUniversidad del Atlántico, Facultad de ciencias básicas, Carrera 30 # 8-49, Puerto Colombia, Atlántico, Colombia
First published on 15th July 2021
Abstract
A straightforward continuous flow synthesis of FGIN-1-27, a compound with potent anxiolytic effects, is described from inexpensive and commercially available starting materials. The four-step synthesis includes the direct C–H arylation of an indole with an arenediazonium salt. The continuous flow route was developed thanks to the use of enabling technologies such as real-time in-line benchtop 19F NMR analysis and an optimization algorithm assisting the decision-making process. These enabling technologies increase the process safety and minimize the number of experiments required in optimization campaigns.
Introduction
FGIN-1-27 (1) is a synthetic agonist ligand for the 18 kDa translocator protein (TSPO), a cholesterol transporter from intracellular stores into mitochondria (Fig. 1).1,2 As a TSPO ligand, FGIN-1-27 (1) increases steroidogenesis of neuroactive steroids such as allopregnanolone and produces potent anxiolytic effects.3 FGIN-1-27 (1) is frequently used by biologists for research purposes as an object of study on its own or as a standard in biological tests.3–6 It is commercially available but only in milligram quantities at a relatively prohibitive cost (ca. 2000 € per mmol). While two batch syntheses of FGIN-1-27 (1) have already been reported in the literature,7,8 we reasoned that we could improve the availability of FGIN-1-27 (1) by developing an efficient, straightforward and experimentally simple continuous flow process. Compared to traditional batch approaches, continuous flow processes are more amenable to scale-up thanks to a better control of reaction parameters leading to higher reproducibility and safer operational conditions.9,10 These points are even more relevant if we consider that the synthesis of FGIN-1-27 (1) might be carried out by biologists or biochemists inexperienced in chemical synthesis. Also, from the viewpoint of chemical synthesis, continuous flow processing allows a scalable access to hazardous transformations that would be otherwise difficult to carry out, if not forbidden, in conventional batch syntheses.11–16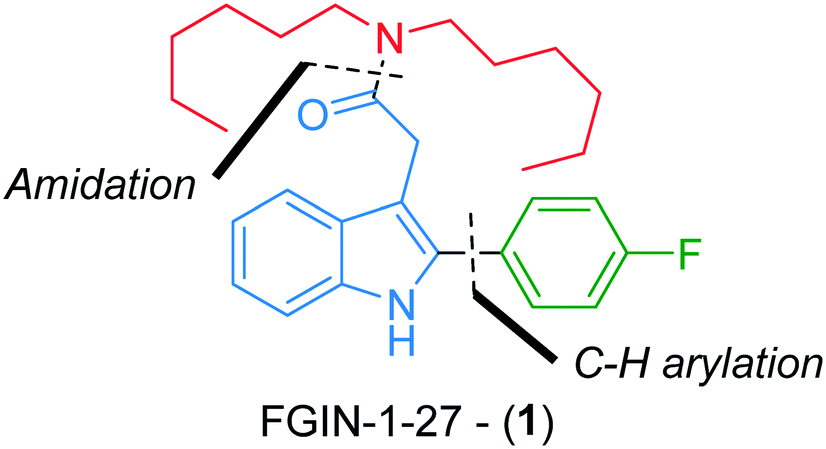 | ||
| Fig. 1 Structure of FGIN-1-27 (1). | ||
Recently, we reported a robust and high yielding method for the palladium-catalyzed C–H arylation of indole-3-acetic acid derivatives with arenediazonium salts in flow.17 We capitalized on this previous work to report, herein, a straightforward flow synthesis of FGIN-1-27 mainly through two assemblies, i.e., C–H arylation of indole and an amidation step (Fig. 1). This work features the use of groundbreaking technologies such as real-time in-line benchtop 19F NMR spectroscopy and an optimization algorithm assisting the decision-making process.
Results and discussion
Our flow strategy to access FGIN-1-27 (1) started with the search of suitable flow conditions for the synthesis of 4-fluorobenzene diazonium tosylate 2. We recently described the palladium-catalyzed direct C–H arylation of indole-3-acetic acid derivatives with arenediazonium salts in a mixture of MeOH–DMF–EtOAc as a solvent.17 Building on these recent results, we reasoned that 4-fluorobenzene diazonium tosylate 2 should be ideally prepared in MeOH with the subjacent idea of anticipating a prospective diazotization-coupling telescoped process. A few years ago, we validated a two-step telescoped flow process approach involving the diazotization of anilines in MeOH, followed by palladium-catalyzed Heck coupling with methyl acrylate.18 With these studies in mind, we were relatively confident about succeeding in preparing diazonium salt 2 in MeOH with the two-stream flow device depicted in Scheme 1. The two inlets were equipped with 0.5 mL injection loops. The first loop was loaded with aniline 3 (0.2 M), p-toluenesulfonic acid (PTSA, 0.22 M) and C6F6 (0.2 M) in MeOH, while the second loop was filled with a solution of t-BuONO (0.3 M) in MeOH. The two streams were mixed in a T-shaped mixer and the resulting mixture was reacted in a PEEK coil reactor (5 mL) maintained at the desired temperature. The outlet of the reactor was connected to the flow cell of a benchtop NMR spectrometer.19–21 The NMR conversion was determined through 1D 19F experiments at 40.88 MHz in stop-flow mode. The advantages of monitoring the formation of 2 by 19F experiments compared to more traditional 1H experiments is three fold.22–24 First, the large range of chemical shifts, typically ranging from −300 to 100 ppm, limits unwanted signal overlapping of aromatic protons which is one of the main issues for 1H experiments conducted on low-field benchtop spectrometers. Second, the 19F nucleus is 100% naturally abundant, allowing fast measurements with 83% of the 1H nucleus receptivity. Third, the integrated field-frequency fluorine lock system of the NMR spectrometer allows the use of non-deuterated solvents. Hexafluorobenzene (C6F6) was used to calibrate the 19F chemical shifts with fluorine atoms resonating at −164.9 ppm. Note that to optimize data acquisition, a small 19F spectral width was chosen between −20 and −140 ppm (see Fig. 2 and 3). Such conditions led to controlled folding of the C6F619F peak at −164.9 ppm to −39 ppm (see Fig. 2 and 3), which allowed the use of the folded peak as a chemical shift reference. The main limitations of the in-line NMR analysis are those mainly associated with the use of a medium magnetic field that results in a relatively low sensitivity (limit of detection in the mM range for abundant nuclei).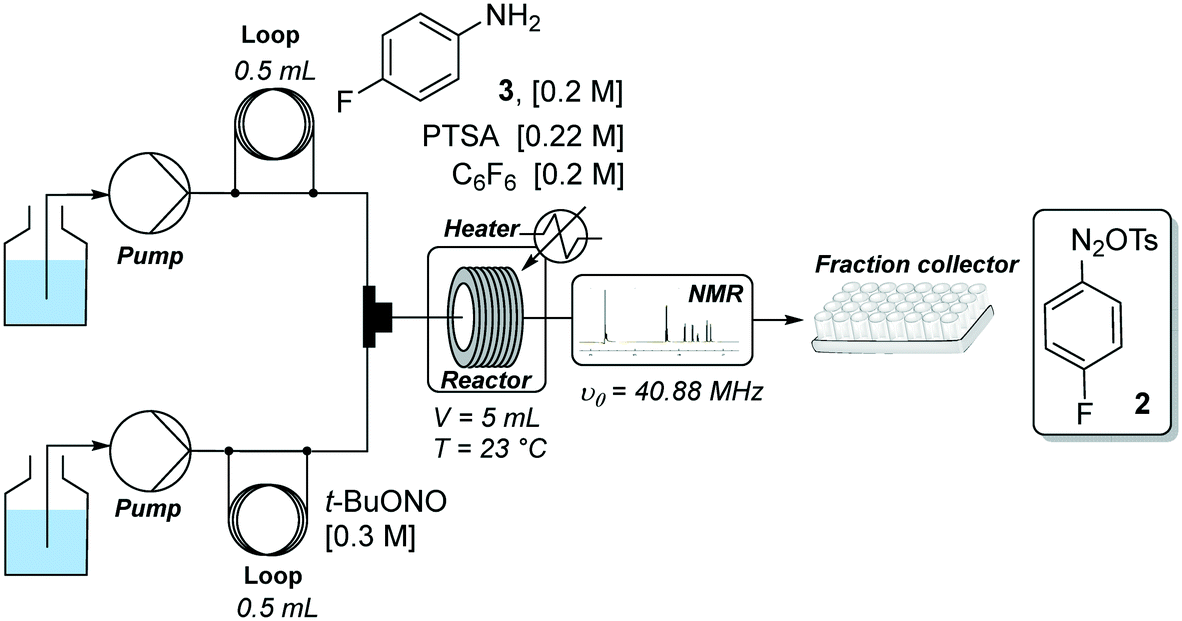 | ||
| Scheme 1 Two-stream flow device for the optimization of the diazonium salt formation. | ||
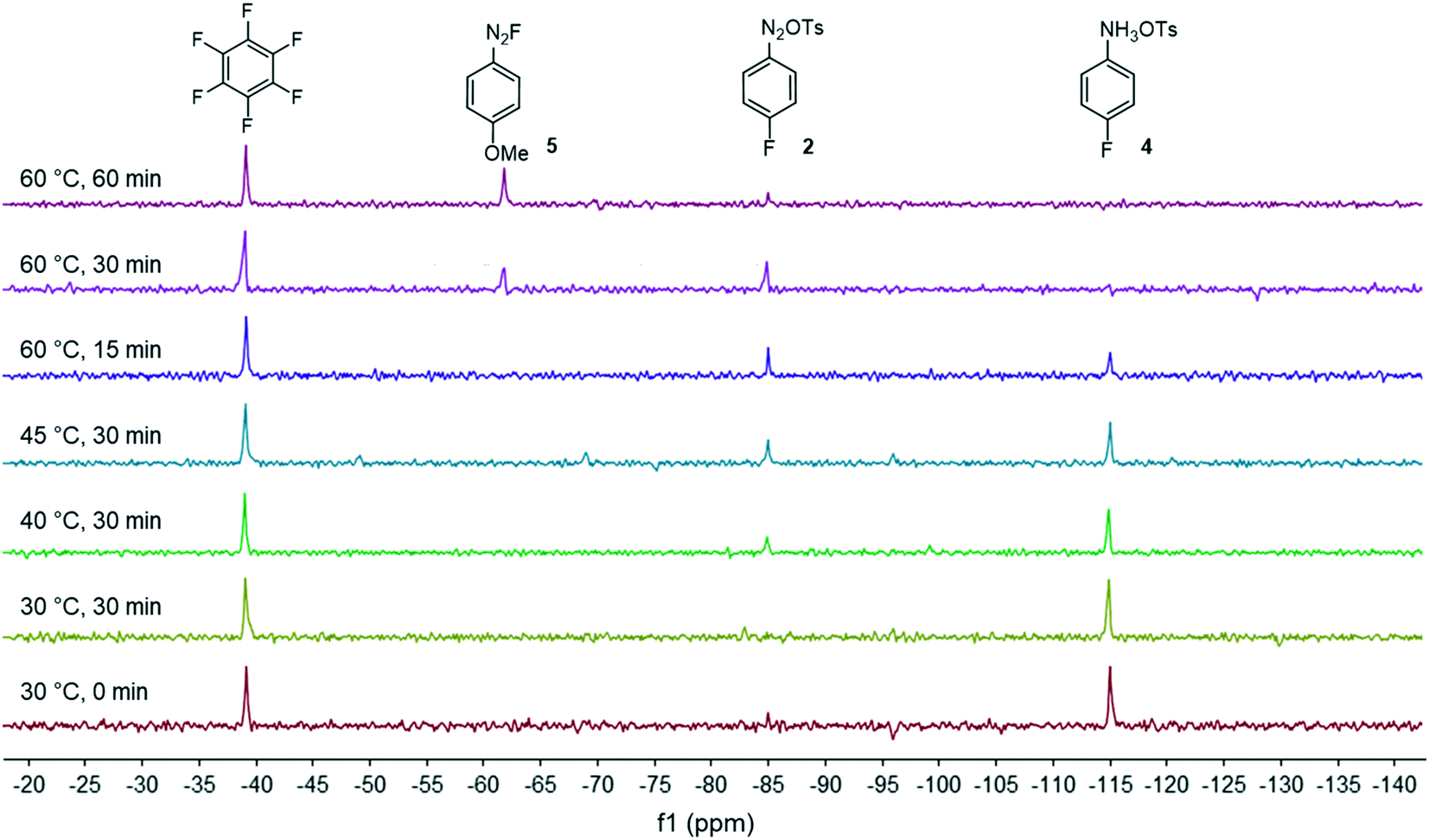 | ||
| Fig. 2 Monitoring of the diazonium salt formation in MeOH by in-line 19F NMR analysis at 40.88 MHz. | ||
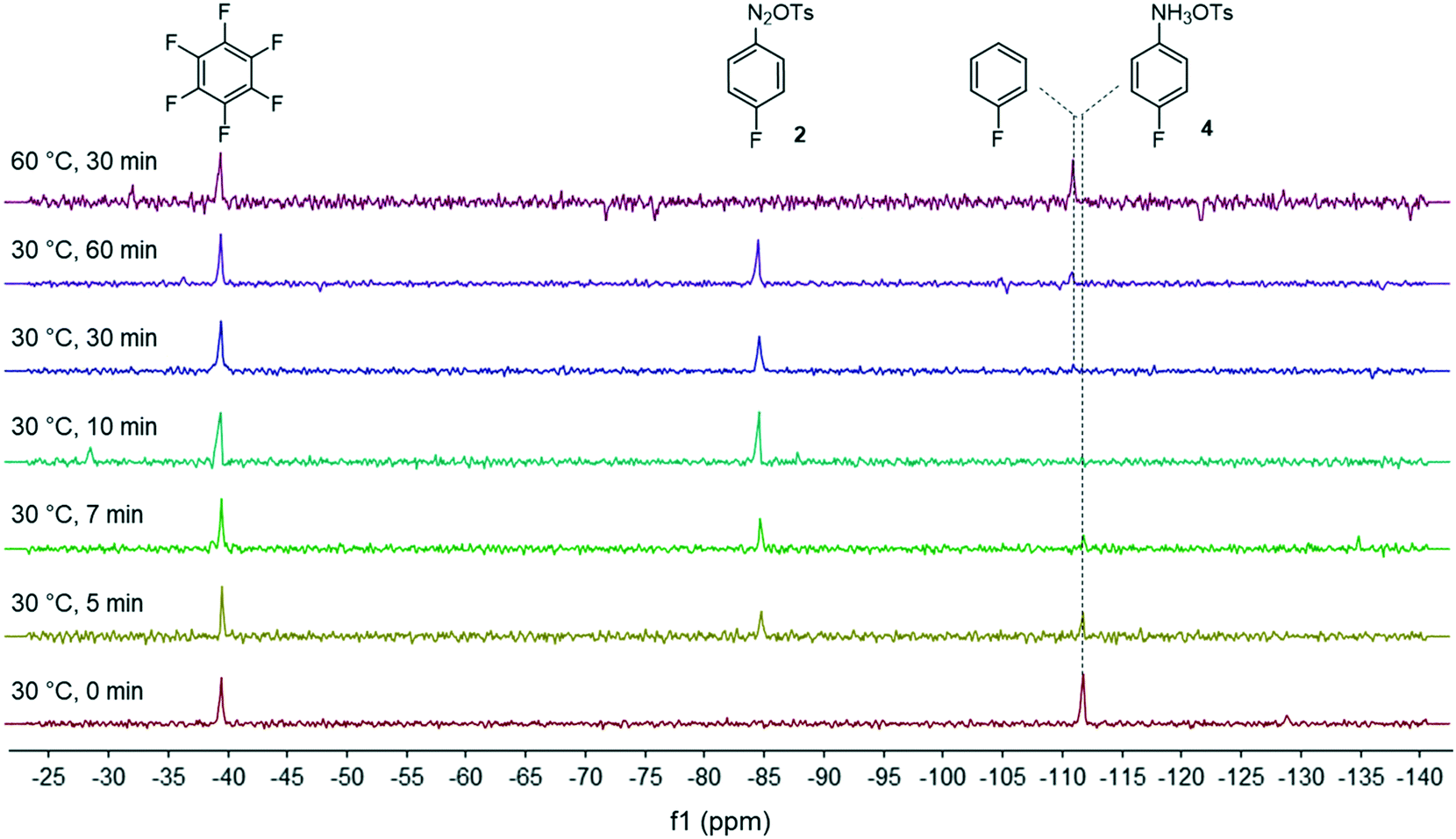 | ||
| Fig. 3 Monitoring of the diazonium salt formation in THF by in-line 19F NMR analysis at 40.88 MHz. | ||
Fig. 2 shows the spectra of crude mixtures recorded in MeOH within 320 seconds (32 scans) corresponding to experiments conducted at a temperature ranging from 30 to 60 °C and within 15 to 30 minutes of residence time. To our surprise, no formation of the diazonium salt 2 could be detected at 30 °C and only a low conversion was observed at 40–45 °C after 30 minutes of residence time. This result was unanticipated since most anilines are usually diazotized within a few minutes under similar flow conditions in MeOH.18 Increasing the temperature to 60 °C allowed, after only 15 minutes of residence time, a substantial conversion which became complete within 30 minutes as evidenced by the disappearance of the starting anilinium 4 (−115 ppm). Unfortunately, together with the formation of the expected diazonium salt 2 (−85 ppm), we observed the appearance of another singlet at −63 ppm assigned to the unanticipated diazonium salt 5. The latter was formed by the nucleophilic aromatic substitution of the fluorine atom by methanol followed by counter-anion metathesis. The strong electron-withdrawing properties of the diazonium function strongly activates the aromatic ring toward the nucleophilic attack of MeOH which likely proceeds through a standard addition–elimination mechanism.25–27 After 60 minutes of residence time at 60 °C, the targeted diazonium salt 2 almost completely disappeared in favor of the unwanted diazonium salt 5.
Facing these unexpected results, we reasoned that the high lability of the fluorine atom of diazonium salt 2 required the use of a non-nucleophilic solvent as recently demonstrated by Schmidt et al.28 In this frame, we reevaluated our flow process by switching the solvent from MeOH to THF which is known to be a suitable solvent for diazonium salt synthesis (Fig. 3).29 We ruled out the use of DMF as the solvent since preliminary experiments suggested that DMF favored the diazonium salt decomposition via a dediazotization pathway.30 We were satisfied to see that in THF, the kinetics of the diazonium formation greatly increased since the starting anilinium 4 was completely consumed after only 10 minutes of residence time. Upon prolonging the residence time (>30 min) or at elevated temperature (60 °C), we observed the progressive degradation of the diazonium salt 2 which underwent a protodediazotisation pathway leading to fluorobenzene.
With the robust flow synthesis of 4-fluorobenzene diazonium tosylate 2 in hand, we continued our studies with the key direct C–H arylation step. In the first attempt, we transposed the experimental flow conditions we recently used for the coupling of indoles with a variety of diazonium salts,17i.e., 44 °C, 74 min residence time and 1.6 equiv. of diazonium, to the coupling of indole 6 with diazonium salt 2 using the two-stream flow setup depicted in Table 1, entry 1. The first injection loop (0.5 mL) was loaded with a solution of 4-fluorobenzene diazonium tosylate 2 in MeOH (0.16 M) which was pumped at 62.5 μL min−1 (pump 1) with MeOH as a carrier solvent. A solution of indole 6 (0.1 M), Pd(OAc)2 (0.01 M) and naphthalene (0.1 M), as the internal standard, in a mixture of DMF/EtOAc (1/4, v/v) was loaded in the second injection loop (0.5 mL) and pumped at 62.5 μL min−1 (pump 2). Both streams met in a T-shaped piece and the reaction occurred in a stainless steel coil reactor (10 mL, 135 μL min−1). The crude mixture was collected in a test tube and analyzed by at-line HPLC. Under such experimental conditions, the expected fluorinated indole 7 was formed with a modest yield (47%) and was accompanied by trace amounts, ca. 2%, of the undesired arylated indole 8 (Table 1). The latter resulted from the coupling of indole 6 with 4-methoxyphenyl diazonium salt 5 which was formed in situ by the nucleophilic aromatic substitution of the fluorine atom with methanol (vide supra). With the aim of improving the reaction outcome leading to 7 and eventually suppressing the formation of unwanted indole 8, we explored the possibility of modifying the solvent composition of the reaction media. Regarding the first line delivering the diazonium salt, the substitution of MeOH for EtOH or DMF lowered the reaction yield of indole 7 to only 5–10% (entries 2 and 4), while the use of i-PrOH was hampered by the low solubility of diazonium salt 2 (entry 3). We also modified the solvent mixture of the second stream, unfortunately, without success since the substitution of DMF/EtOAc for DMF or CH3CN and the decrease of the volume of MeOH were detrimental to the reaction outcome (entries 5–7). Surprisingly, the movement of MeOH from pump 1 to pump 2 completely inhibited the reactivity, suggesting a specific interaction of MeOH with diazonium salt 2 that deserves further clarification (entry 8).
|
|
|||
|---|---|---|---|
| Entry | Solvent 1 | Solvent 2 | Yield (%)a7/8 |
| a Yields determined by HPLC. b The diazonium salt precipitated in i-PrOH. | |||
| 1 | MeOH | DMF/EtOAc (1/4) | 47/2 |
| 2 | EtOH | DMF/EtOAc (1/4) | 10/— |
| 3 | i-PrOH | DMF/EtOAc (1/4) | —b |
| 4 | DMF | DMF/EtOAc (1/4) | 5/— |
| 5 | MeOH | DMF | 44/1 |
| 6 | MeOH | CH3CN | 4/<1 |
| 7 | MeOH/DMF (1/1) | DMF/EtOAc (1/4) | 11/0 |
| 8 | DMF | MeOH/DMF (1/4) | 0/0 |
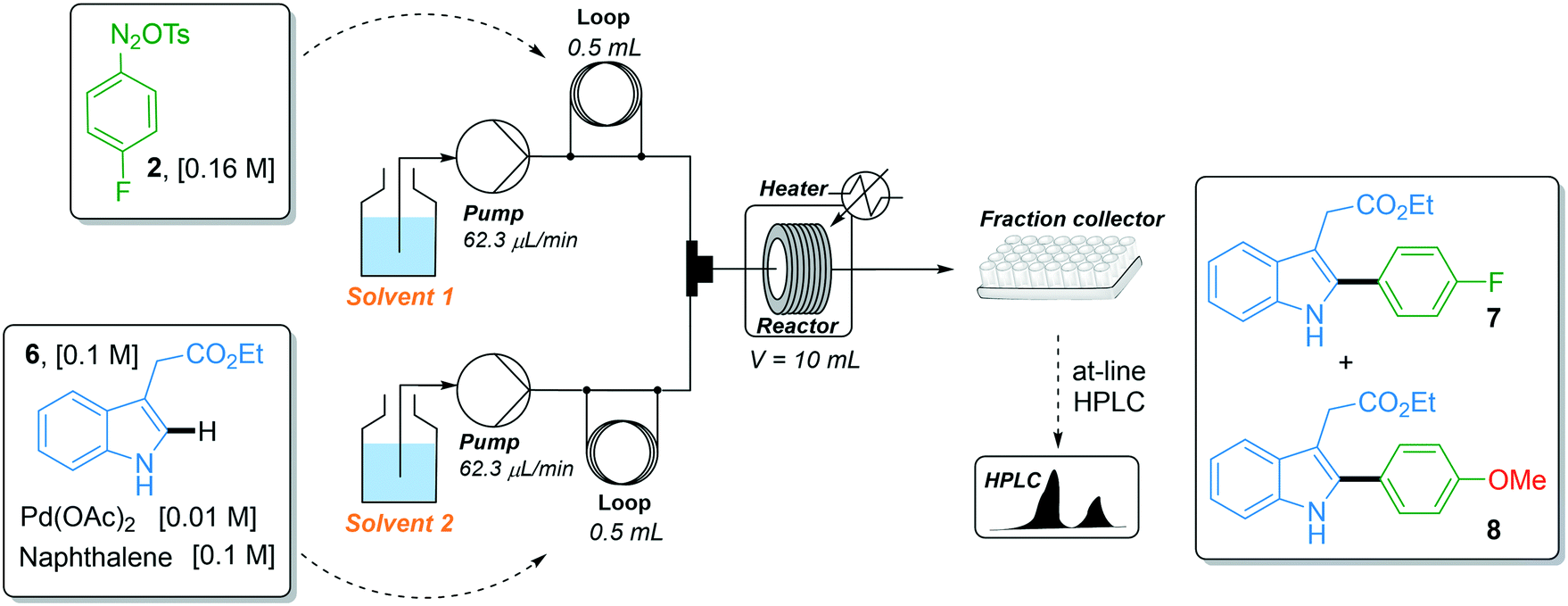
With this short solvent screening, we learned that the combination of solvents initially developed in our previous studies remained the most effective one. The chemical reactivity for the C–H arylation was critically hampered by the absence of MeOH and the stability of 4-fluorobenzene diazonium tosylate 2 was compromised in CH3CN or when the content of DMF was increased, likely due to extensive undesired dediazotization pathways.30 However, the modest reaction yield obtained under the experimental conditions of entry 1 (47%) associated with the long residence time required (74 min) compromised any scaling experiment. Considering that the peculiar reactivity of 4-fluorobenzene diazonium tosylate 2 might not be representative of the other diazonium salts screened in our previous studies,17 we embarked on a meticulous optimization campaign assisted by an optimization algorithm. Our algorithm-assisted optimization strategy was based on the use of a profoundly modified Nelder–Mead method for which a priori and gradient information on the reaction studied was not required since chemical reactions are treated as a black-box. The Nelder–Mead method converts input continuous variables (e.g., temperature, pressure, time, equivalents, etc.) into an output variable to be optimized (yield, cost, productivity, etc.). The n-dimensional chemical space of the objective function is explored through convex polytopes of n + 1 vertices, also called simplexes. Each ranked vertex represents an experiment and the algorithm progresses toward an optimum by replacing the worst vertex (experiment) by a new and (always) better vertex. We and others already demonstrated the suitability of the Nelder–Mead method for chemical problems even in the presence of experimental noise, while traditional gradient-based optimization methods can be severely impacted if experimental failure occurs during the determination of the gradient.19,31–40 However, the Nelder–Mead method only converges to local optima whose quality highly depends on human-biased initialization and lengthy convergence is generally observed when optimizing large dimensional problems. The modified Nelder–Mead version we used in this study addressed these issues as it i/ offers the possibility of temporarily reducing the dimensionality of the search to speed-up the exploration of a subspace, ii/ associates the Golden search method when the dimensionality reduction leads to 1-D optimizations, iii/ includes either automated or human-assisted mechanisms to escape from unsatisfactory local optima and iv/ uses multiple stopping criteria to limit the total number of experiments.41
The C–H arylation of indole 6 with 4-fluorobenzene diazonium tosylate 2 was optimized using the two-stream flow setup depicted in Scheme 2. A solution of 4-fluorobenzene diazonium tosylate 2 in MeOH (0.1 M) was loaded in the first injection loop (2 mL) and flowed at the required flow rate with MeOH as a carrier solvent. A solution of indole 6 (0.1 M), Pd(OAc)2 (0.01 M) and naphthalene (0.1 M) in a mixture of DMF/EtOAc (1/4, v/v) was loaded in the second injection loop (0.5 mL) and flowed with a mixture of DMF/EtOAc (1/4) at the required flow rate. Both streams met in a T-shaped piece and the reaction occurred in a stainless steel coil reactor (5 mL). The crude mixture was collected in test tubes and analyzed by at-line HPLC. The optimization algorithm was fed with the reaction yield determined by HPLC and a new set of experimental conditions was proposed. In this study, we did not use an automated flow device for two reasons. First, the use of an autonomous system is fully justified when it can be continuously used without human interception. Yet, we observed that diazonium salt 2 cannot be stored in MeOH at room temperature for more than 4 hours, precluding the use of an autonomous system pumping a mother solution of diazonium 2 in MeOH for several hours. Second, the release of nitrogen gas accompanying the reaction of indole 6 with diazonium 2 complicated the use of an automated online injection in the HPLC system.
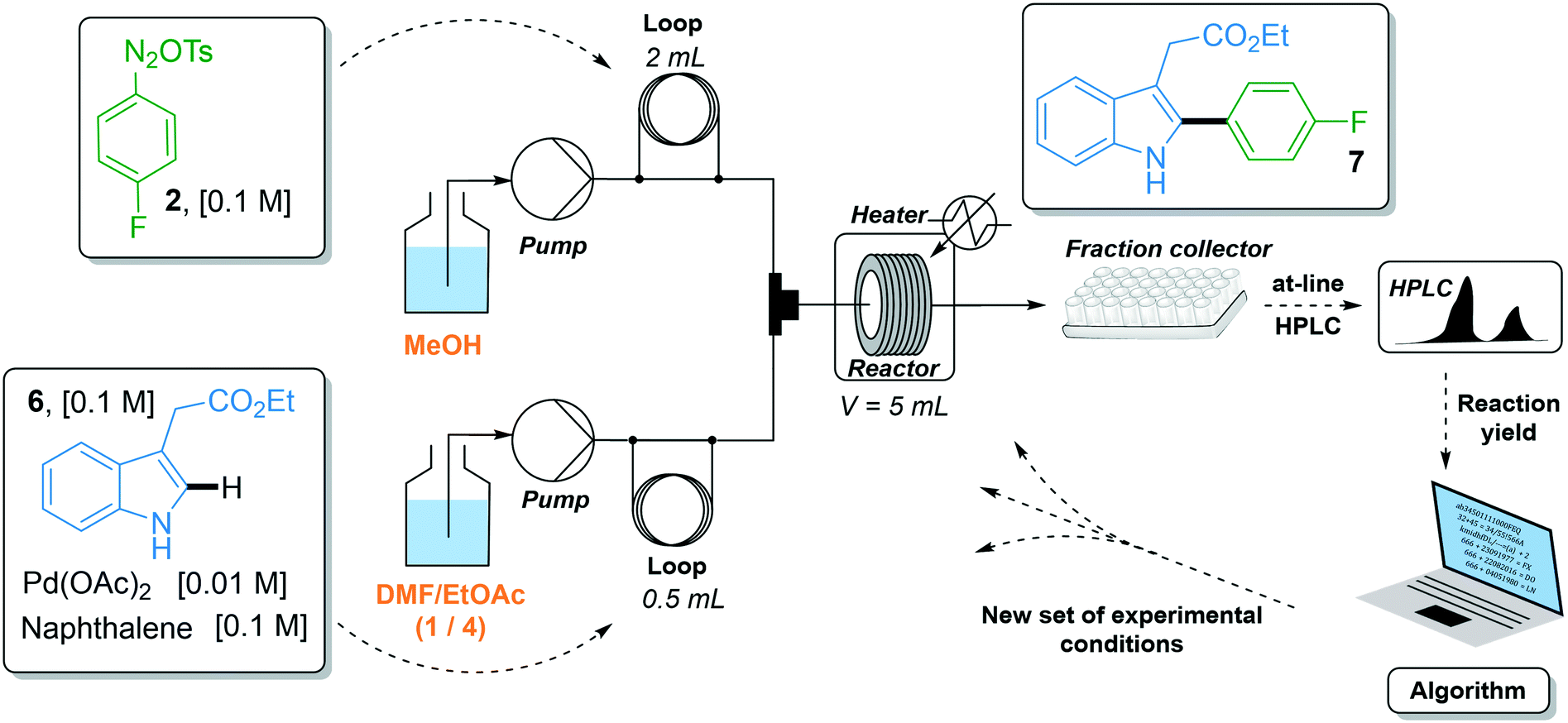 | ||
| Scheme 2 Reaction setup for the optimization of indole 6. | ||
The reaction yield was optimized in a four-dimensional space where the residence time, temperature, equivalents of diazonium 2 and loading of palladium catalyst were the four considered input variables n in the restricted search space of 10–30 min, 25–60 °C, 1–2 equiv. and 1–5 mol%, respectively. The initial starting experiment X0 was fixed at 10 min of residence time, 25 °C, 1 equiv. of diazonium 2 and 1 mol% Pd(OAc)2 with d values of 4 min, 7 °C, 0.2 equiv. and 0.8 mol%, respectively (Fig. 4a and b). Unfortunately, the first simplex, consisting of n + 1 experiments, failed to give the expected arylated indole 7. In this situation, a specific mechanism included in the optimization algorithm allows the proposal of a new random starting experiment X0 while keeping the initial fixed d values. The new X0 experiment proposed by the machine was 25 min of residence time, 39 °C, 1.7 equiv. of diazonium 2 and 4 mol% Pd(OAc)2. With this restart simplex, the reaction yield spectacularly increased, up to 48% in experiment 10. The algorithm further progressed until experiment 18 where it located an optimum (1.7 equiv. diazonium salt, 4.1 mol% Pd, 59 °C and 23 min residence time) corresponding to 80% yield (see Fig. S1 in the ESI†). Being unable to locate a better optimum, the algorithm stopped at experiment 23 after five consecutive rejected simplexes (stopping criterion). The experimental conditions of the 23 experiments can be found in Table S1 in the ESI.† As the optimization was not conducted with an automated flow device, all reactions were conducted twice in order to exclude any potential negative or positive false results. Regarding the random restart simplex proposed by the machine (experiments 6–10), we need to admit that the random simplex proposed inevitably influenced the following experiments. However, as discussed above, the modifications made to our simplex algorithm minimize the impact of human-biased or random initialization on both the rapidity and the quality of the convergence.
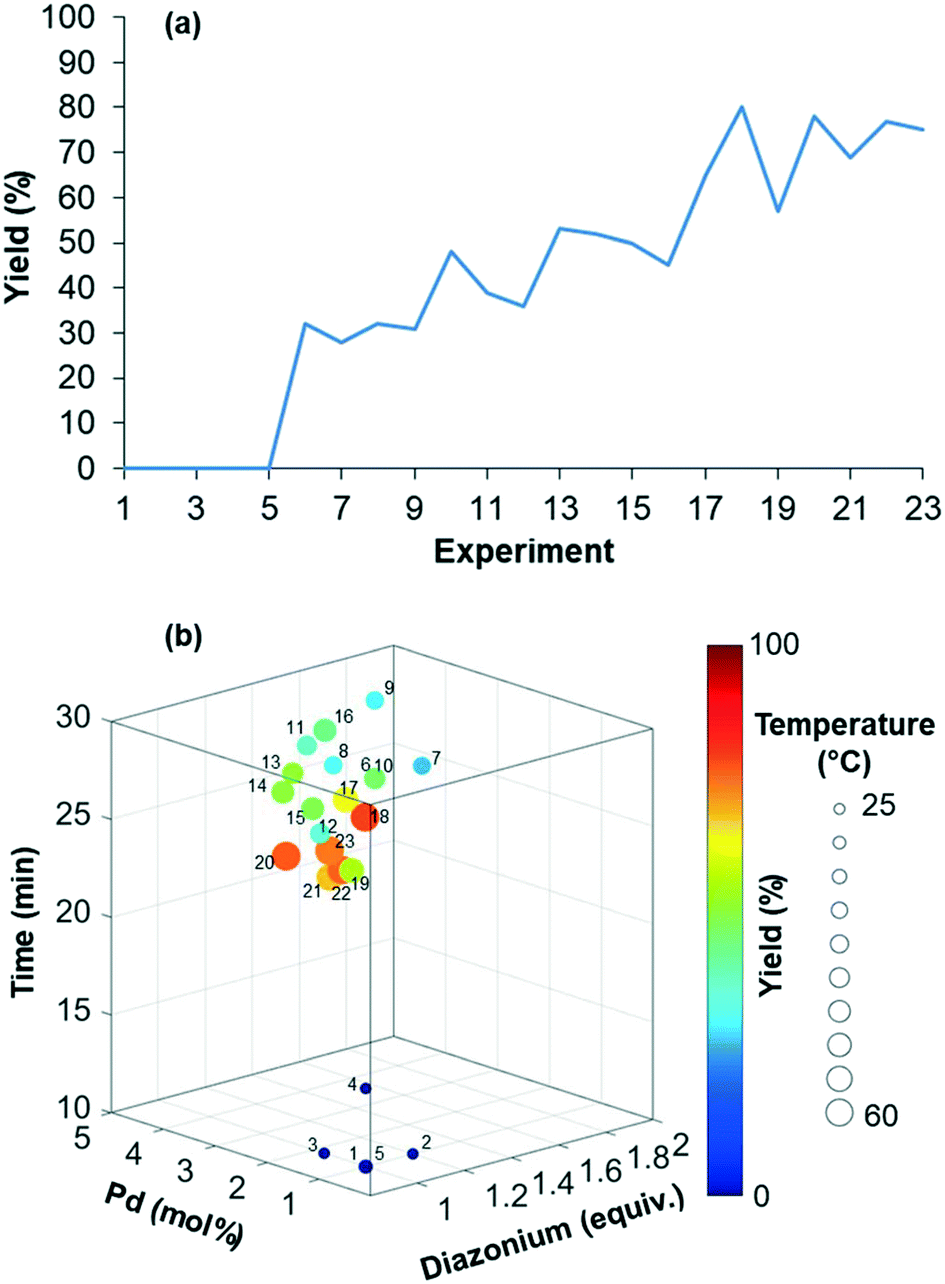 | ||
| Fig. 4 (a) Maximization of the yield of indole 7. (b) Representation of the four-dimensional experimental conditions for the maximization of the yield of indole 7. | ||
In order to install the required dihexylamide group through an amidation step, hydrolysis of the ethyl ester was performed under basic conditions using a two-stream flow setup (Scheme 3a). In the first line, a 1 mL injection loop was loaded with a solution of indole 7 (0.3 M) in THF while in the second line, a solution of 2 M aqueous KOH in MeOH (4/6) was continuously pumped. Both streams met in a T-shaped piece and reacted at 70 °C in a stainless steel coil reactor (5 mL) at a flow rate of 166 μl min−1 (30 min residence time). The resulting mixture was collected in vials and purified by flash chromatography to give the corresponding acid 9 with 97% yield. The amidation of acid 9 with dihexylamine was the last step of the synthetic sequence to obtain FGIN-1-27 (1). Amidation is among the most frequently used reactions in medicinal chemistry,42 and numerous batch procedures and coupling agents have been developed to address specific needs for mild conditions, reaction rates, efficiency and safety.43 By contrast, examples of solution-phase amidation in flow are scarce especially because many coupling agents suffer from either a hazardous profile, limited solubility or the formation of by-products which complicate the product isolation.44–46 For the amidation of acid 9 with dihexylamine, we opted for the combination of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) with 2-hydroxypyridine oxide (HOPO).46 This combination is attractive due to their low thermal hazard,47 fast kinetics and water soluble by-products. A two-stream flow setup was elaborated to proceed with the amidation of acid 9 with dihexylamine (Scheme 3b). In the first stream, a 1 mL injection loop was loaded with a solution of acid 9 (0.23 M), EDC·HCl (0.64 M) and HOPO (0.22 M) in a mixture of THF/H2O/acetone (1/2/4) while in the second stream, a 2 mL injection loop was loaded with a solution of dihexylamine (0.25 M) and Et3N (1.04 M) in a mixture THF/H2O (9/1). Both streams, flowing at 167 μL min−1, met in a T-shaped piece and reacted at 23 °C in a stainless steel coil reactor (10 mL) for a total of 30 min of residence time. The resulting collected mixture was purified by flash chromatography to give FGIN-1-27 (1) with 95% yield.
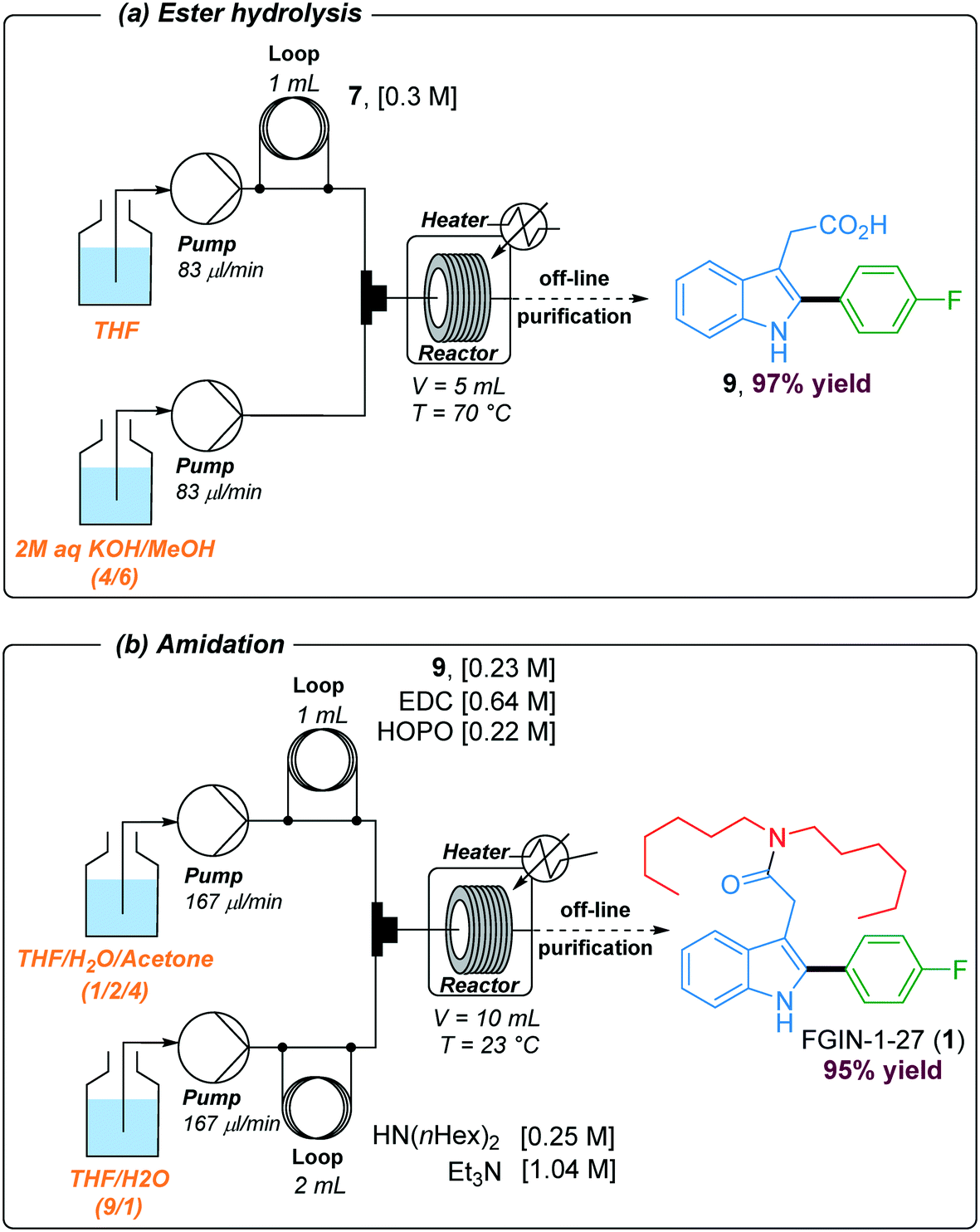 | ||
| Scheme 3 Reaction setups used for preparing acid 9 and FGIN-1-27 (1). | ||
Experimental section
General information
All commercially available chemicals were used as received unless otherwise noted. High-field 1H and 13C NMR spectra were recorded at 300 or 400 and 75 or 100 MHz, respectively. 1H and 13C NMR spectra were referenced to the residual signal of the internal deuterated solvent (CDCl3) at 7.26 and 77.16 ppm, respectively, and coupling constants were measured in hertz. 19F spectra were recorded at 40.88 MHz with a 1 T-benchtop spectrometer (Spinsolve, Magritek) equipped with a flow cell (4 mm id). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad. FT-IR spectra were recorded in ATR mode. Wavelengths of maximum absorbance (νmax) are quoted in wave number (cm−1). Flash column chromatography was performed using silica gel 60 (40–63 μm). In order to remove any trace of waste (Pd particles, inorganic salts…) which progressively deposits on the wall of the reactor coil, the tubing was washed every 100 hours of use with an aqueous solution of nitric acid (1 M, 50 mL), followed by thorough washing with water (100 mL) and CH3CN (100 mL).Details of the experimental flow setup
HPLC pumps (JASCO PU4185) were employed to flow the solution through the system. The reaction yields were determined by HPLC using the following conditions: Agela Promosil C18 column (3.5 mm × 150 mm, 5 μm), solvent: MeOH/H2O (70/30), isocratic mode, flow rate 0.5 mL min−1, UV detection (254 nm).1H-Indole-3-acetic acid ethyl ester 6
Note that 1H-indole-3-acetic acid ethyl ester 6 is commercially available but it can be prepared as well from the corresponding acid by esterification following a modified published procedure.48 Indole acetic acid (10 g, 57.14 mmol) was dissolved in dry EtOH (125 mL) in a 250 mL round bottom flask fitted with a reflux condenser and a calcium chloride guard tube. Sulfuric acid (10 mL, 0.19 mol) was added dropwise at 0 °C for 15 min and the resulting mixture was refluxed for 16 h at 75 °C. The volume of the reaction mixture was reduced to ca. 1/6 of its initial volume under vacuum and diluted with cold water (25 mL). The aqueous layer was extracted with EtOAc (5 × 50 mL). The combined organic layers were successively washed with H2O (25 mL), saturated NaHCO3 solution (2 × 25 mL), H2O (2 × 25 mL) and saturated NaCl solution (2 × 25 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure to give 6 (10.6 g, 91%) as a pale yellow solid which was used in the next step without further purification. mp 40 °C [Lit.49 42–45 °C]. IR (ATR) ν 3353, 2984, 1719, 1456, 1335, 1239, 1172, 1026, 735, 665, 598, 565. 1H NMR (CDCl3, 300 MHz) δ 8.12 (br s), 7.64 (dm, 1H, J = 7.7 Hz), 7.34 (dm, 1H, J = 7.8 Hz), 7.21 (app dt, 1H, J = 1.3, 7.0 Hz), 7.17–7.13 (m, 2H), 4.18 (q, 2H, J = 7.1 Hz), 3.79 (s, 2H), 1.28 (t, 3H, J = 7.1 Hz). 13C NMR (CDCl3, 75 MHz,) δ 172.3, 136.2, 127.3, 123.2, 122.3, 119.7, 119.0, 111.3, 108.6, 60.9, 31.5, 14.4. HRMS (ESI+) m/z [M + Na]+ calcd for C12H13NO2Na 226.0844; found: 226.0847.General experimental setup for the synthesis of 4-fluorobenzene diazonium tosylate 2
The experimental setup consisted of two streams as depicted in Scheme 1. The two inlets were equipped with PEEK injection loops (0.5 mL, 0.76 mm id). The first loop was loaded with 4-fluoroaniline 2 (0.2 M), PTSA (0.22 M) and C6F6 (0.2 M) in MeOH, while the second loop was filled with a solution of t-BuONO (0.3 M) in MeOH. The two streams were merged in a PEEK T-shaped piece (internal volume: 11.4 μL) and the resulting mixture flowed in a PEEK coil reactor (5 mL, 0.76 mm id, 30 °C) at a flow rate of 0.5 mL min−1. The outlet of the reactor was connected to the flow cell of a benchtop NMR spectrometer. The NMR conversion was determined through 1D 19F experiments at 40.88 MHz in stop-flow mode. A preparative experiment, without an in-line NMR spectrometer, was conducted with 5 mL sample loops and 4-fluorobenzene diazonium tosylate 2 was isolated by precipitation in Et2O as a pale yellow solid (99 mg, 89%). mp 131 °C. IR (ATR) ν 3050, 2296, 1576, 1475, 1216, 1189, 1032, 1008, 848, 679, 564, 523 cm−1. 1H NMR (MeOD, 400 MHz) δ 8.77–8.72 (m, 2H), 7.76–7.70 (m, 2H), 7.67 (d, 2H, J = 8.2 Hz), 7.22 (d, 2H, J = 7.9 Hz), 2.36 (s, 3H). 19F NMR (MeOD, 376 MHz) δ −86.4.General experimental setup for the optimization of indole 7
The experimental setup consisted of two streams as depicted in Scheme 2. The first stream, equipped with a stainless steel injection loop (0.5 mL, 0.76 mm id) loaded with a solution of indole 6 (0.1 M), Pd(OAc)2 (see Table S1†) and naphthalene (0.1 M) in DMF/EtOAc (1/4), met in a stainless steel T-shaped piece (internal volume: 0.57 μL) the second stream consisting of a solution of arenediazonium tolylate 2 in MeOH (0.1 M) loaded in the second stainless steel injection loop (2 mL, 1 mm id). The merged streams entered a stainless steel reactor coil (5 mL, 1 mm id) at the required flow rate (see Table S1†) and the resulting indole 7 was collected in vials and analyzed by HPLC to determine the reaction yield. An analytical sample of ethyl 2-(2-(p-tolyl)-1H-indol-3-yl)acetate 7 was obtained under the experimental conditions of experiment 18 after purification by flash chromatography (10% AcOEt–petroleum ether) as a white solid (58 mg, 78%). mp 109 °C. IR (ATR) ν 3354, 2988, 1711, 1453, 1438, 1314, 1270, 1221, 1178, 1034, 840, 744, 566 cm−1. 1H NMR (CDCl3, 300 MHz) δ 8.15 (br s, 1H), 7.70–7.61 (m, 3H), 7.35 (d, 1H, J = 7.5 Hz), 7.25–7.14 (m, 4H), 4.18 (q, 2H, J = 7.2 Hz), 3.80 (s, 2H), 1.27 (t, 3H, J = 7.2 Hz). 13C NMR (CDCl3, 75 MHz,) δ 172.4, 164.4, 161.1, 135.8, 135.4, 130.2, 130.1, 129.0, 128.6, 128.6, 122.8, 120.2, 119.4, 116.2, 115.9, 111.0, 61.1, 31.2, 14.4. HRMS (ESI+) m/z [M + H]+ calcd for C18H17O2NF 298.1243; found 298.1255.2-(4-Fluorophenyl)-1H-indole-3-acetic acid 9
The experimental setup consisted of two streams as depicted in Scheme 3a. In the first stream, a solution of indole 7 (0.3 M) in THF was loaded in an injection loop (1 mL, 0.76 mm id) while a solution of 2 M aqueous KOH in MeOH (4/6) was continuously pumped in the second stream. Both streams, each flowing at 83 μl min−1, met in a stainless steel T-shaped piece (internal volume: 0.57 μL) and reacted in a stainless steel reactor coil (5 mL, 1 mm id) at a flow rate of 166 μl min−1 (30 min residence time). The resulting acid 9 was collected in vials and neutralized with 2.5 M aqueous H3PO4. The biphasic mixture was extracted three times with Et2O, washed with brine, dried over MgSO4 and concentrated under reduced pressure. The crude mixture was purified by flash chromatography (20% AcOEt – 80% cyclohexane) to give acid 9 as a white solid (79 mg, 97%). mp 179 °C. IR (ATR) ν 3424, 1698, 1500, 1432, 1311,1217, 1184, 1155, 934, 839, 757, 623, 469 cm−1.1H NMR (MeOD, 300 MHz) δ 10.74 (br s, 1H), 7.74–7.68 (m, 2H), 7.57 (dd, 1H, J = 0.8, 7.9 Hz), 7.37 (app dt, 1H, J = 0.8, 8.0 Hz), 7.25–7.20 (m, 2H), 7.16–7.11 (m, 1H), 7.07–7.02 (m, 1H), 3.78 (s, 2H). 13C NMR (MeOD, 75 MHz,) δ 176.4, 163.8 (d, 1C, 1JCF = 244 Hz), 137.6, 136.5, 131.2 (d, 2C, 3JCF = 7.5 Hz), 130.5, 130.5, 130.2, 123.0, 120.3, 119.7, 116.5 (d, 2C, 2JCF = 21 Hz), 112.0, 106.1, 31.5. HRMS (ESI−) m/z [M − H]− calcd for C16H11NO2F 268.0774; found: 268.0774.FGIN-1-27 (1)
The experimental setup consisted of two streams as depicted in Scheme 3b. In the first stream, an injection loop (1 mL, 0.76 mm id) was loaded with a solution of acid 9 (0.23 M), EDC·HCl (0.64 M) and HOPO (0.22 M) in a mixture of THF/H2O/acetone (1/2/4) while in the second stream, a 2 mL injection loop (0.76 mm id) was loaded with a solution of dihexylamine (0.25 M) and Et3N (1.04 M) in a mixture of THF/H2O (9/1). Both streams, flowing at 167 μL min−1, met in a T-shaped piece (internal volume: 0.57 μL) and reacted at 23 °C in a stainless steel coil reactor (10 mL, 1 mm id) for a total of 30 min of residence time. The resulting collected mixture was diluted with water (10 mL) and washed three times with dichloromethane (3 × 10 mL). The collected organic extracts were washed with brine (15 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by flash chromatography (8% AcOEt–cyclohexane to 15% AcOEt–cyclohexane) to give FGIN-1-27 (1) as a white solid (93 mg, 95%). mp 97 °C. IR (ATR) ν 3213, 2927, 2854, 1618, 1497, 1451, 1371, 1342, 1226, 1155, 1011, 836, 744, 563 cm−1. 1H NMR (CDCl3, 300 MHz) δ 8.27 (s, 1H), 7.65 (d, 1H, J = 7.6 Hz), 7.54–7.48 (m, 2H), 7.29–7.27 (m, 1H), 7.18–7.07 (m, 4H), 3.84 (s, 2H), 3.30 (t, 2H, J = 7.5 Hz), 3.13 (t, 2H, J = 7.9 Hz), 1.51–1.35 (m, 4H), 1.26–1.10 (m, 10H), 1.06–0.97 (m, 2H), 0.88–0.83 (m, 6H). 13C NMR (CDCl3, 75 MHz,) δ 170.7, 162.6 (d, 1C, 1JCF = 246 Hz), 136.1, 134.7, 130.2 (d, 2C, 3JCF = 7.5 Hz), 129.1, 129.0 (d, 2C, 4JCF = 3.8 Hz), 122.6, 120.1, 119.6, 116.0 (d, 2C, 2JCF = 21.8 Hz), 110.9, 107.3, 48.4, 46.4, 31.8, 31.6, 31.0, 29.2, 27.8, 26.8, 26.6, 22.7, 22.7, 14.2, 14.1. HRMS (ASAP+) m/z [M + H]+ calcd for C28H38N2OF 437.2968; found: 437.2961.Conclusion
In summary, we developed a full flow synthesis of translocator protein ligand FGIN-1-27 (1) in 4 steps and with a 64% overall yield from inexpensive and commercially available starting materials using the palladium-catalyzed direct C–H arylation of 1H-indole-3-acetic acid ethyl ester 6 with 4-fluorobenzene diazonium tosylate 2 as the key step. We also demonstrated the power of flow reactors integrating an in-line benchtop NMR spectrometer to accurately monitor in quasi real-time the reaction progress in optimization stages. For instance, the formation of 4-fluorobenzene diazonium tosylate 2 through the diazotization of the corresponding aniline 3 was unusually and advantageously followed by 19F NMR experiments which addressed the signal overlap issues observed with more traditional 1H NMR analysis. For the more complex multi-dimensional optimization of the direct C–H arylation of 1H-indole-3-acetic acid ethyl ester 6 with 4-fluorobenzene diazonium tosylate 2, the use of a feedback optimization algorithm, assisting chemists in the decision-making process, minimized the number of experiments required to locate an optimum in a short time frame. Through this contribution, we demonstrated that process analytical technologies and optimization algorithms are powerful tools to improve processing times, safety and reaction yields.Author contributions
FXF conceived the project and coordinated the efforts of the research team. NV designed the initial route for FGIN-1-27 and conducted NMR experiments. ECA and EB conducted chemical experiments in flow. EW and DCB developed the optimization algorithm. PG and JF supervised the NMR experiments. MRZ supervised the chemical experiments. FXF wrote the paper with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the University of Nantes, the “Centre National de la Recherche Scientifique” (CNRS) and the “Région des Pays de la Loire” in the framework of the SmartCat project for financial support. N. Vasudevan thanks the UBL for a grant. The assistance of Dr. Shrikant Kunjir (CEISAM, University of Nantes) for the recording of 19F NMR spectra is greatly appreciated. Mr. Romain De Luca (University of Nantes) is gratefully acknowledged for preliminary experiments. We acknowledge Julie Hémez (CEISAM, University of Nantes) for HRMS analyses.References
- E. Romeo, J. Auta, A. P. Kozikowski, D. Ma, V. Papadopoulos, G. Puia, E. Costa and A. Guidotti, J. Pharmacol. Exp. Ther., 1992, 262, 971–978 CAS
.
- E. Romeo, S. Cavallaro, A. Korneyev, A. P. Kozikowski, D. Ma, A. Polo, E. Costa and A. Guidotti, J. Pharmacol. Exp. Ther., 1993, 267, 462–471 CAS
- M. G. Lima-Maximino, J. Cueto-Escobedo, J. F. Rodríguez-Landa and C. Maximino, Pharmacol., Biochem. Behav., 2018, 171, 66–73 CrossRef CAS
- S. M. Petralia and C. A. Frye, Psychopharmacology, 2005, 178, 174–182 CrossRef CAS PubMed
- J. Lv, S. Jiang, Y. Yang, X. Zhang, R. Gao, Y. Cao and G. Song, Front. Pharmacol., 2020, 11, 602889 CrossRef CAS PubMed
- A. Singh, M. Dashnyam, B. Chim, T. M. Escobar, A. E. Dulcey, X. Hu, K. M. Wilson, P. P. Koganti, C. A. Spinner, X. Xu, A. Jadhav, N. Southall, J. Marugan, V. Selvaraj, V. Lazarevic, S. A. Muljo and M. Ferrer, Sci. Rep., 2020, 10, 3766 CrossRef CAS PubMed
- A. P. Kozikowski, D. Ma, E. Romeo, J. Auta, V. Papadopoulos, G. Puia, E. Costa and A. Guidotti, Angew. Chem., Int. Ed. Engl., 1992, 31, 1060–1062 CrossRef
- T. Opatz and D. Ferenc, Org. Lett., 2006, 8, 4473–4475 CrossRef CAS
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed
- R. Gérardy, N. Emmanuel, T. Toupy, V.-E. Kassin, N. N. Tshibalonza, M. Schmitz and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2018, 2018, 2301–2351 CrossRef
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS
- B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298–2308 CrossRef CAS
- N. Oger, E. Le Grognec and F.-X. Felpin, Org. Chem. Front., 2015, 2, 590–614 RSC
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC
- B. Gutmann and O. C. Kappe, J. Flow Chem., 2017, 7, 65–71 CrossRef CAS
- N. Oger, M. d'Halluin, E. Le Grognec and F.-X. Felpin, Org. Process Res. Dev., 2014, 18, 1786–1801 CrossRef CAS
- N. Vasudevan, E. Wimmer, E. Barré, D. Cortés-Borda, M. Rodriguez-Zubiri and F.-X. Felpin, Adv. Synth. Catal., 2021, 363, 791–799 CrossRef CAS
- N. Oger, E. Le Grognec and F.-X. Felpin, J. Org. Chem., 2014, 79, 8255–8262 CrossRef CAS
- V. Sans, L. Porwol, V. Dragone and L. Cronin, Chem. Sci., 2015, 6, 1258–1264 RSC
- B. Picard, B. Gouilleux, T. Lebleu, J. Maddaluno, I. Chataigner, M. Penhoat, F.-X. Felpin, P. Giraudeau and J. Legros, Angew. Chem., Int. Ed., 2017, 56, 7568–7572 CrossRef CAS
- B. Ahmed-Omer, E. Sliwinski, J. P. Cerroti and S. V. Ley, Org. Process Res. Dev., 2016, 20, 1603–1614 CrossRef CAS
- P. Giraudeau and F.-X. Felpin, React. Chem. Eng., 2018, 3, 399–413 RSC
- T. H. Rehm, C. Hofmann, D. Reinhard, H.-J. Kost, P. Lob, M. Besold, K. Welzel, J. Barten, A. Didenko, D. V. Sevenard, B. Lix, A. R. Hillson and S. D. Riegel, React. Chem. Eng., 2017, 2, 315–323 RSC
- B. Musio, E. Gala and S. V. Ley, ACS Sustainable Chem. Eng., 2018, 6, 1489–1495 CrossRef CAS
- I. K. Barben and H. Suschitzky, J. Chem. Soc., 1960, 2735–2739 RSC
- R. Khan, S. Boonseng, P. D. Kemmitt, R. Felix, S. J. Coles, G. J. Tizzard, G. Williams, O. Simmonds, J.-L. Harvey, J. Atack, H. Cox and J. Spencer, Adv. Synth. Catal., 2017, 359, 3261–3269 CrossRef CAS
- O. Fischer and M. R. Heinrich, Chem. – Eur. J., 2021, 27, 5417–5421 CrossRef CAS PubMed
- A. Krause, E. Sperlich and B. Schmidt, Org. Biomol. Chem., 2021, 19, 4292–4302 RSC
- M. P. Doyle and W. J. Bryker, J. Org. Chem., 1979, 44, 1572–1574 CrossRef CAS
- K. S. Nalivela, M. Tilley, M. A. McGuire and M. G. Organ, Chem. – Eur. J., 2014, 20, 6603–6607 CrossRef CAS PubMed
- J. P. McMullen, M. T. Stone, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 7076–7080 CrossRef CAS PubMed
- R. A. Bourne, R. A. Skilton, A. J. Parrott, D. J. Irvine and M. Poliakoff, Org. Process Res. Dev., 2011, 15, 932–938 CrossRef CAS
- D. N. Jumbam, R. A. Skilton, A. J. Parrott, R. A. Bourne and M. Poliakoff, J. Flow Chem., 2012, 2, 24–27 CrossRef CAS
- Z. Amara, E. S. Streng, R. A. Skilton, J. Jin, M. W. George and M. Poliakoff, Eur. J. Org. Chem., 2015, 6141–6145 CrossRef CAS
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, Org. Process Res. Dev., 2016, 20, 386–394 CrossRef CAS
- K. Poscharny, D. C. Fabry, S. Heddrich, E. Sugiono, M. A. Liauw and M. Rueping, Tetrahedron, 2018, 74, 3171–3175 CrossRef CAS
- H.-W. Hsieh, C. W. Coley, L. M. Baumgartner, K. F. Jensen and R. I. Robinson, Org. Process Res. Dev., 2018, 22, 542–550 CrossRef CAS
- E. Wimmer, D. Cortés-Borda, S. Brochard, E. Barré, C. Truchet and F.-X. Felpin, React. Chem. Eng., 2019, 4, 1608–1615 RSC
- D. Cortés-Borda, E. Wimmer, B. Gouilleux, E. Barré, N. Oger, L. Goulamaly, L. Peault, B. Charrier, C. Truchet, P. Giraudeau, M. Rodriguez-Zubiri, E. Le Grognec and F.-X. Felpin, J. Org. Chem., 2018, 83, 14286–14299 CrossRef
- E. C. Aka, E. Wimmer, E. Barré, N. Vasudevan, D. Cortés-Borda, T. Ekou, L. Ekou, M. Rodriguez-Zubiri and F.-X. Felpin, J. Org. Chem., 2019, 84, 14101–14112 CrossRef CAS PubMed
- D. Cortés-Borda, K. V. Kutonova, C. Jamet, M. E. Trusova, F. Zammattio, C. Truchet, M. Rodriguez-Zubiri and F.-X. Felpin, Org. Process Res. Dev., 2016, 20, 1979–1987 CrossRef
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed
- F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS
- T. D. White, K. D. Berglund, J. M. Groh, M. D. Johnson, R. D. Miller and M. H. Yates, Org. Process Res. Dev., 2012, 16, 939–957 CrossRef CAS
- C. S. Polster, K. P. Cole, C. L. Burcham, B. M. Campbell, A. L. Frederick, M. M. Hansen, M. Harding, M. R. Heller, M. T. Miller, J. L. Phillips, P. M. Pollock and N. Zaborenko, Org. Process Res. Dev., 2014, 18, 1295–1309 CrossRef CAS
- B. Li, G. A. Weisenburger and J. C. McWilliams, Org. Process Res. Dev., 2020, 24, 2311–2318 CrossRef CAS
- J. B. Sperry, C. J. Minteer, J. Tao, R. Johnson, R. Duzguner, M. Hawksworth, S. Oke, P. F. Richardson, R. Barnhart, D. R. Bill, R. A. Giusto and J. D. Weaver, Org. Process Res. Dev., 2018, 22, 1262–1275 CrossRef CAS
- D. Kumar, N. M. Kumar, B. Noel and K. Shah, Eur. J. Med. Chem., 2012, 55, 432–438 CrossRef CAS PubMed
- C. M. Griffiths-Jones and D. W. Knight, Tetrahedron, 2011, 67, 8515–8528 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00220a |
| This journal is © The Royal Society of Chemistry 2021 |