
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulation of up-conversion emission in NaYF4 core@shell nanoparticles doped by Er3+, Tm3+, or Yb3+ ions by excitation wavelength—three ions—plenty of possibilities†
Tomasz
Grzyb
 *a,
Piotr
Kamiński
a,
Dominika
Przybylska
a,
Artur
Tymiński
a,
F.
Sanz-Rodríguez
b and
Patricia
Haro Gonzalez
b
*a,
Piotr
Kamiński
a,
Dominika
Przybylska
a,
Artur
Tymiński
a,
F.
Sanz-Rodríguez
b and
Patricia
Haro Gonzalez
b
aDepartment of Rare Earths, Faculty of Chemistry, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: tgrzyb@amu.edu.pl
bFluorescence Imaging Group, Departamento de Física de Materiales e Instituto Nicolás Cabrera, Facultad de Ciencias, Universidad Autónoma de Madrid, Campus de Cantoblanco, Madrid 28049, Spain
First published on 13th April 2021
Abstract
Nanoparticles (NPs) based on host compound NaYF4 with core@shell structures were synthesised by the precipitation reaction in high-boiling point octadecene/oleic acid solvent. Four laser wavelengths were used (808, 975, 1208, or 1532 nm) for excitation of the obtained NPs. The resulting emission and mechanisms responsible for spectroscopic properties were studied in detail. Depending on NP compositions, i.e. type of doping ion (Er3+, Tm3+, or Yb3+) or presence of dopants in the same or different phases, adjustable up-conversion (UC) could be obtained with emission peaks covering the visible to near-infrared range (475 to 1625 nm). The presented results demonstrated multifunctionality of the prepared NPs. NaYF4:2%Tm3+@NaYF4 NPs exhibited emission at 700 and 1450 nm under 808 nm laser excitation or 800 and 1625 nm emission under 1208 nm laser radiation, as a result of ground- and excited-state absorption processes (GSA and ESA, respectively). However, NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs showed the most interesting properties, as they can convert all studied laser wavelengths due to the absorption of Tm3+ (808, 1208 nm) or Er3+ ions (808, 975, 1532 nm), revealing a photon avalanche process under 1208 nm laser excitation, as well as GSA and ESA at other excitation wavelengths. The NaYF4:2%Tm3+@NaYF4:5%Er3+ NPs revealed the resultant emission properties, as the dopant ions were separated within core and shell phases. The NaYF4:18%Yb3+,2%Tm3+@NaYF4 and NaYF4:18%Yb3+,2%Tm3+@NaYF4:5%Er3+ samples showed the brightest emission, around 800 nm, under 975 nm excitation, though other laser wavelengths allowed for observation of luminescence, as well, especially in NPs with Er3+ in the outer shell, capable of UC under 1532 nm. The presented results highlight the unique and universal properties of lanthanide ions for designing luminescent NPs for a variety of potential applications, such as confocal microscopy.
1. Introduction
Upconversion (UC) processes have been investigated intensively in recent years because of their many advantages over typical luminescence.1–8 This anti-Stokes phenomenon can be observed after excitation of materials by long-wavelength radiation and usually generates short-wavelength emissions. The growing interest around UC started after its discovery by Auzel in the 1960s and has significantly increased with the development of nanoscience.6–18 Today, UC is known to be efficient in lanthanide (Ln3+)-doped materials, thanks to their spectroscopic properties, such as well-defined energy levels, the possibility of energy transfer (ET) between them, and narrow spectral bands covering ultraviolet (UV) to near-infrared (NIR) wavelengths. Nanoparticles (NPs) showing UC luminescence (UCNPs) are intensively studied, as they are promising for biomedical applications.19 Currently, novel theranostics benefit from the UC process, allowing for cancer diagnostics and treatment with the possibility of NP excitation in the NIR range.20–23 Other research areas, such as temperature sensors, photocatalysis, solar energy conversion, forensics, anti-counterfeit applications, etc., have also been influenced by the development of UC processes.5,24–30Studies of Ln3+-doped UC materials have continued since the 1960s and expanded in the last 15 years to include nanomaterials, providing insight into mechanisms responsible for luminescence and its enhancement.8,11,31 UC in Ln3+-doped materials is possible via absorption of two or more photons occurring within one type of ion (ground and excited state absorption, GSA or ESA, respectively) and emission of single-photons with higher energies than absorbed.11 Similar excitation pathways are possible in double-doped materials, with the ET process occurring between them. In energy transfer up-conversion (ETU) and cooperative energy transfer (CET) mechanisms, sensitizing ions, typically Yb3+, transfer absorbed energy to activator ions, most often Eu3+, Tb3+, Ho3+, Er3+, and Tm3+ ions.11,32 Photon avalanche (PA) is another UC generation pathway, allowing for “looping” of the absorbed energy through Ln3+ dopant properties. In the PA process, GSA is very weak; however, above appropriate excitation density thresholds, ESA becomes strong, yielding intense UC with higher nonlinearity than other mechanisms.33
UC emission, in the majority of published results, is based on the properties of Yb3+ ions, being excellent absorbers around 975 nm. Usually, doping with Yb3+ and Er3+, Ho3+, or Tm3+ ions yields UC in the visible range.18 With UCNP biological applications, systems based on Nd3+ ions were intensively developed, as they can convert radiation around 808 nm to visible or NIR light.34 The application of Nd3+ ions as sensitizers for UC was possible in core@shell systems, where Nd3+ ions are usually in a different phase than Ln3+ emitters. Another factor yielding interest for Nd3+-doped systems is the increased transparency of biological systems to 808 nm radiation compared to 975 nm radiation.
Excitation at wavelengths longer than 1000 nm has been recently studied, mostly in Er3+-doped NPs, e.g. α-NaErF4 doped with Mn2+, in which a colour shift from green to red was observed under 1530 nm laser excitation;35 with NaGdF4:20%Yb3+,2%Er3+ showing temperature-dependent UC under 980 and 1530 nm excitation;36 NaErF4@NaYF4 NPs with intense emission under 1532 nm;37 or Er3+:NaYF4@NaLuF4 NPs showing UC, highly-dependent on the shell thickness.38 Other published studies also reported UC under different wavelengths, such as 1150 nm used for excitation of LiYF4:Er/Ho@LiYF4 NPs39 or 1157 nm applied to obtain UC from NaYF4:10%Er3+@NaYF4@NaYF4:10%Ho3+@NaYF4.40 Excitation of NPs around 1208 nm has been reported only four times: (1) NaYF4:10%Er3+@NaYF4@NaYF4:10%Ho3+@NaYF4@NaYF4:1%Tm3+@NaYF4 NPs showed 800 nm emission under 1213 nm laser excitation; (2) UC was induced in Tm3+-doped yttrium orthoaluminate nanoperovskite, with a major emission peak at around 810 nm upon 1210 nm pulsed laser excitation;41 (3) NaErF4@NaYF4:10% Tm3+@NaYF4 NPs were used in the time-gated fluorescence imaging system, showing emission under 1208 nm; and (4) most recently, we presented UC under 1208 nm excitation in YPO4:20%Yb3+,1%Er3+,1%Tm3+ NPs, showing emission at 800 and 980 nm.42
Excitation by wavelengths above 1000 nm is still a very rare subject of research. Most of the published studies report UC under excitation within the 1450–1550 nm range, where Er3+ ions can be excited via GSA and ESA mechanisms.37,43 Only five articles demonstrated UC under excitation around 1200 nm (four in nanomaterials).25,40–42,44 The possibility of UC above 1000 nm is useful in conversion of solar light, which is extensively studied. In particular, dye-sensitized solar cells, as well as devices based on perovskites, can benefit from properties of NPs up-converting light from excitation above 1000 nm.45,46 However, biomedicine can also make use of conversion processes with 1208 or 1532 nm radiation, as these wavelengths lie within optical transparency windows.47,48 The selection of appropriate emission, as well as excitation wavelength, is crucial for UCNP applications, e.g. in theranostics, where the emission and excitation must be in the biological windows (NIR-I 700–900 nm, NIR-II 1000–1700 nm).47,49 Excitation and emission within biological windows provide high signal-to-background ratios due to low tissue scattering and minimal autofluorescence.49
In this report, we present intense UC in core@shell NPs under 808, 975, 1208, or 1532 nm excitation, utilizing properties of Tm3+ as sensitizers for 808 and 1208 nm, Er3+ for 808, 975, and 1532 nm, or Yb3+ ions for excitation via 975 nm. With appropriate dopants, as well as core@shell architectures, emission ranging from 475 to 1650 nm can be obtained. The wide energy conversion possibilities of our NPs can be applied in many areas, including bioimaging, security markers, and solar cells.
2. Experimental
2.1. Chemicals
All reagents were used as follows, without further purification. Rare earth oxides Er2O3 (99.99%), Tm2O3 (99.99%), Y2O3 (99.99%), and Yb2O3 (99.99%) were purchased from Stanford Materials (United States). Acetic acid (≥99%), ammonium fluoride (99%, ACS Reagent), and n-hexane (≥99%) were purchased from Honeywell (Poland). The solvents, 1-octadecene (ODE, 90%) and oleic acid (OA, 90%), were purchased from Alfa Aesar (Germany). Sodium oleate (82%) was purchased from Sigma Aldrich (Poland) and ethanol (99.8%) from POCh (Poland). Aqueous solutions of acetic acid (40% and 80%) were prepared by dissolution of acetic acid (≥99%), from POCh (Poland), in demineralised water. Fluoride syntheses were performed under nitrogen flow from Linde (Poland), which was 99.99% pure.2.2. Synthesis of α-NaYF4 and α-NaYF4:Ln3+ NPs
A mixture of OA and 1-ODE (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was poured into a three-neck round bottom flask connected to a reflux condenser and outgassed using a Schlenk line at 100 °C for 1 h under low pressure (<10−1 mbar). Then, the mixture was cooled to room temperature, and the rare earth acetates (see the ESI† for the synthesis procedures) were added, in powder form, under nitrogen flow. The precursors in the solution were then heated to 100 °C under low pressure (<10−1 mbar), and the mixture was kept at this temperature for 1 h.
:1) was poured into a three-neck round bottom flask connected to a reflux condenser and outgassed using a Schlenk line at 100 °C for 1 h under low pressure (<10−1 mbar). Then, the mixture was cooled to room temperature, and the rare earth acetates (see the ESI† for the synthesis procedures) were added, in powder form, under nitrogen flow. The precursors in the solution were then heated to 100 °C under low pressure (<10−1 mbar), and the mixture was kept at this temperature for 1 h.
Next, sodium oleate, in powder form, was added to the solution at 100 °C under the flow of nitrogen. The mixture was outgassed for 20 min, until dissolution of sodium oleate, and, then, ammonium fluoride, in powder form, was added to the synthesis mixture under nitrogen flow. Then, nitrogen was outgassed in three cycles (the total time of this outgassing was around 1 min), and the solution was heated to 200 °C for 1 h under the flow of nitrogen.
Next, the solution was cooled to room temperature and centrifuged (at 9000 rpm for 3 min). The collected supernatant was applied to the isolation of fluoride NPs. The NPs from the supernatant were precipitated by the addition of ethanol (applied as a volume ratio, 1 ml of supernatant: 1 ml of ethanol). The colloidal solution was centrifuged (9000 rpm for 3 min), and the NPs were dissolved in n-hexane (assuming that 1 mmol of NPs was dissolved in 5 ml of n-hexane). Then, the NPs were precipitated, again, by the addition of ethanol (volume ratio: 1 ml of n-hexane to 1 ml of ethanol). The final material was obtained following centrifugation (9000 rpm for 3 min). The obtained precipitate was applied to the synthesis of β-NaYF4:Ln3+ or core@shell NPs. The following materials were obtained: α-NaYF4; α-NaYF4:2%Tm3+; α-NaYF4:5%Er3+,2%Tm3+; and α-NaYF4:18%Yb3+,2%Tm3+.
2.3. Synthesis of β-NaYF4:Ln3+ NPs
The NP synthesis is based on recrystallization of the α-core precursor at higher temperature than that at which it was obtained, and the purity of the α-core precursor before synthesis was checked using thermogravimetric analysis (TGA). The α-core precursor was dissolved in a mixture of organic solutions (1 mmol of the α-core precursor, 3.72 ml of ODE, 3.72 ml of OA) and outgassed at 100 °C for 2 h under low pressure (<10−1 mbar). Then, the mixture was heated to 300 °C (at a heating rate of 5 °C min−1) under nitrogen flow and was kept at this temperature for 55 min. Next, the synthesis mixture was cooled to room temperature. The NPs from the solution were precipitated by the addition of ethanol (applied as a 1:1 volume ratio, e.g. 1 ml of supernatant and 1 ml of ethanol). The colloidal solution was centrifuged (9000 rpm for 3 min), and the NPs were dissolved in n-hexane (assuming that 1 mmol of NPs was dissolved in 5 ml of n-hexane). Then, the NPs were precipitated again by the addition of ethanol (volume ratio: 1 ml of n-hexane to 1 ml of ethanol), and the final material was obtained after centrifugation. The following materials were prepared: β-NaYF4:2%Tm3+; β-NaYF4:5%Er3+,2%Tm3+; and β-NaYF4:18%Yb3+,2%Tm3+.
2.4. Synthesis of core@shell NPs
The synthesis of core@shell NPs was similar to the synthesis of β-NaYF4:Ln3+ NPs. The main difference of this synthesis was the assumed ratio; for each 1 mmol of β-NaYF4:Ln3+ or α-NaYF4 NPs, 2 ml of ODE and 2 ml of OA were used. Purity of the core@shell NP precursors was verified using TGA prior to synthesis. β-NaYF4:Ln3+ obtained by the procedure described in section 2.3 and α-NaYF4 NPs, synthesised according to section 2.2, were mixed in 1:7 ratio, dissolved in the mixture of organic solutions and outgassed at 100 °C for ∼2 h under low pressure (<10−1 mbar). Then, the mixture was heated to 300 °C (at a rate of 5 °C min−1) under nitrogen flow and was held at this temperature for 135 min. The mixture was, then, cooled to room temperature and centrifuged (9000 rpm for 3 min). The collected supernatant was applied to the isolation of core@shell NPs.
2.5. Preparation of ligand-free NPs
The ligand-free NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs used for bioimaging were prepared following the method reported by Bogdan et al. with slight modifications.50 5 ml of oleate-capped NPs dissolved in hexane with a concentration of 20 mg ml−1 were mixed with 5 ml of 2 M HCl solution in water under vigorous stirring at room temperature. Next, the mixture was stirred for 2 h and then ultrasonicated for 5 min. The product was collected by centrifugation and washed three times with a mixture of water and ethanol in the molar ratio of 1:1. The ligand-free NPs were dispersed in 10 mL of distilled water and stored at 4 °C.
2.6. Characterisation of materials
The materials were characterised using analytical methods. TGA was performed using a Thermogravimetric Analyser TGA 4000 (PerkinElmer). X-ray diffraction (XRD) measurements were carried out on a Bruker AXS D8 Advance diffractometer with Cu Kα radiation (λ = 0.154 nm) and a step size of 0.05° in the wide-angle range (10–80°). The XRD patterns of prepared NPs were measured for fresh powder samples, and the obtained diffraction patterns were compared with patterns reported in the Inorganic Crystal Structure Database (ICSD).For transmission electron microscopy (TEM) measurements, the powders were deposited on copper grids covered with a holey carbon film and transferred to a JEOL 1400 electron microscope or Hitachi HT7700 electron microscope operating at 80–120 kV. Fluoride solutions in n-hexane were spotted on grids, prepared directly after their synthesis. Dynamic Light Scattering (DLS) and zeta potential measurements were performed using a Malvern Zetasizer Nano ZS instrument. The measurements were with the hexane solutions of lanthanide fluorides, which were prepared directly after synthesis and purification.
Photoluminescence properties were studied at room temperature using a PIXIS:256E Digital CCD Camera equipped with an SP-2156 Imaging Spectrograph (Princeton Instruments) and QuantaMaster™ 40 spectrophotometer (Photon Technology International), equipped with a H10330C-75 photomultiplier (Hamamatsu). A continuous multiwavelength, 2 W CW, diode laser (CNI) was used as the excitation source at 808, 975, 1208, and 1532 nm, coupled to a 200 μm optical fibre and collimator (from Thorlabs). All spectra were corrected for the spectral response of the equipment. The beam size and laser powers were determined using a 10A-PPS power meter (Ophir Photonics).
2.7. Cell incubation and imaging
HeLa tumoral cells (obtained from ATCC) were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Gibco, Paisley, Scotland, UK), supplemented with foetal calf serum (FCS 10%, Gibco) and 0.5% antibiotics (penicillin G, 10000 U ml−1 and streptomycin sulphate, 10000 mg ml−1; Gibco). Cells were grown in a MIDI40 cell incubator (Thermo Scientific) with a 5% CO2 atmosphere, 95% relative humidity, and a constant temperature of 37 °C. For experiments, cells were plated onto coverslips placed into 6 well plates. After 48 h, cells were incubated at 37 °C for 6 h with NPs. After that, cells were washed 3 times with phosphate-buffered saline (PBS) and fixed in formaldehyde (1:10) in PBS for 20 min at 4 °C. Finally, cells were, again, washed 3 times with PBS and kept cold until analysis.
After incubation, cells were placed under a modified confocal microscope. The laser beam, generated by a single mode fibre coupled diode, was focused onto the sample using a 50× long working distance infrared objective with a numerical aperture of 0.55. The luminescence was collected by the same objective and, after passing through several filters and apertures, was spectrally analysed with a high sensitivity Si charge-coupled device camera (Synapse, Horiba) attached to a monochromator (iH320, Horiba). The substrate containing the cells was mounted on a XY piezo stage (PI Hera, from PI Instruments), providing a displacement range of 250 × 250 μm2 with sub-micrometric resolution.
3. Results and discussion
3.1. Structure and morphology
The synthesis for NPs reported here was adopted from a method developed previously by the Haase group.51 In this approach, rare earth acetates were used as ion precursors, yielding NPs with core@shell structures, narrow size distributions, and, most importantly, high emission intensities and UC quantum yields.XRD measurements were performed to estimate the crystal phase (cubic or hexagonal) of the prepared NPs. The reflections in XRD patterns (see Fig. S1† for all patterns and Fig. 1 for XRD of core@shell NPs) were indexed as the crystal phase of cubic α-NaYF4 (ICSD no. 77099) or hexagonal β-NaYF4 (ICSD no. 51916) crystals, depending on the synthesis conditions. Typically, NPs prepared at 200 °C (section 2.2) were of the cubic crystal structure, with the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. Cubic NPs can be converted to hexagonal crystals with the P
m space group. Cubic NPs can be converted to hexagonal crystals with the P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) space group by recrystallization at 300 °C (section 2.3). The excellent separation of selected reflections for all core@shell samples evidenced the highly ordered crystal phases and suggested that the NPs are characterised by a hexagonal structure.
space group by recrystallization at 300 °C (section 2.3). The excellent separation of selected reflections for all core@shell samples evidenced the highly ordered crystal phases and suggested that the NPs are characterised by a hexagonal structure.
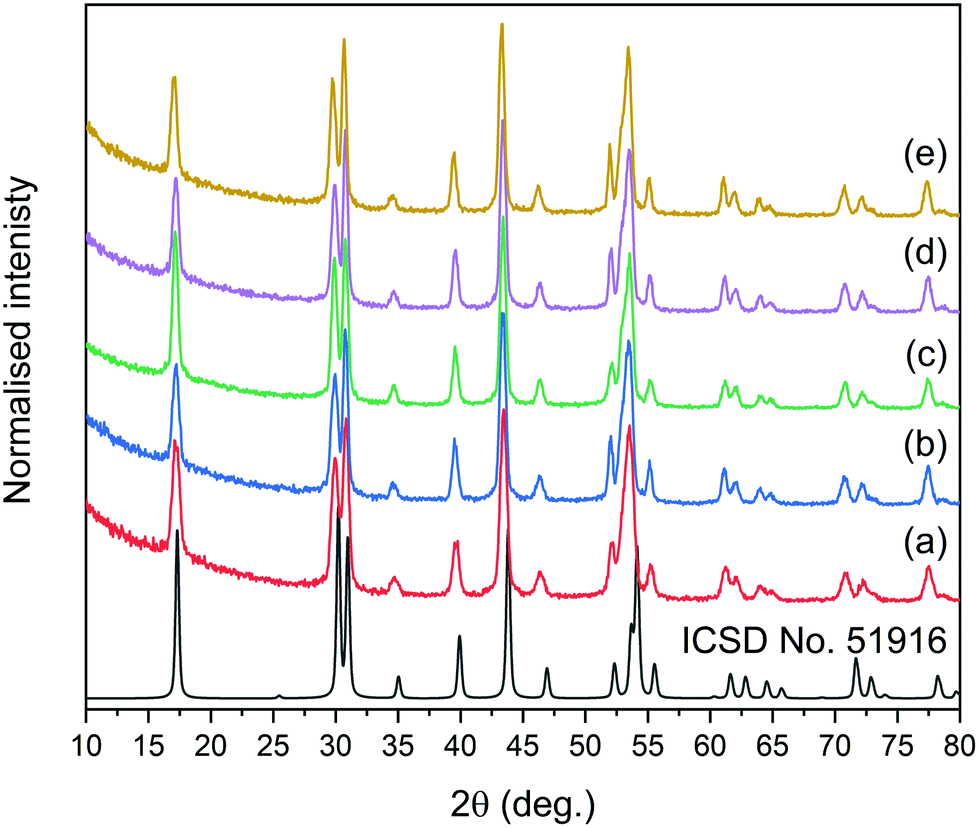 | ||
| Fig. 1 XRD patterns of synthesised core@shell NPs: (a) NaYF4:2%Tm3+@NaYF4, (b) NaYF4:2%Tm3+,5%Er3+@NaYF4, (c) NaYF4:2%Tm3+@NaYF4:5%Er3+, (d) NaYF4:18%Yb3+,2%Tm3+@NaYF4, and (e) NaYF4:18%Yb3+,2%Tm3+@NaYF4:5%Er3+. The ICSD reference represents the XRD pattern of β-NaYF4 with a hexagonal structure. | ||
Based on XRD analysis, the average crystallite sizes were calculated using the Scherrer formula and are presented in Table S1.† All materials described as β-core fluorides were characterised by average crystallite sizes between 8.4 ± 1.1 and 9.5 ± 1.4 nm. The modification of β-core fluorides by shell deposition increased the average crystallites to sizes between 12.3 and 17.8 nm. The NP size distributions, estimated using TEM images (Fig. 2 and S2†) and presented in Table S1,† provided more accurate data and indicated that the β-core NPs were smaller than core@shell NPs, with sizes between 5.4 ± 0.8 nm in the case of NaYF4:2%Tm3+,5%Er3+ and 10.0 ± 1.4 nm for NaYF4:2%Tm3+ NPs. Core@shell NP sizes were between 12.2 ± 3.8 nm for NaYF4:2%Tm3+@NaYF4:5%Er3+ and 19.4 ± 2.9 nm for NaYF4:2%Tm3+,5%Er3+@NaYF4 NPs. DLS analysis, performed for n-hexane colloids (see Table S1 and Fig. S3†), indicated higher NP sizes than expected (between 11.7 ± 2.8 and 12.4 ± 3.0 nm for core NPs or between 11.6 ± 2.8 and 26.8 ± 5.7 nm for core@shell NPs).
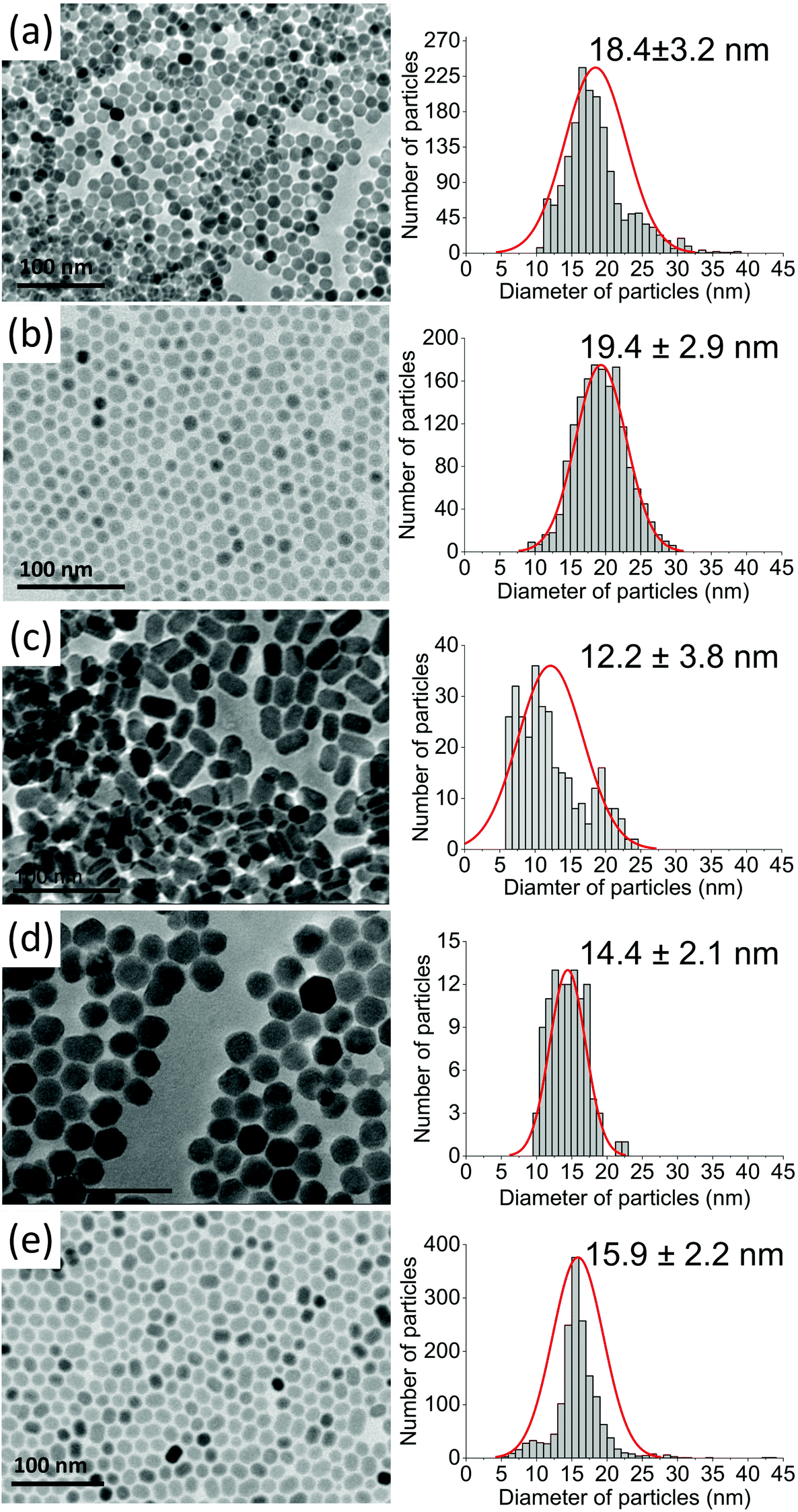 | ||
| Fig. 2 TEM images recorded for (a) NaYF4:2%Tm3+@NaYF4, (b) NaYF4:2%Tm3+,5%Er3+@NaYF4, (c) NaYF4:2%Tm3+@NaYF4:5%Er3+, (d) NaYF4:18%Yb3+,2%Tm3+@NaYF4, and (e) NaYF4:18%Yb3+,2%Tm3+@NaYF4:5%Er3+NPs. | ||
TEM images in Fig. 2 and S2† show well-separated NPs of uniform sizes, typical of syntheses in OA/ODE solvents. Cores in core@shell NPs were small and did not exceed 10 nm in diameter. With the exception of Er3+-doped NPs, cores were of rounded shape with a tendency to form hexagonal nanocrystals. NPs with NaYF4 shells were rather hexagonal-shaped (Fig. 2a, b, and d). However, the addition of Er3+ into the shell resulted in elongated core@shell NPs (Fig. 2c and e). The enlargement of NPs in the presence of Er3+ ions was a result of anisotropic growth. The lattice parameter mismatch between the core and shell led to compressive strain in the epitaxial layer and may be responsible for the anisotropic growth.52,53
3.2. Spectroscopic properties
The unique properties of Ln3+ ions can be utilized in designing materials with various luminescence characteristics, depending on their final applications.24,54–57 Due to the 4f–4f electronic transitions, the emission bands are usually narrow, and their maxima are rather independent of material. Because 4f electrons are shielded by outer-lying 5s and 5p shells, the energies of the ground and excited states of Ln3+ are well-defined and relatively independent of the surrounding environment. In UCNPs described here, we used the properties of Er3+ and Tm3+ ions, showing, in a few examples, that simply by selecting appropriate compositions and excitation wavelengths, NP emission can cover the UV to NIR spectrum (see Fig. 3). To improve emission intensity, NPs were covered by a shell. Comparison of core (Fig. S4–S7†) and core@shell (Fig. 4–7) emissions demonstrates the positive effect of shells, with emission intensities around 100× higher after shelling the NPs. | ||
| Fig. 3 Comparison of the normalised emission peaks observed in the synthesised core@shell NPs under excitation by 808, 975, 1208, or 1532 nm laser wavelengths. | ||
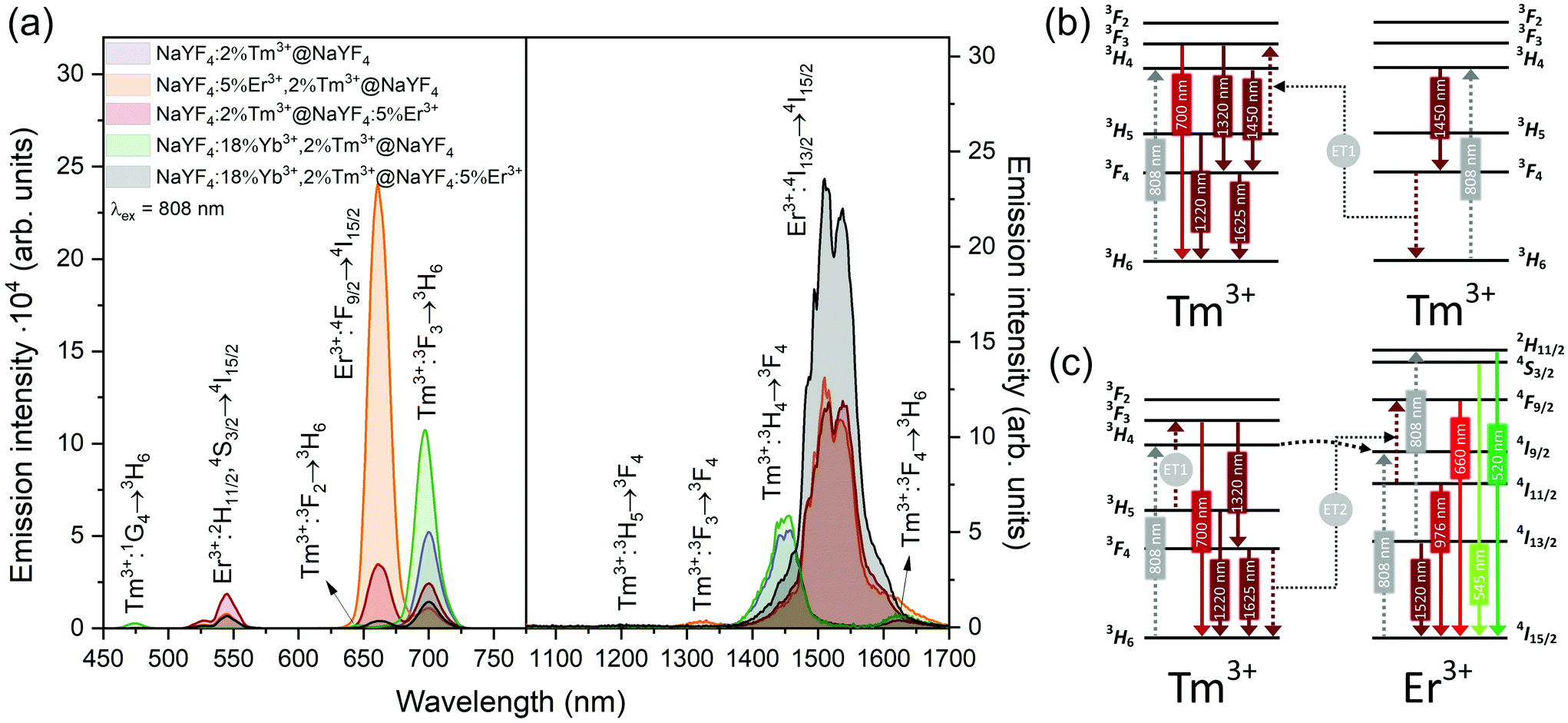 | ||
| Fig. 4 (a) Emission of core@shell NPs in the Vis and NIR range under excitation by 808 nm laser radiation (15 W cm−2). On the left, spectra were measured using a CCD camera, whereas spectra on the right used an NIR PMT (b and c) possible energy transfer schemes in NPs doped with (b) Tm3+or (c) Er3+ and Tm3+ ions under 808 nm laser excitation. | ||
 | ||
| Fig. 5 (a) Emission of core@shell NPs under excitation by 975 nm laser radiation (15 W cm−2). Left spectra were measured with a CCD camera, whereas right spectra used a NIR PMT. (b and c) Mechanisms responsible for the observed emission in UCNPs doped with (b) Er3+ and Tm3+ or (c) Yb3+ and Tm3+ lanthanide ions under 975 nm laser excitation. | ||
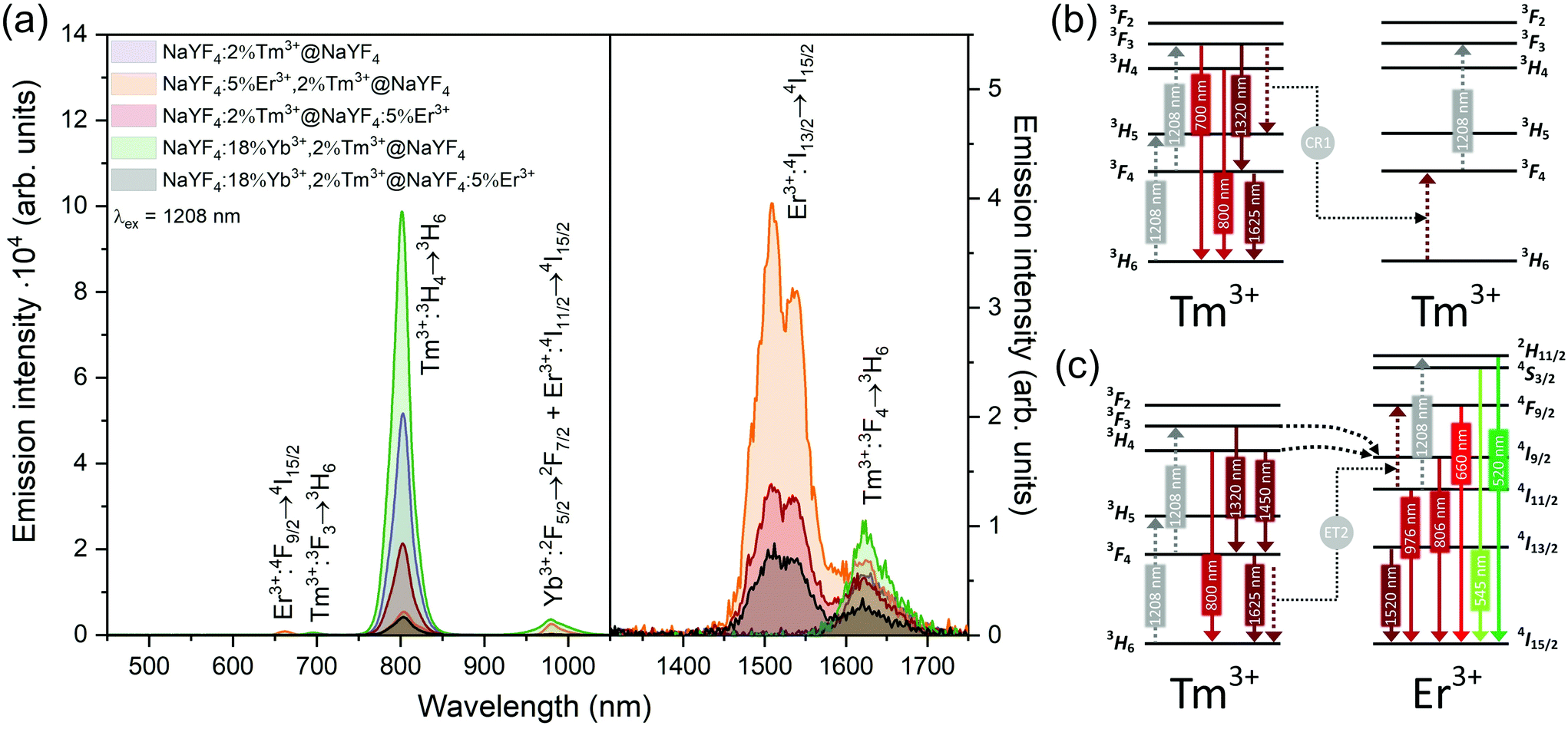 | ||
| Fig. 6 (a) Emission of core@shell NPs collected under excitation by 1208 nm laser radiation (15 W cm−2). Left spectra were measured using a CCD camera and right spectra used a NIR PMT. (b and c) Simplified UC mechanisms responsible for the observed emission of NPs doped with (b) Tm3+ or (c) Er3+ and Tm3+ ions under 1208 nm laser excitation. | ||
 | ||
| Fig. 7 (a) Emission of synthesised core@shell NPs under 1532 nm laser excitation (15 W cm−2). (b) Possible energy transfers in NPs doped with Er3+ and Tm3+ ions under 1532 nm laser excitation. | ||
Emission from the 1G4 excited state was observed in NaYF4:18%Yb3+,2%Tm3+@NaYF4 and NaYF4:18%Yb3+,2%Tm3+@NaYF4:5%Er3+ (very weak) NPs, suggesting the role of Yb3+ in the excitation process. Slope values, determined from laser power dependencies (Fig. S8 and S12†), for the 1G4 → 3H6 transition at 476 nm were higher than one (1.6 and 1.8), indicating a two-photon process responsible for this transition. Yb3+ ions may depopulate Tm3+ from their 3H4 excited state via Tm3+(3H4) → Yb3+(2F7/2) ET affecting, therefore, the ESA process in Tm3+ ions, i.e. increasing the possibility of Tm3+ ion excitation to the 1G4 excited state from the 3H5 state (see Fig. S16a†). There is also a possibility of back ET to Tm3+ ions (ET3), resulting in excitation of Tm3+ ions from the 3H4 state (populated after GSA) to the 1G4 excited state.
NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs showed the brightest UC under 808 nm from all samples. Er3+ ions can effectively absorb 808 nm, undergoing GSA and ESA processes, shown in Fig. 4c. The two-photon process (for power dependencies, see Fig. S12†) allows for excitation of Er3+ to 2H11/2 and 4S3/2 excited states and emission at 520, 545, and 660 nm. Besides typical Er3+ emission, Tm3+ emission bands were also observed under 808 nm excitation. However, in comparison with the emission of NaYF4:2%Tm3+@NaYF4 NPs, quenching of the emission bands at 700, 1450, and 1625 nm was observed. Also, when Er3+ and Tm3+ were present in the same phase, i.e. in NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs, a significant enhancement of the Er3+ emission at 660 nm was observed. These observations indicate ET between Er3+ and Tm3+ ions: ET from the 3H4 excited state of Tm3+ to Er3+, connected to emission quenching at 1450 nm (3H4 → 3F4) and ET from Tm3+ at the 3F4 excited state to Er3+ in the 4I11/2 excited state. The latter transition is responsible for the enhancement of Er3+ red emission when both ions are in a single phase (effect also visible under different excitation wavelengths, see Fig. 8).58
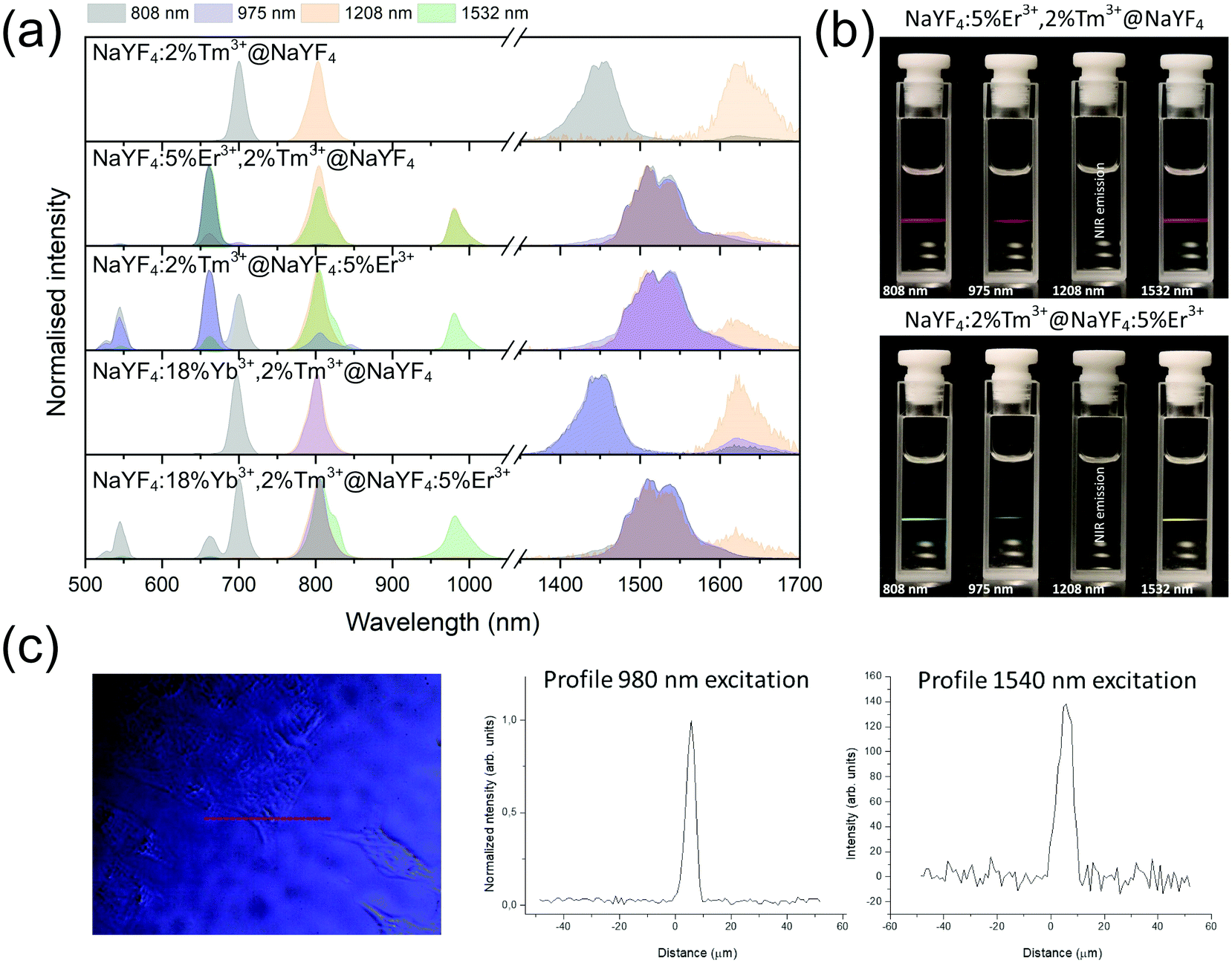 | ||
| Fig. 8 (a) Comparison of core@shell NPs emission spectra under 808, 975, 1208, or 1532 nm excitation, normalised to the most intense band. (b) Emission of NaYF4:5%Er3+, 2%Tm3+@NaYF4 and NaYF4:2%Tm3+@NaYF4:5%Er3+NPs under various excitations in the form of hexane colloids (1 mg cm−3). (c) Confocal microscopy scans under two different excitation wavelengths with cells incubated with NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs. | ||
NIR to NIR/red UC is a highly demanded phenomenon because of its applications in bioimaging and photodynamic therapy.59 In comparison with Vis light, red radiation and NIR radiation exhibit larger penetration depths in biological tissue. Excitation within the NIR-I biological window, by 808 nm, minimises the risk of tissue overheating, which is typical of 975 nm laser light.23 As we have proven here, Er3+ and Tm3+ can be alternatives for Nd3+ ions for sensitization at 808 nm, without any necessity of separation of sensitizers from emitting ions, as in the case of Nd3+-doped materials.60
Addition of Er3+ ions into the shell of NaYF4:18%Yb3+,2%Tm3+@NaYF4:5%Er3+ NPs dramatically reduced UC intensity. Er3+ effectively absorbed 975 nm excitation radiation, limiting the penetration depth in the Yb3+/Tm3+-doped cores. ET between Er3+ in the shell and Tm3+ ions in the core is rather limited. The emission spectrum of NaYF4:2%Tm3+@NaYF4:5%Er3+ NPs showed a different green to red ratio than NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs, where ET between Tm3+ and Er3+ leads to an increased 4F9/2 population of Er3+ ions and shift to red emission (ET2 in Fig. 5b).54 Also, the lack of emission at 1220 nm from Tm3+ was observed when Tm3+ and Er3+ ions are separated between the core and shell (see the right inset in Fig. 5a).
Er3+ ions can also absorb 975 nm radiation due to the 4I15/2 → 4I11/2 transition. Because of its more complex electronic structure than Yb3+ ions and the possibility of cross-relaxation (CR) between multiple levels, the use of Er3+ ions as sensitizers for 975 nm is less effective. However, they are excellent sensitizers for 1532 nm radiation. Still, under 975 nm excitation, UC can be observed from only Er3+-doped NPs as a three-photon process, due to GSA and ESA (see Fig. 5a and b, S13a and b†).
Interesting and unusual UC properties were determined for NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs under 1208 nm excitation. ET between Tm3+ and Er3+ ions was observed, where the first ions acted as sensitizers for excitation radiation, resulting in emission of Er3+ ions at 660, 976, and 1520 nm (Fig. 6a). The emission in the Vis range was relatively weak; however, the 1520 nm emission peak was the most intense from all samples. Er3+ ions cannot undergo GSA under 1208 nm; therefore, only ET between these two dopant ions can explain the observed emission. UC emission from Er3+ ions was also detected in NaYF4:2%Tm3+@NaYF4:5%Er3+. However, intensity of the typical Er3+ peaks was less than that for NPs with Tm3+ and Er3+ in the cores. Analysis of the power dependencies (Fig. S10†) revealed another property of NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs. Above a particular power density threshold, around 40 W cm−2, the slope values connected with 4F9/2 → 4I15/2 (660 nm), 4I9/2 → 4I15/2 (826 nm, mixed with the emission of Tm3+ ions), and 4I11/2 → 4I15/2 (976 nm), increase from 1.8–2.0 to 3.3–4.4 (Fig. S14†). Such behaviour suggests a PA mechanism of UC, where efficient CR induces an ESA mechanism.63 The Er3+/Tm3+ system is complex, and a full explanation of the observed high slope values requires further research. However, we propose a simplified mechanism responsible for the spectroscopic properties of NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs in Fig. 6c.
ET between Tm3+ and Er3+ is possible in both directions, as these ions possess multiple excited states, and some of them are similar in energy.64–66 Emission of Tm3+ ions at 700 and 800 nm is quenched in the presence of Er3+ ions, suggesting that Tm3+(3F3,3H4) → Er3+(4I15/2) ET. Er3+, in the 4I9/2 excited state, can undergo nonradiative relaxation to the 4I11/2 excited state and, after ESA, yields ions in the 2H11/2 or 4S3/2 excited states. Another possibility is ET from Tm3+, resulting in excitation to the 4F9/2 level and enhancing red emission of Er3+ ions, as observed for 808 and 975 nm excitation (Fig. 4 and 5).
When Tm3+ and Er3+ ions are separated between the core and shell, as in NaYF4:2%Tm3+@NaYF4:5%Er3+ NPs, the emission can shift from green (520, 545 nm) and red (660 nm) when NPs are excited at 808 nm or from 975 to 800 nm, when 1208 or 1532 nm lasers are used. The images presented in Fig. 8b show a shift in the Vis range from green, under 808 and 975 nm excitation, to yellow, when 1532 nm laser radiation was used. Additionally, besides the major peak at 1520 nm, excitation by 1208 nm resulted in emission at 1625 nm, due to the 3F4 → 3H6 transition of Tm3+ ions.
NaYF4:18%Yb3+,2%Tm3+@NaYF4 NPs were capable of converting 808 nm excitation light to 700 nm or 975 nm and 1208 nm excitation wavelengths to emission at 800 nm. The latter excitation also allowed for emission at 1625 nm. Addition of Er3+ ions into the NaYF4:18%Yb3+,2%Tm3+@NaYF4:5%Er3+ NP shell resulted in additional peaks at 520 and 545 nm, in addition to the most intense peak at 700 nm, when a 808 nm laser source was used for excitation. Excitation via 975 or 1208 nm radiation resulted in emission at 800 nm or 800 and 976 nm when a 1532 nm laser was used. All studied excitation wavelengths resulted in emission at 1520 nm or, additionally, at 1620 nm in the case of 1208 nm excitation.
The effects of double-wavelength excitation on the emission of the prepared NPs were also analysed. The emission spectra (see Fig. S17†) measured under 975 + 808 nm, 975 + 1208 nm and 975 + 1532 nm laser wavelengths revealed only slight changes in the emission characteristics in comparison with the spectra recorded under single-wavelength excitation. In general, the collected spectra were resultants of emission from NPs under two excitation wavelengths. However, there were also additional effects observed, such as enhanced emission of NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs excited via 975 + 808 nm wavelengths in comparison with 975 + 1208 nm and 975 + 1532 nm excitation. Additional irradiation of NaYF4:2%Tm3+@NaYF4:5%Er3+ NPs by 1208 nm laser wavelength caused an increase of the emission peak at 800 nm in comparison with 975 nm, single-wavelength excitation. Another interesting observation was quenching of the 475 nm emission peak, when NaYF4:18%Yb3+,2%Tm3+@NaYF4 NPs were irradiated by 1208 or 1532 nm simultaneously with 975 nm.
As a proof of concept, the chosen NPs, i.e. NaYF4:5%Er3+,2%Tm3+@NaYF4, were incubated with HeLa cells, and their application as luminescent labels was tested by confocal microscope under two selected laser wavelengths. As shown in Fig. 8c, a 1540 nm laser beam was used for NPs excitation, and the signal was still detectable. However, the microscope used for these measurements was not adapted for use with 1540 nm excitation; therefore, the applied excitation radiation only partially passed through the optics. However, even without optimised equipment, excitation above 1000 nm was used as an alternative to the most frequently studied UC under excitation around 975 nm.
4. Conclusions
NPs with core@shell structures allow not only for bright photon UC but also the possibility of designing final spectroscopic properties. Due to the unique properties of Ln3+ ions, chemists receiving NPs have a wide spectrum of possibilities that interfere with wavelengths these NPs can absorb but, also, with the wavelength of maximum emission. In this article, we showed differences between NPs doped with Er3+ and Tm3+ in the same phase (NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs), dopants separated between the core and shell phases (NaYF4:2%Tm3+@NaYF4:Er3+) and NPs without Er3+ ions (NaYF4:2%Tm3+@NaYF4). Additionally, Yb3+ ions were used as the third co-dopant in NaYF4:18%Yb3+,2%Tm3+@NaYF4 and NaYF4:18%Yb3+,2%Tm3+@NaYF4:5%Er3+ NPs.The prepared NPs showed selective emission properties, adjustable by the laser wavelength. As a result of the emission properties of Er3+ or Tm3+ ions, NPs exhibited emission from both ions or dominant emission from one of them, depending on laser excitation wavelength. In the same way, by simple manipulation of NP composition, as well as changing the excitation laser wavelength between 808, 975, 1208, or 1532 nm, we showed that emission peaks across the entire spectrum could be achieved (475, 520, 545, 660, 700, 800, 976, 1220, 1450, 1520, or 1625 nm).
The studied NP emission revealed complex mechanisms responsible for their excitations. We showed that Tm3+ ion can be used as sensitizers for 808 and 1208 nm, transferring absorbed energy to Er3+ or Yb3+ ions. Moreover, Er3+ ions allowed for sensitization of NPs for 808, 975, and 1532 nm. The presented results indicated that Er3+ and Tm3+ could be good alternatives for Nd3+ sensitizers for 808 nm excitation. NPs with Er3+ and Tm3+ present in the cores showed ET from Tm3+ to Er3+, resulting in increased red emission of Er3+ ions at 660 nm. Furthermore, thanks to specific properties of the Er3+/Tm3+ system, the NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs exhibited a PA phenomenon under 1208 nm laser radiation, responsible for the emission of Er3+ ions, showing no GSA under such excitation.
The properties of the synthesised NPs can be applied in bio-related applications, where emission and excitation in the optical transparency windows are required. Interestingly, the possibility of light conversion above 1000 nm, described herein, may be useful in converting sunlight to the NIR range in photovoltaic panels that are sensitive only to radiation with wavelengths lower than 1000 nm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this research was provided by the National Science Centre, Poland, under grant no. 2016/22/E/ST5/00016; Ministerio de Ciencia e Innovación, Spain (BIOTRAP—PID2019-105195RA-I00); and Universidad Autónoma de Madrid and Comunidad Autónoma de Madrid (SI1/PJI/2019-00052).References
- Y. Fan, L. Liu and F. Zhang, Nano Today, 2019, 25, 68–84 CrossRef CAS
.
- M. R. Hamblin, Dalton Trans., 2018, 47, 8571–8580 RSC
- C. Chen, C. Li and Z. Shi, Adv. Sci., 2016, 3, 1600029 CrossRef PubMed
- L. Tu, X. Liu, F. Wu and H. Zhang, Chem. Soc. Rev., 2015, 44, 1331–1345 RSC
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed
- A. Gnach and A. Bednarkiewicz, Nano Today, 2012, 7, 532–563 CrossRef CAS
- H. H. Gorris and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 3584–3600 CrossRef CAS PubMed
- B. Zhou, B. Tang, C. Zhang, C. Qin, Z. Gu, Y. Ma, T. Zhai and J. Yao, Nat. Commun., 2020, 11, 1–9 Search PubMed
- F. Auzel, J. Lumin., 1990, 45, 341–345 CrossRef CAS
- F. Auzel, C. R. Acad. Sci., Serie B, 1966, 263, 819–821 Search PubMed
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839–1854 RSC
- L. H. Fischer, G. S. Harms and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2011, 50, 4546–4551 CrossRef CAS PubMed
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC
- D. Hudry, I. A. Howard, R. Popescu, D. Gerthsen and B. S. Richards, Adv. Mater., 2019, 31, 1900623 CrossRef PubMed
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561–1584 RSC
- A. Nadort, J. Zhao and E. M. Goldys, Nanoscale, 2016, 8, 13099–13130 RSC
- H. Li, M. Tan, X. Wang, F. Li, Y. Zhang, L. L. Zhao, C. Yang and G. Chen, J. Am. Chem. Soc., 2020, 142, 2023–2030 CrossRef CAS PubMed
- J. Lan, L. Li, Y. Liu, L. Yan, C. Li, J. Chen and X. Chen, Microchim. Acta, 2016, 183, 3201–3208 CrossRef CAS
- D. Ni, W. Bu, S. Zhang, X. Zheng, M. Li, H. Xing, Q. Xiao, Y. Liu, Y. Hua, L. Zhou, W. Peng, K. Zhao and J. Shi, Adv. Funct. Mater., 2014, 24, 6613–6620 CrossRef CAS
- X. Zhu, W. Feng, J. Chang, Y.-W. Tan, J. Li, M. Chen, Y. Sun and F. Li, Nat. Commun., 2016, 7, 10437 CrossRef CAS PubMed
- D. Yang, P. Ma, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2015, 44, 1416–1448 RSC
-
D. Li and G. Chen, in Dye-Sensitized Solar Cells, Elsevier, 2019, pp. 325–340 Search PubMed
- M. S. Shariatdoust, A. L. Frencken, A. Khademi, A. Alizadehkhaledi, F. C. J. M. van Veggel and R. Gordon, ACS Photonics, 2018, 5, 3507–3512 CrossRef CAS
- S. Hao, Y. Shang, D. Li, H. Ågren, C. Yang and G. Chen, Nanoscale, 2017, 9, 6711–6715 RSC
- M. He, Z. Li, Y. Ge and Z. Liu, Anal. Chem., 2016, 88, 1530–1534 CrossRef CAS PubMed
- M. Wang, M. Li, M. Yang, X. M. Zhang, A. Yu, Y. Zhu, P. Qiu and C. Mao, Nano Res., 2015, 8, 1800–1810 CrossRef CAS PubMed
-
C. D. S. Brites, A. Millán and L. D. Carlos, in Handbook on the Physics and Chemistry of Rare Earths, 2016, vol. 49, pp. 339–427 Search PubMed
- M. Ding, B. Dong, Y. Lu, X. Yang, Y. Yuan, W. Bai, S. Wu, Z. Ji, C. Lu, K. Zhang and H. Zeng, Adv. Mater., 2020, 32, 1–10 Search PubMed
- F. E. Auzel, Proc. IEEE, 1973, 61, 758–786 CAS
- T. Grzyb, A. Gruszeczka, R. J. J. Wiglusz, Z. Śniadecki, B. Idzikowski and S. Lis, J. Mater. Chem., 2012, 22, 22989 RSC
- A. Bednarkiewicz, E. M. Chan, A. Kotulska, L. Marciniak and K. Prorok, Nanoscale Horiz., 2019, 4, 881–889 RSC
- Y.-F. Wang, G.-Y. Liu, L.-D. Sun, J.-W. Xiao, J.-C. Zhou and C.-H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed
- Y. Liu, J. Zhao, Y. Zhang, H. Zhang, Z. Zhang, H. Gao and Y. Mao, J. Alloys Compd., 2019, 810, 151761 CrossRef CAS
- J. Wang, H. Lin, Y. Cheng, X. Cui, Y. Gao, Z. Ji, J. Xu and Y. Wang, Sens. Actuators, B, 2019, 278, 165–171 CrossRef CAS
- T. Sun, Y. Li, W. L. Ho, Q. Zhu, X. Chen, L. Jin, H. Zhu, B. Huang, J. Lin, B. E. Little, S. T. Chu and F. Wang, Nat. Commun., 2019, 10, 1–7 CrossRef CAS PubMed
- H. Lakhotiya, A. Nazir, S. Roesgaard, E. Eriksen, J. Christiansen, M. Bondesgaard, F. C. J. M. van Veggel, B. B. Iversen, P. Balling and B. Julsgaard, ACS Appl. Mater. Interfaces, 2019, 11, 1209–1218 CrossRef CAS PubMed
- X. Cheng, H. Ge, Y. Wei, K. Zhang, W. Su, J. Zhou, L. Yin, Q. Zhan, S. Jing and L. Huang, ACS Nano, 2018, 12, 10992–10999 CrossRef CAS PubMed
- W. Shao, G. Chen, T. Y. Ohulchanskyy, A. Kuzmin, J. Damasco, H. Qiu, C. Yang, H. Ågren and P. N. Prasad, Adv. Opt. Mater., 2015, 3, 575–582 CrossRef CAS
- M. A. Hernández-Rodriguez, A. D. Lozano-Gorrín, V. Lavín, U. R. Rodríguez-Mendoza and I. R. Martín, Opt. Express, 2017, 25, 27845 CrossRef PubMed
- A. Tymiński, I. R. Martín and T. Grzyb, Part. Part. Syst. Charact., 2020, 37, 2000068 CrossRef
- D. Avram, C. Colbea and C. Tiseanu, J. Mater. Chem. C, 2019, 7, 13770 RSC
- H. Zhang, Y. Fan, P. Pei, C. Sun, L. Lu and F. Zhang, Angew. Chem., 2019, 131, 10259–10263 CrossRef
- W. Zheng, P. Huang, Z. Gong, D. Tu, J. Xu, Q. Zou, R. Li, W. You, J. C. G. Bünzli and X. Chen, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed
- D. Li, H. Ågren and G. Chen, Dalton Trans., 2018, 47, 8526–8537 RSC
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC
- M. Yamanaka, H. Niioka, T. Furukawa and N. Nishizawa, J. Biomed. Opt., 2019, 24, 1 Search PubMed
- J. Xu, A. Gulzar, P. Yang, H. Bi, D. Yang, S. Gai, F. He, J. Lin, B. Xing and D. Jin, Coord. Chem. Rev., 2019, 381, 104–134 CrossRef CAS
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835–840 CrossRef CAS PubMed
- C. Homann, L. Krukewitt, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and M. Haase, Angew. Chem., Int. Ed., 2018, 57, 8765–8769 CrossRef CAS PubMed
- N. J. J. Johnson and F. C. J. M. van Veggel, ACS Nano, 2014, 8, 10517–10527 CrossRef CAS PubMed
- S. Fischer, N. J. J. Johnson, J. Pichaandi, J. C. Goldschmidt and F. C. J. M. van Veggel, J. Appl. Phys., 2015, 118, 193105 CrossRef
- H. Rabie, Y. Zhang, N. Pasquale, M. J. Lagos, P. E. Batson and K.-B. Lee, Adv. Mater., 2019, 31, 1806991 CrossRef PubMed
- M. Wang, M. Li, A. Yu, Y. Zhu, M. Yang and C. Mao, Adv. Funct. Mater., 2017, 27, 1606243 CrossRef PubMed
- K. Binnemans and P. T. Jones, J. Rare Earths, 2014, 32, 195–200 CrossRef CAS
- C. C. Lin and R.-S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268–1277 CrossRef CAS PubMed
- H. Dong, L.-D. Sun and C.-H. Yan, Chem. Soc. Rev., 2015, 44, 1608–1634 RSC
- G. Tian, W. Ren, L. Yan, S. Jian, Z. Gu, L. Zhou, S. Jin, W. Yin, S. Li and Y. Zhao, Small, 2013, 9, 1929–1938 CrossRef CAS PubMed
- D. Przybylska and T. Grzyb, J. Alloys Compd., 2020, 831, 154797 CrossRef CAS
- G. Boulon, J. Alloys Compd., 2008, 451, 1–11 CrossRef CAS
- U. R. Rodríguez-Mendoza and F. Lahoz, J. Lumin., 2016, 179, 40–43 CrossRef
- M.-F. Joubert, S. Guy, B. Jacquier and C. Linarés, Opt. Mater., 1994, 4, 43–49 CrossRef CAS
- S. Tanabe, K. Suzuki, N. Soga and T. Hanada, J. Lumin., 1995, 65, 247–255 CrossRef CAS
- M. Gupta, M. Adnan, R. Nagarajan and G. V. Prakash, ACS Omega, 2019, 4, 2275–2282 CrossRef CAS PubMed
- J. P. Jouart, M. Bouffard, T. Duvaut and N. M. Khaidukov, Chem. Phys. Lett., 2002, 366, 62–66 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr07136f |
| This journal is © The Royal Society of Chemistry 2021 |