
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of P-chiral tertiary phosphine oxides with an ethynyl group via Cu(I)-catalyzed azide–alkyne cycloaddition†
Ren-Yi
Zhu
a,
Long
Chen
a,
Xiao-Si
Hu
a,
Feng
Zhou
 ab and
Jian
Zhou
*abc
ab and
Jian
Zhou
*abc
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, China. E-mail: jzhou@chem.ecnu.edu.cn
bShanghai Key Laboratory of Green Chemistry and Chemical Process, East China Normal University, Shanghai 200062, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Shanghai 200032, China
First published on 6th November 2019
Abstract
We report the highly enantioselective synthesis of P-chiral tertiary phosphine oxides featuring an ethynyl group via Cu(I)-catalyzed azide–alkyne cycloaddition. Newly developed chiral pyridinebisoxazolines (PYBOX) bearing a bulky C4 shielding group play an important role in achieving excellent enantioselectivity while suppressing side bis-triazoles formation in desymmetrizing prochiral diethynylphosphine oxides. Notably, by tuning the size of the C4 shielding group, it is possible to achieve excellent remote enantiofacial control in desymmetrizing phosphole oxide-diynes with the prochiral P-center farther from the ethynyl group by four covalent bonds. Time-dependent enantioselectivity is observed for these desymmetric CuAAC reactions, suggesting a synergic combination of a desymmetrization and a kinetic resolution, and our ligands prove to be better than unmodified PYBOX in both steps. This finding contributes to a highly enantioselective kinetic resolution of racemic ethynylphosphine oxides. The resulting chiral ethynylphosphine oxides are versatile P-chiral synthons, which can undergo a number of diversifying reactions to enrich structural diversity.
Introduction
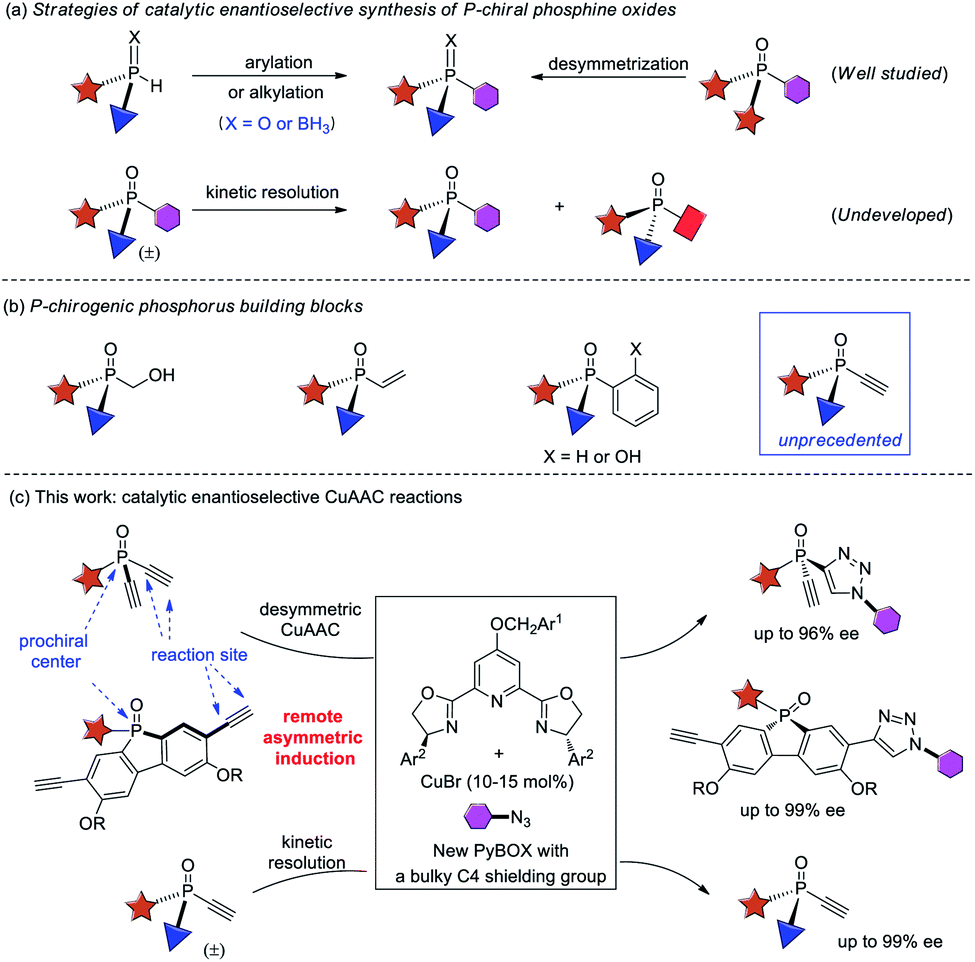
P-chiral phosphorus compounds have widespread applications in many areas such as agrochemistry, biology and pharmacy.1 In particular, they are regarded as a class of promising ligands or organocatalysts, because they can organize the chirality proximate to the catalytic center.2,3 However, their application is rather limited, as compared with the more readily available phosphines with axial, spiro, planar, or carbon-centered chirality. In view of the crucial role of phosphorus ligands4 and organocatalysts5 in asymmetric catalysis, it is important to develop efficient protocols for the facile access of P-stereogenic phosphorus molecules.Conventional syntheses of P-chiral phosphines require using a stoichiometric amount of chiral starting materials or chiral reagents.6 However, recent progress in the field of asymmetric catalysis has provided some elegant protocols1a,7 that are mainly based on two synthetic strategies, the arylation or alkylation of secondary phosphine oxides8 or secondary phosphines9 and the desymmetrization of prochiral phosphorus compounds10 (Scheme 1a). Despite much progress, catalytic enantioselective synthesis of versatile P-chiral phosphorus building blocks is still very limited.
 | ||
| Scheme 1 Access to P-stereogenic compounds. | ||
Because the electronic and steric properties of tertiary phosphines can be readily tuned over a very wide range by varying their substituents,4,5,11 a library of P-chiral phosphines with high structural diversity is very helpful for reaction development. Therefore, P-chiral building blocks featuring a versatile synthetic handle, capable of undergoing various diversifying reactions to enrich structure diversity, are much sought after. They also offer the promise of developing new chiral ligands or organocatalysts via modular combination with other functionalities. Although P-chiral synthons with a hydroxymethyl or vinyl group are known,1a those with an ethynyl group are unprecedented.12 As an acetylene group has many possibilities for elaboration,13 the resulting optically active P-chiral synthons are very useful, but their enantioselective catalytic synthesis is very difficult, due to the shortage of efficient methods to form stereocenters bearing an acetylene group.14
On the other hand, while kinetic resolution is a fundamental strategy to access chiral materials,15 catalytic kinetic resolution of racemic P-chiral molecules is undeveloped. In principle, it is a promising strategy to access two distinct P-chiral phosphine derivatives (Scheme 1a). To our knowledge, only two elegant protocols have been disclosed, the dynamic kinetic resolution of phospholene oxides by Hayashi et al.,16 and the kinetic resolution of phosphinic amides by Cramer et al.17 Herein, we report a highly enantioselective synthesis of diverse P-chiral tertiary phosphine oxides with an acetylene group via Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) by desymmetrization and kinetic resolution (Scheme 1c).
Results and discussion
Desymmetric CuAAC of prochiral diethynyl-phosphine oxides
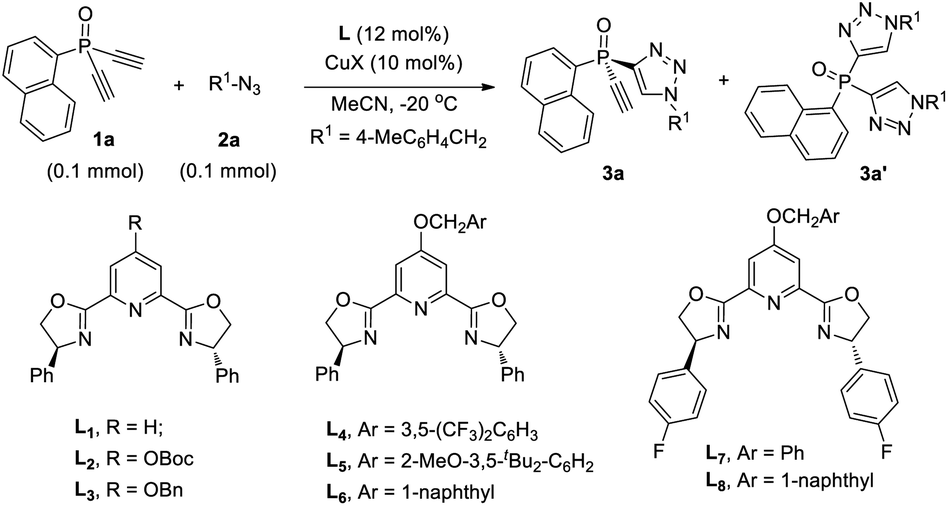
The CuAAC reaction, concurrently discovered by the groups of Meldal18 and Sharpless,19 has found application in many areas.20 However, its application to enantioselective catalysis has met with limited success.21 In 2005, Fokin and Finn pioneered this study, and demonstrated that it was possible to develop enantioselective CuAAC via desymmetrization or kinetic resolution.22 Eight years later, we developed the first highly enantioselective CuAAC, by desymmetrizing oxindole-diynes.23a Uozumi and Xu also disclosed nice desymmetric CuAAC reactions of prochiral dialkynes.23c–f In general, a challenge in developing desymmetric CuAAC is how to suppress side bis-triazoles formation while achieving excellent enantiocontrol, because most known protocols afford substantial amounts of achiral bis-triazoles, with few substrates able to achieve excellent ee values.22–24 On the other hand, the application of CuAAC for the kinetic resolution of racemic alkynes or azides is in its infancy,22,25 although Topczewski most recently made a notable advance with an elegant dynamic kinetic resolution of allylic azides.26 We speculate that asymmetric CuAAC is a promising approach to access P-chiral synthons featuring an acetylene group, by desymmetrization or kinetic resolution (Scheme 1c). The advantages of this method include: (1) easy access to di- and monoethynyl-phosphine oxides 1 and 4 (one-pot, 2–3 steps, see ESI†); and (2) the usefulness of the P-chiral phosphine oxides 3 and 4. They may act as organocatalysts5b,d as well as precursors to various P-chiral phosphine derivatives. Notably, P-chiral phosphines bearing a triazole moiety are unknown, the properties of which are interesting to explore.We began by attempting the desymmetrizing CuAAC of diyne 1a and azide 2a (Table 1). It is worth mentioning that the desymmetrization of diethynylphosphine oxides is unprecedented, although several desymmetrizing reactions of dialkynylphosphine oxides have been reported since the seminar work of Tanaka et al.27 Prochiral diynes bearing terminal alkynes are very difficult substrates for the intermolecular desymmetric reactions, because their linear shape makes it difficult to achieve good enantioselectivity and to suppress side difunctionalization.28 As expected, it was hard to achieve high enantioselectivity and 3a/3a′ ratio in this research. The reaction was first conducted in the condition we optimized for the desymmetric CuAAC of oxindole-diynes, by using catalyst PYBOX L1/CuCl and 2,5-hexanedione as the solvent.23a Unfortunately, product 3a was obtained in only 8% ee, with a poor 2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 3a/3a′ (entry 1). Further screenings revealed that if running in MeCN, the reaction could give 3a in 63% yield and 83% ee, with 3a/3a′ ratio up to 6.8:1 (entry 2).
:1 ratio of 3a/3a′ (entry 1). Further screenings revealed that if running in MeCN, the reaction could give 3a in 63% yield and 83% ee, with 3a/3a′ ratio up to 6.8:1 (entry 2).
| a Determined by 1H NMR. b NMR yield by using 1,3,5-trimethoxybenzene as the internal standard. c Determined by chiral HPLC analysis. d Reaction at 0 °C, 36 h. e Dione = 2,5-hexanedione. f Yield of the isolated product 3a. | ||||||
|---|---|---|---|---|---|---|
|
||||||
| Entry | L | Solvent | CuX | 3a/3a′a | Yield of 3ab (%) | ee of 3ac (%) |
| 1d | L1 | Dionee | CuCl | 2.0:1 |
33 | 8 |
| 2 | L1 | MeCN | CuCl | 6.8:1 |
63 | 83 |
| 3 | L2 | MeCN | CuCl | 5.1:1 |
51 | 84 |
| 4 | L3 | MeCN | CuCl | 10.4:1 |
71 | 90 |
| 5 | L4 | MeCN | CuCl | 6.4:1 |
60 | 84 |
| 6 | L5 | MeCN | CuCl | 12.0:1 |
79 | 91 |
| 7 | L6 | MeCN | CuCl | 13.9:1 |
80f | 93 |
| 8 | L7 | MeCN | CuCl | 11.4:1 |
77 | 91 |
| 9 | L8 | MeCN | CuCl | 9.3:1 |
74 | 89 |
| 10 | L6 | MeCN | CuBr | 13.9:1 |
80f | 95 |
To suppress the formation of the side bis-triazoles 3a′ while improving the enantioselectivity, we tried modifying L1 by a C4 shielding group to improve its chiral pocket, to prevent the interaction of the alkynyl group of monotriazole 3a with the copper center. It was postulated that a C4 group may cooperate with the substituent at the chiral centers of the ligand, to enhance the enantiotopic group discrimination, and to prevent the ethynyl group of chiral monotriazoles 3 from interacting with the copper for a further CuAAC, leading to better mono- and bis-triazoles (M/D) ratio. Several modified PYBOX L2–8 were accessed in three or four steps (see ESI†) and subjected to the model reaction. Gratifyingly, the presence of a suitable C4 shielding group could indeed bring about beneficial effects. While ligand L2 with a tert-butoxycarbonyl group failed to improve the 3a/3a′ ratio (entry 3), ligand L3 (ref. 29) with a flexible OBn group raised the 3a/3a′ ratio to over 10:1, giving 3a in 90% ee (entry 4). This encouraged us to vary the benzyl group to other bulkier substituents. Ligand L4 with an electron-deficient phenyl group led to a poor result (entry 6), but L5 with an electron-rich substituent increased both the enantioselectivity and the 3a/3a′ ratio (entry 6). Ligand L6 with a 1-naphthylmethoxy group further enhanced the 3a/3a′ ratio to 13.9/1, giving 3a in 80% yield and 93% ee (entry 7). The variation of the substituent at the chiral center of the ligand affected the result as well, as shown by the performance of ligands L7,8 (entry 8 vs. 4, 9 vs. 7). Further varying CuCl to CuBr increased the ee to 95%, with the 3a/3a′ ratio unchanged (entry 10).
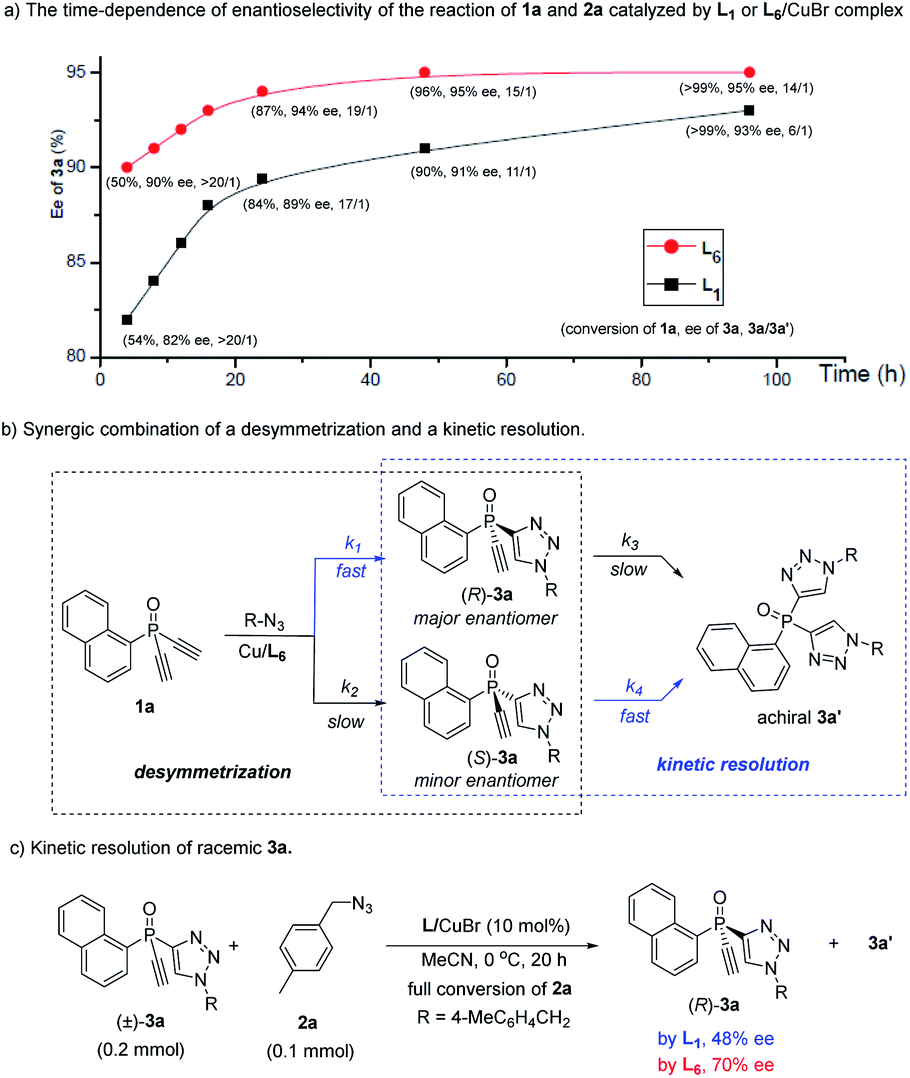
The high 3a/3a′ ratio and ee value achieved by ligand L6 were very impressive. For a better understanding of the role of L6, we evaluated the time-dependent enantioselectivity of the reaction of 1a and 2a, because Uozumi et al. previously showed that the desymmetric CuAAC of diynes bearing prochiral biaryls was a synergic combination of a desymmetrization and a kinetic resolution.30a As shown in Scheme 2a, whether using L1 or L6 as the ligand, the enantioselectivity of 3a gradually improved with increasing levels of conversion of the reaction, while the 3a/3a′ ratio decreased. This suggests that the formation of the undesired achiral 3a′ was beneficial for the ee value of 3a. In the presence of the chiral catalyst, the consumption of the minor enantiomer (S)-3a, generated in the initial desymmetric CuAAC, was faster than that of the major enantiomer (R)-3a (Scheme 2b). Therefore, the reaction of 1a and 2a was also a synergic merger of a desymmetrization and a kinetic resolution,30 where k1> k2 and k4 > k3, representing a favorable scenario to obtain (R)-3a with high ee value.
 | ||
| Scheme 2 Reaction profiles. | ||
Notably, our ligand L6 was superior to L1 in both the desymmetrization and the kinetic steps. Whereas similar conversion of 1a was found with a time of 4 h, with 3a/3a′ ratio over 20:1 in both cases, the use of L6 gave 3a with a clearly higher ee than by using L1 (90 vs. 82%), suggesting that L6 could achieve better enantiotopic group discrimination. On the other hand, when L1 was used, the 3a/3a′ ratio decreased to a greater extent as the reaction proceeded. This implied that the minor enantiomer of 3a was consumed more in the kinetic resolution, and that the high ee value obtained using L1 was at the expense of the chemical yield of product 3a. Furthermore, ligand L6 was also better than L1 in the kinetic resolution of racemic monotriazole 3a (Scheme 2c), in terms of the ee of recovered (R)-3a (70% vs. 48%). This result also implied that our new PYBOX ligands might be promising to develop kinetic resolution shown in Scheme 1.
Now that we have a better understanding of the superiority of our newly developed PYBOX ligand L6 over the parental L1 in the desymmetric CuAAC reaction of 1a and 2a, we next evaluated the scope of this desymmetric CuAAC with respect to differently substituted dialkynylphosphine oxides 1 and azides 2 under the optimized condition (Table 2). All reactions were run in MeCN at −20 °C, using 10 mol% of the chiral catalyst and a 1/2 ratio of 1.0/1.0. The substituent of dialkynylphosphine oxides obviously influenced the reaction. Dialkynylphosphine oxides 1a–d with 1-naphthyl or 2-substituted phenyl group worked well to afford the monotriazoles 3a–d in good yield and excellent ee (entries 1–4). However, dialkynylphosphine oxides 1e–f, with a 3-MeO or 3-bromophenyl group gave the corresponding products 3e–f in lower M/D ratios (entries 5, 6). Oxide 1g bearing a 4-tert-butylphenyl group afforded product 3g in 90% ee and an M/D ratio of 10:1 (entry 7). Unfortunately, tert-butyl substituted oxide 1h gave adduct 3h in diminished 75% ee and an M/D ratio of 3:1 (entry 8). A variety of aliphatic azides 2b–h all worked well to give adducts 3i–o in good yield, excellent ee and a high M/D ratio (entries 9–15). The absolute configuration of product 3b is assigned by X-ray analysis.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | 2 | 3 | 3/3′a | Yield of 3b (%) | ee of 3c (%) |
|
a Determined by 1H NMR analysis.
b Yield of the isolated products 3.
c Determined by chiral HPLC analysis.
d
1:2 = 1.05:1.
e
1:2 = 1:1.05.
|
||||||
| 1 | 1a: R = 1-naphthyl | 2a | 3a | 14:1 |
80 | 95 |
| 2 | 1b: R = 2-MeC6H4 | 2a | 3b | 18:1 |
81 | 94 |
| 3 | 1c: R = 2-BrC6H4 | 2a | 3c | 11:1 |
77 | 95 |
| 4d | 1d: R = 2-EtC6H4 | 2a | 3d | 12:1 |
72 | 92 |
| 5e | 1e: R = 3-MeOC6H4 | 2a | 3e | 7:1 |
65 | 92 |
| 6e | 1f: R = 3-BrC6H4 | 2a | 3f | 4:1 |
60 | 83 |
| 7e | 1g: R = 4-t-BuC6H4 | 2a | 3g | 10:1 |
80 | 90 |
| 8 | 1h: R = t-Bu | 2a | 3h | 3:1 |
51 | 75 |
| 9 | 1b | 2b | 3i | 20:1 |
85 | 96 |
| 10 | 1b | 2c | 3j | 20:1 |
83 | 95 |
| 11 | 1b | 2d | 3k | 16:1 |
84 | 91 |
| 12 | 1b | 2e | 3l | 14:1 |
77 | 90 |
| 13 | 1b | 2f | 3m | 23:1 |
80 | 93 |
| 14 | 1b | 2g | 3n | 13:1 |
72 | 91 |
| 15 | 1b | 2h | 3o | 16:1 |
73 | 90 |
Kinetic resolution of monoethynylphosphine oxide
Since ligand L6 could achieve better result in the kinetic resolution of racemic monotriazole 3a (Scheme 2c), we next tried the kinetic resolution of alkynylphosphine oxide 4a using azide 2a under the same condition. To our delight, the use of 0.5 equiv. of azide 2a relative to 4a allowed the reaction to work well, affording monotriazole 5a in 44% yield and 85% ee, with (S)-4a recovered with 80% ee and 44% yield.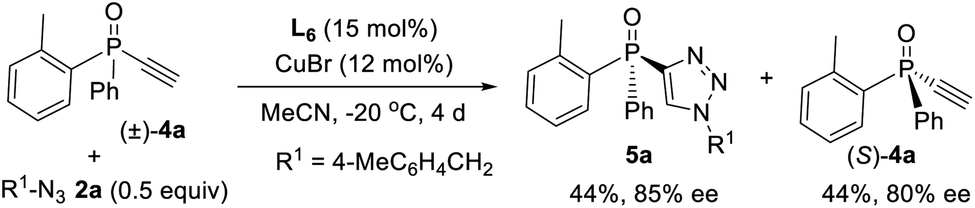
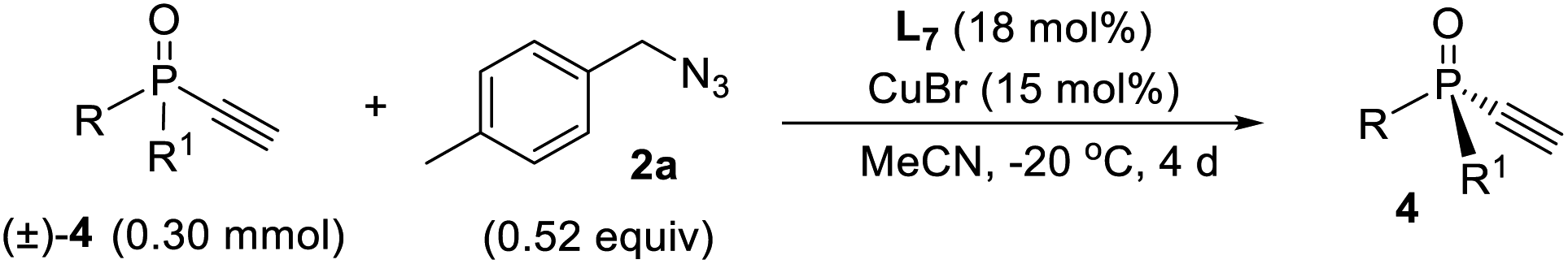
Further optimization provided a condition for the kinetic resolution of alkynylphosphine oxide 4, by using L7 as the ligand and 0.52 equiv. of azide 2a (Table 3). Accordingly, chiral o-methylphenyl-substituted phosphine oxides 4a–g featuring an acetylene group were accessed in 85–99% ee values, regardless of whether the R1 group was a substituted phenyl, 2-thienyl, cyclohexyl, or isopropyl (entries 1–7). On varying the 2-methylphenyl group to an 2-bromophenyl or a 1-naphthyl group, the corresponding alkynylphosphine oxides 4h and 4i were also resolved in 44–45% yields and 95–97% ee (entries 8, 9). The racemic ethynylphosphine oxides 3a, 3b, with a triazole group, could also be readily resolved to afford chiral 3a and 3b in good recovery and ee values (entries 10 and 11). With the acetylene group, these P-chiral phosphine oxides could undergo different diversifying reactions to enhance structural diversity. The bromophenyl group in adducts 4b, 4d, and 4h also offered the promise for further modification. The absolute configuration of product 4a was assigned by X-ray analysis.
|
|
||||
|---|---|---|---|---|
| Entry | 4 (R, R1) | Recoverya (%) | eeb (%) | s factor |
| a The recovery of 4. b Determined by chiral HPLC analysis. c s = ln[(1 − C)(1 − ee)]/ln[(1 − C)(1 + ee)]; C refers to the conversion of (±)-4, [1-(recovery of 4)]. d 0.55 equiv. of 2a was used at −10 °C for 4 d. | ||||
| 1 | 4a: (2-MeC6H4, Ph) | 42 | 96 | 21 |
| 2 | 4b: (2-MeC6H4, 3-BrC6H4) | 47 | 91 | 29 |
| 3 | 4c: (2-MeC6H4, 3-MeOC6H4) | 43 | 85 | 12 |
| 4 | 4d: (2-MeC6H4, 4-BrC6H4) | 42 | 94 | 18 |
| 5 | 4e: (2-MeC6H4, 2-thienyl) | 48 | 99 | 116 |
| 6 | 4f: (2-MeC6H4, cyclohexyl) | 44 | 94 | 23 |
| 7 | 4g: (2-MeC6H4, isopropyl) | 43 | 90 | 16 |
| 8 | 4h: (2-BrC6H4, Ph) | 44 | 95 | 25 |
| 9 | 4i: (1-Naphthyl, Ph) | 45 | 97 | 36 |
| 10d | 3a | 42 | 93 | 17 |
| 11d | 3b | 44 | 92 | 20 |
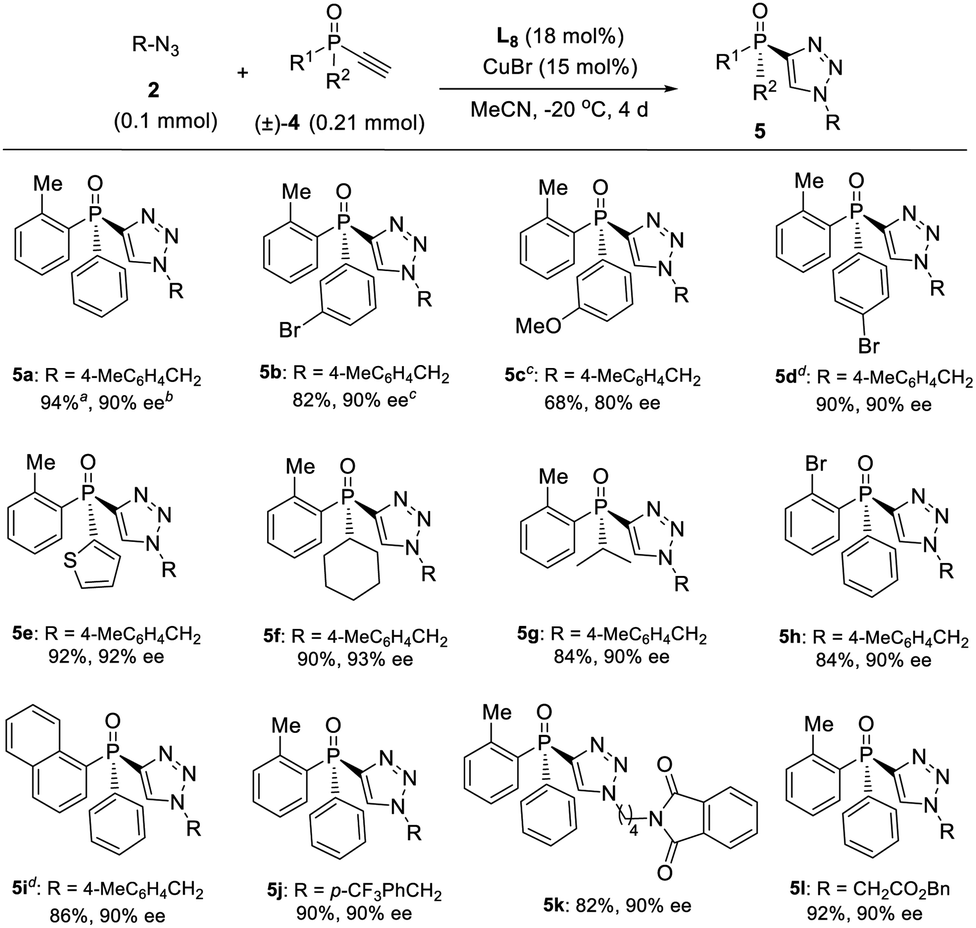
On the other hand, a highly enantioselective CuAAC of ethynylphosphine oxide 4 to P-chiral phosphine oxides 5 featuring a 1,2,3-triazole moiety is also developed by slightly optimizing the condition (Table 4). By using ligand L8 and adjusting the ratio of azides 2 to alkynylphosphine oxide 4, a range of different P-chiral P-substituents could be tolerated, including substituted phenyl, 2-thienyl, 1-napththyl, and aliphatic groups, to afford interesting P-chiral phosphine oxides bearing a 1,2,3-triazole moiety.
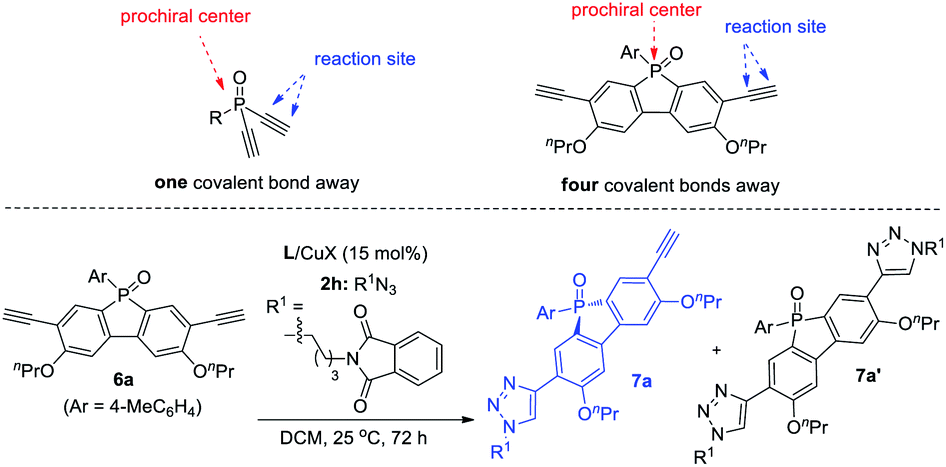
Remote desymmetrization
Achieving remote enantiofacial control is still challenging in asymmetric catalysis. If prostereogenic centers are located farther from the reaction sites, it is very difficult to develop a highly enantioselective remote desymmetrization reaction because of diminished chiral bias.28,31 Most protocols are based on substrates with the prostereogenic center being separated from the functionality by up to three covalent bonds.32 Remote intermolecular desymmetrization at a distance of four or more covalent bonds separation is rare.33 Now that our PYBOX ligands L6 showed superiority over parental L1 in suppressing side reactions and improving enantioselectivity, we tried varying the size of the C4 shielding groups of these ligands to develop desymmetric CuAAC of phosphole oxide-diynes 6, whose ethynyl group is four covalent bonds from phosphine.It is worth mentioning that phosphole oxide-based π-conjugated systems34 have drawn great attention because of their unique electronic properties, such as low-lying LUMO resulting from effective σ*–π* orbital interaction.35 While the diverse synthesis of new phosphole oxide derivatives is of current interest, chiral analogues of this π-system are unknown. In light of this, the study of asymmetric CuAAC of phosphole oxide-diyne 6 not only acts as a testing ground to evaluate the performance of our ligands L5–L8 in remote enantiofacial control, but also affords chiral phosphole oxide derivatives of potential use. The desymmetrization of diyne 6a indeed proved to be difficult. The best result obtained using ligand L1 is to use CH2Cl2 as the solvent, providing 7a in 88% ee, albeit in 45% yield due to the poor ratio of 7a/7a′ (entry 1, Table 5). Gratifyingly, our ligands L5,6 with a bulky shielding group afforded improved results. Ligand L5, with the bulkiest group, gave 7a in 62% yield, 96% ee, and 4.6:1 ratio of 7a/7a′ (entry 2), but ligand L6 gave 7a in lower 93% ee and 3.5:1 of 7a/7a′ (entry 3). This further suggested that by tuning the size of the C4 shielding group of PYBOX ligands, it is possible to develop highly enantioselective desymmetric CuAAC reactions of different prochiral systems. The subsequent optimization showed that L5/CuBr could afford 7a in 98% ee, with a 7a/7a′ ratio up to 6.6:1 (entry 4). By further changing the ratio of 6a and 2h from 1.0:1 to 1.2:1, the 7a/7a′ ratio jumped to 12:1 without the erosion of ee value (entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L | CuX | 6a/2h | 7a/7a′a | Yield of 7ab (%) | ee of 7ac (%) |
| a Determined by 1H NMR analysis. b NMR yield using anisole as the internal standard. c Determined by chiral HPLC analysis. | ||||||
| 1 | L1 | CuCl | 1.0:1 |
1.9:1 |
45 | 88 |
| 2 | L5 | CuCl | 1.0:1 |
4.6:1 |
62 | 96 |
| 3 | L6 | CuCl | 1.0:1 |
3.5:1 |
57 | 93 |
| 4 | L5 | CuBr | 1.0:1 |
6.6/1 | 66 | 98 |
| 5 | L5 | CuBr | 1.2:1 |
12.0/1 | 81 | 98 |
Next, the generality of the desymmetric CuAAC reaction of phosphole-diynes 6 was tested by performing the reaction in CH2Cl2 at 25 °C, using 18 mol% L5 and 15 mol% CuBr, as shown in Table 6. Both aryl and alkyl P-substituents could be tolerated, giving the desired products 7a–i in good yield, with high 7/7′ ratio. Diynes with different ether groups all gave adducts 7j–l with good results. Various azides also worked well to furnish monotriazoles 7m–t in good yield and high 7/7′ ratio. Notably, chiral phospholes 7 were all obtained in >90% ee. The absolute configuration of product 7° was assigned by X-ray analysis. The remote desymmetrization of diynes with ethynyl group five covalent bonds away from P-prochiral central was also attempted, but the enantioselectivity was unsatisfactory.36
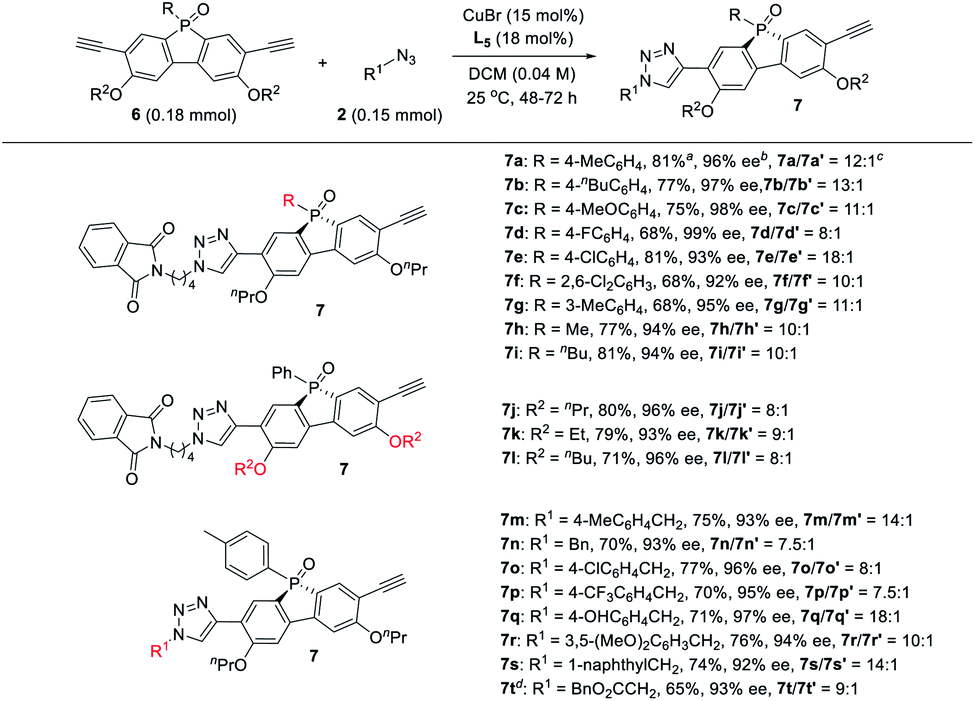
Product elaboration
The thus obtained P-chiral tertiary phosphine oxides featuring an ethynyl group are useful for the diverse synthesis of P-chiral phosphine derivatives. For example, a Gram-scale synthesis gave product 3a in 95% ee with 3a/3a′ ratio of 11/1. A Sonogashira reaction followed by a reduction readily converted 3a to tertiary phosphine 9 without erosion of ee value. Interestingly, phosphine 9 can react with alkenyl ester to give phosphine oxide 11 in 95% ee, with the triazole moiety being replaced. Similarly, tertiary phosphine 12 obtained from 5a also underwent such a replacement reaction to furnish product 13 in 89% ee.37 The structure of the major diastereomer of 14, accessed from the reduction of 13, was confirmed by X-ray analysis. Based on the absolute configuration of the phosphine of 14, it turned out that in the replacement reaction, the configuration of tertiary phosphine 9 or 12 was reversed (Scheme 3).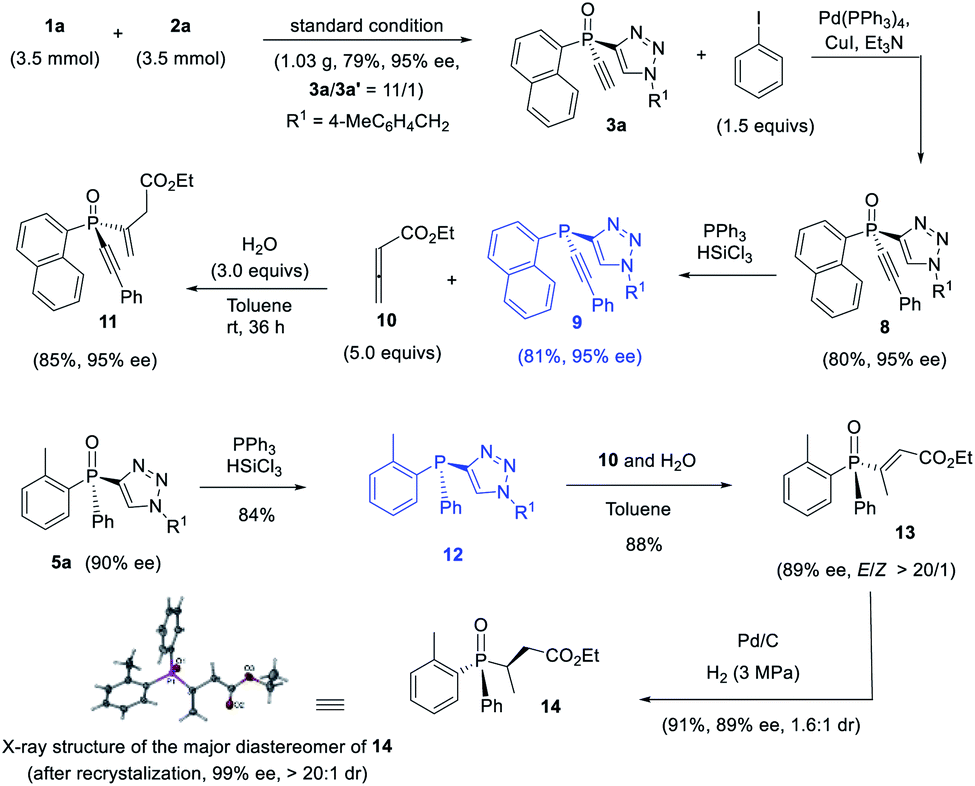 | ||
| Scheme 3 The elaboration of P-chiral phosphine oxides 3a and 5a with a triazole substituent. | ||
The P-chiral monoethynylphosphine oxides 4 can be used to develop a new P-chiral organocatalysts. For instance, via a two-step transformation, P-chiral monophosphine 16 can be readily accessed from 4 with undiminished ee value. Initially, the capacity of these phosphines was evaluated in the [3 + 2] cycloaddition of chalcone and 10 that Fu developed by using an axially chiral monophosphine catalyst,38 with up to 84:16 er for product 17a being obtained. This suggests the potential of P-chiral monoethynylphosphine oxides 4 for developing P-chiral ligand or organocatalysts. Notably, with an methyl or bromo group on the ortho position of phenyl ring, the resulting chiral monophosphines could be readily modified to increase the structural diversity (Scheme 4).3d
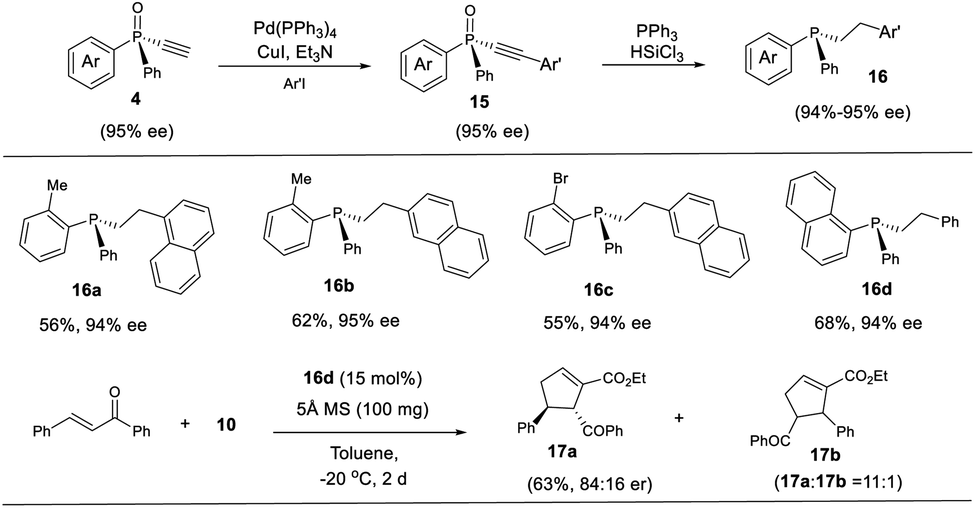 | ||
| Scheme 4 The synthesis of P-chiral monophosphine 16 and their application. | ||
The versatility of monoethynylphosphine oxides 4 as a P-chiral phosphorus building block is further demonstrated by a diastereodivergent39 Mannich reaction with chiral imines 18 derived from enantiopure tert-butylsulfinamide. Because both (R)- and (S)-4a can be readily obtained in excellent ee values via the above established kinetic resolution, it is convenient to take the alkyl–imine addition reaction40 to modularly access four isomers of compounds 19 by using either (R)- or (S)-18. The resulting multifunctional P-chiral phosphine oxide 19 contains three different chiral centers, one carbon and two heteroatom chiralities, which is an attractive framework to develop new chiral ligands and organocatalysts (Scheme 5).41
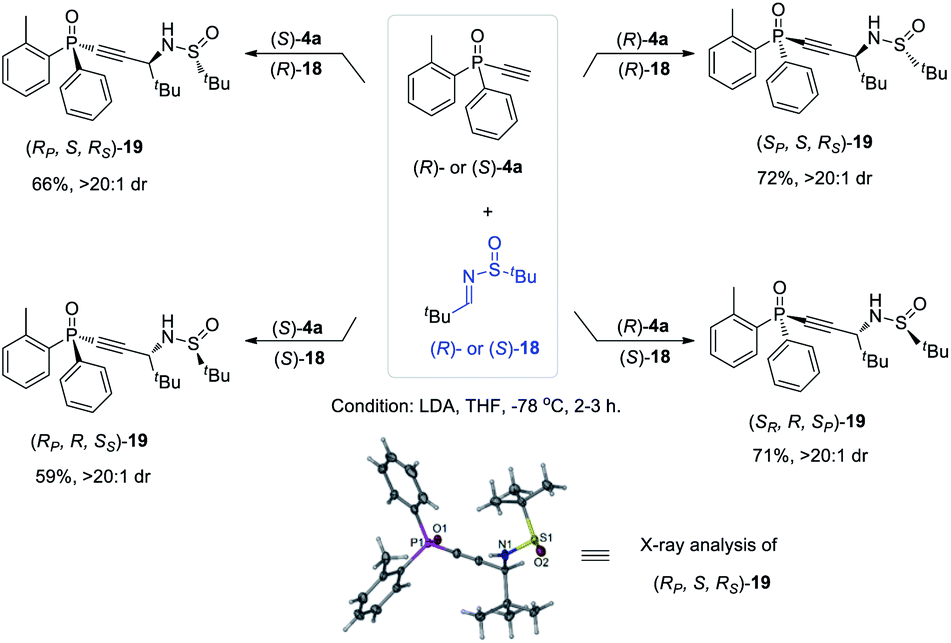 | ||
| Scheme 5 Diastereodivergent synthesis of P-chiral tertiary phosphine oxides sulfinamide 19. | ||
Furthermore, P-chiral monoethynylphosphine oxides 4 can undergo sequential Glaser coupling and reduction to give 4-bis((R)-dialkynylphosphine)butane 20, which could form a digold Au(I) complex, the structure of which was confirmed by X-ray analysis (Scheme 6).
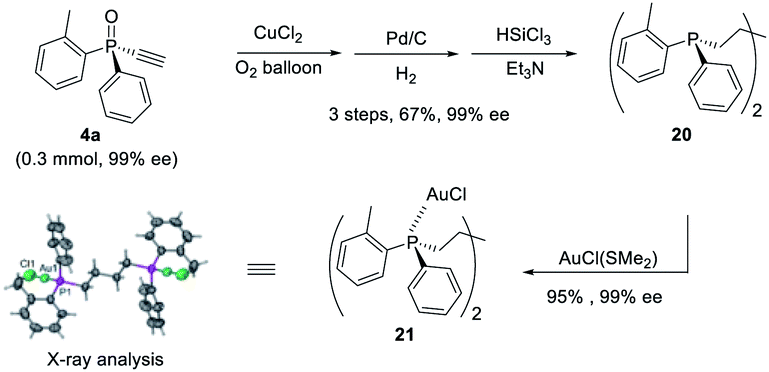 | ||
| Scheme 6 The synthesis of digold Au(I) complex 21. | ||
The optically active phosphole oxides 7 are also intriguing targets for optoelectronic studies. The extension of the π-plane of phosphole oxides is known to be beneficial for π–π stacking and electron-spin delocalization, and may tune the electron affinity of the π-systems. The presence of a triazole moiety should result in an extended π-plane to bring about some beneficial effects. In addition, compounds 7 may be further elaborated by manipulating the acetylene group. For example, an unprecedented phosphole oxide-based chiral platinum(II) acetylide 22 was readily accessed from enantiopure 7a, which might be interesting for the studies in the areas of organometallic gels, solar cells and luminescent materials, in view of the importance of Pt–acetylide as functional units.42 Starting from 7a, a Sonogashira coupling gave an extended π-system 23 in 57% yield, without the erosion of ee value. Chiral P-sulfide 24 was obtained from 23 upon treating with Lawesson's reagent. We initially checked the optical properties of compounds 6a, 7a, 23 and 24, with absorption and emission data shown below. As compared with 6a, the UV/vis absorption and emission band maxima of compound 7a are slightly red-shifted, but that of product 23 with extended π-system is obviously red-shifted. In addition, the quantum yield (QY) of 7a was significantly higher than that of 6a (0.40 vs. 0.14) and the chiral P-sulfide 24 showed lower QY than that of 23. These results showed that the properties of chiral phosphole 7 could be readily tuned for optoelectronic applications (Scheme 7).
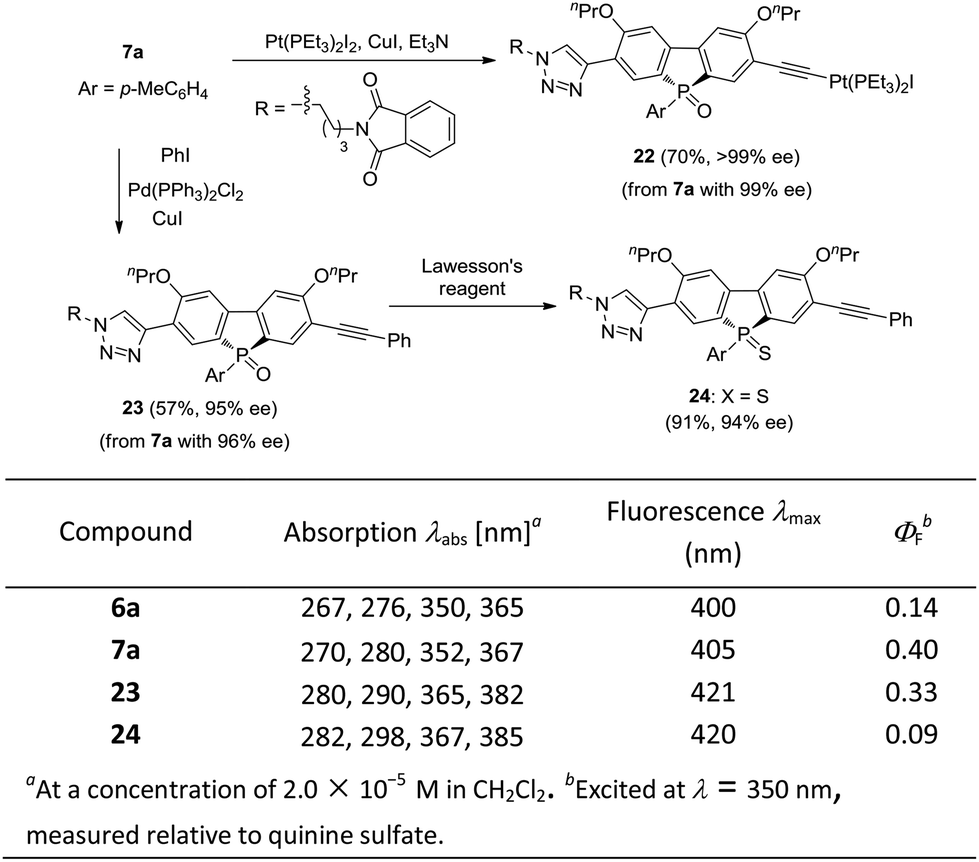 | ||
| Scheme 7 The Synthesis of 22–24 and their photophysical properties. | ||
We also examined the CD spectra of 7a, and found (R)-7a showed an obvious positive first (λ = 270 nm) and negative second (λ = 235 nm) Cotton effect peak (Fig. 1). (S)-7a showed mirror image with (R)-3a in the 230–300 nm region. In the region of 300–400 nm, (R)/(S)-7a also showed symmetry CD spectrum, however, no Cotton effect peak were observed. The highest optical anisotropy factor was observed at 235 nm (gabs = 3 × 10−4) for both (R)-7a and (S)-7a, this value was in the region of most chiral organic molecule (from 10−5 to 10−2). Based on these data, we tried to measure the circularly polarized luminescence (CPL) of (R)/(S)-7a with CPL-200. Unfortunately, due to the low chiral optical activity of these compound and measurement limit (glum ∼ 10−4), we failed to collect high quality CPL spectrum.
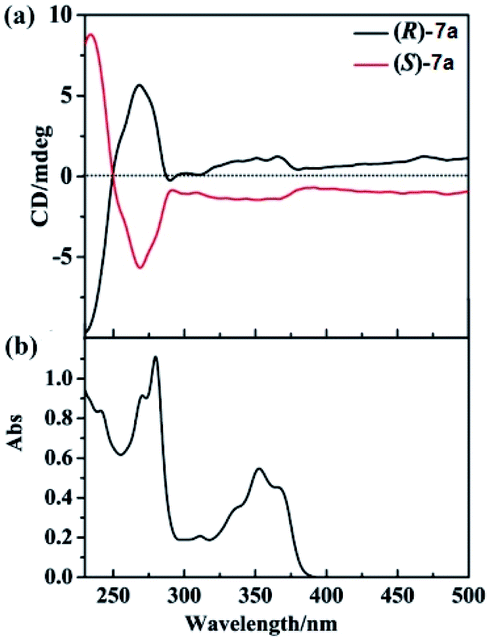 | ||
| Fig. 1 (a) CD spectra of (R)-7a (black line) and (S)-7a (red line) at 2 × 10−5 M (10 mm path length) in CH2Cl2 at 25 °C. (b) UV-vis spectra of (R)-7a in CH2Cl2 at 25 °C. | ||
Conclusions
In conclusion, we have developed highly enantioselective CuAAC reactions for the synthesis of P-chiral phosphorus synthons featuring a versatile ethynyl group, which can undergo various diversifying reactions to access structurally diverse P-chiral phosphine derivatives. Importantly, newly developed PYBOX-type ligands featuring a C4 bulky shielding group offer a flexible solution for the development of enantioselective CuAAC: by varying the C4 shielding group,43 the desymmetrization of diethynylphosphine oxides 1, the kinetic resolution of monoethynylphosphine oxides 4, and the remote desymmetrization of phosphole-diynes 7 is developed, affording the corresponding P-chiral phosphine derivatives in excellent enantioselectivity. The exploitation of new PYBOX ligands bearing various types of C4 shielding groups to develop asymmetric CuAAC reactions for the synthesis of optically active alkynes or azides, as well as the application of the resulting P-chiral synthons for the development of new ligands and catalysts is ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely thank Prof. Zhiyong Tang and Dr Lin Shi of National Center for Nanoscience and Technology for the kind help in evaluating the optical properties of phosphole oxide derivatives. The financial support from NSFC (21672068, 21725203), 973 program (2015CB856600), the Ministry of Education (PCSIRT) and the Fundamental Research Funds for the Central Universities is highly appreciated.Notes and references
- (a) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771 RSC; (b) Phosphorus Chemistry I Asymmetric synthesis and Bioactive Compounds, ed. J.-L. Montchamp, Topics in Current Chemistry, Springer, 2015, vol. 360 Search PubMed.
- As ligands: (a) L. Horner, H. Siegel and H. Büthe, Angew. Chem., Int. Ed., 1968, 7, 942 CrossRef CAS; (b) W. S. Knowles and M. J. Sabacky, Chem. Commun., 1968, 22, 1445 RSC; (c) B. D. Vineyard, W. S. Knowles, M. J. Sabacky, G. L. Bachman and D. J. Weinkauff, J. Am. Chem. Soc., 1977, 99, 5946 CrossRef CAS; (d) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183 CrossRef CAS; (e) W. Tang and X. Zhang, Angew. Chem., Int. Ed., 2002, 41, 1612 CrossRef CAS; (f) W. Tang, W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 943 CrossRef CAS; (g) A. M. Taylor, R. A. Altman and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 9900 CrossRef CAS; (h) T. Imamoto, K. Tamura, Z. Zhang, Y. Horiuchi, M. Sugiya, K. Yoshida, A. Yanagisawa and I. D. Gridnev, J. Am. Chem. Soc., 2012, 134, 1754 CrossRef CAS PubMed; (i) G. Liu, X. Liu, Z. Cai, G. Jiao, G. Xu and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 4235 CrossRef CAS; (j) J. Jia, Z. Ling, Z. F. Zhang, K. Tamur, I. D. Gridnev, T. Imamoto and W. B. Zhang, Adv. Synth. Catal., 2018, 360, 738 CrossRef CAS; (k) B. Li, J. Chen, Z. Zhang, I. D. Gridnev and W. Zhang, Angew. Chem., Int. Ed., 2019, 58, 7329 CrossRef CAS PubMed.
- For a recent review on the use of P-chirogenic compounds in asymmetric organocatalysis, see: (a) E. Rémond and S. Jugé, Chem. Today, 2014, 32, 49 Search PubMed See for examples: (b) S. A. Shaw, P. Aleman and E. Vedejs, J. Am. Chem. Soc., 2003, 125, 13368 CrossRef CAS; (c) M. Sampath and T.-P. Loh, Chem. Sci., 2010, 1, 739 RSC; (d) E. Rémond, J. Bayardon, S. Takizawa, Y. Rousselin, H. Sasai and S. Jugé, Org. Lett., 2013, 15, 1870 CrossRef; (e) S. Takizawa, E. Rémond, F. A. Arteaga, Y. Yoshida, V. Sridharan, J. Bayardon, S. Jugé and H. Sasai, Chem. Commun., 2013, 49, 8392 RSC; (f) C. E. Henry, Q. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, J. Am. Chem. Soc., 2014, 136, 11890 CrossRef CAS.
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) J.-H. Xie and Q.-L. Zhou, Acc. Chem. Res., 2008, 41, 581 CrossRef CAS PubMed; (c) A. Börner, Phosphorus Ligands in Asymmetric Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed; (d) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef.
- (a) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC. Chiral tertiary phosphine oxides are also useful Lewis base catalysts: (b) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS; (c) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS; (d) M. Benaglia and S. Rossi, Org. Biomol. Chem., 2010, 8, 3824 RSC; (e) W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657 RSC; (f) Y. Xiao, H. Guo and O. Kwon, Aldrichimica Acta, 2016, 49, 3 CAS.
- (a) E. Bergin, C. T. ƠConnor, S. B. Robinson, E. M. McGarrigle, C. P. ƠMahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566 CrossRef CAS; (b) T. León, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2011, 133, 5740 CrossRef; (c) D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728 CrossRef CAS; (d) O. Berger and J.-L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377 CrossRef CAS; (e) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J.-N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474 CrossRef CAS; (f) K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906 CrossRef CAS; (g) S. Rast, B. Mohar and M. Stephan, Org. Lett., 2014, 16, 2688 CrossRef CAS; (h) Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734 CrossRef CAS.
- For reviews, see ref. 1a and (a) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1 CrossRef CAS; (b) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477 RSC.
- For examples: (a) X. Fu, W.-T. Loh, Y. Zhang, T. Chen, T. Ma, H. Liu, J. Wang and C.-H. Tan, Angew. Chem., Int. Ed., 2009, 48, 7387 CrossRef CAS; (b) H. Zhang, Y.-M. Sun, L. Yao, S.-Y. Ji, C.-Q. Zhao and L.-B. Han, Chem.–Asian J., 2014, 9, 1329 CrossRef CAS; (c) P. Z. Xie, L. Guo, L. L. Xu and T.-P. Loh, Chem.–Asian J., 2016, 11, 1353 CrossRef CAS; (d) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183 CrossRef CAS; (e) J.-P. Wang, S.-Z. Nie, Z.-Y. Zhou, J.-J. Ye, J.-H. Wen and C.-Q. Zhao, J. Org. Chem., 2016, 81, 7644 CrossRef CAS; (f) J.-Y. Du, Y.-H. Ma, R.-Q. Yuan, N. Xin, S.-Z. Nie, C.-L. Ma, C.-Z. Li and C.-Q. Zhao, Org. Lett., 2018, 20, 477 CrossRef CAS PubMed.
- For examples: (a) V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786 CrossRef CAS PubMed; (b) C. Scriban and D. S. Glueck, J. Am. Chem. Soc., 2006, 128, 2788 CrossRef CAS; (c) C. Scriban, D. S. Glueck, J. A. Golen and A. L. Rheingold, Organometallics, 2007, 26, 1788 CrossRef CAS; (d) V. S. Chan, M. Chiu, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021 CrossRef CAS; (e) Y. Huang, Y. Li, P.-H. Leung and T. Hayashi, J. Am. Chem. Soc., 2014, 136, 4865 CrossRef CAS PubMed.
- (a) J. S. Harvey, S. J. Malcolmson, K. S. Dunne, S. J. Meek, A. L. Thompson, R. R. Schrock, A. H. Hoveyda and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 762 CrossRef CAS PubMed; (b) Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS; (c) Z.-Q. Lin, W.-Z. Wang, S.-B. Yan and W.-L. Duan, Angew. Chem., Int. Ed., 2015, 54, 6265 CrossRef CAS; (d) L. Liu, A.-A. Zhang, Y. Wang, F. Zhang, Z. Zuo, W.-X. Zhao, C.-L. Feng and W. Ma, Org. Lett., 2015, 17, 2046 CrossRef CAS; (e) Z. Huang, X. Huang, B. Li, C. Mou, S. Yang, B.-A. Song and Y. R. Chi, J. Am. Chem. Soc., 2016, 138, 7524 CrossRef CAS PubMed; (f) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364 CrossRef CAS; (g) Y.-S. Jang, M. Dieckmann and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 15088 CrossRef CAS; (h) Z. Wang and H. Tamio, Angew. Chem., Int. Ed., 2018, 57, 1702 CrossRef CAS PubMed; (i) Y.-S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS; (j) Y. Zheng, L. Guo and W. Zi, Org. Lett., 2018, 20, 7039 CrossRef CAS; (k) G.-H. Yang, Y. Li, X. Li and J.-P. Cheng, Chem. Sci., 2019, 10, 4322 RSC.
- J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS.
- Ethynylphosphine oxides have been used as building blocks, but no enantioselective synthesis is reported. (a) E. P. Kyba, S. P. Rines, P. W. Owens and S.-S. P. Chou, Tetrahedron Lett., 1981, 22, 1875 CrossRef CAS; (b) B. Stephane, G. Mihaela, F. Louis and M. Max, J. Org. Chem., 1999, 64, 4920 CrossRef PubMed.
- (a) F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) B. M. Trost and A. H. Weiss, Adv. Synth. Catal., 2009, 351, 963 CrossRef CAS PubMed; (c) I.-T. Trotus, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761 CrossRef CAS PubMed.
- Known methods, either reactions of substrates with an acetylene group substituted prochiral center or the desymmetrization of prochiral bisalkynes, are limited in substrate scope and require the tedious synthesis of starting materials. For known reports: (a) Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342 CrossRef CAS; (b) A. K. Mourad, J. Leutzow and C. Czekelius, Angew. Chem., Int. Ed., 2012, 51, 11149 CrossRef CAS PubMed; (c) X.-P. Zeng, Z.-Y. Cao, X. Wang, L. Chen, F. Zhou, F. Zhu, C.-H. Wang and J. Zhou, J. Am. Chem. Soc., 2016, 138, 416 CrossRef CAS PubMed.
- J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5 CrossRef CAS.
- K. M.-H. Lim and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 8122 CrossRef CAS PubMed.
- Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981 RSC.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed; (c) C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200 CrossRef CAS PubMed; (d) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC; (e) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (f) E. Lallana, R. Riguera and E. Fernandez-Megia, Angew. Chem., Int. Ed., 2011, 50, 8794 CrossRef CAS PubMed.
- For a highlight, see: W. D. G. Brittain, B. R. Buckley and J. S. Fossey, ACS Catal., 2016, 6, 3629 CrossRef CAS.
- J.-C. Meng, V. V. Fokin and M. G. Finn, Tetrahedron Lett., 2005, 46, 4543 CrossRef CAS.
- (a) F. Zhou, C. Tan, J. Tang, Y.-Y. Zhang, W.-M. Gao, H.-H. Wu, Y.-H. Yu and J. Zhou, J. Am. Chem. Soc., 2013, 135, 10994 CrossRef CAS PubMed; (b) G. R. Stephenson, J. P. Buttress, D. Deschamps, M. Lancelot, J. P. Martin, A. I. G. Sheldon, C. Alayrac, A.-C. Gaumont and P. C. B. Page, Synlett, 2013, 24, 2723 CrossRef CAS; (c) T. Osako and Y. Uozumi, Org. Lett., 2014, 16, 5866 CrossRef CAS PubMed; (d) T. Song, L. Li, W. Zhou, Z.-J. Zheng, Y. Deng, Z. Xu and L.-W. Xu, Chem.–Eur. J., 2015, 21, 554 CrossRef CAS PubMed; (e) M.-Y. Chen, T. Song, Z.-J. Zheng, Z. Xu, Y.-M. Cui and L.-W. Xu, RSC Adv., 2016, 6, 58698 RSC; (f) M.-Y. Chen, Z. Xu, L. Chen, T. Song, Z.-J. Zheng, J. Cao, Y.-M. Cui and L.-W. Xu, ChemCatChem, 2018, 10, 280 CrossRef CAS.
- Mechanistic studies revealed that bis-triazoles formation is a severe problem in CuAAC of diynes or diazides systems, see: V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210 CrossRef CAS PubMed.
- (a) W. D. G. Brittain, B. R. Buckley and J. S. Fossey, Chem. Commun., 2015, 51, 17217 RSC; (b) J. R. Alexander, A. A. Ott, E.-C. Liu and J. J. Topczewski, Org. Lett., 2019, 21, 4355 CrossRef CAS PubMed.
- E.-C. Liu and J. J. Topczewski, J. Am. Chem. Soc., 2019, 141, 5135 CrossRef CAS PubMed.
- (a) G. Nishida, K. Noguchi, M. Hirano and K. Tanaka, Angew. Chem., Int. Ed., 2008, 47, 3410 CrossRef CAS PubMed; (b) Y.-K. Tahara, T. Sato, R. Matsubara, K. S. Kanyiva and T. Shibata, Heterocycles, 2016, 93, 685 CrossRef CAS; (c) Y. Zhang, F. Zhang, L. Chen, J. Xu, X. Liu and X. Feng, ACS Catal., 2019, 9, 4834 CrossRef CAS.
- X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330 CrossRef CAS PubMed.
- M. Nandita, S. William and W. Marcus, J. Mol. Catal. A: Chem., 2011, 334, 1 CrossRef.
- (a) T. Osako and Y. Uozumi, Synlett, 2015, 26, 1475 CrossRef CAS. For the same phenomenon observed in other desymmetric reactions: (b) K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964 CrossRef CAS PubMed; (c) C. Roux, M. Candy, J.-M. Pons, O. Chuzela and C. Bressy, Angew. Chem., Int. Ed., 2014, 53, 766 CrossRef CAS PubMed.
- J. Y. Lee, Y. S. You and S. H. Kang, J. Am. Chem. Soc., 2011, 133, 1772 CrossRef CAS PubMed.
- For reviews on asymmetric desymmetrization, see ref. 28, and (a) M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 1765 RSC; (b) A. J. Metrano and S. J. Miller, Acc. Chem. Res., 2019, 52, 199 CrossRef CAS PubMed.
- To the best of our knowledge, there is only one example of remote intermolecular desymmetrization at a distance of five covalent bonds separation, and no example of four covalent bonds separation, see: (a) C. A. Lewis, A. Chiu, M. Kubryk, J. Balsells, D. Pollard, C. K. Esser, J. Murry, R. A. Reamer, K. B. Hansen and S. J. Miller, J. Am. Chem. Soc., 2006, 128, 16454 CrossRef CAS PubMed. In some intramolecular desymmetric reactions, the distance between one reaction site and the prochiral center is up to 4 covalent bonds, but that of the other is only two or three, see for example: (b) F. Zhou, G.-J. Cheng, W. Yang, Y. Long, S. Zhang, Y.-D. Wu, X. Zhang and Q. Cai, Angew. Chem., Int. Ed., 2014, 53, 9555 CrossRef CAS PubMed.
- For reviews: (a) Y. Matano and H. Imahori, Org. Biomol. Chem., 2009, 7, 1258 RSC; (b) Y. Ren and T. Baumgartner, Dalton Trans., 2012, 41, 7792 RSC. For recent examples: (c) M. Stolar, J. Borau-Garcia, M. Toonen and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 3366 CrossRef CAS PubMed; (d) E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539 CrossRef CAS PubMed; (e) C. Reus, M. Stolar, J. Vanderkley, J. Nebauer and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 11710 CrossRef CAS PubMed; (f) V. Quint, F. Morlet-Savary, J.-F. Lohier, J. Lalevée, A.-C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436 CrossRef CAS PubMed; (g) N. M.-W. Wu, M. Ng, W. H. Lam, H.-L. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 15142 CrossRef CAS PubMed.
- Y. Matano, A. Saito, T. Fukushima, Y. Tokudome, F. Suzuki, D. Sakamaki, H. Kaji, A. Ito, K. Tanaka and H. Imahori, Angew. Chem., Int. Ed., 2011, 50, 8016 CrossRef CAS PubMed.
- As kindly suggested by one of reviewers, we tried the desymmetric CuAAC of diyne 25, whose ethynyl group is five covalent bonds from the prochiral phosphine, to examine the potency of our newly developed PYBOX ligands bearing a bulky shielding group. Not surprisingly, only 10% ee for the desired chiral monotrizaole 26 was obtained by using PYBOX L5, with a M/D ratio of 4:1
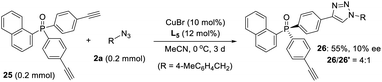 .
. - P.-Y. Lee and L.-C. Liang, Inorg. Chem., 2008, 47, 749 CrossRef CAS PubMed.
- J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426 CrossRef CAS PubMed.
- For reviews, see: (a) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC; (b) L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 6464 CrossRef CAS PubMed; (c) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534 CrossRef CAS; (d) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS PubMed; (e) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed.
- (a) W.-J. Yoo, L. Zhao and C.-J. Li, Aldrichimica Acta, 2011, 44, 43 CAS; (b) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790 RSC.
- (a) Z.-M. Zhang, B. Xu, S. Xu, H.-H. Wu and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 6324 CrossRef CAS PubMed; (b) L. B. Schenkel and J. A. Ellman, Org. Lett., 2003, 5, 545 CrossRef CAS PubMed; (c) X.-F. Bai, T. Song, Z. Xu, C.-G. Xia, W.-S. Huang and L.-W. Xu, Angew. Chem., Int. Ed., 2015, 54, 5255 CrossRef CAS PubMed; (d) L. Du, P. Cao, J. Xing, Y. Lou, L. Jiang, L. Li and J. Liao, Angew. Chem., Int. Ed., 2013, 52, 4207 CrossRef CAS PubMed.
- (a) J. A. Whiteford, C. V. Lu and P. J. Stang, J. Am. Chem. Soc., 1997, 119, 2524 CrossRef CAS; (b) W. Wang, L.-J. Chen, X.-Q. Wang, B. Sun, X.-P. Li, Y.-Y. Zhang, J.-M. Shi, Y.-H. Yu, L. Zhang, M.-H. Liu and H.-B. Yang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5597 CrossRef CAS PubMed; (c) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed.
- We provided a tentative model to explain the role of the C4 shielding group of PYBOX ligands, as well as the observed enantiofacial control of the desymmetrization of diynes 1 based on the absolute configuration of products 3. Please see page S75 and S76 of the ESI.†.
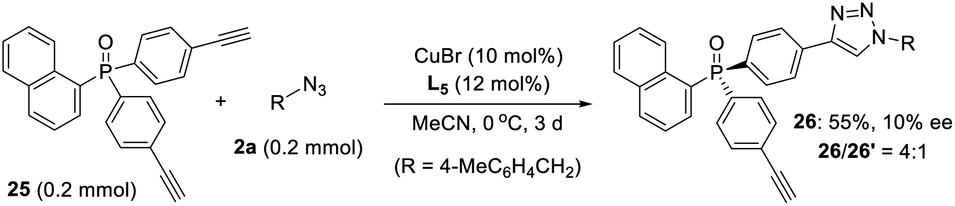
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data. CCDC 1508003, 1548228, 1450187, 1552231, 1920853, 1920855 and 1826888. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04938j |
| This journal is © The Royal Society of Chemistry 2020 |