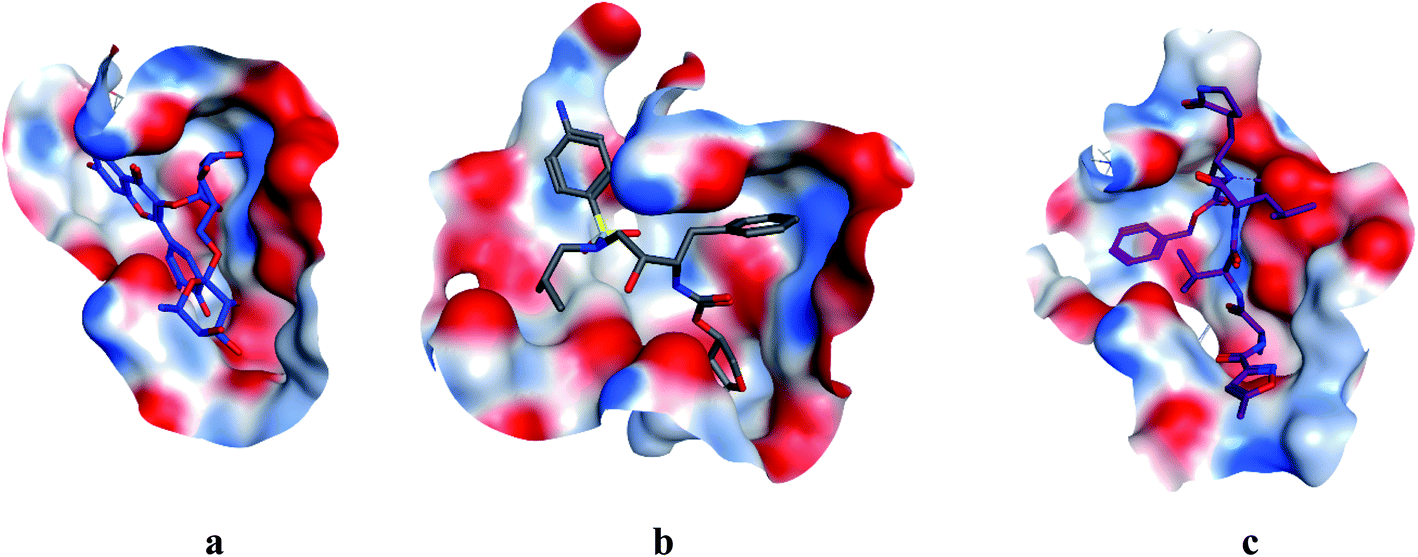DOI:
10.1039/D0RA03582C
(Paper)
RSC Adv., 2020,
10, 19570-19575
Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease
Received
21st April 2020
, Accepted 14th May 2020
First published on 21st May 2020
Abstract
In December 2019, an outbreak of coronavirus disease 2019 (COVID-19) commenced in Wuhan, China and affected around 210 countries and territories in a matter of weeks. It has a phylogenetic similarity to SARS-CoV and it was named coronavirus 2 (SARS-CoV-2) and caused severe acute respiratory syndrome that could lead to death. One of the promising therapeutic strategies for virus infection is the search for enzyme inhibitors among natural compounds using molecular docking in order to obtain products with minimal side effects. COVID-19 virus main protease plays a vital role in mediating viral transcription and replication, introducing it as an attractive antiviral agent target. Metabolic profiling of the aqueous extract of Salvadora persica L. (Salvadoraceae) aerial parts dereplicated eleven known flavonol glycosides using LC-HRESIMS. All the annotated flavonoids exhibited significant binding stability at the N3 binding site to different degrees, except isorhamnetin-3-O-β-D-glucopyranoside, when compared with the currently used COVID-19 main protease inhibitor, darunavir. Structural similarity between the identified flavonoids enabled the study of the relationship between their structure and interactions with the receptor in the N3 binding site of the COVID-19 main protease. The results indicate that the basic flavonol nucleus possesses activity itself. Moreover, the presence of a rutinose moiety at the 3 position of ring C and absence of an O-methyl group in ring B of the flavonol structure could increase the binding stability. This study provides a scientific basis for the health benefits of the regular use of S. persica as it leaches bioactive flavonoids in the aqueous saliva.
1. Introduction
In December 2019, coronavirus disease 2019 (COVID-19) commenced in Wuhan city, rapidly spread throughout China and affected around 210 countries and territories in a matter of weeks. The pathogen was identified as a novel enveloped RNA betacoronavirus that has a phylogenetic similarity to SARS-CoV and it was named coronavirus 2 (SARS-CoV-2).1 Patients with the infection suffer from severe acute respiratory syndrome that may result in death due to massive alveolar damage and progressive respiratory failure.2 According to World Health Organization (WHO) situation report – 103 on 2nd May 2020, the virus had caused 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 267184 infections and 229971 deaths all over the world. Therefore, a global response is desperately needed to find effective drugs against this unprecedented pandemic disease. To date, no specific treatment has been approved against COVID-19 disease however several protocols were tested such as chloroquine derivatives,3 azithromycin4 and convalescent plasma.5 One of the promising targets of treatment is COVID-19 main protease (Mpro). Recently published high-resolution structure of COVID-19 protease6,7 created the opportunity to develop its inhibitor and gave an essential key to control virus transcription and replication.8,9 By comprehensive proteolytic processing, the functional polypeptides are liberated from the polyproteins, mainly by a 33.8 kDa main protease (Mpro) which is also known as 3C-like protease. Starting with the autolytic cleavage of Mpro itself from pp1a and pp1ab, this enzyme can digest the polyprotein at not less than eleven conserved sites.10 The significance of Mpro in coronavirus life cycle, and the absence of similar homologues in humans, introduce Mpro as an interesting target for antiviral drug design.11
267184 infections and 229971 deaths all over the world. Therefore, a global response is desperately needed to find effective drugs against this unprecedented pandemic disease. To date, no specific treatment has been approved against COVID-19 disease however several protocols were tested such as chloroquine derivatives,3 azithromycin4 and convalescent plasma.5 One of the promising targets of treatment is COVID-19 main protease (Mpro). Recently published high-resolution structure of COVID-19 protease6,7 created the opportunity to develop its inhibitor and gave an essential key to control virus transcription and replication.8,9 By comprehensive proteolytic processing, the functional polypeptides are liberated from the polyproteins, mainly by a 33.8 kDa main protease (Mpro) which is also known as 3C-like protease. Starting with the autolytic cleavage of Mpro itself from pp1a and pp1ab, this enzyme can digest the polyprotein at not less than eleven conserved sites.10 The significance of Mpro in coronavirus life cycle, and the absence of similar homologues in humans, introduce Mpro as an interesting target for antiviral drug design.11
Natural products are considered as the most consistently successful source of drug leads.12 Chewing stick or Salvadora persica L. (Salvadoraceae) mostly known as Arak or Miswak in Arabic,13 is one of the most popular medicinal plants between Muslims.14 It is used for teeth cleaning and as oral hygiene by leaching out important bioactive compounds such as flavonoids, alkaloids, vitamin C, in saliva contributing to its diverse biological activities such as antimicrobial and anticariogenic actions, in addition to its effects on dental plaque, gingival health, and periodontal status.13,15 Interestingly, it was reported to possess antiviral activity against herpes simplex virus-1.16 To the best of authors' knowledge, there's no previous report about its action on COVID-19 virus.
Metabolomics is an important comprehensive analytical mean that is used to study the metabolite profiles of different biological systems especially those with complex chemical diversity such as plants.17 LC-HRESIMS is one of the promising tools in metabolic profiling that can detect/dereplicate an extensive range of metabolites simultaneously without tedious isolation process.12
The present study was undertaken to evaluate the efficacy of medicinal plant-based bioactive compounds, identified by metabolic profiling from S. persica against COVID-19 protease using molecular docking study. In addition to, comparison of the binding stability of these compounds with that of the currently used COVID-19 main protease inhibitor, darunavir.
2. Materials and methods
2.1. Plant material
S. persica aerial parts (stem and leaves) were collected on Jan. 2018 from El-Orman Botanical Garden, Giza, Egypt. It was kindly identified by Eng. Trease Labib, former-Head of El-Orman Botanical Garden and Prof. Nasser Barakat, Professor of Plant Ecology, Botany and Microbiology Department, Faculty of Science, Minia University, Egypt. A voucher specimen (2018-BuPD 70) was deposited at the Department of Pharmacognosy, Faculty of Pharmacy, Beni-Suef University, Egypt.
2.2. Preparation of plant extract and metabolic profiling
Air-dried and finely powdered S. persica aerial parts (3 g each) were exhaustively defatted using petroleum ether (3 × 15 mL) at room temperature and concentrated under vacuum to afford 40 mg crude extracts. The crude extract was subjected to successive fractionation using dichloromethane, ethyl acetate and finally distilled water (each; 3 × 15 mL) to afford 21, 10, 239 mg respectively after evaporation. The aqueous extract was subjected to LC-HRESIMS analysis according to Elmaidomy, Mohammed et al. 2019.12 An Acquity Ultra-Performance Liquid Chromatography (UPLC) system coupled to a Synapt G2 HDMS quadrupole time-of-flight hybrid mass spectrometer (Waters, Milford, MA, USA) was used for LC-HRESIMS analysis. Chromatographic separation was performed using a BEH C18 column (2.1 × 100 mm, 1.7 μm particle size) and a guard column (2.1 × 5 mm, 1.7 μm particle size). A protocol of binary linear solvent gradient elution was carried out with the use of 0–100% eluent B, for 6 min, at 0.3 mL min−1 flow rate, and 0.1% formic acid in water (v/v) as solvent A and acetonitrile as solvent B. The column temperature was adjusted at 40 °C and the injection volume was 2 μL. Detection of the metabolites was performed after chromatographic separation using mass spectrometry by electrospray ionization (ESI) in positive mode and the source was set to 120 °C. The ESI capillary voltage was operated at 0.8 kV while the sampling cone voltage was adjusted to 25 V. Nitrogen (flow rate of 800 L h−1, at 350 °C) was used as the desolvation gas and the cone gas (flow rate of 30 L h−1). The mass range for TOF-MS was set from m/z 50–1200. The raw data was imported in MZmine 2.12 through selection of the ProteoWizard converted positive files in mzML format. Mass ion peaks were detected, followed by chromatogram builder and chromatogram deconvolution. Local minimum search algorithm was applied, and the isotopes were identified via isotopic peaks grouper. Detection of the missing peaks was performed using gap-filling peak finder. An adduct search, in addition to complex search were applied. Then, the processed data set was subjected to molecular formula prediction as well as, peak identification. The positive and negative ionization mode data sets from plant extract were dereplicated against Dictionary of Natural Products (DNP) database.
2.3. Docking studies
2.3.1. Target compounds optimization. The identified compounds were constructed in the form of a 3D model. After checking their structures and the formal charges on atoms by 2D depiction, the target compounds were subjected to a conformational search. All conformers were subjected to energy minimization. All the minimizations were performed until a RMSD gradient of 0.01 kcal mol−1 and RMS distance of 0.1 Å with MMFF94X force-field. The partial charges were automatically calculated. The obtained database was saved in the form of MDB file to be used in the docking calculations.
2.3.2. Optimization of the enzymes active site. The X-ray crystallographic structure of Mpro complexed with N3 was obtained from the Protein Data Bank through the internet (http://www.rcsb.org/, PDB code 6LU7).6 To prepare the enzyme for docking studies, hydrogen atoms were added to the system with their standard geometry. Automatic correction was applied to check for any errors in the atom's connection and type. Selection of the receptor and its atoms potential were fixed. Site Finder was used for the active site search in the enzyme structure using all default items. Dummy atoms were created from the site finder of the pocket.
2.3.3. Docking of the target molecules to viral main protease binding site. Docking of conformation database of the target compounds was performed. The following methodology was generally applied: the enzyme active site file was loaded, and the dock tool was initiated. The program specifications were adjusted so that dummy atoms as the docking site, alpha triangle as the placement methodology and London dG as the scoring methodology. The latter was adjusted to its default values. The MDB file of the ligand was loaded and dock calculations were run automatically. The obtained poses were studied, and the poses showing the best ligand–enzyme interactions were selected and stored for energy calculations.
3. Results and discussion
3.1. Metabolomic analysis
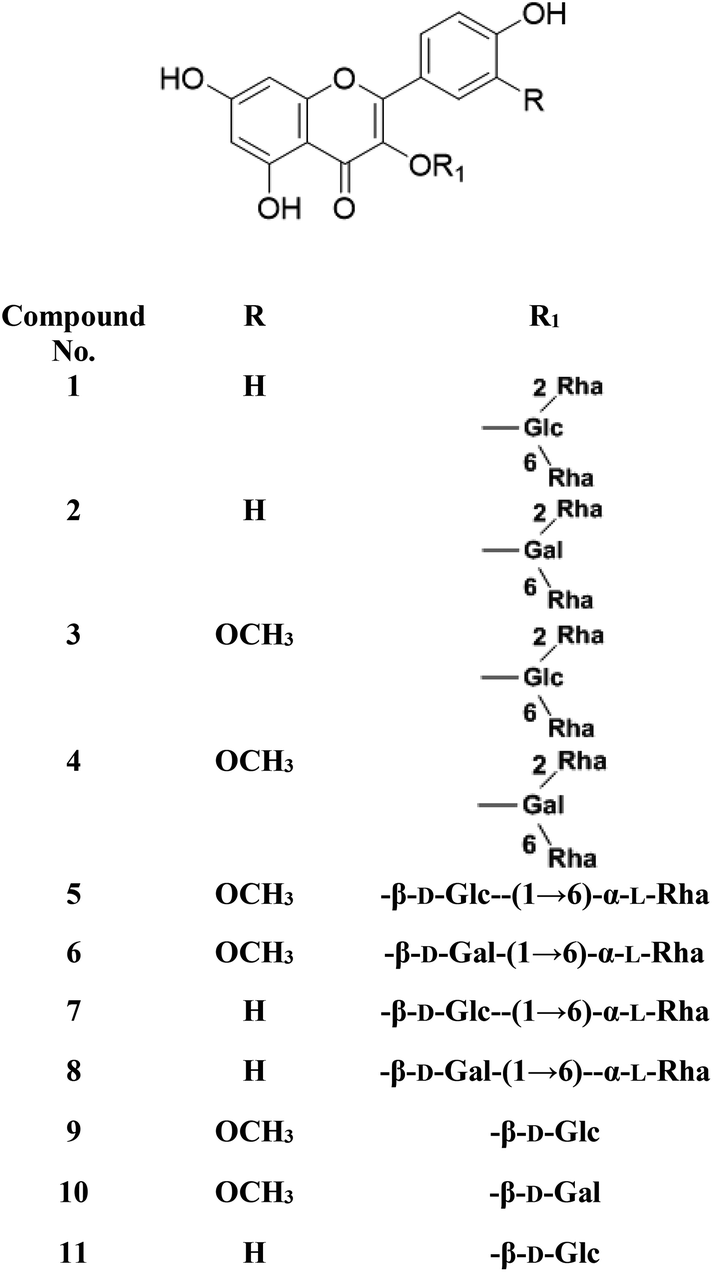
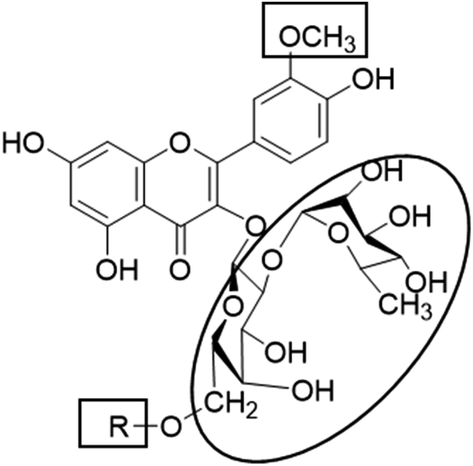
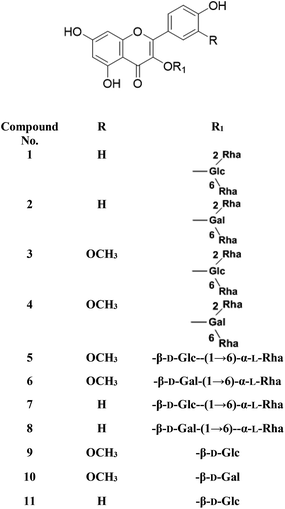
Metabolic profiling of the secondary metabolites of the S. persica aerial parts using LC-HRESIMS for dereplication purposes, resulted in the annotation of 11 compounds (Table 1, Fig. 1), all of them was flavonoid glycosides. Metabolites identification was based on employing macros and algorithms that coupled MZmine with online and in-house DNP databases. All the eleven identified flavonoid glycoside was previously isolated from S. persica leaves.18 These metabolites present as glycosides that belong to flavonol subclass (kaempferol 1, 2, 7, 8, 11) and its O-methylated derivative (isorhamnetin 3, 4, 5, 6, 9, 10). The glycosides were from triglycoside 1, 2, 3, 4, diglycoside 5, 6, 7, 8 and monoglycoside (9, 10, 11) types.
Table 1 The LC-HR-ESIMS dereplication results of the aqueous extract of Salvadora persica aerial parts
| No. |
Metabolite name |
Molecular formula |
RT (min.) |
m/z |
| 1 |
Kaempferol-3-O-α-L-rhamnopyranosyl(1→6)-O-[α-L-rhamnopyranosyl(1→2)]-O-β-D-glucopyranoside |
C33H40O19 |
2.20 |
741.2248 |
| 2 |
Kaempferol-3-O-α-L-rhamnopyranosyl(1→6)-O-[α-L-rhamnopyranosyl(1→2)]-O-β-D-galactopyranoside (mauritianin) |
C33H40O19 |
2.20 |
741.2248 |
| 3 |
Isorhamnetin-3-O-α-L-rhamnopyranosyl(1→6)-O-[α-L-rhamnopyranosyl(1→2)]-O-β-D-glucopyranoside |
C34H42O20 |
2.32 |
793.2169 |
| 4 |
Isorhamnetin-3-O-α-L-rhamnopyranosyl(1→6)-O-[α-L-rhamnopyranosyl(1→2)]-O-β-D-galactopyranoside |
C34H42O20 |
2.32 |
793.2169 |
| 5 |
Isorhamnetin-3-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside (narcissin) |
C28H32O16 |
2.81 |
647.1789 |
| 6 |
Isorhamnetin-3-O-α-L-rhamnopyranosyl-(1→6)-β-D-galactopyranoside |
C28H32O16 |
2.81 |
647.1789 |
| 7 |
Kaempferol-3-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside |
C27H30O15 |
2.91 |
617.1483 |
| 8 |
Kaempferol-3-O-α-L-rhamnopyranosyl-(1→6)-β-D-galactopyranoside |
C27H30O15 |
2.91 |
617.1483 |
| 9 |
Isorhamnetin-3-O-β-D-glucopyranoside |
C22H22O12 |
4.47 |
501.1003 |
| 10 |
Isorhamnetin-3-O-β-D-galactopyranoside |
C22H22O12 |
4.47 |
501.1003 |
| 11 |
Kaempferol-3-O-β-D-glucopyranoside (astragalin) |
C21H20O11 |
4.91 |
471.0904 |
 |
| | Fig. 1 Structures of the dereplicated metabolites from the aqueous extract of Salvadora persica aerial parts. | |
From the metabolomics data, the mass ion peak with m/z 741.2248 for the predicted molecular formula C33H40O19, was dereplicated as the flavonoid glycoside, kaempferol-3-O-rhamnosylrutinoside 1 and kaempferol-3-O-rhamnosylrobinobioside (mauritianin) 2, respectively. While the peak at m/z 793.2169 that in agreement with the suggested molecular formula C34H42O20, was identified as isorhamnetin-3-O-rhamnosylrutinoside 3 and isorhamnetin-3-O-rhamnosylrobinobioside 4, respectively. Whereas the mass ion peak at m/z 647.1789, corresponding to the suggested molecular formula C28H32O16, was dereplicated as isorhamnetin-3-O-rutinoside (narcissin) 5 and isorhamnetin-3-O-robinobioside 6, respectively. Moreover, the mass ion peak at m/z 617.1483, with the suggested as molecular formula C27H30O15, was dereplicated as kaempferol-3-O-rutinoside 7 and kaempferol-3-O-robinobioside 8, respectively. Additionally, the peak at m/z 501.1003, with the molecular formula C22H22O12, was distinguished as isorhamnetin-3-O-glucoside 9 and isorhamnetin-3-O-galactoside 10, respectively. Likewise, the monoglycoside with the molecular formula C21H20O11 was characterized as kaempferol-3-O-glucoside (astragalin) 11 from the mass ion peak at m/z 471.0904.
3.2. Docking study
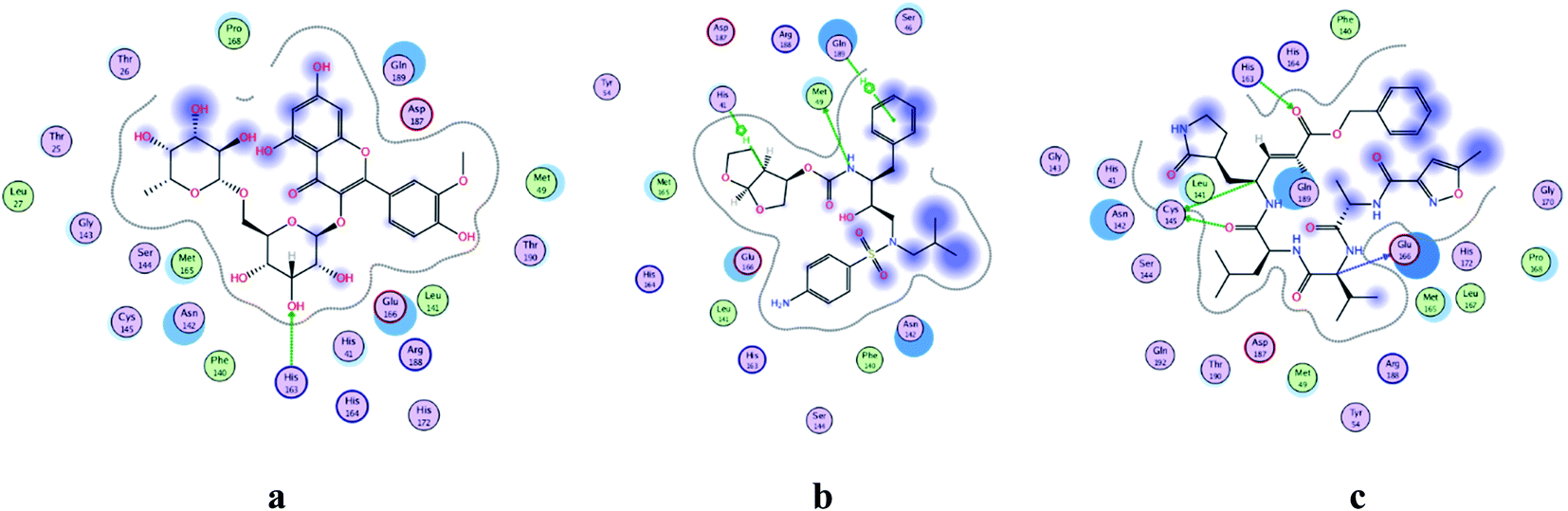
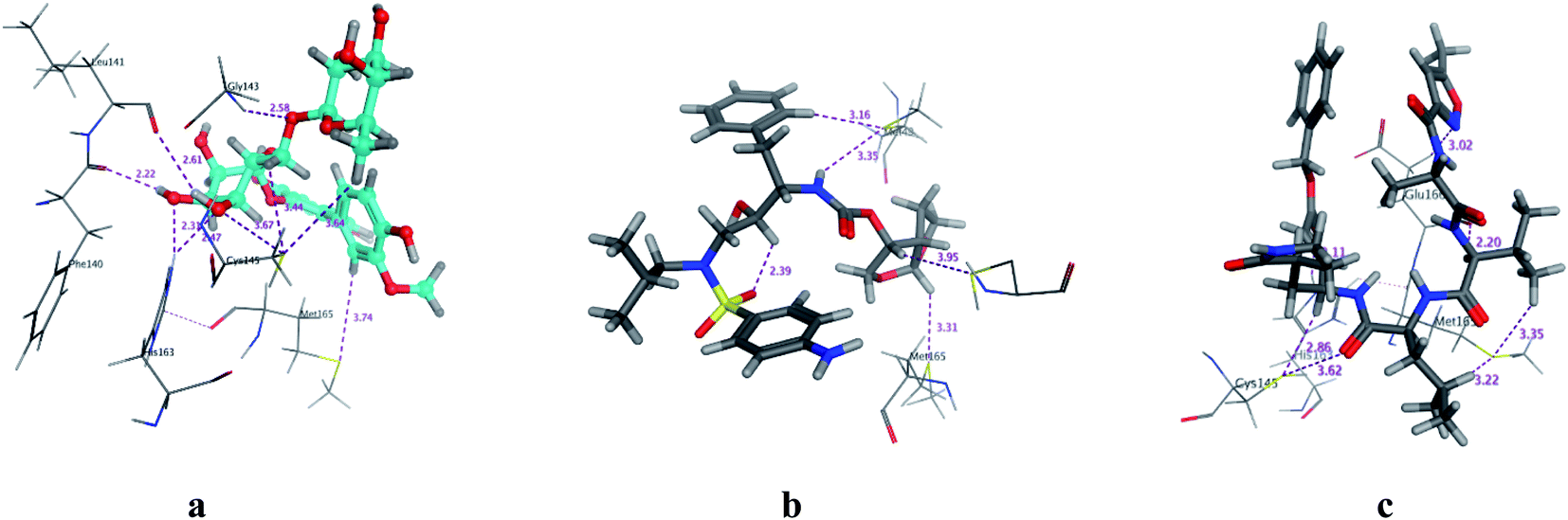
COVID-19 virus Mpro has a Cys–His catalytic dyad, and the substrate-binding site is located in a cleft between Domain I and II. N3 is fitted inside the substrate-binding pocket of COVID-19 virus Mpro showing asymmetric unit containing only one polypeptide. All compounds showed nearly bindings like N3. Results of interaction energies with Mpro are shown in Table 2. Molecular docking simulation of identified flavonoid compounds 1–11, darunavir and N3 into Mpro active site was done. They got stabilized at the N3-binding site of Mpro by variable several electrostatic bonds (Fig. 2–4). The order of strength of binding was as follows:
| N3 > 5 > 7 > 3 > 4 > 2 > 6 > 10 > 1 > 8 > 11 > darunavir > 9 |
Table 2 Receptor interaction with identified flavonoids, darunavir and N3 into the N3 binding site in the COVID-19 main protease
| Compound |
dG kcal mol−1 |
Receptor amino acid |
| 1 |
−7.5332 |
Glu 166, Thr 190, Met 165, Ser 46 and Met 49 |
| 2 |
−7.8617 |
Asn 142, Phe 140, Glu 166 and Met 165 |
| 3 |
−8.0848 |
Met 149, Thr 190, Gly 143, Asn 142, Glu 166 and Met 165 |
| 4 |
−8.0810 |
Cys 145, Leu 141, His 163 and Phe 140 |
| 5 |
−8.2530 |
Cys 145, Gly 143, Leu 141, His 163 and Met 165 |
| 6 |
−7.6093 |
Cys 145, Phe 140, Met 49, Thr 29 and Gly 143 |
| 7 |
−8.1203 |
Cys 145, His 163, Asn 142, Glu 166 and Met 165 |
| 8 |
−7.4654 |
Cys 145, His 163, Phe 140 and Glu 166 |
| 9 |
−6.6389 |
Cys 145, Phe 140 and Met 149 |
| 10 |
−7.5771 |
Cys 145, His 163, Asn 142, Met 165 and Glu 166 |
| 11 |
−7.0959 |
Cys 145, Met 165 and Thr 190 |
| Darunavir |
−7.0415 |
Cys 145, Met 49, Gln 189 and His 41 |
| N3 |
−8.3963 |
Cys 145, Met 165 and His 163 |
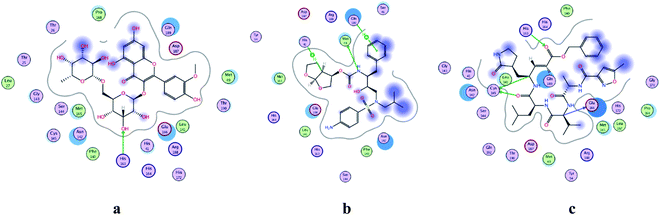 |
| | Fig. 2 2D representation of docking of compounds: flavonoid 5 (a), darunavir (b), and N3 (c) into the N3 binding site of the COVID-19 main protease. | |
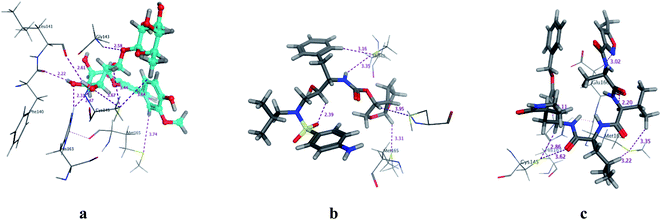 |
| | Fig. 3 3D Docking poses of compounds: flavonoid 5 (a), darunavir (b), and N3 (c) into the N3 binding site of the COVID-19 main protease. | |
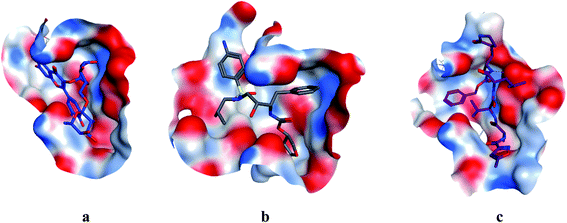 |
| | Fig. 4 Surface and maps of compounds: flavonoid 5 (a), darunavir (b), and N3 (c) docked into N3 binding site of the COVID-19 main protease. | |
All identified flavonoid compounds 1–11 except 9 were better than the currently used COVID-19 main protease inhibitor darunavir. Flavonoids 5, 7, 3 and 4 showed the highest stability in the N3 binding site.
3.3. Comparative structural evaluation of receptor interaction between identified flavonoids into N3 binding site in COVID-19 main protease
In the current study, all the identified flavonols 1–11 had an excellent binding stability in the N3 binding site in different degrees when compared with the currently used covid-19 main protease inhibitor darunavir (Table 2), except for flavonol 9 which exhibited lesser binding stability. This suggesting that the basic flavonol nucleus (Fig. 5) possesses activity itself. By investigating sugar moiety in the tested flavonoids, it was concluded that the presence of disaccharide rutinose (α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranose) at position no. 3 is essential for activity. That could be explained from receptor interaction of rutinosides 5, 7 which had the highest binding stability while monoglucosides 9, 11, having the same aglycone moiety but lacking rutinose moiety, were the least active. This was confirmed in robinobiosides 6, 8 in which glucose moiety of rutinose is replaced by galactose and the activity of both of them was less than that of rutinosides 5, 7. Moreover, the presence of another rhamnose group in addition to rutinose moiety decrease the binding stability as indicated in rutinoside 5, 7 which were more active than the corresponding rhamnosyl rutinosides 3, 1. Concerning the aglycone part of the characterized flavonoids, it was concluded that the presence of O-methyl in ring B of the flavonol skeleton decreases the binding stability. This was indicated by studying the binding stability of flavonoids having the same sugar moiety but differ in the aglycone moiety such as isorhamnetin monoglucoside 9 which was less active than kaempferol monoglucoside 11. Fig. 5 shows the important structure features that affect the receptor interaction of flavonoids into the N3 binding site in the COVID-19 main protease.
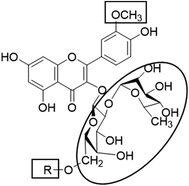 |
| | Fig. 5 Structure elements of flavonoids that increase (circle) or decrease (rectangle) binding stability into N3 binding site in the COVID-19 main protease. | |
4. Conclusion
COVID-19 virus is the causative agent of 2019–2020 pandemic pneumonia attack that commenced in Wuhan. Finding out known natural drug that inhibit COVID-19 virus main protease (Mpro) will result in a pivotal role in controlling its replication and transcription. Eleven known compounds were identified from the aqueous fraction of S. persica aerial parts using LC-HRESIMS for dereplication purposes; all of them were flavonol glycosides and isolated before from the leaves. Identification of structurally related flavonoids enabled to test the relationship between their structure and interactions with receptor in the N3 binding site in the COVID-19 main protease. All the tested flavonoids exhibited significant binding stability except isorhamnetin-3-O-β-D-glucopyranoside when compared with the currently used covid-19 main protease inhibitor, darunavir. The presence of rutinose moiety at 3 position of ring C and absence of O-methyl group in ring B of the flavonol structure could increase the binding stability. This study provides scientific basis for the health benefits of S. persica as it leaks the bioactive flavonoids in the aqueous saliva during its regular use. More studies are required to compare the observed effect of the active flavonoids using in vitro and in vivo testing and more efforts are needed to investigate the actions of other flavonols with different substitutions. Furthermore, meaningful assessments are needed to report the duration and frequency of S. persica use to prevent COVID-19 viral infection.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
No acknowledgments were reported by the authors.
References
- W. Guan, Z. Ni, Y. Hu, W. Liang, C. Ou, J. He, L. Liu, H. Shan, C. Lei and D. S. Hui, N. Engl. J. Med., 2020, 382, 1708–1720 CrossRef CAS PubMed.
- Z. Xu, L. Shi, Y. Wang, J. Zhang, L. Huang, C. Zhang, S. Liu, P. Zhao, H. Liu and L. Zhu, Lancet Respir. Med., 2020, 8, 420–422 CrossRef CAS PubMed.
- J. Gao, Z. Tian and X. Yang, BioSci. Trends, 2020, 14, 72–73 CrossRef CAS PubMed.
- P. Gautret, J.-C. Lagier, P. Parola, L. Meddeb, M. Mailhe, B. Doudier, J. Courjon, V. Giordanengo, V. E. Vieira and H. T. Dupont, Int. J. Antimicrob. Agents, 2020, 105949, DOI:10.1016/j.ijantimicag.2020.105949.
- L. Chen, J. Xiong, L. Bao and Y. Shi, Lancet Infect. Dis., 2020, 20, 398–400 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang and C. Peng, bioRxiv, 2020 DOI:10.1101/2020.02.26.964882.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- O. M. Aly, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12061302.v1.
- K. Anand, G. J. Palm, J. R. Mesters, S. G. Siddell, J. Ziebuhr and R. Hilgenfeld, EMBO J., 2002, 21, 3213–3224 CrossRef CAS PubMed.
- A. Hegyi and J. Ziebuhr, J. Gen. Virol., 2002, 83, 595–599 CrossRef PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- A. H. Elmaidomy, R. Mohammed, H. M. Hassan, A. I. Owis, M. E. Rateb, M. A. Khanfar, M. Krischke, M. J. Mueller and U. Ramadan Abdelmohsen, Metabolites, 2019, 9, 223 CrossRef CAS PubMed.
- H. S. Halawany, Saudi Dent J., 2012, 24, 63–69 CrossRef PubMed.
- D. J. Mabberley, Mabberley’s Plant-Book: A Portable Dictionary of Plants, Their Classification and Uses, Cambridge University Press, NewYork, NY, USA, 3rd edn, 2008 Search PubMed.
- M. M. Ghoneim, W. M. Afifi, M. Ibrahim, M. Elagawany, M. T. Khayat, M. H. Aboutaleb and A. M. Metwaly, Pharmacogn. Mag., 2019, 15, 232 CrossRef CAS.
- M. Y. Taha, Al-Rafidain Dental Journal, 2008, 8, 50–55 CrossRef.
- J. L. Rambla, M. López-Gresa, J. Bellés and A. Granell, in Plant Functional Genomics, Springer, 2015, pp. 221–235 Search PubMed.
- A. Ali, M. Assaf, M. El-Shanawany and M. Kamel, Bull. Pharm. Sci., Assiut Univ., 1997, 20, 181–186 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Marwa S. El-Hawaryb,
Dalia El Amira,
Omar M. Alyc,
Usama Ramadan Abdelmohsenbd and
Mohamed S. Kamelbd
*a,
Marwa S. El-Hawaryb,
Dalia El Amira,
Omar M. Alyc,
Usama Ramadan Abdelmohsenbd and
Mohamed S. Kamelbd