
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward ideal carbon dioxide functionalization
Yang
Yang
 and
Ji-Woong
Lee
*
and
Ji-Woong
Lee
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, Copenhagen Ø, 2100, Denmark. E-mail: jiwoong.lee@chem.ku.dk
First published on 20th February 2019
Abstract
This Perspective recapitulates recent developments of carbon dioxide utilization in carbon–carbon bond formation reactions, with an intention of paving a way toward sustainable CO2-functionalization and its tangible applications in synthetic chemistry. CO2 functionalization reactions possess intrinsic drawbacks: the high kinetic inertness and thermodynamic stability of CO2. Numerous procedures for CO2 utilization depend on energy-intensive processes (i.e. high pressure and/or temperature), often solely relying on reactive substrates, hampering its general applications. Recent efforts thus have been dedicated to catalytic CO2-utilization under ambient reaction conditions, however, it is still limited to a few activation modes and the use of reactive substrates. Herein, ideal CO2-functionalization with particular emphasis on sustainability will be discussed based on the following sub-categories; (1) metal-catalyzed ‘reductive’ carboxylation reaction of halides, olefins and allyl alcohols, (2) photochemical CO2-utilization, (3) redox-neutral CO2-functionalization, and (4) enantioselective catalysis incorporating CO2 to form C–CO2 bonds (excluding strain mediated reactions with epoxide- and aziridine-based substrates). Recent progress in these fields will be discussed with the proposed reaction mechanisms and selected examples, highlighting redox-neutral, umpolung, and asymmetric carboxylation to postulate ideal CO2 functionalization reactions to be developed in the near future.
 Yang Yang | Yang Yang (left) was born in 1989 in Hubei Province, P. R. China. He received his BSc (2013) and MSc (2016) in chemistry under the supervision of Prof. Wei Wang at the State Key Laboratory of Applied Organic Chemistry (SKLAOC), Lanzhou University. He joined the Lee group in 2017 as a PhD student. His current research interest focuses on asymmetric CO2-functionalization promoted by organocatalysts. Ji-Woong Lee (right) received his BSc and MSc in chemistry from Sungkyunkwan University under the guidance of Prof. Choong Eui Song (2009). In 2013 he obtained his PhD under the supervision of Prof. Dr Benjamin List at the Max-Planck-Institute fur Kohlenforschung. After postdoctoral research at the Weizmann Institute of Science with Prof. Rafal Klajn and at UC-Berkeley with Prof. Jeffrey R. Long, he has been an assistant professor at the University of Copenhagen since 2016. His team is working on desalination, water purification, materials chemistry, new methodology development in CO2-functionalization and asymmetric catalysis. |
1. Introduction
Carbon is an essential element for all living organisms, and is present in carbohydrates, amino acids, proteins, and lipids. These biomolecules are synthesized with specific selectivities controlled by the natural molecular foundry – enzymes – to sustain forms of life. The sustainability of bio- and chemical networks in living organisms is powered by the seemingly unlimited solar energy. Owing to the evolution of cyanobacteria and their photosynthesis,1 our planet became a unique biosphere where water was split into oxygen and hydrogen, while consuming (or fixating) CO2 to generate reduced organic matter.Photosynthesis and CO2 fixation operate under ambient conditions; artificial photosynthesis is yet to be realized,2 and can ensure sustainable growth of the human civilization. The challenge lies in overcoming the thermodynamic stability and kinetic inertness of CO2, which possesses the highest oxidation state of carbon. Therefore, it is inevitable to employ reducing reagents (reactive metals, H2, electricity, and highly reducing chemicals) to overcome the intrinsic reaction barrier of CO2-activation, particularly to enable the reactions to be operative under mild reaction conditions.
Recently, the global society has raised concerns related to excessive energy consumption and uncontrollable anthropogenic CO2 emission.3 Although CO2 functionalization can provide ideal solutions, chemical reactions with CO2 currently suffer from low efficiency, making it impossible to mitigate the overwhelmingly large quantity of accumulated CO2 in the atmosphere at low concentrations.4 Yet, chemical recycling of carbon dioxide has been recognized as a promising supplement to the natural carbon cycle,5 while producing value-added fine chemicals.6 In this context, CO2 can serve as an inexpensive and non-toxic renewable C1-building block.4,7 For example, light hydrocarbons and C1- or C2-units (i.e. carbon monoxide, formic acid, formaldehyde, methanol, and oxalic acid) are accessible from CO2, mostly catalyzed by heterogeneous materials (semiconductors,8 zeolites,9 COFs,10 MOFs,11 and g-C3N4 (ref. 12)). On the other hand, homogeneous catalysis has shown remarkable potential in C–C bond formation reactions, via formal insertion of CO2 at C–H bonds. The utility of carboxylic acids and their derivatives is certainly applicable with broad interest in organic synthesis13 and pharmaceutical chemistry.14
As categorized in Table 1, catalytic CO2-functionalization reactions have been reviewed, particularly transition-metal catalyzed C–C bond formation reactions,15 carboxylation reactions catalyzed by palladium,16 silver,17 copper18 or copper–NHC (N-heterocyclic carbene) complexes,19 and nickel/iron20 catalysts, asymmetric CO2-functionalization reactions21 and photocatalytic CO2-functionalization.22 Other types of reactions are also tabulated to guide the readers for further reading in specific topics of interest. For example, carbonate formation reactions with epoxides and ring-strain mediated reactions,23 catalytic alkylation with CO2,24etc., will not be discussed in this Perspective.
| Year (ref.) | Title | Keywords |
|---|---|---|
| 2014 (ref. 3a) | Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2 | CO2 emission and utilization |
| 2014 (ref. 3b) | Porous inorganic membranes for CO2 capture: present and prospects | CO2 capture |
| 2001 (ref. 3c) | Catalysis research of relevance to carbon management: progress, challenges, and opportunities | CO2 emission and utilization |
| 2007 (ref. 4) | Transformation of carbon dioxide | CO2 conversion |
| 2018 (ref. 7) | Sustainable conversion of carbon dioxide: an integrated review of catalysis and life cycle assessment | Catalysis, carbon life cycle assessment |
| 2018 (ref. 8a) | Cocatalysts in semiconductor-based photocatalytic CO2 reduction: achievements, challenges, and opportunities | Photocatalytic CO2 reduction |
| 2013 (ref. 8b) | Photocatalytic reduction of CO2 on TiO2 and other semiconductors | |
| 2014 (ref. 8c) | Photocatalytic conversion of CO2 into renewable hydrocarbon fuels: state-of-the-art accomplishment, challenges, and prospects | |
| 2017 (ref. 11a) | The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion | MOFs in CO2 utilization |
| 2017 (ref. 11b) | Metal organic framework based catalysts for CO2 conversion | |
| 2015 (ref. 12a) | A review on g-C3N4 for photocatalytic water splitting and CO2 reduction | g-C3N4 in CO2 utilization |
| 2018 (ref. 15a) | Transition metal-catalyzed carboxylation reactions with carbon dioxide | Metal-catalyzed carboxylation |
| 2016 (ref. 15b) | Metal-catalyzed carboxylation of organic (pseudo)halides with CO2 | |
| 2018 (ref. 15c) | Transition metal-catalyzed carboxylation of unsaturated substrates with CO2 | |
| 2018 (ref. 16) | Recent advances in palladium-catalyzed carboxylation with CO2 | |
| 2016 (ref. 17) | Silver-catalyzed carboxylation | |
| 2016 (ref. 18) | Copper-catalyzed carboxylation reactions using carbon dioxide | |
| 2013 (ref. 19) | N-heterocyclic carbene (NHC)–copper-catalysed transformations of carbon dioxide | |
| 2016 (ref. 20) | Ni- and Fe-catalyzed carboxylation of unsaturated hydrocarbons with CO2 | |
| 2015 (ref. 23a) | Recent advances in the catalytic preparation of cyclic organic carbonates | Cyclic organic carbonates |
| 2018 (ref. 23b) | Catalytic strategies for the cycloaddition of pure, diluted, and waste CO2 to epoxides under ambient conditions | |
| 2015 (ref. 23c) | Synthesis of cyclic carbonates from epoxides and carbon dioxide by using organocatalysts | |
| 2018 (ref. 24a) | Catalytic reductive N-alkylations using CO2 and carboxylic acid derivatives: recent progress and developments | Catalytic alkylation |
| 2017 (ref. 24b) | Utilization of CO2 as a C1 building block for catalytic methylation reactions | |
| 2017 (ref. 21a) | Enantioselective incorporation of CO2: status and potential | Asymmetric functionalization |
| 2016 (ref. 21b) | CO2-mediated formation of chiral fine chemicals | |
| 2018 (ref. 22a) | Photoredox catalysis as a strategy for CO2 incorporation: direct access to carboxylic acids from a renewable feedstock | Photocatalytic carboxylation using CO2 |
| 2017 (ref. 22b) | Photochemical carboxylation of activated C(sp3)–H bonds with CO2 | |
| 2017 (ref. 85a) | Reversible hydrogenation of carbon dioxide to formic acid and methanol: Lewis acid enhancement of base metal catalysts | Formic acid and methanol derivatives |
| 2015 (ref. 85b) | CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction | |
| 2014 (ref. 85c) | Recycling of carbon dioxide to methanol and derived products-closing the loop |
The purpose of this Perspective is the following: providing a general concept of catalytic CO2-functionalization by exemplifying recent progress (up to 2018). Section 2 will discuss transition-metal catalysis with a hint of sustainability. Sections 3 and 4 will explore recently reported photochemical redox catalysis by utilizing synthetic dyes with the aid of pre-established transition metal catalysis, and single-electron reduction of CO2via a redox-neutral mechanism. Section 5 will focus on a handful but remarkable examples of asymmetric C–C bond formation reactions by the action of metal–chiral ligand complexes. The future perspective on ideal CO2-functionalization will also be discussed in the context of umpolung carboxylation, redox-neutral photochemistry and asymmetric CO2-activation to reduce the prevailing energy input or highly reactive species. This discussion will lead to an alternative platform for sustainable CO2 recycling, to mimick the natural carbon cycle by utilizing the combined knowledge in organic, inorganic, photo- and materials chemistry, and enzymatic engineering for improved carbon fixation as well.
2. Metal-catalyzed reductive carboxylation with halides, olefins and allyl alcohols
The catalytic application of transition metals for carboxylation with CO2 was triggered by the seminal work by Nobile25 (Scheme 1, eqn (1)) and Osakada26 (Scheme 1, eqn (2)), where stoichiometric Ph–Ni(L)–Br (L = 2,2′-bipyridine (bpy)) participated in CO2 insertion at the Ph–Ni bond, affording benzoic acid as the final product.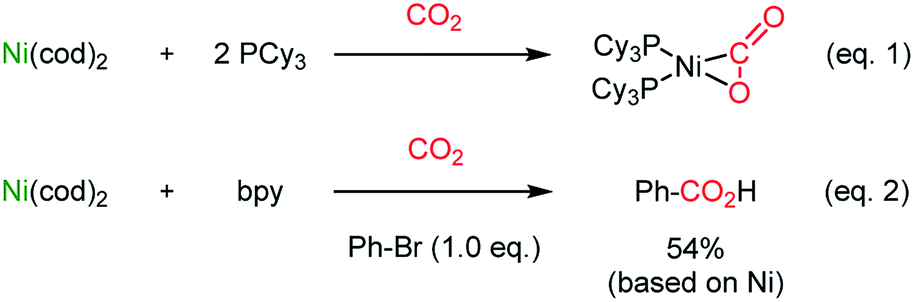 | ||
| Scheme 1 Stoichiometric CO2-functionalization using Ni(0). | ||
The Martin group employed a Pd(II)–Pd(0) cycle in the catalytic carboxylation reaction of aryl bromides using ZnEt2 as a terminal reducing reagent.27 This methodology was further expanded to abundant Ni(II) catalysis by the Tsuji group,28 realizing carboxylation of aryl chloride with Mn powder as a reducing reagent. New reductive carboxylation reactions were developed later by the Martin group with a broad range of substrate scope, including organic halides,29 sulfonates,29b esters,30 benzylic ammonium salts31 (Scheme 2a), allyl acetates,32 allyl alcohols33 (Scheme 2b), and unsaturated hydrocarbons (Scheme 2c and d).34 The facile insertion of CO2 into R–Ni was tested with olefin substrates, enabling olefin activation without an apparent hydride donor (Scheme 2). These protocols provided a broad substrate scope and high functional group tolerance. However, it is necessary to use (over)stoichiometric amounts of reducing reagents (i.e. Mn, Zn, ZnR2, and etc.) to complete the catalytic cycle.
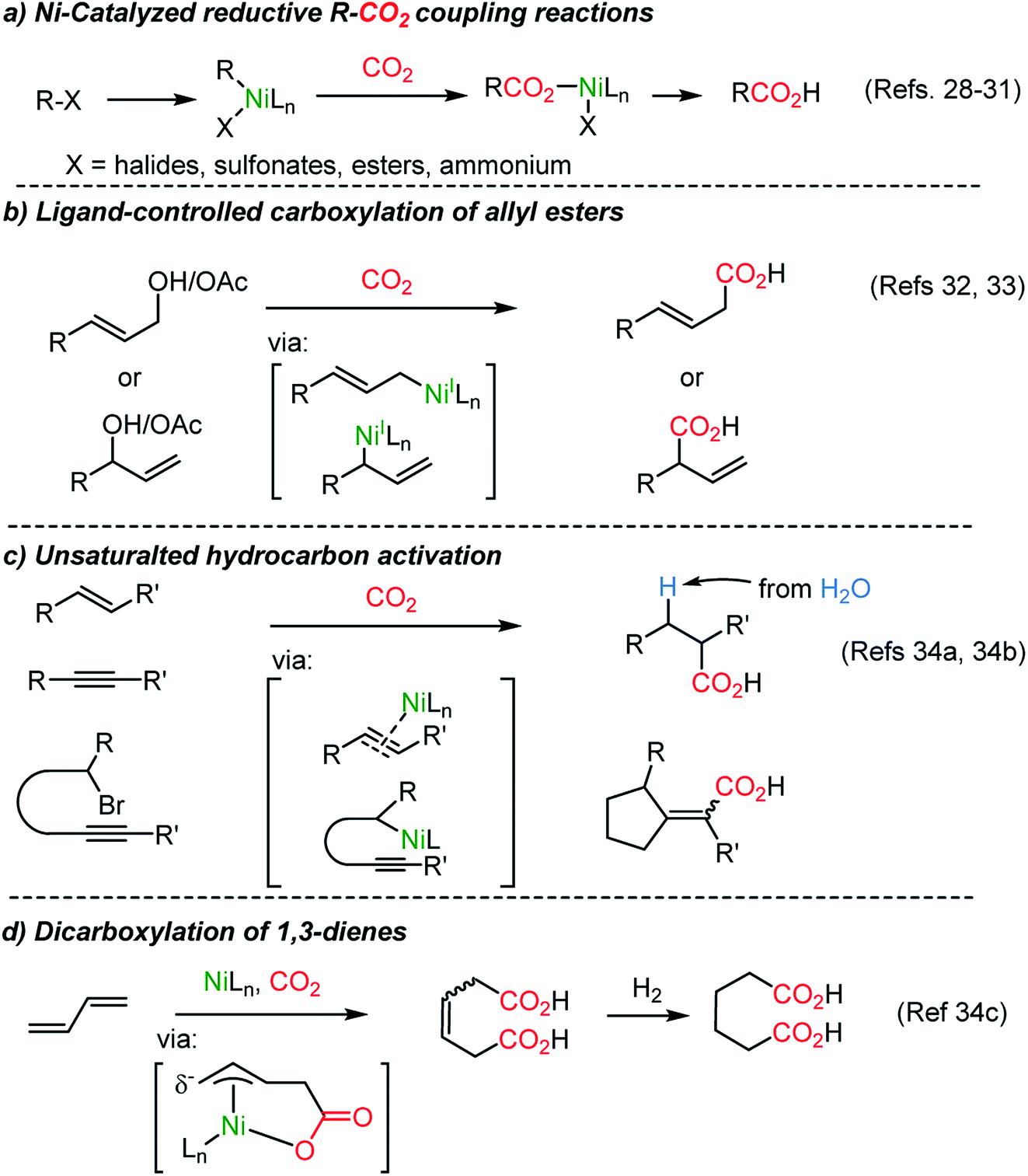 | ||
| Scheme 2 Ni-catalyzed reductive CO2-functionalization reactions. | ||
In 2017, a breakthrough CO2-functionalization was reported by the Martin group proposing a ‘chain-walking’ mechanism with catalytic Ni–H species (Scheme 3).35 Although the β-hydride elimination is undesired in transition metal-catalyzed coupling reactions,36 in the proposed reaction mechanism, a chain-walking process was key to generate thermodynamically more stable species, thus contributing to the high regio- and chemoselectivity of the targeted insertion reactions.37 For carboxylation reactions with CO2, the Martin group showed temperature-controlled site-selectivity affording linear and branched carboxylated products (l![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :b ratios). The authors suggested a Curtin–Hammett scenario, where the reaction proceeded through common intermediates or transition states under fast equilibrium (Scheme 3a). More strikingly, the chain-walking mechanism was translated to a useful method starting from a mixture of alkyl bromides – expanding the utility of the protocol significantly. Regardless of regioisomers, linear alkanes were smoothly converted to carboxylated products under a bromination/carboxylation reaction sequence (1 atm of CO2). The iterative reversible β-hydride elimination/insertion reactions occurred, converging regioisomers of alkyl bromides into a single carboxylated product (Scheme 3b).
:b ratios). The authors suggested a Curtin–Hammett scenario, where the reaction proceeded through common intermediates or transition states under fast equilibrium (Scheme 3a). More strikingly, the chain-walking mechanism was translated to a useful method starting from a mixture of alkyl bromides – expanding the utility of the protocol significantly. Regardless of regioisomers, linear alkanes were smoothly converted to carboxylated products under a bromination/carboxylation reaction sequence (1 atm of CO2). The iterative reversible β-hydride elimination/insertion reactions occurred, converging regioisomers of alkyl bromides into a single carboxylated product (Scheme 3b).
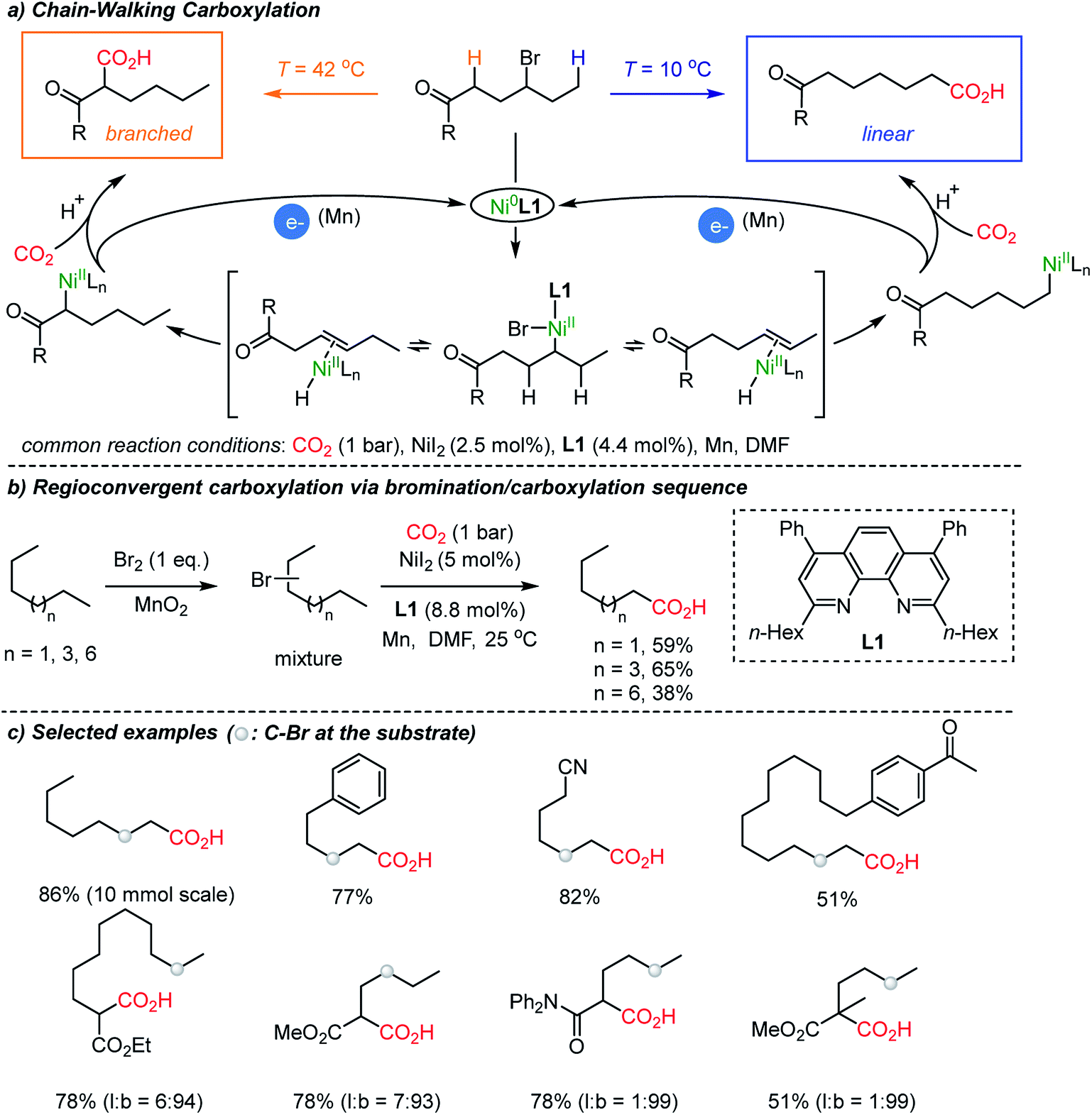 | ||
| Scheme 3 Chain-walking carboxylation of halogenated hydrocarbons. | ||
The proposed chain-walking process with high site-selectivity represents a significant potential toward fatty-acid syntheses from bulk petroleum raw materials. In this context, the same group extended the methodology with olefin substrates, enabling carboxylation reactions in the presence of water as a proton source.34b In the case of alkenes, water served as a way to access metal-hydride species,38 namely Ni–H species, which in turn can participate in the above-mentioned chain-walking mechanism. Indeed, a linear carboxylic acid was the main product with high selectivity (b:l = 1:99) even from an unrefined mixture of olefin isomers (Scheme 4a). As for alkynes, however, only a branched carboxylation product was obtained (Scheme 4b). The authors proposed that the Ni-L2 complex favored the formation of a thermodynamically more stable α,β-unsaturated nickelalactone (Ni-1) with internal alkynes in a CO2 environment. Therefore, a branched carboxylic acid was obtained with high selectivity (b:l = 99:1) after reduction with H2 and Pd/C. The ‘uni-directional’ chain-walking mechanism highlights the potential application of this process in producing added value chemicals from CO2 and crude industrial feedstock.
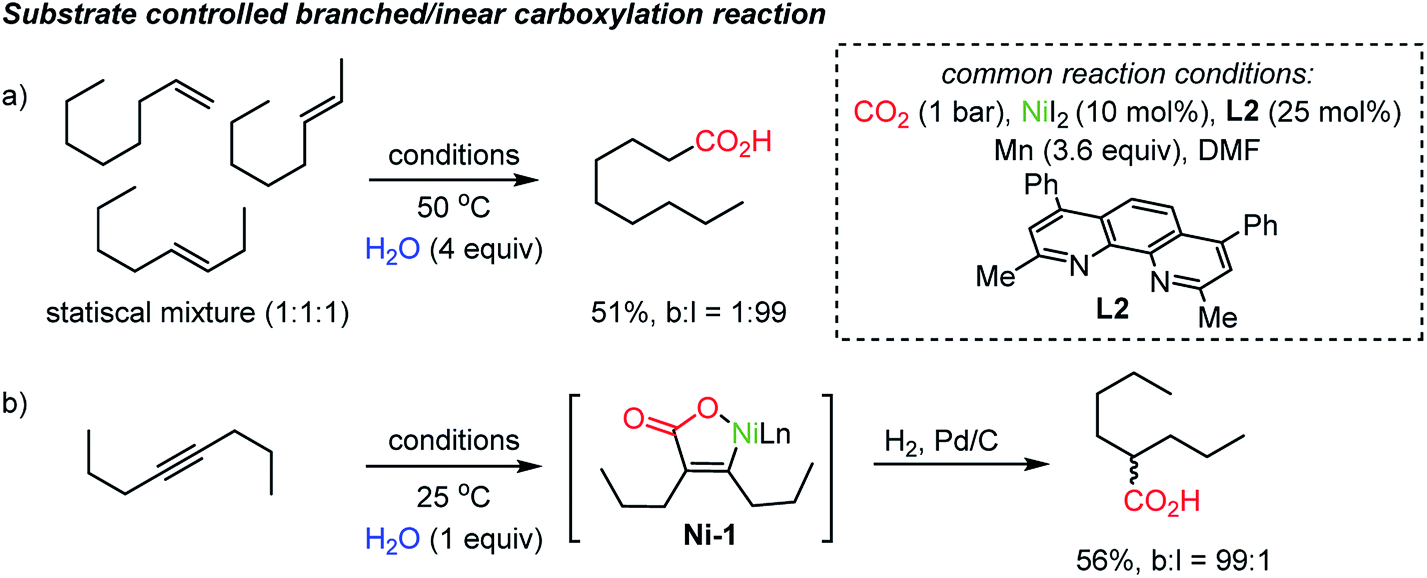 | ||
| Scheme 4 Site-selective carboxylation dictated by the degree of unsaturation. | ||
It is noteworthy that the variation of the ligand is critical in Ni-catalyzed reactions. The substituent adjacent to the nitrogen atoms in bidentate ligands (L1 and L2), such as bipyridine and phenanthroline, differentiates the site-selectivity of the carboxylation reaction. High site-selectivity is a pre-requisite for many organic transformations, for example in allylic substitution reactions. Catalytic metal–ligand complexes govern chemo-, regio- and even enantioselectivity.39 Allyl alcohol is a substrate class with high accessibility yet low chemical utility for allylation reactions due to the apparently low leaving group ability of the hydroxide. It has been proved that in situ activation of allylic alcohol with ‘activating reagents’ can mediate various types of transformation,40 shortening the synthetic steps avoiding the preparation of activated substrates41 (like amines,41a ammonium salts,41b carbamates,41c carbonates,41d esters,41e ethers,41f nitro compounds,41g phosphates,41h and sulfones41i). For example, CO2 was involved in the asymmetric Pd-catalyzed direct α-allylation of ketones.40a The use of CO2 as a catalyst is noticeable although only a ‘catalytic’-amount of it would be necessary for the process.
The Martin group employed CO2 as an activating reagent as well as a C1 source for the carboxylation of allylic alcohols to afford β,γ-unsaturated carboxylic acids (Scheme 5a).33 Once again, ligand-controlled selectivity was observed starting from linear or branched allylic alcohols affording high yields of linear and branched carboxylation products (Scheme 5b). The former resulted from CO2 insertion between the α-carbon and Ni(I) center. Alternatively, α-branched acids were obtained when the tridentate ligand L3 was employed. The critical role of the ligands was rationalized by stoichiometric studies of active NiL2 or NiL3 species in the absence of Zn metal (yields: linear, 0%; branched 73%). The transition-state, Ni-2, was proposed for the nucleophilic attack from the γ-carbon of an η1-allyl Ni(II) intermediate to CO2. Also, a six-membered cyclic conformation can be suggested, similar to the reported nucleophilic addition of Pd-(π)allyl intermediates to CO2 or carbonyl substrates.42,43 The utility of the reaction was further verified by producing useful intermediates for the synthesis of γ-lactone-based bioactive compounds.44
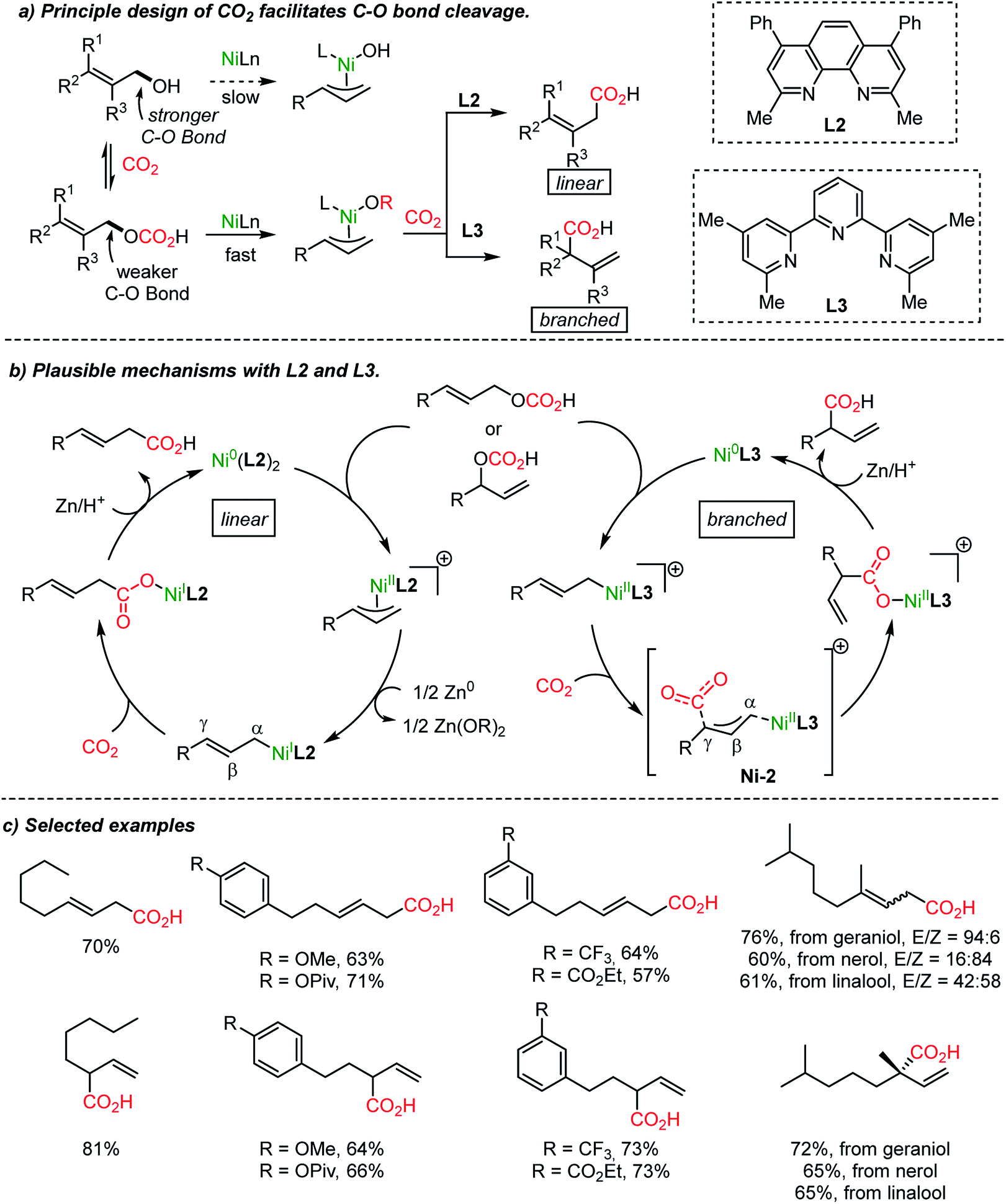 | ||
| Scheme 5 Site-selective catalytic carboxylation of allylic alcohols. | ||
Dienes, abundant and accessible chemical feedstocks, have the same oxidation states as allylic alcohols. However, activation of dienes and conjugated olefins poses a great challenge. Recently, Ni-based catalysts were evaluated for a catalytic carboxylation reaction of dienes toward carboxylated or dicarboxylated products in stoichiometric amounts of a Ni(0) complex.45 Although limited only to activated substrates, alkynes46 and silylallenes47 were transformed to the desired dicarboxylated products. The Martin group successfully implemented a catalytic dicarboxylation reaction for 1,3-dienes with high site-selectivity (up to 90%), to furnish diesters (Scheme 6a).34c Various functional groups were tolerated including heterocycles, organotin, nitrile, and esters. Structurally simple dienes such as butadiene, isoprene, and piperylene – major byproducts of steam cracking in ethylene production plants – were converted to the corresponding terminal diacids with excellent site-selectivity in moderate yields (up to 65% yield, 99:1 selectivity, Scheme 6b). Single crystal structure analysis determined the formation of monocarboxylated η3-Ni nickelalactone (Ni-3). The corresponding dicarboxylation product could be obtained when Ni-3 was treated with CO2 under optimized reaction conditions (also see Mori group's work45c), shedding some light on the reaction mechanism (Scheme 6c).
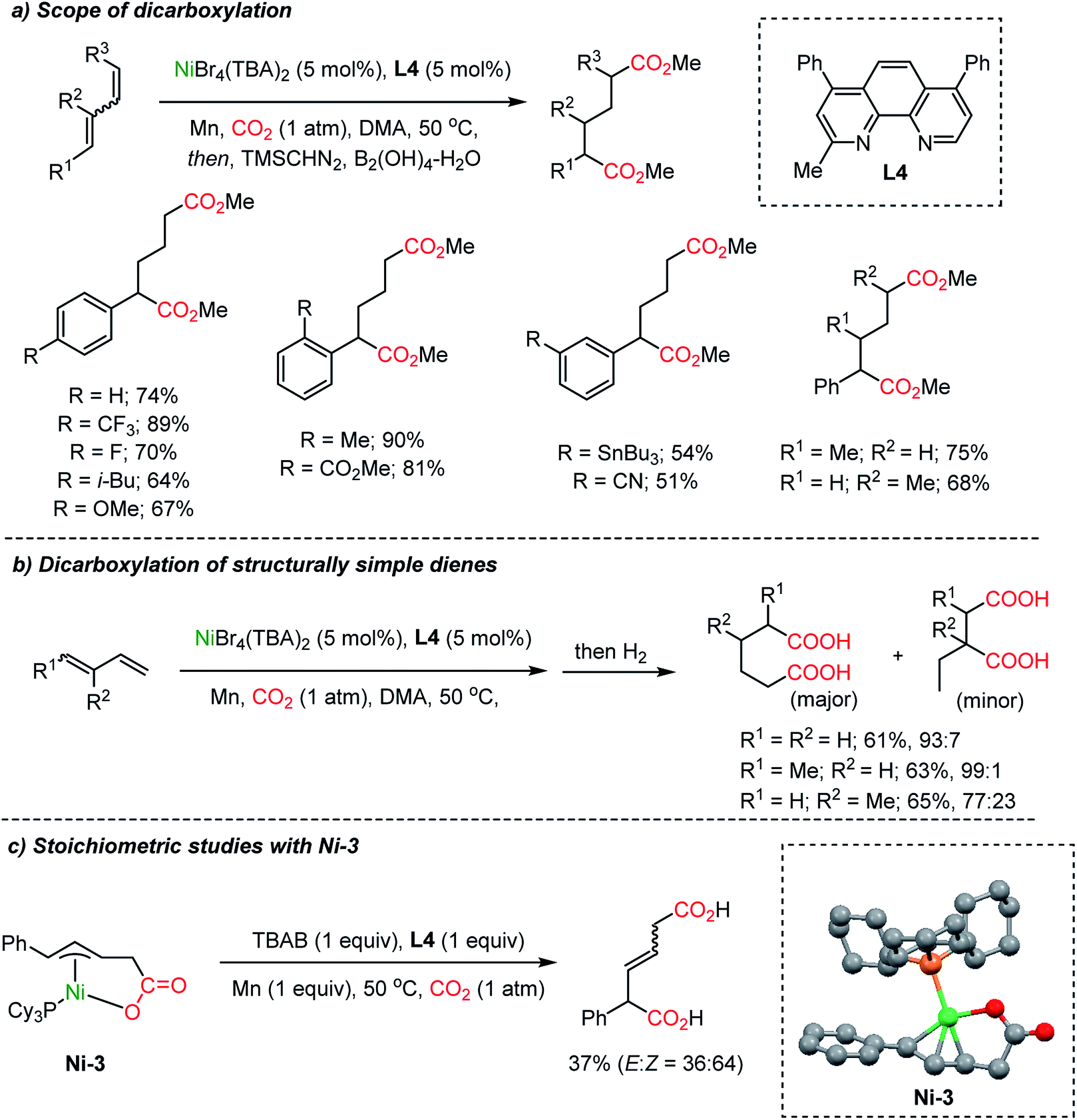 | ||
| Scheme 6 Ni-catalyzed dicarboxylation of 1,3-dienes and a mechanistic study. | ||
The transition metal-catalyzed carboxylation reactions of the above-mentioned recent examples showed unprecedented catalytic performances with a variety of substrates, yet they require stoichiometric reducing reagents to sustain the catalytic cycle. Certain improvements have been attempted by utilizing insoluble reducing reagents (Mn, and Zn powder) replacing highly reactive RMgX, Et2Zn, or AlMe3. In an ideal CO2 functionalization process, a redox-neutral mechanism would be more desirable,48 where no additional oxidants or reductants are required. In this context, the next two section will describe reactions utilizing photocatalysts, demonstrating sustainable light-induced chemical reduction reactions, mimicking photosynthesis.
3. Photocatalytic carboxylation with CO2
Photosynthesis is the master process in the realm of CO2-functionalization, as it is called CO2-fixation. This ideal process operates via multi-step electron transfer and chemical transformation reactions,49 resulting in somewhat limited CO2-fixation efficiency, constraining the capacity of nature's carbon cycle (Fig. 1 highlighted in green).50 Recent efforts in enzymatic engineering in chemical biology for in vitro CO2 fixation51 would potentially lead to enhanced photosynthesis.52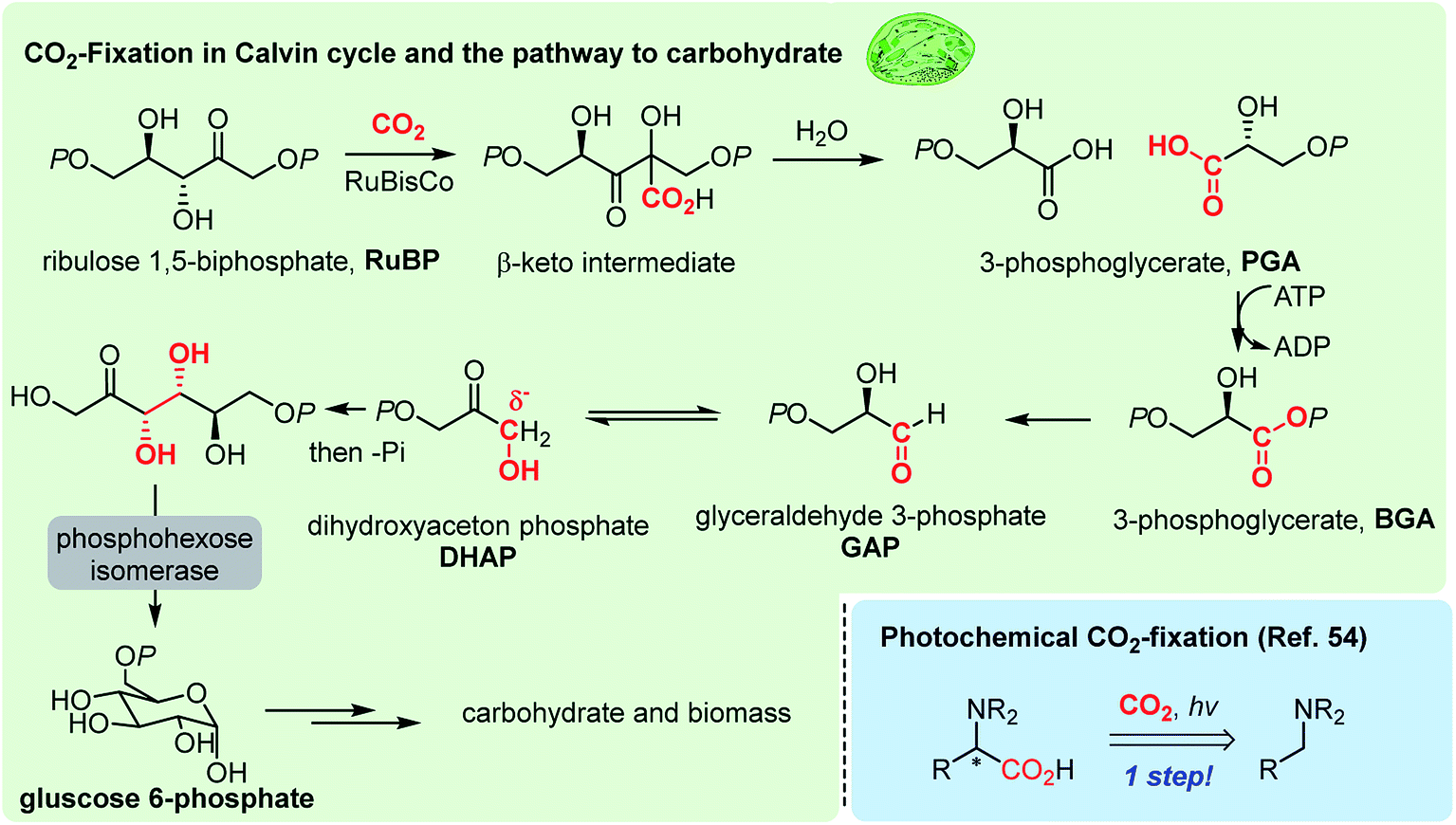 | ||
| Fig. 1 Natural photosynthesis and an example of artificial photosynthesis. | ||
Solar energy obviously represents one of the most promising and limitless energy sources, which can be harnessed using a photosensitizer. In the late 1970s, seminal studies were reported regarding photocatalytic CO2 reduction by the Tazuke, Fujishima, Honda, and Lehn groups,53 which formed the basis of the modern photoredox activation of CO2. Further developments in photocatalysts played a significant role in CO2 reduction reactions mainly targeting industrial feedstock molecules, such as carbon monoxide, methanol, methane and formic acid.8 In this regard, artificial CO2 functionalization reactions have shown elegant modes of action in C–C bond formation reactions.22 For example, a photochemical CO2-fixation provided α-amino acid derivatives in a one-step reaction (Fig. 1 bottom right54). The key to the success of this field will be to maintain mild reaction conditions to conserve complex molecular structures of products, while providing appropriate reduction potential for the reductive CO2-functionalization.
The Iwasawa group demonstrated a dual catalytic system with a Pd/Ir-photocouple for carboxylation reactions of aryl halides in the absence of metallic reducing reagents (Scheme 7).55 Hünig's base (3 eq.) served as a sacrificial electron donor in photoredox cycles, generating Pd(0)-complexes in the proposed catalytic carboxylation cycles (Scheme 10b). Although Ar–Pd(II)–Br(XPhos) possesses a high reduction potential (−2.28 V, vs. Fc/Fc+), a new peak at −1.4 V was observed from cyclic voltammetry (CV) measurements. The coordination of CO2 on Pd might influence the redox chemistry of the metal complex, therefore reducing the required reduction potential. In addition to the common insertion of CO2 into the active Pd(II)–C bond, the authors suggested the formation of two intermediates, a Pd(I)- or Pd(II)–CO2 complex (Scheme 7b, path b). After methylation with TMSCHN2, various carboxylic acid esters were obtained including a sterically hindered acid (i.e. 2,4,5-triisopropyl carboxylic acid methyl ester).
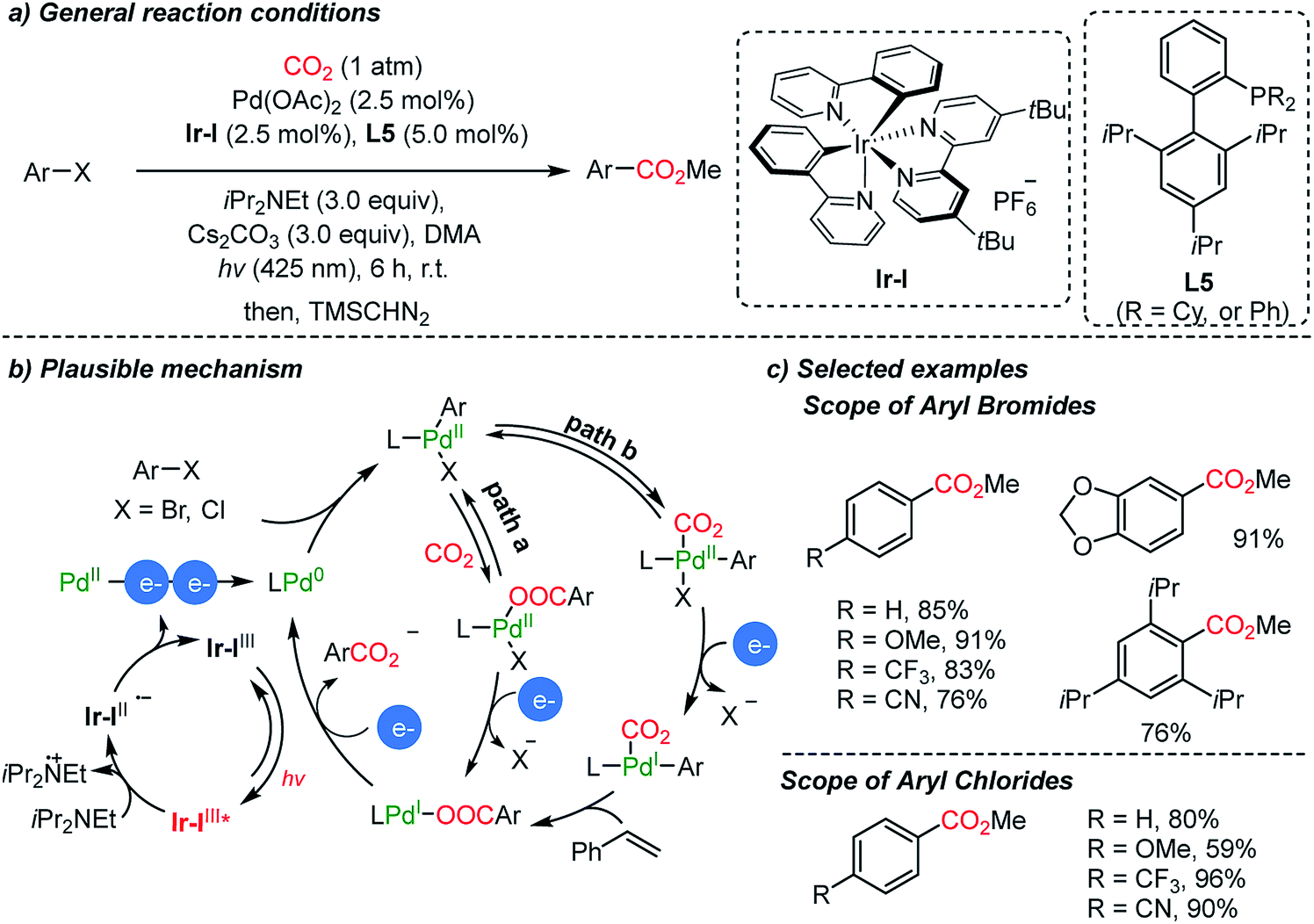 | ||
| Scheme 7 Carboxylation of aryl halides by Pd/Ir dual catalysis. | ||
Starting from simple feedstocks, Ar–Br and alkyl–Br, the König group reported visible light-induced carboxylation mediated by nickel catalysts (Scheme 8).56 The plausible reaction mechanism could be divided into two distinct catalytic cycles. The first one involved a one electron delivery to a Ni(II) or Ni(I) complex from the anion radical (4CzIPN˙−). Hantzsch ester (HEH, 2 equiv. required) was used as a terminal reducing agent in the presence of a reducing excited sensitizer (4Czlpn*) and light (left circle, Scheme 8b). Second, the oxidative addition to a Ni(0) complex was suggested, which undergoes reduction and then an insertion reaction with CO2 (right circle, Scheme 8b). The catalytically active Ni(0) species can be regenerated from Ni(I) with electron sources produced from the left circle.
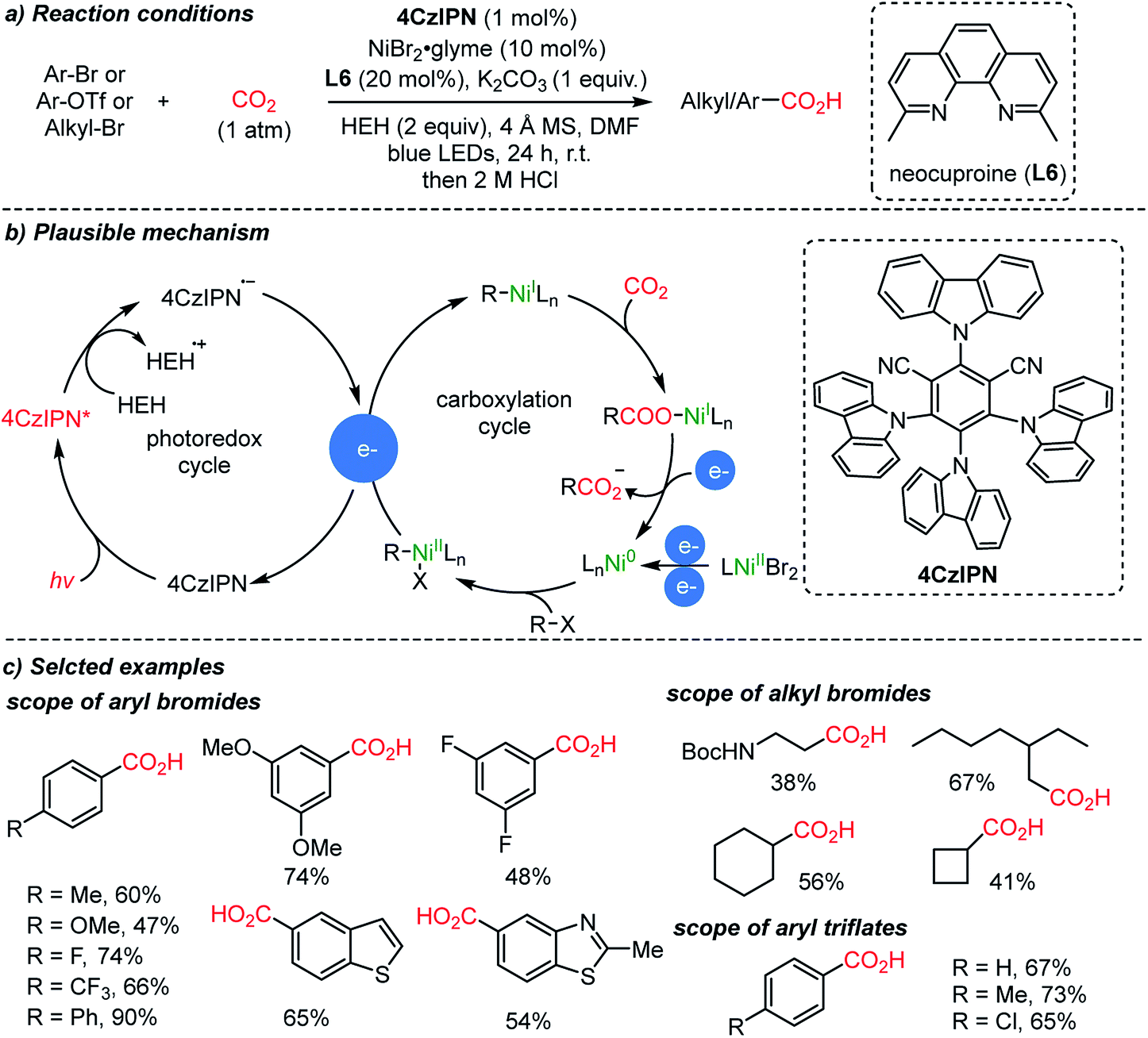 | ||
| Scheme 8 Photoredox cycles and carboxylation cycles in a co-catalysis system. | ||
The same group expanded the dual catalysis strategy to the carboxylation of styrenes,57 affording Markovnikov (branched) or anti-Markovnikov (linear) products selectively controlled by the choice of the ligand (Scheme 9). The suggested reaction mechanism explained that the observed chemoselectivity (branched/linear) was controlled by the different ligands (L6, neocuproine and L7, dppb). According to DFT calculations, Ni(0) species with the more sterically demanding ligand dppb (L7) tend to coordinate with CO2, forming a 5-membered nickelalactone (Ni-4) with styrene. With the less hindered neocuproine ligand (L6), the reaction proceeds via hydrometalation of styrene to afford Ni(II), which is subsequently reduced by the catalytic action of the photosensitizer (4CzIPN, Scheme 9b). The electrons generated from the photocatalytic cycle are used to reduce Ni(II) or Ni(I) to Ni(0), which can diverge to the hydrometalation step (left) and CO2 activation step (right) to generate branched and linear products respectively while completing the catalytic cycles.
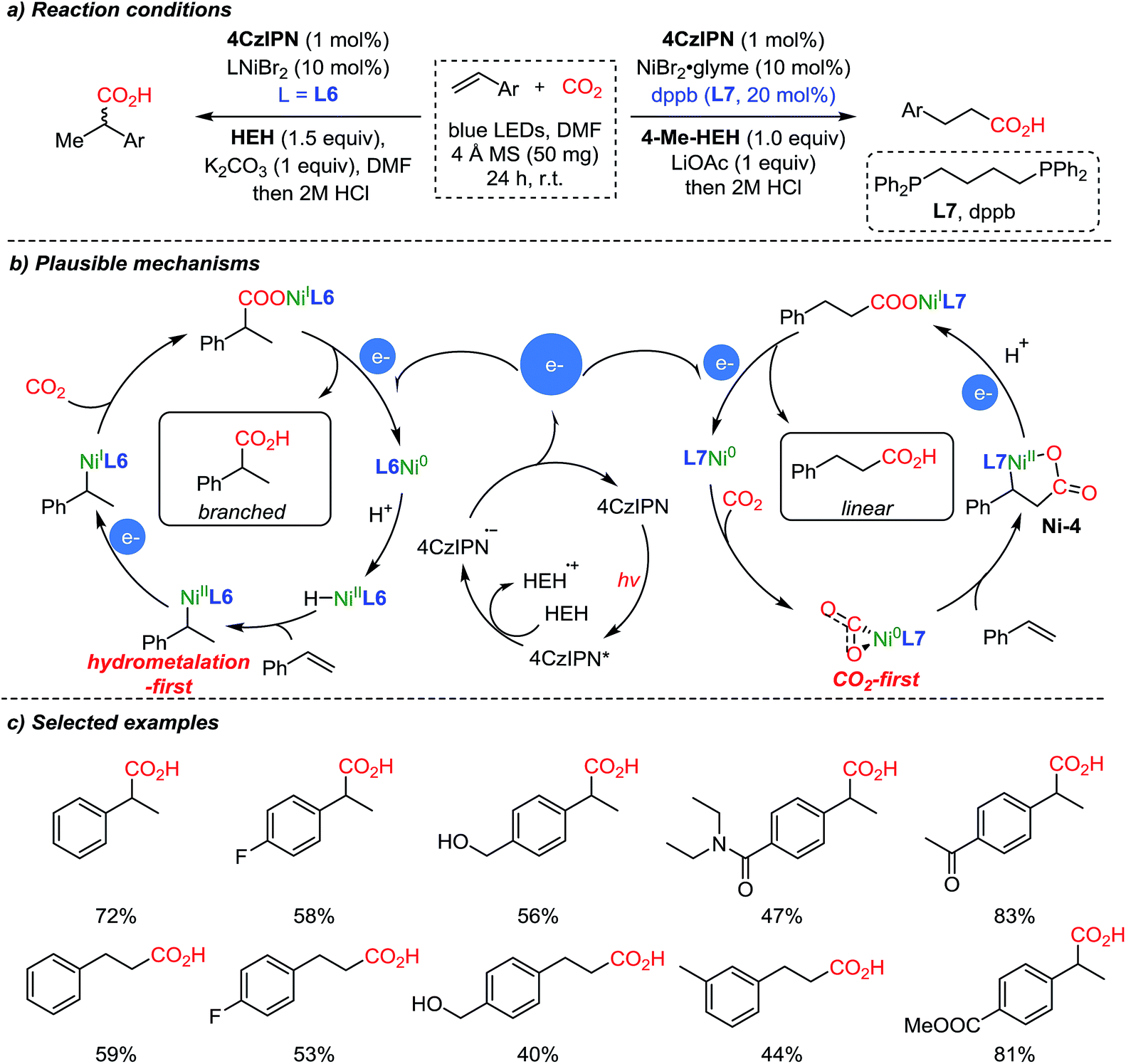 | ||
| Scheme 9 Site-selective photocatalytic carboxylation controlled by ligands. | ||
The Jamison group reported styrene functionalization reactions in a CO2 atmosphere to generate β-aryl carboxylic acids (Scheme 10).58 In this case PMP (1,2,2,6,6-pentamethylpiperidine) was employed as a sacrificial organic electron donor, while utilizing water as an additive under modified reaction conditions. Although it is unclear, the addition of water induced high selectivity toward the mono-carboxylated product compared to other tested hydride or proton donors. The suggested reaction mechanism shows that the carboxylation with CO2˙− results in the formation of a stabilized benzylic radical (E0 = from −1.82 to −0.71 V vs. SCE). Therefore, further reduction is feasible leading to the generation of carboxylated benzylic anion species, which could be protonated upon addition of water.
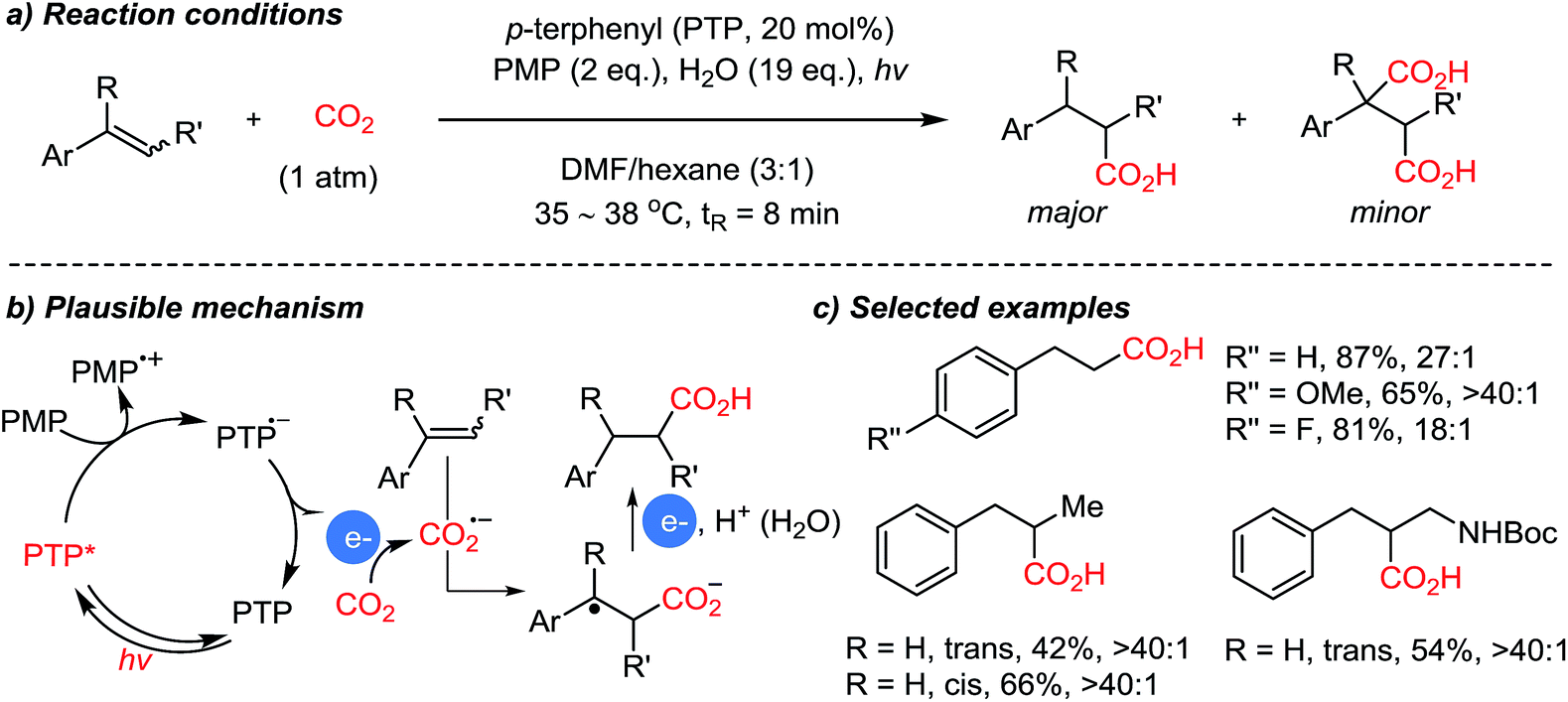 | ||
| Scheme 10 Photocatalytic direct β-selective hydrocarboxylation of styrenes. | ||
Photoactivation of organic substrates has been a successful transformation with high chemoselectivity to produce Markovnikov (branched) or anti-Markovnikov (linear) carboxylic acids. Also, the Murakami group reported a carboxylation reaction with α-alkyl ketones and CO2via a Norrish type II activation mechanism.59 The carboxylation reactions at toluenyl carbon were also conducted in natural sunlight at ambient temperature with good isolated yields of the desired products. The authors suggested an energetically feasible [4 + 2]cycloaddition reaction by DFT calculations, which was determined by the thermal reaction of benzocyclobutenol to generate an o-quinodimethane intermediate.
Recently, Hou et al. reported carboxylation reactions of internal and terminal alkynes promoted by Co/Ir dual catalysis (Scheme 11).60 The authors proposed that the reaction proceeded via functionalization of alkynes to generate an (E)-Co–CO2 complex which is an intermediate for various products – carboxylic acids, pyrones, α,β-unsaturated γ-lactones, coumarins, and 2-quinolones, by sharing a common intermediate, (E)-int (Scheme 11a). Pyrones were formed through a formal [2 + 2+ 2] cycloaddition with terminal alkynes (R1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H). In the case of internal alkynes, pharmaceutically vital heterocycles such as coumarins and 2-quinolones were obtained with high selectivity. The suggested mechanism proceeded via intramolecular cyclization of acrylic acid intermediates. The E/Z isomerization of acrylic acid was confirmed by control experiments with (or without) the Ir-photoredox catalyst under irradiation (or in the dark). Also, this newly developed carboxylation/acyl-migration cascade reaction is feasible for alkyne difunctionalization, highlighting its utility in the field of light-driven CO2-fixation.
H). In the case of internal alkynes, pharmaceutically vital heterocycles such as coumarins and 2-quinolones were obtained with high selectivity. The suggested mechanism proceeded via intramolecular cyclization of acrylic acid intermediates. The E/Z isomerization of acrylic acid was confirmed by control experiments with (or without) the Ir-photoredox catalyst under irradiation (or in the dark). Also, this newly developed carboxylation/acyl-migration cascade reaction is feasible for alkyne difunctionalization, highlighting its utility in the field of light-driven CO2-fixation.
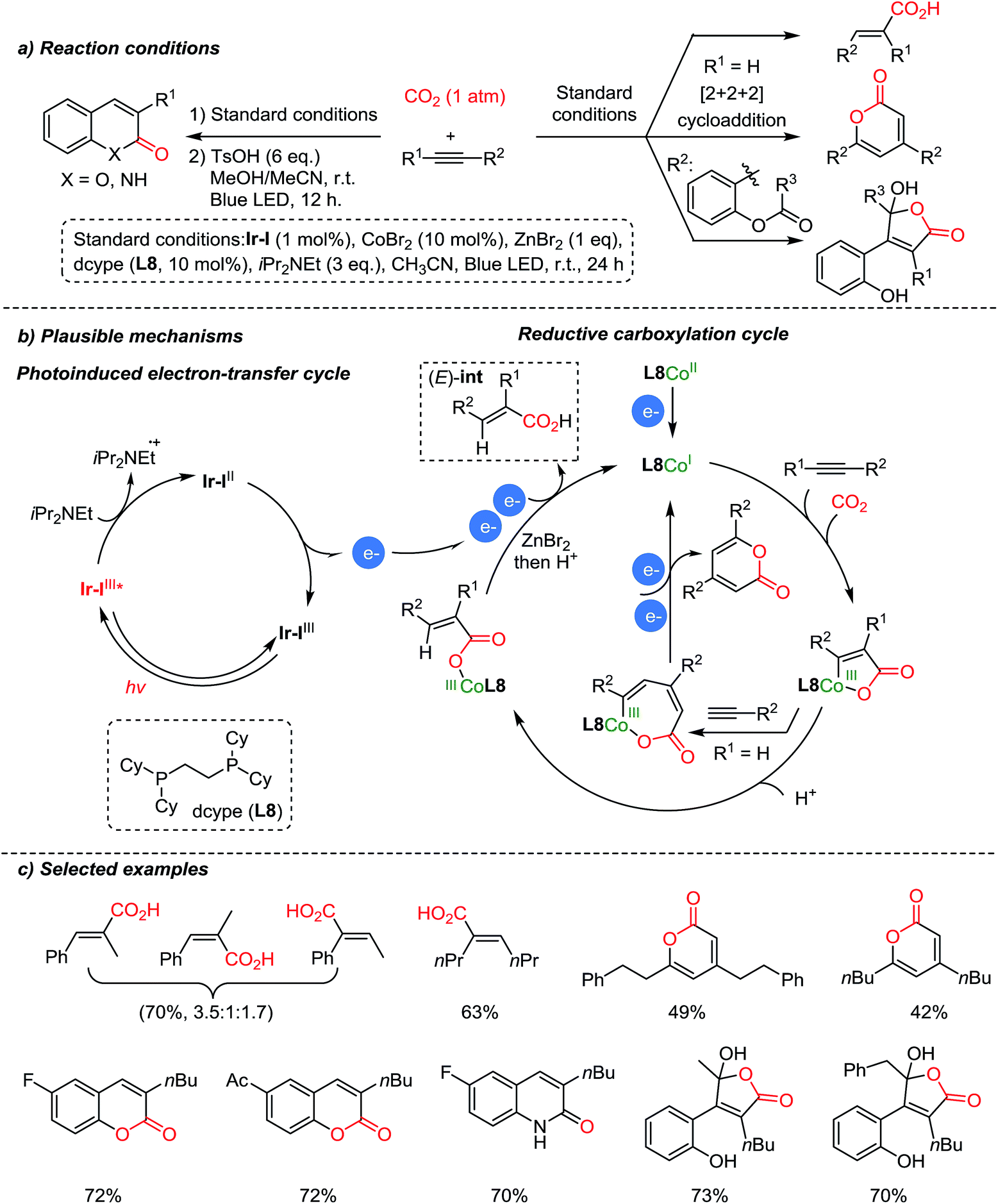 | ||
| Scheme 11 Carboxylation of alkynes by Co/Ir dual catalysis. | ||
The Yu group reported the photocatalytic hydrocarboxylation of enamides and imines to afford α-amino acids with excellent chemo- and regio-selectivity (Scheme 12).61 The pre-equilibrium of enamides and imines was combined with photocatalytic reduction. Despite the inherent nucleophilicity of enamides, kinetic studies indicated that the imines underwent the desired hydrocarboxylation faster than the competitive β-carboxylation reaction. The authors proposed an umpolung reaction of the α-amino carbanion under metal-free conditions. The carboxylated products were obtained with a broad substrate scope regardless of the electronic and steric properties of substituents. In addition, the enamide and imine starting materials were equally effective, confirming the fast pre-equilibrium before the reduction/carboxylation steps.
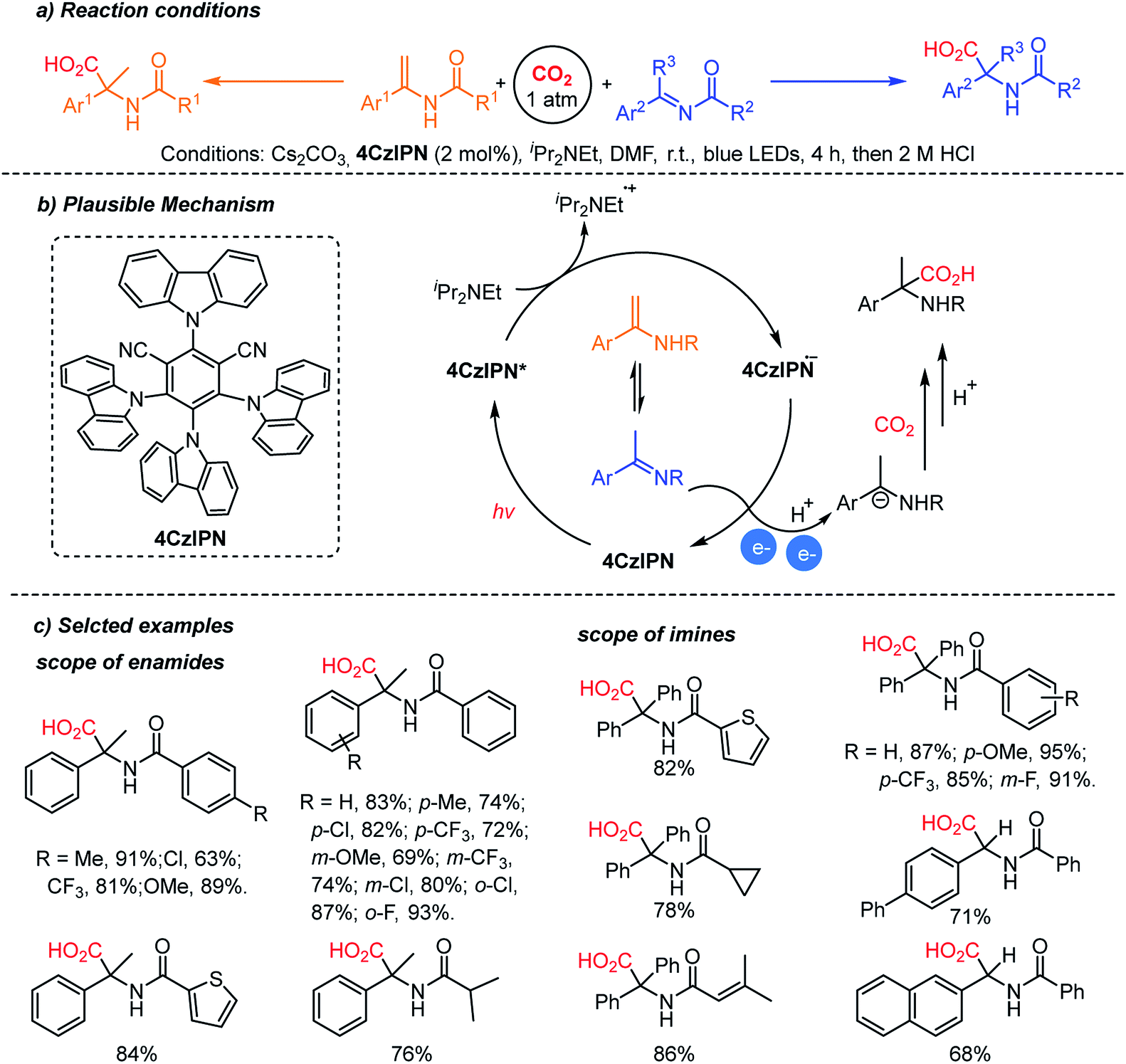 | ||
| Scheme 12 Photocatalytic hydrocarboxylation of enamides and imines. | ||
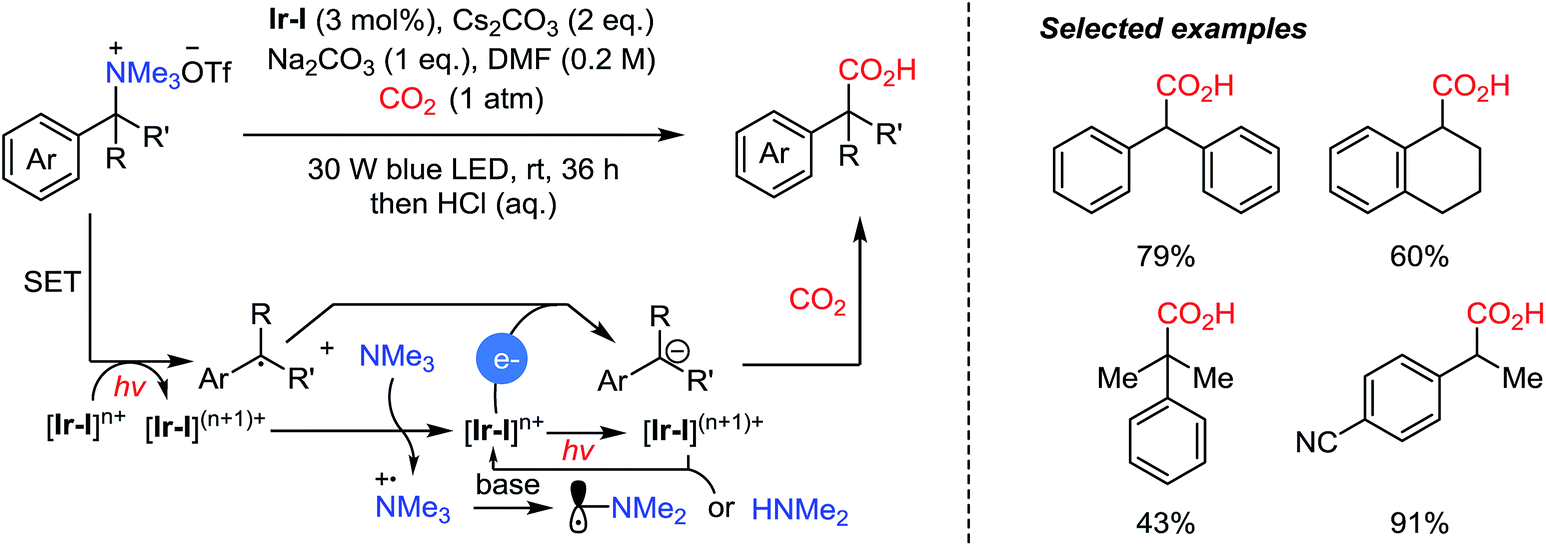
Very recently, the Walsh group presented photocatalytic carboxylation of benzophenone-derived ketimines by employing an Ir-complex (Ir-I) under mild conditions (Scheme 13).62 The radical anion was generated by single electron transfer (SET) from [Ir-I]* to ketimines, which was facilitated by the coordination between the imine and Cy2MeN˙+.63 Spin density calculation was carried out to evaluate the radical anions (A, B) suggesting that the carbon atom was more negatively charged than the nitrogen atom (spin density, radical probability on C: 0.05–0.18 and N: 0.37).64 Subsequently, the more reactive N-centered radical species abstracts a hydrogen atom from Cy2MeN˙e to form an α-amino carbanion and an iminium cation [Cy2NCH2]+via an umpolung reactivity (Scheme 13b). The carbanion then undergoes nucleophilic addition to CO2 affording the desired carboxylation product. The obtained α,α-disubstituted α-amino acid shows potential application of the protocol in asymmetric synthesis to generate quaternary stereogenic centers, which are often difficult to control.65
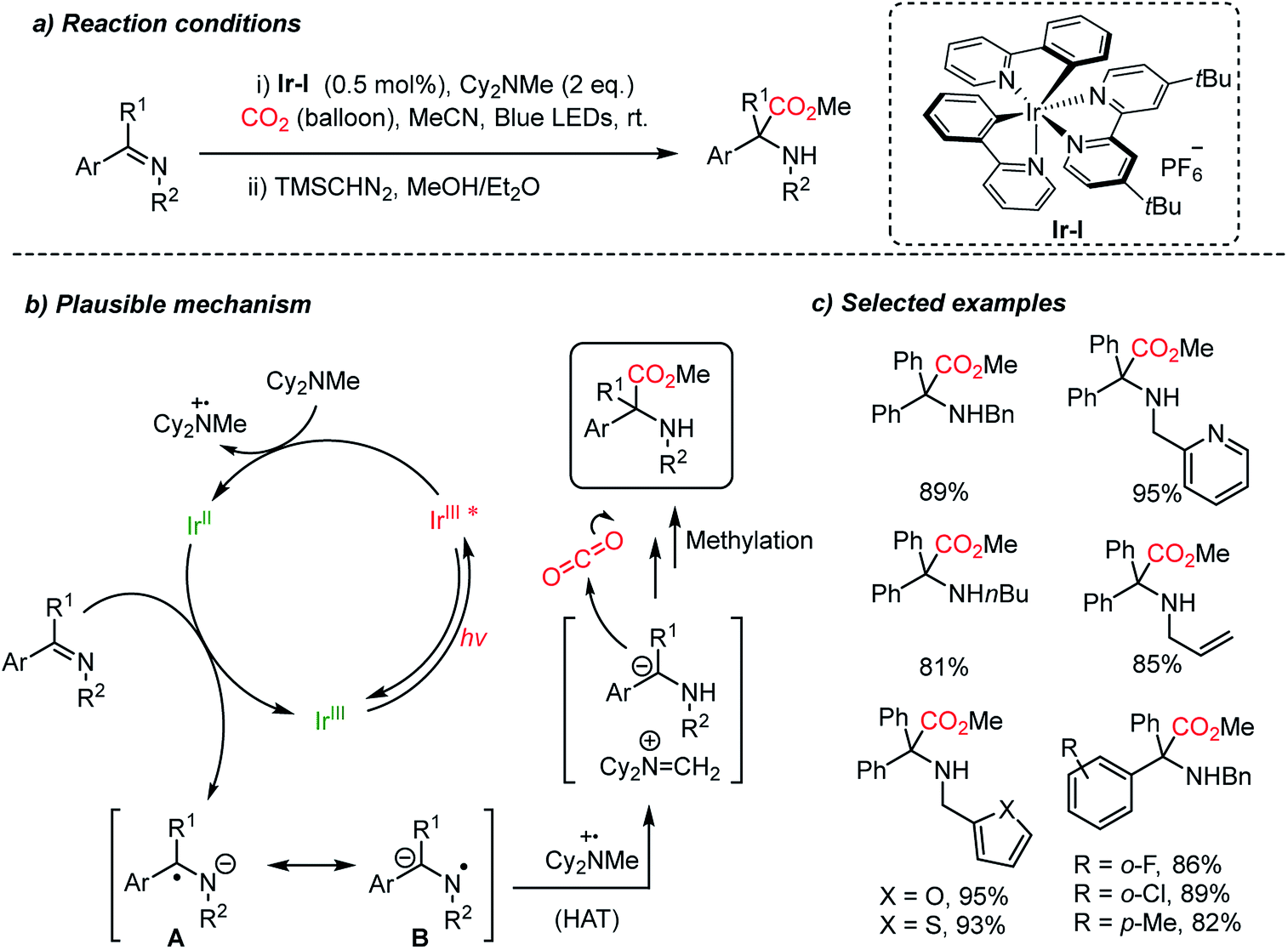 | ||
| Scheme 13 Photocatalytic carboxylation of ketimines. | ||
Direct carboxylation of imines and amines with CO2 represents a very promising pathway to afford α-amino acids, especially those promoted by photoredox catalysts as shown above (Schemes 12, 13 and 17). Compared to tertiary amines, however, α-functionalization (i.e. α-carboxylation) of primary amines still remains a great challenge due to the lower reactivity of the α-C–H bond. Besides carboxylation reactions, CO2 has been used as an activating group,33,40 a directing group66 and a protecting group67 in organic synthesis. Ye et al. recently reported the photocatalytic α-alkylation of primary amines to yield γ-lactams with CO2 as a temporary activator and as a protecting group (Scheme 14).68 Various α,β-unsaturated esters were tolerated in the presence of an Ir-II photosensitizer. Quinuclidine was employed as a sacrificial electron donor. According to the suggested reaction mechanism, CO2 was regenerated after releasing lactam products via an intramolecular cyclization reaction. The in situ carbamate formation reaction suppressed the reactivity of primary amines while increasing the reactivity of α-C–H bonds according to the computational studies. The generation of the α-radical of the substrate is highly intriguing due to the potential applications toward various electrophiles and radical–radical coupling reactions. Furthermore, the use of tertiary amines as a base will enable a potential asymmetric catalysis to afford enantioenriched products.
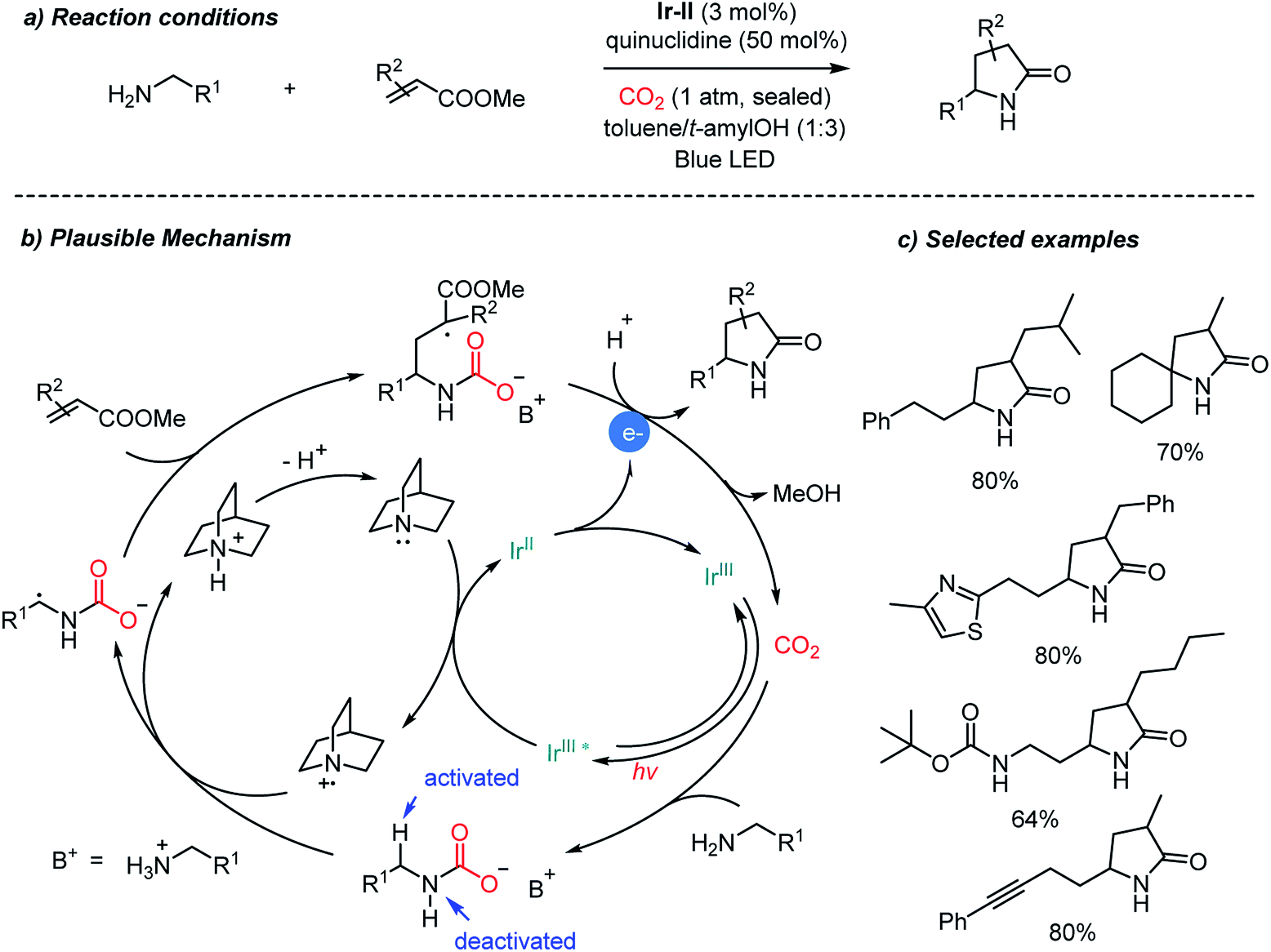 | ||
| Scheme 14 Catalytic application of CO2 for photocatalytic α-alkylation of primary amines. | ||
This section summarizes recent progress in photo-CO2-functionalization without strong metallic reducing agents. Instead, an organic sacrificial electron source or a reducing reagent was employed (i.e. triethylamine, piperidine, Hünig's base and Hantzsch esters) in the presence of photocatalysts with an appropriate reduction potential to complete the catalytic cycles. Various types of substrates underwent C–CO2 bond formation reactions to provide unique molecular structures under ambient photosynthetic conditions (low CO2 pressure, and accessible light sources). However, there is still plenty of room to develop more elegant methodologies in terms of sustainability. The next section will discuss redox-neutral carboxylation without external reductants.
4. Recent developments in redox-neutral CO2-functionalization
It is thought that catalytic carboxylation of non-activated organic substrates would be an ideal approach to CO2-utilization, avoiding reactive organometallic reagents (RMgX, RLi, R2Zn, R4Sn, etc.). For example, solar energy provides chemical reduction potential to enable CO2 conversion in the Calvin cycle, where actual CO2-fixation and C–CO2 bond formation reaction occur under mild conditions via an α-ketol rearrangement (Fig. 2).50,69 This “enantioselective” CO2-fixation process generates a new C–C bond while creating additional stereogenic center(s) via a redox-neutral pathway. Accordingly, recent progress in photo-redox catalysis offers a promising platform to develop sustainable CO2 utilization reactions under mild conditions in the absence of additional reducing reagents.54,70 When combined with practicability and scalability, redox-neutral CO2-functionalization strategies will provide a tangible scenario of sustainable artificial carbon fixation.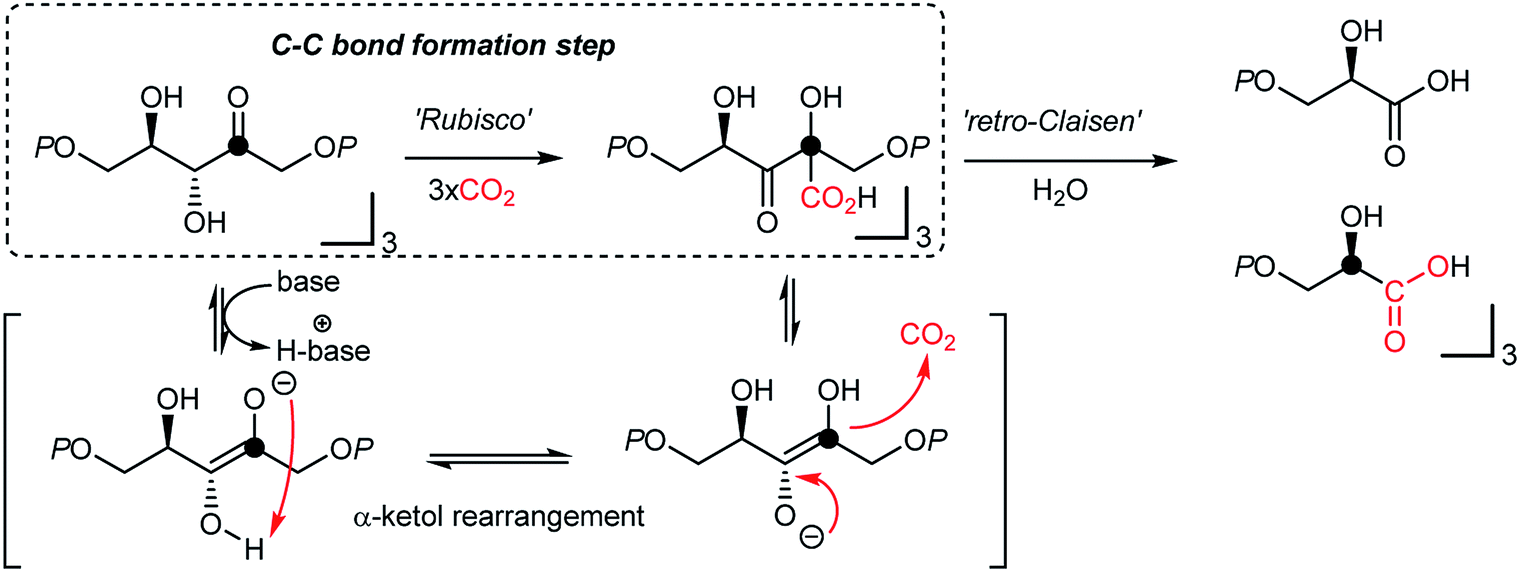 | ||
| Fig. 2 Calvin–Benson–Bassham (CBB) cycle at the RubisCo enzyme reaction center. Note that the C–CO2 H bond formation reaction occurs at the ketone functional group, and the two glyceric acid products are enantiomerically enriched. | ||
The following examples in this section represent their redox-neutral reaction profile in terms of the proposed reaction mechanisms – no terminal reducing or oxidizing reagents. Despite the fact that these reactions require activated substrates or radical initiators or a strong base, the generation of C–CO2 bonds with CO2 is a remarkable step toward truly ideal CO2-functionalization. Keeping in mind that solar energy might be the only and truly sustainable energy source, a few examples of redox-neutral photocatalytic CO2-functionalization reactions are also highlighted in this section.
The Sato group recently reported a direct carboxylation reaction at the allylic C(sp3)–H bond (Scheme 15).71 The use of the AlMe3 – non-nucleophilic base – was ascribed to the initial generation of catalytically active Co(I) species, therefore the catalytic cycle is free from an external reducing reagent. The carboxylation reaction of allylarenes and 1,4-dienes was proven to be effective with a nucleophilic η1-allyl-Co(I) catalyst after intensive screening of transition metal catalysts such as Cr(II), Mn(I), Fe(III), Rh(I), Ir(I), Ni(II) and Cu(I). The role of the ligand was critical; Xantphos (L9) showed high selectivity without the formation of isomerization or methylation byproducts by the use of AlMe3. Various terminal alkenes were smoothly converted to β,γ-unsaturated acids with excellent functional group tolerance, including amides, esters, and ketones. The authors suggested that the presence of the low-valent Co(I)-complex was the key to the successful carboxylation reaction with high selectivity. This protocol expands upon the scope of carboxylation to C(sp3)–H bonds, which represents atom- and step-economic approaches to construct molecular complexity by incorporating CO2.
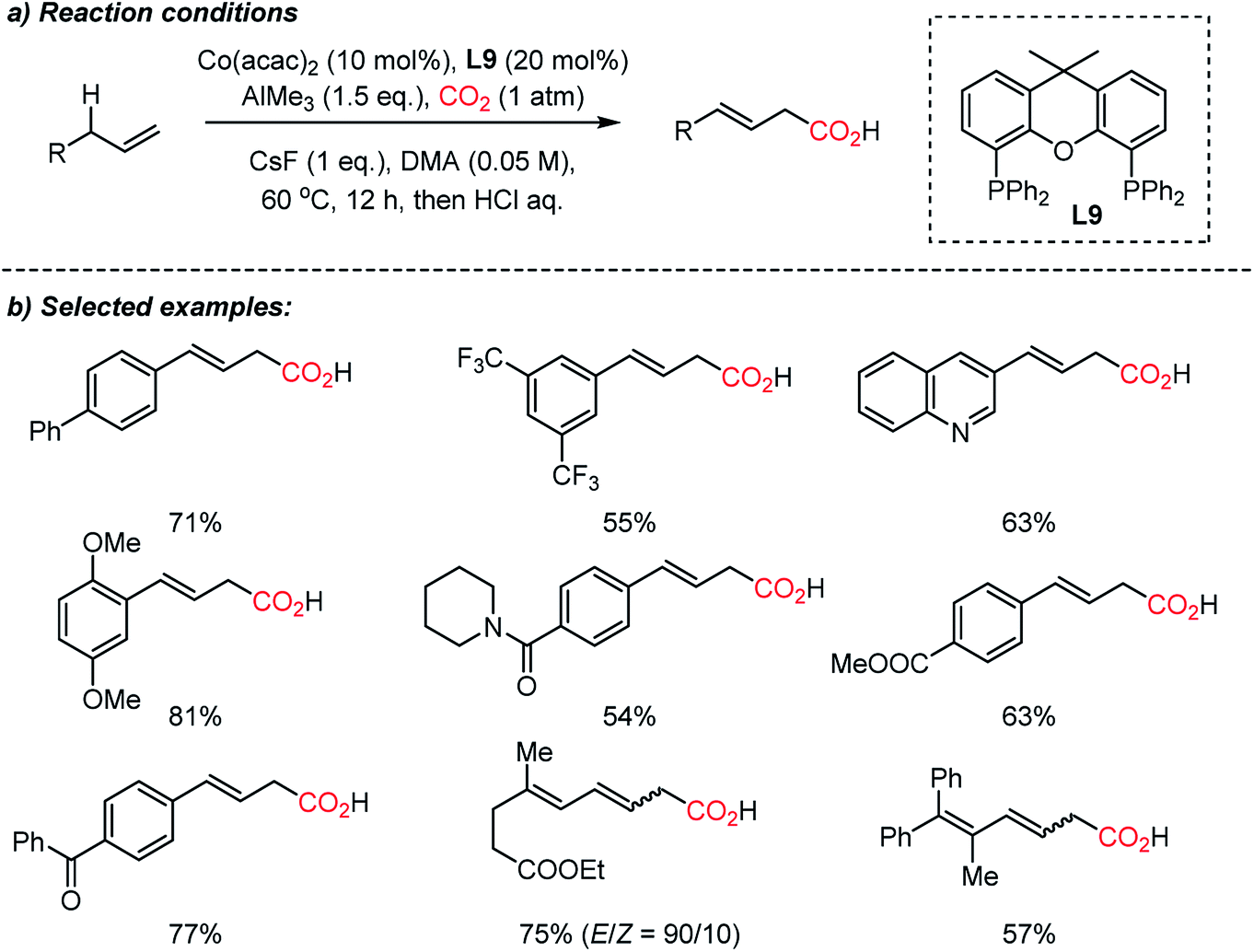 | ||
| Scheme 15 Cobalt-catalyzed direct carboxylation of allylic C(sp3)–H bonds. | ||
Very recently, the Yu group reported photocatalytic carboxylation of tetraalkyl ammonium salts via C–N bond cleavage (Scheme 16).72 Trimethylamine was generated in situ by single-electron transfer (SET) from the excited Ir-I to the substrates. In turn, the resulting active Ir-I species could be reduced by the tertiary amine. Afterwards, carbanions undergo a carboxylation reaction after another SET step between the excited photoredox catalyst and the alkyl radical. The authors suggested that the oxidized trimethylamine was transformed to amine species, like α-radical [Me2NCH2˙], or dimethylamine after hydrolysis. As electron donors, trimethylamine and dimethylamine accounted for 2 equivalents of reducing reagents required to complete the catalytic cycle. This built-in reductant was generated and demonstrated carboxylation reactions without additional reducing reagents, compared to Ni-catalyzed reductive carboxylation of benzylic C–N bonds.31
 | ||
| Scheme 16 Photocatalytic carboxylation with a built-in reductant as the electron donor. | ||
In the above-mentioned cases, organic amines act as sacrificial electron donors, where the resulting radical cation trialkyl amines have dramatically reduced pKa at the α-protons.73 In the presence of a base, a deprotonation reaction would generate an amine with an α-radical, which can couple with other reactive species. The single-electron reduction of CO2 to CO2˙− is in general a rate-determining step due to the high reduction potential (−2.21 V vs. SCE (saturated calomel electrode) in DMF (N,N-dimethylformamide)).74 A viable C–C bond formation reaction with CO2˙− and amine based α-radicals would afford α-amino acids as the product. This was realized by the Jamison group demonstrating a metal-free photoredox conversion of CO2 (Scheme 17).54 An organic sensitizer, p-terphenyl, mediated single electron transfer reactions (reduction potential: −2.63 V vs. SCE in DMF) to perform the suggested one-electron reduction of CO2, providing α-amino acids in the absence of additional reducing reagents. Various aryl-substituted α-amino acids were prepared in good to excellent yields. The convenience of continuous flow chemistry75 was an added benefit of the photocatalysis to provide essential synthetic building blocks from carbon dioxide. The generation of CO2-radical anion is highly attractive, considering its vast application potential in organic synthesis for carboxylation reactions. This photocatalysis mediated by p-terphenyl showed promise toward metal-free CO2-functionalization via a single-electron reduction mechanism in terms of atom-economy (redox-neutral), feasibility (continuous flow setups), and utility of the final products (α-amino acids) containing stereogenic centers.
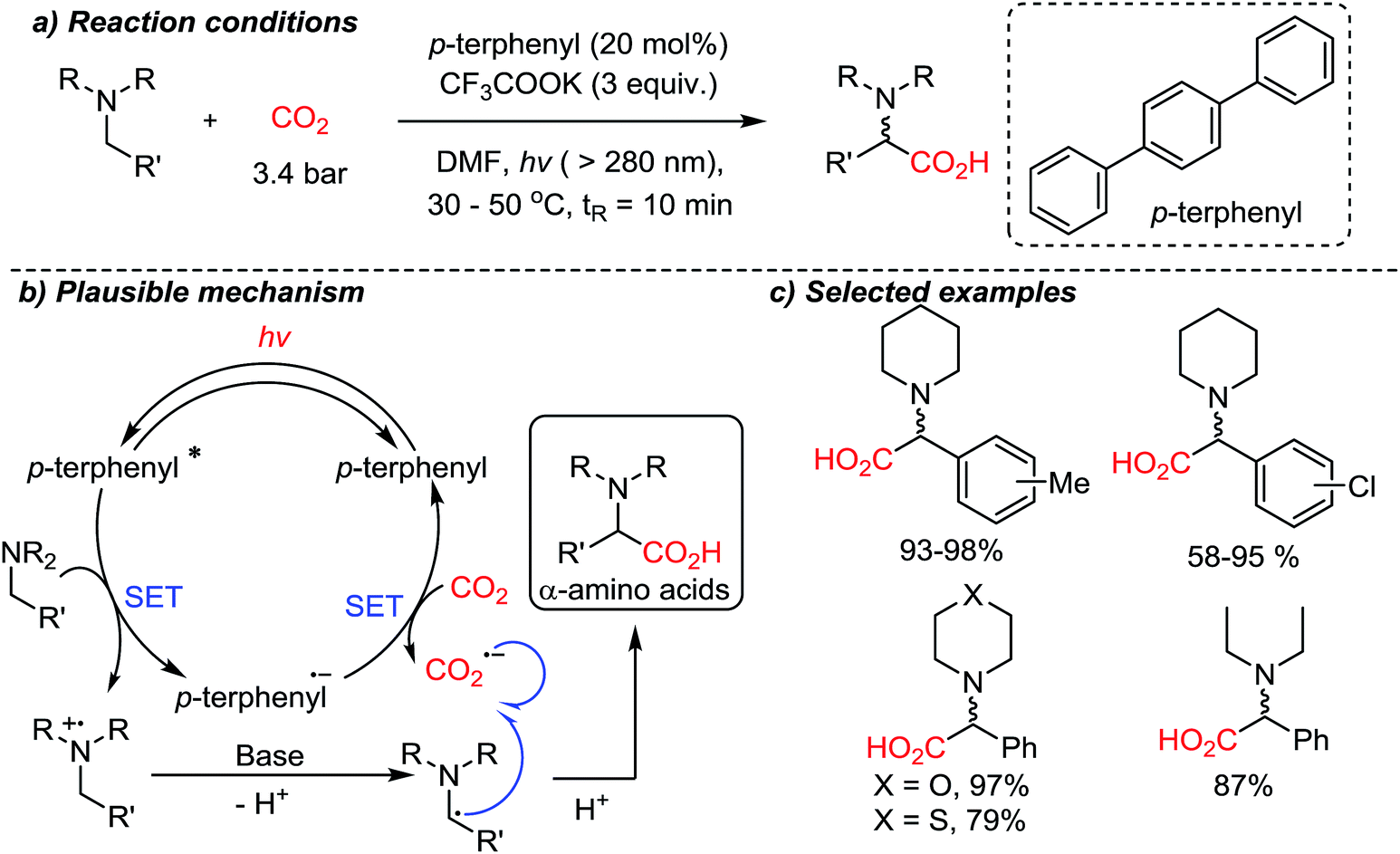 | ||
| Scheme 17 Photoredox CO2-activation to access α-amino acids using a p-terphenyl photosensitizer. | ||
Owing to the recent developments in organic photosynthesis and photosensitizers,22,76 unprecedented reactivity patterns were achieved with CO2 as a C1 source. For example, the Martin group showed photocatalytic dicarbofunctionalization of styrene derivatives initiated by radicals under mild reaction conditions, where stabilized benzyl carbanions react with CO2 (Scheme 18).70a Various radical initiators, such as trifluoro- and difluorosulfonates, and trifluoroborate salts, were proven to be effective under photochemical reaction conditions. The photocatalytic redox cycle was mediated by an Ir-complex (Ir-II). This protocol provides two new C–C bonds with a stereogenic center in the absence of additional stoichiometric reducing reagents. Trisubstituted alkenes were also employed to afford carboxylic acids with a quaternary stereogenic center. The convenient introduction of the (di)trifluoromethyl group highlights potential applications of radical carboxylation reactions in drug discovery and pharmaceutical industry.77
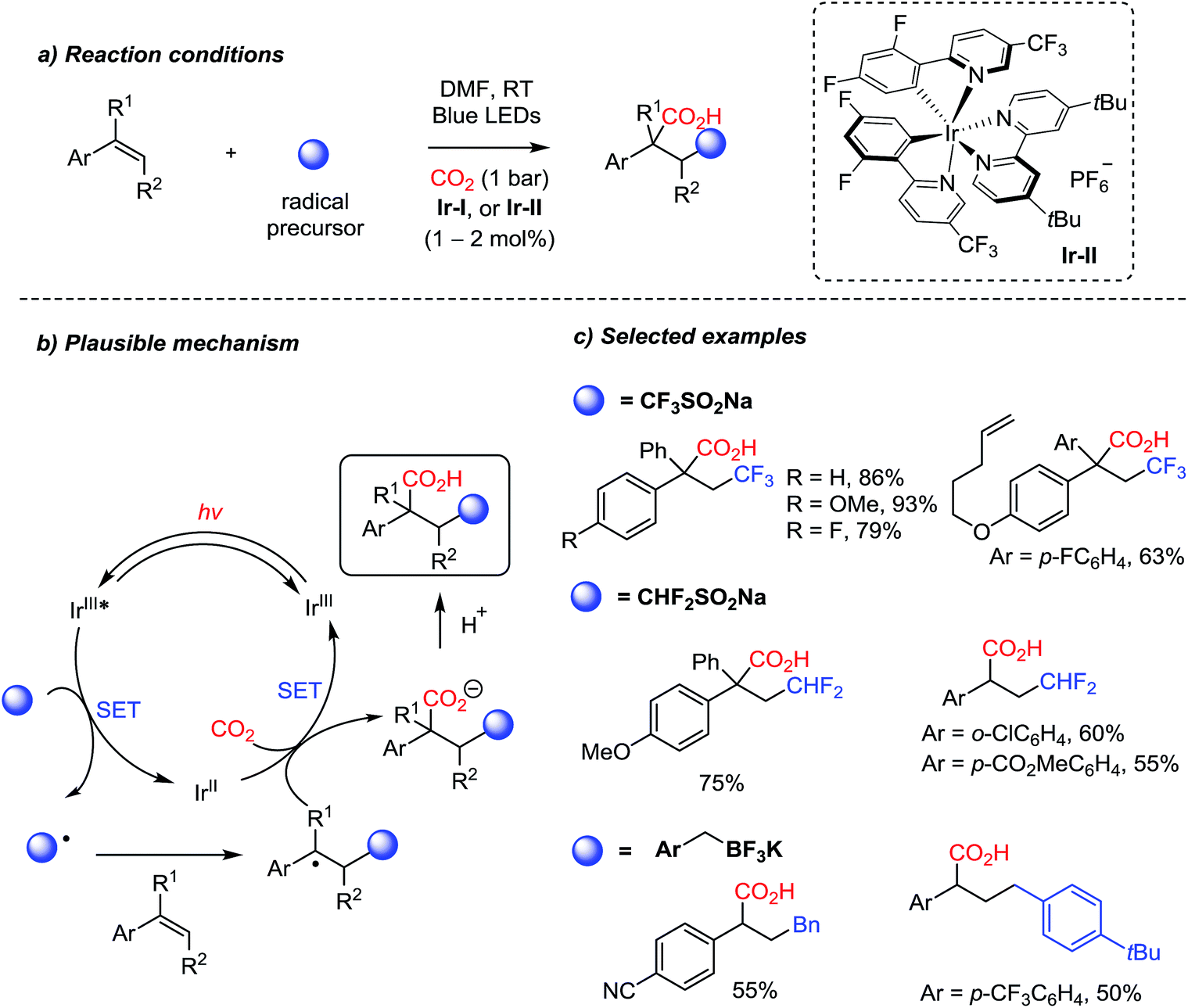 | ||
| Scheme 18 Structural diversity of carboxylation with radical initiators. | ||
The Yu group developed the first thiocarboxylation of styrenes by using an Fe/S complex as the photosensitizer (Scheme 19).70b Various β-thioacids were synthesized selectively with different regioselectivities from the previous protocol (Scheme 18). Mechanistic studies revealed that single-electron reduction of CO2 can be initiated by the excited Fe/S complex, yielding the CO2 radical anion (CO2˙−). This radical intermediate was trapped subsequently by an alkene substrate to generate a stabilized alkyl radical, which led to anti-Markovnikov regioselectivity. Thiolation of alkyl radicals was mediated by the [Fe/S] radical cation, highlighting the application potential of the methodology in the synthesis of β-thioacids – an intermediate for the antidepressant drug thiazensim.78 Also, considering the Fe- and S-rich environment in the prebiotic era, the presented reaction could help us to rethink the CO2 chemistry in the primordial soup, potentially affording complicated photoredox reactions with CO2 to furnish chiral molecules.
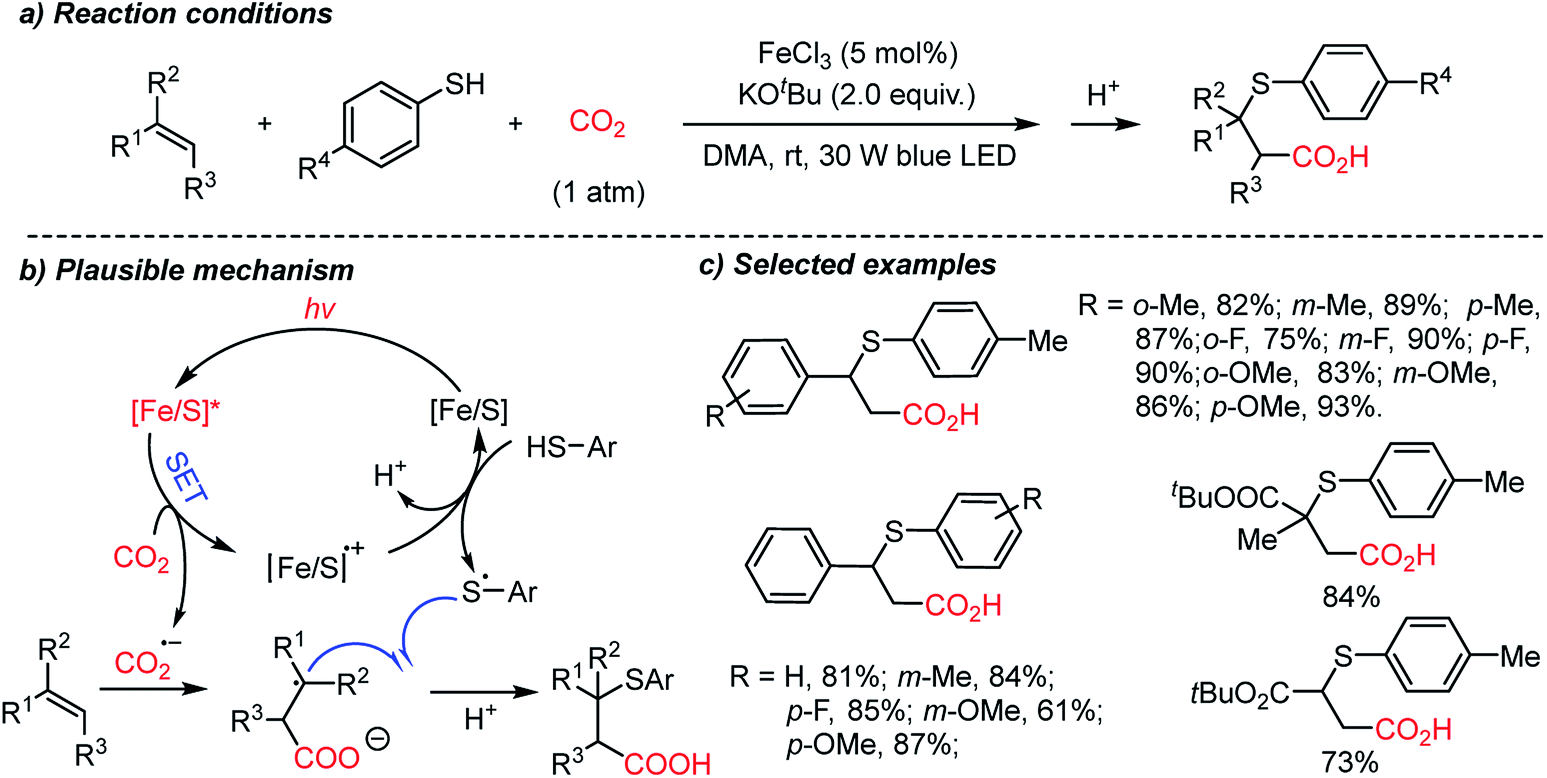 | ||
| Scheme 19 Fe–S catalyzed thiocarboxylation induced by visible light. | ||
The progress in redox-neutral CO2 functionalization showed elegant reaction mechanisms operating under mild conditions, for example, via CO2 insertion into metal–carbon bonds or CO2˙− captured by activated substrates. This represents a promising and ideal mode of action, whereby no additional sacrificial redox agents were applied to construct multiple C–C, and C–X bonds. Thus, high atom economy and step-efficiency are expected in constructing molecules with CO2 as a non-toxic C1 source, boosting research in CO2-utilization from recently developed dicarbofunctionalization.79 Meanwhile, the structural diversity of recent CO2-functionalization reactions shows the significant potential of CO2 in asymmetric synthesis and catalysis. Further investigations on the asymmetric activation of CO2 and its utilization in CO2-functionalization will allow us to achieve higher values of products while recycling CO2.
5. Asymmetric catalytic carboxylation with CO2
Besides the asymmetric synthesis of cyclic carbonates or polycarbonates with epoxides or diols,21b,80 the construction of enantioselective C–CO2 bonds using CO2 has been a formidable challenge under the influence of chiral catalysts or chiral environments. This is due to the high stability of CO2, limiting the scope of reaction partners; highly reactive organometallic species and/or harsh reaction conditions are necessary thus low stereoselectivity is in general expected.21a In 2004, the Mori group reported the carboxylative cyclization reaction of bis-1,3-dienes catalyzed by a Ni catalyst (Scheme 20).81 The authors performed facile 5-membered ring formation reactions in the presence of excess amounts of dialkyl zinc (4.5 equiv.). The obtained products possess three consecutive stereogenic centers with absolute diastereoselectivity with good yield and excellent enantioselectivity.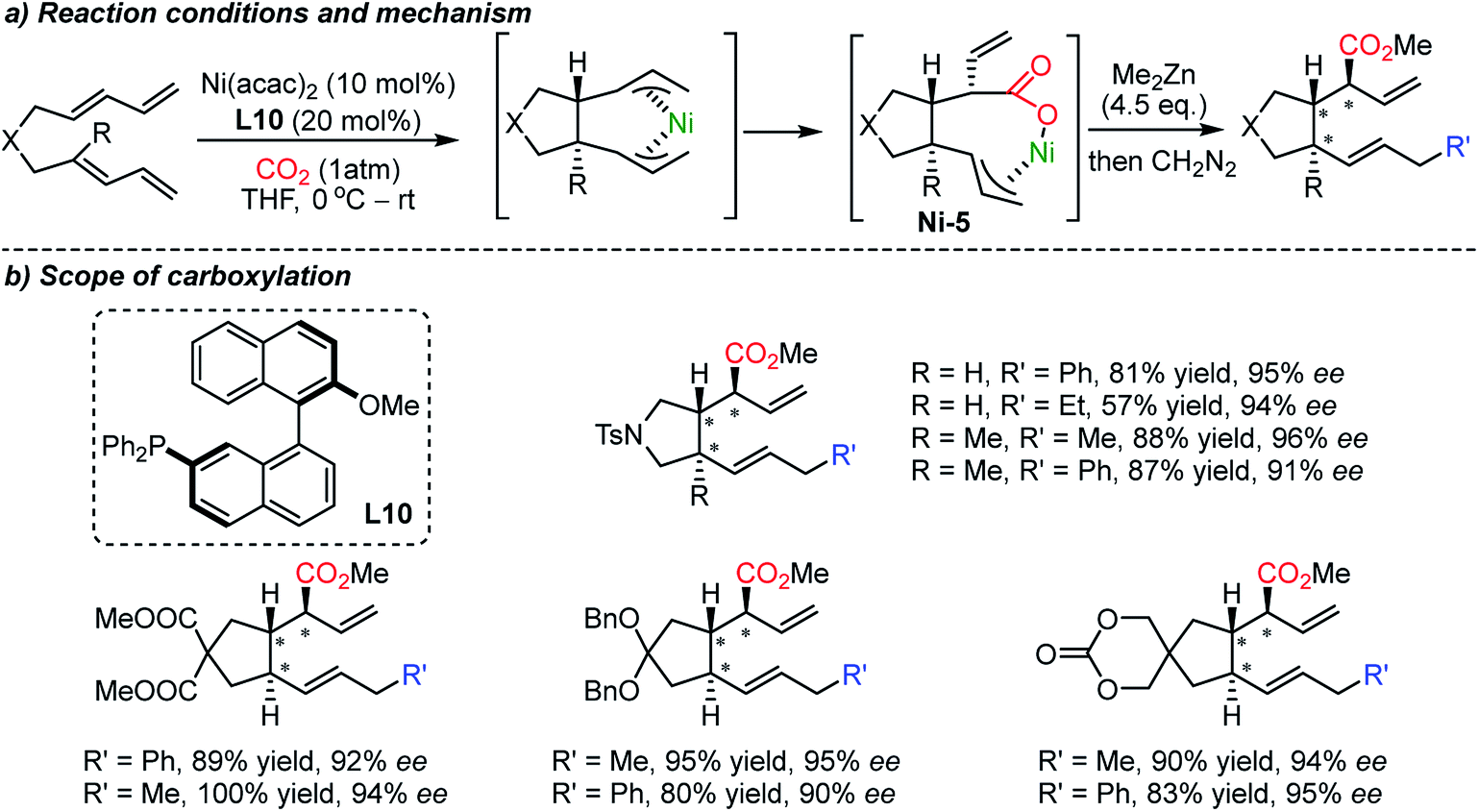 | ||
| Scheme 20 Asymmetric carboxylative cyclization of bis-1,3-dienes. | ||
In 2017, the Marek group developed an enantioselective Cu-catalyzed carbomagnesiation reaction of cyclopropenes, which could be selectively carboxylated with CO2 as an electrophile (Scheme 21).82 High diastereoselectivity was observed which is not fully understood yet based on the control experiment without the copper catalyst (racemic but moderate diastereoselectivity, 9:1 dr). Other electrophiles such as iodine, bromine and allylbromide were smoothly incorporated to furnish the desired products. Although Grignard reagents are reactive nucleophiles, the sequential addition of the alkene and CO2 prevented direct attack of these nucleophiles on CO2 at low reaction temperature (0 °C) in the presence of a copper catalyst. The observed stereoselectivity was attributed to the stability of the stereogenic center at the carbon–Cu moiety, explaining the cis geometry between the nucleophile and electrophilic CO2.
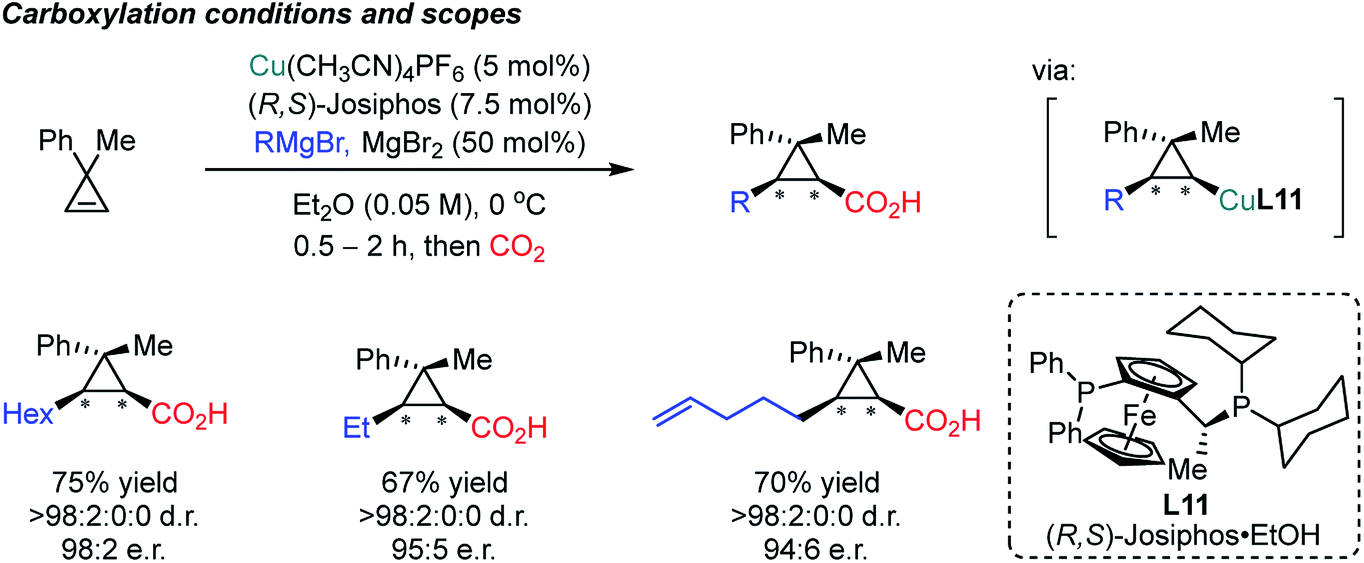 | ||
| Scheme 21 Asymmetric carbomagnesiation/carboxylation of cyclopropenes. | ||
The Yu group83 recently reported a highly regio- and enantioselective copper-catalyzed CO2-functionalization reaction of olefins owing to enantioselective Cu–H catalysis84 (Scheme 22). Inspired by the CO2 reduction reaction to methanol85 and other higher alcohols,86 the authors developed the sequential enantioselective Cu–H addition, carboxylation and reduction reactions to achieve hydroxymethylation of olefins. A preliminary mechanistic study revealed that the L12 Cu–R (C) species showed no reactivity toward reduced CO2 (R3Si–OCOH) (dashed arrow), indicating the direct carboxylation of C in the chiral environment to ensure the obtained high enantioselectivity. Furthermore, the developed methodology was applied to 1,3-dienes, affording (Z)-selective homoallylic alcohols with good enantioselectivities. Further derivatization of the hydroxymethylation products afforded elegant syntheses of enantioenriched (R)-(−)-curcumene87 and (S)-(+)-ibuprofen, starting from CO2 as a C1 building block.
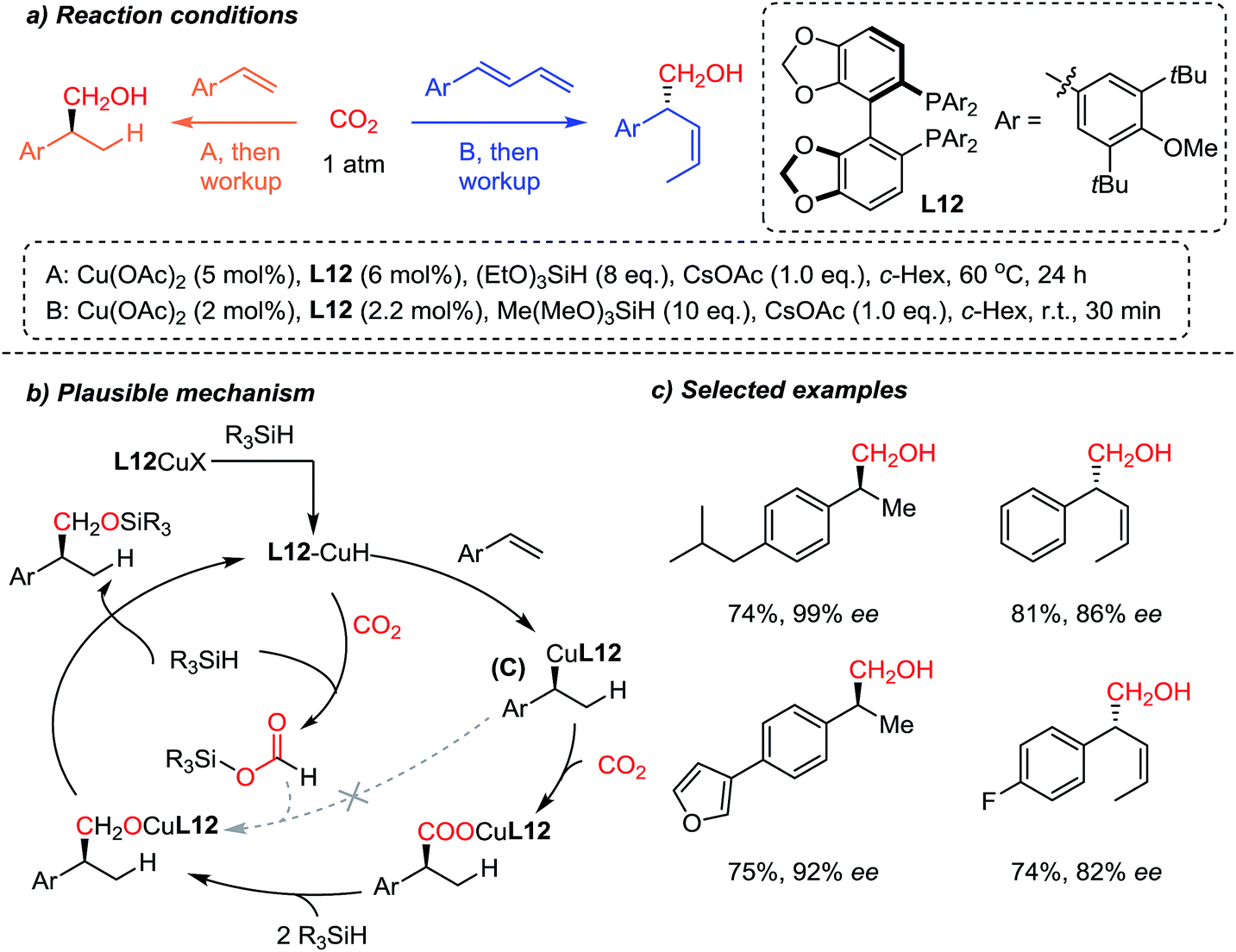 | ||
| Scheme 22 Enantioselective incorporation of CO2via hydrocarboxylation of styrene derivatives and dienes. | ||
Although asymmetric catalytic C–C bond formation has achieved relatively considerable progress,88 only a few methodologies have been reported with CO2 as a sustainable C1 source while creating stereogenic center(s) with high stereoselectivity. Considering that the carbon fixation process produces carbohydrates and biomass with absolute enantioselectivity, it is a logical extension to implement asymmetric carboxylation reactions in artificial CO2 fixation. Chemical synthesis offers various synthetic pathways and tools that can be easily tested, potentially providing a playground for facile screening and method development. For example, photochemical reactions with chiral catalysts including a chiral iridium catalyst89 or Lewis-acid assisted photocatalysis90 for CO2-functionalization are seemingly feasible methods to be developed. Considering the mode of action of RubisCo enzyme, redox-inactive metals and ligands (e.g. Mg–biotin complex) would be critical to improve the availability of CO2 in organic reactions.91 On the other hand, it could be inferred that chiral CO2-complexes may play a significant role in CO2-activation via bifunctional asymmetric catalysts.92 It would be exciting to see the development of CO2-functionalization, with foreseeable sustainability and increased utility of the final products in organic synthesis.
6. Conclusion and outlook: umpolung reactivities towards CO2
It is a formidable challenge to define an “ideal” carbon dioxide functionalization considering that many factors – environmental impact, atom-economy, sustainability, utility of products, and reaction conditions – are involved in designing reaction processes. Harnessing the full capacity of CO2-functionalization can be envisaged with sustainable and accessible chemical feed stocks, catalysts, and reaction conditions.Victor Grignard, in 1912, stated this in his Nobel Lecture – “Willstatter in fact recognized that …organic magnesium compounds must form and that the absorption of CO2gas by chlorophyll would in every way be comparable to the Grignard reaction”. The mode of action of magnesium compounds in chlorophyll differs from what Grignard speculated, however, one of the earliest umpolung reactions with CO2 and Grignard reagents paved a way for modern CO2 functionalization to date. Considering the formation of Grignard reagents, an umpolung process utilized polarized bonds, Cδ+–Xδ−, by inverting the electronic nature of the carbon to nucleophilic by forming Cδ−–Mg–X.
In this context, recent developments in umpolung carboxylation reactions have shown unprecedented reaction patterns (Scheme 23):61,62,93 for example, umpolung reactivity has been implemented to functionalize CO2 for an aldehyde carboxylation reaction through a redox-neutral mechanism (Scheme 23a).93b The obtained product, α-keto acid, was smoothly converted to the corresponding α-amino acid under reductive amination reaction conditions mimicking the biosynthesis of various amino acids. The use of nitrogen-containing nucleophiles offers direct synthesis of α-amino acid derivatives (Scheme 23c–e). By employing cyanohydrin, hydrazone, photocatalysts, and a base, in situ generated umpolung species were transformed to the desired carboxylated products under mild reaction conditions, with or without reducing agents. This is particularly interesting to hypothesize the evolution of α-amino acids from the CO2-rich prebiotic environment. It has been postulated that cyanide is an abundant source of a carbon nucleophile in the synthesis of biologically active molecules in the primordial soup.94 The use of aldehydes, cyanide, and CO2 in synthesizing biologically active molecules is a promising step toward answering the important question: what is the origin of life? Was there an involvement of photochemical CO2-activation? Was it promoted by an optically pure component to induce homochirality? The forthcoming ideal CO2-functionalization may answer these conundrums.
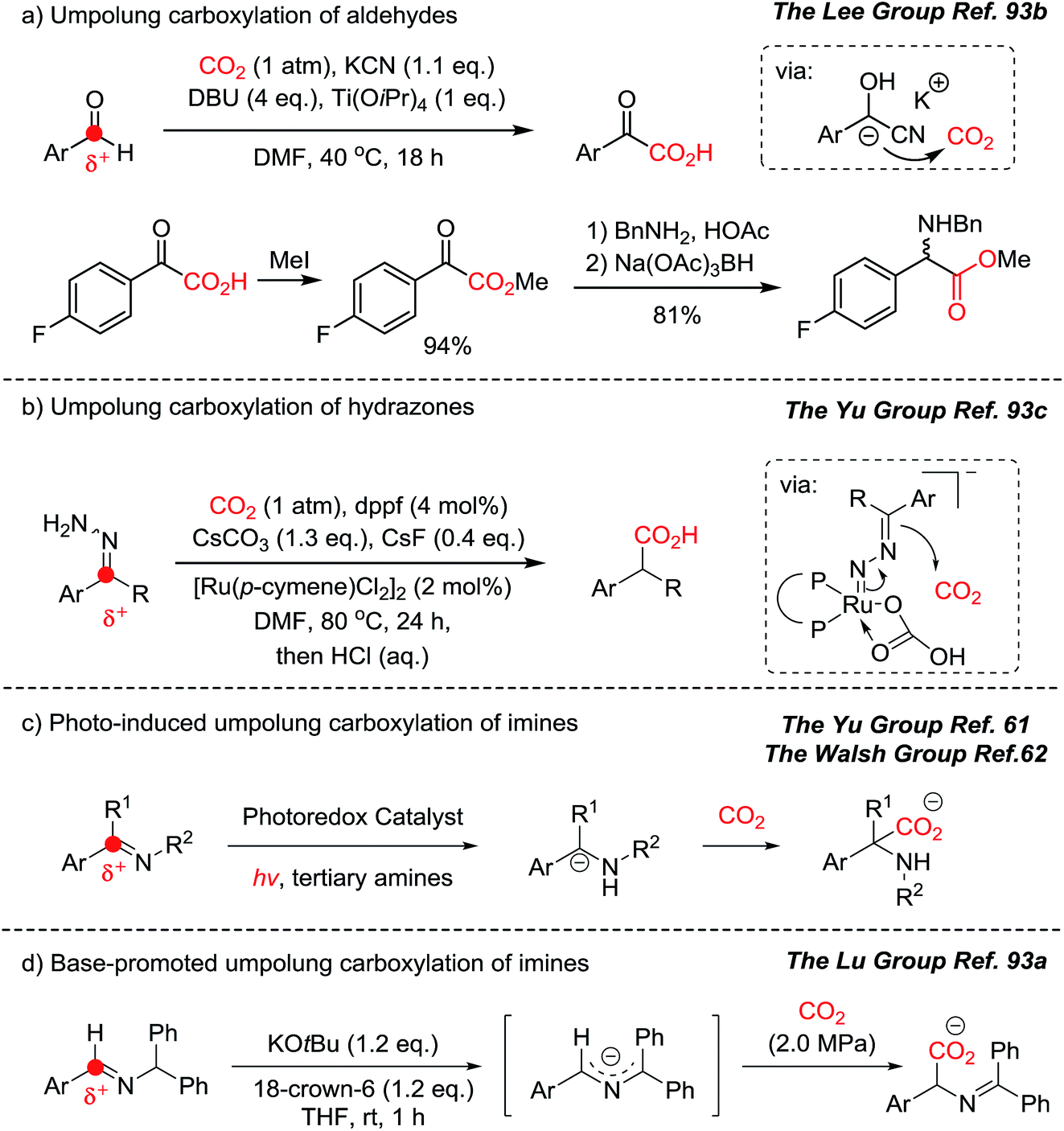 | ||
| Scheme 23 Direct umpolung carboxylation. (a) Umpolung carboxylation of aldehyde and reductive amination of α-keto acids to α-amino acids; (b) umpolung carboxylation of hydrazones; (c) photo-induced and (d) base-promoted umpolung carboxylation of imines. | ||
In summary, this Perspective collects the recent literature in CO2 functionalization and groups it into four categories: (1) metal catalyzed direct carboxylation, (2) photocatalytic carboxylation reactions, (3) redox-neutral carboxylation, and (4) asymmetric introduction of catalytic CO2 for C–C bond formation reactions. Even a broad scope of substrates and remote site functionalization were achieved; transition metal-catalyzed reductive carboxylation is mostly limited to CO2 insertion reaction with (over)stoichiometric amounts of reducing reagents. However, photoredox catalysts present promising access to more diversified CO2 reactions, like dicarbofunctionalization, single-electron reduction and radical coupling via a redox-neutral mechanism. Thanks to these developments of methodologies, as discussed at the end of Section 5, more examples in challenging enantioselective C–CO2 bond formation will be realized in the near future. Although enzymatic CO2 functionalization reactions are not covered in this Perspective, they have shown their very promising application in artificial carbon recycling processes.51,95 Synergetic and interdisciplinary CO2 fixation with biological and chemical catalysts will be particularly interesting in (asymmetric) photocatalytic conversion of CO2, truly mimicking photosynthesis to provide ideal CO2 functionalization reactions.
Conflicts of interest
There are no conflicts to declare.References
- (a) G. Drews, in Bioenergetic Processes of Cyanobacteria: From Evolutionary Singularity to Ecological Diversity, ed. G. A. Peschek, C. Obinger and G. Renger, Springer Netherlands, Dordrecht, 2011, p. 265 Search PubMed; (b) R. M. Soo, J. Hemp, D. H. Parks, W. W. Fischer and P. Hugenholtz, Science, 2017, 355, 1436 CrossRef CAS PubMed.
- (a) D. Gust and T. A. Moore, Science, 1989, 244, 35 CrossRef CAS PubMed; (b) X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924 CrossRef CAS PubMed.
- (a) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (b) M. Pera-Titus, Chem. Rev., 2014, 114, 1413 CrossRef CAS PubMed; (c) H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953 CrossRef CAS PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881 CrossRef CAS PubMed.
- (a) O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934 CrossRef CAS PubMed; (b) C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187 CrossRef CAS PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434 CrossRef CAS PubMed.
- (a) J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed; (b) S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed; (c) W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607 CrossRef CAS PubMed.
- (a) P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019 CrossRef CAS PubMed; (b) J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed; (c) J. Wang, A. Zhang, X. Jiang, C. Song and X. Guo, J. CO2 Util., 2018, 27, 81 CrossRef CAS.
- (a) S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed; (b) S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614 CrossRef CAS PubMed; (c) Z.-J. Mu, X. Ding, Z.-Y. Chen and B.-H. Han, ACS Appl. Mater. Interfaces, 2018, 10, 41350 CrossRef CAS PubMed.
- (a) C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS; (b) J. W. Maina, C. Pozo-Gonzalo, L. Kong, J. Schütz, M. Hill and L. F. Dumée, Mater. Horiz., 2017, 4, 345 RSC.
- (a) S. Ye, R. Wang, M.-Z. Wu and Y.-P. Yuan, Appl. Surf. Sci., 2015, 358, 15 CrossRef CAS; (b) S. Yin, J. Han, T. Zhou and R. Xu, Catal. Sci. Technol., 2015, 5, 5048 RSC.
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed.
- (a) E. Proschak, P. Heitel, L. Kalinowsky and D. Merk, J. Med. Chem., 2017, 60, 5235 CrossRef CAS PubMed; (b) H. Maag, in Prodrugs: Challenges and Rewards Part 1, ed. V. J. Stella, R. T. Borchardt, M. J. Hageman, R. Oliyai, H. Maag and J. W. Tilley, Springer New York, New York, NY, 2007, p. 703 Search PubMed.
- (a) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS PubMed; (b) M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739 CrossRef PubMed; (c) S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439 CrossRef CAS.
- J. Song, Q. Liu, H. Liu and X. Jiang, Eur. J. Org. Chem., 2018, 2018, 696 CrossRef CAS.
- K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524 RSC.
- S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666 RSC.
- L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC.
- F. Juliá-Hernández, M. Gaydou, E. Serrano, M. van Gemmeren and R. Martin, Top. Curr. Chem., 2016, 374, 45 CrossRef PubMed.
- (a) J. Vaitla, Y. Guttormsen, J. K. Mannisto, A. Nova, T. Repo, A. Bayer and K. H. Hopmann, ACS Catal., 2017, 7, 7231 CrossRef CAS; (b) X.-B. Lu, Top. Organomet. Chem., 2016, 53, 171 CrossRef CAS.
- (a) Y. Charles, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201806285; (b) Y.-Y. Gui, W.-J. Zhou, J.-H. Ye and D.-G. Yu, ChemSusChem, 2017, 10, 1337 CrossRef CAS PubMed.
- (a) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef; (b) R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419 CrossRef CAS; (c) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436 CrossRef CAS PubMed.
- (a) J. R. Cabrero-Antonino, R. Adam Ortiz and M. Beller, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201810121; (b) Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077 CrossRef CAS.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636 RSC.
- K. Osakada, R. Sato and T. Yamamoto, Organometallics, 1994, 13, 4645 CrossRef CAS.
- A. Correa and R. Martín, J. Am. Chem. Soc., 2009, 131, 15974 CrossRef CAS PubMed.
- T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106 CrossRef CAS PubMed.
- (a) T. León, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221 CrossRef PubMed; (b) Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212 CrossRef CAS PubMed; (c) T. Moragas and R. Martin, Synthesis, 2016, 48, 2816 CrossRef CAS; (d) M. Börjesson, T. Moragas and R. Martin, J. Am. Chem. Soc., 2016, 138, 7504 CrossRef PubMed.
- A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed.
- T. Moragas, M. Gaydou and R. Martin, Angew. Chem., Int. Ed., 2016, 55, 5053 CrossRef CAS PubMed.
- T. Moragas, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 17702 CrossRef CAS PubMed.
- M. van Gemmeren, M. Börjesson, A. Tortajada, S. Z. Sun, K. Okura and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 6558 CrossRef CAS PubMed.
- (a) X. Wang, Y. Liu and R. Martin, J. Am. Chem. Soc., 2015, 137, 6476 CrossRef CAS PubMed; (b) M. Gaydou, T. Moragas, F. Juliá-Hernández and R. Martin, J. Am. Chem. Soc., 2017, 139, 12161 CrossRef CAS PubMed; (c) A. Tortajada, R. Ninokata and R. Martin, J. Am. Chem. Soc., 2018, 140, 2050 CrossRef CAS PubMed.
- F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed.
- (a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed; (b) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC; (c) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- (a) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209 CrossRef CAS PubMed; (b) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153 CrossRef CAS PubMed.
- O. V. Ozerov, Chem. Soc. Rev., 2009, 38, 83 RSC.
- (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (b) J. Thomas and F. Peter, Chem.–Eur. J., 2009, 15, 9632 CrossRef PubMed; (c) J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387 CrossRef; (d) B. M. Trost, Acc. Chem. Res., 1980, 13, 385 CrossRef CAS.
- (a) G. Pupo, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 6099 CrossRef CAS PubMed; (b) S. Masato, S. Isao and Y. Akio, Bull. Chem. Soc. Jpn., 1996, 69, 1065 CrossRef; (c) S. B. Lang, T. M. Locascio and J. A. Tunge, Org. Lett., 2014, 16, 4308 CrossRef CAS PubMed; (d) I. Starý, I. G. Stará and P. Kočovský, Tetrahedron Lett., 1993, 34, 179 CrossRef; (e) I. Starý, I. G. Stará and P. Kočovský, Tetrahedron, 1994, 50, 529 CrossRef; (f) R. Kumareswaran and Y. D. Vankar, Tetrahedron Lett., 1997, 38, 8421 CrossRef CAS.
- (a) K. E. Atkins, W. E. Walker and R. M. Manyik, Tetrahedron Lett., 1970, 11, 3821 CrossRef; (b) T. Hirao, N. Yamada, Y. Ohshiro and T. Agawa, J. Organomet. Chem., 1982, 236, 409 CrossRef CAS; (c) I. Minami, Y. Ohashi, I. Shimizu and J. Tsuji, Tetrahedron Lett., 1985, 26, 2449 CrossRef CAS; (d) B. M. Trost and M. H. Hung, J. Am. Chem. Soc., 1983, 105, 7757 CrossRef CAS; (e) B. M. Trost and T. R. Verhoeven, J. Am. Chem. Soc., 1980, 102, 4730 CrossRef CAS; (f) J. Tsuji, Y. Kobayashi, H. Kataoka and T. Takahashi, Tetrahedron Lett., 1980, 21, 1475 CrossRef CAS; (g) R. Tamura and L. S. Hegedus, J. Am. Chem. Soc., 1982, 104, 3727 CrossRef CAS; (h) Y. Tanigawa, K. Nishimura, A. Kawasaki and S.-I. Murahashi, Tetrahedron Lett., 1982, 23, 5549 CrossRef CAS; (i) B. M. Trost, N. R. Schmuff and M. J. Miller, J. Am. Chem. Soc., 1980, 102, 5979 CrossRef CAS.
- H.-W. Suh, L. M. Guard and N. Hazari, Chem. Sci., 2014, 5, 3859 RSC.
- N. Solin, J. Kjellgren and K. J. Szabó, J. Am. Chem. Soc., 2004, 126, 7026 CrossRef CAS PubMed.
- (a) H. Takahata, Y. Uchida and T. Momose, J. Org. Chem., 1995, 60, 5628 CrossRef CAS; (b) Y. Zhang, X. Wang, M. Sunkara, Q. Ye, L. V. Ponomereva, Q.-B. She, A. J. Morris and J. S. Thorson, Org. Lett., 2013, 15, 5566 CrossRef CAS PubMed.
- (a) H. Hoberg and B. Apotecher, J. Organomet. Chem., 1984, 270, c15 CrossRef CAS; (b) H. Hoberg, K. Jenni, C. Krüger and E. Raabe, Angew. Chem., Int. Ed., 1986, 25, 810 CrossRef; (c) M. Takimoto and M. Mori, J. Am. Chem. Soc., 2001, 123, 2895 CrossRef CAS PubMed.
- T. Fujihara, Y. Horimoto, T. Mizoe, F. B. Sayyed, Y. Tani, J. Terao, S. Sakaki and Y. Tsuji, Org. Lett., 2014, 16, 4960 CrossRef CAS PubMed.
- M. Takimoto, M. Kawamura, M. Mori and Y. Sato, Synlett, 2005, 2005, 2019 Search PubMed.
- J. Xie, A. H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193 RSC.
- (a) M. Hervás, J. A. Navarro and M. A. De la Rosa, Acc. Chem. Res., 2003, 36, 798 CrossRef PubMed; (b) S. Nishitani, N. Kurata, Y. Sakata, S. Misumi, A. Karen, T. Okada and N. Mataga, J. Am. Chem. Soc., 1983, 105, 7771 CrossRef CAS.
- N. J. Claassens, Microb. Biotechnol., 2017, 10, 31 CrossRef PubMed.
- T. Schwander, L. Schada von Borzyskowski, S. Burgener, N. S. Cortina and T. J. Erb, Science, 2016, 354, 900 CrossRef CAS PubMed.
- (a) D. R. Ort, S. S. Merchant, J. Alric, A. Barkan, R. E. Blankenship, R. Bock, R. Croce, M. R. Hanson, J. M. Hibberd, S. P. Long, T. A. Moore, J. Moroney, K. K. Niyogi, M. A. J. Parry, P. P. Peralta-Yahya, R. C. Prince, K. E. Redding, M. H. Spalding, K. J. van Wijk, W. F. J. Vermaas, S. von Caemmerer, A. P. M. Weber, T. O. Yeates, J. S. Yuan and X. G. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8529 CrossRef CAS PubMed; (b) D. C. Ducat and P. A. Silver, Curr. Opin. Chem. Biol., 2012, 16, 337 CrossRef CAS PubMed.
- (a) S. Tazuke and H. Ozawa, J. Chem. Soc., Chem. Commun., 1975, 237 RSC; (b) S. Tazuke and N. Kitamura, Nature, 1978, 275, 301 CrossRef CAS; (c) T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637 CrossRef CAS; (d) J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701 CrossRef CAS PubMed; (e) J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2016, 9, 453 CrossRef PubMed.
- K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 9467 CrossRef CAS PubMed.
- Q.-Y. Meng, S. Wang and B. König, Angew. Chem., Int. Ed., 2017, 56, 13426 CrossRef CAS PubMed.
- Q.-Y. Meng, S. Wang, G. S. Huff and B. König, J. Am. Chem. Soc., 2018, 140, 3198 CrossRef CAS PubMed.
- H. Seo, A. Liu and T. F. Jamison, J. Am. Chem. Soc., 2017, 139, 13969 CrossRef CAS PubMed.
- Y. Masuda, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2015, 137, 14063 CrossRef CAS PubMed.
- J. Hou, A. Ee, W. Feng, J.-H. Xu, Y. Zhao and J. Wu, J. Am. Chem. Soc., 2018, 140, 5257 CrossRef CAS PubMed.
- T. Ju, Q. Fu, J.-H. Ye, Z. Zhang, L.-L. Liao, S.-S. Yan, X.-Y. Tian, S.-P. Luo, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2018, 57, 13897 CrossRef CAS PubMed.
- X. Fan, X. Gong, M. Ma, R. Wang and P. J. Walsh, Nat. Commun., 2018, 9, 4936 CrossRef PubMed.
- S. Humbel, I. Côte, N. Hoffmann and J. Bouquant, J. Am. Chem. Soc., 1999, 121, 5507 CrossRef CAS.
- M. Li, O. Gutierrez, S. Berritt, A. Pascual-Escudero, A. Yeşilçimen, X. Yang, J. Adrio, G. Huang, E. Nakamaru-Ogiso, M. C. Kozlowski and P. J. Walsh, Nat. Chem., 2017, 9, 997 CrossRef CAS PubMed.
- (a) K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234 CrossRef CAS; (b) S. Fustero, M. Sánchez-Roselló, C. Báez, C. del Pozo, J. L. García Ruano, J. Alemán, L. Marzo and A. Parra, Amino Acids, 2011, 41, 559 CrossRef CAS PubMed; (c) C. Spino and C. Gobdout, J. Am. Chem. Soc., 2003, 125, 12106 CrossRef CAS PubMed.
- M. Kapoor, D. Liu and M. C. Young, J. Am. Chem. Soc., 2018, 140, 6818 CrossRef CAS PubMed.
- (a) V. Laserna, G. Fiorani, C. J. Whiteoak, E. Martin, E. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2014, 53, 10416 CrossRef CAS PubMed; (b) A. Peeters, R. Ameloot and D. E. De Vos, Green Chem., 2013, 15, 1550 RSC.
- J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037 CrossRef CAS PubMed.
- H. Wu, R. Andres, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 499 CrossRef CAS PubMed.
- (a) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915 CrossRef CAS PubMed; (b) J.-H. Ye, M. Miao, H. Huang, S.-S. Yan, Z.-B. Yin, W.-J. Zhou and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15416 CrossRef CAS PubMed.
- K. Michigami, T. Mita and Y. Sato, J. Am. Chem. Soc., 2017, 139, 6094 CrossRef CAS PubMed.
- L.-L. Liao, G.-M. Cao, J.-H. Ye, G.-Q. Sun, W.-J. Zhou, Y.-Y. Gui, S.-S. Yan, G. Shen and D.-G. Yu, J. Am. Chem. Soc., 2018, 140, 17338 CrossRef CAS PubMed.
- X. Zhang and F. G. Bordwell, J. Org. Chem., 1992, 57, 4163 CrossRef CAS.
- (a) E. Lamy, L. Nadjo and J. M. Saveant, J. Electroanal. Chem. Interfacial Electrochem., 1977, 78, 403 CrossRef CAS; (b) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed.
- (a) K. Booker-Milburn, Nat. Chem., 2012, 4, 433 CrossRef CAS PubMed; (b) S. V. Ley, D. E. Fitzpatrick, R. M. Myers, C. Battilocchio and R. J. Ingham, Angew. Chem., Int. Ed., 2015, 54, 10122 CrossRef CAS PubMed; (c) J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891 RSC.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (d) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed.
- S. Y. Dike, D. H. Ner and A. Kumar, Bioorg. Med. Chem. Lett., 1991, 1, 383 CrossRef CAS.
- (a) J.-H. Ye, L. Zhu, S.-S. Yan, M. Miao, X.-C. Zhang, W.-J. Zhou, J. Li, Y. Lan and D.-G. Yu, ACS Catal., 2017, 7, 8324 CrossRef CAS; (b) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS PubMed.
- (a) M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129 CrossRef CAS PubMed; (b) Y. Liu, W.-M. Ren, K.-K. He and X.-B. Lu, Nat. Commun., 2014, 5, 5687 CrossRef PubMed.
- M. Takimoto, Y. Nakamura, K. Kimura and M. Mori, J. Am. Chem. Soc., 2004, 126, 5956 CrossRef CAS PubMed.
- L. Dian, D. S. Müller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783 CrossRef CAS PubMed.
- Y.-Y. Gui, N. Hu, X.-W. Chen, L. L. Liao, T. Ju, J.-H. Ye, Z. Zhang, J. Li and D.-G. Yu, J. Am. Chem. Soc., 2017, 139, 17011 CrossRef CAS PubMed.
- (a) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS PubMed; (b) M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48 CrossRef CAS PubMed.
- (a) W. H. Bernskoetter and N. Hazari, Acc. Chem. Res., 2017, 50, 1049 CrossRef CAS PubMed; (b) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936 CrossRef CAS PubMed; (c) A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995 RSC.
- Y. Tani, K. Kuga, T. Fujihara, J. Terao and Y. Tsuji, Chem. Commun., 2015, 51, 13020 RSC.
- A. Gualandi, P. Canestrari, E. Emer and P. G. Cozzi, Adv. Synth. Catal., 2014, 356, 528 CrossRef CAS.
- (a) P. Renaud and P. Leong, Science, 2008, 322, 55 CrossRef CAS PubMed; (b) T. Yasukawa, H. Miyamura and S. Kobayashi, Chem. Soc. Rev., 2014, 43, 1450 RSC; (c) T. Tetsu, I. Takanori and K. Shū, Angew. Chem., Int. Ed., 2013, 52, 6590 CrossRef PubMed; (d) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed; (e) R. Mahrwald, in Enantioselective Organocatalyzed Reactions II: Asymmetric C–C Bond Formation Processes, Springer Netherlands, Dordrecht, 2011 Search PubMed.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100 CrossRef CAS PubMed.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391 CrossRef CAS.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303 CrossRef CAS PubMed.
- (a) J.-A. Ma and D. Cahard, Angew. Chem., Int. Ed., 2004, 43, 4566 CrossRef CAS PubMed; (b) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187 CrossRef CAS PubMed.
- (a) C.-X. Guo, W.-Z. Zhang, H. Zhou, N. Zhang and X.-B. Lu, Chem.–Eur. J., 2016, 22, 17156 CrossRef CAS PubMed; (b) M. Juhl and J.-W. Lee, Angew. Chem., Int. Ed., 2018, 57, 12318 CrossRef CAS PubMed; (c) S.-S. Yan, L. Zhu, J.-H. Ye, Z. Zhang, H. Huang, H. Zeng, C.-J. Li, Y. Lan and D.-G. Yu, Chem. Sci., 2018, 9, 4873 RSC.
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301 CrossRef CAS PubMed.
- J. Martin, L. Eisoldt and A. Skerra, Nat. Catal., 2018, 1, 555 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |