
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Terpene- and terpenoid-based polymeric resins for stereolithography 3D printing†
Andrew C.
Weems
,
Kayla R.
Delle Chiaie
 ,
Joshua C.
Worch
,
Connor J.
Stubbs
and
Andrew P.
Dove
*
,
Joshua C.
Worch
,
Connor J.
Stubbs
and
Andrew P.
Dove
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: a.dove@bham.ac.uk
First published on 9th September 2019
Abstract
Thiol–ene ‘click’ reactions utilizing terpenes and a four-arm thiol were employed to produce thermoset 3D printed structures using vat photopolymerisation. Five terpenes were characterized for reactivity using both 1H NMR spectroscopy and photorheology, allowing for screening of both monomers and prepolymer oligomers as possible candidates for stereolithographic 3D printing. The time to crosslinking for limonene- and linalool-based resins was found to be approximately 5 s while nerol- and geraniol-based resins crosslinked over the course of 1 h, under the 3D printing conditions. The materials produced from photo-crosslinking displayed a range of thermomechanical behaviours, with varied post-printing thermal curing cycles utilized to alter thermomechanical behaviour from a brittle elastomer with strains at failure of ca. 50% (Young's modulus of ∼0.4 MPa) to more traditional engineering thermoplastic behaviours with elastic moduli above 20 MPa and strains to failure of 180%. The relationship between material properties and surface energy was elucidated through the use of thermomechanical characterizations (differential scanning calorimetry, dynamic mechanical analysis, thermogravimetric analysis), and select compositions are demonstrated to be suitable for printing into complex 3D shapes through additive manufacturing techniques.
Introduction
Photopolymer resins are widely used in a range of applications including coatings, lithography and more recently 3D printing.1–3 The application of light allows for liquid materials to be processed, typically by crosslinking, into a desired shape or geometry thus being easier to process rapidly compared with more conventional machining of solid material.1,4,5 Printing conditions that include reaction rate, mechanical resilience of the crosslinked material, and the environmental conditions must be considered for all but the most specialized printing assemblies, as these factors will determine the final part suitability as well as its ability to be processed by the selected printing technique.1,2,4–7 For vat-curing photopolymer systems, radical polymerisations of vinyl or unsaturated monomers/oligomers/polymers and epoxide ring-opening polymerisations are the most common on account of the rapid reaction rates.7 A high rate of crosslinking is necessary to transition the material from a relatively low viscosity liquid resin into a solid part capable of supporting subsequent solid layers, which has limited the available monomer library of suitable materials to date.The use of bio-sourced molecules for polymer synthesis and manufacturing processes has risen steadily, and is beginning to see exponential growth as a result of global green movements, public interest in more environmentally friendly processes, and attempts to reduce waste through recycling and biodegradation of materials. Despite this interest, the application of sustainably-sourced monomers in photo-crosslinkable systems has been limited. The use of neolignans such as magnolol and honokiol has been demonstrated in the synthesis of thermosetting materials, where the two honokiol isomers have been used to produce both polymeric and monomeric units for materials possessing tunable thermomechanical properties that are entirely dependent upon the linkage utilized during synthesis.8 The synthesis of benzoxazine from the same biosourced products was also demonstrated, where performance polymers with glass transition temperatures (Tg) above 300 °C and thermal stabilities exceeding 440 °C.9 Other examples of biosourced monomers include various sugars used in polycondensations and ring opening polymerisations,10,11 dihydrolevoglucosenone and syrinaldehyde that have been used as a precursor for methacrylic monomers and in subsequent free radical polymerisations,12,13 vanillin in cross-conjugated pyrrole-based polymers as well as in electrochemical reductive polymerisation to realise polyvanillin,14,15 furfurylamine, which has been used to modify magnolol to synthesize benzoxazine for use as a thermosetting resin through ring opening polymerisation,9 alloocimene polymerised through a redox emulsion process,16 as well as carene-8 and pinene-derived lactams for anionic polymerisation of polyamides,17 and phellandrene-derived polyols used in polyurethane foam synthesis.18
Of the aforementioned monomer classes and species, terpenes and terpenoids are some of the most promising and most commonly used, with utility in fragrances and perfumes, food, cosmetics, pharmaceuticals and even cigarettes and other inhalation products.19–22 This class of monomer is widely available, and may be found in essential oils, tree sap and materials, citrus fruit, and even as by-products from other manufacturing processes, such as the paper industry's Kraft process, or simply through the processing of citrus fruits and other renewable resources.23–26 The inherent double bonds present in mono-, di-, tri-, and other polyterpenes make them excellent candidates for the creation of sustainably-sourced photo-crosslinkable resins, yet to date only a few studies have investigated their application beyond utilizing appended (meth)acrylate or epoxide functionalities for (controlled) radical polymerisation27,28–30 modified polysaccharides,31 polycarbonates,32 and even polysilanes,33 with processing still being limited in such studies. Herein, we report our study of the application of five terpenes: limonene, terpinene, geraniol, nerol and linalool (Fig. 1) for direct photocuring using radical thiol–ene addition chemistry and demonstrate their translation into resins for 3D printing, in an attempt to overcome these limitations.
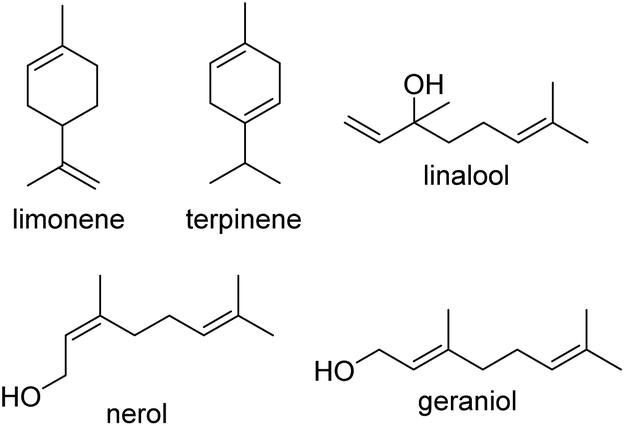 | ||
| Fig. 1 Monomer structures used in synthesis and 3D printing of terpene-based resins. | ||
Methods and materials
General considerations
All chemicals were commercially available (purchased from Sigma-Aldrich unless otherwise stated) and used without further purification (1H NMR spectroscopy spectrum ESI Fig. S1–S7†). Solvents were of ACS grade or higher. NMR spectra (400 MHz for 1H and 125 MHz for 13C) were recorded on a Bruker 400 spectrometer and processed using MestReNova v9.0.1 (Mestrelab Research, S.L., Santiago de Compostela, Spain). Chemical shifts were referenced to residual solvent peaks at δ = 7.26 ppm (1H) and δ = 77.16 ppm (13C) for CDCl3.General prepolymer synthesis
As a general case, prepolymer synthesis for limonene is described, with other synthetic protocols included in the ESI.† Limonene (1.0 g, 7.3 mmol) and pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) (0.76 g, 1.6 mmol) were added to a 50 mL round bottom flask in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and dissolved in 10 wt% acetone. To this solution was added 1.5 wt% photoinitiator (Irgacure 819), dissolved by stirring and then irradiated with 365 nm light for 4 h at room temperature. Aliquots were taken using a pipette at various time intervals for 1H NMR spectroscopic analysis until the mixture became too viscous to sample. Upon completion, the solution was concentrated, dissolved in ethyl acetate, and washed with 1 M HCl. The prepolymer was isolated in vacuo and used as an oligomer for resin synthesis. 1H NMR (400 MHz, chloroform-d) δ 5.38 (m, 1H), 4.70 (s, 1H), 4.22–4.14 (s, 4H), 3.64–3.50 (m, 1H), 2.86–2.55 (m, 11H), 2.43–2.27 (m, 1H), 2.17 (dt, J = 3.9, 1.8 Hz, 5H), 2.00 (s, 1H, acetone), 1.95 (m, 2H), 1.84–1.54 (m, 5H), 1.26 (m, 1H), 1.03–0.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 120.60, 120.54, 108.36, 62.23, 62.15, 60.48, 51.43, 51.25, 41.06, 38.32, 38.23, 37.68, 37.58, 37.51, 37.42, 36.81, 35.01, 34.68, 30.95, 30.76, 30.60, 29.93, 29.53, 27.89, 27.49, 27.44, 27.30, 27.12, 25.23, 23.47, 20.83, 20.42, 19.68, 19.61, 16.14, 15.67.
:1 ratio and dissolved in 10 wt% acetone. To this solution was added 1.5 wt% photoinitiator (Irgacure 819), dissolved by stirring and then irradiated with 365 nm light for 4 h at room temperature. Aliquots were taken using a pipette at various time intervals for 1H NMR spectroscopic analysis until the mixture became too viscous to sample. Upon completion, the solution was concentrated, dissolved in ethyl acetate, and washed with 1 M HCl. The prepolymer was isolated in vacuo and used as an oligomer for resin synthesis. 1H NMR (400 MHz, chloroform-d) δ 5.38 (m, 1H), 4.70 (s, 1H), 4.22–4.14 (s, 4H), 3.64–3.50 (m, 1H), 2.86–2.55 (m, 11H), 2.43–2.27 (m, 1H), 2.17 (dt, J = 3.9, 1.8 Hz, 5H), 2.00 (s, 1H, acetone), 1.95 (m, 2H), 1.84–1.54 (m, 5H), 1.26 (m, 1H), 1.03–0.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 120.60, 120.54, 108.36, 62.23, 62.15, 60.48, 51.43, 51.25, 41.06, 38.32, 38.23, 37.68, 37.58, 37.51, 37.42, 36.81, 35.01, 34.68, 30.95, 30.76, 30.60, 29.93, 29.53, 27.89, 27.49, 27.44, 27.30, 27.12, 25.23, 23.47, 20.83, 20.42, 19.68, 19.61, 16.14, 15.67.
General synthesis of resins from terpenes
The monomeric terpene (limonene, 1.0 g, 7.3 mmol) and PETMP (1.719 g, 3.6 mmol) were dissolved in acetone (5 mL). Irgacure 819 (40.5 mg, 1.5 wt%, 0.1 mmol) was added to the mixture in the absence of ambient light and left without stirring for 12 h. To this solution, photoinhibitor (Kalsec Durabrite® Oleoresin Paprika Extract NS, 1 mL) was added and mixed until homogenized.General synthesis of resins from terpene prepolymers
To the prepolymer resin was added the remaining half measure of thiol. As an example, nerol prepolymer (1.76 g) and PETMP (0.76 g, 1.6 mmol) were dissolved in acetone (0.28 g). To this solution was added Irgacure 819 (0.113 g, 0.3 mmol) photoinitiator (1 mL). The solution was stirred overnight in a brown glass vial before any testing. Aliquots were taken using a pipette at various time intervals for 1H NMR spectroscopic analysis until the mixture became too viscous to sample.Post-polymerisation treatment
Solid polymers were cured for 12 h at 30 °C, 80 °C or 120 °C prior to testing.34 Samples were photocured for 30 min under ambient conditions and then placed in an isothermal oven at standard atmospheric pressure and moisture. After the cure cycle, samples were removed and allowed to cool to room temperature over 12 h prior to additional testing.Photorheology
The crosslinking kinetics of the resins were examined as a function of gelation time by measuring the dampening or phase ratio (tanδ), storage moduli, loss moduli, complex viscosity, and film thickness by photorheology. An Anton Paar rheometer (Anton Paar USA Inc, Ashland, VA, USA) fitted with a detachable photoillumination system (Exfo OmniCure 1500s light source, broadband Hg-lamp) with two parallel plates (10 mm disposable aluminum hollow shaft plate, Anton Paar). Resin samples were sheared between two parallel plates, one made of glass and transparent, at 1 Hz for 50 s without irradiation. After this time, the light source was switched on and measurements were taken every 0.2 s over the course of 2 min. The inflection points of the moduli plots, and the peak tanδ values, were used to determine the time to gelation of the resin. Sample shrinkage was determined by measuring the distance between the plates at the same sampling rate as the other metrics.
Gelation calculation
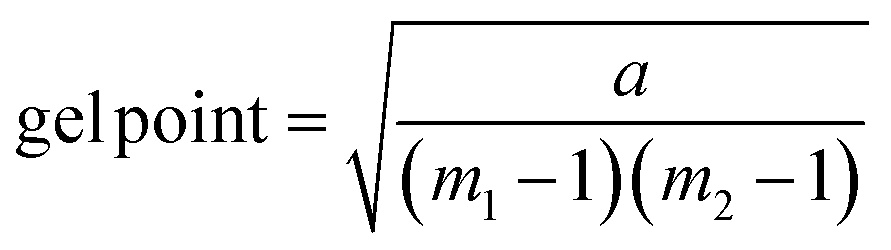
| (1) |
Mechanical testing
Dog bones (ASTM Type IV) were cut from cast films and tested using uniaxial tensile testing (Testometric MCT-350, 100 kgf load cell, Testometric Company Ltd, Rochdale, United Kingdom) at ambient moisture and temperature. Samples were placed in the tension clamps and allowed to vibrationally equilibrate for 10 min, at which point each sample was extended at 5 mm min−1 until failure. Seven samples were run per composition.Dynamic mechanical analysis
Rectangular dynamic mechanical analysis samples (2.0 cm × 0.5 cm × 0.2 cm) were prepared via 3D printing. Samples were analysed in tension mode using autotension mode, with a frequency of 1 Hz, a preload force of 1 N, and a static force of 0.1 N (DMA; Mettler-Toledo TT-DMA system (Mettler-Toledo AG, Schwerzenbach, Switzerland)). The measurements were analysed using Mettler-Toledo STARe v.10.00 software. Three samples were used in each analysis. Thermal sweeps were conducted at 2 °C min−1, starting at −80 °C and ending at 160 °C before cooling to ambient conditions at an average initial rate of 10 °C min−1 to 60 °C, followed by 2 °C min−1 to 20 °C, at which point the scaffold was cycled again for 15 cycles. The peak ratio between the loss and storage moduli (E′′/E′, tanδ) was defined as the glass transition temperature, Tg. This method was also used to determine curing kinetics of the films.
Differential scanning calorimetry (DSC)
Thermal analysis by DSC (DSC3+, Mettler Toledo, AG, Schwerzenbach, Switzerland) was conducted on approximately 1 mg samples hermetically sealed in aluminium pans and placed in the thermal cell. Samples were chilled from room temperature to −80 °C before being heated to 200 °C at a cooling/heating rate of 10 °C min−1, cycled twice to obtain three heating cycles. The half-height transition of the pseudo-second order transition of the enthalpy measurement was taken as the Tg, with analysis performed in StarAnalysis (Mettler Toledo, AG, Schwerzenbach, Switzerland).Contact angle
Surface energy calculations were estimated by contact angle measurements performed using a Krüss DSA25S drop-shape analyser (Hamburg, Germany) at 20 °C. Spin-coated polymer films were subjected to 2 μL sessile drops of two solvents (deionised water and diiodomethane) and measured using Youngs–Laplace fitting. Surface energy was split into dispersive and polar contributions from Fowkes theory and calculated from the Owens equation. Measurements were performed in triplicate and standard deviation is shown as uncertainty.3D printing
Scaffolds based upon previously reported geometries were printed from resins using varied conditions dependent upon composition. Resins were added in 100 mL quantities to the resin tray, allowing for complete and even coverage of the optical window and the surface of the printing plate. Printing parameters were individually determined for each resin composition through optimization of irradiance, irradiation time, resulting film thickness, and semi-quantified feature resolution (percentage of theoretical resolution), and were further optimized in the printing vat as necessary. Porous scaffolds were printed by applying the photomask (MiiCraft 50×, BURMS, Jena, Germany) with a λ = 365 nm light source. Per slice time was varied according to photorheology experiments to produce robust solid structures which could support subsequent polymer layers. The final structures were rinsed with acetone to remove residual resin and photoinhibitor, as denoted by colour removal.Results and discussion
Resin synthesis and crosslinking
The thiol–ene ‘click’ reaction between limonene and various thiols has been discussed in a number of different publications previously.31 In our hands, however, while the 1H NMR spectroscopic analysis of the product of the reaction between PETMP and limonene confirmed the preferential addition of the thiol to the exocyclic alkene (in preference to the endocyclic alkene), in contrast to the observation of chemoselective addition to the exocyclic alkene observed by Firdaus et al.,30 our results indicate that about 27% of the endo-alkene moieties react with the thiol compared to 93% of the exo-alkene species (ESI Table 1†). Extension of this study to other terpenes revealed that in linalool, the addition of thiols was more evenly distributed between the alkenes with 93% tri-substituted alkene and 80% di-substituted alkene consumed (ESI Table S2, Fig. S13†). Similarly, for nerol and geraniol, approximately 45% of the stereogenic alkene was consumed while 31% of the non-stereogenic alkene consumed at similar rates between both monomers (ESI Tables S3 and S4, Fig. S13†). Terpinene was the least reactive of the examined monomers, with less than 10% total alkene conversion after 8 h of irradiation.A limitation of our spectroscopic probing is the high conversion of the endo alkene to a thioether bond at equimolar concentrations of thiol and alkene in the crosslinking limonene and linalool systems; previous studies have indicated that higher concentrations of thiols are necessary to achieve conversions above 50%.30 In this study, gelation was found to occur (and thereby limit our ability to probe subsequent crosslinking with 1H NMR spectroscopy after gelation for the limonene and linalool monomers) within 60 s, while the nerol and geraniol systems could be examined over the course of 2 h.
The spectroscopic analysis of thiol addition was supported by photorheological studies in which monomer systems underwent rapid phase transitions (formation of a solid polymer film from the liquid resin) and exponential increases in storage moduli over the course of irradiation (Table 1). For limonene and linalool, the phase transition took place within 4 and 10 s respectively (ESI Fig. S14†), with steady state mechanical behaviours achieved by 12 s for both systems (Fig. 2A), which indicates great promise for the two monomer systems with regards to their printability in vat polymerisation processes. The sluggish reaction between thiols and the alkene units in nerol, geraniol, and terpinene as observed by 1H NMR spectroscopy also manifested in slower crosslinking reactions. Only after prolonged irradiation times were increases in stiffness of the resins observed, with nearly 30 min necessary for the onset of storage modulus increase and phase transition for the geraniol, and approximately 112 min until a steady state storage modulus value was achieved. The nerol system was even less reactive, with an initial marginal (yet reproducible) increase in storage modulus immediately upon irradiation, followed by the phase transition peak occurring at ca. 92.6 min and a steady state storage modulus at 111.9 min. The limonene and linalool materials display storage moduli of 167 kPa and 149 kPa, respectively, compared to 15 kPa and 107 kPa for nerol and geraniol respectively. This indicates that not only are the isomer networks slower to react, but that the nerol- and geraniol-based networks may not possess sufficient mechanical stability for printing. Terpinene was found to not gel within the examined time period of 4 h, and while the steady state storage modulus value was achieved within 3.2 s, the magnitude was still within the qualitative realm of a liquid resin (0.415 kPa), indicating that the resin does not undergo sufficient gelation to produce a solid network.
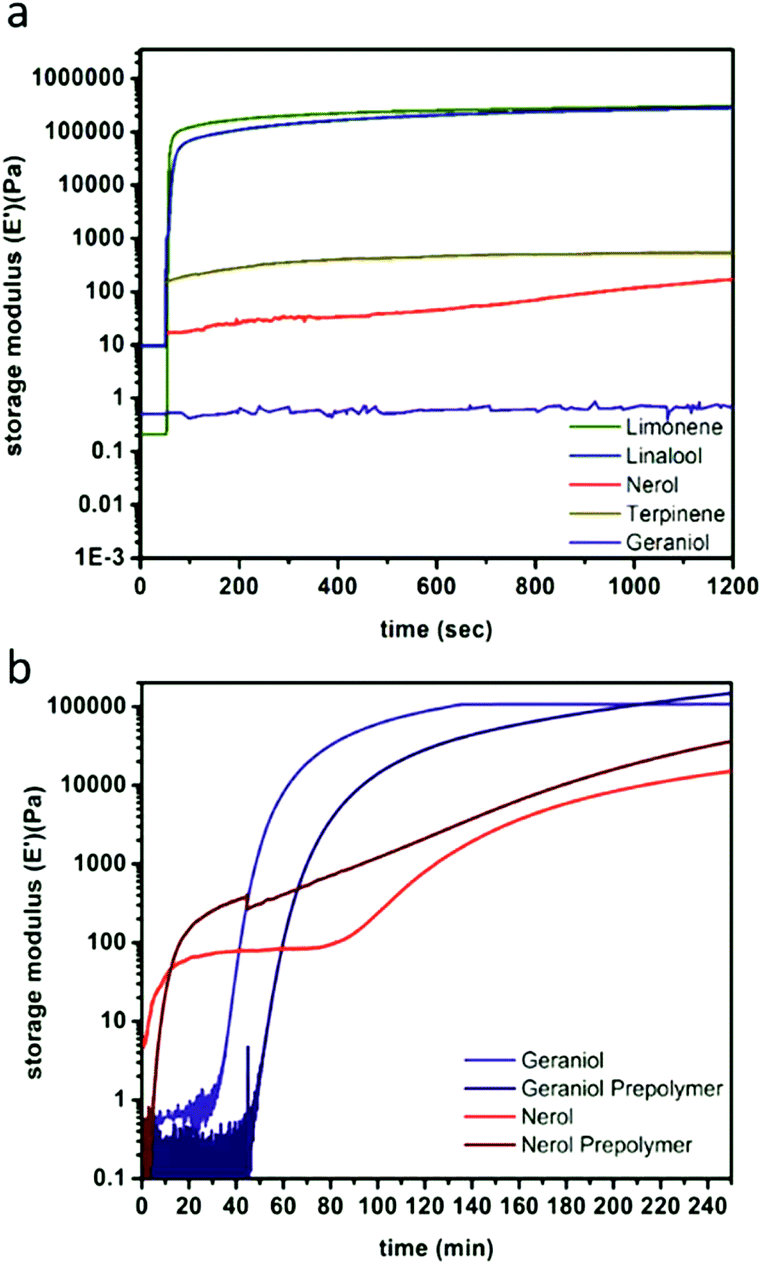 | ||
| Fig. 2 Representative photorheology of monomer-derived resins over 1200 s (a) and comparing monomer and prepolymer nerol and geraniol over 3.5 h (b) as determined by network storage moduli. | ||
δ) and storage modulus steady state, along with the storage modulus of the network after gelation
The limitations of the nerol, geraniol, and terpinene monomer systems, as well as the low viscosities of the limonene system (linalool displayed a slightly higher viscosity attributed to the hydroxyl group), inspired us to increase resin viscosity through the use of a prepolymer, with the hope of expanding the possible library of useful 3D printable terpene materials. Initial prepolymer networks were synthesised by reacting half of the thiol groups with the same quantity of alkenes used earlier, followed by irradiation over the course of 4 h at λ = 365 nm. The limonene prepolymer was formed after the consumption of around 50% of the alkenes from a mixture of 2:1 alkene:thiol, preferentially consuming the exo-alkene groups in limonene to produce a slightly viscous prepolymer that contained residual alkene moieties. The addition of further thiol (to reach 1:1 thiol:alkene stoichiometry) led to solidification that ultimately resulted in decreased times to gelation as measured by photorheology (Fig. 2B, ESI Fig. S14 & S15†).
Photorheology of the nerol and geraniol systems indicated that monomer stereochemistry affects reactivity, as the Z-alkene isomer (nerol) proceeds through two distinct reaction stages (Fig. 2B) as indicated by the rapid increase in storage modulus over the initial 18.3 min followed by a plateau until the second increase beginning at 1.3 h. 1H NMR spectroscopy indicated that the E-alkene monomer (geraniol) is more reactive compared to the nerol, but that irradiation at λ = 365 nm also drives isomerization of the stereogenic alkene towards an equilibrium (Fig. 3, ESI Fig. S13a†). Both isomers are interconverted, although the E-alkene isomer is preferred and reaches an equilibrium of 75:25 E:Z for the remaining, unreacted alkene. This behaviour agrees with what was displayed by photorheological examinations of the prepolymer and the monomers. This observation means that (despite the different reactivity of E and Z isomers to undergo thiol–ene addition) under irradiation, the more rapidly consumed geraniol isomerizes to a mixture of the less reactive Z-alkene monomer (nerol) during the prepolymer synthesis which in turn reduces the rate of phase transition and increases the time to the steady state mechanical behaviour (Fig. 2C). Conversely, nerol isomerizes to a mixture possessing the more reactive E-alkene isomer, thereby increasing its reactivity.
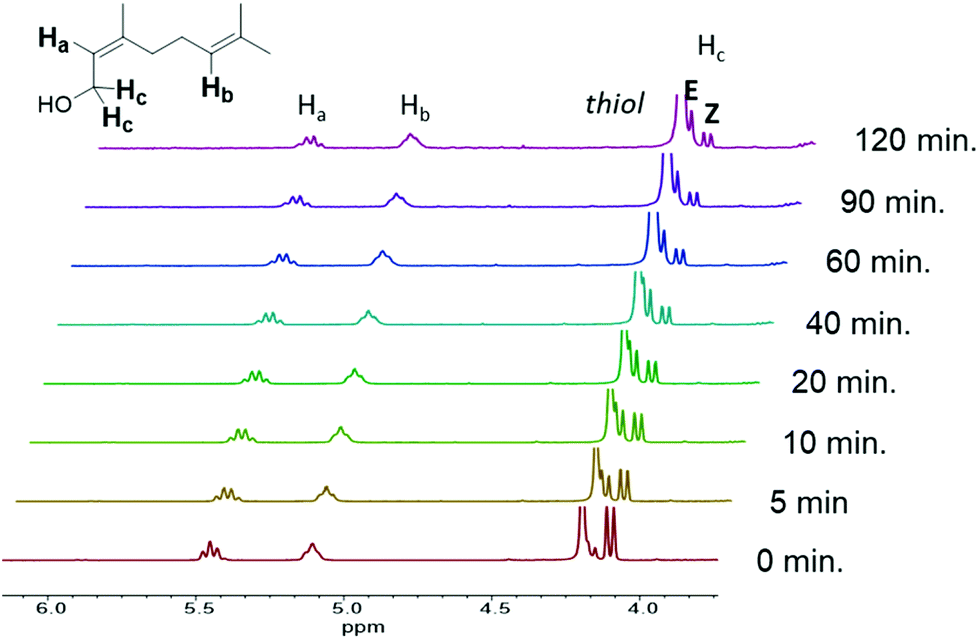 | ||
| Fig. 3 1H NMR spectrum of nerol prepolymer, with the terpene reacting with PETMP under irradiation at λ = 365 nm, displaying monomer isomerization over the course of 120 min. | ||
Unlike the limonene system, in which ca. 50% of the available alkenes are consumed during prepolymer synthesis (the extent of the alkene consumption is determined by the stoichiometry of the thiol; for the prepolymer system a ratio of 2:1 alkene:thiol was used to restrict network synthesis and prevent premature gelation, ESI Table S5†) or the aforementioned nerol and geraniol prepolymers (ESI Tables S6 and S7†), the linalool system displayed nearly 80% alkene conversion by 1H NMR spectroscopy for prepolymers that were expected to display just 50% conversion (ESI Table S8†).
This is most likely a result of free-radical polymerisation side reactions that produce oligomeric units which could then terminate chain ends by thiol–ene reactions and/or undergo subsequent crosslinking reactions at the remaining alkenes. While myrcene (a triterpene) polymerizes through a different mechanism, similar post-polymerisation techniques have been leveraged for its functionalization.35 Photorheology over a 12 h period supports the NMR spectroscopic findings (ESI Fig. S16†), with a nearly two orders of magnitude increase in storage modulus observed for the exposure of only linalool monomer and photoinitiator (1.5 wt%, no thiol). Despite the higher initial storage modulus achieved by the limonene prepolymer, the linalool prepolymer displayed similar storage modulus values after steady-state mechanical behaviour was reached. Additionally, the initial prepolymer viscosity was approximately an order of magnitude higher (80 and 951 mPa s, respectively).
Formation of the terpinene prepolymer was again not successful as determined by a number of factors, including resin component miscibility and resin reactivity. The two prepolymer components were found to be immiscible upon concentration even after 48 h of irradiation at room temperature, and crosslinking reactions were only possible after 3 days at 120 °C under curing conditions. The low activity of this crosslinking reaction meant that terpinene was not suitable for 3D printing applications, although the films were processed for further examination of their properties.
Thermomechanical testing
Examination of the stress–strain behaviour of the photo-crosslinked materials revealed that they displayed typical elastomeric behaviours with the exception of the limonene which behaved as a more traditional engineering plastic. The displayed strains at break are relatively large for highly crosslinked thermosetting materials, particularly for the limonene system after post-curing at high temperatures.34,36,37 Strain at break was found to correlate with the post-polymerisation post-cure temperature (ESI Fig. S17†), with limonene displaying approximately 180% strain before break after post-curing at 120 °C (after irradiation), but after a 30 °C post-curing step of the same duration, fracture strain was only 77.3% (55.9% strain for the prepolymer) (Fig. 4A, Table 2). The elastomeric behaviour of the samples cured at or below 80 °C was found to be relatively consistent, approximately 0.8 MPa, although toughness (2740 J m−3) and ultimate strength (24.4 MPa) were found to increase with increasing cure temperature. 120 °C for 12 h was found to be the ideal cure conditions, and was used for curing of the other materials prior to examination. When cured under these same conditions, linalool, nerol and geraniol display elastic moduli ranging from 0.4 to 0.5 MPa, with similar strains at break (92 to 110%) ultimate tensile strengths of 2.4 to 2.8 MPa and similar toughness (102 to 140 J m−3). Even with this decreased perfomance relative to the limonene series, these materials still display superior strain at failure compared to contemporary materials that explore the use of other renewable monomers such as myrcene, isosorbide, and eugenol where strains at break are typically 50% or less.38–40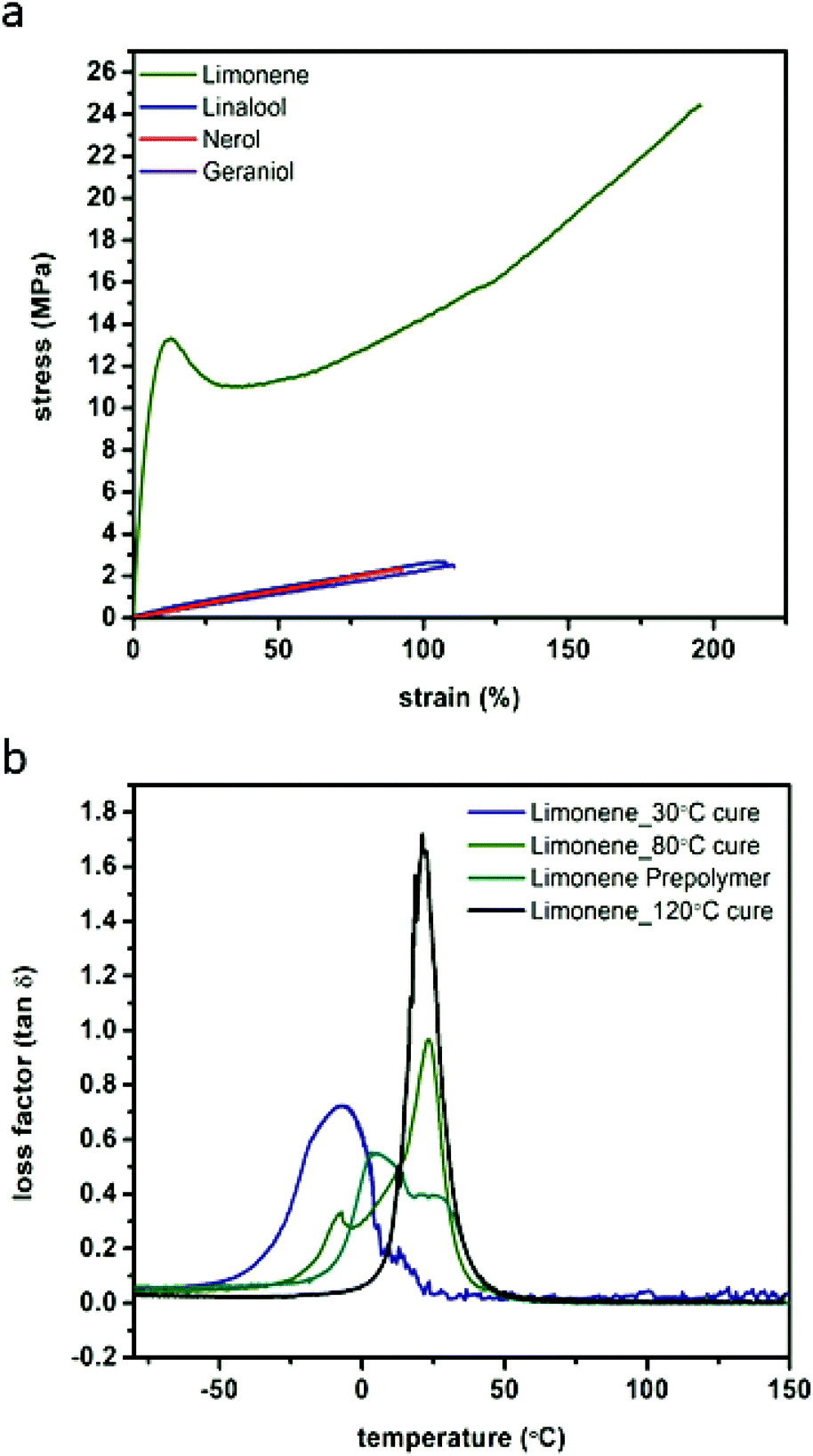 | ||
| Fig. 4 Representative tensile analysis curves performed at ambient conditions comparing monomer structure (a) and DMA loss factor curves comparing curing conditions of limonene networks (b). | ||
Interestingly, the behaviour of the other monomer systems was found to be similar to the uncured limonene (Fig. 4B, ESI Fig. S17†).
Only the limonene was able to display the more characteristic engineering thermoplastic curve shape, with a defined plastic region as opposed to the more elastomeric behaviour of the others. This behaviour is attributed to the limonene polymer network behaviour, which is glassier compared to the other networks as determined by DMA and DSC (ESI Fig. S18 & S19†). For the linalool, nerol, and geraniol, the first loss factor peak (tanδ) located between 11.9 and 15.1 °C (DMA) and half-height transition (DSC) ranging from −3.3 to 2.7 °C, indicate the Tg of the polymers is approximately 20 °C below ambient conditions, the typically accepted threshold for assumed steady-state thermomechanical behaviour as well as outside the full-width half maximum (FWHM) of the loss factor peak as determined by DMA. At ambient conditions, however, limonene has not yet achieved a complete phase transition and therefore will display characteristics of a glassy polymer rather than a fully rubbery polymer as displayed by nerol, geraniol, and linalool. DMA analysis indicates that the FWHM of the tanδ measurements decreased with a post-photo polymerisation thermal cure (Fig. 4B, ESI Table S9†), further supporting the choice of post-cure conditions. Terpinene networks were found to exhibit similar thermal properties (ESI Table S9†), as well. Thermal degradation (ESI Fig. S19b†) is expected to take place at the ester linkages in the PETMP monomer, resulting in the formation of the dicarboxylic acid substituent for nerol, geraniol, limonene, terpinene, and linalool. All of the terpene monomers possess low boiling points, and it was expected that along with pentaerythritol, which displays a boiling point at approximately 275 °C, the thermal degradation products will rapidly be volatized upon reaching 300 °C. This was reproducibly found in the TGA thermograms where a very rapid and abrupt mass loss takes place in the aforementioned region.
Contact angle
Contact angle measurements of the film surfaces indicate that the limonene is the most hydrophobic of the materials, as determined by both water contact angle (Fig. 5) and by diiodomethane contact angle. This indicates a low degree of possible wetting for these materials (ESI Table S10†) which agrees with a qualitative assessment of the monomer structures. Linalool, nerol and geraniol all display greater hydrophilic tendencies as a result of the hydroxyl groups that are present. The lower water contact angles displayed by these materials indicate that the surfaces are more hydrophilic (assuming consistent surface roughness as a result of similar processing conditions). Ultimately, the increased total surface energy of the monomers relative to the limonene is attributed again to polymer mobility, in the same manner as the thermomechanical behaviour. With the linalool, nerol and geraniol being tested well above their Tg, the polymer surfaces are more mobile and therefore display more liquid-like behaviour (as determined from steady-state loss moduli values determined from photorheology and DMA). The outlier behaviour of the terpinene is attributed to the difficulties in fully processing the material, as opposed to an unusual hydrophilic interaction displayed by the material (as indicated by the increased total surface energy compared with the other monomer systems) (ESI Fig. S20†). | ||
| Fig. 5 Representative water contact angle images of terpene polymer spin-coated film surfaces. | ||
3D printing
Resins for microstereolithography 3D printing were made using stoichiometric amounts of alkene and thiol (2:1, terpene:PETMP), along with 1.5 wt% photoinitiator and 1.5 wt% photoinhibitor. Based on the previously discussed photorheology study and the characterized relationships between irradiance, gelation/solidification, and resolution (ESI Fig. S21†), similar times were used for exposures of each 50 μm slice. Qualitatively, linalool was the most effective monomer for printing due the combination of its low viscosity and its reactivity, with limonene requiring additional exposure times to induce photo-crosslinking at a sufficient degree to provide structure support between layers during the peeling process of DLP. In fact, the limonene monomer system was only utilized for producing pseudo-3D structures (Fig. 6A) (2D designs with a height component but minimal part complexity in the z-axis), while linalool resins were suitable for producing porous Hart's cubes (Fig. 6B, ESI Fig. S22†).
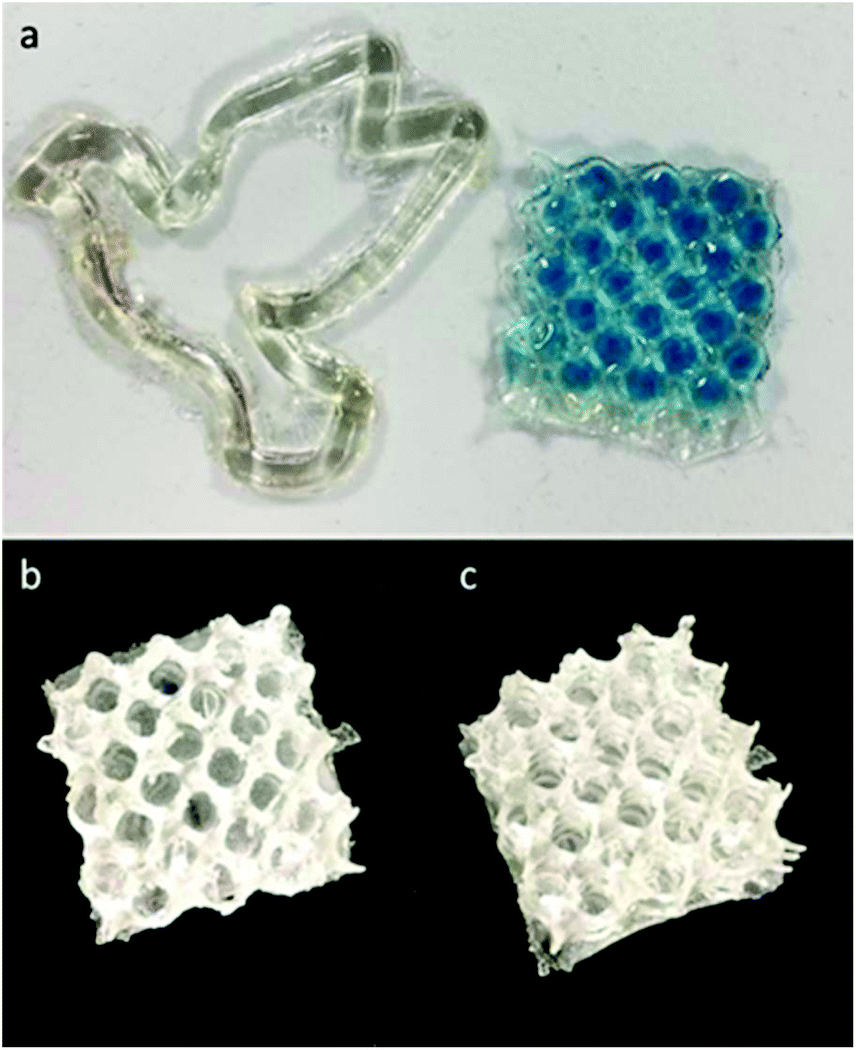 | ||
| Fig. 6 Examples of 3D printed structures displaying a “2D” dove and porous mesh (a) and the corresponding 3D printed Hart cubes from linalool (b) and limonene prepolymer (c) resins. | ||
There are two considerations for these materials when addressing terpene monomer/prepolymer suitability for 3D printing: reactivity and viscosity. As such, we postulated that the reduced viscosity of the limonene may cause more resin flow during the print pulling process, reducing structural integrity and preventing part solidification. Hence, to achieve 3D printable resins from limonene, the limonene prepolymer was applied. This could be successfully printed (Fig. 6C) into the same porous structure as the linalool resin. Nerol and geraniol, as well as the prepolymers, were not successfully printed into 3D structures as a consequence of their slow photo-crosslinking behaviour.
While the nerol, geraniol, and terpinene monomers or prepolymers were not suitable for direct printing, the low viscosity of the terpene in combination with its reactivity could make them suitable as additives in resin inks. As such, the use of nerol and geraniol as diluents was further exploited in order to reduce part shrinking. While unreactive diluents such as propylene carbonate may cause severe part shrinking and therefore cracking, application of either nerol or geraniol as reactive diluents (up to 10 wt% were examined) led to reduced part shrinking to less than 1%, as determined both through rheology measurements during photocuring of thin films (measured by the offset of the parallel plates) and through measurements of the final printed parts (bulk metrology). Additional considerations for these materials are extensive, as their 3D printing opens up avenues into 3D printable antimicrobial agents, recyclable printing resins, optically transparent printed parts, and even biomaterials with tunable thermomechanical and surface properties.41–43
Conclusions
Overall, novel resins for photopolymerisation 3D printing were obtained from naturally-derived monomer sources. The crosslinking of terpenes with PETMP by radical thiol–ene addition enabled their ready solidification under irradiation with UV light. These materials were successfully 3D printed in a vat polymerisation process, with minimal shrinkage but with a wide spectrum of thermomechanical behaviours possible through the tuning of the material composition and thermal curing profiles utilized. Ultimately, these materials demonstrate great potential as 3D printing materials, as the strains at failure exceeding 170% are far superior to contemporary examinations, and possible further avenues of exploration indicates that these materials have potential as biomedical or commercial materials with environmentally-friendly qualities.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The authors gratefully acknowledge funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curia grant agreement (ACW MCEF #793247, JCW #751150).References
- S. A. M. Tofail, E. P. Koumoulos, A. Bandyopadhyay, S. Bose, L. O'Donoghue and C. Charitidis, Mater. Today, 2018, 21, 22–37 CrossRef.
- D. Rejeski, F. Zhao and Y. Huang, Addit. Manuf., 2018, 19, 21–28 CrossRef.
- D. P. Eyers and A. T. Potter, Comput. Ind., 2017, 92–93, 118–136 Search PubMed.
- V. S. D. Voet, T. Strating, G. H. M. Schnelting, P. Dijkstra, M. Tietema, J. Xu, A. J. J. Woortman, K. Loos, J. Jager and R. Folkersma, ACS Omega, 2018, 3, 1403–1408 CrossRef CAS PubMed.
- F. P. Melchels, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 6121–6130 CrossRef CAS PubMed.
- S. M. Oskui, G. Diamante, C. Liao, W. Shi, J. Gan, D. Schlenk and W. H. Grover, Environ. Sci. Technol. Lett., 2015, 3, 1–6 Search PubMed.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mulhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- P. N. Stockmann, D. L. Pastoetter, M. Woelbing, C. Falcke, M. Winnacker, H. Strittmatter and V. Sieber, Macromol. Rapid Commun., 2019, 40, e1800903 CrossRef PubMed.
- N. Teng, S. Yang, J. Dai, S. Wang, J. Zhao, J. Zhu and X. Liu, ACS Sustainable Chem. Eng., 2019, 7, 8715–8723 CrossRef CAS.
- A. A. Wroblewska, S. Stevens, W. Garsten, S. M. A. De Wildeman and K. V. Bernaerts, ACS Sustainable Chem. Eng., 2018, 6, 13504–13517 CrossRef CAS PubMed.
- G. L. Gregory, E. M. Hierons, G. Kociok-Köhn, R. I. Sharma and A. Buchard, Polym. Chem., 2017, 8, 1714–1721 RSC.
- P. Ray, T. Hughes, C. Smith, M. Hibbert, K. Saito and G. P. Simon, Polym. Chem., 2019, 10(24), 3334–3341 RSC.
- R. Kakuchi, S. Yoshida, T. Sasaki, S. Kanoh and K. Maeda, Polym. Chem., 2018, 9, 2109–2115 RSC.
- L. V. Kayser, E. M. Hartigan and B. A. Arndtsen, ACS Sustainable Chem. Eng., 2016, 4, 6263–6267 CrossRef CAS.
- A. S. Amarasekara, B. Wiredu and A. Razzaq, Green Chem., 2012, 14(9), 2395–2397 RSC.
- P. Sahu, P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2017, 5, 7659–7669 CrossRef CAS.
- M. Winnacker and J. Sag, Chem. Commun., 2018, 54, 841–844 RSC.
- N. Elbers, C. K. Ranaweera, M. Ionescu, X. Wan, P. K. Kahol and R. K. Gupta, J. Renewable Mater., 2017, 5, 74–83 CrossRef.
- M. Winnacker, Angew. Chem., Int. Ed., 2018, 57, 14362–14371 CrossRef CAS PubMed.
- P. A. Tierney, C. D. Karpinski, J. E. Brown, W. Luo and J. F. Pankow, Tob. Control, 2016, 25, e10–e15 CrossRef PubMed.
- J. Meehan-Atrash, W. Luo and R. M. Strongin, ACS Omega, 2017, 2, 6112–6117 CrossRef CAS PubMed.
- C. H. Ho, W. H. Griest and M. R. Guerin, Anal. Chem., 2002, 48, 2223–2226 CrossRef PubMed.
- M. Tsujimura, M. Tsuji, H. Kofujita and T. Ohira, J. Wood Sci., 2015, 61, 308–315 CrossRef CAS.
- M. Tsujimura, M. Goto, M. Tsuji, Y. Yamaji, T. Ashitani, K.-i. Kimura, T. Ohira and H. Kofujita, J. Wood Sci., 2019, 65, 19 CrossRef.
- K. S. Cho, Y. R. Lim, K. Lee, J. Lee, J. H. Lee and I. S. Lee, Toxicol. Res., 2017, 33, 97–106 CrossRef CAS PubMed.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- A. Stamm, M. Tengdelius, B. Schmidt, J. Engström, P. O. Syrén, L. Fogelström and E. Malmström, Green Chem., 2019, 21, 2720–2731 RSC.
- M. R. Thomsett, J. C. Moore, A. Buchard, R. A. Stockman and S. M. Howdle, Green Chem., 2019, 21, 149–156 RSC.
- L. Peña Carrodeguas, C. Martín and A. W. Kleij, Macromolecules, 2017, 50, 5337–5345 CrossRef.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- M. H. Alves, H. Sfeir, J. F. Tranchant, E. Gombart, G. Sagorin, S. Caillol, L. Billon and M. Save, Biomacromolecules, 2014, 15, 242–251 CrossRef CAS PubMed.
- N. Kindermann, À. Cristòfol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- F. V. Drozdov, G. V. Cherkaev and A. M. Muzafarov, J. Organomet. Chem., 2019, 880, 293–299 CrossRef CAS.
- K. T. Wacker, A. C. Weems, S. M. Lim, S. Khan, S. E. Felder, A. P. Dove and K. L. Wooley, Biomacromolecules, 2019, 20, 109–117 CrossRef CAS PubMed.
- A. Matic and H. Schlaad, Polym. Int., 2018, 67, 500–505 CrossRef CAS.
- A. C. Weems, J. K. Carrow, A. K. Gaharwar and D. J. Maitland, Macromolecules, 2018, 51, 9078–9087 CrossRef CAS.
- A. C. Weems, K. T. Wacker and D. J. Maitland, J. Appl. Polym. Sci., 2019, 136(35), 47857 CrossRef.
- T. Modjinou, D.-L. Versace, S. Abbad-Andallousi, N. Bousserrhine, J. Babinot, V. Langlois and E. Renard, ACS Sustainable Chem. Eng., 2015, 3, 1094–1100 CrossRef CAS.
- J.-T. Miao, L. Yuan, Q. Guan, G. Liang and A. Gu, ACS Sustainable Chem. Eng., 2018, 6, 7902–7909 CrossRef CAS.
- T. Liu, L. Sun, R. Ou, Q. Fan, L. Li, C. Guo, Z. Liu and Q. Wang, Chem. Eng. J., 2019, 368, 359–368 CrossRef CAS.
- Y. Zhou, Y. Ye, W. Zhang, S. Li, J. Chen, S. Wang, D. Li and C. Mu, Carbohydr. Polym., 2016, 154, 13–19 CrossRef CAS PubMed.
- Y. Lyu, H. Ren, M. Yu, X. Li, D. Li and C. Mu, Carbohydr. Polym., 2017, 174, 1095–1105 CrossRef CAS PubMed.
- J. Dai, S. Ma, L. Zhu, S. Wang, L. Yang, Z. Song, X. Liu and J. Zhu, Polymer, 2017, 108, 215–222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00950g |
| This journal is © The Royal Society of Chemistry 2019 |