
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ electron microscopy observation of the redox process in plasmonic heterogeneous-photo-sensitive nanoparticles†
Diego
Muraca
 *a,
Lucia B.
Scaffardi
bc,
Jesica M. J.
Santillán
b,
David
Muñetón Arboleda
b,
Daniel C.
Schinca
bc and
Jefferson
Bettini
*d
*a,
Lucia B.
Scaffardi
bc,
Jesica M. J.
Santillán
b,
David
Muñetón Arboleda
b,
Daniel C.
Schinca
bc and
Jefferson
Bettini
*d
aInstituto de Física ‘Gleb Wataghin’, Universidade Estadual de Campinas, Campinas, Brazil. E-mail: dmuraca@ifi.unicamp.br
bCentro de Investigaciones Ópticas (CIOp) (CONICET La Plata-CIC-UNLP), Gonnet, La Plata, Buenos Aires, Argentina
cFacultad de Ingeniería, UNLP, La Plata, Buenos Aires, Argentina
dElectron Microscopy Laboratory, Brazilian National Nanotechnology Laboratory/CNPEM, Brazil. E-mail: jefferson.bettini@lnnano.cnpem.br
First published on 5th August 2019
Abstract
Observation of relevant phenomena related with dynamical redox process in a plasmonic heterogeneous-photocatalyst system composed by silver nanoparticles (NPs) around and in contact with amorphous silver chloride NPs are reported by in situ transmission electron microscopy. During this process, nanobubbles are initially produced inside the silver chloride NPs, which immediately begin to move within the amorphous phase. Besides, silver atoms inside the silver chloride NPs start to migrate out the occupied volume leaving a space behind, which is filled by crystalline regions of silver chloride located between the pre-existing silver NPs. During the observation time, fast-nucleation, movement, growth, and fast-dissolution of silver NPs take place. Specific space correlation with silver mass loss (or gain) when a new NP is formed (or dissolved), was detected in different regions during the reaction. This mass loss (or gain) takes place on certain places of pre-existing silver NPs. All these phenomena were observed for a configuration comprising at least two silver NPs separated few nanometers apart by a silver chloride NP.
Introduction
Understanding the catalytic properties of nanosystems requires extensive investigation by multidisciplinary techniques to collect information at different scales of their chemical, structural and electronic properties.1–3 During the last years, nanoplasmonic probes with catalytic materials used as an efficient plasmonic photocatalyst have gained relevant interest due to the improvement of their photocatalytic properties as well as their stability by the coupling between the material and plasmonic NPs.4–6 Several examples of the enhancement of photocatalytic activity by plasmonic NPs suggest that they are the critical point for developing an efficient plasmonic photocatalyst as well as a photovoltaic nanostructure.5,7–12Catalytic reactions observed by in situ transmission electron microscopy may provide a unique understanding of the involved processes due to the resolution attained with this technique.3 This understanding is essential for the development of catalysts, which in turn are of fundamental importance for energy and environmental technologies.13–15 Effects of intense electron probes, such as material accumulation and hole formation, that depend on the induced surface diffusion or sputtering mechanisms, were reported using field-emission transmission electron microscope.16,17 However, when in situ transmission electron microscopy is performed on a nanosystem composed by NPs with a specific configuration and an electron-beam sensitive material (as the previously mentioned), the resulting interaction gives rise to new phenomena. Herein, we report the experimental and direct observation of redox–driven reaction (AgCl + 1e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Ags + Cl−) of photo-sensitive and catalytic material composed of metallic silver NPs (Ag-NPs) and silver chloride nanoparticle (AgCl-NP). The electron dose used during all processes was about 2000 e Å−2 s during movie acquisition (five movies) with a vacuum environment of 1.5 × 10−5 Pa.
Ags + Cl−) of photo-sensitive and catalytic material composed of metallic silver NPs (Ag-NPs) and silver chloride nanoparticle (AgCl-NP). The electron dose used during all processes was about 2000 e Å−2 s during movie acquisition (five movies) with a vacuum environment of 1.5 × 10−5 Pa.
Two articles were published recently regarding in situ electron microscopy studies on Ag–AgCl NPs systems.13,14 The first one, published by Wu et al.,13 reported on the oxidation dynamics of an Ag/AgCl heterostructure using in situ environmental TEM microscopy in combination with DFT theory, working with a temperature controlled flow nanoreactor to independently change temperature and oxygen pressure. Their results show that sputtering of Cl atoms under the impact of incident electrons is much more likely than that of Ag atoms, thus leaving behind Ag atoms which oxidize to Ag2O or AgO depending on the low or high (atmospheric) O2 pressure in the flowing nanoreactor. The authors did not report any “mass transport” phenomena.
The other recent article published is a letter of S. Du et al.,14 where they reported in situ electron microscopy studies on Ag–AgCl NPs systems. Although the studied systems are very similar to ours, they focused on the mentioned lattice fluctuations and the structural evolution of AgCl–Ag during electron microscopy experiments. Du et al. also observed nucleation and fast growth of Ag NP within the AgCl domain. The cited work those not describe completely the ion disassembling and diffusion, neither how the charge distribution on the particles evolves.
Among other things, our work aims to clarify some unclearly understood phenomena. Here, we analyze in detail the redox reaction of Ag–AgCl–Ag nanosystem at different stages, from the beginning when nanobubbles are initially produced inside the silver chloride NPs and immediately begin to move. We also recorded (more than once) and analyze fast-nucleation and fast-dissociation of small silver NP on the crystalline AgCl phase. From our analysis, we corroborate that the new NP nucleation involves a fast mass transport correlated with specific places in the original Ag-NPs and, when the small NP dissolves, the observed correlation persists on a cyclic-type basis behaviour. In addition, we showed not only the nucleation of small Ag nanoparticle on the AgCl nanoparticle, but also movement of atoms in detail. We observed that any portion of the smaller silver NPs in contact with AgCl can be partially or completely dissolved, while the major one grows significantly. Hence, we proved that the AgCl is the key for Ag NP dissolution. We verify that the processes of nucleation, movement and dissolution of small Ag NP within the AgCl NP require a specific configuration consisting of at least two silver NPs separated few nanometers apart by a silver chloride NP and in the capture zone.18 Finally, we noted that AgCl defects are important not only for Ag growth at the Ag–AgCl interface, but also for playing an essential role as nucleating points for the growth of new small Ag NPs.19
A Beginning of the reaction
Initially, the system is mainly composed by Ag-NPs (always more than one) and a metastable AgCl-NP between and in contact with the Ag-NPs (Movie 1 of the ESI†), as shown on the temporal evolution of Fig. 1. The AgCl-NP is composed by a majority of amorphous phase with a minor crystalline phase, as well as with some possible chlorine nanobubbles moving inside. Although a crystalline phase is more stable than an amorphous one, the volumetric evolution from amorphous to crystalline phase in AgCl-NP was not directly observed in the experiments, instead, other effects were seen and analyzed. At the first stage of the reaction, we noticed two main processes over time: the growth of the bigger Ag-NP with the dissociation of AgCl-NP followed by the formation, increase and movement of nanobubbles inside the AgCl-NP. | ||
| Fig. 1 This figure corresponds to different times of Movie 1 (of the ESI†) at the initial stage of the reaction with 3 Ag NPs and AgCl NP in between them. The corresponding times are shown on each image. Darkest contrasts correspond to the Ag NP, whereas less dark contrast corresponds to the AgCl phase. The insets inside of each figure correspond to a fast Fourier transform (FFT) of the region inside the red box for each frame. The FFTs show weak spots indicating the existence of a minority crystalline phase with a majority of amorphous phase. Two nanometer sized nanobubbles (brighter contrast) are present on the system within the AgCl-NP at 9.8 s. Yellow arrows indicate places with presence of nanobubbles. | ||
The growth of the larger Ag-NP from the dissociation of AgCl-NP may be attributed to different possible effects, like inelastic energy transfer to the AgCl-NP, plasmons produced by the electron beam and sputtering effects.13,16,17 The inelastic energy transferred to the AgCl-NP can break the chemical bonding, generating Ag+ and Cl− ions inside and mainly on the surface of AgCl-NP. In turn, Cl− ions may form Cl2 molecules inside and on the surface of AgCl-NP. The Cl2 molecules on the surface can be ejected due to the weak bonding with the surface, leaving remnant Ag+ ions inside and on the surface of AgCl-NP. Another possible effect is the sputtering of Ag and Cl atoms, which can occur at the AgCl-NP surface.19
Related to sputtering effects, the results obtained by the majority of the authors referred to crystalline materials. However, sputtering phenomena can present some similar behavior in our material due to the observed crystalline phase. During the scattering of an electron, the energy transfer to the nucleus will depend on the mass of the scattering atom (M), the kinetic energy of the incident electron (E0), and electron rest mass (m0) according to
| E ≈ 2E0[(2E0 + 2m0c2)/Mc2]sin2(θ/2), | (1) |
| Emax ≈ 2E0[(2E0 + 2m0c2)/Mc2]. | (2) |
For 200 keV (E0 experimental condition) the maximum energy per electron that can be transferred to Ag and Cl atoms is 4.86 eV and 14.80 eV respectively, and the electron dose used during all process was kept near 2000 e Å−2 s during the experiments. The surface binding (considering the difference an atom or molecule in gas and bulk phase) energy to remove an atom on the AgCl surface is 2.53 eV and 4.07 eV for Ag and Cl atom respectively, and the minimum incident electron energy required for surface sputtering is 116.6 eV for Ag atom and 61.7 eV for Cl atom.13,17 Hence, Raymond F. Egerton verifies the higher probability of sputtering Cl atoms than Ag atoms from the surface of an AgCl, indicating a higher sputtering rate for Cl than for Ag atoms.17 Then, Yimin et al. showed that the sputtering process creates an excess of Ag+ ions on the surface, while the Cl− ions are forming a gas phase.13 The Ag+ ions of the AgCl-NP generated by the elastic and inelastic processes involved in electron collisions acquire enough momentum and kinetic energy transferred from the electron beam, allowing diffusing interstitially on the surface to the Ag-NP.13,20
Apart from the mentioned processes, electron-flux (from the electron beam) that induced plasmon excitation on the particles must be regarded as the cause of some phenomena during the reaction. It has been reported that electrons can interact with metallic nanostructures like NPs, films and nanowires, inducing the presence of localized surface plasmons.19 Plasmons in electron microscopy experiments can either decay radiatively or via inelastic interactions allowing electronic excitations like e–h pair creation and secondary electron emission near the surface, similar to SPR-mediated charge injection from metal to semiconductor on photon SPR-mediated reaction.5,6,22,23 Hence, plasmons can induce hot-electron generation on the interface between a metal NP and the AgCl semiconductor.10 We could expect that during the initial stage of the reaction, the chlorine ions diffuse on the AgCl, reducing and forming chlorine gas which, in turn, form bubbles. The remaining silver ions diffuse to the silver NP. Finally, the Ag+ ions can be reduced on the border of pre-existing Ag-NPs accepting an electron from them and possibly assisted by surface plasmons induced by the electron beam.
During the formation and movement of nanobubbles inside the AgCl-NP, Cl2 molecules formed by the inelastic and elastic processes can diffuse through holes generated by the lack of silver atoms and form the nanobubbles by aggregation.24 After nanobubbles formation they begin to move slowly, limited by the AgCl-NP during more than 160 s as can be clearly observed in Movie 2 (ESI†). Growth dynamics, nucleation and migration of nanobubbles were previously observed by electron microscopy on other materials as graphene and lead-iron.24,25
The assumption of nanobubbles existence (initially with a spherical shape) is based on the lower contrast observed in some parts of the AgCl-NP (see also Movie 2, Fig. S1 and S2 on ESI†). Images of scanning transmission electron microscopy (STEM) acquired during the in situ energy dispersive spectroscopy corroborated the presence of nanobubbles during the reaction of the analogous system (ESI, Fig. S2†). Comparing the size of AgCl-NPs at the beginning and the end of the reaction, the initial thickness of AgCl-NP is estimated to be 14.2 nm, assuming a lamellar shape. Therefore, the size of the nanobubbles fits inside of AgCl-NP. Suggesting the possible existence of a solid shell with an electrical potential on the AgCl-NP surface (FFT insets on Fig. 1 and ESI Fig. S1†). The electron beam can induce phase transformation from amorphous to crystalline by energy transfer.26–28 The interface between the AgCl-NP and vacuum can reduce the thermodynamic barrier for the crystallization process like heterogeneous nucleation, where the weak spots observed in the FFTs can be correlated with the crystalline phase. The sputtering process on the surface may avoid the volumetric phase transition from amorphous to crystalline preventing nanobubbles (also with a net charge inside) to reach the border by diffusion. The number and size of the nanobubbles increase as AgCl-NP thickness is reduced, enabling the AgCl-NP to keep approximately the same 2D surface size (not the thickness). In brief, nanobubbles movement may be related to the rate of the mentioned processes and the number of nanobubbles can be related to the thickness of AgCl-NP during the reaction.
B Nucleation and dissolution of silver nanoparticles
When the thickness of AgCl-NP reaches a critical value (see ESI: Movie 1 from 486.6 s, Movie 2 from 897.6 and Movie 3 from 0 s and Fig. S3†), the region with the darkest contrasts (Movie 1, ESI†) in the AgCl-NP starts to decrease in size from the top-right edge, although not uniformly. Then, “nucleation” and “dissolution” of small silver NPs occur very fast during the changes of the AgCl-NP crystalline regions, and undoubtedly, silver NPs nucleation and AgCl-NP crystalline structure are both interconnected with each other. While the reaction proceeds, new Ag-NP with a diameter smaller than 5 nm “nucleates” in between the other pre-existing Ag-NPs. This “nucleation” happened more than once at different places and at different times (see ESI: Movie 1 from 486.6 s, Fig. S4 to S6†). Each nucleated NP also dissolves, sometimes very fast, with a typical time <0.2 s. Making accurate analyses in the positions where the NPs were formed. It was possible to correlate their formation with the existence of AgCl crystalline phase. We noticed that the atoms that are forming this new NP have a correlation with at least two different and specific places (apart from the ones that could come from the AgCl-NP), denoted by an arrow and the red box of Fig. 2, 585.6 s. A difference in the number of atoms can be evidenced by the dark contrast shown in the inserted image, which corresponds to atoms correlation from the corner of bottom-right NPs surface (see Experimental section for more details of the images analysis). After 0.2 s, the size of the new NP increases. It was possible to show that a certain amount of the atoms responsible for the NP size increase is related with mass loss from the upper left part of the larger pre-existing Ag-NP, as shown in the inset of Fig. 2 (585.8 s), which was obtained by image subtraction. Hence, in the first part of nucleation, the atoms at the upper left corner are allowed to “diffuse”. Then, the new NP increases again mainly by mass loss from the left side corner of the large Ag-NP, confirming again the correlation between some parts on the edge of larger Ag-NP and the new NPs formed on AgCl. It is appropriate to point out here that our experiments are limited by a frame time of 0.2 seconds. Considering that the atoms cluster “diffuse” from one place to another distant 30 nm in 0.2 seconds and considering the relation| (DT)1/2 = d, | (3) |
 | ||
| Fig. 2 Time sequence of the reaction for 0.8 seconds of the Movie 1.† The corresponding times are shown on each image. 585.4 s: shows the initial condition of the time sequence with three original silver NPs and a small silver nanoparticle formed on the AgCl, between the Ag NPs. 585.6 s: small Ag-NP is formed, dark contrast on the small box evidence mass loss from the Ag-NP at the bottom right denoted by red box. The mass difference was obtained by subtraction of figure at 585.6 s from 585.4 s. White arrow shows other place with different mass before and after the nanoparticle formation. 585.8 s: small formed nanoparticle increases its size, dark contrast on the small box evidence mass loss on the Ag-NP at the top left denoted by the red box. The mass difference was obtained by subtraction of figure at 585.8 s from 585.6 s. 586.2: small formed nanoparticle is completed dissolved, bright contrast on the small box evidences mass gain on the Ag-NP at the up left denoted by the red box. The mass difference was obtained by subtraction of figure at 586.2 s from 585.8 s. | ||
As shown in Fig. 2, at an elapsed time of 0.4 s (from 585.8 s to 586.2 s), the small nanoparticle is completely dissolved and a mass gain was recorded at the original place on the outer surface of the pre-existing NP (shown as white contrast in the box on Fig. 2, 586.2 s). From these facts, it can be stated that the formed NPs are in a metastable state. This usually does not happen in particles of the same composition and size isolated on the same experimental conditions. In general, after NP nucleation, it should grow due to diffusion or coalescence mechanisms. These phenomena of NP nucleation (N), dissolution (D), growth (G) and reduction (R) processes between the Ag-NPs were observed several times during one reaction as is shown on Movie 1† listed above: N1-487.6 s, D1-547.4 s, D2-554.0 s, G1-554.4 s, G2-555.6 s, G3-557.6 s, R1-558.0 s, R2-5559.2 s, D2-561.6 s, N2-568.8 s, D3-569.6 s, N3-573.6 s, D4-574.4 s, N4-585.0 s, D5-585.4 s, N5-585.6 s, D6-586.2 s, N6-587.0 s, N7-594.0 s and D7-594.8 s, and on several experiments (see ESI Fig. S4 to S6†). During our experiments, the transport dynamics as well as nucleation, dissolution, growth and reduction observed in situ microscopy experiments cannot be fully described considering classical diffusion mechanisms, i.e., only regarding Frenkel, Schottky defects and/or Fick's first law, nor related theories or the classical mechanism of chemical nucleation and growth of NPs.29,30
C Silver chloride as key for silver nanoparticle dissolution
It was corroborated in our experiments that AgCl is the key component that allows the dissolution of the silver NP. To support this fact, different Ag-NPs of similar sizes in contact and isolated from AgCl were subjected to the same experimental conditions and showed different behaviors. Movie 3 (ESI†) illustrates a configuration where an Ag-NP can or cannot be dissolved during the reaction on in situ TEM experiments. While the Ag-NP isolated from the AgCl remains stable, the ones in contact can be dissolved (see ESI: Fig. S7 and S8†). In addition, this phenomenon was not observed when only one silver nanoparticle is in contact with silver chloride (see ESI: Fig. S9†).Among many physical phenomena involved in this system, two of them could be mainly associated with the dissolution of Ag-NPs. The first one is the reaction between Ag-NP surface and chloride ions (Cl−) forming AgCl on the surface of Ag-NP, and consequent dissolution by an electron beam, silver ions (Ag+) migrates to the largest Ag-NP and chloride ions forming Cl2 gas, which evaporates in the microscope. The second one is the charging and electric field effect, in a junction of metal and dielectric (insulator material) surface charges can be concentrated at the interface between conductor and insulator material. Due to the nature of Ag-NP, a large charge can be accumulated at some edge of Ag-NP in contact with the AgCl, and the charge accumulation can locally heat the surface enough to melt the Ag-NP locally. The melting temperature of NPs decreases when the size is reduced. Therefore, the Ag-NP could systematically (edge by edge) be melted due to charge accumulation effects. It has been reported that electric field, which implies charge accumulation, was used to dissolve silver and gold NPs in glass.31,32
One more evidence of Ag-NP dissolution on the AgCl-NP phase is shown in Fig. 3, which corresponds to time composition variation of the system followed by energy dispersive spectroscopy (EDS) in equal sample and with similar nanoparticle configuration. During the STEM experiment (see Movie 4, Fig. S2 and S3, ESI†) the movement of a nanobubble in the AgCl-NP, detected by dark contrast, was acquired at different times. Also, after some time (Movie 4,† 1858 s), a dark contrast begins to increase on the upper right edge with a square shape, related to the AgCl crystalline shell structure of the AgCl NP and in accordance with results described and observed on Movie 1 (ESI†).
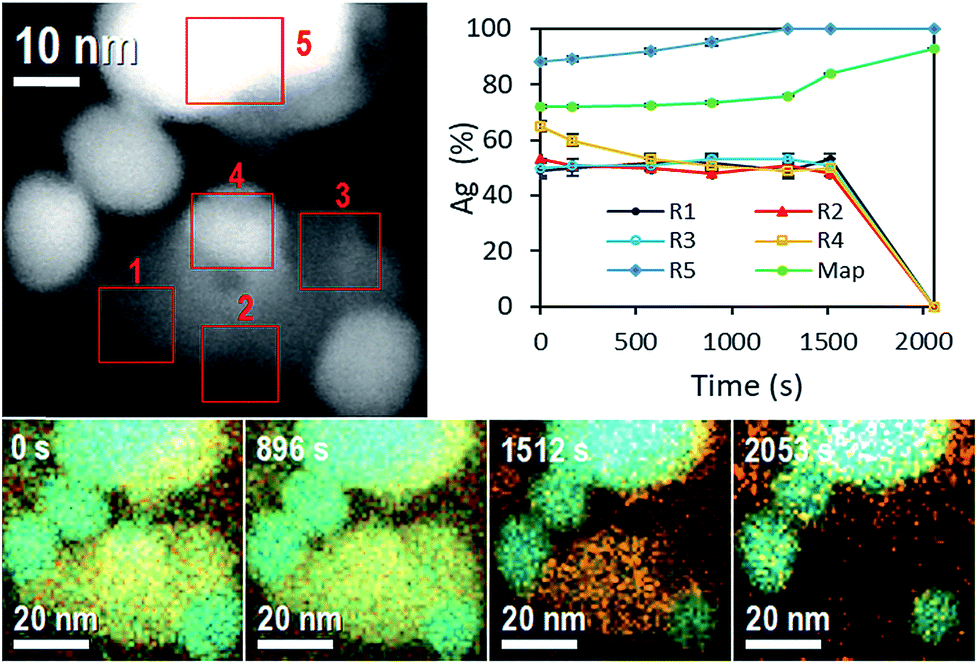 | ||
| Fig. 3 Top left: STEM image of in situ energy dispersive spectroscopy showing different regions (R1 to R5) where the atomic composition spectrum was acquired. Top right: atomic composition evolution obtained by means of different regions R1 to R5 and the full map composition. Bottom: in situ energy dispersive spectroscopy showing composition variation for 2053 s. Cyan color corresponds to silver atoms and orange color corresponds to chlorine atom. | ||
On EDS experiment, seven temporal and accumulative composition mapping were obtained from the full image shown in at the top left of Fig. 3, in five different selected regions (R). Regions R1, R2 and R3 correspond only to the mapping of AgCl-NP, and the average of Ag atomic content oscillates around 50% ± 1%, pointing out the Cl2 gas is not trapped significantly inside those regions of the AgCl-NP (the obtained compositions are reported in Tables 1 and 2).
| Ag (% at.) | Cl (% at.) | Time (s) |
|---|---|---|
| 72 ± 0.6 | 28 ± 1.2 | 0 |
| 72 ± 0.5 | 28 ± 1.0 | 169 |
| 73 ± 0.4 | 27 ± 0.8 | 576 |
| 74 ± 0.4 | 26 ± 0.9 | 896 |
| 76 ± 0.3 | 24 ± 0.9 | 1295 |
| 84 ± 0.2 | 16 ± 1.2 | 1512 |
| 93 ± 0.1 | 7 ± 2.2 | 2053 |
| Time (s) | R1 (% at.) | R2 (% at.) | R3 (% at.) | R4 (% at.) | R5 (% at.) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ag | Cl | Ag | Cl | Ag | Cl | Ag | Cl | Ag | Cl | |
| 0 | 49 ± 3 | 51 ± 5 | 53 ± 3 | 47 ± 5 | 50 ± 3 | 50 ± 4 | 65 ± 2 | 35 ± 4 | 89 ± 1 | 11 ± 4 |
| 169 | 50 ± 3 | 49 ± 4 | 51 ± 2 | 49 ± 3 | 51 ± 2 | 48 ± 3 | 60 ± 2 | 40 ± 3 | 88 ± 1 | 11 ± 3 |
| 576 | 52 ± 2 | 48 ± 3 | 50 ± 2 | 50 ± 3 | 51 ± 2 | 49 ± 3 | 53 ± 2 | 47 ± 2 | 92 ± 1 | 8 ± 3 |
| 896 | 52 ± 2 | 48 ± 3 | 48 ± 3 | 52 ± 3 | 53 ± 2 | 47 ± 3 | 51 ± 2 | 49 ± 3 | 95 ± 1 | 4 ± 3 |
| 1295 | 49 ± 3 | 51 ± 4 | 51 ± 3 | 49 ± 3 | 53 ± 2 | 47 ± 3 | 49 ± 2 | 51 ± 3 | 100 ± 1 | 0 ± 3 |
| 1512 | 53 ± 2 | 49 ± 4 | 48 ± 3 | 52 ± 4 | 51 ± 4 | 49 ± 5 | 50 ± 3 | 50 ± 4 | 100 ± 0 | 0 ± 4 |
| 2053 | 00 ± 0 | 00 ± 0 | 00 ± 0 | 00 ± 0 | 00 ± 0 | 00 ± 0 | 00 ± 0 | 00 ± 0 | 100 ± 0 | 0 ± 5 |
However, in R2 a reduction of Ag atomic content was observed twice (from 49.2 to 46.6%, and 50.8 to 45.7%) from 576 s to 896 s and from 1295 s to 1512 s, some amount of Cl2 gas can be trapped inside of AgCl-NP during the acquisitions, see Movie 4 (ESI: Fig. S2 and S3†); the R2 also shows a darkening closely at the same time variation, from 1170 s to 1502 s. Initially, the R4 shows a silver NP on the AgCl-NP surface and Ag composition of 65%. The Ag content decreases to 50% at 576 s due to the dissolution of Ag-NP (during all experiments, Ag-NPs were utterly dissolved when they were entirely on the AgCl-NP surface). No enrichment of Ag content was observed in R1, R2, and R3, indicating no absorption of Ag atoms by the AgCl-NP, supporting the hypothesis that Ag atoms migrate to other Ag-NP, preferably to the large one. The R5 shows enrichment of Ag content over the time due to the redox process of AgCl-NP, where the final measured content was 98% at. of Ag. For the full sample mapping, the silver content increases slightly (72.1% at. to 76.1% at.) from 0 to 1295 s and rate of Ag content (76.1% at. to 92.7% at.) increases from 1295 s to 2053 s (see Table 2). This time corresponds roughly to the moment at which the white contrast of AgCl-NP starts to reduce in size in Movie 4.† Other EDS experiments are reported on ESI (Fig. S10 and Table S1†).
D Silver chloride defects as preferential nucleation's points
The reasons for specific places on the preexisting NPs showed mass loss/gain when the new NPs are formed/dissolved, and what guides the NP movement after its formation, could be due to different causes. It was mentioned before that silver NPs nucleation and AgCl-NP crystalline structure should be linked by a certain interrelationship. Recently J. S. Du et al., observed lattice oscillation on AgCl during a redox reaction on electron microscopy experiments on equal Ag–AgCl system.14 We also observed this lattice oscillation during our experiments (not shown here). These lattice dynamics could locally stretch or stress favoring a specific place for nanoparticle nucleation. Hence, to prove the possible relation between AgCl phase and Ag nucleation and dissolution, the AgCl phase was analized before and after each nucleation event during the experiment. Fig. 4 shows in more detail the analysis of the nucleation process shown previously in Fig. 2. The FFT (Fig. 4(b)) from the red box shows some regions marked with orange circles with higher intensity than the background of the FFT in the same radial distance, and corresponds to (220) AgCl planes distance (0.20 nm). | ||
| Fig. 4 (a) Image before the nucleation. Red box denotes the region where nucleation occurs (585.4 s), (b) FFT of the region denoted by red box; inset white box denotes 2× zoom of region marked with orange circles. (c) Inverse FFT of the same region (2× zoom in), see also ESI Fig. S11† (d) inverse FFT with the background removed (2× zoom in) with red lines showing stressed planes (0.24 nm) and green lines showing stretched planes (0.32 nm), (e) inverse FFT with the background removed and coloured with orange colour, (f) image with the nucleated particle and same region marked by red box, (g) image of the marked red box in panel (f) with 2× zoom in (585.6 s), (h) FFT of the region denoted by red box (585.6 s) with the (200) Ag planes marked with a blue mask on (h), (i) inverse FFT from the points marked with a blue mask, (200) Ag planes and (j) composite image with coloured orange and blue images. | ||
The four spots with the highest intensities correspond to (200) AgCl planes distance (0.28 nm) and the weakest one corresponds to (220) AgCl planes distance (0.20 nm), making 45 degrees with the (200) AgCl planes distance. Therefore, the AgCl planes are oriented in the [001] zone axis. However, the (220) planes distance denoted by orange circles are at 60, 70 and 90 degrees with the (200) AgCl planes respectively (white box denotes 2× zoom of region marked with orange circles), note also the complementary side in the FFT to better observe these regions. The (200) and (220) at 90 degrees correspond to [011] zone axis orientation, and in this orientation, the (111) planes can appear and have 0.32 nm distance and produce some strain in this region. In fact, the Fig. 4(d) shows some (200) AgCl planes stretched and stressed with 0.32 nm and 0.24 nm distance (0.32 + 0.24 = 2 × 0.28) inside the white circle. (111) plane is stretching the (200) AgCl plane and as a compensation some planes are stressed and showing 0.24 nm distance. Red lines for stressed planes (0.24 nm) and green lines for stretched planes (0.32 nm) mark these regions. This defect could create a region with some charge or field gradient, attracting Ag ions to nucleate in this region, as was observed in other materials.33,34 However, more studies need to be done to fully support this claim. Fig. 4(g) shows the region of the nucleated Ag-NP and Fig. 4(h) the corresponding FFT. In order to see the matching of the nucleated particle and the (200) defects regions, Ag spots were selected by a mask and the inverse FFT shown in Fig. 4(i) was colored blue. The Fig. 4(j) shows the composite image obtained from Fig. 4(e) and (i).
The sequential images on Fig. 5(a) to (f) correspond to the growth of nucleated Ag-NP, i.e., in this specific case we are not analysing the nucleation (shown previously in Fig. 4). The FFT from the red box, Fig. 5(b) shows regions with the intensity higher than the background of the FFT denoted by orange circles. The plane distance of these regions is 0.255 nm, and the average angle between them is 57 degrees. The (111) Ag planes have 0.236 nm (fcc-structure), and two (111) Ag planes are only observable when the NP is oriented in the [110] zone axis. However, the angle between the (111) Ag planes in the [110] zone axis is 70.5 degrees, the angle measured in the Fig. 5(b) is close to the angle (54.7 degrees) between the (200) and (111) Ag planes in the [110] zone axis. The distance of (200) Ag planes is 0.204 nm. The ambiguous relation between angle and distances of the regions marked in the orange circles can produce stretched (200) Ag planes between (111) Ag planes. Indeed, Fig. 5(d) shows inside the white circle some parts of (111) AgCl planes with stretched (200) Ag planes. Red lines for stretched (200) Ag planes (0.215 nm) and green lines for (111) Ag planes mark these regions.
 | ||
| Fig. 5 This figure corresponds to the growth of nucleated Ag-NP showed during the Movie 1 (ESI†). From left to right, from up to down, (a) image before the growth of nucleated Ag-NP with the region where the growth occurs denoted by a red box (577.6 s), the inset image correspond to the FFT of the region also marked with a red box and indicated by the red arrow, (b) the FFT of the region denoted by red box, (c) the inverse FFT of the same region (2× zoom in), (d) the inverse FFT with the background removed (2× zoom in); see also ESI Fig. S11† (e) the inverse FFT with the background removed and colored with orange color, (f) the image with the Ag-NP grown and same region marked by red box, (g) the image marked by red box with 2× zoom in (577.8 s), (h) the FFT of the region denoted by red box (577.8 s), (i) the image obtained from the subtraction of images (g) and (c) and (j) the composite image with image (i) and colored orange image. | ||
Fig. 5(d) shows two arrows, the bottom one indicates a vacancy defect and the top one shows a change in the angle of (111–200) Ag planes generating highly stressed Ag plane, very close to the arrow, with 0.148 nm. Again, these defects can create a region with some field gradient, attracting the Ag ions, and explain the growth of Ag-NP observed in the Fig. 5(f). Fig. 5(g) shows the region marked with a red box in the Fig. 5(f) with 2× zoom in. The red box on Fig. 5(a), indicated by the red arrow, shows the FFT region in the Fig. 5 (577.6 s). This region corresponds to the primary place from where the mass of Ag atoms is reduced when the growth of Ag-NP accurs. The (200) and (111) spots showed in the FFT has almost the same orientation of the places circled in Fig. 5(b), showing a possible relationship between Ag-NP planes orientation and the crystal orientation of the pre-existing NPs. Fig. 5(h) shows the FFT of the region marked with a red box, showing two clear spots with 0.231 nm distance and 54.5 degrees between them, suggesting that the relation between angle and distances between (111) Ag planes is not the only reason evidencing stretched (200) Ag planes. However, on the next sequential time image (not shown here) a reduction in the Ag-NP particle size is observed, corroborating that the growth can be related to the stretched (200) Ag planes. Fig. 5(i) shows the results of the difference between the Fig. 5(g) and (e). Fig. 5(j) shows the composite image acquired from Fig. 5(e) and (i). Hence, before nanoparticle nucleation, during NPs size increase and after its dissolution, crystal strain in this region was detected, expressed locally on stretched and stressed of the AgCl phase (see also ESI Fig. S12†).
These AgCl defects can explain the possible existence of preferential space regions for the news NP nucleation and growth. However, they would not explain the correlation of Ag atoms loss/gain mass places when the NPs are formed/dissolved. Even more, it was observed (and can be clearly observed on Movie 1 of ESI†), that the new formed NPs can show an unexpected behavior after its formation, “moving” between the other metallic NPs for several seconds (see ESI Movie 1† from 551.8 s to 559.8 s, for example). This ion cluster or NP moves at a higher velocity than the bubbles at the initial stage of the reaction and seems to be attracted by the metallic NPs. In the experiments, we never observed similar phenomena for preexisting Ag-NPs smaller than 7 nm, or the distance among them smaller than 2–3 nm.
E Plasmons and induced fields
As it is known and, electrons passing near a surface metallic can generate different electric field contributions (other than dipolar).5,6,21 In the simplest approach, small metallic sphere with specific permittivity immersed in a material with different permittivity exhibits plasmon modes at frequencies determined by multipolar order excitation and both permitivities.35 As was mentioned previously, on the contact region between the Ag-NP and the AgCl-NP, these plasmon modes will allow electron transfer, promoting the chemical reduction of the silver ions on the silver chloride and in the edge of the silver NP. After some time, most of the AgCl-NP will have a larger amount of crystalline AgCl phase than the amorphous phase, i.e., the amount of available chlorine and silver for the redox reaction is strongly lowered, and the excited electron from the silver surface cannot be injected to the AgCl-NP at the same rate. Hence, near-field electromagnetic mechanisms will persist, and the semiconductor experiences localized electric field effects near the metallic phase that can affect the semiconductor properties.21 Often, these fields are of high intensity (higher than fields generated by photon/electron at the sample position) and non-uniform in space, allowing an increase of e–h rate formation on specific places.10,21 In other words, this can be understood considering that the metallic nanostructures act as space-localized amplifiers leading to an increment of e–h pair formation near the plasmonic nanostructure.36,37 Then, surface plasmon resonance (SPR) electrons leads to energy accumulation, which will be concentrated in the regions where the metallic NP is not in contact with the AgCl outer surface, allowing the atoms there to diffuse. Simultaneously, plasmon modes of two or more NPs can interact with each other leading electromagnetically hybridized electromagnetic states and a non-uniform electric field between the NPs.20,38,39 Hence in our Ag–AgCl nanoparticle configurations (two or more Ag NPs separated few nanometers apart with a AgCl semiconductor or dielectric with electrochemistry properties) the electrons from the electron beam will electrically polarize the NPs surface and will possibly generate a time-dependent electric potential between them mediated by the dielectric allowing to the new NP experiment a net and variable force. Specific electric field configuration on the plasmonic NPs and/or the AgCl matrix can induce “hot regions” that may contribute to the NP movements or dissolution. It must also be considered that the local charge environment changes where new NP is formed. Plasmons may be generated on this new NP, thus redistributing energy and inducing its possible dissolution, together with AgCl structure modification.Conclusions
In summary, we report by in situ electron microscopy observation different stages and new phenomena of the dynamical redox process in plasmonic heterogeneous-photo-sensitive NPs. Initially with nanobubbles formation, growth and movement. Then we register nucleation and dissolution of small silver NPs. Was verify that silver chloride NP is the key for the silver NPs dissolution, i.e., the presence of silver chloride matrix is a necessary condition for silver NP dissolution. Also, it was corroborated that defects on silver chloride NP acts as nucleation points to new silver NPs, during the reaction. We verify, as well, space correlations places, between the Ag and AgCl NPs, during this dynamical process, when a NP is nucleated or dissolved.The nucleation, dissolution, growth and reduction events observed in our experiments, together with the previous discussion leads us to think that plasmons and the electric field configuration between small metallic NPs are influencing the reaction.
These discoveries could contribute to improving the design of materials with photocatalytic properties, not necessarily restricted to AgCl material.
Experimental section
A Materials synthesis
Silver colloidal suspensions were generated using a 120 fs pulse width Ti:Sapphire chirped pulse amplification system from Spectra Physics, centred at 800 nm wavelength, 1 kHz repetition rate and a maximum output pulse energy of 1 mJ. A solid silver disk (99.99%) placed at the bottom of a 4 mL beaker filled with Dihydrated Sodium Citrate (DSC) aqueous stabilizer solution at a concentration of 1 mM and containing 30 mg kg−1 of ion chloride, was used as ablation target. Ablation was performed by focusing the laser beam using a 5 cm focal length lens on the Ag disk producing a fluence of 5.9 J cm−2. An XYZ motorized micrometric stage was used to move the target to ablate always on a fresh surface of the Ag sample. After about 10 minutes, a typical yellowish colour suspension indicating the presence of silver NPs was obtained.B Electron microscopy equipment, settings and methods
In situ TEM analysis was performed on a JEOL JEM 2100F with a field emission gun (FEG) operating at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kV. The samples were prepared by a simple colloid dropping of the as-prepared colloids on amorphous carbon film supported on copper grids. In situ energy dispersive spectroscopy (EDS) was also acquired during the reaction using an Oxford SDD 100 mm2, X-MAX model and AZTEC software for EDS data acquisition and processing. High-angle annular dark-field imaging (HAADF) JEOL SIOD digital scanning system with HAADF detector was used. All the experiments were performed at room temperature. The images were acquired using an SC600 CCD camera (Gatan) in camera view mode (live mode). Each image was constructed with a time frame of 0.2 s and binning 2 to increase the intensity (contrast) of the image. The movies were acquired using AutoScreenRecorder software (screen capture software) with an acquisition time of 0.2 s.
kV. The samples were prepared by a simple colloid dropping of the as-prepared colloids on amorphous carbon film supported on copper grids. In situ energy dispersive spectroscopy (EDS) was also acquired during the reaction using an Oxford SDD 100 mm2, X-MAX model and AZTEC software for EDS data acquisition and processing. High-angle annular dark-field imaging (HAADF) JEOL SIOD digital scanning system with HAADF detector was used. All the experiments were performed at room temperature. The images were acquired using an SC600 CCD camera (Gatan) in camera view mode (live mode). Each image was constructed with a time frame of 0.2 s and binning 2 to increase the intensity (contrast) of the image. The movies were acquired using AutoScreenRecorder software (screen capture software) with an acquisition time of 0.2 s.
For EDS analysis, we assume the whole AgCl NP area in Fig. 3 (isolated from the part with that Ag-NP is in contact or on AgCl) as our standard sample (50–50%). We also used the first three measures of whole AgCl NP to obtain an average standard sample with Ag 50% and Cl 50%. Therefore, we calculated the EDX quantifications with their respective errors. They were obtained from Poison error, and two sigma error bars was considered to describe the uncertainty for any measurement. The mass gain/loss image analysis process is detailed in ESI Fig. S15.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank FAPESP, C2NANO-Brazilian Nanotechnology National Laboratory (LNNano) at Centro Nacional de Pesquisa em Energía e Materiais (CNPEM)/MCT (Research Proposals #16976, #19927 and #18425). This work was Granted by PIP 0280 of CONICET, MINCyT-PME 2006-00018, 11/I197, Facultad de Ingeniería of Universidad Nacional de La Plata, Argentina. Fabrication of NPs by Ultrafast Pulse Laser Ablation was carried out at CIOp (CONICET, CICBA and UNLP), La Plata, Argentina. We would like to acknowledge Prof. Edson R. Leite, Scientific Director of Brazilian Nanotechnology National Laboratory, for useful discussion during the manuscript production.Notes and references
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek and J. Fernández Sanz, et al. , Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butche, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef CAS PubMed.
- F. F. Tao and M. Salmeron, Science, 2011, 331, 171–174 CrossRef CAS PubMed.
- E. M. Larsson, C. Langhammer, I. Zorić and B. Kasemo, Science, 2009, 326, 1091–1094 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- D. Chen, T. Li, Q. Chen, J. Gao, B. Fan, J. Li, X. Li, R. Zhang, J. Sun and L. Gao, Nanoscale, 2012, 4, 5431–5439 RSC.
- M. Long and W. Cai, Nanoscale, 2014, 6, 7730–7742 RSC.
- C. G. Silva, R. Juárez, T. Marino, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef PubMed.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- C. An, J. Wang, W. Jiang, M. Zhang, X. Ming, S. Wang and Q. Zhang, Nanoscale, 2012, 4, 5646–5650 RSC.
- A. Wu Yimin, L. Li, L. Zheng, A. Kinaci, M. K. Y. Chan, Y. Sun, J. R. Guest, I. McNulty, T. Rajh and Y. Liu, ACS Nano, 2016, 10, 3738–3746 CrossRef CAS PubMed.
- S. J. Du, J. Park, Q. H. Kim, W. Jhe, V. P. Dravid, D. Yang and D. A. Weitz, J. Phys. Chem. Lett., 2017, 8, 5853–5860 CrossRef PubMed.
- N. Divins, I. Angurell, C. Escudero, V. Pérez-Dieste and J. Llorca, Science, 2014, 346, 620–623 CrossRef CAS PubMed.
- H. Yoshida, Y. Kuwauchi, J. R. Jinschek, K. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta and S. Takeda, Science, 2012, 335, 317–319 CrossRef CAS PubMed.
- R. F. Egerton, F. Wang and P. A. Crozier, Microsc. Microanal., 2006, 12, 65–71 CrossRef CAS PubMed.
- R. A. Puglisi, G. Nicotra, S. Lombardo, C. Spinella, G. Ammendola, M. Bileci and C. Grardi, Surf. Sci., 2004, 550(10), 119–126 CrossRef CAS.
- N. Itoh, Nucl. Instrum. Methods Phys. Res., Sect. B, 1987, 27, 155–166 CrossRef.
- A. L. Koh, K. Bao and I. Khan, ACS Nano, 2009, 3, 3015–3022 CrossRef CAS PubMed.
- F. J. García de Abajo, Rev. Mod. Phys., 2009, 82, 209–275 CrossRef.
- Y. Tang, Z. Jiang, G. Xing, A. Li, P. D. Kanhere, Y. Zhang, T. C. Sum, S. Li, X. Chen, Z. Dong and Z. Chen, Adv. Funct. Mater., 2013, 23, 2932–2940 CrossRef CAS.
- H. Petek and S. Ogawa, Annu. Rev. Phys. Chem., 2002, 53, 507–531 CrossRef CAS PubMed.
- K. Niu, T. Frolov, H. L. Xin, J. Wang, M. Asta and H. Zheng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 42 CrossRef PubMed.
- D. Shin, J. Bo Park, Y.-J. Kim, S. J. Kim, J. H. Kang, B. Lee, S.-P. Cho, B. H. Hong and K. S. Novoselov, Nat. Commun., 2015, 6, 6068 CrossRef PubMed.
- Z. Huang, J. Qi, L. Zhou, Z. Feng, X. Yu, Y. Gong, M. Yang, Q. Shi, N. Wei and T. Lu, J. Appl. Phys., 2015, 118(21), 214901 CrossRef PubMed.
- H. Inui, H. Mori, T. Sakata and H. Fujita, J. Non-Cryst. Solids, 1990, 116(1), 1–15 CrossRef CAS.
- X. Qua and Q. Deng, RSC Adv., 2017, 7, 37032–37038 RSC.
- H. Mehrer, Diffusion in Solids, Springer Berlin Heidelberg, New York, 2007 Search PubMed.
- W. D. Compton and R. J. Maurer, J. Phys. Chem. Solids, 1956, 1, 191–199 CrossRef.
- A. Podlipensky, A. Abdolvand, G. Seifert, H. Graener, O. Deparis and P. G. Kazansky, J. Phys. Chem. B, 2004, 108, 17699–17702 CrossRef CAS.
- Z. Zou, Q. Wang, X. Chen and S. Qu, J. Appl. Phys., 2009, 105, 103114 CrossRef.
- R. Ishikawa, S. D. Findlay, T. Seki, G. Sánchez-Santolino, Y. Kohno, Y. Ikuhara and N. Shibata, Nat. Commun., 2018, 9, 3878 CrossRef PubMed.
- S. Fang, Y. Wen, C. S. Allen, C. Ophus, G. G. D. Han, A. I. Kirkland, E. Kaxiras and J. H. Warner, Nat. Commun., 2019, 10, 1127 CrossRef PubMed.
- C. Kittel, Quantum Theory of Solids, Wiley, New York, 1963 Search PubMed.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga and H. Murakami, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS PubMed.
- Z. Liu, W. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Nano Lett., 2011, 11, 1111–1116 CrossRef CAS PubMed.
- B. H. Kim, J. Yang, D. Lee, B. K. Choi, T. Hyeon and J. Park, Adv. Mater., 2017, 1703316 Search PubMed.
- S. KadKhodazadeh, J. Rosenkrantz de Larsson, M. Beleggia, H. Kneipp, J. B. Wagner and K. Keipp, J. Phys. Chem. C, 2014, 118, 5478–5485 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Movies description (Movie 1, Movie 2, Movie 3, Movie 4), supplementary information (Fig. S10 and S11), Table S1 and image data processing details (Fig. S11 to S13). See DOI: 10.1039/c9na00469f |
| This journal is © The Royal Society of Chemistry 2019 |