
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly soluble fluorine containing Cu(I) AlkylPyrPhos TADF complexes†
Jasmin M.
Busch
 a,
Daniel M.
Zink
a,
Patrick
Di Martino-Fumo
b,
Florian R.
Rehak
c,
Pit
Boden
b,
Sophie
Steiger
b,
Olaf
Fuhr
d,
Martin
Nieger
e,
Wim
Klopper
*c,
Markus
Gerhards
*b and
Stefan
Bräse
*af
a,
Daniel M.
Zink
a,
Patrick
Di Martino-Fumo
b,
Florian R.
Rehak
c,
Pit
Boden
b,
Sophie
Steiger
b,
Olaf
Fuhr
d,
Martin
Nieger
e,
Wim
Klopper
*c,
Markus
Gerhards
*b and
Stefan
Bräse
*af
aInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Karlsruhe, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
bChemistry Department, TU Kaiserslautern and Research Center Optimas, Erwin-Schrödinger-Straße 52, 67663 Kaiserslautern, Germany. E-mail: gerhards@chemie.uni-kl.de
cInstitute of Physical Chemistry – Theoretical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany. E-mail: klopper@kit.edu
dKarlsruhe Institute of Nanotechnology (INT) and Karlsruhe Nano-Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: olaf.fuhr@kit.edu
eDepartment of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtasen aukio 1), 00014 Helsinki, Finland. E-mail: martin.nieger@helsinki.fi
fInstitute of Toxicology and Genetics (ITG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 21st August 2019
Abstract
Luminescent Cu(I) AlkylPyrPhos complexes with a butterfly-shaped Cu2I2 core and halogen containing ancillary ligands, with a special focus on fluorine, have been investigated in this study. These complexes show extremely high solubilities and a remarkable (photo)chemical stability in a series of solvents. A tunable emission resulting from thermally activated delayed fluorescence with high quantum yields was determined by luminescence and lifetime investigations in solvents and solids. Structures of the electronic ground states were analyzed by single crystal X-ray analysis. The structure of the lowest excited triplet state was determined by transient FTIR spectroscopy, in combination with quantum chemical calculations. With the obtained range of compounds we address the key requirement for the production of organic light emitting diodes based on solution processing.
Introduction
Organic light emitting diodes (OLEDs) are nowadays a very hot topic for the display and lighting technology, because of their energy efficiency, colour richness and brightness.1,2 Considering the process of OLED production, solution processed techniques are very appealing because of reduced production costs in comparison to the expensive vacuum techniques.3 Therefore the solubility of the materials is one of the most important aspects. Focusing on the emissive layer, luminescent organic compounds often have poor solubilities compared to metal-based emitters, which are in many cases smaller and more flexible in solution. Besides the examples of iridium4 and platinum5,6 complexes as phosphorescent emitters, many highly luminescent complexes with earth-abundant copper have been described in the literature.7–9 Several Cu(I) complexes possess the widely investigated thermally activated delayed fluorescence (TADF), harvesting almost all excitons via the singlet state already at room temperature through reverse intersystem crossing to reach quantum efficiencies of up to 100%.10–18 After the early stages of this technology, including the copper complexes bearing phenanthroline (phen)19 and bis(pyrazol-1-yl)biphenylborate,20 a large class of dinuclear Cu(I) complexes bridged by a N,P-unit, expanded to two ancillary phosphine ligands, came up. A broad range of copper complexes with 2-(diphenylphosphino)pyridine and its derivatives as bridging ligands (Cu(I) PyrPhos complexes) have been described already in the literature.21–28 Especially the complexes with a methylated bridging ligand (MePyrPhos) have been investigated extensively due to higher solubility and a blue-shifted emission of the corresponding complexes. Another approach was to use alkyl-, alkoxy-, mixed or substituted arylphosphines as ancillary ligands to tune the solubility of the Cu(I) complexes.21 Related to these studies the influence of halide containing arylphosphines as ancillary phosphines was investigated in the present study to benefit of the solubility enhancement of especially trifluoromethyl groups and fluorine atoms.29 The corresponding phosphines have only been rarely described for emitting materials based on copper so far.30–36 Often the fluorine based ligands are described for pharmaceutical applications37–41 or in catalysis.42–44 Recently tris(4-trifluoromethylphenyl)phosphine has been reported in a luminescent tetra- and heptanuclear copper iodide cluster.45,46This work presents Cu(I) AlkylPyrPhos TADF complexes including tris(4-fluorophenyl)- (b), tris(4-trifluoromethylphenyl)- (c) and tris(4-chlorophenyl)phosphine (d) as ancillary ligands in combination with a selection of alkylated pyridine bridging ligands, including the novel 2-(diphenylphosphino)-4-tert-butylpyridine (3) as well as the previously described (4-methyl-2-(diphenylphosphino)pyridine47 (2), 4-(cyclopentylmethyl)-2-(diphenylphosphino)pyridine23 (4), and the commercially available 2-(diphenylphosphino)pyridine (1) (Fig. 1).
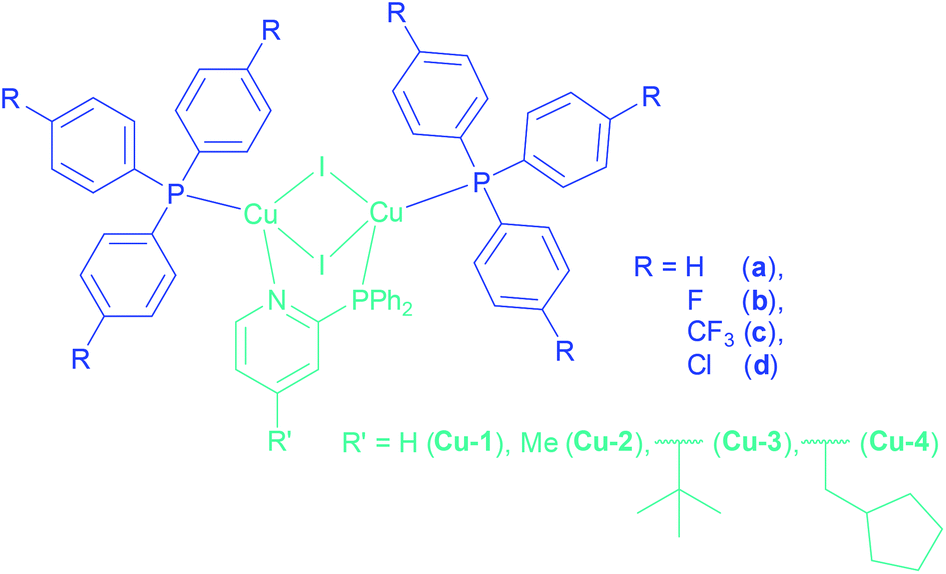 | ||
| Fig. 1 Structures of the highly soluble halide containing Cu(I) AlkylPyrPhos complexes. | ||
Results and discussion
Synthesis
The copper complexes of this work are named according to their corresponding ligands, Cu-LP. Hereby, the number for L indicates the type of bridging ligand and the letter P the ancillary ligand as marked in Fig. 1. The complexes Cu-1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and Cu-2a21,22,24 have already been reported earlier.
22 and Cu-2a21,22,24 have already been reported earlier.
The synthesis of the Cu(I) complexes in this study were performed by stoichiometrically controlled reactions according to literature known procedures (Scheme 1).21,47
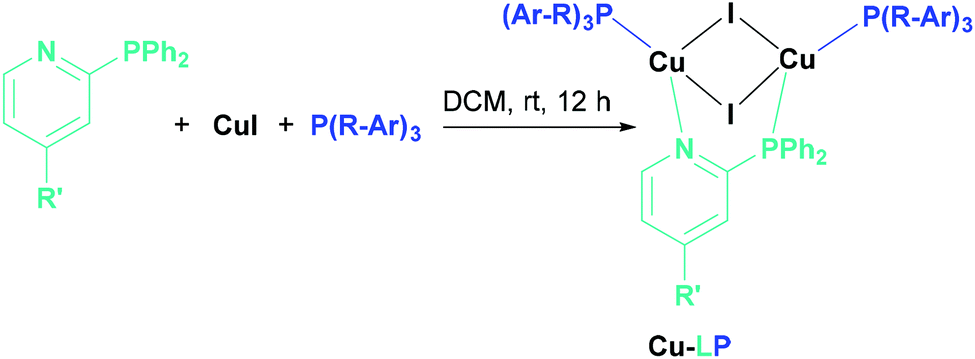 | ||
| Scheme 1 Synthesis of Cu(I) AlkylPyrPhos complexes. | ||
Even ancillary ligands bearing the strong electron-withdrawing fluorine substituent lead to the corresponding Cu(I) complexes of the ratio [Cu2I2LP2]. The synthesis only failed for tris-(pentafluorophenyl)phosphine as ancillary ligand. Probably the donor properties of the lone pair of the phosphine are reduced due to the strong electron-withdrawing properties of the fluorines, leading to the tetranuclear structure, a Cu4I4 unit bridged two times by the PyrPhos ligand, described already by Chen et al.48
Crystallography
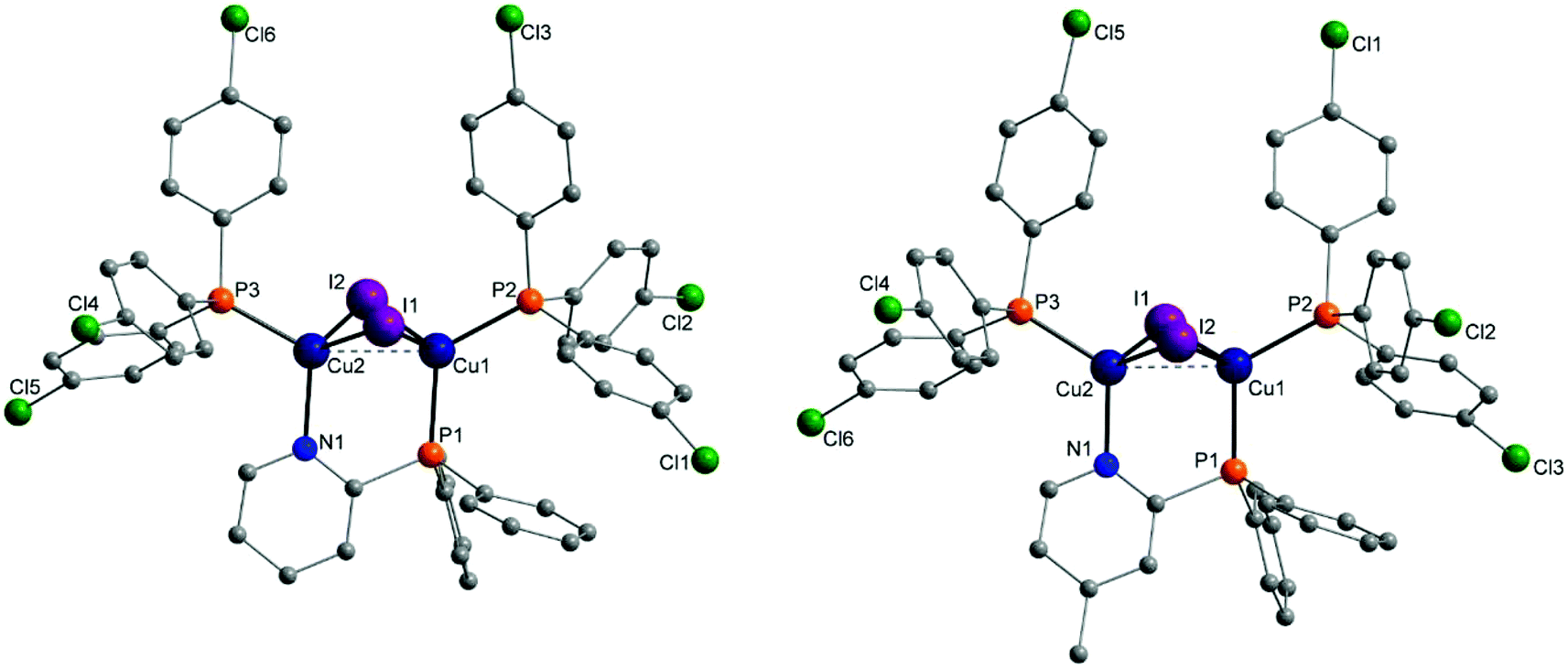
Molecular structures of almost every complex of this work were obtained. For the crystallisation the layering method with dichloromethane (DCM) and n-pentane was used. The structures of the dinuclear copper complexes bearing tris(4-fluorophenyl)phosphine (b) as ancillary ligands and the whole range of AlkylPyrPhos as bridging ligand (R′ = H (1), Me (2), tertBu (3), cyclopentylmethyl (4)), Cu-1b, Cu-2b, Cu-3b and Cu-4b, are shown in Fig. 3. A structure with tris(4-trifluoromethylphenyl)phosphine as monodentate ligand (c) was obtained for Cu-2c. The trifluoromethyl groups in this molecular structure are disordered as expected. For clarity only one isomer of complex Cu-2c is shown in Fig. 2. In 4-position chloro substituted triphenylphosphines serve as ancillary ligands in the structures Cu-1d and Cu-2d (Fig. 4).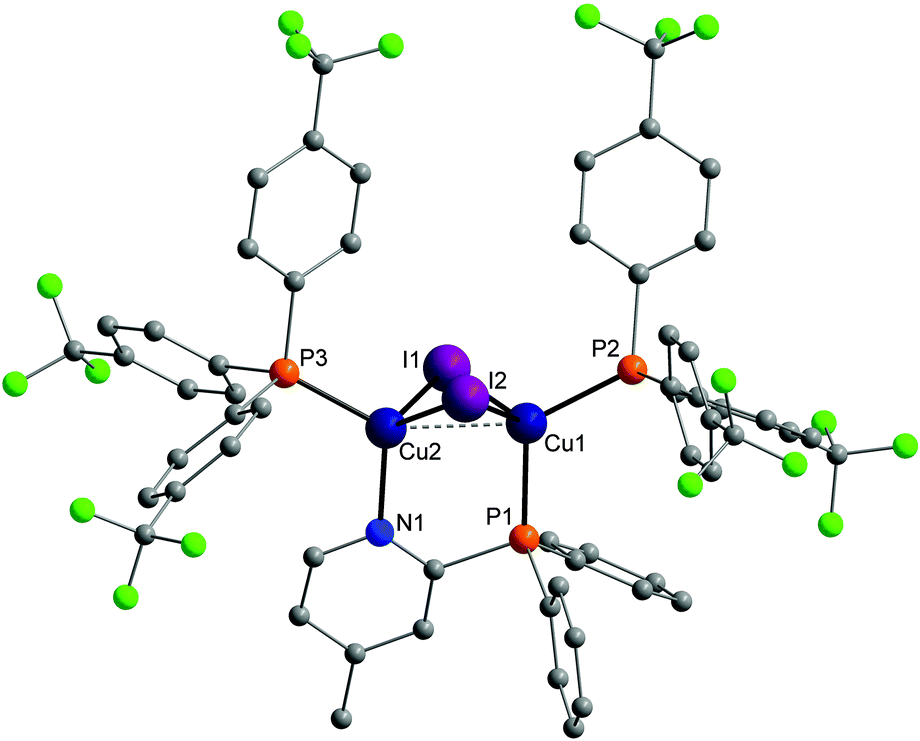 | ||
| Fig. 2 Molecular structure of complex Cu-2c bearing trifluoromethyl containing ancillary ligands. Hydrogen atoms and the disorder of the trifluoromethyl groups were omitted for clarity. | ||
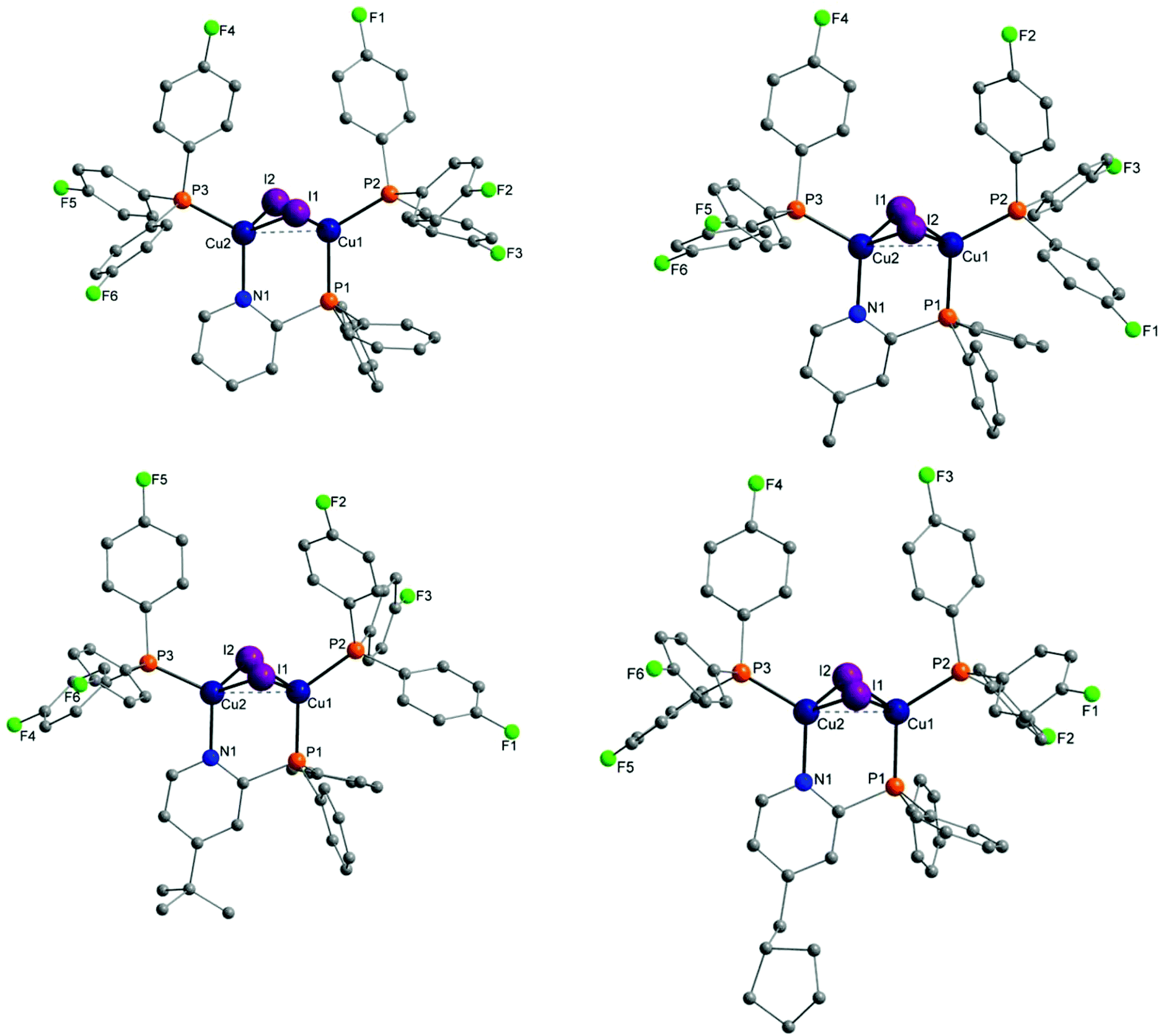 | ||
| Fig. 3 Molecular structures of complexes Cu-1b, Cu-2b (top), Cu-3b and Cu-4b (bottom) bearing tris(4-fluorophenyl)phosphine as ancillary ligands. Hydrogen atoms and solvent molecules were omitted for clarity. | ||
 | ||
| Fig. 4 Molecular structures of complexes Cu-1d and Cu-2d bearing tris(4-chlorophenyl)phosphine as ancillary ligands. Hydrogen atoms and solvent molecules were omitted for clarity. | ||
The most important bond lengths and angles (I–Cu–I, P–Cu–P) of the Cu(I) complexes are listed in Table 1. In all complexes the copper halide core Cu2I2 is butterfly shaped and the copper centres are coordinated in a tetrahedral geometry as already described previously for dinuclear PyrPhos Cu(I) complexes.21,49–51 The average distance between the two copper atoms is 2.70 Å over all obtained structures of the complexes. The shortest Cu⋯Cu distance was found in complex Cu-2c with 2.66 Å and is slightly shorter than described for the known PyrPhos complexes.21
| Cu-1b | Cu-1d | Cu-2b | Cu-2c | Cu-2d | Cu-3b | Cu-4a | Cu-4b | |
|---|---|---|---|---|---|---|---|---|
| Lengths | ||||||||
| Cu–Cu | 2.7158(5) | 2.709(2) | 2.7219(18) | 2.6609(9) | 2.6797(4) | 2.7435(8) | 2.6854(5) | 2.6824(14) |
| CuP–I | 2.6865(4) | 2.677(2) | 2.6722(14) | 2.6661(7) | 2.6603(3) | 2.6644(7) | 2.7071(4) | 2.6800(12) |
| 2.6821(4) | 2.6732(16) | 2.6880(15) | 2.6798(7) | 2.6582(3) | 2.6989(6) | 2.6827(4) | 2.6740(11) | |
| CuP–PNP | 2.2447(8) | 2.246(3) | 2.247(3) | 2.2531(13) | 2.2556(6) | 2.2439(11) | 2.2511(7) | 2.242(2) |
| CuN–N | 2.094(2) | 2.103(9) | 2.091(7) | 2.088(4) | 2.0832(17) | 2.098(3) | 2.094(2) | 2.088(6) |
| CuN–PP | 2.2530(8) | 2.231(3) | 2.249(3) | 2.2375(13) | 2.2384(6) | 2.2542(12) | 2.2373(7) | 2.245(2) |
| CuP–PP | 2.2482(8) | 2.260(3) | 2.247(3) | 2.2428(13) | 2.2469(6) | 2.2557(11) | 2.2493(8) | 2.244(2) |
| Angles | ||||||||
| Cu–I–Cu | 59.856(13) | 61.05(5) | 61.08(4) | 59.51(2) | 60.384(8) | 61.200(19) | 59.759(12) | 59.84(3) |
| 59.531(13) | 60.12(5) | 60.58(4) | 59.44(2) | 60.555(8) | 61.590(19) | 60.410(12) | 60.22(3) | |
| P–Cu–P | 120.32(3) | 121.61(12) | 120.21(10) | 118.12(5) | 118.70(2) | 125.18(4) | 126.94(3) | 122.15(8) |
Figures of the molecular structure of complex Cu-4a and the tertBuPyrPhos ligand 3 are provided in the ESI† of this study (Fig. S18 and S16†). Exact parameters of the single crystal X-ray analysis are given in the ESI.† The full data sets for the following CCDC-numbers are available at the Cambridge Crystallographic Data Centre and can be downloaded for free. CCDC 1919266 (ligand 3), 1919267 (complex Cu-1b), 1919268 (complex Cu-1d), 1919269 (complex Cu-2b), 1919270 (complex Cu-2c), 1918364 (complex Cu-2d), 1919271 (complex Cu-3b), 1919272 (complex Cu-4a) and 1919273 (complex Cu-4b).†
Solubility study
The solubility of the luminescent copper complexes plays an essential role for the solution processing of OLEDs. Therefore, the solubility was tested in n-hexane, toluene, ethanol (EtOH), chlorobenzene and dichloromethane (Table 2). The whole range of complexes showed extremely high solubilities (at least 20 mg mL−1) in for OLED production established solvents, chlorobenzene and toluene, besides dichloromethane, which was used for the synthesis of the complexes. The solubility of complex Cu-2c was outstanding in comparison to the other complexes. This complex was dissolvable in every of the tested solvents. The trifluoromethyl groups on the ancillary ligands showed to mediate and increase the solubility (to some extent Cu-1c as well). These are therefore very interesting candidates for an orthogonal deposition strategy on the OLED substrate because most of the common materials used in the other layers are not soluble in n-hexane or ethanol.| n-Hexane | Toluene | EtOH | Chlorobenzene | DCM | |
|---|---|---|---|---|---|
| a The solubility of the Cu(I) complexes was classified as follows: ++ (20 mg mL−1), +(10 mg mL−1), −(1 mg mL−1) and − − for lower solubility. | |||||
| Cu-1b | − − | − − | − − | − − | + |
| Cu-1c | − | ++ | − | ++ | ++ |
| Cu-1d | − − | ++ | − − | ++ | ++ |
| Cu-2b | − − | ++ | − − | ++ | ++ |
| Cu-2c | ++ | ++ | ++ | ++ | ++ |
| Cu-2d | − − | ++ | − − | ++ | ++ |
| Cu-3a | − − | ++ | − − | ++ | ++ |
| Cu-3b | − − | ++ | − − | ++ | ++ |
| Cu-4a | − − | ++ | − − | ++ | ++ |
| Cu-4b | − − | ++ | − − | ++ | ++ |
19F NMR and 31P NMR measurements
Comparing the shifts of the Cu(I) complexes Cu-1b, Cu-2b, Cu-3b and Cu-4b in the 19F NMR spectra with the 19F NMR shift for tris(4-fluorophenyl)phosphine b, the difference was only 1.5 ppm (Table 3). Measuring an average value of −114.8 ppm the 19F NMR resonance of the copper complex is slightly shifted to the downfield. In contrast, for the trifluoromethyl compounds Cu-1c, Cu-2c and ligand c, which are located further in the downfield, a shift to the highfield was observed, −65.9 to −66.1 ppm. No influence of the bridging ligands was found in the 19F NMR spectra.| Compound | 19F NMR δ [ppm] | 31P NMR δ [ppm] |
|---|---|---|
| (F–Ph)3P | −116.35 (d) | −10.40 (q) |
| Cu-1b | −114.83 | −12.43 (bs) |
| Cu-2b | −114.87 | −14.13 (bs) |
| Cu-3b | −114.82 | −14.47 (bs) |
| Cu-4b | −114.79 | −12.90 (bs) |
| (CF3–Ph)3P | −65.87 | −6.68 |
| Cu-1c | −66.08 | −12.65 (bs) |
| Cu-2c | −66.07 | −12.71 (bs) |
The comparison of the 31P NMR shifts of the copper complexes and their corresponding ancillary ligand showed a highfield shift. Depending on the electron-donating strength of the group on the bridging ligand in the complex, this effect on the 31P NMR shift was stronger.
Absorption spectra
Of all copper complexes Cu-1b–Cu-4b absorption spectra were recorded in dichloromethane at ambient temperature with concentrations of 7 × 10−6 mol L−1. The plot of the corresponding molar extinction coefficients is shown in Fig. 5. All spectra are very broad and have no defined structure, only some are showing visibly broad maxima (Cu-1c, Cu-2c). The pattern of the absorption spectra corresponds to the one previously described for Cu(I) PyrPhos systems.21,23,47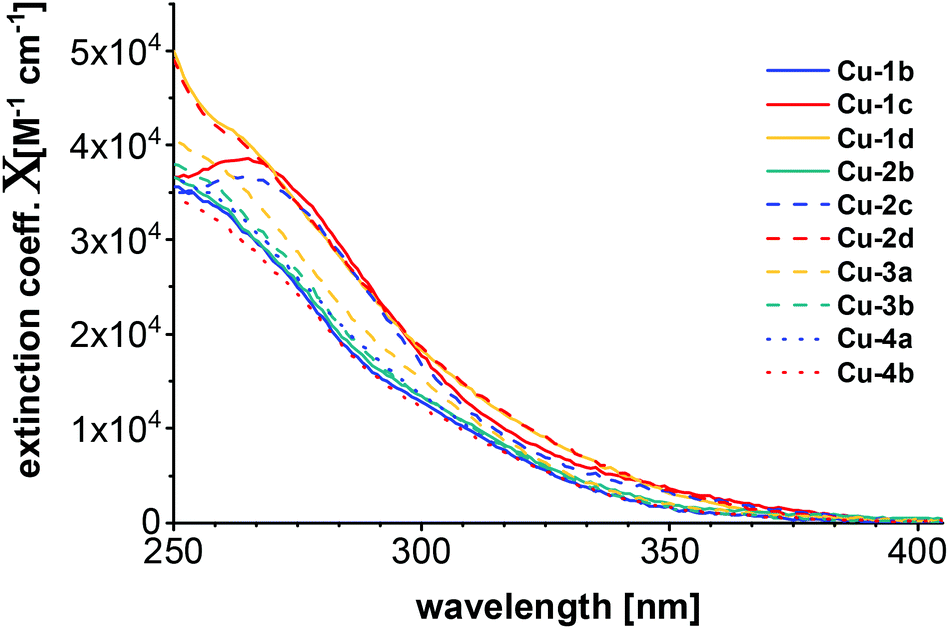 | ||
| Fig. 5 Absorption spectra of all copper complexes Cu-1b–Cu-4b measured in DCM (7 × 10−6 M) at room temperature. | ||
Individual plots of the absorption spectra of the copper complexes Cu-1b–Cu-4b and their corresponding bridging and ancillary ligands are given in the ESI† of this study (Fig. S25–S29†) as well as the UV/Vis spectra recorded of the complexes Cu-1b, Cu-2b and Cu-2c in ethanol (Fig. S30†) and neat films of PMMA, DPEPO and mCBP (Fig. S31†).
Quantum chemical calculations
All calculations were performed with the TURBOMOLE program package.52,53 The ground and excited state structures were obtained with PBE54/def2-TZVP for copper and iodine, and def2-SV(P) for the ligands. UV/Vis spectra were computed at the level of eigenvalue-only self-consistent GW55 (evGW) with the correlation-kernel-augmented Bethe–Salpeter equation56,57 (cBSE) using the def2-TZVP basis set (and def2-SV(P) for hydrogen atoms) and PBE058 as density functional. Additionally, def2-ecp pseudopotentials were applied for iodine. The resolution of identity was used within evGW with the contour deformation technique to compute the HOMO–LUMO gap.59 For all computed spectra, Gaussian broadening with a full width at half maximum of 2500 cm−1 was used. The length representation was chosen for calculating the oscillator strength. See ESI† for more computational details. The computed UV/Vis spectra (Fig. 6) can reproduce some of the experimental observations. The complexes Cu-1d and Cu-2d (chlorinated ancillary ligands) are shifted towards longer wavelengths with respect to experiment (where they are presumably located below 250 nm) and show the strongest absorptions.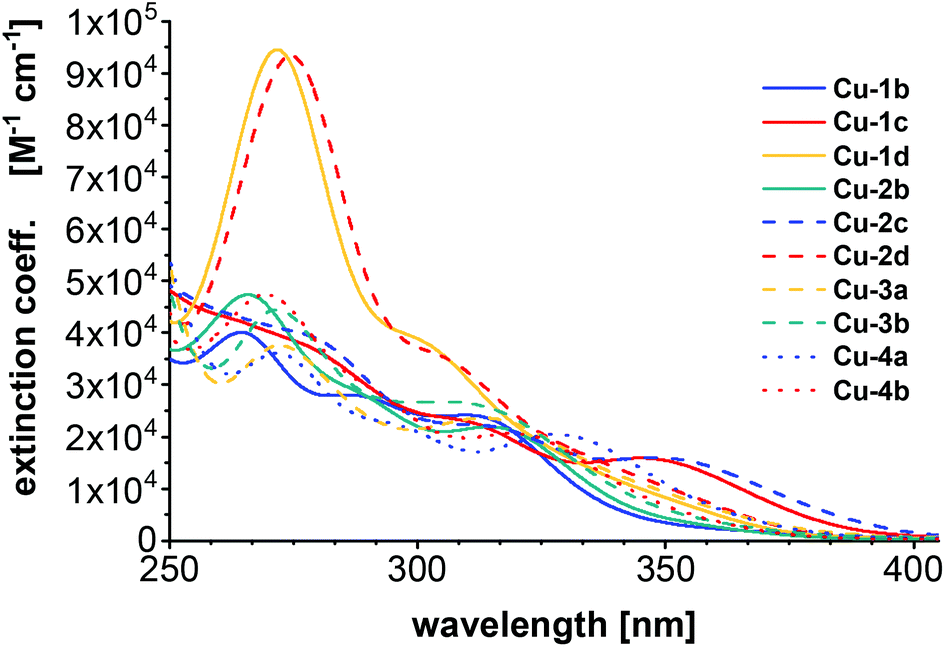 | ||
| Fig. 6 UV/Vis spectra computed using evGW/cBSE with the def2-TZVP basis (def2-SV(P) for hydrogen) and the PBE0 functional. | ||
The previously mentioned broad maxima of Cu-1c and Cu-2c are not reproduced but instead Cu-2b, Cu-3b and Cu-4b are showing maxima around 260 nm. The UV/Vis spectra were also computed with the same computational setting using evGW/BSE57,59 using PBE0 with Kohn–Sham orbitals on the one hand and CAM-B3LYP60 on the other (see ESI Fig. S32†). Compared to evGW/cBSE the evGW/BSE approach gives qualitatively the same result which is slightly red-shifted while CAM-B3LYP predicts most of the maxima around 230 nm – blue-shifted compared to the experiment. The evGW/cBSE approach shows qualitatively the best agreement with the experimental spectra and to gain further insight into the UV/Vis spectra, the natural transition orbitals of Cu-2b for the lowest singlet and triplet excitation at the T1 structure were calculated, see Fig. 7. For both the hole mainly lies at the copper–iodine centre while the electron is located on the pyridine group of the bridging ligand.
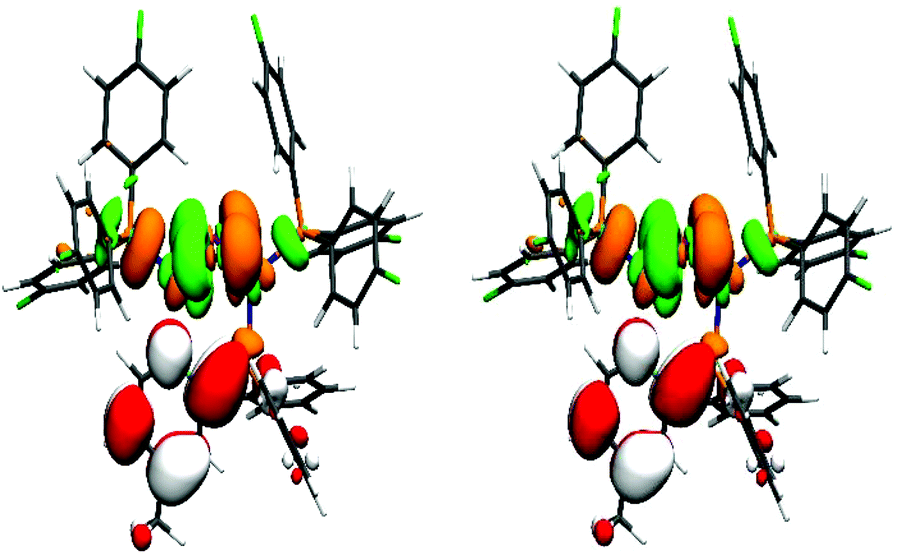 | ||
| Fig. 7 Natural transition orbitals of Cu-2b for the lowest singlet (left) and triplet (right) excitation at the T1 structure. The colours green/orange indicate the hole while red/white correspond to the electron. (Isovalue: ±0.03a−3/20, Cu = blue, P = orange, I = purple, N = cyan, F = green, C = grey, H = white). | ||
The vertical singlet–triplet energy gap (ΔEvST) shown in Table 4 was determined as the difference of the first singlet and triplet excitation based on the singlet ground state (ΔEvST(S0)), first triplet excited state (ΔEvST(T1)) or between the first singlet and triplet excited state electronic structures (ΔEvST(S1/T1)), respectively. For ΔEvST(S0) of Cu-1b–Cu-2c, and Cu-4a and Cu-4b a clear trend is observed, ancillary ligands with electron-withdrawing groups such as fluorine or trifluoromethyl are strongly increasing ΔEvST. However, Cu-2d with chlorine containing ancillary ligands falls out of this trend as it has a higher ΔEvST(S0) compared to its fluorinated counterpart Cu-2b. The first triplet excitation of Cu-2c at the S0 structure (ESI, Fig. S24,† top) does however not correspond to an excitation from the copper–iodine centre to the pyridine part of the bridging ligand – in contrast to the other complexes – but rather to an excitation to one of the ancillary ligands. Hence, the second triplet excitation was analysed and the electron was mainly found at the pyridine part of the bridging ligand (ESI, Fig. S24,† bottom) and ΔEvST(S0) decreased from 0.149 to 0.089 eV. The previously discussed trend has less impact but is still present for ΔEvST(T1), ΔEvST increases from Cu-2b to Cu-2c and Cu-4a to Cu-4b, and again Cu-2d falls out of the trend. For ΔEvST(S1/T1) a different result is found, ΔEvST remains constant for Cu-2b to Cu-2c with 0.047 eV while increasing for Cu-4a to Cu-4b from 0.046 to 0.049 eV. Additionally, the predicted gap for Cu-2d of 0.050 eV is higher compared to the fluorinated counterpart Cu-2b with 0.047 eV. The values for ΔEvST(T1) and ΔEvST(S1/T1) can be understood as upper and lower limits for the energy barrier of the reverse intersystem crossing. The experimental gaps of Cu-2b and Cu-1b with 0.019 and 0.027 eV (see next section) are in accordance with the calculated values. Based on ΔEvST(T1) and ΔEvST(S1/T1) the preliminary conclusion can be made that the ancillary ligands with electron-withdrawing groups have no significant impact on ΔEvST.
Photophysical properties
For all Cu(I) AlkylPyrPhos complexes emission spectra (Fig. 8) and photoluminescence quantum yields were measured and the excited state lifetimes were determined of the powder samples (Table 5) as well as in films by time-correlated single-photon counting (TCSPC) (Fig. 10 and Table 6).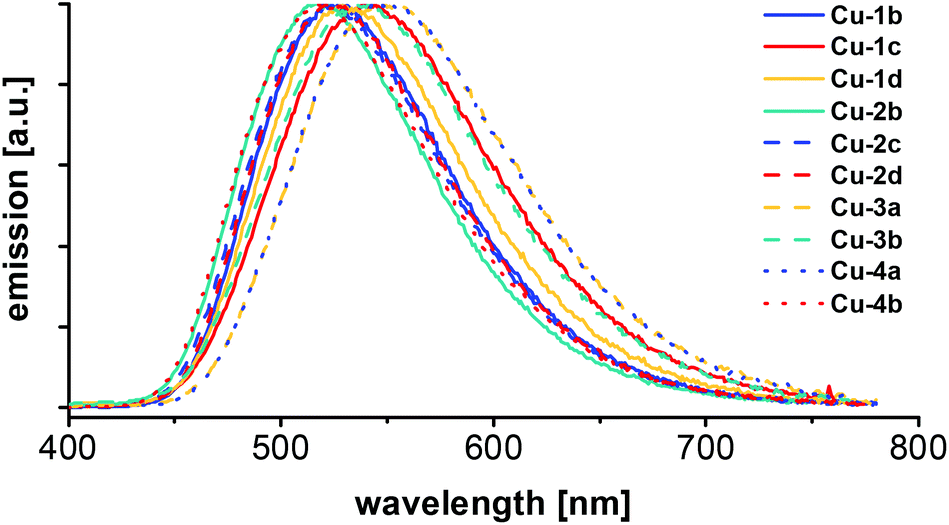 | ||
| Fig. 8 Emission spectra of the series of Cu(I) AlkylPyrPhos complexes, measured of the powder samples at room temperature with 350 nm excitation wavelength. | ||
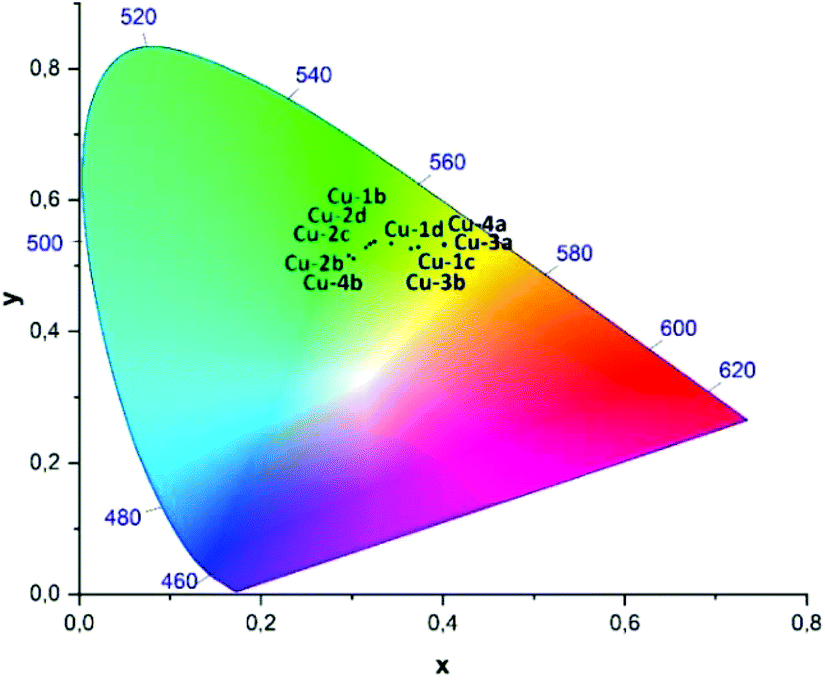 | ||
| Fig. 9 CIE-diagram (1931) showing the x,y-coordinates of the photoluminescence measured in powder at room temperature for the complexes Cu-1b–Cu-4b, cf. Table 5. | ||
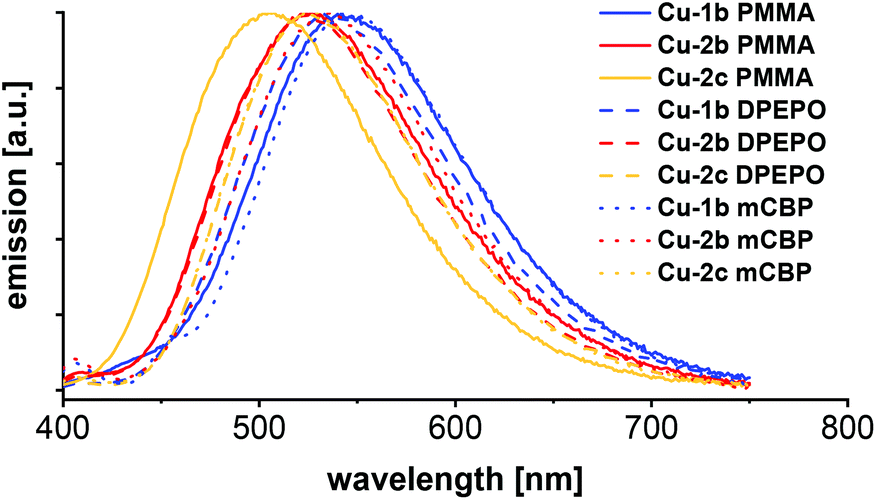 | ||
| Fig. 10 Emission spectra of PMMA, DPEPO and mCBP films with 10 wt% of the complexes Cu-1b, Cu-2b and Cu-2c respectively, measured at room temperature with 350 nm excitation wavelength. | ||
| Complex | λ PL [nm] | Φ PL [%] | τ [μs] | CIE X | CIE Y |
|---|---|---|---|---|---|
| Cu-1b | 524 | 93 | 5.8 | 0.33 | 0.54 |
| Cu-1c | 541 | 70 | 5.5 | 0.37 | 0.53 |
| Cu-1d | 528 | 80 | 10.2 | 0.34 | 0.53 |
| Cu-2b | 519 | 89 | 5.5 | 0.30 | 0.52 |
| Cu-2c | 524 | 90 | 5.5 | 0.32 | 0.53 |
| Cu-2d | 524 | 76 | 6.8 | 0.32 | 0.53 |
| Cu-3a | 549 | 73 | 5.1 | 0.40 | 0.53 |
| Cu-3b | 539 | 73 | 7.3 | 0.37 | 0.53 |
| Cu-4a | 547 | 79 | 5.5 | 0.40 | 0.53 |
| Cu-4b | 519 | 88 | 6.3 | 0.30 | 0.51 |
| Complex | Host | λ PL [nm] | Φ PL [%] | τ [μs] | CIE X | CIE Y |
|---|---|---|---|---|---|---|
| Cu-1b | PMMA | 542 | 59 | 9.5 | 0.39 | 0.53 |
| DPEPO | 532 | 64 | 7.2 | 0.36 | 0.51 | |
| mCBP | 544 | 54 | 7.3 | 0.38 | 0.52 | |
| Cu-2b | PMMA | 529 | 78 | 7.8 | 0.35 | 0.52 |
| DPEPO | 526 | 65 | 6.9 | 0.32 | 0.52 | |
| mCBP | 529 | 60 | 6.7 | 0.36 | 0.53 | |
| Cu-2c | PMMA | 505 | 77 | 10 | 0.27 | 0.44 |
| DPEPO | 526 | 69 | 8.3 | 0.32 | 0.52 | |
| mCBP | 526 | 60 | 8.4 | 0.32 | 0.52 |
The maxima of emission wavelengths range from 519 nm for the fluorinated copper complexes (Cu-2b and Cu-4b) to 549 nm for complex Cu-3a with triphenylphosphine as ancillary ligands. The complexes Cu-3b and Cu-4b bearing fluorinated ancillary phosphine ligands possess a blue-shifted emission in comparison to their analogues with triphenylphosphine ligands (Cu-3a and Cu-4a), probably due to packing effects in the powder.
In comparison with the literature known complexes Cu-1a and Cu-2a this trend could not be confirmed. An emission wavelength of 514 nm22 was reported for complex Cu-1a and 515 nm22 and 510 nm24 were found for complex Cu-2a in powder measurements previously, which points in the direction of packing effects influencing the emission wavelengths slightly. The corresponding CIE X and Y coordinates of the Cu(I) complexes Cu-1b–Cu-4b measured in powder are shown in the CIE-diagram (1931) in Fig. 9 and are all located in the yellow greenish (Cu-3a, Cu-4a) to turquoise area (Cu-2b, Cu-4b).
The highest photoluminescence quantum yield (PLQY) for this series of copper complexes was determined for complex Cu-1b with 93% and is located in the top range of Cu(I) PyrPhos complexes described previously in literature. 86%22 PLQY was reported for complex Cu-1a and 88%22 PLQY were described for Cu-2a. Comparing the values for the PLQYs of the fluorine bearing Cu(I) complexes in this study with their standards with triphenylphosphine, PLQYs were equal or slightly higher in most cases (88% PLQY for Cu-4b, 79% PLQY for Cu-4a).
Regarding the excited state lifetimes of the powder measurements (Table 5), all values were found in the microsecond range (5.1–10.2 μs). No significant difference was observed for the complexes with halide bearing ligands (Cu-1b, Cu-1c, Cu-1d, Cu-2b, Cu-2c, Cu-2d, Cu-3b and Cu-4b) compared to the complexes with triphenylphosphine (Cu-3a and Cu-4a) and the previously described compounds (2.8 μs22 for Cu-1a and 3.8 μs22 and 1.9 μs24 for Cu-2a). All copper complexes described in this work (Cu-1b–Cu-4b) possess TADF. The microsecond lifetimes indicate an emission only via the singlet state as TADF.
Besides photophysical measurements of the powder, selected copper complexes Cu-1b, Cu-2b and Cu-2c (all bearing fluorinated ancillary ligands) were studied in neat films of 10 wt% in PMMA, DPEPO and mCBP as host materials. The corresponding emission spectra are shown in Fig. 10 and the photophysical data are given in Table 6. The matrix of the film has almost no influence on the photophysical properties, the emission spectra of the films are in general only slightly red shifted compared to the powder measurements (542/532/544 nm (PMMA/DPEPO/mCBP) compared to 524 nm (powder) for complex Cu-1b, 529/526/529 nm (PMMA/DPEPO/mCBP) compared to 519 nm (powder) for complex Cu-2b).
As expected, the quantum yields of the films were lower than in powder, because of the quenching effects with the host material. For example, complex Cu-1b had 64% PLQY in DPEPO and 54% PLQY in mCBP, while the quantum yield was 93% in powder. In general, the quantum yields of the DPEPO films with copper complexes were higher than in the mCBP films.
Temperature dependent luminescence spectra were recorded of KBr pellets (Cu-1b, Cu-2b) at 290 and 20 K (Fig. 11, Table S4†). The emission maxima at 290 K are similar to the values obtained from powder samples (Fig. 8) and neat films (Fig. 10), which is also valid for the measured lifetimes. Upon cooling to 20 K the emission is red-shifted by about 150 cm−1 (0.019 eV) (Cu-2b) and 215 cm−1 (0.027 eV) (Cu-1b), respectively, compared to the emission at 290 K. This results from the inhibition of the TADF mechanism at 20 K so that emission occurs only via the T1 state. The red-shifts correspond to the singlet–triplet energy gaps (Table S5†) and are in accordance with the calculated range for the energy gaps from ΔEvST(T1) and ΔEvST(S1/T1) (Table 4), obtained by applying the Bethe–Salpeter equation. It should further be mentioned that by application of the TD-DFT method (B3LYP-D3(BJ)/def2-TZVP) similar energy gaps of 161 and 222 cm−1 were calculated which are in excellent agreement with the experimental values. Both theoretical methods predict a slightly larger energy gap for Cu-1b. The almost exclusive observation of phosphorescence at 20 K is confirmed by the luminescence lifetimes which are at 20 K about six times higher compared to the values at 290 K. All excited state lifetimes obtained from TCSPC measurements at 290 and 20 K are listed in the ESI (Table S4†).
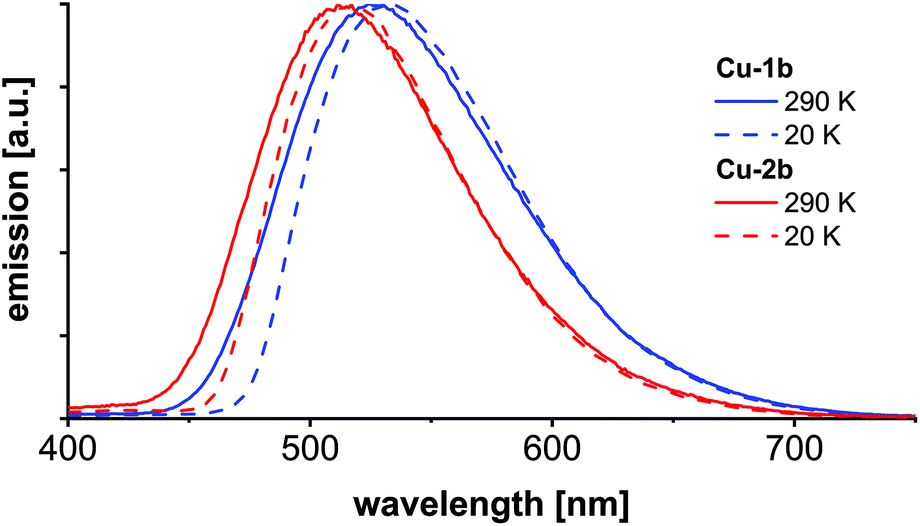 | ||
| Fig. 11 Emission spectra of Cu-1b and Cu-2b measured in the KBr matrix at 290 K and 20 K with 380 nm excitation wavelength. | ||
Photoluminescence investigations in solution were conducted for the three chosen complexes Cu-1b, Cu-2b and Cu-2c to evaluate their (photo)chemical stability in a series of solvents. In the chlorinated solvents dichloromethane and chlorobenzene the complexes Cu-1b and Cu-2b exhibit two emission bands (Fig. 12 and Fig. S37†). A broad emission peak was observed at 554–577 nm with a lifetime of 0.2–0.4 μs, indicating delayed fluorescence and thus TADF in solution, and a short-lived (≤2 ns) fluorescence band around 388 nm similar to the emission spectrum of the free tris(4-fluorophenyl)phosphine ligand b. Thus, degradation of Cu-1b and Cu-2b occurs at least on a minute time scale in dichloromethane and chlorobenzene.
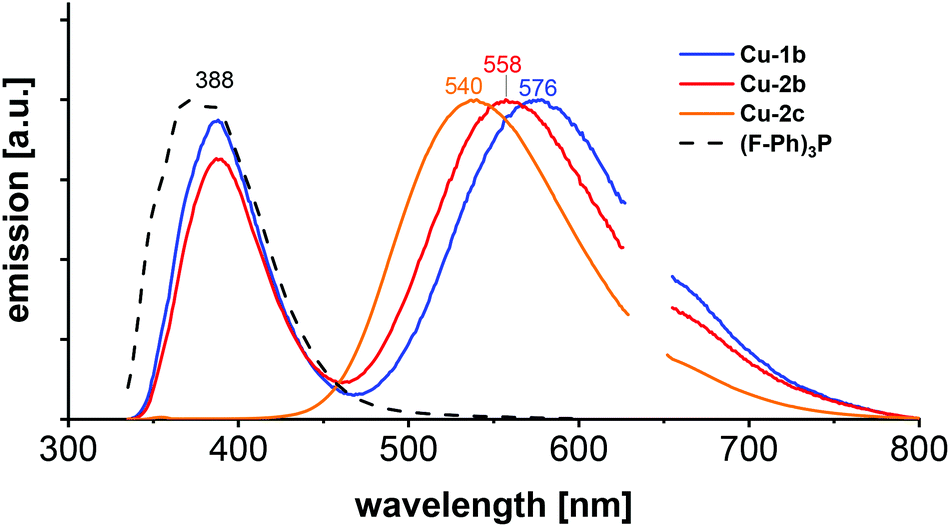 | ||
| Fig. 12 Emission spectra of Cu-1b, Cu-2b, Cu-2c and (F–Ph)3P (b) in dichloromethane, measured at room temperature with 320 nm excitation wavelength. | ||
In the case of a solution of Cu-2c in chlorobenzene the relative intensity of the ligand-shaped fluorescence is much smaller compared to the TADF emission on the same time regime (Fig. S37†), which is a hint for an increased stability. Interestingly, only one emission band (540 nm) was observed for Cu-2c in dichloromethane. The absence of any blue-shifted fluorescence illustrates the stability of the complex. The luminescence spectrum remains unchanged after 24 h when stored in the dark, so decomposition can be excluded even on larger time scales for Cu-2c. Hence, the ancillary tris(4-trifluoromethylphenyl)phosphine ligand c not only assures a high solubility but also allows a high stability in chlorinated solvents. However, solutions of Cu-2b and Cu-2c in dichloromethane and chlorobenzene showed a decrease of luminescence by about 50% of the integrated TADF emission after 3 to 4 min of UV irradiation (300 nm). Further stability studies were performed in ethanol, where complex Cu-2b showed a poor stability with a strong fluorescence band resembling the emission of the free tris(4-fluorophenyl)phosphine ligand b. However, only a minor degradation was observed on a minute time scale for a solution of Cu-1b, whereas Cu-2c again turned out as the most stable complex, showing no decomposition (Fig. S37†).
An important difference between the behaviour in chlorinated solvents and in ethanol is that complexes Cu-2b and Cu-2c are photochemically much more stable in the latter case with a 50% drop of the TADF emission after 45 and 60 min of irradiation (300 nm), respectively. Only poor (photo)chemical stabilities were observed for Cu-1b, Cu-2b and Cu-2c in hexane and toluene. The results of the stability studies are summarized in Table 7.
| a The stability of the Cu(I) complexes was classified as follows: ++ (very high) to − − (very low). Black: chemical stability, red: photochemical stability according to fluorescence spectra. |
|---|
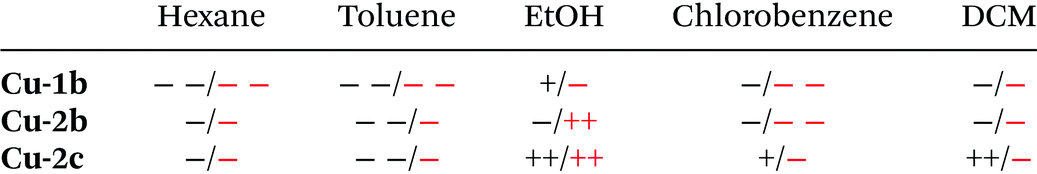
|
The emission maxima in solution are mainly red-shifted compared to the results obtained from solid state measurements, as described in earlier works on Cu(I) AlkylPyrPhos complexes.21,22,24 This behaviour results from a higher degree of freedom in solution affecting the structural relaxation.21,22,24 Only very few lifetime studies in solution have been reported on this type of Cu(I) complexes up to now.27,28 TCSPC measurements were performed in dichloromethane, chlorobenzene and ethanol for the complexes Cu-1b, Cu-2b and Cu-2c. Almost all lifetimes were located in the sub-microsecond regime (0.2–0.4 μs, cf. Table S6†) and are significantly higher than the values of a few nanoseconds reported in an earlier work.27 The increased values for the investigated complexes with fluorinated ancillary ligands may be explained by weaker interactions with solvent molecules leading to a smaller contribution of ultrafast non-radiative deactivation processes.
Most surprising is the lifetime of the compound Cu-1b in ethanol with a very long lifetime of 4.9 μs assigned to phosphorescence. The latter one might result from an increased energy gap between T1 and S1 in EtOH, which would suppress reversed intersystem crossing at room temperature.
Step-scan FTIR spectroscopy
For further characterization step-scan (transient) FTIR spectroscopy was applied to the complexes Cu-1b and Cu-2b to yield further information on the nature of the long-lived electronically excited states. The FTIR ground state spectrum of Cu-2b (Fig. 13), measured as a KBr pellet at 20 K, agrees well with the theoretical S0 spectrum, calculated by B3LYP-D3(BJ)/def2-TZVP. The only significant discrepancy is the overestimation of the intensity of the band at 1231 cm−1. The sample was then excited with a 355 nm laser pulse leading to metal-to-ligand (MLCT) and halide-to-ligand (XLCT) charge transfer transitions from the copper(I) iodide core to the π systems of the AlkylPyrPhos ligand. A step-scan difference spectrum with positive and negative bands was obtained after electronic excitation (Fig. 13). The negative peaks are bleach bands that correlate with the vibrations in the ground state and result from the depopulation of the electronic ground state. The positive bands are assigned to the IR absorption of the populated long-lived electronically excited states. It has to be mentioned that some vibrations in the electronic ground state (cf. positions at 1589, 1231 and 1160 cm−1) show deviations from the corresponding bleach band in the step-scan spectrum. This results from a spectral overlap of the negative and positive bands in the difference spectrum. Most positive peaks are red-shifted as the respective bonds are elongated in the electronically excited states. However, the ground state vibration at 1231 cm−1, assigned to the C–F stretching vibration, is blue-shifted and thus strengthened in the excited state. Interestingly, three pronounced new bands appear at 1574, 1339 and 1063 cm−1 in the difference spectrum, which are not present in the ground state and are very important for the assignment of the electronically excited state (cf. following paragraph).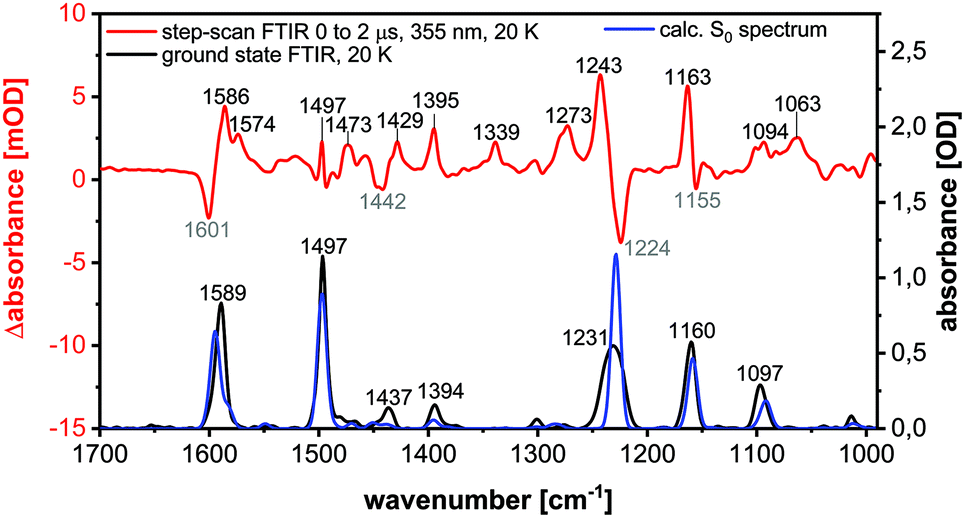 | ||
| Fig. 13 FTIR ground state spectrum at 20 K (black), calculated S0 spectrum (B3LYP-D3(BJ)/def2-TZVP, FWHM = 8 cm−1, Gaussian profile, scaled by 0.975) (blue) and step-scan difference spectrum 0 to 2 μs after excitation (λEX = 355 nm) (red) of Cu-2b. | ||
The peaks in the step-scan difference spectrum decrease with ongoing time after laser excitation, resulting from the repopulation of the electronic ground state. The time traces of the eleven most significant positive and negative bands with a global biexponential decay fit are shown in the ESI (Fig. S40†). Two time constants of 1053 ± 49 ns (contribution 11%) and 15447 ± 494 ns (contribution 89%) were obtained. The two decay times may be described by a relaxation in the triplet manifold and the phosphorescence lifetime respectively.27 The significantly increased lifetime compared to room temperature results from the suppression of the TADF mechanism at 20 K, confirming the TCSPC results. The discrepancies between the time constants obtained by the TCSPC and step-scan techniques may be explained by the contribution of non-radiative processes in the latter case.
The excited state spectrum shown in Fig. 14 was generated by adding 1.5% of the intensity of the ground state spectrum to the step-scan difference spectrum, so that the negative bands disappear and only the excited state absorption peaks are seen in the spectrum. The bands at 1063, 1339 and 1574 cm−1 are not observed in the electronic ground state and are important for the identification of the excited state. The vibration at 1574 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations in the phenyl rings, whereas the peaks at 1339 and 1063 cm−1 result from C–H bending motions. For further assignments of, in general, not localized vibrations, see Table S9.† The excited state spectrum matches very well with the calculated spectrum of the T1 state. All the three abovementioned characteristic excited state vibrations are observed in the theoretical spectrum, so that the excited state can be assigned to the T1 state. The observed phosphorescence lifetime is another evidence for the T1 state.27,28 A description of the most important geometrical parameters of the calculated S0 and T1 states (B3LYP-D3(BJ)/def2-TZVP) is given in the ESI (Fig. S42 and Table S8†).
C stretching vibrations in the phenyl rings, whereas the peaks at 1339 and 1063 cm−1 result from C–H bending motions. For further assignments of, in general, not localized vibrations, see Table S9.† The excited state spectrum matches very well with the calculated spectrum of the T1 state. All the three abovementioned characteristic excited state vibrations are observed in the theoretical spectrum, so that the excited state can be assigned to the T1 state. The observed phosphorescence lifetime is another evidence for the T1 state.27,28 A description of the most important geometrical parameters of the calculated S0 and T1 states (B3LYP-D3(BJ)/def2-TZVP) is given in the ESI (Fig. S42 and Table S8†).
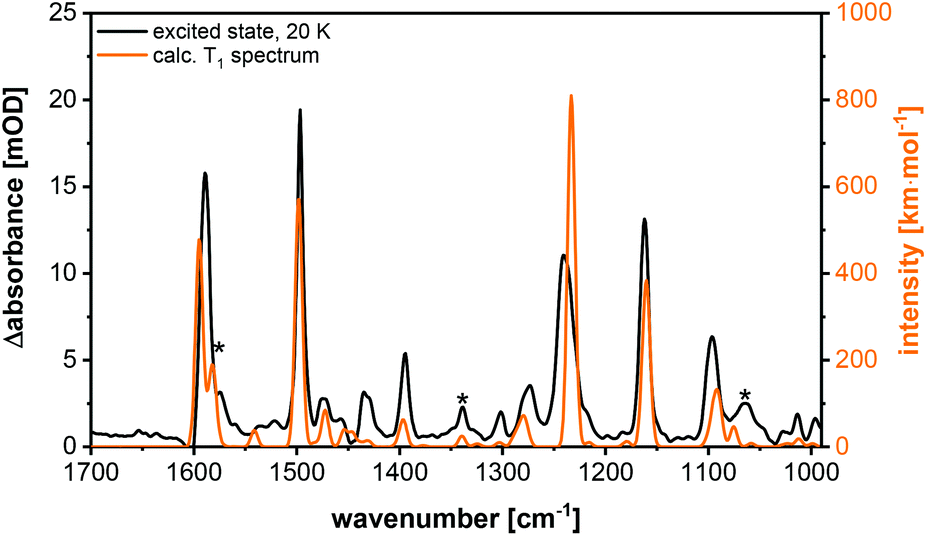 | ||
| Fig. 14 Excited state spectrum (black) and calculated T1 spectrum (orange) (B3LYP-D3(BJ)/def2-TZVP, FWHM = 8 cm−1, Gaussian profile, scaled by 0.975) of Cu-2b. The T1 specific bands are marked with asterisks. | ||
For comparison, step-scan measurements were performed at 290 K. The measured excited state spectra at 290 K and at 20 K are compared in Fig. S38.† These are very similar, so that temperature has no significant influence on the structure of the electronically excited state. Thus, the T1 state should mainly contribute to the step-scan FTIR spectrum at 290 K, as reported earlier for similar MePyrPhos Cu(I) complexes.27,28
The time traces of six pronounced bands where considered in a global biexponential decay fit, where the time constants of 139 ± 16 ns (contribution 13%) and 3083 ± 209 ns (contribution 87%) were obtained. These lifetimes can be assigned to internal conversion in the triplet regime and TADF respectively (Table S7†). The TADF time constant is in accordance with the rate constant obtained by TCSPC for the KBr sample at 290 K.
Additionally, step-scan FTIR investigations were performed on the complex Cu-1b. The corresponding excited state spectrum is shown in Fig. 15, additional spectra are depicted in the ESI (Fig. S43–S45†). The complexes Cu-1b and Cu-2b show almost identical ground state spectra, but an additional band is observed in the excited state spectrum of Cu-2b (Fig. S47†). This difference is confirmed by the calculations for the T1 state, so that the band could be assigned to an in-plane C–H bending vibration of the pyridine moiety. Finally, it should be mentioned that in contrast to investigations in solution no degradation of the investigated complexes is observed in the solid state (KBr pellets).
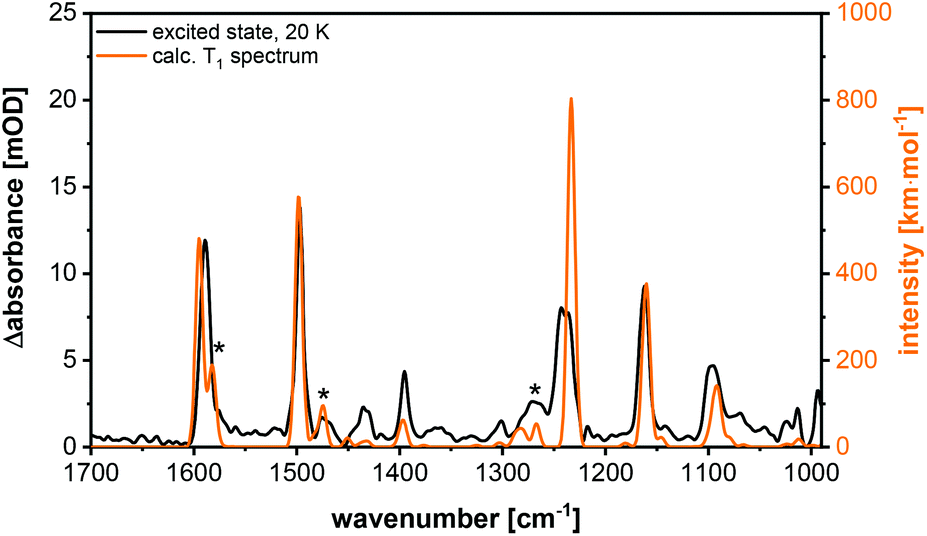 | ||
| Fig. 15 Excited state spectrum (black) and calculated T1 spectrum (B3LYP-D3(BJ)/def2-TZVP, FWHM = 8 cm−1, Gaussian profile, scaled by 0.975) (orange) of Cu-1b. The T1 specific bands are marked with asterisks. | ||
Conclusions
In this work we presented a new class of halide bearing Cu(I) AlkylPyrPhos complexes, which showed remarkable high solubility in many solvents, especially the complexes Cu-1c and Cu-2c with trifluoromethyl groups. The molecular structures of almost all AlkylPyrPhos copper complexes in this study were confirmed by X-ray analysis and showed the same butterfly shaped structure as described for previous Cu(I) PyrPhos complexes.Absorption spectra in dichloromethane were recorded for all copper complexes (Cu-1b–Cu4b) and were compared with the computed UV/Vis spectra using evGW/cBSE with the def2-TZVP (def2-SV(P) for hydrogen) basis. A good reproduction of the experimental observations was achieved. An extensive photophysical characterisation of all Cu(I) complexes was done by powder measurements and measurements in neat films of 10 wt% Cu(I) complex in PMMA, DPEPO and mCBP as host materials. Further detailed photophysical studies especially of the selected fluorinated compounds Cu-1b and Cu-2b were performed in KBr and in solution. The complexes possessed very high luminescence quantum yields in powder, up to 93% and 64% in the DPEPO film (Cu-1b), while emitting in the yellow greenish (Cu-3a, Cu-4a) to turquoise area (Cu-2b, Cu-4b). In addition, stability studies in solution of complex Cu-2c revealed a high (photo)chemical stability in a variety of solvents. The calculations demonstrated that all the presented complexes, showing TADF in the solid state and in solution, have similar vertical singlet–triplet energy gaps (ΔEvST), independently of the ancillary ligands. However, the medium (e.g. film, solution, KBr pellet) turned out to have a significant influence on the emission wavelength. The lowest lying T1 state could be characterised by time-resolved step-scan FTIR spectroscopy.
Combining extremely high solubility and high quantum yields also in neat films, the Cu(I) AlkylPyrPhos TADF complexes are excellent candidates for solution-processed OLEDs.
Experimental
All experiments were performed under Schlenk conditions. Solvents were purchased from Fisher in p.a. quality for general use and unstabilized in HPLC grade for drying them in the solvent purification system MB-SPS-800 from MBraun with special drying columns. Tetrahydrofuran of the SPS system was degassed for 20 min with argon before usage. Copper iodide was purchased in 99.999% trace metals basis quality from Sigma Aldrich and was used without any further purification as well as all the other precursors and ancillary ligands, which were purchased also partly from Fisher and abcr. DMSO-d6 for NMR measurements was purchased in a 10 mL vial with septum and was also degassed before usage.The experimental set-ups for the photophysical investigations are described in detail in the ESI.†
Synthesis of the ligands
While 2-(diphenylphosphino)pyridine (1) was purchased from abcr, 4-methyl-2-(diphenylphosphino)pyridine (2) and (4-(cyclopentylmethyl)-2-(diphenylphosphino)pyridine (4) were synthesized according to the literature known procedures.23,47 The experimental data of ligand 3 is given in the ESI.†General procedure for the synthesis of the Cu(I) complexes Cu-1b–Cu-4b
For the synthesis of the Cu(I) AlkylPyrPhos complexes a 20 mL crimp vial was charged with the corresponding bridging ligand (0.70 mmol, 1.00 eq.), ancillary ligand (1.40 mmol, 2.00 eq.) and copper iodide (1.40 mmol, 2.00 eq.). The material was dissolved in 15 mL dry dichloromethane under argon and the suspension was degassed with argon for five minutes. After stirring for 12 h at room temperature in the dark the solvent of the reaction mixture was reduced to 5 mL and the mixture was poured in 100 mL of n-pentane. The precipitate was filtered off, washed with n-pentane and diethyl ether and was dried in vacuum. Crystals were grown in 5 mL Vials via the layering approach, using dichloromethane to dissolve the compound and n-pentane was applied on top.The experimental data for the complexes Cu-1b, Cu-2b and Cu-2c is given below and the experimental data for all other copper complexes can be found in the ESI.†
1
H NMR (400 MHz, DMSO-d6) δ [ppm] = 8.70 (bs, 1H, HPyr), 7.91 (t, 3JHH = 7.8 Hz, 1H, HPyr), 7.57 (bs, 1H, HPyr), 7.53–7.15 (m, 35H). – 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 164.5 (s), 162.0 (s), 135.9 (q, J = 15.6 Hz, J = 8.6 Hz), 133.4 (d, J = 13.0 Hz), 130.1 (s), 129.0 (d, J = 28.2 Hz), 128.6 (d, J = 7.8 Hz), 115.9 (dd, J = 21.2 Hz, J = 8.8 Hz). – 31P NMR (162 MHz, DMSO-d6) δ [ppm] = −5.29 (bs, 1P, PPyrPhos), −12.43 (bs, 2P, P(F–Ph)3P). – 19F NMR (376 MHz, DMSO-d6) δ [ppm] = −114.83 (s, 6F, F(F–Ph)3P). – MS (FAB, 3-NBA) m/z [%] = 1337 (4) [M + Cu]+, 1021 (5) [Cu3I2LP]+, 884 (19) [Cu2IP2]+, 831 (16) [Cu2ILP]+, 705 (43) [Cu3I2L]+, 695 (100) [CuP2]+, 642 (52) [CuLP]+, 589 (27) [CuL2]+, 568 (23) [Cu2IP]+, 515 (76) [Cu2IL]+. – IR (ATR) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) [cm−1] = 3043 (vw), 1585 (m), 1494 (m), 1451 (w), 1433 (w), 1393 (w), 1301 (vw), 1224 (m), 1157 (m), 1093 (w), 1013 (w), 826 (m), 759 (w), 742 (w), 693 (m), 634 (w), 518 (m), 508 (m), 488 (w), 470 (m), 442 (m), 430 (m). – Anal. calcd for C53H38Cu2F6I2NP3 (1274.8): C 49.86, H 3.00, N 1.10; found: C 49.91, H 3.09, N 1.17. A molecular structure of the complex was obtained.
[cm−1] = 3043 (vw), 1585 (m), 1494 (m), 1451 (w), 1433 (w), 1393 (w), 1301 (vw), 1224 (m), 1157 (m), 1093 (w), 1013 (w), 826 (m), 759 (w), 742 (w), 693 (m), 634 (w), 518 (m), 508 (m), 488 (w), 470 (m), 442 (m), 430 (m). – Anal. calcd for C53H38Cu2F6I2NP3 (1274.8): C 49.86, H 3.00, N 1.10; found: C 49.91, H 3.09, N 1.17. A molecular structure of the complex was obtained.
1
H NMR (400 MHz, DMSO-d6) δ [ppm] = 8.51 (bs, 1H, HPyr), 7.47–7.20 (m, 36H), 2.27 (s, 3H, HMe). – 13C NMR (400 MHz, DMSO-d6) δ [ppm] = 164.4 (s), 162.0 (s), 136.0 (q, J = 15.7 Hz, J = 8.4 Hz), 133.4 (d, J = 13.3 Hz), 130.0 (s), 129.0 (d, J = 28.3 Hz), 128.6 (d, J = 7.6 Hz), 115.9 (dd, J = 21.2 Hz, J = 9.4 Hz), 20.6 (s, 1C, CMe). – 31P NMR (162 MHz, DMSO-d6) δ [ppm] = −4.98 (bs, 1P, PMePyrPhos), −14.13 (bs, 2P, P(F–Ph)3P). – 19F NMR (376 MHz, DMSO-d6) δ [ppm] = −114.87 (s, 6F, F(F–Ph)3P). – MS (FAB, 3-NBA) m/z [%] = 1351 (4) [M + Cu]+, 1161 (2) [M − I]+, 1074 (1) [Cu3I2P2]+, 1035 (2) [Cu3I2LP]+, 996 (6) [Cu3I2L2]+, 884 (8) [Cu2IP2]+, 845 (13) [Cu2ILP]+, 719 (19) [Cu3I2L]+, 695 (23) [CuP2]+, 568 (5) [Cu2IP]+, 529 (35) [Cu2IL]+, 378 (4) [CuP]+, 340 (38) [CuL]+, 278 (13) [L + H]+. – IR (ATR) [cm−1] = 3053 (vw), 2958 (vw), 2923 (vw), 2856 (vw), 1585 (m), 1494 (m), 1433 (w), 1393 (w), 1300 (w), 1225 (w), 1158 (m), 1093 (w), 1013 (w), 825 (m), 742 (w), 693 (w), 635 (vw), 518 (m), 494 (w), 463 (w), 442 (w), 430 (w). – Anal. calcd for C54H40Cu2F6I2NP3 (1288.9): C 50.25, H 3.12, N 1.09; found: C 50.59, H 3.21, N 1.27. A molecular structure of the complex including one molecule of n-pentane was obtained.
1
H NMR (400 MHz, DMSO-d6) δ [ppm] = 8.51 (bs, 1H), 7.72 (bs, 24H), 7.50 (bs, 1H), 7.43–7.26 (m, 11H), 2.27 (s, 3H, HMe). – 13C-NMR (101 MHz, DMSO-d6) δ [ppm] = 137.1 (d, J = 23.6 Hz), 134.5 (d, J = 14.1 Hz), 133.3 (d, J = 13.2 Hz), 131.0 (s), 130.5 (d, J = 30.1 Hz), 130.1 (s), 130.0 (d, J = 5.0 Hz), 128.6 (d, J = 7.7 Hz), 127.9 (s), 125.5 (bs), 125.2 (s), 122.4 (s), 119.7 (s), 20.6 (s, 1C, CMe). – 31P NMR (162 MHz, DMSO-d6) δ [ppm] = −4.40 (bs, 1P, PMePyrPhos), −12.71 (bs, 2P, P(CF3−Ph)3P). – 19F NMR (376 MHz, DMSO-d6) δ [ppm] = −66.07 (s, 24F, F(CF3−Ph)3P). – MS (FAB, 3-NBA) m/z [%] = 1651 (2) [M + Cu]+, 1461 (1) [M − I]+, 1374 (3) [Cu3I2P2]+, 1184 (4) [Cu2IP2]+, 995 (5) [Cu2ILP]+, 806 (8) [CuLP]+, 718 (9) [Cu2IP]+, 528 (11) [CuP]+, 467 (10) [P + H]+, 340 (100) [CuL]+, 278 (17) [L + H]+. – IR (ATR) [cm−1] = 3049 (vw), 1606 (w), 1437 (vw), 1397 (w), 1319 (s), 1163 (m), 1120 (m), 1059 (m), 1015 (m), 831 (m), 742 (w), 694 (m), 634 (vw), 597 (m), 517 (m), 496 (w), 463 (w), 413 (w). – Anal. calcd for C60H40Cu2F18I2NP3 (1590.7): C 45.30, H 2.53, N 0.88; found: C 45.39, H 2.38, N 1.00. A molecular structure of the complex was obtained.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Andreas Wenz and Yelda Nur Demirdögen (BSc.) for their synthetic support. This work was supported by the Karlsruhe Institute of Technology (KIT). We acknowledge the financial support of the 3MET projects T1, C1 and C2 (SFB/TRR 88) by the Deutsche Forschungsgemeinschaft (DFG) as well as cynora GmbH. We also thank the Karlsruhe School of Optics and Photonics (KSOP) for their support.Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- S. A. Kumar, J. S. Shankar, B. K. Periyasamy and S. K. Nayak, Polym.-Plast. Technol. Mater., 2019, 1–28, DOI:10.1080/25740881.2018.1563133.
- R.-P. Xu, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2016, 4, 9116–9142 RSC.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- T. Fleetham, G. Li and J. Li, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed.
- E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS.
- C. Vogler, H.-D. Hausen, W. Kaim, S. Kohlmann, H. E. A. Kramer and J. Rieker, Angew. Chem., 1989, 101, 1734–1735 CrossRef CAS.
- D. Felder, J.-F. Nierengarten, F. Barigelletti, B. Ventura and N. Armaroli, J. Am. Chem. Soc., 2001, 123, 6291–6299 CrossRef CAS PubMed.
- M. Nishikawa, S. Sawamura, A. Haraguchi, J. Morikubo, K. Takao and T. Tsubomura, Dalton Trans., 2015, 44, 411–418 RSC.
- M. J. Leitl, F. R. Kuchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117, 11823–11836 CrossRef CAS PubMed.
- H. Ohara, A. Kobayashi and M. Kato, Dalton Trans., 2014, 43, 17317–17323 RSC.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- X. Hong, B. Wang, L. Liu, X.-X. Zhong, F.-B. Li, L. Wang, W.-Y. Wong, H.-M. Qin and Y. H. Lo, J. Lumin., 2016, 180, 64–72 CrossRef CAS.
- T. Hasegawa, A. Kobayashi, H. Ohara, M. Yoshida and M. Kato, Inorg. Chem., 2017, 56, 4928–4936 CrossRef CAS PubMed.
- L. Lin, D.-H. Chen, R. Yu, X.-L. Chen, W.-J. Zhu, D. Liang, J.-F. Chang, Q. Zhang and C.-Z. Lu, J. Mater. Chem. C, 2017, 5, 4495–4504 RSC.
- C. H. Huang, M. Wen, C. Y. Wang, Y. F. Lu, X. H. Huang, H. H. Li, S. T. Wu, N. F. Zhuang and X. L. Hu, Dalton Trans., 2017, 46, 1413–1419 RSC.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- C. Bizzarri, F. Hundemer, J. Busch and S. Bräse, Polyhedron, 2018, 140, 51–66 CrossRef CAS.
- G. Blasse and D. R. McMillin, Chem. Phys. Lett., 1980, 70, 1–3 CrossRef CAS.
- R. Czerwieniec, J. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed.
- D. Volz, D. M. Zink, T. Bocksrocker, J. Friedrichs, M. Nieger, T. Baumann, U. Lemmer and S. Bräse, Chem. Mater., 2013, 25, 3414–3426 CrossRef CAS.
- D. M. Zink, D. Volz, T. Baumann, M. Mydlak, H. Flügge, J. Friedrichs, M. Nieger and S. Bräse, Chem. Mater., 2013, 25, 4471–4486 CrossRef CAS.
- D. M. Zink, M. Bachle, T. Baumann, M. Nieger, M. Kuhn, C. Wang, W. Klopper, U. Monkowius, T. Hofbeck, H. Yersin and S. Bräse, Inorg. Chem., 2013, 52, 2292–2305 CrossRef CAS PubMed.
- D. Volz, M. Wallesch, S. L. Grage, J. Gottlicher, R. Steininger, D. Batchelor, T. Vitova, A. S. Ulrich, C. Heske, L. Weinhardt, T. Baumann and S. Bräse, Inorg. Chem., 2014, 53, 7837–7847 CrossRef CAS PubMed.
- D. Volz, Y. Chen, M. Wallesch, R. Liu, C. Flechon, D. M. Zink, J. Friedrichs, H. Flugge, R. Steininger, J. Gottlicher, C. Heske, L. Weinhardt, S. Bräse, F. So and T. Baumann, Adv. Mater., 2015, 27, 2538–2543 CrossRef CAS PubMed.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed.
- F. Bappler, M. Zimmer, F. Dietrich, M. Grupe, M. Wallesch, D. Volz, S. Bräse, M. Gerhards and R. Diller, Phys. Chem. Chem. Phys., 2017, 19, 29438–29448 RSC.
- M. Zimmer, F. Dietrich, D. Volz, S. Bräse and M. Gerhards, ChemPhysChem, 2017, 18, 3023–3029 CrossRef CAS PubMed.
- L. H. He, Y. S. Luo, B. S. Di, J. L. Chen, C. L. Ho, H. R. Wen, S. J. Liu, J. Y. Wang and W. Y. Wong, Inorg. Chem., 2017, 56, 10311–10324 CrossRef CAS PubMed.
- V. W.-W. Yam, W.-K. Lee and K.-K. Cheung, Dalton Trans., 1996, 2335–2339 RSC.
- V. W.-W. Yam, C.-H. Lam and N. Zhu, Inorg. Chim. Acta, 2002, 331, 239–245 CrossRef CAS.
- K. J. Lotito and J. C. Peters, Chem. Commun., 2010, 46, 3690–3692 RSC.
- G. A. Bowmaker, J. V. Hanna, S. P. King, F. Marchetti, C. Pettinari, A. Pizzabiocca, B. W. Skelton, A. N. Sobolev, A. Tăbăcaru and A. H. White, Eur. J. Inorg. Chem., 2014, 2014, 6104–6116 CrossRef CAS.
- C. L. Chan, K. L. Cheung, W. H. Lam, E. C. Cheng, N. Zhu, S. W. Choi and V. W. Yam, Chem. - Asian J., 2006, 1, 273–286 CrossRef CAS PubMed.
- G. A. Crosby, G. R. Gamble and K. J. Jordan, Electronic Excited States Of Copper(I) Substituted-1,10-Phenanthroline And Substituted-Phosphine Mixed-Ligand Complexes, Proc. SPIE 1054, Fluorescence Detection III, 1989 DOI:10.1117/12.951561.
- L.-L. Hu, C. Shen, W.-K. Chu, J. Xiang, F. Yu, G. Xiang, Y. Nie, C.-L. Kwok, C.-F. Leung and C.-C. Ko, Polyhedron, 2017, 127, 203–211 CrossRef CAS.
- C. Marzano, M. Pellei, S. Alidori, A. Brossa, G. G. Lobbia, F. Tisato and C. Santini, J. Inorg. Biochem., 2006, 100, 299–304 CrossRef CAS PubMed.
- V. Gandin, F. Tisato, A. Dolmella, M. Pellei, C. Santini, M. Giorgetti, C. Marzano and M. Porchia, J. Med. Chem., 2014, 57, 4745–4760 CrossRef CAS PubMed.
- A. Bykowska, R. Starosta, J. Jezierska and M. Jeżowska-Bojczuk, RSC Adv., 2015, 5, 80804–80815 RSC.
- K. H. Mashat, B. A. Babgi, M. A. Hussien, M. Nadeem Arshad and M. H. Abdellattif, Polyhedron, 2019, 158, 164–172 CrossRef CAS.
- H. Liu, W. Yang, W. Zhou, Y. Xu, J. Xie and M. Li, Inorg. Chim. Acta, 2013, 405, 387–394 CrossRef CAS.
- L. J. Gooßen, N. Rodríguez, F. Manjolinho and P. P. Lange, Adv. Synth. Catal., 2010, 352, 2913–2917 CrossRef.
- Y. Ma, S. Qing, N. Li, L. Zhang, S. Li, Z. Gao, H. Li, W. Eli and T. Wang, Int. J. Chem. Kinet., 2015, 47, 621–628 CrossRef CAS.
- Y. C. Yuan, H. B. Yang, X. Y. Tang, Y. Wei and M. Shi, Chem. – Eur. J., 2016, 22, 5146–5150 CrossRef CAS PubMed.
- B. Huitorel, H. El Moll, R. Utrera-Melero, M. Cordier, A. Fargues, A. Garcia, F. Massuyeau, C. Martineau-Corcos, F. Fayon, A. Rakhmatullin, S. Kahlal, J. Y. Saillard, T. Gacoin and S. Perruchas, Inorg. Chem., 2018, 57, 4328–4339 CrossRef CAS PubMed.
- H. El Moll, M. Cordier, G. Nocton, F. Massuyeau, C. Latouche, C. Martineau-Corcos and S. Perruchas, Inorg. Chem., 2018, 57, 11961–11969 CrossRef CAS PubMed.
- D. Volz, T. Baumann, H. Flügge, M. Mydlak, T. Grab, M. Bächle, C. Barner-Kowollik and S. Bräse, J. Mater. Chem., 2012, 22, 20786 RSC.
- K. Chen, J. Shearer and V. J. Catalano, Inorg. Chem., 2015, 54, 6245–6256 CrossRef CAS PubMed.
- D. Volz, M. Nieger, J. Friedrichs, T. Baumann and S. Bräse, Langmuir, 2013, 29, 3034–3044 CrossRef CAS PubMed.
- D. M. Zink, T. Baumann, J. Friedrichs, M. Nieger and S. Bräse, Inorg. Chem., 2013, 52, 13509–13520 CrossRef CAS PubMed.
- D. Volz, A. F. Hirschbiel, D. M. Zink, J. Friedrichs, M. Nieger, T. Baumann, S. Bräse and C. Barner-Kowollik, J. Mater. Chem. C, 2014, 2, 1457 RSC.
- TURBOMOLE V7.4 2019, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007; TURBOMOLE GmbH: since 2007; available from http://www.turbomole.com.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- X. Blase, C. Attaccalite and V. Olevano, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115103 CrossRef.
- C. Holzer and W. Klopper, J. Chem. Phys., 2018, 149, 101101 CrossRef PubMed.
- K. Krause and W. Klopper, J. Comput. Chem., 2017, 38, 383–388 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- X. Gui, C. Holzer and W. Klopper, J. Chem. Theory Comput., 2018, 14, 2127–2136 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1919266–1919270, 1918364 and 1919271–1919273. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02447f |
| This journal is © The Royal Society of Chemistry 2019 |