
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembled orthoester cryptands: orthoester scope, post-functionalization, kinetic locking and tunable degradation kinetics†
Henrik
Löw
,
Elena
Mena-Osteritz
and
Max
von Delius
 *
*
Institute of Organic Chemistry and Advanced Materials, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: max.vondelius@uni-ulm.de
First published on 27th April 2018
Abstract
Dynamic adaptability and biodegradability are key features of functional, 21st century host–guest systems. We have recently discovered a class of tripodal supramolecular hosts, in which two orthoesters act as constitutionally dynamic bridgeheads. Having previously demonstrated the adaptive nature of these hosts, we now report the synthesis and characterization – including eight solid state structures – of a diverse set of orthoester cages, which provides evidence for the broad scope of this new host class. With the same set of compounds, we demonstrated that the rates of orthoester exchange and hydrolysis can be tuned over a remarkably wide range, from rapid hydrolysis at pH 8 to nearly inert at pH 1, and that the Taft parameter of the orthoester substituent allows an adequate prediction of the reaction kinetics. Moreover, the synthesis of an alkyne-capped cryptand enabled the post-functionalization of orthoester cryptands by Sonogashira and CuAAC “click” reactions. The methylation of the resulting triazole furnished a cryptate that was kinetically inert towards orthoester exchange and hydrolysis at pH > 1, which is equivalent to the “turnoff” of constitutionally dynamic imines by means of reduction. These findings indicate that orthoester cages may be more broadly useful than anticipated, e.g. as drug delivery agents with precisely tunable biodegradability or, thanks to the kinetic locking strategy, as ion sensors.
Introduction
The development of new macro(bi)cyclic compounds has been a key driving force of progress in supramolecular chemistry.1 The discovery of pillararenes,2 for instance, has opened up new avenues in supramolecular polymer chemistry,3 and the rational design of cyanostar4 and triazolophane macrocycles5 enabled remarkable binding affinities for hard-to-bind anions such as PF6−, as well as unprecedented rotaxane syntheses4a,6 and fundamental insights on the nature of hydrogen bonding.7 In this article, we follow up on our discovery of a new class of self-assembled macrobicyclic host8 and report comprehensive data on the scope, structure and properties of these compounds (Scheme 1).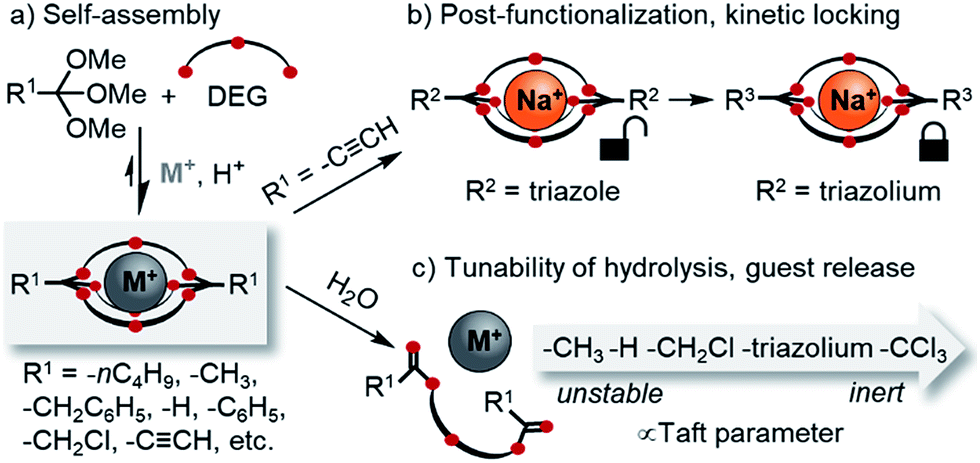 | ||
| Scheme 1 Overview on the scope of this study. (a) Template-directed self-assembly of orthoester cryptates: scope of orthoesters (R1) investigated in this contribution; (b) post-functionalization (R2) and kinetic locking (R3) of orthoester cryptates; (c) tunability of the degradation kinetics. DEG: diethylene glycol. M+ in this work: Na+ or Li+. | ||
Dynamic combinatorial chemistry9 has been used extensively for generating macrocycles and cages from smaller subcomponents,9g,10 yet prior to our discovery of orthoester cryptands,8 there were only few reports on the self-assembly of purely organic dynamic covalent cages suitable to encapsulate single11 cationic12 or anionic13 guests. Self-assembled macrobicyclic hosts beyond dynamic covalent chemistry have been reported, most notably metallosupramolecular cryptates14 as well as clathrochelates.15 However, these compounds share with conventional cryptands the feature that their host framework is no longer dynamic and stimuli-responsive once the self-assembly process is complete. This is different for orthoester cryptands, which under anhydrous acidic conditions are adaptive to their environment,16 which allowed us to demonstrate that a wide range of metal ions selectively directs the self-assembly of their thermodynamically preferred host.17
Another interesting feature of orthoester cryptands is that, in contrast to conventional cryptands featuring nitrogen bridgeheads, their synthesis requires only a single step whose yield can be as high as 92%, as we demonstrate in this work (Scheme 1a). Moreover, orthoester bridgeheads possess a substituent (R1, Scheme 1) that should allow modulating the stability, solubility and lipophilicity of the cages and provide a convenient handle for adding further functionality. However, prior to this work, only three such substituents (R1 = –CH3, –CH2CH3, –H) have been explored.8
When considering applications in the emerging field of supramolecular medicinal chemistry,18 the most important feature of orthoester cryptands is their inherent tendency to hydrolyze19 under general acid catalysis to ring-opened esters, which leads to the irreversible release of the encapsulated guest. Although earlier studies have investigated the kinetics of orthoester hydrolysis and its pH dependency,20 a systematic study on a wide range of orthoester substituents (R1, Scheme 1) is still elusive, and the kinetics of orthoester exchange as a new addition to the toolbox of dynamic covalent chemistry21 remain unexplored.
Herein, we report a comprehensive study on the scope and generality of self-assembled orthoester cryptands (Scheme 1a) as well as their post-functionalization and post-synthetic stabilization against hydrolysis or exchange reactions (“kinetic locking”, Scheme 1b). Moreover, we explored the question to which extent the kinetics of orthoester exchange and hydrolysis can be tuned as a function of different orthoester substituents and whether the observed trends can be rationalized on the basis of linear free energy relationships (Scheme 1c).22
Results and discussion
Scope of orthoester cryptand self-assembly
The systematic exploration of the scope and generality of the synthesis of orthoester cryptands was the first objective of this study, because previous investigations had been limited to orthoformates (R1 = H), orthoacetates (R1 = CH3) and orthopropanoates (R1 = CH2CH3), and the self-assembly had only been successful for the two latter starting materials.8,17 We therefore chose five commercial and six non-commercial23 orthoesters with diverse functional groups ranging from electron-donating (e.g. R1 = –nC4H9) to aromatic (e.g. R1 = –C6H6) and strongly electron-withdrawing (e.g. R1 = –CF3). Furthermore, an alkynyl substituted orthoester was prepared in view of a possible post-functionalization of the self-assembled host. As shown in Scheme 2, these eleven orthoesters were subjected to standard reaction conditions for orthoester exchange. Specifically, these reaction conditions entail the use of diethylene glycol (DEG; grey box in Scheme 2) as a ligand for cation binding and substrate for orthoester/alcohol exchange, as well as trifluoroacetic acid (TFA) as catalyst. In addition, a suitable alkali metal template (e.g. NaBArF) and molecular sieves (5 Å) are required to provide a driving force for cryptate self-assembly and to maintain anhydrous reaction conditions.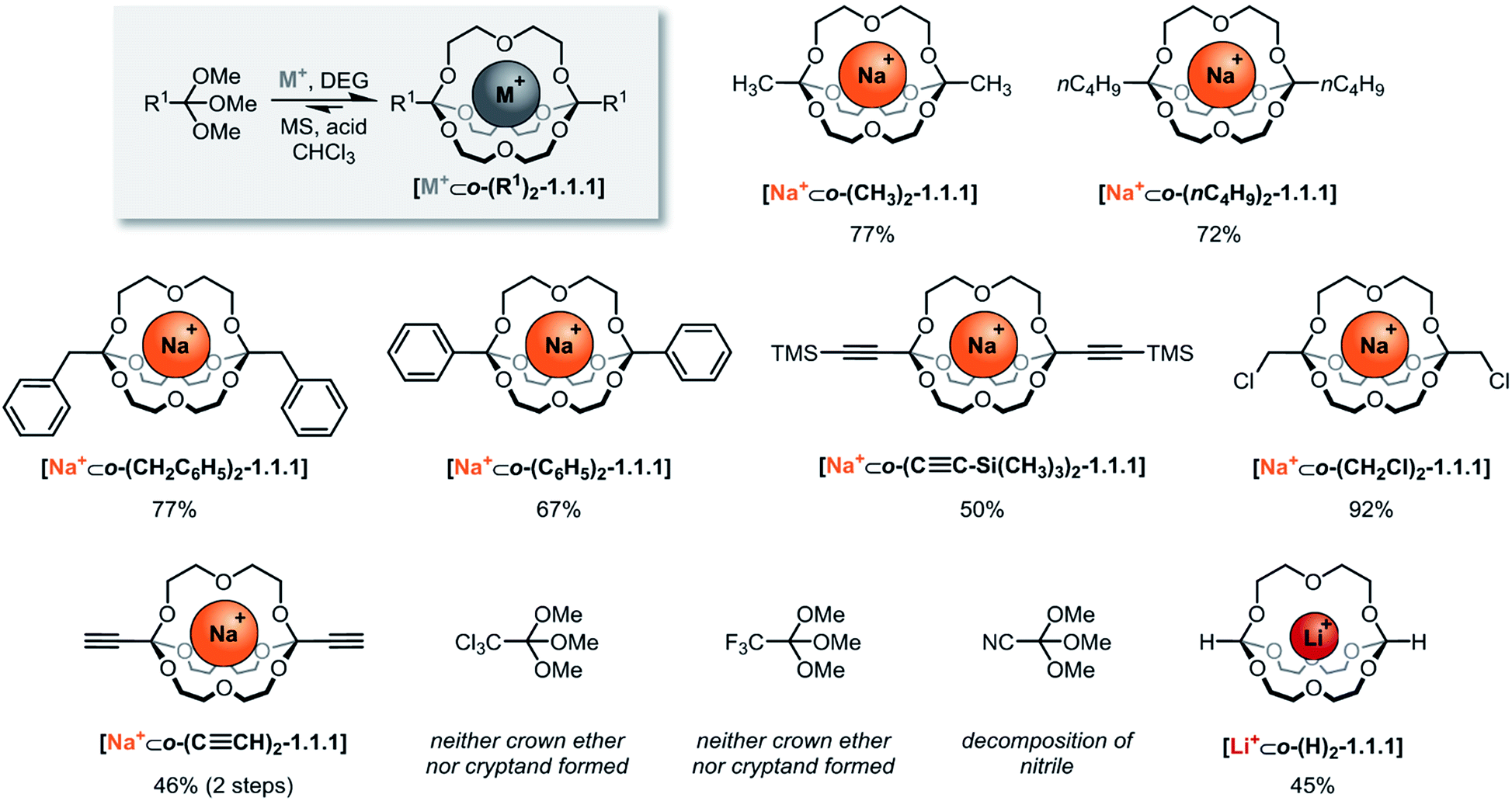 | ||
| Scheme 2 Scope of self-assembled orthoester cryptands. Reaction conditions: 60 μmol sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF) or LiBArF, 180 μmol diethylene glycol, 120 μmol orthoester, 1.2 μmol TFA (R1 = –CH2Cl: 12 μmol TFA) in CHCl3, 7–13 days (for further details, see ESI†). Percent values indicate isolated yields. M+: Na+; Li+. DEG: diethylene glycol. MS: 5 Å molecular sieves. | ||
By optimizing our procedures for preparing anhydrous starting materials (see ESI†), we were able to minimize the undesired hydrolysis of orthoesters to esters and to improve the isolated yield of previously reported orthoacetate cryptate [Na+⊂o-(CH3)2-1.1.1]8 from 67% to 77%. Yields in the range of 50% to 92% were obtained for five new orthoester cryptates (R1 = –nC4H9, –CH2C6H5, –C6H5, –C≡C–Si(CH3)3 and –CH2Cl; Scheme 2), demonstrating that alkyl, benzyl and phenyl substituted orthoester cryptates can be accessed in a straight-forward manner. The reaction times were relatively long (7 to 13 days), and we had previously speculated that this is mainly due to the slow uptake of methanol by molecular sieves, which presumably provides an additional entropic driving force for self-assembly.24
Our study on the kinetics of orthoester exchange supports this hypothesis, by showing that the self-assembly reactions require significantly more time than simpler exchange reactions of the same orthoester substrates (vide infra). Purification of the crude cryptates was possible by passing a chloroform solution through a short plug of silica gel. The highly efficient synthesis of a chloromethyl-substituted cage shows that mildly electron-withdrawing substituents are tolerated, even though their presence presumably destabilizes the oxonium ion intermediate of the exchange reaction. In fact, the observed isolated yield of 92% might well be a consequence of this effect, because, for the same reason, this cryptate is particularly stable against hydrolysis (vide infra), which minimizes any losses during purification.
The valuable alkynyl-substituted cryptate [Na+⊂o-(C≡CH)2-1.1.1] could be obtained in 46% yield over two steps after deprotection of the corresponding TMS-alkynyl-substituted cryptate. We were unable to self-assemble this compound directly from the corresponding orthoester (R1 = C≡CH), which is likely a result of the stronger electron-withdrawing effect of the free alkyne, in comparison to the silyl-protected analogue.‡ In line with this reasoning, we did not observe any evidence for cryptand formation starting from even more electron-deficient orthoesters (R1 = –CCl3, –CF3, –CN). It should be noted that, in model experiments (vide infra), we were able to induce orthoester exchange in two of these substrates, but the required strong acid catalysts (e.g., trifluoromethanesulfonic acid) appear to be too harsh to allow for template-directed cryptate self-assembly.
Arguably the most intriguing case in this scope study is the reaction of trimethyl orthoformate (R1 = H) with diethylene glycol. In our initial communication on cryptate [Na+⊂o-(CH3)2-1.1.1] we had reported that trimethyl orthoformate (H–C(OCH3)3) under standard conditions gives rise to the self-assembly of crown ether complexes [Na+⊂o-(H)2-(OCH3)2-16-crown-6], but not to the corresponding cryptands. This was a surprising finding, because the cryptate self-assembly would simply require the addition of one additional DEG bridge to the crown ether complex. We have now discovered that an orthoformate cryptate [Li+⊂o-(H)2-1.1.1] can in fact be obtained, but only if a lithium, instead of a sodium template is used.
To better understand this unexpected result, we isolated empty cryptand o-(H)2-1.1.1 by treatment of [Li+⊂o-(H)2-1.1.1] with anion exchange resin (Cl form; for further details, see ESI†) to study the thermodynamics of lithium and sodium binding. Therefore, we performed 1H NMR titrations with NaBArF and LiBArF in acetonitrile at different concentrations (1 to 10 mM) and fitted the resulting binding isotherms with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric model on the website http://supramolecular.org (original data is available for download in the spirit of “open-science”,26 see ESI†). The quality of the data was evaluated based on triplicate titrations (95% confidence interval, Table 1) and statistical analyses of the goodness of fit (see ESI†). The resulting association constants show that lithium binds to o-(H)2-1.1.1 about 200 times more strongly than sodium (see Table 1). A comparison with data on methyl-substituted cryptand o-(CH3)2-1.1.1 (ref. 25) (Table 1, right column) reveals that the substitution of methyl for hydrogen in o-(H)2-1.1.1 leads to stronger binding of lithium and weaker binding of sodium. These Ka values provide a compelling explanation for our unexpected observation that cryptand o-(H)2-1.1.1 can be obtained with a lithium template, but not with a sodium template, even though titrations were performed in acetonitrile and self-assembly reactions are carried out in chloroform,§ where the relative magnitudes of ion–dipole interactions are typically more pronounced than in acetonitrile. It will be interesting to address the question why cryptand o-(H)2-1.1.1 exhibits such outlier thermodynamic behavior, but this will likely require high-level computational studies that are beyond the scope of this contribution.
:1 stoichiometric model on the website http://supramolecular.org (original data is available for download in the spirit of “open-science”,26 see ESI†). The quality of the data was evaluated based on triplicate titrations (95% confidence interval, Table 1) and statistical analyses of the goodness of fit (see ESI†). The resulting association constants show that lithium binds to o-(H)2-1.1.1 about 200 times more strongly than sodium (see Table 1). A comparison with data on methyl-substituted cryptand o-(CH3)2-1.1.1 (ref. 25) (Table 1, right column) reveals that the substitution of methyl for hydrogen in o-(H)2-1.1.1 leads to stronger binding of lithium and weaker binding of sodium. These Ka values provide a compelling explanation for our unexpected observation that cryptand o-(H)2-1.1.1 can be obtained with a lithium template, but not with a sodium template, even though titrations were performed in acetonitrile and self-assembly reactions are carried out in chloroform,§ where the relative magnitudes of ion–dipole interactions are typically more pronounced than in acetonitrile. It will be interesting to address the question why cryptand o-(H)2-1.1.1 exhibits such outlier thermodynamic behavior, but this will likely require high-level computational studies that are beyond the scope of this contribution.
| Entry | o-(H)2-1.1.1 | o-(CH3)2-1.1.1 (ref. 25) | |
|---|---|---|---|
| a Titrations were performed using BArF salts in acetonitrile at different host concentrations (1 to 10 mM). Association constants were determined by 1H NMR titrations and fitting of isotherms on http://supramolecular.org. Estimates of uncertainty reflect 95% confidence intervals (triplicate titrations). For further details, see ESI. | |||
| 1 | Li+ | 13000 ± 1000 M−1 |
1700 M−1 |
| 2 | Na+ | 60 ± 20 M−1 | 1300 M−1 |
Solid state structures
To gain structural insights into the prepared macrobicyclic compounds, we attempted to crystallize all new orthoester cryptates by slow diffusion of hexane into chloroform solutions. To our delight, we were able to obtain single crystals suitable for X-ray crystallography of eight new orthoester cryptates (see Chart 1). All sodium-based architectures, including previously reported [Na+⊂o-(CH3)2-1.1.1],8 feature the encapsulation of the alkali metal ion at the centre of the cavity, held in place by nine Na–O bonds. The only exception to this mode of binding is [Na+⊂o-(C6H5)2-1.1.1], in which only eight Na–O bonds are observed. One orthoester oxygen is more distant (3.53 Å) and two are closer to the metal ion (2.32, 2.40 Å) than in all other sodium-containing structures (2.43–2.92 Å). Due to this slight distortion, caused most likely by packing effects, the three chain oxygen atoms are closer to the metal ion (2.37–2.46 Å) than in the other seven sodium cryptates (2.48–2.67 Å; see Chart 1i for a statistical analysis of bond lengths). Most of the orthoester cryptates crystallize in a triclinic crystal system (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), but monoclinic (P21/n or P21/c) and orthorhombic (Pbca) systems were also observed.
), but monoclinic (P21/n or P21/c) and orthorhombic (Pbca) systems were also observed.
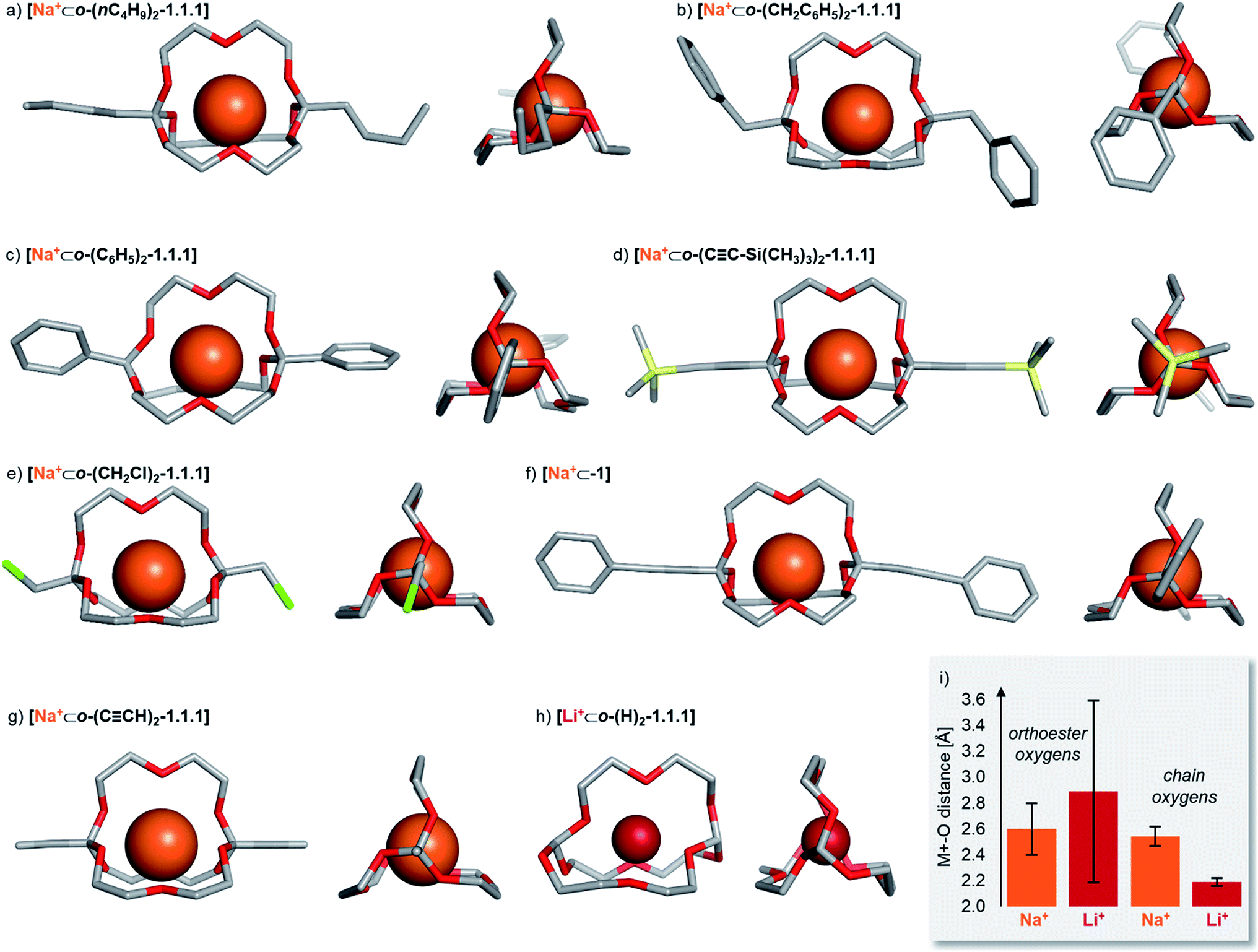 | ||
| Chart 1 Solid-state structures of eight orthoester cryptands. Single crystals were obtained by the layering method (hexane/chloroform). Hydrogen atoms, anions, solvent and disorder (where applicable) are omitted for clarity. Metal ions are displayed at 100% of effective ionic radius.27 (a) Crystal system: triclinic. Na–O distance (orthoester oxygen): 2.45–2.92 Å. Na–O distance (chain oxygen): 2.48–2.53 Å. (b) Crystal system: monoclinic. Na–O distance (orthoester oxygen): 2.47–2.76 Å. Na–O distance (chain oxygen): 2.51–2.57 Å. (c) Crystal system: orthorhombic. Na–O distance (orthoester oxygen): 2.32–3.53 Å. Na–O distance (chain oxygen): 2.37–2.46 Å. (d) Crystal system: triclinic. Na–O distance (orthoester oxygen): 2.48–2.59 Å. Na–O distance (chain oxygen): 2.54–2.67 Å. (e) Crystal system: monoclinic. Na–O distance (orthoester oxygen): 2.47–2.89 Å. Na–O distance (chain oxygen): 2.48–2.57 Å. (f) Crystal system: triclinic. Na–O distance (orthoester oxygen): 2.43–2.69 Å. Na–O distance (chain oxygen): 2.56–2.61 Å. (g) Crystal system: triclinic. Na–O distance (orthoester oxygen): 2.46–2.66 Å. Na–O distance (chain oxygen): 2.53–2.67 Å. (h) Crystal system: orthorhombic. Li–O distance (orthoester oxygen): 1.96–3.67 Å. Li–O distance (chain oxygen): 2.16–2.23 Å. (i) Comparison of average Na–O distance of all sodium-based solid-state structures with average Li–O distance in [Li+⊂o-(H)2-1.1.1], including standard deviation. For further details, see ESI.† | ||
We were also able to obtain X-ray crystallographic data on a cryptate encapsulating a lithium ion for the first time. Numerous previous attempts at crystallizing [Li+⊂o-(CH3)2-1.1.1] had been met with failure, presumably due to the relatively low binding constant (Table 1) and (degenerate) binding dynamics in this host.25 In stark contrast to the sodium-based structures, cryptate [Li+⊂o-(H)2-1.1.1] features only five Li–O bonds. This binding mode leads to rather short Li–O bonds of the three chain oxygen atoms (2.16–2.23 Å) and even shorter contacts for the two orthoester oxygen atoms that are involved in Li–O bonds (1.96 Å). Hence, the remaining four orthoester oxygen atoms are significantly more distant (2.94–3.67 Å) than in the sodium-containing solid state structures (see Chart 1i). The crystal system observed for [Li+⊂o-(H)2-1.1.1] is orthorhombic (P212121).
Kinetics of orthoester exchange and hydrolysis
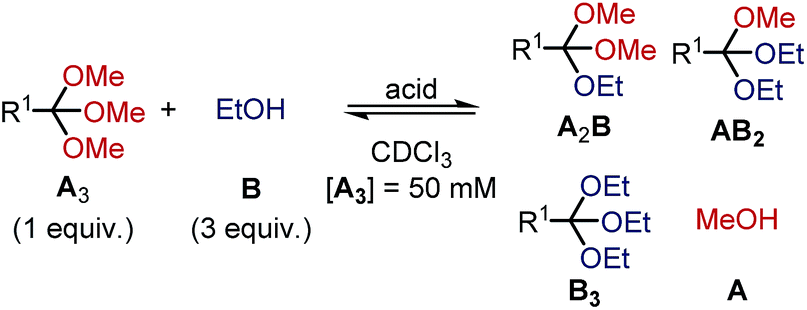
To shed light on the differences in reactivity and stability of orthoesters and their corresponding cryptands, we decided to investigate the kinetics of the exchange reactions of a diverse set of orthoesters with ethanol (see Table 2). To this end, 13 trimethyl orthoesters (A3) were treated with three equivalents of ethanol (B) and acid catalyst under standardized conditions and the reaction progress was monitored for up to 16 hours by 1H NMR spectroscopy. To account for the vast differences in reactivity between the orthoesters, both the amount of acid, as well as the type of acid had to be varied. For example, the CCl3– and CF3–substituted orthoesters were found to be so inert that even large quantities of trifluoroacetic acid did not lead to discernible exchange products. Hence, whenever an orthoester was found to be too inert to react within 16 h under mild reaction conditions, the next harsher set of reaction conditions was investigated (and so on). Besides this step-wise enhancement of acid catalysis, the second crucial parameter in Table 2 is the equilibration time t, which we defined as the first data point at which ≥99% conversion to the equilibrium distribution was reached based on 1H NMR integration. The kinetic data allowed us to order the 13 studied orthoesters from most reactive to most inert (top to bottom in Table 2).|
|
||||
|---|---|---|---|---|
| Acid | R1 | t [min] | Product ratio (A3:A2B:AB2:B3) |
Taft22 |
| a Reaction conditions: orthoester (A3, 37.5 μmol, 1.0 equiv.), alcohol (B, 112.5 μmol, 3.0 equiv.), internal standard (9.41 μmol) and acid catalyst (0.01 to 100 mol%) were added to the reaction vessel from stock solutions. CDCl3 was added to obtain a total volume of 750 μL. The reaction was monitored by 1H NMR spectroscopy. b t: equilibration time, defined as data point when 99% conversion to plateau-level of methanol A was exceeded for the first time; n.d.: not determined due to peak overlap in NMR spectrum. Estimated error: ±5%. c 1-Benzyl-4-(trimethoxymethyl)-1H-1,2,3-triazole. d 1-Benzyl-3-methyl-4-(trimethoxymethyl)-1H-1,2,3-triazol-3-ium. | ||||
| 0.01% TFA | –nC4H9 | 10 | n.d. | −0.13 |
| 0.01% TFA | –CH3 | 280 | 26:44:24:6 |
0.00 |
| 0.1% TFA | –CH2C6H5 | 650 | 23:41:29:6 |
0.22 |
| 1% TFA | –H | 40 | 19:43:31:7 |
0.49 |
| 1% TFA | –C6H5 | 50 | 23:42:27:7 |
0.60 |
| 1% TFA | –C≡C–TMS | 90 | 22:41:29:7 |
— |
| 1% TFA | –Triazolec | 130 | n.d. | — |
| 1% TFA | –CH2Cl | 640 | 16:42:32:10 |
1.05 |
| 10% TFA | –C≡CH | 300 | 32:35:26:6 |
2.18 |
| 50% TfOH | –CCl3 | 330 | n.d. | 2.65 |
| 50% TfOH | –Triazoliumd | 490 | 16:39:33:11 |
— |
| 100% TfOH | –CF3 | >1000 | n.d. | 2.61 |
| 100% TfOH | –CN | Decomposition | 3.30 | |
A comparison of the equilibration times reveals that electron-rich orthoesters are vastly more reactive than electron-deficient orthoesters and that the reactivity of orthoesters can be fine-tuned over a remarkably wide range. For instance, trimethyl orthovalerate (R1 = –nC4H9, see Table 2) equilibrates with ethanol in only 13 min with 0.01% TFA, whereas the reaction of 1,1,1-trifluoro-2,2,2-trimethoxyethane (R1 = –CF3, see Table 2) did not reach equilibrium after 16 h, even with 100 mol% trifluoromethanesulfonic acid (TfOH).
Because the results summarized in Table 2 represent the first account of a reactivity sequence for orthoester exchange, we attempted to identify a linear free energy relationship (LFER) that would correlate with the observed trend. We quickly noticed that the Hammett substituent parameter (σ) and variations thereof are a poor fit, which is not too surprising given that the Hammett reference reaction is rather unlike orthoester exchange. As can be deduced from Table 2, the Taft parameter for the R1 group does, however, correlate very well with the observed reactivity trend with the only exception (CCl3vs. CF3, see Table 2) being based on a very narrow margin and not relevant to templated cryptate self-assembly. From a mechanistic perspective, this finding makes good sense, because the Taft LFER is based on the acid-catalysed hydrolysis of aliphatic esters, which proceeds via the same oxonium intermediates as orthoester exchange.28 Because many orthoester derivatives are commercially available, this correlation with the Taft LFER will be very valuable for designing and optimizing future self-assemblies based on orthoester exchange.
In respect to the scope of self-assembled orthoester cryptates (vide supra), these results imply that there is a threshold where self-assembly is no longer possible, because the R1 substituent is too strongly electron-withdrawing and that this threshold lies between –CH2Cl (Taft 1.05, see Table 2) and –C≡CH (Taft 2.18, see Table 2). Nevertheless, we would like to emphasize that cryptands equipped with functional groups with Taft values > 1 are accessible via post-functionalization methods (e.g. R1 = –C≡CH by deprotection or –triazolium by methylation, vide infra).
When considering the thermodynamic, rather than kinetic implications of these results, it is evident from Table 2 that the observed equilibrium compositions (A3:A2B:AB2:B3) are mostly unaffected by the R1 substituent and the acid catalyst. Yet to our surprise, when plotting the evolution of individual species over time, we found that the thermodynamically least favoured product (B3) generally reaches its plateau level first. This finding can be rationalized by conflicting steric and electronic effects: the sterically more demanding ethyl group makes B3 the thermodynamically least stable product, yet the oxonium ion that leads to the formation of B3 is the most stable intermediate, which means that the conversion of A2B into AB2 is slower than the conversion of AB2 into B3. See ESI† for further details and kinetic simulations of this interesting “bottleneck” in the evolution of the chemical network over time,29 including the exchange reaction with 2-chloroethanol, which confirms the above reasoning.
In view of possible applications of orthoester cryptates in drug delivery,19,30 we turned our attention towards the hydrolytic degradation of orthoesters and the corresponding self-assembled cryptands (Table 3). Six representative orthoesters were dissolved in phosphate buffer solutions of varying pH. The reactions were monitored by 1H NMR spectroscopy to determine the half-life t1/2 and the kinetic rate kobs of the hydrolysis reaction under pseudo first order conditions. As expected, the kinetics of orthoester hydrolysis follow the same trend that was observed for orthoester exchange. While electron-rich orthoesters (e.g., R1 = –CH3, see Table 3, entry 1) hydrolyze readily even at neutral pH, more electron-deficient orthoesters (e.g., R1 = –triazolium, –CCl3, see Table 3, entries 5 and 6) are remarkably stable: even at pH 1 their half-lives are beyond 16 h. In the context of drug delivery, we believe that orthoformates (R1 = –H; t1/2 70 min at pH 6, see Table 3, entry 2) and chloromethyl-substituted orthoesters (R1 = –CH2Cl; t1/2 121 min at pH 5 and 34 min at pH 4, see Table 3, entry 3) offer the most appealing hydrolysis profiles.¶
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | pH 8 | pH 7 | pH 6 | pH 5 | pH 4 | pH 3 | pH 1 | |
| a Reaction conditions: orthoester (A3, 37.5 μmol) or cryptand (o-(CH3)2-1.1.1, 18.75 μmol) and internal standard (12.3 μmol) were added to the reaction vessel. Buffer solution was added to obtain a total volume of 750 μL. The reaction was monitored by 1H NMR spectroscopy. t1/2: half-life of starting material, defined as point when 50% of starting material were consumed. Estimated error: ±4%. b 1-Benzyl-4-(trimethoxymethyl)-1H-1,2,3-triazole. c 400 μL DMSO added to increase solubility of starting material. Comparison of measurements in pure buffer and with addition of DMSO revealed that kobs is decreased by ca. one order of magnitude upon addition of the co-solvent (for further details, see ESI). d 1-Benzyl-3-methyl-4-(trimethoxymethyl)-1H-1,2,3-triazol-3-ium. | |||||||||
| 1 | –CH3 | t 1/2 [min], kobs [s−1] | 60, 1.8 × 10−4 | 10, 1.3 × 10−3 | 2, 6.1 × 10−3 | <1 | |||
| 2 | –H | t 1/2 [min], kobs [s−1] | >1000 | 510, 2.5 × 10−5 | 70, 1.6 × 10−4 | 20, 8.8 × 10−4 | 7, 2.0 × 10−3 | <1 | |
| 3 | –CH2Cl | t 1/2 [min], kobs [s−1] | >1000 | 660, 1.7 × 10−5 | 120, 8.6 × 10−5 | 30, 2.9 × 10−4 | <1 | ||
| 4 | –Triazoleb | t 1/2 [min] | <1c | ||||||
| 5 | –Triazoliumd | t 1/2 [min], kobs [s−1] | Inert | >10000, 1.6 × 10−6 |
|||||
| 6 | –CCl3 | t 1/2 [min] | >1000c | ||||||
| 7 | o-(CH3)2-1.1.1 | t 1/2 [min], kobs [s−1] | 30, 3.5 × 10−4 | ||||||
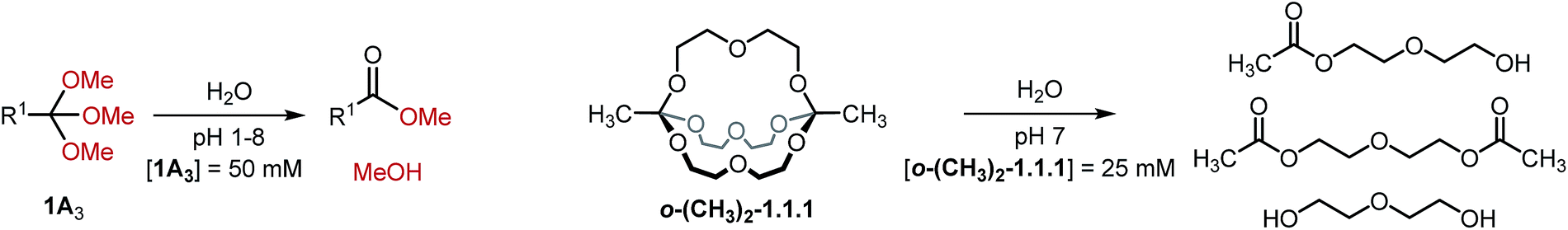
Finally, we monitored the hydrolysis of cryptand o-(CH3)2-1.1.1 under comparable reaction conditions (see Table 3, entry 7). As anticipated,8 the observed hydrolysis rate for cryptand o-(CH3)2-1.1.1 is somewhat lower than that for the simple orthoester but in the same range, indicating that our kinetic findings are transferable from orthoesters to orthoester cryptands.
Post-functionalization and kinetic locking of orthoester cryptands
Having established the broad scope and the kinetic limitations of the self-assembly reaction, we turned our attention towards further diversifying orthoester cryptates by post-functionalization (see Scheme 3a).31 Cryptate [Na+⊂o-(C≡CH)2-1.1.1], containing two functional alkyne handles, was subjected to a Sonogashira reaction with iodobenzene to furnish cryptand 1 in 94% yield. The basicity of the reaction conditions had the advantageous effect that no orthoester hydrolysis was observed, but also led to decomplexation, thus requiring a trivial second step to reintroduce the sodium guest, thus furnishing cryptate [Na+⊂1]. We believe that this type of reaction will be particularly useful for the preparation of intriguing degradable 1D polymers and for applications in ion sensing, because the aromatic substituents are in conjugation with the orthoester bridgeheads. The introduction of conjugated chromophores or fluorophores should result in pronounced differences in optical output depending on the nature of guest ions, and could also provide a theranostic readout32 for cage degradation and concomitant guest release. | ||
| Scheme 3 Post-functionalization of orthoester cryptands. (a) Reaction conditions: (i) [Na+⊂o-(C≡CH)2-1.1.1] (11.8 μmol, 1.0 equiv.), iodobenzene (23.6 μmol, 2.0 equiv.), NEt3 (35.4 μmol, 3.0 equiv.), CuI (0.12 μmol, 0.01 equiv.), Pd(PPh3)4 (0.12 μmol, 0.01 equiv.), THF, 40 °C, 4 d, 94%. (ii) 1 (9.28 μmol, 1.0 equiv.), NaBArF (9.28 μmol, 1.0 equiv.), CH3CN, r.t., 5 min, quant. (iii) [Na+⊂o-(C≡CH)2-1.1.1] (11.0 μmol, 1.0 equiv.), benzyl azide (22.0 μmol, 2.0 equiv.), Cu(MeCN)4PF6 (0.11 μmol, 0.01 equiv.), TBTA (0.11 μmol, 0.01 equiv.), MeOH, 70 °C, 24 h, 92%. (iv) [Na+⊂2] (3.9 μmol, 1.0 equiv.), MeI (excess), MeCN, 70 °C, 3 d, 83%. (b) Partial 1H NMR stacked plot (500 MHz, 298 K, CD3CN). | ||
An alternative method for post-functionalization was investigated by the CuAAC “click” reaction between [Na+⊂o-(C≡CH)2-1.1.1] and benzyl azide. This reaction proceeded in protocol, the product cryptate [Na+⊂2] was obtained in excellent yield (92%), and in contrast to the Sonogashira reaction without decomplexation of the metal guest. Although electronic communication between substituents and cage is likely weak in such bis-triazoles,33 we believe that this type of functionalization will prove valuable for installing functional moieties, for example for modulating lipophilicity, targeting biological binding motifs34 or stimuli-responsive triggering of cage decomposition.35
Encouraged by the efficient post-functionalization reactions and the observed differences in hydrolysis rates (Table 3), we wondered whether we could drastically stabilize compound [Na+⊂2] by methylation of the triazoles.36 At least in principle, the electron-withdrawing effect of the resulting triazolium on the orthoester bridgehead could lead to significantly reduced orthoester reactivity and pave the way towards applications of orthoester cryptands that require stability in water. As shown in Scheme 3a, treatment of [Na+⊂2] with an excess of iodomethane furnished the corresponding bis-triazolium cryptate [Na+⊂3] in 83% yield (Scheme 3a, bottom). In proof-of-principle experiments, the corresponding triazolium-substituted orthoester indeed proved to be inert towards hydrolysis at pH 3 (Table 3, entry 5) and underwent orthoester exchange only with 50% triflic acid (Table 2). At pH 1 the half-life for hydrolysis was beyond 16 h, whereas all other orthoesters suitable to prepare cryptates hydrolyzed rapidly under these conditions. The extrapolated half-life of the triazolium–orthoester in fact exceeded 10000 min, whereas the half-life of the triazole–orthoester is below one minute, which accounts for an increase in stability of at least four orders of magnitude (Table 3, entries 4 and 5). As such, this post-stabilization method is akin to the kinetic locking of imines by means of reduction to the corresponding amines, which has found widespread use in the synthesis of mechanically interlocked molecules, organic cages, porous materials and other applications of dynamic covalent chemistry.37
Conclusions
In this work, we have demonstrated that orthoester cryptands equipped with a broad range of substituents are accessible in one step via efficient, metal-templated self-assembly reactions. Cryptands substituted with strongly electron-withdrawing groups could not be obtained, but this limitation was circumvented by highly efficient post-functionalization methodologies based on a Sonogashira or CuAAC reaction of the bis-alkynyl cryptate. Methylation of a triazole-substituted cryptate furnished a supramolecular host that can be considered kinetically locked in respect to orthoester exchange and hydrolysis, but still binds effectively to cationic guests. In future studies, it could be beneficial to reverse this “locking” by site-selective N-dealkylation, which appears to be possible under relatively mild basic conditions.38 We also carried out detailed, comparative kinetic studies on orthoester exchange and hydrolysis, firmly establishing that the tunability of orthoester degradation makes these architectures interesting candidates for the (topical) delivery of ionic drugs, which strictly requires the biodegradability of otherwise toxic supramolecular hosts.39 Further studies on (an)ion encapsulation, sensing, transport and release are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (Emmy-Noether grant DE1830/2-1). We would like to thank Tobias Bachmann for experimental work carried out during his master thesis. Bernhard Müller is acknowledged for assistance with X-ray crystallography.Notes and references
- (a) Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC; (b) P. A. Gale and J. W. Steed, Supramolecular chemistry: from molecules to nanomaterials, Wiley, 2012 Search PubMed.
- T. Ogoshi, S. Kanai, S. Fujinami and T. Yamagishi, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- (a) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed; (b) T. Ogoshi, T. A. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed; (c) N. L. Strutt, H. Zhang, M. A. Giesener, J. Lei and J. F. Stoddart, Chem. Commun., 2012, 48, 1647–1649 RSC; (d) B. Shi, D. Xia and Y. Yao, Chem. Commun., 2014, 50, 13932–13935 RSC; (e) W. Xia, M. Ni, C. Yao, X. Wang, D. Chen, C. Lin, X. Y. Hu and L. Wang, Macromolecules, 2015, 48, 4403–4409 CrossRef CAS; (f) T. Ogoshi, H. Kayama, D. Yamafuji, T. Aoki and T. Yamagishi, Chem. Sci., 2012, 3, 3221–3226 RSC; (g) S. Sun, X.-Y. Hu, D. Chen, J. Shi, Y. Dong, C. Lin, Y. Pan and L. Wang, Polym. Chem., 2013, 4, 2224–2229 RSC; (h) B. Xia, B. Zheng, C. Han, S. Dong, M. Zhang, B. Hu, Y. Yu and F. Huang, Polym. Chem., 2013, 4, 2019–2024 RSC; (i) X. Wang, K. Han, J. Li, X. Jia and C. Li, Polym. Chem., 2013, 4, 3998–4003 RSC.
- (a) S. Lee, C. H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed; (b) E. M. Fatila, M. Pink, E. B. Twum, J. A. Karty and A. H. Flood, Chem. Sci., 2018, 9, 2853–3070 RSC.
- (a) Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed; (b) Y. Li and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 12111–12122 CrossRef CAS PubMed.
- B. Qiao, Y. Liu, S. Lee, M. Pink and A. H. Flood, Chem. Commun., 2016, 52, 13675–13678 RSC.
- Y. Liu, A. Sengupta, K. Raghavachari and A. H. Flood, Chem, 2017, 3, 411–427 CAS.
- R.-C. Brachvogel, F. Hampel and M. von Delius, Nat. Commun., 2015, 6, 7129 CrossRef PubMed.
- (a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (b) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed; (c) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634 RSC; (d) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Curr. Opin. Chem. Biol., 2002, 6, 321–327 CrossRef CAS PubMed; (e) R.-C. Brachvogel and M. von Delius, Eur. J. Org. Chem., 2016, 2016, 3662–3670 CrossRef CAS; (f) Q. Ji, R. C. Lirag and O. Š. Miljanić, Chem. Soc. Rev., 2014, 43, 1873–1884 RSC; (g) A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- (a) N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS PubMed; (b) A. González-Álvarez, I. Alfonso, F. López-Ortiz, Á. Aguirre, S. García-Granda and V. Gotor, Eur. J. Org. Chem., 2004, 2004, 1117–1127 CrossRef; (c) B. Fuchs, A. Nelson, A. Star, J. F. Stoddart and S. Vidal, Angew. Chem., Int. Ed., 2003, 42, 4220–4224 CrossRef CAS PubMed; (d) G. R. L. Cousins, R. L. E. Furlan, Y.-F. Ng, J. E. Redman and J. K. M. Sanders, Angew. Chem., Int. Ed., 2001, 40, 423–428 CrossRef CAS; (e) G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC; (f) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- (a) O. W. Howarth, G. G. Morgan, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 1999, 2097–2102 RSC; (b) Q. Lu, J.-M. Latour, C. J. Harding, N. Martin, D. J. Marrs, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 1994, 1471–1478 RSC; (c) D. MacDowell and J. Nelson, Tetrahedron Lett., 1988, 29, 385–386 CrossRef CAS; (d) J. Jazwinski, J.-M. Lehn, D. Lilienbaum, R. Ziessel, J. Guilhem and C. Pascard, J. Chem. Soc., Chem. Commun., 1987, 1691–1694 RSC.
- (a) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS PubMed; (b) H. Y. Au-Yeung, F. B. L. Cougnon, S. Otto, G. D. Pantoş and J. K. M. Sanders, Chem. Sci., 2010, 1, 567 RSC; (c) J. A. Berrocal, R. Cacciapaglia and S. Di Stefano, Org. Biomol. Chem., 2011, 9, 8190 RSC; (d) Y. Zhang, Y. M. Legrand, A. Van Der Lee and M. Barboiu, Eur. J. Org. Chem., 2016, 2016, 1825–1828 CrossRef CAS.
- (a) Z. Rodriguez-Docampo, E. Eugenieva-Ilieva, C. Reyheller, A. M. Belenguer, S. Kubik and S. Otto, Chem. Commun., 2011, 47, 9798 RSC; (b) E. A. Katayev, G. D. Pantos, A. D. Reshetova, V. N. Khrustalev, V. M. Lynch, Y. A. Ustynyuk and J. L. Sessler, Angew. Chem., Int. Ed., 2005, 44, 7386–7390 CrossRef CAS PubMed.
- (a) R. W. Saalfrank, A. Dresel, V. Seitz, S. Trummer, F. Hampel, M. Teichert, D. Stalke, C. Stadler, J. Daub, V. Schunemann and A. X. Trautwein, Chem.–Eur. J., 1997, 3, 2058–2062 CrossRef CAS; (b) R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8795–8824 Search PubMed.
- (a) Y. Z. Voloshin, O. A. Varzatskii, A. I. Stash, V. K. Belsky, Y. N. Bubnov, I. I. Vorontsov, K. A. Potekhin, M. Y. Antipin and E. V. Polshin, Polyhedron, 2001, 20, 2721–2733 CrossRef CAS; (b) M. D. Wise, J. J. Holstein, P. Pattison, C. Besnard, E. Solari, R. Scopelliti, G. Bricogne and K. Severin, Chem. Sci., 2015, 6, 1004–1010 RSC; (c) S. M. Jansze, G. Cecot, M. D. Wise, K. O. Zhurov, T. K. Ronson, A. M. Castilla, A. Finelli, P. Pattison, E. Solari, R. Scopelliti, G. E. Zelinskii, A. V. Vologzhanina, Y. Z. Voloshin, J. R. Nitschke and K. Severin, J. Am. Chem. Soc., 2016, 138, 2046–2054 CrossRef CAS PubMed; (d) E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J.-M. Saveéant, J. Am. Chem. Soc., 2012, 134, 6104–6107 CrossRef PubMed; (e) M. D. Wise, A. Ruggi, M. Pascu, R. Scopelliti and K. Severin, Chem. Sci., 2013, 4, 1658 RSC.
- (a) J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed; (b) C. W. Hsu and O. Š. Miljanić, in Dynamic Covalent Chemistry Reactions, Principles, and Applications, ed. W. Zhang and Y. Jin, John Wiley & Sons Ltd., 1st edn, 2017, pp. 253–286 Search PubMed.
- O. Shyshov, R.-C. Brachvogel, T. Bachmann, R. Srikantharajah, D. Segets, F. Hampel, R. Puchta and M. von Delius, Angew. Chem., Int. Ed., 2017, 56, 776–781 CrossRef CAS PubMed.
- (a) P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC; (b) J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC.
- (a) J. N. Brönsted and W. F. K. Wynne-Jones, Trans. Faraday Soc., 1929, 25, 59–76 RSC; (b) E. H. Cordes and H. G. Bull, Chem. Rev., 1974, 74, 581–603 CrossRef CAS; (c) Y. Chiang, A. J. Kresge, M. O. Lahti and D. P. Weeks, J. Am. Chem. Soc., 1983, 105, 6852–6855 CrossRef CAS; (d) P. Deslongchamps, Y. L. Dory and S. Li, Tetrahedron, 2000, 56, 3533–3537 CrossRef CAS; (e) S. L. Repetto, J. F. Costello, C. P. Butts, J. K. W. Lam and N. M. Ratcliffe, Beilstein J. Org. Chem., 2016, 12, 1467–1475 CrossRef CAS PubMed.
- R.-C. Brachvogel and M. von Delius, Chem. Sci., 2015, 6, 1399–1403 RSC.
- J. A. Dean and N. A. Lange, Lange's Handbook of Chemistry, McGraw-Hill, New York, 15th edn, 1999 Search PubMed.
- (a) R. H. DeWolfe, Synthesis, 1974, 3, 152–172 Search PubMed; (b) S. Kirchmeyer, A. Mertens, M. Arvanaghi and G. A. Olah, Synthesis, 1983, 6, 498–500 CrossRef; (c) K. Yamamoto, S. Abrecht and R. Scheffold, Chimia, 1991, 45, 86–88 CAS; (d) R. L. Talbott, Fluorocarbon Orthoesters, US Pat., 3415847A, 1968.
- M. von Delius, Synlett, 2015, 27, 177–180 CrossRef.
- R.-C. Brachvogel, H. Maid and M. von Delius, Int. J. Mol. Sci., 2015, 16, 20641–20656 CrossRef CAS PubMed.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- (a) R. W. Taft, J. Am. Chem. Soc., 1952, 74, 2729–2732 CrossRef CAS; (b) R. W. Taft, Steric Effects in Organic Chemistry, Wiley, New York, 1956 Search PubMed.
- A. G. Orrillo, A. La-Venia, A. M. Escalante and R. L. E. Furlan, Chem.–Eur. J., 2018, 24, 3141–3146 CrossRef CAS PubMed.
- (a) S. Ulrich and P. Dumy, Chem. Commun., 2014, 50, 5810–5825 RSC; (b) E. Bartolami, Y. Bessin, V. Gervais, P. Dumy and S. Ulrich, Angew. Chem., Int. Ed., 2015, 54, 10183–10187 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- S. Potratz, A. Mishra and P. Bäuerle, Beilstein J. Org. Chem., 2012, 8, 683–692 CrossRef CAS PubMed.
- M. Srinivasarao and P. S. Low, Chem. Rev., 2017, 117, 12133–12164 CrossRef CAS PubMed.
- T. Nakashima, K. Tsuchie, R. Kanazawa, R. Li, S. Iijima, O. Galangau, H. Nakagawa, K. Mutoh, Y. Kobayashi, J. Abe and T. Kawai, J. Am. Chem. Soc., 2015, 137, 7023–7026 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, T. N. T. Phan, D. Gigmes and E. Drockenmuller, ACS Macro Lett., 2014, 3, 658–662 CrossRef CAS.
- (a) C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705 RSC; (b) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003 RSC; (c) J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC; (d) Y. W. Wu, S. Te Tung, C. C. Lai, Y. H. Liu, S. M. Peng and S. H. Chiu, Angew. Chem., Int. Ed., 2015, 54, 11745–11749 CrossRef CAS PubMed; (e) F. R. Bowler, J. J. Diaz-Mochon, M. D. Swift and M. Bradley, Angew. Chem., Int. Ed., 2010, 49, 1809–1812 CrossRef CAS PubMed.
- Z. Monasterio, A. Irastorza, J. I. Miranda and J. M. Aizpura, Org. Lett., 2016, 18, 2511–2514 CrossRef CAS PubMed.
- M. von Delius and R.-C. Brachvogel, Novel Crown ether complexes and methods for producing the same, EP14186189.8, 2014.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization and spectral data, crystallographic data. CCDC 1823126, 1823728, 1823730, 1823774, 1823819, 1823845, 1824092 and 1832928. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01750f |
| ‡ In principle, oxonium ions of alkynyl-substituted orthoesters could undergo 1,4-addition with alcohol nucleophiles, thus generating tetrasubstituted allene derivatives. However, our results suggest that such side reactions do not occur. For instance, the orthoester with R1 = –C≡CH undergoes clean exchange in the presence of 10% TFA (Table 2). |
| § NMR titrations had to be carried out in acetonitrile to guarantee fast exchange on the NMR timescale, as well as sufficient solubility of all starting materials. |
| ¶ The toxicity profile of all possible hydrolysis products will need to be considered for applications in drug delivery. For instance, DEG will likely have to be replaced with a less toxic derivative (proof-of-principle reported in ref. 17) and R1 = CH2Cl might pose a major liability due to the potential formation of toxic chloroacetic acid. |
| This journal is © The Royal Society of Chemistry 2018 |