
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescence properties of heterotrinuclear Zn(II)–M(II) (M = Ca, Sr and Ba) bis(salamo)-type complexes†
Li Wang,
Xiao-Yan Li,
Qing Zhao,
Li-Hong Li and
Wen-Kui Dong *
*
School of Chemical and Biological Engineering, Lanzhou Jiaotong University, Lanzhou, Gansu 730070, P. R. China. E-mail: dongwk@126.com
First published on 17th October 2017
Abstract
A series of hetero-trinuclear Zn(II) complexes, [Zn2Ca(L)(OAc)2]·CHCl3 (1), [Zn2Sr(L)(OAc)2] (2) and [Zn2Ba(L)(OAc)2] (3) with a bis(salamo)-type tetraoxime ligand H4L were synthesized and characterized by elemental analyses, IR, UV-vis spectra etc. Spectral titrations and X-ray crystallography clearly show that the stoichiometry of the heterotrinuclear complexes are all 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 (ligand/Zn(II)/M(II)). The different natures of the N2O2 and O6 sites of the ligand H4L lead to the site-selective introduction of two different kinds of metal(II) atoms. All the Zn(II) atoms are penta-coordinated with distorted square pyramidal geometries. The coordination numbers of Ca(II), Sr(II) and Ba(II) atoms in the O6 environment are all 8, and they have slightly distorted square antiprism geometries. Furthermore, ion competitive experiments show that the coordinating capability in the central O6 site is in the order of Ca(II) > Sr(II) > Ba(II).
:2:1 (ligand/Zn(II)/M(II)). The different natures of the N2O2 and O6 sites of the ligand H4L lead to the site-selective introduction of two different kinds of metal(II) atoms. All the Zn(II) atoms are penta-coordinated with distorted square pyramidal geometries. The coordination numbers of Ca(II), Sr(II) and Ba(II) atoms in the O6 environment are all 8, and they have slightly distorted square antiprism geometries. Furthermore, ion competitive experiments show that the coordinating capability in the central O6 site is in the order of Ca(II) > Sr(II) > Ba(II).
1. Introduction
Transition metal complexes containing salen-type N2O2 ligands have received considerable attention in the past few decades,1 because they are important materials for biological fields,2 catalysis,3 magnetic materials,4 supramolecular structures,5 luminescence,6 electrochemical fields,7 molecular recognition8 and other aspects. Compared to salen-type ligands, two salicylaldehyde molecules were ligated with a bridging unit as a linker with a structural configuration (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O–(CH)n–O–NCH–) in the structure of salamo-type ligand.9 It can be seen that the oxygen atoms are added to a salen-type ligand, finally forming a salamo-type ligand.10
N–O–(CH)n–O–NCH–) in the structure of salamo-type ligand.9 It can be seen that the oxygen atoms are added to a salen-type ligand, finally forming a salamo-type ligand.10
It is worth noting that a large number of studies have been devoted to single salamo ligands for the synthesis of single, double or multinuclear transition metal(II) complexes.11 However, there are a few studies on bis(salamo) ligands and their complexes.9 In this article, three heterotrinuclear complexes [Zn2Ca(L)(OAc)2]·CHCl3 (1), [Zn2Sr(L)(OAc)2] (2) and [Zn2Ba(L)(OAc)2] (3) with a naphthalenediol-based bis(salamo) ligand H4L have been synthesized and structurally characterized. When the two salamo moieties of H4L are metalated, six oxygen atoms are fixed in an acyclic, C-shaped arrangement.12 The resultant 18-crown-6-like recognition site would be suitable for alkaline earth metal ion recognition. Meanwhile, a naphthalenediol ring was introduced, expected to enhance the fluorescence properties of the resultant heterotrinuclear Zn(II)–M(II) (M = Ca, Sr and Ba) complexes with the naphthalenediol-based bis(salamo)-type ligand.
2. Experimental
2.1 Materials and instruments
3-Ethoxy salicylaldehyde was purchased from Tokyo Chemical Industry and was used without further purification. Other reagents and solvents were of analytical grade supplied by Tianjin Chemical Reagent Factory. C, H and N analyses were performed by using a GmbH VarioEL V3.00 automatic elemental analysis instrument. Elemental analysis for metals was detected by an IRIS ER/S WP-1 ICP atomic emission spectrometer. FT-IR spectra were recorded on a VERTEX70 FT-IR spectrophotometer, with samples prepared as KBr (400–4000 cm−1) pellets. Melting points were obtained by the use of a microscopic melting point apparatus made by the Beijing Taike Instrument Limited Company. 1H NMR spectra were determined by a German Bruker AVANCE DRX-400 spectrometer. UV-vis absorption spectra were recorded on a Hitachi U-3900H spectrometer. Fluorescent spectra were taken on an LS-55 fluorescence photometer. X-ray single crystal structure determinations were carried out on a Bruker Smart Apex CCD diffractometer.2.2 Syntheses of H4L and its complexes
:2)], λmax (nm) [2.5 × 10−5 M]: 263, 341, 359, 378.
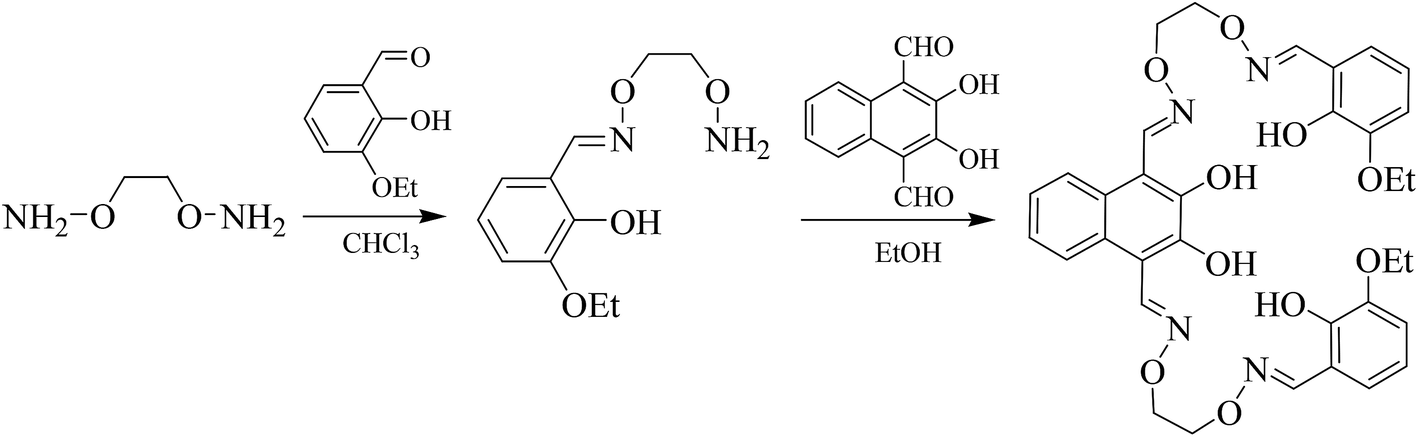 | ||
| Scheme 1 Synthetic route to bis(salamo)-type tetraoxime ligand H4L. | ||
Complex 1, clear yellow crystals. Yield: 3.6 mg, 67.2%. Anal. calc. for C39H39CaCl3N4O14Zn2 (%): C, 43.99; H, 3.69; N, 5.26; Ca, 3.76; Zn, 12.28. Found (%): C, 43.82; H, 3.73; N, 5.34; Ca, 3.52; Zn, 12.26. Selected IR bands (KBr pellet, cm−1): 2977 (m), 1591 (w), 1461 (m), 1317 (s), 1082 (m).
Complex 2, dark yellow crystals. Yield: 3.0 mg, 60.8%. Anal. calc. for C38H38N4O14SrZn2 (%): C, 45.96; H, 3.86; N, 5.64; Sr, 8.82; Zn, 13.17. Found (%): C, 45.98; H, 3.79; N, 5.71; Sr, 8.76; Zn, 13.24. Selected IR bands (KBr pellet, cm−1): 2983 (m), 1585 (w), 1461 (m), 1317 (s), 1075 (m).
Complex 3, clear yellow crystals. Yield: 3.4 mg, 65.3%. Anal. calc. for C38H38BaN4O14Zn2 (%): C, 43.77; H, 3.67; N, 5.37; Ba, 13.17; Zn, 12.54. Found (%): C, 43.82; H, 3.54; N, 5.27; Ba, 13.29; Zn, 12.62. Selected IR bands (KBr pellet, cm−1): 2935 (m), 1588 (w), 1457 (m), 1316 (s), 1075 (m).
2.3 X-ray crystallographic analysis
The crystallographic data for complexes 1, 2 and 3 are summarized in Table 2. X-ray single-crystal diffraction data were collected on a graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) instrument at 295(1), 296(2) and 295(1) K for complexes 1, 2 and 3, respectively. Complexes 1, 2 and 3 are all crystallized in the triclinic crystal system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The structures were solved by using the program SHELXS-97 (ref. 14) and Fourier difference techniques, and refined by a full-matrix least-squares method on F2 using SHELXL-2014.15 H atoms were included at the calculated positions and constrained to ride on their parent atoms. The crystallographic data are summarized in Table 1. CCDC – 1564436 (1), 1564435 (2) and 1564434 (3) contain the supplementary crystallographic data for this article.†
. The structures were solved by using the program SHELXS-97 (ref. 14) and Fourier difference techniques, and refined by a full-matrix least-squares method on F2 using SHELXL-2014.15 H atoms were included at the calculated positions and constrained to ride on their parent atoms. The crystallographic data are summarized in Table 1. CCDC – 1564436 (1), 1564435 (2) and 1564434 (3) contain the supplementary crystallographic data for this article.†
| Complex | 1 | 2 | 3 |
|---|---|---|---|
| a R1 = Σ‖Fo| − |Fc‖/Σ|Fo|.b wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2, w = [σ2(Fo2) + (0.0784P)2 + 1.3233P]−1, where P = (Fo2 + 2Fc2)/3.c GOF = [Σw(Fo2 − Fc2)2/(nobs − nparam)]1/2. | |||
| Empirical formula | C39H39CaCl3N4O14Zn2 | C38H38SrN4O14Zn2 | C38H38BaN4O14Zn2 |
| Formula weight | 1064.910 | 993.08 | 1042.80 |
| Temperature (K) | 295(1) | 296(2) | 295(1) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | P |
P |
P |
| a (Å) | 12.4374(17) | 12.5470(19) | 12.6093(11) |
| b (Å) | 12.9596(18) | 13.3336(19) | 13.4496(12) |
| c (Å) | 13.3757(19) | 15.453(2) | 15.4036(14) |
| a (°) | 84.531(12) | 111.202(5) | 111.422(8) |
| β (°) | 80.178(12) | 101.075(5) | 100.715(7) |
| γ (°) | 86.928(11) | 100.367(5) | 99.989(7) |
| Volume (Å3) | 2113.1(5) | 2275.7(6) | 2304.7(4) |
| Z | 2 | 2 | 2 |
| Calculated density (g cm−3) | 1.674 | 1.449 | 1.503 |
| θ range for data collection (°) | 3.268 to 26.022 | 2.406 to 25.499 | 3.410 to 26.021 |
| F (000) | 1088 | 1008 | 1044 |
| h/k/l (min, max) | −14, 15/−15, 15/−16, 15 | −15, 15/−16, 16/−18, 13 | −15, 15/−17, 16/−18, 19 |
| Crystal size (mm) | 033 × 0.27 × 0.25 | 0.28 × 0.25 × 0.22 | 0.27 × 0.13 × 0.10 |
| Reflections collected | 14901/8331 [Rint = 0.0606] |
17211/8409 [Rint = 0.0353] |
16711/8995 [Rint = 0.0787] |
| Independent reflection | 8331 | 8409 | 8995 |
| Completeness to 26.32 (%) | 99.80 | 99.20 | 99.00 |
| Data/restraints/parameters | 8331/0/572 | 8409/29/186 | 8995/0/536 |
| Final R indices [I > 2σ (I)]a | R1 = 0.0584, wR2 = 0.0900 | R1 = 0.0665, wR2 = 0.2241 | R1 = 0.0858, wR2 = 0.2080 |
| R indices (all data)b | R1 = 0.1055, wR2 = 0.1163 | R1 = 0.0875, wR2 = 0.2376 | R1 = 0.1460, wR2 = 0.2558 |
| Goodness-of-fit for (F2)c | 1.040 | 1.066 | 1.017 |
3. Results and discussion
3.1 IR spectrum analysis
The obtained IR spectra show that the free ligand H4L and its corresponding complexes 1, 2 and 3 exhibit various bands in the region of 4000–400 cm−1 (Fig. 1). The O–H stretching band of the free ligand H4L is observed at 3391 cm−1. This band disappears in the spectra of complexes 1, 2 and 3, indicating that the phenolic and naphthalenediol hydroxyl groups of H4L have been completely deprotonated and coordinated with the metal atoms. The characteristic band of the CN stretching vibration of the free ligand H4L is located at 1605 cm−1. This band is shifted by 14, 20 and 17 cm−1 in the low wavenumber direction in complexes 1, 2 and 3, indicating that the Zn(II) atoms are coordinated with the N2O2 atoms of the ligand.16
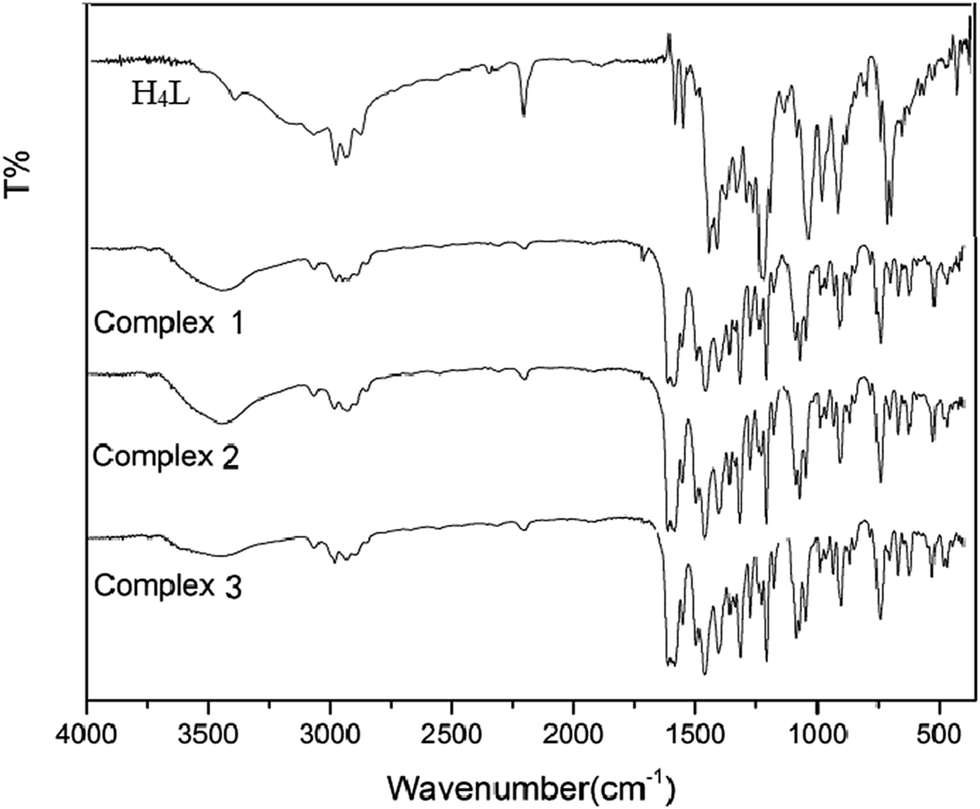 | ||
| Fig. 1 IR spectra of the ligand H4L and its corresponding complexes 1–3. | ||
3.2 UV-vis titration spectroscopy analysis
The UV spectrum of the ligand H4L is shown in Fig. 2(a). There are four consecutive absorption peaks at ca. 268, 341, 359 and 379 nm. The presence of these four peaks indicates that there is a large conjugate structure in the ligand H4L.17 | ||
| Fig. 2 (a) UV-vis spectrum of the free ligand in CHCl3/CH3OH; (b) the changes in the Zn(II)–L complex upon addition of Ca(OAc)2·H2O; (c) the changes in the Zn(II)–L complex upon addition of Sr(OAc)2·H2O; (d) the changes in the Zn(II)–L complex upon addition of Ba(OAc)2. | ||
In the solution of chloroform:methanol (v/v = 2:3), the coordination ratio of the [Zn3(L)]2+ complex with alkaline earth metal ions was measured by UV-vis spectroscopy. (Cligand = 2.5 × 10−5 mol L−1, Cmetal salt = 1.5 × 10−2 mol L−1). The results are shown in Fig. 2(b–d).
In the titration experiments of complex 1, the titration curves changed over two ranges. The first was in the range 360–380 nm, where the titration curves were on the rise. The second was in the range 326–303 nm, where the titration curves were decreasing, and the isoabsorptive point is at 303 nm. We selected the experimental values of the curves at 370 nm to make the concentration scale scatter plot, assuming 1:1 stoichiometry between Ca2+ and [Zn3(L)]2+. Similar changes also appeared in complexes 2 and 3, which gave the same conclusions.
3.3 Description of crystal structures of complexes 1–3
The structures of the heterotrinuclear complexes 1–3 were determined by single-crystal X-ray diffraction. Selected bond lengths and angles of complexes 1–3 are listed in Table 2. The corresponding hydrogen bonds of complexes 1–3 are summarized in Table 3.| Complex 1 | Complex 2 | Complex 3 | |||
|---|---|---|---|---|---|
| Ca1–O1 | 2.650(3) | Sr1–O1 | 2.739(6) | Ba1–O1 | 2.845(8) |
| Ca1–O2 | 2.400(3) | Sr1–O2 | 2.531(5) | Ba1–O2 | 2.704(7) |
| Ca1–O5 | 2.398(3) | Sr1–O5 | 2.541(4) | Ba1–O5 | 2.667(6) |
| Ca1–O6 | 2.415(3) | Sr1–O6 | 2.533(4) | Ba1–O6 | 2.685(7) |
| Ca1–O9 | 2.382(3) | Sr1–O9 | 2.546(5) | Ba1–O9 | 2.716(7) |
| Ca1–O10 | 2.603(3) | Sr1–O10 | 2.745(5) | Ba1–O10 | 2.853(7) |
| Ca1–O11 | 2.389(4) | Sr1–O11 | 2.578(6) | Ba1–O11 | 2.719(7) |
| Ca1–O14 | 2.384(4) | Sr1–O14 | 2.549(5) | Ba1–O13 | 2.723(7) |
| O1–Ca1–O2 | 61.44(10) | O1–Sr–O2 | 59.29(16) | O1–Ba1–O2 | 56.7(2) |
| O1–Ca1–O5 | 121.72(10) | O1–Sr–O5 | 116.91(16) | O1–Ba1–O5 | 113.4(2) |
| O1–Ca1–O6 | 154.46(12) | O1–Sr–O6 | 146.68(18) | O1–Ba1–O6 | 145.8(2) |
| O1–Ca1–O9 | 105.92(11) | O1–Sr–O9 | 114.26(16) | O1–Ba1–O9 | 122.8(2) |
| O1–Ca1–O10 | 68.94(11) | O1–Sr–O10 | 79.05(19) | O1–Ba1–O10 | 89.2(2) |
| O1–Ca1–O11 | 76.33(11) | O1–Sr–O11 | 74.91(17) | O1–Ba1–O11 | 113.0(2) |
| O1–Ca1–O14 | 115.66(12) | O1–Sr–O14 | 116.62(19) | O1–Ba1–O13 | 79.5(2) |
| O2–Ca1–O5 | 66.06(10) | O2–Sr–O5 | 63.75(14) | O2–Ba1–O5 | 61.1(2) |
| O2–Ca1–O6 | 129.88(11) | O2–Sr–O6 | 123.40(15) | O2–Ba1–O6 | 118.3(2) |
| O2–Ca1–O9 | 164.52(11) | O2–Sr–O9 | 172.49(15) | O2–Ba1–O9 | 179.2(2) |
| O2–Ca1–O10 | 103.24(10) | O2–Sr–O10 | 114.11(15) | O2–Ba1–O10 | 123.5(2) |
| O2–Ca1–O11 | 104.01(12) | O2–Sr–O11 | 106.22(17) | O2–Ba1–O11 | 75.1(2) |
| O2–Ca1–O14 | 78.72(12) | O2–Sr–O14 | 77.27(17) | O2–Ba1–O13 | 108.0(2) |
| O5–Ca1–O6 | 63.85(10) | O5–Sr–O6 | 60.56(13) | O5–Ba1–O6 | 58.91(19) |
| O5–Ca1–O9 | 129.09(11) | O5–Sr–O9 | 123.74(14) | O5–Ba1–O9 | 119.1(2) |
| O5–Ca1–O10 | 149.29(13) | O5–Sr–O10 | 151.84(17) | O5–Ba1–O10 | 147.7(2) |
| O5–Ca1–O11 | 93.96(12) | O5–Sr–O11 | 97.83(18) | O5–Ba1–O11 | 69.1(2) |
| O5–Ca1–O14 | 75.14(12) | O5–Sr–O14 | 72.38(17) | O5–Ba1–O13 | 99.5(2) |
| O6–Ca1–O9 | 65.42(10) | O6–Sr–O9 | 64.05(14) | O6–Ba1–O9 | 61.7(2) |
| O6–Ca1–O10 | 120.48(11) | O6–Sr–O10 | 119.41(15) | O6–Ba1–O10 | 114.4(2) |
| O6–Ca1–O11 | 78.43(12) | O6–Sr–O11 | 72.80(17) | O6–Ba1–O11 | 95.8(2) |
| O6–Ca1–O14 | 89.85(12) | O6–Sr–O14 | 94.89(18) | O6–Ba1–O13 | 70.1(2) |
| O9–Ca1–O10 | 62.25(10) | O9–Sr–O10 | 59.29(14) | O9–Ba1–O10 | 56.8(2) |
| O9–Ca1–O11 | 79.89(12) | O9–Sr–O11 | 74.26(18) | O9–Ba1–O11 | 105.6(2) |
| O9–Ca1–O14 | 100.97(12) | O9–Sr–O14 | 103.93(17) | O9–Ba1–O13 | 71.2(2) |
| O10–Ca1–O11 | 116.74(12) | O10–Sr–O11 | 109.12(19) | O10–Ba1–O11 | 81.0(2) |
| O10–Ca1–O14 | 74.51(12) | O10–Sr–O14 | 79.74(18) | O10–Ba1–O13 | 107.5(2) |
| O11–Ca1–O14 | 166.76(12) | O11–Sr–O14 | 167.2(2) | O11–Ba1–O13 | 165.4(3) |
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠D–H⋯A | Symmetry |
|---|---|---|---|---|---|
| Complex 1 | |||||
| C10–H10B⋯O13 | 0.97 | 2.37 | 3.308(6) | 163 | |
| C11–H11A⋯O13 | 0.97 | 2.60 | 3.433(7) | 145 | 2 − x, −y, −z |
| C18–H18⋯O3 | 0.93 | 2.37 | 3.195(6) | 147 | −1 + x, y, z |
| C25–H25B⋯O12 | 0.97 | 2.60 | 3.524(6) | 160 | |
|
|||||
| Complex 2 | |||||
| C1–H1B⋯O14 | 0.96 | 2.50 | 3.264(19) | 137 | |
| C10–H10A⋯O12 | 0.97 | 2.41 | 3.339(9) | 160 | |
| C22–H22⋯O8 | 0.93 | 2.47 | 3.233(9) | 139 | −1 + x, y, z |
| C25–H25B⋯O13 | 0.97 | 2.46 | 3.380(10) | 159 | |
|
|||||
| Complex 3 | |||||
| C1–H1C⋯O13 | 0.96 | 2.43 | 3.373(19) | 167 | |
| C10–H10B⋯O12 | 0.97 | 2.40 | 3.326(15) | 160 | |
| C21–H21⋯O8 | 0.93 | 2.48 | 3.251(13) | 140 | 1 + x, y, z |
| C25–H25B⋯O14 | 0.97 | 2.35 | 3.284(14) | 160 | |
| C33–H33A⋯O11 | 0.96 | 2.42 | 3.32(2) | 156 | |
The crystal structure, atom numberings and the coordination polyhedra for Zn(II) and Ca(II) atoms in complex 1 are shown in Fig. 3. Complex 1 crystallizes in the triclinic crystal system, space group P. From the structure of complex 1, we can see that the coordination ratio of ligand (L)4− to metal atoms (Zn(II) and Ca(II)) in the heteronuclear complex 1 is 1:2:1. Two Zn(II) atoms (Zn1 and Zn2) are located in the N2O2 coordination environment of a completely deprotonated (L)4− unit, and two μ-acetate groups bridge three metal(II) atoms. The distances between the two Zn(II) atoms (Zn1 and Zn2) and the four phenoxy oxygen atoms of the (L)4− unit range from 1.986(2) to 2.123(1) Å, and the distances between the Ca(II) atom and the four phenolic oxygen atoms (O2, O5, O6 and O9) on the (L)4− unit are 2.382(3) to 2.415(3) Å, which are shorter than the distances between the Ca(II) atoms and the ethoxy groups (Ca–O1 2.650(3) and Ca–O10 2.603(3)). The distances between the metal atoms and the coordination atoms indicate that the coordination cavity of the Ca(II) atom is larger than the coordination cavity in which the Zn1 and Zn2 atoms are located. The dihedral angles between the naphthalene ring and the two benzene rings are 44.88(4)° (C1–C6) and 11.96(4)° (C27–C32), respectively. The two μ-acetate groups are located on both sides of the naphthalene ring.
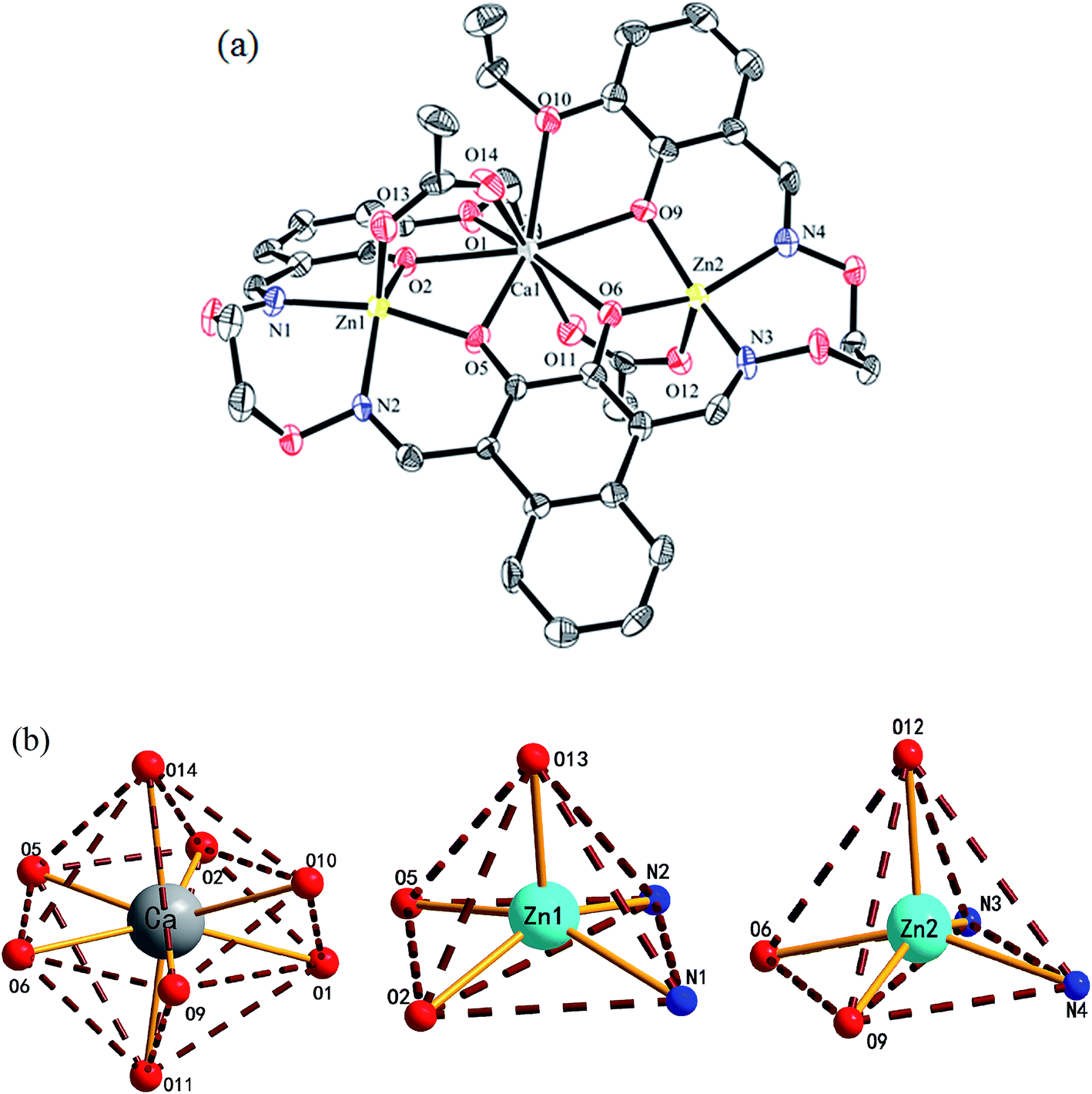 | ||
| Fig. 3 (a) View of the molecular structure of complex 1 (hydrogen atoms and solvent molecules are omitted for clarity, and thermal ellipsoids are drawn at the 30% probability level). (b) Coordination polyhedra for the Zn(II) and Ca(II) atoms of complex 1. | ||
In complex 1, the coordination pattern of the two Zn(II) atoms (Zn1 and Zn2) are penta-coordinated, and the τ values are 0.17 and 0.10,18 respectively. Zn1 and Zn2 atoms both have distorted square pyramidal geometries,19 while the central Ca(II) atom is octa-coordinated with a distorted square antiprism geometry.
In complex 1, there are two intramolecular hydrogen bonds, C10–H10B⋯O13 and C25–H25B⋯O12.20 As shown in Fig. 4, it is worth noting that the donor of both hydrogen bonds is located in the salamo unit and the receptors are oxygen atoms on the bridged μ-acetate groups. Thus, there are two kinds of intermolecular weak interactions existing in complex 1. Among them, C25–H25A⋯Cg1 (C1–C2–C3–C4–C5–C6) and C10–H10A⋯Cg2 (C13–C14–C15–C16–C17–C18) form the intermolecular C–H⋯π interactions, and C18–H18⋯O3 and C11–H11A⋯O13 form the intermolecular C–H⋯O hydrogen bonds. As shown in Fig. 5, they finally form an infinite 2D supramolecular network along the ac plane.21
 | ||
| Fig. 4 View of the intramolecular hydrogen bonds of complex 1. | ||
 | ||
| Fig. 5 View of an infinite 2D supramolecular network of complex 1. | ||
The crystal structures and atom numberings of complexes 2 and 3 are shown in Fig. S1 and S2,† respectively. The crystal structures of complexes 2 and 3 are similar to that of complex 1. As shown in Fig. S3 and S4,† there are some intramolecular hydrogen bond interactions in complexes 2 and 3. The infinite 2D supramolecular networks of complexes 2 and 3 are formed by abundant intermolecular C–H⋯O and C–H⋯π hydrogen bond interactions, as shown in Fig. S5 and S6.†
3.4 Fluorescence spectra
The fluorescence properties of H4L and its corresponding complexes 1–3 were investigated at room temperature (Fig. 6). The ligand exhibits an intense emission peak at 508 nm. Compared with H4L, the absorption peaks of complexes 1–3 are bathochromically-shifted, which is due to the intraligand π–π* transition.22 It is obvious that the Ca(II), Sr(II) and Ba(II) enhance the fluorescence intensity. In order to explore the differences in coordination capabilities of Ca(II), Sr(II) and Ba(II) ions, ion competitive experiments were investigated, as shown in Fig. 6. We have studied the fluorescence changes of complex 2 solution (1.0 × 10−5 mol L−1) in the presence of Ca(II) and Ba(II), respectively. As shown in Fig. 6(a), a dramatic increase in emission intensity is induced by the addition of Ca(II) to the Sr(II) complex solution, suggesting that the Sr(II) is replaced by Ca(II). When Ba(II) was added, the fluorescence intensity showed no change, indicating that the coordination ability of Sr(II) was stronger than that of Ba(II). Similarly, as shown in Fig. 6(b), when Ca(II) was added to the solution of complex 3 (1.0 × 10−5 mol L−1), the fluorescence intensity increased sharply, indicating that the coordination ability of Ca(II) was stronger than that of Ba(II). As a result, the coordinating capability in the central O6 site is in the order of Ca(II) > Sr(II) > Ba(II).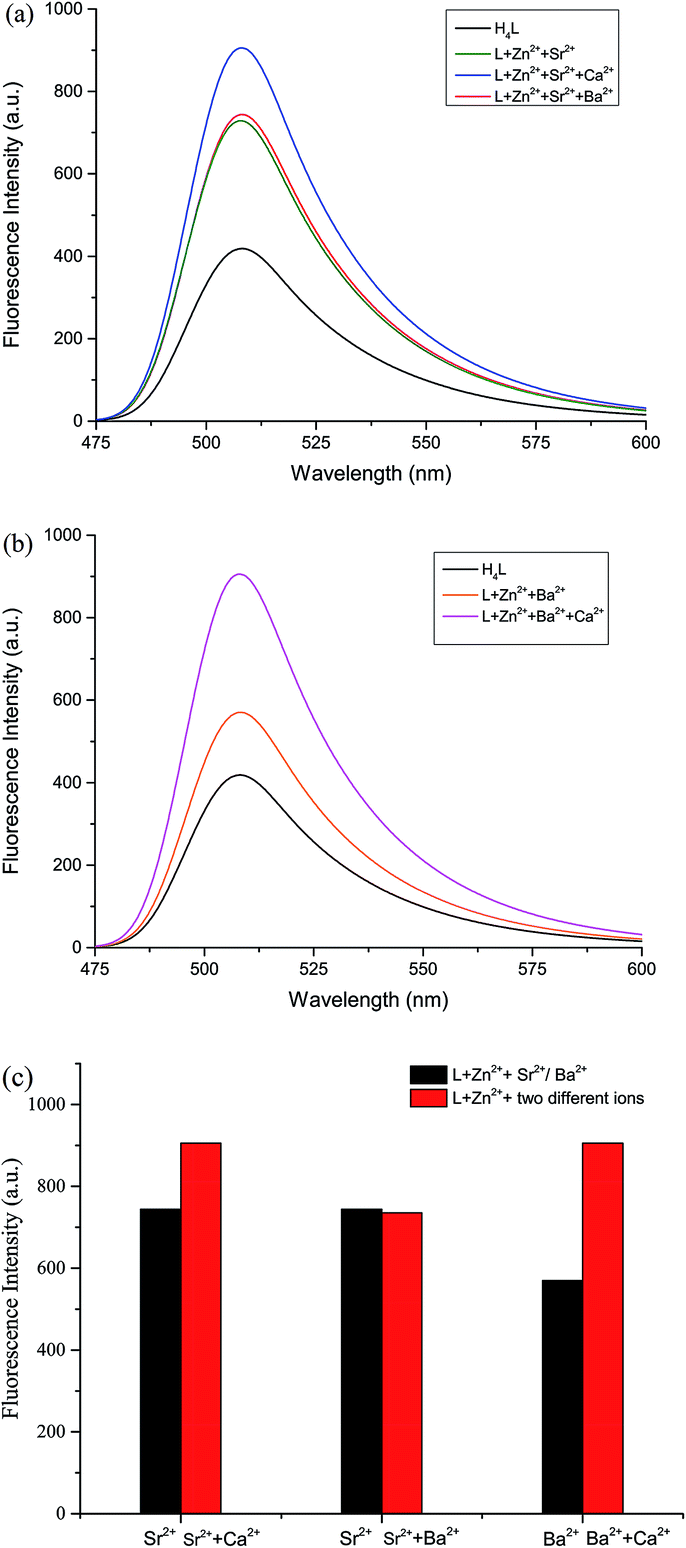 | ||
| Fig. 6 (a) Fluorescence spectra of complex 2 solution (1.0 × 10−5 mol L−1) in the absence and presence of various metal ions (1.0 equiv. of Ca(II) and Ba(II)). (b) Fluorescence spectra of complex 3 solution (1.0 × 10−5 mol L−1) in the absence and presence of various metal ions (1.0 equiv. of Ca(II)). (c) Fluorescence response of L–Zn(II) solution (1.0 × 10−5 mol L−1) to various metal ions. ex = 367 nm, em = 508 nm. | ||
4. Conclusion
Three heteronuclear Zn(II)–M(II) (M = Ca, Sr and Ba) complexes with a naphthalenediol-based bis(salamo)-type tetraoxime ligand H4L were designed and synthesized. The fluorescence spectra of complexes 1, 2 and 3 were also studied. The coordination ratio of Zn(II) to H4L was 2:1, and that of M(II) (M = Ca, Sr and Ba) to H4L was 1:1. X-ray crystal structures reveal that the different natures of the N2O2 and O6 sites of H4L lead to the site-selective introduction of two different kinds of metal(II) atoms. The coordination numbers of Ca(II), Sr(II) and Ba(II) atoms in the O6 environment are all 8, and they have slightly distorted square antiprism geometries. As a result, the coordinating capability in the central O6 site is in the order of Ca(II) > Sr(II) > Ba(II). The supramolecular structures of complexes 1, 2 and 3 are formed via intermolecular C–H⋯O and C–H⋯π hydrogen bond interactions, leading to a self-assembly infinite 2D network structure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21361015 and 21761018) and the Program for Excellent Team of Scientific Research in Lanzhou Jiaotong University (201706), which are gratefully acknowledged.References
- (a) S. S. Sun, C. L. Stern, S. T. Nguyen, J. T. Hupp and J. T. Hupp, J. Am. Chem. Soc., 2004, 126, 6314–6326 CrossRef CAS PubMed; (b) L. Zhao, X. T. Dang, Q. Chen, J. X. Zhao and L. Wang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 1–6 CrossRef CAS; (c) L. Zhao, L. Wang, Y. X. Sun, W. K. Dong, X. L. Tang and X. H. Gao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 1303–1308 CrossRef CAS; (d) P. Wang and L. Zhao, Spectrochim. Acta, Part A, 2015, 135, 342–350 CrossRef CAS PubMed; (e) Y. X. Sun, S. T. Zhang, Z. L. Ren, X. Y. Dong and L. Wang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 995–1000 CrossRef CAS; (f) X. Y. Dong, Y. X. Sun, L. Wang and L. Li, J. Chem. Res., 2012, 36, 387–390 CrossRef CAS; (g) Y. X. Sun and X. H. Gao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2011, 41, 973–978 CrossRef CAS; (h) Y. X. Sun, L. Xu, T. H. Zhao, S. H. Liu, G. H. Liu and X. T. Dong, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 509–513 CrossRef CAS.
- (a) H. L. Wu, Y. C. Bai, Y. H. Zhang, Z. Li, M. C. Wu, C. Y. Chen and J. W. Zhang, J. Coord. Chem., 2014, 67, 3054–3066 CrossRef CAS; (b) C. Y. Chen, J. W. Zhang, Y. H. Zhang, Z. H. Yang, H. L. Wu, G. L. Pan and Y. C. Bai, J. Coord. Chem., 2015, 68, 1054–1071 CrossRef CAS; (c) H. L. Wu, G. L. Pan, Y. C. Bai, Y. H. Zhang, H. Wang, F. R. Shi, X. L. Wang and J. Kong, J. Photochem. Photobiol., B, 2014, 135, 33–43 CrossRef CAS PubMed; (d) H. L. Wu, H. Wang, X. L. Wang, G. L. Pan, F. R. Shi, Y. H. Zhang, Y. C. Bai and J. Kong, New J. Chem., 2014, 38, 1052–1061 RSC; (e) H. L. Wu, G. L. Pan, Y. C. Bai, H. Wang, J. Kong, F. R. Shi, Y. H. Zhang and X. L. Wang, J. Coord. Chem., 2013, 66, 2634–2646 CrossRef CAS.
- (a) T. K. Chin, S. Endud, S. Jamil, S. Budagumpi and H. O. Lintang, Catal. Lett., 2013, 143, 282–288 CrossRef CAS; (b) X. Y. Li, L. Chen, L. Gao, Y. Zhang, S. F. Akogun and W. K. Dong, RSC Adv., 2017, 7, 35905–35916 RSC.
- (a) W. K. Dong, J. C. Ma, Y. J. Dong, L. C. Zhu and Y. Zhang, Polyhedron, 2016, 115, 228–235 CrossRef CAS; (b) P. P. Liu, L. Sheng, X. Q. Song, W. Y. Xu and Y. A. Liu, Inorg. Chim. Acta, 2015, 434, 252–257 CrossRef CAS; (c) X. Q. Song, P. P. Liu, Z. R. Xiao, X. Li and Y. A. Liu, Inorg. Chim. Acta, 2015, 438, 232–244 CrossRef CAS; (d) Y. A. Liu, C. Y. Wang, M. Zhang and X. Q. Song, Polyhedron, 2017, 127, 278–286 CrossRef CAS.
- (a) L. Q. Chai, G. Wang, Y. X. Sun, W. K. Dong, L. Zhao and X. H. Gao, J. Coord. Chem., 2012, 65, 1621–1631 CrossRef CAS; (b) L. Q. Chai, J. J. Huang, H. S. Zhang, Y. L. Zhang, J. Y. Zhang and Y. X. Li, Spectrochim. Acta, Part A, 2014, 131, 526–533 CrossRef CAS PubMed; (c) L. Q. Chai, G. Liu, J. Y. Zhang, J. J. Huang and J. F. Tong, J. Coord. Chem., 2013, 66, 3926–3938 CrossRef CAS; (d) L. Q. Chai, L. J. Tang, L. C. Chen and J. J. Huang, Polyhedron, 2017, 122, 228–240 CrossRef CAS; (e) L. Q. Chai, K. Y. Zhang, L. J. Tang, J. Y. Zhang and H. S. Zhang, Polyhedron, 2017, 130, 100–107 CrossRef CAS.
- (a) Y. J. Dong, X. Y. Dong, W. K. Dong, Y. Zhang and L. S. Zhang, Polyhedron, 2017, 123, 305–315 CrossRef CAS; (b) W. K. Dong, J. C. Ma, Y. J. Dong, L. Zhao, L. C. Zhu, Y. X. Sun and Y. Zhang, J. Coord. Chem., 2016, 69, 3231–3241 CrossRef CAS; (c) H. L. Wu, C. P. Wang, F. Wang, H. P. Peng, H. Zhang and Y. C. Bai, J. Chin. Chem. Soc., 2015, 62, 1028–1034 CrossRef CAS.
- (a) W. K. Dong, J. C. Ma, L. C. Zhu, Y. Zhang and X. L. Li, Inorg. Chim. Acta, 2016, 445, 140–148 CrossRef CAS; (b) S. Ömer, Ö. Ö. Ümmühan, S. Nurgul, A. Burcu, S. Musa, T. Tuncay and S. Zeynel, Tetrahedron, 2016, 72, 5843–5852 CrossRef.
- (a) W. K. Dong, X. L. Li, L. Wang, Y. Zhang and Y. J. Dong, Sens. Actuators, B, 2016, 229, 370–378 CrossRef CAS; (b) W. K. Dong, S. F. Akogun, Y. Zhang, Y. X. Sun and X. Y. Dong, Sens. Actuators, B, 2017, 238, 723–734 CrossRef CAS; (c) B. J. Wang, W. K. Dong, Y. Zhang and S. F. Akogun, Sens. Actuators, B, 2017, 247, 254–264 CrossRef CAS; (d) L. M. Pu, S. F. Akogun, X. L. Li, H. T. Long, W. K. Dong and Y. Zhang, Polyhedron, 2017, 134, 356–364 CrossRef CAS.
- (a) S. Akine, T. Taniguchi and T. Nabeshima, Inorg. Chem., 2008, 47, 3255–3264 CrossRef CAS PubMed; (b) S. Akine, T. Taniguchi, T. Saiki and T. Nabeshima, J. Am. Chem. Soc., 2005, 127, 540–541 CrossRef CAS PubMed; (c) S. Akine, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2006, 128, 15765–15774 CrossRef CAS PubMed.
- S. Akine, T. Taniguchi, W. K. Dong, S. Masubuchi and T. Nabeshima, J. Org. Chem., 2005, 70, 1704–1711 CrossRef CAS PubMed.
- (a) X. Y. Dong, S. F. Akogun, W. M. Zhou and W. K. Dong, J. Chin. Chem. Soc., 2017, 64, 412–419 CrossRef CAS; (b) Y. J. Dong, X. L. Li, Y. Zhang and W. K. Dong, Supramol. Chem., 2017, 29, 518–527 CrossRef CAS; (c) W. K. Dong, J. Zhang, Y. Zhang and N. Li, Inorg. Chim. Acta, 2016, 444, 95–102 CrossRef CAS; (d) W. K. Dong, G. Li, Z. K. Wang and D. X. Yong, Spectrochim. Acta, Part A, 2014, 133, 340–347 CrossRef CAS PubMed.
- (a) L. Chen, W. K. Dong, H. Zhang, Y. Zhang and Y. X. Sun, Cryst. Growth Des., 2017, 17, 3636–3648 CrossRef CAS; (b) W. K. Dong, S. S. Zheng, J. T. Zhang, Y. Zhang and Y. X. Sun, Spectrochim. Acta, Part A, 2017, 184, 141–150 CrossRef CAS PubMed; (c) S. S. Zheng, W. K. Dong, Y. Zhang, L. Chen and Y. J. Ding, New J. Chem., 2017, 41, 4966–4973 RSC.
- (a) S. Akine, T. Tadokoro and T. Nabeshima, Inorg. Chem., 2012, 51, 11478–11486 CrossRef CAS PubMed; (b) S. Akine, S. Sairenji, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2013, 135, 12948–12951 CrossRef CAS PubMed; (c) H. A. Tran, J. Collins and P. E. Georghiou, New J. Chem., 2008, 32, 1175–1182 RSC.
- G. M. Sheldrick, SHELXS 97, Program for crystal structure solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL 97, Program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- Y. J. Dong, J. C. Ma, L. C. Zhu, W. K. Dong and Y. Zhang, J. Coord. Chem., 2017, 70, 103–115 CrossRef CAS.
- (a) L. Xu, L. C. Zhu, J. C. Ma, Y. Zhang, J. Zhang and W. K. Dong, Z. Anorg. Allg. Chem., 2015, 641, 2520–2524 CrossRef CAS; (b) W. K. Dong, J. T. Zhang, Y. J. Dong, Y. Zhang and Z. K. Wang, Z. Anorg. Allg. Chem., 2016, 642, 189–196 CrossRef CAS; (c) W. K. Dong, J. C. Ma, L. C. Zhu, Y. X. Sun, S. F. Akogun and Y. Zhang, Cryst. Growth Des., 2016, 16, 6903–6914 CrossRef CAS.
- (a) A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 7, 1349–1356 RSC; (b) T. Konno, K. Tokuda, J. Sakurai and K. I. Okamoto, Bull. Chem. Soc. Jpn., 2000, 73, 2767–2773 CrossRef CAS.
- G. L. Paraginski, M. Hörner, D. F. Back and J. Beck, J. Mol. Struct., 2016, 1104, 79–84 CrossRef CAS.
- (a) L. Zhao, X. T. Dang, Q. Chen, J. X. Zhao and L. Wang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 1241–1246 CrossRef CAS; (b) L. Q. Chai, Y. X. Li, L. C. Chen, J. Y. Zhang and J. J. Huang, Inorg. Chim. Acta, 2016, 444, 193–201 CrossRef CAS; (c) Y. X. Sun, C. Y. Li, C. J. Yang, Y. Y. Zhao, J. Q. Guo and B. Yu, Chin. J. Inorg. Chem., 2016, 32, 327–335 CAS; (d) Y. X. Sun, L. Wang, X. Y. Dong, Z. L. Ren and W. S. Meng, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 599–603 CrossRef CAS.
- (a) P. Wang and L. Zhao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 1095–1101 CrossRef CAS; (b) Y. X. Sun, Y. Y. Zhao, C. Y. Li, B. Yu, J. Q. Guo and J. Li, Chin. J. Inorg. Chem., 2016, 32, 913–920 CAS; (c) L. Q. Chai, K. H. Mao, J. Y. Zhang, K. Y. Zhang and H. S. Zhang, Inorg. Chim. Acta, 2017, 457, 34–40 CrossRef CAS; (d) L. Q. Chai, J. J. Huang, J. Y. Zhang and Y. X. Li, J. Coord. Chem., 2015, 68, 1224–1237 CrossRef CAS; (e) X. Q. Song, G. Q. Cheng, X. R. Wang, W. Y. Xu and P. P. Liu, Inorg. Chim. Acta, 2015, 425, 145–153 CrossRef CAS.
- W. K. Dong, J. C. Ma, L. C. Zhu, Y. X. Sun, S. F. Akogun and Y. Zhang, New J. Chem., 2016, 40, 6998–7010 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1564434–1564436. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08789f |
| This journal is © The Royal Society of Chemistry 2017 |