DOI:
10.1039/C7RA09351A
(Paper)
RSC Adv., 2017,
7, 48723-48729
First graphene oxide promoted metal-free nitrene insertion into olefins in water: towards facile synthesis of activated aziridines†
Received
23rd August 2017
, Accepted 29th September 2017
First published on 17th October 2017
Abstract
A facile metal-free graphene oxide (GO)-catalyzed synthesis of tosylaziridines using PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NTs as the nitrene source is reported. The reaction involves nitrene insertion into a variety of styrene/nitrostyrene derivatives in the presence of iodine at room temperature and in water. The envisaged process is highly green, operationally simple, it employs metal-free catalysis and it affords excellent yields (85–92%) and high diastereoselectivity (95–98%) of the Z-isomer of the product. The catalyst used could be recycled for further use in other reactions.
NTs as the nitrene source is reported. The reaction involves nitrene insertion into a variety of styrene/nitrostyrene derivatives in the presence of iodine at room temperature and in water. The envisaged process is highly green, operationally simple, it employs metal-free catalysis and it affords excellent yields (85–92%) and high diastereoselectivity (95–98%) of the Z-isomer of the product. The catalyst used could be recycled for further use in other reactions.
Aziridines have been synthetic targets as well as scaffolds since Gabriel’s discovery in 1888. They represent an important structural motif in drugs and natural products (Fig. 1) and are highly useful as feedstock in organic synthesis to provide functionalized carbon chains due to the inherent strain in small ring heterocycles.1 In terms of synthetic transformations, the utility of aziridines arises from their selective ring-opening reactions using various nucleophiles, which often form the basis of more complex target syntheses.2,3 Owing to their chemical and biological interest, numerous methods are reported for their synthesis,4–14 including the aziridination of olefins,4–6 the addition of carbene and ylide to imines,7–9 and different cyclization protocols.10–14 However, the direct transfer of nitrogen atoms to alkenes has been the most practical and useful method for the synthesis of aziridines.15 Although transition-metal catalyzed aziridination of alkenes has been well reported in the literature, metal-free aziridination using different nitrogen sources is still an attractive strategy from practical, economical and, of course, environmental points of view.16–20 The nitrene transfer reaction,21,22 and the use of iminoiodinanes23 as nitrene precursors and thus the efficient conditions for catalytic aziridination of olefins using iodine(III) oxidants are reported.24 Despite previous work, the development of more environmentally benign nitrene transfer reactions for the construction of diverse aziridines is still in great demand. Thus, inspired by these reports and in continuation of our interest in developing efficient and green catalytic routes25 for bioactive molecules,26 we decided to undertake the aziridination of olefins using PhINTs as the nitrene source.27 Recently, we have reported the synthesis of functionalized graphene oxide,28 which could easily be dispersed in water. Keeping the use of water as a reaction medium in mind, we anticipated using the reported Fe-decorated GO as a catalyst for the present investigation. To our surprise, we were successful in isolating the target aziridine but the yield was not satisfactory. Then we turned our attention to employing a metal-free catalytic process and tried graphene oxide only as a catalyst in the presence of PhINTs and I2 and the reaction was successful in terms of yield of pure product. Herein, we disclose a metal-free aziridination of styrenes as well as nitrostyrenes using an equimolar amount of PhINTs in the presence of I2 using GO as the catalyst in an aqueous medium and at ambient reaction conditions (Scheme 1). To the best of our knowledge, the aziridination of olefins through graphene oxide-catalyzed nitrene insertion reactions under metal-free conditions using water as a solvent has not been reported so far.
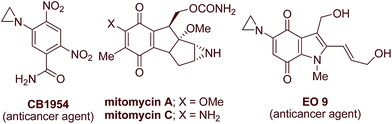 |
| | Fig. 1 Drugs and natural products containing aziridine. | |
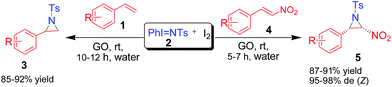 |
| | Scheme 1 Synthesis of the GO-catalyzed aziridination of olefins. | |
In our preliminary experimentation, we first examined equimolar mixtures of styrene 1a and PhINTs 2 with 10 mol% of I2 in water at room temperature (Table 1, entry 1), but we did not isolate product 3a without the use of a catalyst, even upon changing the solvent system (MeOH, MeCN, THF and 1,4-dioxane) (Table 1, entries 2–5). Next, the screening of the catalytic and solvent systems was undertaken, as given in Table 1. Initially, we used our recently reported catalyst,28 Fe–GO (15 mg) using water as a solvent, and we successfully isolated the corresponding aziridine product 3a in moderate yield (Table 1, entry 6). Encouraged by the result, we tried several graphene-based materials as catalysts in the present synthetic protocol (Table 1, entries 6–9). However, GO was found to be the most effective among GO, rGO, graphite and Fe–GO, while rGO and graphite were ineffective for the conversion (Table 1, entries 8 and 9). The optimum catalyst loading for graphene oxide was found to be 15 mg. When the amount of catalyst decreased from 15 mg to 10 mg, the yield of product 3a reduced (Table 1, entry 15) but the use of 20 mg of the catalyst showed the same yield (Table 1, entry 16). Solvent screening for the synthesis of 3a was also undertaken using various solvents such as MeCN, DCM, 1,4-dioxane, THF, MeOH and water (Table 1, entries 7, 10–14). When the reaction was carried out in aprotic solvents such as MeCN, DCM, 1,4-dioxane and THF, it afforded the aziridine 3a in trace amounts (Table 1). However, when using polar protic solvents, the yield was improved (Table 1, entry 14) and therefore, water was found to be the best choice of solvent in the present optimization process.
Table 1 Screening of catalysts and solvents for the aziridination of styrene 1aa
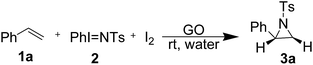
|
| Entry |
Catalyst (mg) |
I2 (mol%) |
Solvent |
Timeb (h) |
Yieldc,d (%) |
| Reaction conditions: a mixture of 1a (1 equi.), 2 (1 equi.), I2 (15 mol%) and GO (15 mg) in 5 mL of water was stirred at room temperature. The stirring time at room temperature. Yield of isolated and purified product. Characterized by spectral (IR, 1H NMR, 13C NMR and EIMS) data. The reaction was carried out in the dark. |
| 1 |
— |
10 |
Water |
12 |
— |
| 2 |
— |
10 |
MeOH |
12 |
— |
| 3 |
— |
10 |
MeCN |
12 |
— |
| 4 |
— |
10 |
THF |
12 |
— |
| 5 |
— |
10 |
1,4-Dioxane |
12 |
— |
| 6 |
Fe–GO (15) |
10 |
H2O |
12 |
43 |
| 7 |
GO (15) |
10 |
H2O |
10 |
92 |
| 8 |
rGO (15) |
10 |
H2O |
12 |
— |
| 9 |
Graphite |
10 |
H2O |
12 |
— |
| 10 |
GO (15) |
10 |
Benzene |
10 |
— |
| 11 |
GO (15) |
10 |
DCM |
12 |
23 |
| 12 |
GO (15) |
10 |
DMF |
10 |
— |
| 13 |
GO (15) |
10 |
THF |
10 |
29 |
| 14 |
GO (15) |
10 |
MeOH |
10 |
53 |
| 15 |
GO (10) |
10 |
H2O |
10 |
73 |
| 16 |
GO (20) |
10 |
H2O |
10 |
92 |
| 17 |
GO (15) |
5 |
H2O |
10 |
— |
| 18 |
GO (15)e |
10 |
H2O |
10 |
92 |
The role of I2 was also investigated and a reaction of 1a and 2 and GO (15 mg) was performed in the absence of iodine, but no aziridine 3a was isolated (Table 1, entry 17) which is evidence that iodine plays a crucial role in the formation of aziridine rings. When the reaction was performed in the dark it afforded 3a in low yield, showing that the reaction involved a radical which was promoted by ambient room light in the laboratory (Table 1, entry 18).
After optimization of the reaction conditions, we next investigated the substrate scope of the present aziridination of styrene. The substituents on the benzene ring of styrene 1 did not show any specific effect over the product outcome as well as the conversion rate. Substrates with electron-donating and electron withdrawing substituents at the para-position were tolerated to afford the corresponding aziridine 3 in consistently good yields (Table 2). Substituents at the ortho or meta positions on the phenyl ring caused almost no change in reactivity (Table 2, entries 7 and 8). However, when using an aliphatic alkene, a moderate yield of the corresponding aziridine was obtained. The formation of product 3 was confirmed by spectral analysis. In the 1H NMR spectra of 3a (Fig. S1; ESI†), a double doublet at δ 3.78 (J = 7.0, 4.5 Hz) for CH indicates the presence of two vicinal CH2 protons, further confirmed by two doublets at δ 2.99 (J = 7.0 Hz) and δ 2.43 (J = 4.5 Hz). Thus, the present optimized synthesis of stirring an equimolar mixture of styrene 1 and PhINTs 2, iodine (10 mol%) and GO (15 mg) in water for 10–12 h was successful and afforded tosylaziridine 3 in 85–92% yields (Table 2).
Table 2 Substrate scope of styrene 1 in the present aziridination processa
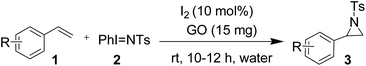
|
| Entry |
1 (R) styrene |
3 (R) product |
Timeb (h) |
Yieldc,d (%) |
| Reaction conditions: a mixture of 1 (0.75 mmol), 2 (0.75 mmol), I2 (0.075 mmol) and GO (15 mg) dispersed in 5 mL of water was stirred at room temperature. The stirring time at room temperature. Yield of isolated and purified product. Characterized by spectral (IR, 1H NMR, 13C NMR and EIMS) data. |
| 1 |
H, 1a |
H, 3a |
10 |
92 |
| 2 |
4-Me, 1b |
4-Me, 3b |
10 |
91 |
| 3 |
4-OMe, 1c |
4-OMe, 3c |
12 |
87 |
| 4 |
4-Cl, 1d |
4-Cl, 3d |
10 |
90 |
| 5 |
4-CN, 1e |
4-CN, 3e |
11 |
91 |
| 6 |
4-CF3, 1f |
4-CF3, 3f |
10 |
85 |
| 7 |
2-Me, 1g |
2-Me, 3g |
10 |
90 |
| 8 |
3-Me, 1h |
3-Me, 3h |
10 |
89 |
| 9 |
4-NO2, 1i |
4-NO2, 3i |
11 |
91 |
| 10 |
4-Br, 1j |
4-Br, 3j |
10 |
89 |
Although 2-nitroaziridines appear to be a scaffold for further transformation, very few reports are available on the synthesis of C-nitroaziridines12,30–32 using nitroalkene or its derivatives with a nitrogen source, namely ethyl[(4-nitrobenzenesulfonyl)oxy]carbamate (NsONHCO2Et)/azide, amine or imine (eqn (1)–(3) of Scheme 2). Keeping these literature reports in mind, we extended our envisaged aziridination process to the synthesis of 2-nitro-1-tosylaziridine 5 employing the optimized reaction conditions in hand. Thus, a mixture of nitrostyrene 4, PhINTs 2, iodine (10 mol%) and GO (15 mg) was stirred at room temperature in water and the corresponding 2-nitro-1-tosylaziridine was synthesized in 5–7 h in good to excellent yield (87–91%) (Table 3). However, nitrostyrenes bearing various types of electron releasing and electron withdrawing groups at the para position were in good agreement with the yield of product 5 (Table 3). Also, the ortho- and meta-substituted phenyl ring afforded approximately the same yield of pure product 5. Increasing the mol% of iodine did not increase the yield. However, decreasing the amount of iodine resulted in substantial suppression of product yield (Table 3, entries 8 and 9). A similar effect was observed when varying the amount of the GO catalyst (Table 3, entries 10 and 11). The 1H NMR spectra of 5a shows two doublets at δ 2.28 and 4.55 for both CHs in aziridine (5a) with the same coupling constant (J = 9.5 Hz) confirming the formation of nitroaziridine 5 (Fig. S2, ESI†). Next, the Z-stereochemistry of aziridines 5 was assigned on the basis of higher J-values (9.5–9.8 Hz) of 2-H and 3-H than those of (E)-aziridines (6.5–6.6 Hz). This conforms with the earlier observations for aziridines reported in the literature.32–35 Furthermore, strong NOEs between 2-H and 3-H in aziridines 5 also confirm that these protons are on the same face of the molecule and thus, prove their Z-stereochemistry (Fig. 2). The plausible mechanism for the formation of aziridines 3 and 5 is given in Scheme 3. The radical pathway is favoured in the light of the homolytic weakness of the N–I bond37 and low conversion rate in the dark using the same reaction conditions (Table 1). Initially, PhINTs 2 reacts with I2 in the presence of GO, generating N,N-diiodotosylamide 6, which is the cornerstone of the present nitrene insertion process. Presumably, homolytic cleavage of the N–I bond37 of 6 generates the amidyl radical 7, which is followed by the intramolecular cyclization of 8, assisted by the iodine free radical to furnish the target molecules 3 and 5 with the regeneration of I2, which is to be reused in the catalytic cycle (Scheme 3).
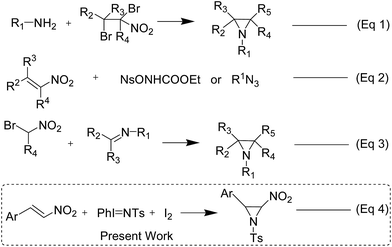 |
| | Scheme 2 Various synthetic routes for the synthesis of nitro-aziridines. | |
Table 3 Synthesis of 2-nitro-1-tosylaziridine 5a
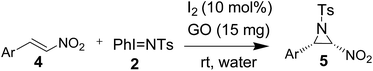
|
| Entry |
4 (Ar) nitrostyrene |
5 (Ar) product |
Timeb (h) |
Yieldc,d (%) |
Z/Ee |
| Reaction conditions: a mixture of 4 (3.5 mmol), 2 (0.750 mmol), I2 (10 mol%) and GO (15 mg) dispersed in 5 mL of water was stirred at room temperature. The stirring time at room temperature. Yield of isolated and purified product. Characterized by spectral (IR, 1H NMR, 13C NMR and EIMS) data. Determined by 1H NMR spectroscopy. Reaction was performed in 15 mol% of I2. Reaction was performed in 5 mol% of I2. Reaction was performed with 20 mg of GO. Reaction was performed with 10 mg GO. |
| 1 |
Ph, 4a |
Ph, 5a |
5 |
89 |
96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :04 :04 |
| 2 |
4-OMeC6H4, 4b |
4-OMeC6H4, 5b |
6 |
87 |
95:05 |
| 3 |
4-ClC6H4, 4c |
4-ClC6H4, 5c |
6 |
88 |
97:03 |
| 4 |
4-MeC6H4, 4d |
4-MeC6H4, 5d |
7 |
91 |
98:02 |
| 5 |
2-ClC6H4, 4e |
2-ClC6H4, 5e |
6 |
88 |
96:04 |
| 6 |
4-NO2C6H4, 4f |
4-NO2C6H4, 5f |
6 |
90 |
97:03 |
| 7 |
3-MeC6H4, 4g |
3-MeC6H4, 5h |
5 |
91 |
97:03 |
| 8f |
Ph, 4a |
Ph, 5a |
5 |
89 |
96:04 |
| 9g |
Ph, 4a |
Ph, 5a |
5 |
68 |
96:04 |
| 10h |
Ph, 4a |
Ph, 5a |
5 |
89 |
96:04 |
| 11i |
Ph, 4a |
Ph, 5a |
5 |
61 |
96:04 |
 |
| | Fig. 2 NOE experiment on nitro-aziridines 5. | |
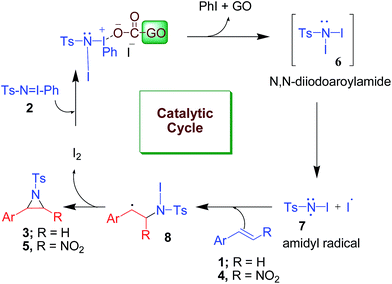 |
| | Scheme 3 Plausible mechanism for the formation of tosylaziridines 3 and 5. | |
Graphene oxide (GO) is a single hexagonal lattice sheet of sp2 and sp3 hybridized C atoms. The oxidation of graphite results in a number of defects or holes on its surface. The presence of numerous –COOH groups at the edges of flakes as well as at the edges of defect sites may be utilized for its functionalization, which makes GO an efficient catalyst (Fig. 3).36 The reusability of GO as a heterogeneous acid catalyst was also examined and it was recovered from the first batch of reactions by centrifugation followed by washing and drying, then it was reused for six subsequent batch reactions. Appreciable conversions were achieved in all recycling experiments carried out at room temperature (Fig. 4). Comparison of the FT-IR spectra of GO before and after use confirmed that the catalyst remained the same after the reaction (Fig. 5).
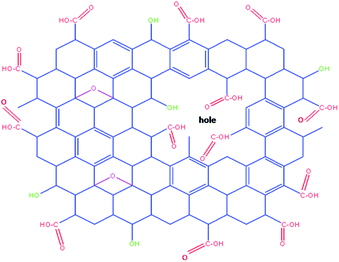 |
| | Fig. 3 Various functional groups of the graphene oxide used as a catalyst. | |
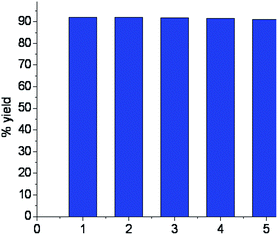 |
| | Fig. 4 Reusability experiment of the GO catalyst. | |
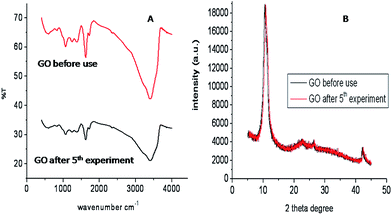 |
| | Fig. 5 IR (A) and XRD (B) of GO before and after use in a reusability experiment. | |
Conclusion
In summary, we have disclosed an unprecedented and efficient route for the synthesis of tosylaziridines using PhINTs via nitrene insertion into olefins in the presence of graphene oxide as a catalyst in water. The envisaged green method has several advantages over existing procedures for the synthesis of activated aziridines, viz., higher yields of products, simple operation, and ambient reaction conditions and would be a better practical alternative to cater to the needs of academia as well as industry.
Experimental
Preparation of graphene oxide
GO was synthesized from the oxidation of graphite powder under oxidising conditions using the modified Hummers method29 followed by exfoliation in an aqueous solution. Graphite flakes (1 g) were slowly added to ice-cold concentrated sulfuric acid (23 mL) then continued stirring was performed, putting the whole mixture in an ice bath along with sodium nitrate (0.5 g) and then vigorously stirring using a magnetic stirrer. After that, potassium permanganate (3 g) was added to the reaction mixture very slowly, keeping the temperature within 0–5 °C. The mixture was allowed to stir at room temperature for 30 min forming a thick paste. It was diluted with distilled water (46 mL) under stirring conditions. The temperature of the solution was raised to about 98 °C and the mixture was allowed to stir for 4 h and was then finally cooled down. Then 100 mL of deionized water was added followed by slow addition of 3 mL H2O2 (30%). The color of the solution changed from dark brown to yellowish brown. The overall solution was exfoliated under stirring followed by centrifugation at 3000 rpm to collect the solid mass at the bottom. The remaining solid material was then washed with 50 mL of warm deionised water and 30 mL of 5% HCl. Finally, the brown mass was collected and dried at 50 °C under vacuum to obtain solid graphene oxide. The formation of GO and the presence of various chemical functionalities on the GO nanosheets have been well characterized and analyzed by TEM, XRD, FTIR and UV-Vis spectroscopy.
TEM analysis of graphene oxide
HR-TEM measurements were performed to determine the morphology of graphene oxide. Bends and wrinkles were observed on the graphene oxide nanosheets at several places (Fig. 6). Graphene oxide nanosheets appear to have a single layer morphology. Well-defined diffraction spots in a selected area electron diffraction (SAED) pattern illustrate that the monolayer graphene oxide formed. However, in the HR-TEM image, the presence of topological features along with an overlapping area of graphene oxide nanosheets reveal that they are highly dispersed in water.
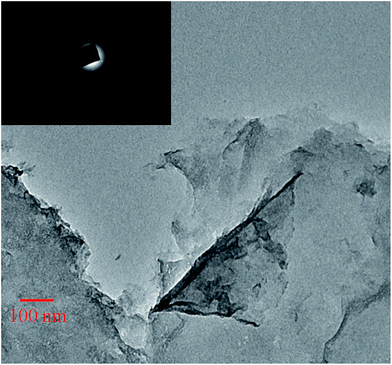 |
| | Fig. 6 High resolution TEM image and diffraction pattern (inset) of the graphene oxide nanosheets. | |
XRD analysis of graphene oxide
XRD analysis was used to characterize the crystalline nature and phase purity of the as-synthesized graphene oxide. Pure graphite has a strong and sharp diffraction peak at 2θ = 23.62°, which corresponds to the (002) plane of the hexagonal graphite structure with an interlayer spacing of 3.76 Å (Fig. S3, ESI†). After chemical oxidation and exfoliation into graphene oxide, the 23.62° peak disappeared and a wide diffraction peak centred at 10.13° appeared, corresponding to the (002) plane and the interlayer spacing of the material was 8.72 Å. An increased interlayer distance between consecutive carbon basal planes is attributed to the intercalation of oxygen functional groups (–COOH, –OH etc.) and water molecules into the carbon layer structure. XRD shows various defects and functional groups carrying sp3 hybridized carbon atoms, which are introduced during the oxidation process.
FT-IR spectrum of graphene oxide (GO)
Fig. S4, ESI† shows the FTIR spectrum of GO, which shows a broad peak at 3403.74 cm−1 in the high-frequency area attributed to the stretching mode of the O–H bond, revealing the presence of hydroxyl groups in graphene oxide. The band observed at 1725.16 cm−1 was assigned to the carboxyl group. The sharp peak found at 1608.79 cm−1 is a resonance peak that can be assigned to the stretching and bending vibrations of the –OH groups from water molecules adsorbed on graphene oxide. The peak at 1381.42 cm−1 arises from the C–OH group. The peak at 1241.65 cm−1 denotes C–O (epoxy group) stretching and the C–O (alkoxy) stretching vibration bands appear at 1061.08 cm−1. FTIR spectra provide the presence of ample numbers of oxygen functionalities such as hydroxyl, epoxide, carboxylate and carbonyl on graphene oxide.
UV-Vis spectrum of graphene oxide
The UV-Vis spectrum of graphene oxide is also shown in Fig. S5, ESI.† According to the absorbance spectra, the main spectrum of graphene oxide has a strong absorption peak at 234.32 nm, attributed to the π → π* transition of the C–C conjugated aromatic domains and a weak absorption (shoulder) at 306.64 nm due to the n → π* transition of the CO bond.
General method for the synthesis of aziridines 3 and 5
Styrene (0.75 mmol) was added to a mixture of iodine (10 mol%), PhINTs (0.750 mmol) and GO (15 mg) in distilled water (5 mL). The reaction mixture was stirred at room temperature for 10 to 12 h. The reaction progress was recorded periodically and after completion of the reaction (using TLC), the reaction mixture was washed sequentially with diethyl ether (10 mL × 3), brine solution (10 mL × 3) and distilled water (10 mL) and it was finally dried over potassium carbonate. Evaporation of the solvent on a rotavapor left the crude solid product which was purified by column chromatography on silica gel (ethyl acetate/n-hexane = 1/9) to provide the pure products 3 and 5. The products were confirmed by 1H NMR and 13C NMR spectroscopy and all the characterization data match those reported in the literature well.15h,32 However, the spectroscopic data of representative compounds is given. 3a: IR (KBr) νmax = 3051, 2838, 1603, 1582, 1455 cm−1. 1H NMR (400 MHz, CDCl3): δ = 2.39 (d, 1H, J = 4.5 Hz, 3-Ha), 2.99 (d, 1H, J = 7.0 Hz, 3-Hb), 2.43 (s, 3H, TsCH3), 3.78 (dd, 1H, J = 4.5, 7.0 Hz, 2-H), 7.87 (d, J = 8.0 Hz, 2Harom), 7.21–7.34 (m, 7Harom) ppm. 13C NMR (100 MHz, CDCl3): δ = 23.7, 34.7, 43.8, 125.1, 126.3, 127.5, 128.3, 129.6, 134.1, 136.5, 142.8 ppm. EIMS: m/z = 273 [M+]. C15H15NO2S: calcd C 65.91, H 5.53, N 5.12; found: C 65.73, H 5.16, N 5.41. 5a: IR (KBr) νmax = 3049, 2835, 1607, 1579, 1509, 1459, 1324 cm−1. 1H NMR (400 MHz, CDCl3): δ = 2.12 (s, 3H, TsCH3), 2.28 (d, 1H, J = 9.5 Hz, 3-H), 4.54 (d, 1H, J = 9.5 Hz, 2-H), 7.20–7.41 (m, 7Harom), 7.89 (d, J = 8.0 Hz, 2Harom) ppm. 13C NMR (100 MHz, CDCl3): δ = 23.9, 42.9, 78.1, 125.9, 126.6, 127.5, 128.4, 129.3, 131.5, 137.9, 143.9 ppm. EIMS: m/z = 318 [M+]. C15H14N2O4S: calcd C 56.59, H. 4.43, N 8.80; found: C 56.88. H 4.26, N 8.47.
Conflicts of interest
There is no conflicts of interest.
Acknowledgements
We sincerely thank SERB, DST, New Delhi, for the INSPIRE Faculty Research Award (IFA12-CH-61), DST PURSE for publication fees support and AIRF, Jawaharlal Nehru University for providing microanalysis and spectra.
Notes and references
-
(a) S. Gabriel, Chem. Ber., 1888, 21, 1049 CrossRef;
(b) D. Tanner, Angew. Chem., Int. Ed., 1994, 33, 599 CrossRef;
(c) Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
-
(a) X. E. Hu, Tetrahedron, 2004, 60, 2701 CrossRef CAS;
(b) H. Stamm, J. Prakt. Chem./Chem.-Ztg., 1999, 341, 319 CrossRef CAS;
(c) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC;
(d) M. A. Graham, A. H. Wadsworth, M. Thornton-Pett and C. M. Rayner, Chem. Commun., 2001, 966 RSC;
(e) R. Maeda, R. Ishibashi, R. Kamaishi, K. Hirotaki, H. Furuno and T. Hanamoto, Org. Lett., 2011, 13, 6240 CrossRef CAS PubMed;
(f) M. Sayyad, Y. Nanaji and M. K. Ghorai, J. Org. Chem., 2015, 80, 12659 CrossRef CAS PubMed;
(g) L. Gao, K. Fu and G. Zheng, RSC Adv., 2016, 6, 47192 RSC;
(h) Z. Jian and J. S. Kalman, Angew. Chem., Int. Ed., 2016, 55, 15026 Search PubMed.
-
(a) I. D. G. Watson, L. Yu and A. K. Yudin, Acc. Chem. Res., 2006, 39, 194 CrossRef CAS PubMed;
(b) S. Dalili, A. Caiazzo and A. K. Yudin, J. Organomet. Chem., 2004, 689, 3604 CrossRef CAS;
(c) I. D. G. Watson and A. K. Yudin, J. Org. Chem., 2003, 68, 5160 CrossRef CAS PubMed;
(d) A. Caiazzo, S. Dalili and A. K. Yudin, Synlett, 2003, 2198 CAS;
(e) A. Caiazzo, S. Dalili and A. K. Yudin, Org. Lett., 2002, 4, 2597 CrossRef CAS PubMed;
(f) X. E. Hu, Tetrahedron, 2004, 60, 2701 CrossRef CAS;
(g) P. Lu, Tetrahedron, 2010, 66, 2549 CrossRef CAS;
(h) V. K. Rai, N. Sharma and A. Kumar, Synlett, 2013, 24, 0097 CrossRef CAS;
(i) A. P. Spork and T. J. Donohoe, Org. Biomol. Chem., 2015, 13, 8545 RSC;
(j) M. Sengoden, R. Irie and T. Punniyamurthy, J. Org. Chem., 2016, 81, 11508 CrossRef CAS PubMed.
-
(a) P. Müller, C. Baud and Y. Jacquier, Tetrahedron, 1996, 52, 1543 CrossRef;
(b) D. A. Evans, M. M. Faul and M. T. Bilodeau, J. Am. Chem. Soc., 1994, 116, 2742 CrossRef CAS;
(c) R. S. Atkinson, Tetrahedron, 1999, 55, 1519 CrossRef CAS.
- A. Y. Rulev, A. R. Romanov, E. V. Kondrashov, I. A. Ushakov, A. V. Vashchenko, V. M. Muzalevskiy and V. G. Nenajdenko, J. Org. Chem., 2016, 81, 10029 CrossRef CAS PubMed.
-
(a) M. Moens, N. De Kimpe and M. D’hooghe, J. Org. Chem., 2014, 79, 5558 CrossRef CAS PubMed;
(b) S. Nicholas, N. S. Dolan, R. J. Scamp, T. Yang, J. F. Berry and J. M. Schomaker, J. Am. Chem. Soc., 2016, 138, 14658 CrossRef PubMed.
- K. B. Hansen, N. S. Finney and E. N. Jacobsen, Angew. Chem., Int. Ed., 1995, 34, 676 CrossRef CAS.
-
(a) K. Juhl, R. G. Hazell and K. A. Jørgensen, J. Chem. Soc., Perkin Trans. 1, 1999, 2293 RSC;
(b) R. Pellicciari, L. Amori, N. Kuznetsova, S. Zlotsky and A. Gioiello, Tetrahedron Lett., 2007, 48, 4911 CrossRef CAS.
-
(a) H. Luo, K. Chen, H. Jiang and S. Zhu, Org. Lett., 2016, 18, 5208 CrossRef CAS PubMed;
(b) B.-N. Lai, J.-F. Qiu, H.-X. Zhang, J. Nie and J.-A. Ma, Org. Lett., 2016, 18, 520 CrossRef CAS PubMed.
- J. E. G. Kemp, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 7, p. 467 Search PubMed.
- A. Padwa and S. S. Murphree, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and T. L. Gilchrist, Pergamon Elsevier Science, Oxford, 2000, vol. 12, ch. 4.1, p. 57 Search PubMed.
-
(a) N. De Kimpe, R. Verheé, L. De Buyck and N. Schamp, Synth. Commun., 1975, 5, 269 CrossRef CAS;
(b) N. De Kimpe, L. Moens, R. Verhé, L. De Buyck and N. Schamp, J. Chem. Soc., Chem. Commun., 1982, 19 RSC;
(c) N. De Kimpe, P. Sulmon, R. Verhé, L. De Buyck and N. Schamp, J. Org. Chem., 1983, 48, 4320 CrossRef CAS;
(d) N. De Kimpe, R. Verhé, L. De Buyck and N. Schamp, J. Org. Chem., 1980, 45, 5319 CrossRef CAS.
- H. M. I. Osborn and J. Sweeney, Tetrahedron: Asymmetry, 1997, 8, 1693 CrossRef CAS.
- L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881 CrossRef CAS PubMed.
-
(a) Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed;
(b) S. Minakata, Y. Takeda and K. Kiyokawa, in Methods and Applications of Cycloaddition Reactions in Organic Syntheses, ed. N. Nishiwaki, Wiley-VCH, Weinheim, Germany, ch. 2, ISBN 978-1-118-29988-3 Search PubMed;
(c) P. Muller and C. Fruit, Chem. Rev., 2003, 103, 2905 CrossRef PubMed;
(d) G. S. Singh, M. D’hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef CAS PubMed;
(e) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831 RSC;
(f) J. W. W. Chang, T. M. U. Ton and P. W. H. Chan, Chem. Rev., 2011, 11, 331 CAS;
(g) D. M. Jenkins, Synlett, 2012, 1267 CrossRef CAS;
(h) K. Kiyokawa, T. Kosaka and S. Minakata, Org. Lett., 2013, 15, 4858 CrossRef CAS PubMed.
- J. M. Mahoney, C. R. Smith and J. N. Johnston, J. Am. Chem. Soc., 2005, 127, 1354 CrossRef CAS PubMed.
-
(a) J. U. Jeong, B. Tao, I. Sagasser, H. Henniges and K. B. Sharpless, J. Am. Chem. Soc., 1998, 120, 6844 CrossRef CAS;
(b) T. Ando, D. Kano, S. Minakata, I. Ryu and M. Komatsu, Tetrahedron, 1998, 54, 13485 CrossRef CAS;
(c) D. Kano, S. Minakata and M. Komatsu, J. Chem. Soc., Perkin Trans. 1, 2001, 3186 RSC;
(d) S. Minakata, D. Kano, Y. Oderaotoshi and M. Komatsu, Angew. Chem., Int. Ed., 2004, 43, 79 CrossRef PubMed;
(e) S. Minakata, Y. Murakami, R. Tsuruoka, S. Kitanaka and M. Komatsu, Chem. Commun., 2008, 6363 RSC;
(f) Y. Murakami, Y. Takeda and S. Minakata, J. Org. Chem., 2011, 76, 6277 CrossRef CAS PubMed;
(g) I. Saikia, B. Kashyap and P. Phukan, Chem. Commun., 2011, 47, 2967 RSC.
-
(a) T. Siu and A. K. Yudin, J. Am. Chem. Soc., 2002, 124, 530 CrossRef CAS PubMed;
(b) J. Li, J.-L. Liang, P. W. Hong Chan and C.-M. Che, Tetrahedron Lett., 2004, 45, 2685 CrossRef CAS;
(c) J. Li, P. W. H. Chan and C.-M. Che, Org. Lett., 2005, 7, 5801 CrossRef CAS PubMed;
(d) Y.-M. Shen, M.-X. Zhao, J. Xu and Y. Shi, Angew. Chem., Int. Ed., 2006, 45, 8005 CrossRef CAS PubMed;
(e) L. B. Krasnova and A. K. Yudin, Org. Lett., 2006, 8, 2011 CrossRef CAS PubMed;
(f) R. D. Richardson, M. Desaize and T. Wirth, Chem.–Eur. J., 2007, 13, 6745 CrossRef CAS PubMed;
(g) A. Yoshimura, K. R. Middleton, C. Zhu, V. N. Nemykin and V. V. Zhdankin, Angew. Chem., Int. Ed., 2012, 51, 8059 CrossRef CAS PubMed.
-
(a) S. Minakata, Y. Morino, Y. Oderaotoshi and M. Komatsu, Chem. Commun., 2006, 3337 RSC;
(b) R. Fan, D. Pu, J. Gan and B. Wang, Tetrahedron Lett., 2008, 49, 4925 CrossRef CAS;
(c) L. Deiana, G.-L. Zhao, S. Lin, P. Dziedzic, Q. Zhang, H. Leijonmarck and A. Cordova, Adv. Synth. Catal., 2010, 352, 3201 CrossRef CAS.
- C. Varszegi, M. Ernst, F. van Laar, B. F. Sels, E. Schwab and D. E. De Vos, Angew. Chem., Int. Ed., 2008, 47, 1477 CrossRef CAS PubMed.
-
(a) M. M. Dia-Requejo, T. R. Belderrain, M. C. Nicasio, S. Trofimenko and P. J. Perez, J. Am. Chem. Soc., 2003, 125, 12078 CrossRef PubMed;
(b) M. R. Fructos, S. Trofimenko, M. M. Dia-Requejo and P. J. Perez, J. Am. Chem. Soc., 2006, 128, 11784 CrossRef CAS PubMed;
(c) A. A. Lamar and K. M. Nicholas, J. Org. Chem., 2010, 75, 7644 CrossRef CAS PubMed;
(d) L. Maestre, W. M. C. Sameera, M. M. Dia-Requejo, F. Maseras and P. J. Perez, J. Am. Chem. Soc., 2013, 135, 1338 CrossRef CAS PubMed.
- H. Kwart and A. A. Kahn, J. Am. Chem. Soc., 1967, 89, 1951 CrossRef CAS.
-
(a) R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728 CrossRef CAS;
(b) D. Mansuy, J.-P. Mahy, A. Dureault, G. Bedi and P. Battioni, Chem. Commun., 1984, 1161 RSC.
-
(a) D. A. Evans, M. M. Faul and M. T. Bilodeau, J. Am. Chem. Soc., 1994, 116, 2742 CrossRef CAS;
(b) P. Dauban, L. Sanière, A. Tarrade and R. H. Dodd, J. Am. Chem. Soc., 2001, 123, 7707 CrossRef CAS PubMed.
-
(a) V. K. Rai, P. K. Rai, S. Bajaj and A. Kumar, Green Chem., 2011, 13, 1217 RSC;
(b) L. D. S. Yadav, S. Singh and V. K. Rai, Green Chem., 2009, 11, 878 RSC;
(c) L. D. S. Yadav, S. Yadav and V. K. Rai, Green Chem., 2006, 8, 455 RSC.
-
(a) F. Verma, P. K. Singh, S. R. Bhardiya, M. Singh, A. Rai and V. K. Rai, New J. Chem., 2017, 41, 4937–4942 RSC;
(b) A. Rai, P. K. Singh, P. Shukla and V. K. Rai, Tetrahedron Lett., 2016, 57, 5084 CrossRef CAS;
(c) V. K. Rai, G. P. Sahu and A. Rai, Tetrahedron Lett., 2015, 56, 2664 CrossRef CAS;
(d) A. K. Singh, R. Chawla, A. Rai and L. D. S. Yadav, Chem. Commun., 2012, 48, 3766 RSC;
(e) A. Rai, V. K. Rai, A. Singh and L. D. S. Yadav, Eur. J. Org. Chem., 2011, 4302 CrossRef CAS.
- Y. Yamada, T. Yamamoto and M. Okawara, Chem. Lett., 1975, 361 CrossRef CAS.
- M. Singh, S. R. Bhardiya, H. Kashyap, F. Verma, V. K. Rai and I. Tiwari, RSC Adv., 2016, 6, 104868–104874 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
-
(a) H. Person and A. Foucaud, Sci. Chim., 1975, 281, 325 CAS;
(b) H. Person and A. Foucaud, Bull. Soc. Chim. Fr., 1976, 7–8, 325 Search PubMed;
(c) H. Person, F. Tonnard, A. Foucaud and C. Fayat, Tetrahedron Lett., 1973, 14, 2495 CrossRef.
- J. P. Edasery and N. H. Cromwell, J. Heterocycl. Chem., 1979, 16, 831 CrossRef CAS.
- L. D. S. Yadav, G. Srivastava and R. Kapoor, Tetrahedron Lett., 2009, 50, 5420 CrossRef CAS.
- U. K. Nadir and A. Arora, Synth. Commun., 1996, 26, 2355 CrossRef CAS.
- V. K. Aggarwal, E. Alonso, M. Ferrara and E. S. Spey, J. Org. Chem., 2002, 67, 2335 CrossRef CAS PubMed.
- V. K. Aggarwal and M. Ferrara, Org. Lett., 2002, 2, 4107 CrossRef.
-
(a) K. S. S. K. Muhamad, F. Mohamed, S. Radiman, A. Hamzah, S. Sarmani, K. K. Siong, M. S. Yasir, I. A. Rahman and N. R. A. M. Rosli, AIP conference proceeding, American Institute of Physics, 2016, vol. 1784, p. 040013, DOI:10.1063/1.4966799;
(b) H. Guo, X. Wang, Q. Qian, F. Wang and X. Xia, A green approach to the synthesis of graphene nanosheets, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS PubMed;
(c) D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288 RSC;
(d) A. M. Dimiev and S. Eigler, A book: fundamental and applications, Wiley publications, 2016, ISBN No-9781-119-06940 Search PubMed.
- S. A. Glover, A. Goosen and R. D. S. Venter, S. Afr. J. Chem., 1978, 31, 33 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization details are given. See DOI: 10.1039/c7ra09351a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Vijai K. Rai
b,
Vijai K. Rai










